Спосіб одержання 3,5-дизаміщених 1,2,4-оксадіазолів (варіанти)
Номер патенту: 114915
Опубліковано: 28.08.2017
Автори: Грехем Чарльз Річард, Браун Девід Луїс, Міллер Уілльям Харольд


















Формула / Реферат
1. Спосіб одержання 3,5-дизаміщеного 1,2,4-оксадіазолу Формули (Іа) або (Іb) або його солі:
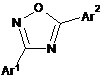 ,
,
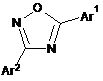 ,
,
(Іа)
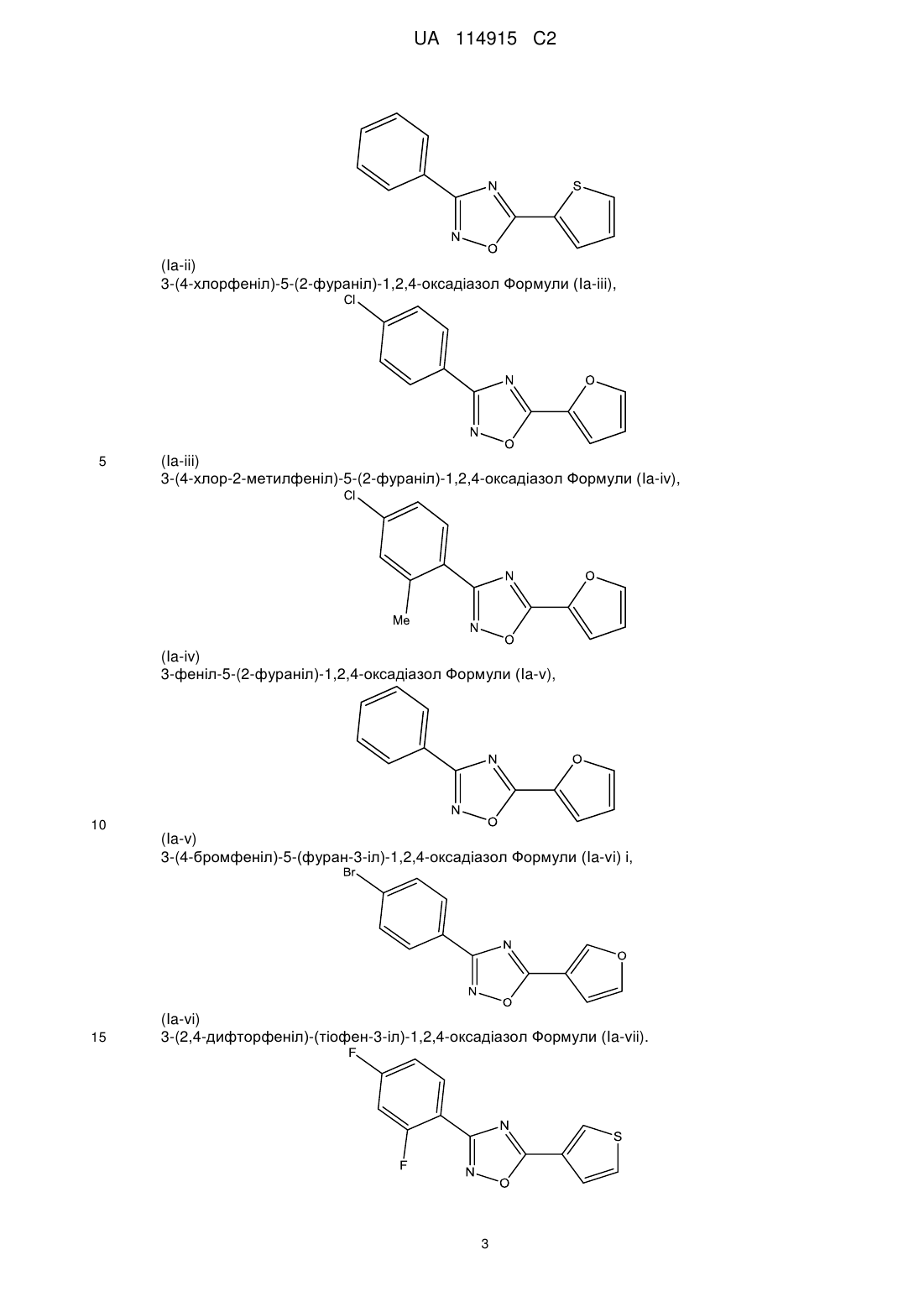
(Іb)
де спосіб включає реакцію N-гідроксіамідину Формули (IIа) або (IIb), відповідно, або його таутомерної форми:
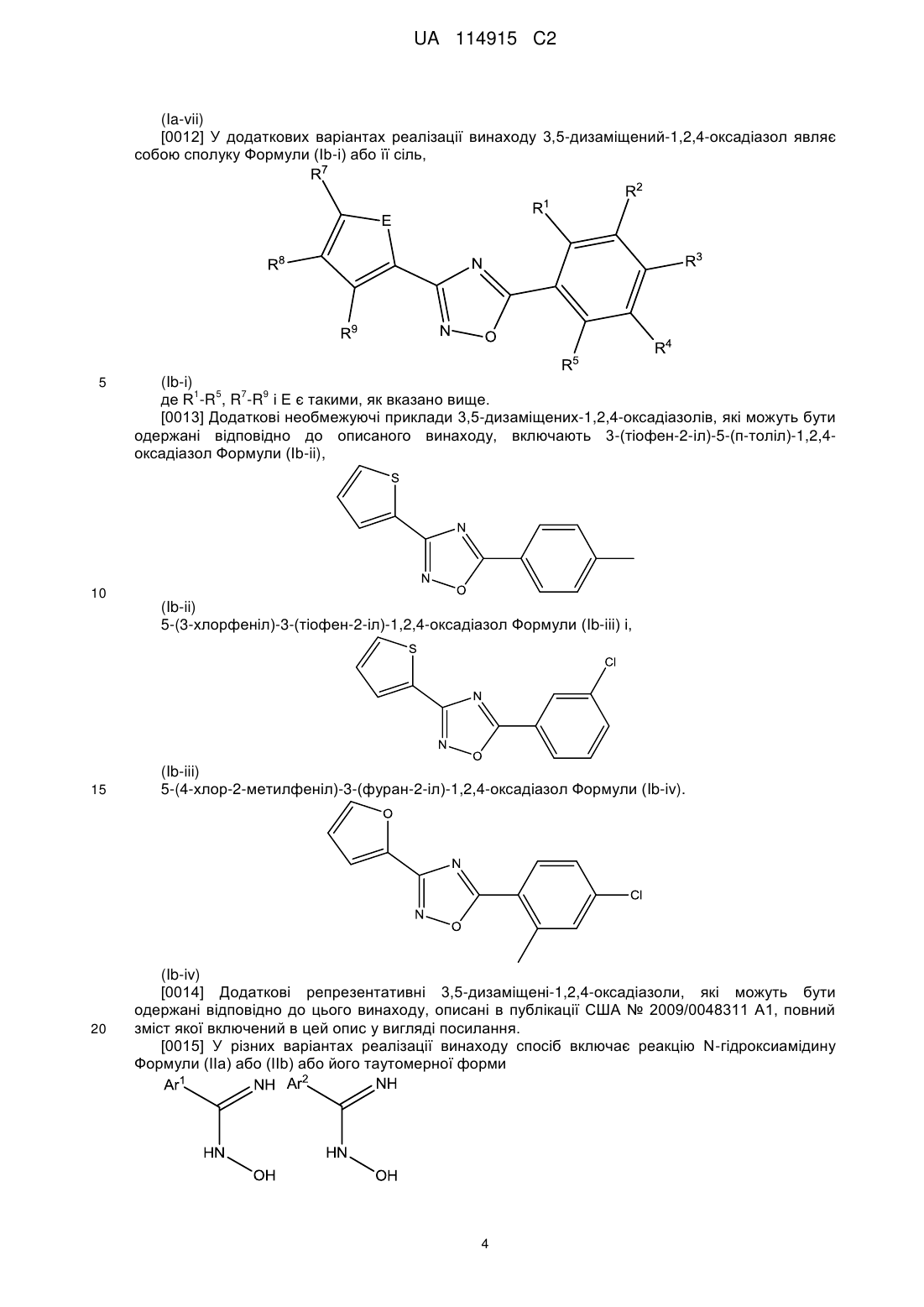
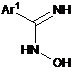 ,
,
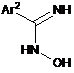 ,
,
(ІIа)
(ІIb)
з хлорангідридом Формули (IIIа) або (IIIb), відповідно:
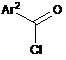 ,
,
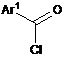 ,
,
(ІIIа)
(ІIIb)
в реакційній суміші, яка містить органічний розчинник, що не змішується з водою, і водну основу, причому температура реакційної суміші не перевищує близько 85 °C;
і де Аr1 вибирають з групи, яка складається з фенілу, піридилу, піразилу, оксазолілу або ізоксазолілу, кожний з яких може бути необов'язково незалежно заміщений одним або більше замісниками, вибраними з групи, яка складається з галогену, CF3, СН3, OCF3, ОСН3, CN і С(Н)О; і
Аr2 вибирають з групи, яка складається з тієнілу, фуранілу, оксазолілу або ізоксазолілу, кожний з яких може бути необов'язково незалежно заміщений одним або більше замісниками, вибраними з групи, яка складається з фтору, хлору, СН3 і OCF3.
