1-(1н-імідазол-4-іл)алкіл-бензаміди, їх хлоргідрати та оптичні ізомери, які мають протиішемічні та антагоністичні властивості alрha2-адренергічних рецепторів
Номер патенту: 12835
Опубліковано: 28.02.1997
Автори: Ерік Коссман, Філіпп Мішель, Жан-П'єр Жерт, Ернст Вульферт, Жан Гобер








Формула / Реферат
(57) 1-(1Н-имидазол-4-ил)алкил-бензамиды, их хлоргидраты и оптические изомеры общей формулы
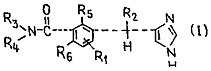
где
1) R1, R2, R3, R4 - каждый означает водород; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата;
2) R1, R3, R4 - каждый означает водород; R2 - метил; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата;
3) R1, R2, R4 - каждый означает водород; R3 метил; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата;
4) R1, R2, R4, R6 - каждый означает водород; R3 - аминогруппа; R5 - гидроксильная группа;
5) R1, R2, R4 - каждый означает водород; R3 - гидроксиэтил; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата;
6) R1, R3, R4 - каждый означает водород; R5 - метоксигруппа; R6 - атом водорода в форме хлоргидрата;
7) R1, R2, R3, R4, R5 - каждый означает водород; R6 - гидроксильная группа;
8) R1, R2, R3, R4 - каждый означает водород; R5 - гидроксильная группа; R6 - гидроксильная группа в форме хлоргидрата;
9) R1 - третбутил; R2, R3, R4, R6 - каждый означает водород, R5 - гидроксильная группа.
10) R1, R2, R3, R4 - каждый означает водород; R5 - гидроксильная группа; R6 - метил в форме хлоргидрата;
11) R1 - метил; R2, R3, R4, R6 - каждый означает водород; R5 - гидроксильная группа;
12) R1, R3, R4 - каждый означает водород; R3 - гидроксильная группа; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата;
13) R1, R3, R4 - каждый означает водород; R2 - метил; R5 - гидроксильная группа; R6 - атом водорода в форме хлоргидрата и оптического изомера,
обладающие антиишемическими и антагонистическими свойствами a2-адренергических рецепторов.
Текст
1-(1 Н-имидазол-4-ил)алкил-бензамиды, их хлоргидраты и оптические изомеры об щей формулы R3 g j* «г (О N и где 1) Ri, R2. R3, R4 - каждый означает водо род; Rs - гидроксильная группа; R6 - атом водорода в форме хлоргидрата; 2) Ri, R3, R4 - каждый означает водород; R2 - метил; Rs - гидроксильная группа; R6 атом водорода в форме хлоргидрата; 3) Ri, R2, R4 - каждый означает водород; R3 метил; Rs - гидроксильная группа; R6 атом водорода в форме хлоргидрата; 4) Ri, R2, R4, R6 - каждый означает водо род; R3 ~ аминогруппа; Rs - гидроксильная группа; 5) Ri, R2, R4 - каждый означает водо род; R3~ гидрокииэтил; Rs~ гидроксильная группа; R6 - атом водорода в форме хлор гидрата; 6) Ri, R3, R4 - каждый означает водород; Rs - метоксигруппа; R6 - атом водорода в форме хлоргидрата; 7) Ri, R2, R3, R4, Rs - каждый означает водород; Re- гидроксильная группа; 8) Ri, R2, R3, R4 - каждый означает водород; Rs - гидроксильная группа; Re гидроксильная группа в форме хлоргид рата; 9) Ri - третбутил; R2, R3, R4, R6 - каждый означает водород, Rs- гидроксильная груп па. 10) Ri, R2, R3, R4 - каждый означает во дород; Rs - гидроксильная группа; R6 - ме тил в форме хлоргидрата; 11) Ri - метил; R2, R3, R4, R6 - каждый означает водород; Rs - гидроксильная груп па; 12) Ri, R2, R4 - каждый означает водо род; R3 - гидроксильная группа; Rs - гидро ксильная группа; R6 - атом водорода в форме хлоргидрата; 13) Ri, R3, R4 - каждый означает водо род; R2 - метил; Rs - гидроксильная группа: R6 ~ атом водорода в форме хлоргидрата и оптического изомера, обладающие антиишемическими и антагонистическими свойствами «2 -адренергических рецепторов. ю 00 со о J 12835 Изобретение относится к новым 1-(1Нимидазол-4-ил)алкил-бензамидам или к их хлоргидратам, или оптическим изомерам. Соединения могут быть использованы в терапевтической области, т.