2. Спосіб за п. 1, який відрізняється тим, що включає реакцію N-гідроксіамідину Формули (IIа) або його таутомерної форми з хлорангідридом Формули (IIIа) з утворенням 3,5-дизаміщеного 1,2,4-оксадіазолу Формули (Іа) або його солі.
3. Спосіб за п. 1, який відрізняється тим, що включає реакцію N-гідроксіамідину Формули (IIb) або його таутомерної форми з хлорангідридом Формули (IIIb) з утворенням 3,5-дизаміщеного 1,2,4-оксадіазолу Формули (Іb) або його солі.
4. Спосіб за будь-яким із пп. 1-3, який відрізняється тим, що реакційна суміш додатково містить каталізатор фазового переносу.
5. Спосіб за будь-яким із пп. 1-4, який відрізняється тим, що органічний розчинник, що не змішується з водою, розчиняє N-гідроксіамідин і 3,5-дизаміщений 1,2,4-оксадіазол.
6. Спосіб за будь-яким з пп. 1-5, який відрізняється тим, що органічний розчинник, що не змішується з водою, утворює азеотроп з водою.
7. Спосіб за будь-яким із пп. 1-6, який відрізняється тим, що органічний розчинник, що не змішується з водою, вибирають з групи, яка складається з 2-метилтетрагідрофурану і бутилацетату.
8. Спосіб за будь-яким із пп. 1-7, який відрізняється тим, що N-гідроксіамідин розчиняють в органічному розчиннику, що не змішується з водою, до додавання хлорангідриду, з утворенням реакційної суміші.
9. Спосіб за будь-яким із пп. 1-8, який відрізняється тим, що додатково включає виділення 3,5-дизаміщеного 1,2,4-оксадіазолу з реакційної суміші у вигляді осаду з водного шару після видалення з реакційної суміші щонайменше частини органічного розчинника, що не змішується з водою.
10. Спосіб за будь-яким із пп. 1-9, який відрізняється тим, що реакція N-гідроксіамідину хлорангідриду приводить до утворення проміжного оксимового естеру Формули (IVa) або (IVb), його солі або його таутомерної форми:
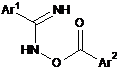 ,
,
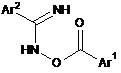 ,
,
(ІVа)
(ІVb)
де Аr1 і Аr2 є такими, як вказано у п. 1.
11. Спосіб за п. 10, який відрізняється тим, що проміжний оксимовий естер виділяють до утворення 3,5-дизаміщеного 1,2,4-оксадіазолу.
12. Спосіб за будь-яким із пп. 1-11, який відрізняється тим, що N-гідроксіамідин являє собою бензамідоксим, одержаний в результаті реакції заміщеного або незаміщеного бензонітрилу і гідроксиламіну, і одержаний бензамідоксим розчиняють в розчиннику, вибраному з групи, яка складається з 2-метилтетрагідрофурану і бутилацетату.
13. Спосіб за п. 12, який відрізняється тим, що реакцію з утворенням бензамідоксиму проводять при температурі від близько 20 °C до близько 75 °C.
14. Спосіб за будь-яким із пп. 1-13, який відрізняється тим, що спосіб включає:
(1) введення в реакційну посудину N-гідроксіамідину у вигляді розчину в органічному розчиннику, що не змішується з водою;
(2) додавання в реакційну посудину частини водної основи і, необов'язково, каталізаторафазового переносу;
(3) додавання в реакційну посудину хлорангідриду з утворенням реакційної суміші; і
(4) додавання до реакційної суміші в реакційній посудині додаткової водної основи.
15. Спосіб за будь-яким із пп. 1, 2 або 4-14, який відрізняється тим, що 3,5-дизаміщений 1,2,4-оксадіазол являє собою 3-феніл-5-(2-тієніл)-1,2,4-оксадіазол.
Текст
Реферат: Пропонуються способи одержання 3,5-дизаміщених 1,2,4-оксадіазолів і їх солей, які включають реакцію N-гідроксіамідину з хлорангідридом в реакційній суміші, що містить органічний розчинник, що не змішується з водою, і водну основу, при відносно низьких температурах реакції. UA 114915 C2 (12) UA 114915 C2 UA 114915 C2 5 10 15 20 25 30 35 40 ГАЛУЗЬ ВИНАХОДУ [0001] У цьому документі описуються способи одержання біологічно активних 3,5дизаміщених-1,2,4-оксадіазолів або їх солей, які застосовують, наприклад, у боротьбі з нематодами. РІВЕНЬ ТЕХНІКИ [0002] Виявляється, що 1,2,4-оксадіазоли, і, зокрема, 3,5-дизаміщені-1,2,4-оксадіазоли, мають біологічну активність в галузі фармацевтики і сільського господарства. Наприклад, 3,5дизаміщені-1,2,4-оксадіазоли розкриваються в якості агентів пригнічення і лікування захворювань (патент США № 6,992,096), в якості терапевтичних засобів для лікування гепатиту C (публікація США № 2005/0075375 A1), а також для боротьби з нематодами в сільському господарстві (публікація США № 2009/0048311 A1). [0003] 3,5-Дизаміщені-1,2,4-оксадіазоли можуть бути одержані за допомогою декількох способів. Одним з них є одержання 3,5-дизаміщених-1,2,4-оксадіазолів за допомогою реакції ариламідоксиму і хлорангідриду (публікація США № 2008/0269236 A1, патент США № 7,678,922, публікація США № 2008/0113961 A1, № JP 2008120794 і Bioorg. Med Chem Lett. 19, 4410). Інший спосіб одержання 3,5-дизаміщених-1,2,4-оксадіазолів являє собою реакцію бензамідоксиму або пропіонамідоксиму з карбоновою кислотою або естером (WO 2011/141326, WO 2012/012477 і WO 2011/085406). Інші способи одержання 3,5-дизаміщених-1,2,4-оксадіазолів включають реакцію бензаміду з амідом Вайнреба (публікація США № 2010/0048648 A1) і реакцію гідроксиламінгалогеніду і нітрилу (патент США № 3,211,742). [0004] Тоді як існують способи одержання 3,5-дизаміщених-1,2,4-оксадіазолів, дуже бажаними є альтернативні способи, які можуть забезпечити більш ефективний синтез. Зокрема, синтетичні способи з меншою кількістю стадій виділення і/або розчинників можуть бути ефективнішими і менш дорогими. Крім того, способи, які зменшують і/або усувають механічні домішки, скорочують час реакції і/або використовують менше проміжних реакцій, можуть значно скоротити капіталовкладення в устаткування при великотоннажному виробництві. І, нарешті, використання м'якших умов реакції (наприклад, нижчих температур) може запобігти розкладанню бажаних проміжних сполук і продуктів, призводячи до меншої кількості небажаних побічних продуктів і, врешті-решт, до кращого профілю чистоти продукту. [0005] Згадування будь-якого посилання, наданого вище, не повинно розглядатись як визнання того, що таке посилання являє собою попередній рівень техніки цієї заявки. СУТЬ ВИНАХОДУ [0006] У цьому документі пропонуються способи одержання 3,5-дизаміщених-1,2,4оксадіазолів і їх солей. Наприклад, способи, описані в цьому документі, використовують для одержання 3,5-дизаміщеного-1,2,4-оксадіазолу Формули (Ia) або (Ib), (Ia) (Ib) 1 де Ar являє собою феніл, піридил, піразил, оксазоліл або ізоксазоліл, кожен з яких може бути необов'язково незалежно заміщений одним або більше замісниками, наприклад, алкілом, 2 галоалкілом, алкокси, галогеналкокси, галогеном, ацилом, естером і нітрилом; і Ar являє собою тієніл, фураніл, оксазоліл, ізоксазоліл або феніл, кожен з яких може бути необов'язково незалежно заміщений одним або більше замісниками, наприклад фтором, хлором, CH 3 і OCF3. В одному варіанті реалізації винаходу спосіб включає реакцію N-гідроксиамідину Формули (IIa) 1 2 або (IIb), відповідно, або його таутомерної форми, де Ar і Ar є такими, як вказано вище, 45 (IIa) (IIb) 1 2 з хлорангідридом Формули (IIIa) або (IIIb), відповідно, де Ar і Ar є такими, як вказано вище, 1 UA 114915 C2 5 10 15 20 25 30 35 40 (IIIa) (IIIb) в реакційній суміші, яка містить органічний розчинник, що не змішується з водою, і водну основу, і температура реакційної суміші не перевищує близько 85 °C. ДЕТАЛЬНИЙ ОПИС СУТІ ВИНАХОДУ [0007] Цей винахід належить до поліпшених способів одержання 3,5-дизаміщених-1,2,4оксадіазолів і їх солей. Різні варіанти реалізації способу забезпечують більшу легкість виробництва, м'якші умови реакції, скорочення тривалості циклів реакції, меншу кількість проміжних реакцій і/або значне зниження вимог до капітального устаткування. [0008] Як правило, способи, описані в цьому документі, використовують для одержання 3,5дизаміщеного-1,2,4-оксадіазолу Формули (Ia) або (Ib) або його солей, (Ia) (Ib) 1 де Ar являє собою феніл, піридил, піразил, оксазоліл або ізоксазоліл, кожен з яких може бути необов'язково незалежно заміщений одним або більше замісниками. В одному варіанті 1 реалізації винаходу Ar є необов'язково незалежно заміщеним одним або більше замісниками, обраними з групи, яка складається з алкілу, галогеналкілу, алкокси, галогеналкокси, галогену, ацилу, естеру і нітрилу, кожен з яких може бути необов'язково незалежно заміщений. В одному 1 варіанті реалізації винаходу Ar є необов'язково незалежно заміщеним одним або більше замісниками, обраними з групи, яка складається з галогену, CF 3, CH3, OCF3, OCH3, CN і C(H)O. 2 Ar являє собою тієніл, фураніл, оксазоліл, ізоксазоліл або феніл, кожен з яких може бути 2 необов'язково незалежно заміщений. В одному варіанті реалізації винаходу Ar є необов'язково незалежно заміщеним одним або більше замісниками, обраними з групи, яка складається з фтору, хлору, СН3 і OCF3. 1 [0009] Наприклад, в деяких варіантах реалізації винаходу Ar являє собою незаміщений 1 феніл. В інших варіантах реалізації винаходу Ar являє собою монозаміщений феніл, в якому 1 замісником є галоген. В інших варіантах реалізації винаходу Ar являє собою дизаміщений 2 хлоралкілфеніл. У деяких варіантах реалізації винаходу Ar являє собою заміщений тієніл або 2 заміщений фураніл. У деяких варіантах реалізації винаходу Ar являє собою незаміщений тієніл або незаміщений фураніл. [0010] У деяких варіантах реалізації винаходу 3,5-дизаміщений-1,2,4-оксадіазол являє собою сполуку Формули (Ia-i) або її сіль, (Ia-i) 1 5 2 4 де R і R незалежно обирають з водню, CH3, F, Cl, Br, CF3 і OCF3; R і R незалежно 3 обирають з водню, F, Cl, Br і CF3; R обирають з водню, CH3, CF3, F, Cl, Br, OCF3, OCH3, CN і 7 8 9 C(H)O; R і R незалежно обирають з водню і фтору; R обирають з водню, F, Cl, CH3 і OCF3; і Е являє собою О, N або S. [0011] Необмежуючі приклади 3,5-дизаміщених-1,2,4-оксадіазолів, які можуть бути отримані відповідно до цього винаходу, включають 3-феніл-5-(2-тієніл)-1,2,4-оксадіазол Формули (Ia-ii), 2 UA 114915 C2 (Ia-ii) 3-(4-хлорфеніл)-5-(2-фураніл)-1,2,4-оксадіазол Формули (Ia-iii), 5 (Ia-iii) 3-(4-хлор-2-метилфеніл)-5-(2-фураніл)-1,2,4-оксадіазол Формули (Ia-iv), (Ia-iv) 3-феніл-5-(2-фураніл)-1,2,4-оксадіазол Формули (Ia-v), 10 (Ia-v) 3-(4-бромфеніл)-5-(фуран-3-іл)-1,2,4-оксадіазол Формули (Ia-vi) і, 15 (Ia-vi) 3-(2,4-дифторфеніл)-(тіофен-3-іл)-1,2,4-оксадіазол Формули (Ia-vii). 3 UA 114915 C2 (Ia-vii) [0012] У додаткових варіантах реалізації винаходу 3,5-дизаміщений-1,2,4-оксадіазол являє собою сполуку Формули (Ib-i) або її сіль, 5 (Ib-i) 1 5 7 9 де R -R , R -R і Е є такими, як вказано вище. [0013] Додаткові необмежуючі приклади 3,5-дизаміщених-1,2,4-оксадіазолів, які можуть бути одержані відповідно до описаного винаходу, включають 3-(тіофен-2-іл)-5-(п-толіл)-1,2,4оксадіазол Формули (Ib-ii), 10 (Ib-ii) 5-(3-хлорфеніл)-3-(тіофен-2-іл)-1,2,4-оксадіазол Формули (Ib-iii) і, 15 20 (Ib-iii) 5-(4-хлор-2-метилфеніл)-3-(фуран-2-іл)-1,2,4-оксадіазол Формули (Ib-iv). (Ib-iv) [0014] Додаткові репрезентативні 3,5-дизаміщені-1,2,4-оксадіазоли, які можуть бути одержані відповідно до цього винаходу, описані в публікації США № 2009/0048311 A1, повний зміст якої включений в цей опис у вигляді посилання. [0015] У різних варіантах реалізації винаходу спосіб включає реакцію N-гідроксиамідину Формули (IIa) або (IIb) або його таутомерної форми 4 UA 114915 C2 (IIa) (IIb) 1 2 де Ar і Ar є такими, як вказано вище, з хлорангідридом Формули (IIIa) або (IIIb), відповідно, 5 (IIIa) (IIIb) 1 2 де Ar і Ar є такими, як вказано вище. Реакцію конденсації N-гідроксиамідину з хлорангідридом проводять в реакційній суміші, що містить органічний розчинник і водну основу. Без зв'язку з певною теорією, у деяких варіантах реалізації винаходу, як вважають, реакція Nгідроксиамідину і хлорангідриду відбувається з одержанням проміжного оксимового естеру Формули (IVa) або (IVb), його солі або його таутомерної форми, 10 15 20 25 30 35 40 45 50 (IVa) (IVb) 1 2 де Ar і Ar є такими, як вказано вище. Як описано детальніше нижче, присутність водної основи полегшує циклізацію оксимового естеру при відносно низьких температурах з одержанням 3,5-дизаміщеного-1,2,4-оксадіазольного продукту. В одному варіанті реалізації винаходу реакції конденсації і/або циклізації дозволено протікати безпосередньо до кінцевого 3,5-дизаміщеного-1,2,4-оксадіазольного продукту, без виділення або очищення проміжного оксимового естеру. В інших варіантах реалізації винаходу проміжний оксимовий етсер або його частина може бути виділена і/або очищена зодержанням 3,5-дизаміщеного-1,2,4оксадіазольного продукту. [0016] Відповідно до різних варіантів реалізації винаходу розчинники, використовувані для одержання реакційної суміші, обирають на основі одного або більше критеріїв, для сприяння спрощенню і загальним економічним показникам способу. У деяких варіантах реалізації винаходу використовуваний органічний розчинник здатен розчинити N-гідроксиамідин Формули (IIa) або (IIb) і 3,5-дизаміщений-1,2,4-оксадіазольний продукт Формули (Ia) або (Ib). У деяких варіантах реалізації винаходу використовуваний органічний розчинник також здатен розчиняти проміжний оксимовий естер Формули (IVa) або (IVb). Використання органічного розчинника для розчинення вихідного матеріалу