к. они проявляют антиишемическую активность и являются агонистами аг-адренергических рецепторов. Цель изобретения - получение новых замещенных 1Н-имидазолов, обладающих 10 кроме характерных для данного ряда свойств, еще и агонистическими свойствами 2-адренергических рецепторов. Поставленная цель достигается новыми 15 соединениями общей формулы N x t> 20 (П Н NИ где Ri, R2 ~ могут быть идентичными или различными, каждый из них представляет собой атом водорода или алкильный радикал; R3 - атом водорода, алкильный или гидроксиалкыльный радикал, амино- или гидроксильиая группа; R4 ~ атом водорода или алкильный радикал, или R3, R4 взятые вместе с атомом азота«с которым они соединены, представляют собой гетероциклический радикал, выбранный из радикалов 35 пирролидино, пиперидине или морфолино; Rs, R6 идентичные или различные, каждый из которых представляет собой атом водорода, гидроксильную группу, алкильный или алкоксирадикал, причем по крайней мере один из радикалов R5, Re имеет иное, чем атом водорода, значение, а все алкил- и алкоксирадикалы имеют 1-4 атомов углерода. Изобретение относится также к их солям с шепотами, нетоксичным и терапевтически допустимым. Если о молекуле имеется один асимметричный атом углерода, то соединения формулы 1 могут быть либо в рацемической форме, либо в форме одного или другого 50 энантиомера. Эти разные формы также входят в рамки настоящего изобретения. Способ получения замещенных 1-(1Нимидазол-4-ил)алкил-бензамидов формулы (I) состоит в реакции алкил-1-{1Н-имидазол- 55 4-ил)алкил-бензоата формулы *5 R. N N СИ) в которой Ri, R2, Rs, Re имеют то же значение, что и выше; R7 - алкильный радикал, имеющий 1-4 атомов углерода с азотсодержащим соединением формулы HIM: (111) где R3. R4 имеют указанное значение. Эту реакцию обычно осуществляют при нормальном давлении или при повышенном давлении в автоклаве либо в спиртовом растворителе, например в этаноле или метаноле, либо при большом избытке азотного соединения, используемого как исходное, при температуре обычно в пределах от комнатной до температуры рефлюкса при необходимости в присутствии катализатора, такого как метилат натрия. В соответствии с частной формой осуществления способа, относящейся к получению замещенных 1-(1Нимидазол-4-ил)-алкил-бензамидов формулы (I) в которых Rs, R6 пред 25 ставляют собой водород, алкильный или алкоксильный радикал, имеющий 1-4 атомов углерода, причем по крайней мере один из радикалов Rs, Re иной, чем водород; проводят реакцию 1-(1Н-имидазол-4-ил)алкил30 бензойной кислоты формулы R2 о R5 N (IV) H но'С ~A$ *6 F H где R2. Ri - атом водорода или алкильный радикал, имеющий 1-4 атомов углерода; Rs, 40 R6 имеют указанное значение, с азотсодержащим соединением формулы (ИО 45 где R3> R4 имеют указанное выше значение. Для проведения этой реакции исходная кислота формулы (IV) предварительно активируется известным способом при помощи подходящего реактива, например алкилга-логеноформиатом, предпочтительно этилхлорформиатом. Эту реакцию обычно