N-гідроксиамідину, а також проміжного оксимового естеру і 3,5дизаміщеного-1,2,4-оксадіазольного продукту знижує або виключає необхідність виділення Nгідроксиамідину і/або оксимового естеру, а також знижує або виключає необхідність заміни розчинника до циклізації з утворенням 3,5-дизаміщеного-1,2,4-оксадіазольного продукту. У різних варіантах реалізації винаходу органічний розчинник переважно може бути використаний для перенесення реагуючого N-гідроксиамідину з однієї реакційної посудини в іншу. У деяких випадках органічний розчинник, що не змішується з водою, використовують для виділення Nгідроксиамідину Формули (IIa) або (IIb) і одержання 3,5-дизаміщеного-1,2,4-оксадіазольного продукту. Використання органічного розчинника, що не змішується з водою, дозволяє розділення водної і органічної фаз реакційної суміші, що забезпечує доцільніше перенесення реагуючого N-гідроксиамідину з однієї посудини в іншу, а також полегшує виділення кінцевого 3,5-дизаміщеного-1,2,4-оксадіазольного продукту, як викладено детальніше нижче. Крім того, в одному варіанті реалізації винаходу, органічний розчинник, що не змішується з водою, утворює азеотроп з водою. Утворення азеотропу полегшує видалення, за допомогою, наприклад, випарювання або перегонки, розчинника з метою виділення проміжного оксимового естеру або 3,5-дизаміщеного-1,2,4-оксадіазольного продукту. В альтернативному варіанті, можна використовувати більше ніж один органічний розчинник, наприклад, розчинник, що не змішується з водою, для одержання проміжного оксимового стеру Формули (IVa) або (IVb), і розчинник, що змішується з водою, для одержання 3,5-дизаміщеного-1,2,4-оксадіазольного продукту. Необмежуючі приклади прийнятних органічних розчинників включають ацетон, 2бутанон, етилацетат, ізопропілацетат, бутилацетат, хлороформ, дихлорметан, діетиловий етер, метил-трет-бутиловий етер, дибутиловий етер, анізол, тетрагідрофуран, 2метилтетрагідрофуран, ксилол і толуол. У різних варіантах реалізації винаходу органічний розчинник включає ацетон, 2-бутанон, тетрагідрофуран, 2-метилтетрагідрофуран або 5 UA 114915 C2 5 10 15 20 25 30 35 40 45 50 55 60 бутилацетат. В одному варіанті реалізації винаходу органічний розчинник являє собою бутилацетат або 2-метилтетрагідрофуран. [0017] Водна основа містить хімічний реагент, що реагує з кислотним побічним продуктом реакції конденсації між N-гідроксиамідином і хлорангідридом. Крім того, без зв'язку з будь-якою конкретною теорією, вважають, що водна основа каталізує циклізацію оксимового естеру Формули (IVa) або (IVb) з утворенням 3,5-дизаміщеного-1,2,4-оксадіазолу Формули (Ia) або (Ib). [0018] "Водна основа" в контексті цього документу означає сильну основу або основу, яка повністю або майже повністю дисоціює у воді. Використання сильної основи може переважно призводити до скорочення часу реакції. У деяких варіантах реалізації винаходу водна основа являє собою лужну основу, яка містить лужний метал або лужноземельний метал, і яка дисоціює з утворенням гідроксильних іонів у водному розчині. Необмежуючі приклади відповідних водних основ включають водні розчини неорганічних основ, таких як гідроксид натрію, гідроксид калію, гідроксид літію, гідроксид кальцію і їх суміші. В одному варіанті реалізації винаходу водний розчин основи, доданий до реакційної суміші, містить основний реагуючий компонент в концентрації від близько 5 % до близько 60 % по масі. В одному ілюстративному варіанті реалізації водна основа являє собою лужний гідроксид, який реагує з кислотним побічним продуктом реакції конденсації між N-гідроксиамідином і хлорангідридом з утворенням води (вода утворюється лише з гідроксидної основи) і солі. [0019] Відповідно до одного з варіантів реалізації винаходу реакційна суміш необов'язково містить каталізатор фазового переносу. Каталізатор фазового переносу являє собою хімічний реагент, що полегшує міграцію реагента з водної фази в органічну фазу або навпаки. Відповідно, використання каталізатора фазового переносу переважно може привести до скорочення часу реакції і до більш високих ступенів перетворення або виходу. [0020] У різних варіантах реалізації винаходу каталізатор фазового переносу являє собою сіль четвертинної амонієвої основи, сіль фосфонію або краун-ефір. Необмежуючі приклади прийнятних каталізаторів фазового переносу включають гідроксид тетрабутиламонію, гідроксид бензилтриметиламонію, бромід тетрабутиламонію, хлорид тетрабутиламонію, йодид тетрабутиламонію, бромід тетрабутилфосфонію і хлорид тетрабутилфосфонію. В одному варіанті реалізації винаходу каталізатор фазового переносу являє собою гідроксид тетрабутиламонію. В одному варіанті реалізації винаходу каталізатор фазового переносу додають до реакційної суміші у вигляді водного розчину (наприклад, 40 % по масі водного розчину каталізатора фазового переносу) так, щоб кінцева концентрація складала близько 0,5-3 моль% в перерахунку на хлорангідрид, прийнятніше – в діапазоні близько 0,5-1,5 моль% або близько 1,5-2 моль% в перерахунку на хлорангідрид. [0021] В одному варіанті реалізації способу N-гідроксиамідин Формули (IIa) або (IIb) розчиняють в органічному розчиннику, що не змішується з водою, і одержаний розчин вводять в реакційну посудину. Воду, щонайменше частину водної основи, і необов'язковий каталізатор фазового переносу додають в реакційну посудину. Це роблять одночасно і/або послідовно. Це призводить до одержання двофазної реакційної суміші, в якій N-гідроксиамідин залишається розчиненим в органічній фазі, що містить органічний розчинник, що не змішується з водою. Хлорангідридний реагент Формули (IIIa) або (IIIb) і будь-яку частину водної основи, що залишилася, додають в реакційну посудину. Хлорангідрид і частина водної основи, що залишилася, можуть бути додані одночасно або послідовно. В одному варіанті реалізації винаходу N-гідроксиамідин додають до реакційної суміші в кількості, яка дорівнює або перевищує кількість хлорангідриду з розрахунку на моль основи. Додаткова кількість води може бути додана до реакційної суміші у будь-який момент часу в ході реакції. [0022] У деяких варіантах реалізації винаходу суміш можна перемішувати. У додаткових варіантах реалізації винаходу реакцію можна проводити безперервно, наприклад, в проточному хімічному реакторі з мішалкою. [0023] Для того, щоб збільшити швидкість реакції і досягти прийнятного часу реакції, концентрація основи у водній фазі реакційної суміші переважно має бути достатньою для досягнення рН щонайменше близько 8, щонайменше близько 9 або щонайменше близько 10, де більш високий рН призводить до збільшення швидкості реакції. Наприклад, в одному варіанті реалізації винаходу, рН від близько 12 до близько 13 являє собою діапазон рН, який призводить до збільшення швидкості реакції. Значення рН водної фази може бути збережене або збільшене в ході процесу за допомогою додавання до реакційної суміші додаткової водної основи. [0024] Температура реакційної суміші складає від близько 25 °C до близько 85 °C. У деяких варіантах реалізації винаходу температурі реакційної суміші дозволено зростати в результаті екзотермічної реакції, і, переважно, від близько 55 °C до близько 75 °C. Може бути дозволено, щоб реакція протікала за відсутності зовнішнього нагрівання, застосованого до реакційної 6 UA 114915 C2 5 10 15 20 25 30 35 40 45 50 55 60 суміші, хоча у разі потреби може застосовуватись нагрівання із використанням зовнішнього джерела, щоб підтримувати бажану температуру і швидкість реакції. [0025] Швидкість додавання хлорангідриду і водної основи, що залишилася, залежить від масштабу реакції і здатності контролювати температуру. Наприклад, швидкість додавання хлорангідриду може складати від 30 хв до 4 годин. Переважно реакційній суміші дозволяють нагріватись внаслідок екзотермічності реакції, як описано вище, і можна підтримувати температуру від близько 55 °C до близько 75 °C впродовж від близько 30 хвилин до близько 2 годин, щоб дозволити реакції завершитися. Після завершення реакції водній і органічній фазам, як правило, дозволяють розділитися ще теплими. У деяких варіантах реалізації винаходу водна фаза може бути необов'язково нейтралізована до нижчого рН (наприклад, рН знижують до менш ніж близько 9), для сприяння подальшому утворенню 3,5-дизаміщеного-1,2,4оксадіазольного продукту. У деяких варіантах реалізації винаходу частину водної фази видаляють з реакційної суміші, і випарюванням або перегонкою видаляють органічний розчинник. Коли рівень розчинника знижений, азеотроп або суміш води і розчинника допомагає відігнати сліди розчинника. Перегонку можна виконувати при атмосферному тиску або у вакуумі. До реакційної суміші може бути додана додаткова кількість води в об'ємі, достатньому для заміни відігнаного розчинника і для полегшення виділення кінцевого продукту. Коли розчинник випаровується, 3,5-дизаміщений-1,2,4-оксадіазольний продукт осаджують у водному шарі,що призводить до суспендування твердих часток у водному шарі. По суті, коли увесь розчинник видалений, твердий 3,5-дизаміщений-1,2,4-оксадіазольний продукт може бути виділений шляхом фільтрації, центрифугування і/або декантування. [0026] 3,5-Дизаміщений-1,2,4-оксадіазольний продукт не може в обов'язковому порядку осідати у вигляді дрібнокристалічної твердої речовини, а може утворювати кульки (наприклад, декілька мм в діаметрі), великі тверді грудки неправильної форми або інкрустовані тверді речовини на стінках реактора і мішалці, що є проблемою для вивантаження з реактора і подальшої обробки. Одним з рішень є невидалення водної фази побічної солі. Проте, високі рівні залишкової солі (наприклад, NaCl) в 3,5-дизаміщеному-1,2,4-оксадіазольному продукті можуть викликати проблеми приформулюванні продукту. [0027] Альтернативний варіант реалізації винаходу полягає в тому, щоб використовувати поверхнево-активну речовину для стабілізації крапель продукту (можливо метастабільного розплаву або дуже концентрованого розчину) і дозволити краплям твердіти у вигляді осаду у водному шарі без коалесціювання у більші краплі, які інакше, врешті-решт, можуть "виходити" у вигляді великих грудок і/або інкрустованих твердих речовин. Прийнятні поверхнево-активні речовини можуть бути обрані з групи, яка складається з аніонних або неіонних диспергуючих речовин, аніонних або неіонних детергентів, аніонних або неіонних поверхнево-активних речовин і їх комбінацій. Необмежуючі приклади прийнятних поверхнево-активних речовин включають Morwet D-425 (аніонна диспергуюча речовина, натрієва сіль конденсату алкілнафталінсульфонату, від AkzoNobel), Morwet D-425 і Greenworks (лаурилсульфат натрію (аніонний детергент), алкілполіглюкозид (неіонна поверхнево-активна речовина), лаураміноксид (неіонний детергент) і гліцерин), Morwet D-425 і Pluronic L-35 (поверхнево-активна речовина, ПЕГ-ППГ-ПЕГ блок-співполімер поліетиленгліколь-поліпропіленгліколь-поліетиленгліколь, Mn≈1900), Pluronic L-35 і Triton X-100 (неіонний детергент, октилфенолетоксилат (n≈10)). В одному варіанті реалізації винаходу поверхнево-активну речовину додають до реакційної суміші, по суті, після завершення реакції і до видалення щонайменше частини органічного розчинника, що не змішується з водою, з реакційної суміші. [0028] У деяких варіантах реалізації винаходу, особливо у великомасштабних процесах або умовах виробництва з великими об'ємами розчинника, розчинник може бути видалений і повернений в повторний цикл для повторного використання в процесі (наприклад, для використання в розчиненні N-гідроксиамідину в подальшому циклі способу). [0029] N-гідроксиамідин Формули (IIa) або (IIb), використовуваний при отриманні 3,5дизаміщеного-1,2,4-оксадіазолу, як описано вище, може бути одержаний з комерційних джерел або може бути одержаний з використанням способів, відомих фахівцеві в цій галузі техніки. Наприклад, ариламідоксим (наприклад, бензамідоксим) або необов'язково незалежно заміщений ариламідоксим може бути одержаний шляхом реакції відповідного арилнітрилу з гідрохлоридом гідроксиламіну і водною основою. Однією з ілюстративних водних основ для цієї мети є гідроксид натрію. [0030] У деяких випадках для одержання вихідного ариламідоксиму, гідрохлорид гідроксиламіну суспендують в спиртовому розчиннику, такому як метанол або етанол, і об'єднують з водною основою еквівалентної молярної маси. В одному варіанті реалізації винаходу арилнітрил додають в такій кількості, щоб гідроксиламін був в надлишку, наприклад, 7 UA 114915 C2 5 10 15 20 25 30 35 40 45 50 55 60 від близько 1,01 до близько 1,25 молярних еквівалентів. Отримана реакція є злегка екзотермічною і одержана реакційна суміш нагрівається до температури від близько 20 °C до близько 75 °C, від близько 20 °C до близько 65 °C, від близько 50 °C до близько 75 °C або від близько 50 °C до близько 60 °C. Коли реакційна суміш нагрівається, додають арилнітрил. Потім, в деяких варіантах реалізації винаходу, дозволено продовжувати нагрівання реакційної суміші до тих пір, поки реакція не завершиться. [0031] Після завершення реакції ариламідоксимний продукт може бути виділений