осуществляют при температуре около 0°С. в инертном растворителе, например в дих-лорметане или ацетонитриле, и в присутствии вспомогательного основания, например триэтиламина. В соответствии с другой формой осуществления изобретения, используемой для 12835 получения замещенных 1-(1Н-имидазол-4ил)алкил-бензамидов формулы (I), где Ri, R3, R4 - водород; Rs гидроксил; R6 - водород или алкильный радикал, имеющий 1-4 атомов углерода, осуществляют гидролиз в кислой 5 среде 2-гидрокси-3[1-(1 Н-имидазол-4-ил)алкил]-бензонитрила формулы (V) 10 где R2, R6 - каждый атом водорода или ал- 15 кильный радикал, имеющий 1-4 атомов углерода. Этот гидролиз обычно осуществляют в водном растворе серной кислоты 80 об.% при 60-75°С в течение нескольких часов. 20 Согласно варианту, этот гидролиз может быть также осуществлен в безводном метиловом спирте, к которому добавлены следы воды и через который пропускают ток газообразной хлористоводородной кислоты. 25 Промежуточный продукт, который образуется in situ, не изолируется и непосредственно превращается в амид путем нагрева. Пример 1. Получение исходных алкил 1(1Н-имидазол-4-ил)алкил-бензоатов фор- 30 мулы (II). А. Этерификация соответствующих кислот (способ а). 1. Хлоргидрат этил-2-гидрокси-5-[(1Нимидазол-4-ил)метил]-бензоата. 35 Током газообразной хлористоводородной кислоты при 0°С насыщают суспензию 3,1 г (12,2 моль) хлоргидрата-2-гидрокси-5-[(1 Нимидазол-4-ил)метил]-бензойной кислоты (приготовлена, как описано ниже в 40 примере 2.С) в 150 мл абсолютного этанола. Затем медленно нагревают до температуры рефлекса и поддерживают так в течение 10 ч. Затем растворитель испаряют до осаждения эфира. Эфир фильтруют промывают ди- 45 этиловым эфиром и затем высушивают. Получают 2,3 г хлоргидрата этил-2-гидро-кси-5-[(1Н-имидазол4-ил)метил]-бензоата. Выход: 68%. Температура плавления 195-198°С. Спектр ЯМР: (ДМСО) Дельта 1,34 50 (ЗН.т.), 4,03 (2Н, S), 4,37 (2Н, q), 6,95 (1Н. d), 7,40 (1Y, S), 7,49 (1Н, dd), 8,70 (1Н, d), 9,12 (1H,S), 10,6 (1H.S). 2. Этил-2-гидрокси-3-[1-(1 Н-имидазол4-ил)этил]-бензоат. 55 Это соединение получают так же, как описано выше в п. 1, но из 2-гидрокси-3-{1(1 Н-имидазол-4-ил)этил]-бензойной кислоты (ее получение описано ниже в примере 2.А.2). В конце реакции реакционную среду нейтрализуют путем добавления концентрированного раствора аммиака, минеральные соли фильтруют и фильтрат выпаривают при пониженном давлении. Полученный остаток очищают хроматографически на силикагеле (элюат: смесь дихлорметан-метанол 8:2 об). Получают этил-2-гидрокси-3-[1-(1 Нимидазол-4-ил)этил]-бензоат с выходом 35%. Соответствующий хлоргидрат плавится при 168°С (этанол-эфир). Анализ для C14H16N2O3 HCI, %: Рассчитано: С 56,66; Н 5,40; N 9,44. Определено: 56,58; 5,40; 9,21. 3. Этил-2-гидрокси-3-{1-(1 Н-имидазол4-ил)пентил]-бензоат. Нагревают обратным холодильником в течение 9 ч 1,18 г (3,8 ммоль) хлоргидрата 2-гидрокси-3-[1-(1Н-имидазол-4-ил)пентил] бензойной кислоты (приготовляется, как описано ниже в примере 2.А.З) в растворе 15 мл триэтил ортоформиата в присутствии 1,2 г безводного монтмориллонита К 10 Затем фильтруют и выпаривают при пониженном давлении. Полученный остаток хроматографически очищают на 150 г окиси кремния (элюант : смесь дихлорметан - метанол аммиак 95:5:0,5 об.). Получают 0,213 г этил2-гидрокси-3-{ 1 -(1 Н-имидазол-4-ил)пентил] бензоата. Спектр ЯМР (ДМСО): Дельта 0,81 (ЗН, т), 1,0-1,29 (2Н, м), 1,34 (ЗН, т), 1,77-2,0 (2Н, м), 4,31-4,41 (ЗН, т+q), 6,77 (Ж, с), 6,86 (1Н, т), 7,46-7,50 (2Н, д+с). 