для використання в способі згідно цього винаходу. Наприклад, спиртовий розчинник може бути видалений шляхом перегонки, внаслідок чого залишається реакційна суміш, яка містить продукт фази розплаву і водну фазу. Доки реакційна суміш ще гаряча, додають органічний розчинник, як описано вище, для розчинення ариламідоксимного продукту в органічній фазі. Потім водна фаза може бути відокремлена і видалена. У різних варіантах реалізації винаходу органічний розчинник задовольняє критеріям органічного розчинника, використовуваного у виробництві 3,5дизаміщеного-1,2,4-оксадіазолу, як викладено детально вище. Таким чином, фаза органічного розчинника, що містить розчинений ариламідоксимний продукт, може бути спрямована безпосередньо в реакцію конденсації/циклізації для одержання 3,5-дизаміщеного-1,2,4оксадіазолу без подальшої обробки. Способи, описані в цьому документі, можуть бути легко адаптовані для одержання інших N-гідроксиамідинових сполук Формули (IIa) або (IIb). [0032] В альтернативному варіанті реалізації винаходу вихідний матеріал ариламідоксиму (наприклад, бензамідоксим) одержують шляхом змішування і реакції відповідного арилнітрилу з водним гідроксиламіном (наприклад, водний розчин гідроксиламіну включає, але не обмежується водним розчином вільної основи гідроксиламіну або водним гідроксиламіном, одержаним з гідрохлориду). При використанні водного гідроксиламіну, вилучається стадія нейтралізації гідрохлориду гідроксиламіну водною основою разом з необхідністю в спиртовому розчиннику. Це забезпечує значну економію капіталу і матеріалів в процесах великомасштабного виробництва завдяки відсутності водної основи, зберігання спирту і устаткування для перегонки, а також завдяки іншим перевагам, таким як спрощення обробки водних відходів шляхом уникнення одержання солі на стадії нейтралізації. У варіанті реалізації винаходу, де вихідний матеріал ариламідоксиму отримують шляхом використання водного гідроксиламіну, розчинник не потрібен. Проте, може бути використаний розчинник, у тому числі спиртовий розчинник, такий як метанол або етанол. Для того, щоб полегшити подальшу обробку, в якості розчинника можна використовувати органічний розчинник, що не змішується з водою, або комбінацію розчинників, обраних на основі одного або більше критеріїв, викладених в деталях вище. Наприклад, органічним розчинником може бути бутилацетат або 2метилтетрагідрофуран. В альтернативному варіанті, якщо вихідний матеріал ариламідоксиму одержують шляхом об'єднання і реакції відповідного арилнітрилу з водним гідроксиламіном за відсутності розчинника, після завершення реакції, одержаний в результаті продукт може бути розчинений в органічному розчиннику, як описано вище, для подальшої обробки. [0033] Хлорангідрид Формули (IIIa) або (IIIb) (наприклад, 2-тіофенкарбонілхлорид або 2фуранкарбонілхлорид), використовуваний в способах одержання 3,5-дизаміщеного-1,2,4оксадіазолу, як описано вище, може бути одержаний з комерційних джерел або може бути одержаний з використанням способів, відомих фахівцеві в цій галузі техніки. Наприклад, тіофен може бути використаний для одержання або 2-ацетилтіофену, або 2-тіофенкарбоксальдегіду. Кожен з них може бути використаний для одержання 2-тіофенкарбонової кислоти, а потім і 2тіофенкарбонілхлориду. Інші способи можуть бути відповідним чином використані для одержання інших хлорангідридів у рамках Формули (III) або (IIIb). [0034] Наступні приклади слід розглядати лише як ілюстративні і вони не призначені для обмеження обсягу цього опису. ПРИКЛАДИ Приклад 1: Одержання Бензамідоксиму [0035] Гідрохлорид гідроксиламіну (8,85 г, 0,126 моль) і метанол (45 мл) завантажували в 100 мл колбу, з подальшим додаванням 51 % гідроксиду натрію (9,82 г, 0,125 моль). До цієї суміші додавали бензонітрил (10,32 г, 0,100 моль), нагрівали реакційну суміш до 55-60 °C і витримували впродовж 2 годин. ВЕРХ продемонструвала, що реакція завершилась, на що вказувало зникнення бензонітрилу. Частину метанолу (30 мл) видаляли шляхом перегонки, додавали деіонізовану (ДІ) воду (22 мл), а метанол, що залишився, відганяли при 300 торр з одержанням двох рідких фаз (розплаву бензамідоксиму і солоної води). Додавали 2метилтетрагідрофуран (20,0 г) і фазуводної солі зливали з одержанням 33,34 г розчину бензамідоксиму в 2-метилтетрагідрофурані, з втратою 39,45 % при сушці і ВЕРХ. Вихід бензамідоксиму складав 96,5 %. 8 UA 114915 C2 5 10 15 20 25 30 35 40 45 50 55 Приклад 2: Одержання 3-Феніл-5-(тіофен-2-іл)-1,2,4-оксадіазолу в 2-Метил-тетрагідрофурані [0036] 39,45 % розчин бензамідоксиму в 2-метилтетрагідрофурані (33,34 г) завантажували в 100 мл колбу і потім додавали воду (9,0 г), 40 % водний розчин гідроксиду тетрабутиламонію (1,02 г) і 51 % гідроксид натрію (3,52 г, 0,045 моль). До цієї суміші одночасно додавали 2тіофенкарбонілхлорид (13,83 г, 0,0936 моль) і 51 % гідроксид натрію (7,33 г, 0,0935 моль) впродовж 30-хвилинного періоду при нагріванні при близько 70 °C. Реакція завершувалась менш, ніж за 80 хвилин. Фазу водної солі видаляли, додавали воду (30 г) і 2метилтетрагідрофуран частково видаляли шляхом перегонки при атмосферному тиску (13,5 мл). Додавали додаткову кількість (32 г) гарячої води і залишок 2-метилтетрагідрофурану відганяли, впродовж цього періоду часу 3-феніл-5-(2-тієніл)-1,2,4-оксадіазольний продукт випадав в осад. Водну суспензію 3-феніл-5-(2-тієніл)-1,2,4-оксадіазолу фільтрували, промивали водою і сушили впродовж ночі у вакуумній печі з одержанням 3-феніл-5-(2-тієніл)-1,2,4оксадіазолу, 20,47 г, чистота 99,1 %, вихід 95,0 %. Приклад 3: Одержання 3-Феніл-5-(тіофен-2-іл)-1,2,4-оксадіазолу в Ацетоні [0037] Бензамідоксим (5,00 г, 36,7 ммоль) розчиняли в ацетоні (50 мл) при 0 °C. Розчин перемішували і додавали 50 % водний розчин гідроксиду натрію (4,5 мл). Слід відмітити, що в реакційній суміші відбувається виділення тепла і утворюється осад. Виділення тепла тривало близько 15 хвилин і суспензія загусла. Розчин нагрівали до 20 °C і додавали зовнішнє охолодження. Впродовж 15 хвилин додавали 2-тіофенкарбонілхлорид (5,36, 36,7 ммоль), підтримуючи температуру реакційної суміші нижче 30 °C. Реакцію витримували при температурі 30 °C впродовж 15 хвилин і суспензія рідшала. Реакційну суміш охолоджували до кімнатної температури і перемішували впродовж ще 20 хвилин. Додавали воду (50 мл) і отримані тверді речовини перемішували впродовж 1 години. Тверді речовини збирали шляхом вакуумної фільтрації, промивали водою (2×50 мл) і сушили на воронці Бюхнера потоком повітря з вакуумного насосу впродовж ночі. Отримували 3-феніл-5-(2-тієніл-1,2,4-оксадіазол (6,94 г, 30,4 ммоль) у вигляді білої твердої речовини (вихід 83 %). Приклад 4: Одержання Бензамідоксиму [0038] Гідрохлорид гідроксиламіну (88,5 г, 1,26 моль) і метанол (450 мл) завантажували в 1000 мл колбу і потім додавали 51 % гідроксид натрію (97,7 г, 1,25 моль). Додавали бензонітрил (103,1 г, 1,00 моль) і нагрівали реакційну суміш при 65-71 °C впродовж 4 годин. ВЕРХ продемонструвала, що реакція завершилась, на що вказувало зникнення бензонітрилу. Частину метанолу (366 мл) видаляли відгонкою, додавали ДІ воду (200 мл), а метанол, що залишився, переганяли при 200 торр з одержанням двох рідких фаз (розплаву бензамідоксиму і солоної води). Додавали бутилацетат (650 мл) і ДІ воду (100 мл) і фазу водної солі зливали. Розчин бутилацетату промивали один раз 100 мл води з одержанням 715,04 г синього каламутного розчину бензамідоксиму у бутилацетаті, з втратою 18,1 % при сушці. Вихід бензамідоксиму складав 95,1 % з 3,4 % виходом бензамідного побічного продукту. Приклад 5: Одержання Бензамідоксиму [0039] Бензонітрил (25,0 г, 242 ммоль) додавали в 250 мл колбу і нагрівали до 50 °C. За допомогою шприцевого насоса із швидкістю 0,17 мл/хв (2 години додаткового часу) додавали водний гідроксиламін (50 %, 20,4 г, 309 ммоль). Після завершення додавання 50 % водного розчину гідроксиламіну, реакційну суміш додатково перемішували впродовж 1 години 30 хвилин при 50 °C. Реакція завершилася і додавали 80 мл 2-метилтетрагідрофурану і 25 мл води. Розчин перемішували впродовж 10 хвилин і шари розділяли. Збирали водний органічний шар білого кольору (96,3 г). Після аналізу органічний шар містив 29,48 м/м % бензамідоксиму (28,4 г, 208 ммоль). Вихід складав 86 %. Приклад 6: Одержання Бензамідоксиму [0040] Бензонітрил (50,0 г, 0,485 ммоль) додавали в 250 мл колбу і нагрівали до 70 °C з перемішуванням. Додавали деіонізовану воду (1,0 г) і температуру вирівнювали при 70 °C. За допомогою шприцевого насоса впродовж 1,5 годин підтримуючи температуру на рівні 70° ± 2 °С додавали водний гідроксиламін (50 %, 37,50 г, 34,8 мл, вміст був 53,3 %, 0,605 моль). ВЕРХ аналіз показав, що перетворення бензонітрилу склало >99 % після близько 2,5 годин реакції. Завантажувану партію нагрівали в цілому впродовж 3,5 годин при 70 °C, потім охолоджували до 60 °C. Завантажувану партію переносили в 500 мл колбу з 2-метилтетрагідрофураном (208,98 г), з подальшим додаванням 20 % (мас.) водного NaCl (40,0 г). Суміш нагрівали до 60 °C і розділяли фази при 60 °C з одержанням розчину бензамідоксиму в 2-метилтетрагідрофурані (281,80 г) і водних відходах (44,61 г). Розчин 2-метилтетрагідрофурану містив 21,73 мас. % бензамідоксиму. Приклад 7: Одержання 3-Феніл-5-(тіофен-2-іл)-1,2,4-оксадіазолу в Бутилацетаті 9 UA 114915 C2 5 10 15 20 25 30 35 40 45 50 55 60 [0041] Розчин бензамідоксиму (18,3 %) у бутилацетаті (27,30 г, 36,7 ммоль) завантажували в 100 мл колбу і потім додавали 40 % водний розчин гідроксиду тетрабутиламонію (0,40 г) і 51 % гідроксид натрію (0,86 г, 11,0 ммоль). До цієї суміші одночасно додавали 2тіофенкарбонілхлорид (5,44 г, 36,8 ммоль) і 51 % гідроксид натрію (2,88 г, 36,7 ммоль) впродовж 30 хвилин при нагріванні при близько 70 °C. Додавали ще 0,66 г 51 % гідроксиду натрію, щоб підвищити рН до 9,75. Реакція завершувалася менш, ніж за 2,5 години (ВЕРХ аналіз). Додавали гарячу деіонізовану воду для розчинення солей(10 мл),після чого розділяли, промивали 10 мл гарячої води і розділяли. Додавали деіонізовану воду (50 мл) і видаляли бутилацетат шляхом азеотропної перегонки при 66-75 °C і 300 торр. 3-Феніл-5-(тіофен-2-іл)-1,2,4-оксадіазол осаджувався при перегонці. Продукт вивантажували, фільтрували через грубу скляну фриту, промивали водою (2×15 мл) і сушили впродовж ночі у вакуумній печі з одержанням 3-феніл-5(тіофен-2-іл)-1,2,4-оксадіазолу (7,50 г, 88,2 %); чистота 98,6 %. Приклад 8: Одержання 4-Хлорбензамідоксиму [0042] Розчин 50 % водного гідроксиду натрію (2,8 г, 36,34 ммоль) змішували в метанолі (20,0 мл). Додавали гідрохлорид гідроксиламіну (2,55 г, 36,34 ммоль) і суміш перемішували впродовж 5 хвилин. Додавали 4-хлорбензонітрил (4,0 г, 29,08 ммоль) і суміш нагрівали до 60 °C. Неоднорідну суміш перемішували при 60 °C впродовж 3 годин. Суміш виливали у воду (30 мл) і тверді речовини розчинялись і з'являвся осад. Метанол видаляли при пониженому тиску і тверді речовини, що залишилися, збирали шляхом вакуумної фільтрації. Тверді речовини промивали водою (2×10 мл). Отримували 4-хлорбензамідоксим (4,38 г, 25,6 ммоль) у вигляді твердої речовини білого кольору. (вихід 88 %). Приклад 9: Одержання 3-(4-Хлорфеніл)-5-(2-тієніл)-1,2,4-оксадіазолу [0043] 4-Хлорбензамідоксим (1,00 г, 5,8 ммоль) розчиняли в 2-метилтетрагідрофурані (10,0 мл) і воді (1,0 мл). Додавали 40 % водний розчин гідроксиду бензилтриметиламонію (100 мкл) і нагрівали розчин до 50 °C. Впродовж 15 хвилинпо краплях одночасно додавали 50 % водний розчин гідроксиду натрію (720 мг, 9,00 ммоль) і 2-тіофенкарбонілхлорид (939,0 мг, 6,4 ммоль). Температуру суміші нагрівали до 70 °C. Суміш охолоджували до кімнатної температури і додавали воду (40 мл). Тверді речовини, що утворилися, і 2-метилтетрагідрофуран видаляли при пониженому тиску. Тверді речовини виділяли шляхом вакуумної фільтрації. Отримували 3(4-хлорфеніл)-5-(2-тієніл)-1,2,4-оксадіазол (1,32 г, 5,0 ммоль) у вигляді білої твердої речовини. (вихід 86 %). Приклад 10: Одержання 3-(4-Хлорфеніл)-5-(2-фураніл)-1,2,4-оксадіазолу [0044] 4-Хлорбензамідоксим (1,24 г, 7,25 ммоль) розчиняли в 2-метилтетрагідрофурані (10,0 мл) і воді (1,0 мл). Додавали 40 % водний розчин гідроксиду бензилтриметиламонію (100 мкл) і 50 % водний розчин гідроксиду натрію (736 мг, 9,20 ммоль). Суміш перемішували і утворювався осад. По краплях додавали фуроїлхлорид (1,24 г, 7,25 ммоль) і відмічали виділення тепла. Суміш перемішували при температурі 70 °C впродовж 1 години і охолоджували до кімнатної температури. Розчин виливали у воду (25 мл) і 2-метилтетрагідрофуран видаляли при пониженому тиску. Отримані тверді речовини виділяли шляхом вакуумної фільтрації. Отримували 3-(4-хлорфеніл)-5-(2-фураніл)-1,2,4-оксадіазол (1,22 г, 4,94 ммоль) у вигляді білої твердої речовини. (вихід 68 %). Приклад 11: Одержання 3-Феніл-5-(2-фураніл)-1,2,4-оксадіазолу [0045] Бензамідоксим (2,00 г, 14,7 ммоль) і 40 % водний розчин гідроксиду бензилтриметиламонію (100 мкл) об'єднували в 2-метилтетрагідрофурані (20 мл) і воді (2 мл). Розчин нагрівали до 50 °C і по краплях одночасно впродовж 10 хв додавали 50 % водний розчин гідроксиду натрію (1,52 г, 19,1 ммоль) і 2-фуроїлхлорид (2,00 г, 15,3 ммоль). Температура в посудині піднімалася до 74 °C. Суміш перемішували при температурі 60 °C впродовж 48 годин і тверді речовини розчинялися. Суміш охолоджували до кімнатної температури і виливали у воду (25 мл). 2-Метилтетрагідрофуран видаляли при пониженому тиску і у водному розчині тверді речовини утворювалися. Тверді речовини збирали шляхом вакуумної фільтрації. Отримували 3-феніл-5-(2-фураніл)-1,2,4-оксадіазол (2,33 г, 9,5 ммоль) у вигляді білої твердої речовини. (вихід 64 %). Приклад 12: Одержання 3-Феніл-5-(тіофен-2-іл)-1,2,4-оксадіазолу [0046] Розчин бензамідоксиму (24,92 %) в 2-метил ТГФ (67,53 г, 124 ммоль) завантажували в 500 мл колбу і потім додавали ДІ воду (20,0 г), 55 % водний гідроксид тетрабутиламонію (0,94 г) і 50 % гідроксид натрію (0,96 г, 12 ммоль). До цієї суміші одночасно впродовж 30 хвилин при нагріванні до близько 70°С додавали 2-тіофенкарбонілхлорид (17,77 г, 120 ммоль) і 50 % гідроксид натрію (9,60 г, 120 ммоль). Реакція завершувалась менш ніж за одну годину. Температуру знижували до 60 °C і рН водної фази нейтралізовували до 7,61 і зливали у відходи. До суміші додавали гарячий розчин 1,25 г Morwet D-425 в 100 мл ДІ води, і 2-фазну 10 UA 114915 C2 5 10 15 суміш нагрівали для відгонки азеотропу 2-метилтетрагідрофуран/вода (температура кипіння 71 °C). В колбу-реактор впродовж близько 15 хвилинзакачували додаткову кількість води (150 мл), після того, як почали відганяти 2-метилтетрагідрофуран. При гарному перемішуванні, температура бака досягала 100,5 °C (температура пари 97 °C). Суспензію охолоджували і фільтрували з використанням грубої скляної фрити. Накип зіскоблювали з мішалки і об'єднували з вологим осадом; накип складав лише близько 3 % загального об'єму продукту. Об'єднаний вологий осад промивали гарячою ДІ водою (3 × 50 мл) і сушили у вакуумі впродовж 5 хвилин з одержанням вологого осаду 3-феніл-5-(тіофен-2-іл)-1,2,4-оксадіазолу, 34,80 г. Втрата при сушці вологого осаду складала 20,13 %. Вміст висушеного 3-феніл-5-(тіофен-2-іл)-1,2,4оксадіазолу складав 96,12 %, а рівень залишкового хлориду складав
ДивитисяДодаткова інформація
Назва патенту англійськоюProcesses for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles
Автори англійськоюMiller, William Harold, Graham, Charles Richard, Brown, David Louis
Автори російськоюМиллер Уилльям Харольд, Грэхэм Чарльз Ричард, Браун Дэвид Луис
МПК / Мітки
МПК: C07D 413/04
Мітки: одержання, спосіб, 1,2,4-оксадіазолів, варіанти, 3,5-дизаміщених
Код посилання
<a href="https://ua.patents.su/20-114915-sposib-oderzhannya-35-dizamishhenikh-124-oksadiazoliv-varianti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 3,5-дизаміщених 1,2,4-оксадіазолів (варіанти)</a>
Спосіб одержання 3,5-дизаміщених 1,2,4-оксадіазолів

Номер патенту: 112887
Опубліковано: 26.12.2016
Автор: Толмачов Андрій Олексійович
МПК: C07D 231/00
Мітки: одержання, 1,2,4-оксадіазолів, 3,5-дизаміщених, спосіб
Формула / Реферат:
1. Спосіб одержання 3,5-дизаміщеного 1,2,4-оксадіазолу формули (І):,в якійR являє собою С1-Салкіл, С3-С10циклоалкіл, С6-С10арил або С2-С9гетероарил, кожен з яких необов'язково заміщений одним або більшою кількістю замісників, незалежно вибраних з С1-С6алкілу, С2-С6алкенілу, С2-С6алкінілу, С1-С6алкокси, С3-С10циклоалкілу, С6-С10арилу, С2-С9гетероарилу,...
Спосіб одержання хіральних 1,4-дизаміщених піперазинів, проміжна сполука (варіанти)

Номер патенту: 79774
Опубліковано: 25.07.2007
Автори: Зелдіс Джозеф, Фейгельсон Грегг Брайан, Джирковскі Іво
МПК: C07D 405/12, C07D 319/00, C07D 295/08, C07D 295/10, C07D 487/10, C07D 213/75, C07C 229/12, C07B 53/00
Мітки: сполука, 1,4-дизаміщених, піперазинів, спосіб, проміжна, хіральних, одержання, варіанти
Формула / Реферат:
1. Спосіб стереоселективного одержання 1,4-дизаміщених піперазинів формули IX, (IX)де R являє собою С1-С3-алкільну групу;Аr являє собою дигідробензодіоксиніл, бензодіоксиніл або феніл, необов'язково заміщений до трикратно замісниками, незалежно один від одного вибраними з галогену, метокси, галогенметилу, дигалогенметилу і тригалогенметилу; і Aryl...
Спосіб одержання 4,6-дизаміщених 2-амінопіридино-3-карбонітрилів

Номер патенту: 102981
Опубліковано: 25.11.2015
Автори: Обушак Микола Дмитрович, Вахула Андрій Романович, Литвин Роман Зіновійович, Горак Юрій Ігорович
МПК: A61K 31/44
Мітки: 4,6-дизаміщених, спосіб, одержання, 2-амінопіридино-3-карбонітрилів
Формула / Реферат:
Спосіб одержання 4,6-дизаміщених 2-амінопіридино-3-карбонітрилів, що ґрунтується на взаємодії ароматичних альдегідів з ароматичними кетонами, малонодинітрилом і ацетатом амонію в органічному розчиннику, який відрізняється тим, що як альдегіди використовують 5-арил-2-фуранкарбальдегіди, а як розчинник - етиловий спирт і одержують сполуки загальної формули,де...
Спосіб одержання 2,6-дизаміщених діетил-4-(5-арил-2-фурил)-1,4-дигідро-3,5-піридиндикарбоксилатів

Номер патенту: 94002
Опубліковано: 27.10.2014
Автори: Вахула Андрій Романович, Литвин Роман Зіновійович, Горак Юрій Ігорович, Обушак Микола Дмитрович, Лаба Євген-Олег Володимирович
МПК: A61K 31/4422
Мітки: спосіб, діетил-4-(5-арил-2-фурил)-1,4-дигідро-3,5-піридиндикарбоксилатів, 2,6-дизаміщених, одержання
Формула / Реферат:
Спосіб одержання 2,6-дизаміщених діетил-4-(5-арил-2-фурил)-1,4-дигідро-3,5-піридиндикарбоксилатів, який полягає на взаємодії альдегідів арилфуранового ряду з 1,3-дикарбонільними сполуками і гідрокарбонатом амонію в органічному розчиннику, який відрізняється тим, що як альдегіди використовують 5-арил-2-фуранкарбальдегіди, як 1,3-дикарбонільні сполуки - ацетооцтовий або фуроїлоцтовий естери, як розчинник - етиловий спирт і одержують сполуки...
Спосіб одержання 3,5-дизаміщених 1,3-тіазол-2(3н)-тіонів

Номер патенту: 82988
Опубліковано: 27.08.2013
Автори: Вакула Володимир Миколайович, Яременко Федір Георгійович, Сова Олександр Миколайович
МПК: C07D 277/08, C07B 35/00
Мітки: 3,5-дизаміщених, спосіб, 1,3-тіазол-2(3н)-тіонів, одержання
Формула / Реферат:
Спосіб одержання 3,5-дизаміщених 1,3-тіазол-2(3Н)-тіонів, який відрізняється тим, що вказані сполуки одержують реакцією дегідратації 5-метилензаміщених 4-гідрокси-1,3-тіазолідин-2-тіонів у кислотному середовищі.
Попередній патент: Спосіб одержання белітового цементу з високою реактивністю і низьким співвідношенням кальцій/силікат
Наступний патент: Торцева пружна муфта
Випадковий патент: Спосіб одержання поліфенольного комплексу з анаболічною дією