7,63 (1Н. д). Полученный таким образом продукт используют как он есть без очистки для получения соответствующего бензамида (пример 4,15). 4. Метил-2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензоат. Это соединение получа ют так, как описано выше в п.2, но исходя их хлоргидрата 2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензойной кислоты (получается, как описано ниже в примере 2.А.1) и мета нола. Температура плавления: 153-154°С. Спектр ЯСР (ДМСО): дельта 3,87 (2Н, м). 3,91 (1Н, с), 6,77 (1Н, с), 6,87 (1Н, т), 7,45 (1Н, дд), 7,58 (1Н, с), 7,69 (1Н, дд), 10 (1Н. с). В. Путем превращения Клайзснэ. 1. Метил-3-[(1 Н-имидазол-4-ил)метил]-2метоксибензоат. 1 .а. Метил-2-(2-хлор-2-пропенилокси)бензоат. В течение 10 ч нагревают с обратным холодильником суспензию из 304 г (2 моль) метил-2-гидроксибензоата, 25 г иодида калия, 69 г (0,5 моль) карбоната калия и 69 г (0,625 моль) 2,3-дихлорпропена в 3 л сухого ацетона. Через 2,5,5 и 7,5 ч реакции каждый раз добавляют 69 г карбоната калия и 69 г 12835 2,3-дихлорпропена. Затем суспензию фильтруют и выпаривают фильтрат под пониженным давлением. Остаток разбавляют этилацетатом и раствор промывают последовательно в насыщенном водном растворе тиосульфата натрия, в воде и окончательно в водном насыщенном растворе хлористого натрия. Органическую фазу высушивают над сульфатом натрия и затем перегоняют при пониженном давлении. Таким образом получают 394 г метил-2-(2-хлор-2-пропенилокси)-бензоат. Выход: 87%. Температура кипения: 119°С/1,3 мбар. 1.6, Метил-3-(2-хлор-2-пропенил)-2-гидроксибензоат. Тщательно дегазируют в аргоне 274,1 г (1,21 моль) метил-2-(2-хлор-2-пропенилоксфбензоата, находящегося в двухлитровой колбе За гем нагревают как можно быстрее до 260°С. При этой температуре резко начинается экзотермическая реакция: температура сама возрастает до 293°С, происходит рефлекс смеси и почернение реакционной смеси. После охлаждения до комнатной температуры отгоняют продукт при пониженном давлении. Получают 241,1 г метил3-(2-хлор-2-пропенил)-2-гидроксибензоата. Выход188%. Температура кипения: 109110°С/1,3 мбар. Спектр ЯМР (СДСІЗ). дельта 3,71 (2Н, с), 3,95 (ЗН, с), 5,17(1Н,м), 5,28 (1Н,м), 6,90(1 Н, м), 7,48 (1Н, дд), 7,85 (1Н, дд), 11,22 (1Н, с). 1 с. Метил-3-(2-хлор-2-пропенил)-2-метоксибензоат. Не превышая температуру 10°С, добавляют порциями по 8,81 г(ЗОбммоль) гидрида натрия к раствору 57,7 г (255 ммоль) метил3-(2-хлор-2-пропенил)-2-гидроксибензоата в 500 мл безводного диметилформамида. Смесь нагревают при 40°С в течение 15 мин. Затем добавляют 43,45 г (306 ммоль) метил иодида в растворе 50 мл толуола, затем смесь выдерживают при 40°С в течение 3 ч. Реакционную смесь осторожно выливают в 5 л воды и производят многократное экстрагирование этилацетатом. Затем органическую фазу концентрируют до объема 500 мл, промывают ее водой и последовательно насыщенным видным раствором хлористого натрия. Раствор высушивают над сульфатом натрия и растворитель отгоняют при пониженном давлении, остаток хроматографически очищают на силикагеле (элюант : дихлорметан : гексан - 50:50 об). Таким образом получают 49,2 г метил-3-(2-хлор-2пропенил)-2-метоксибензоата. Выход: 60%. Температура кипения: 107110°С/0,5 мбар (масло). Спектр ЯМР (СДСІз): дельта 3,73 (2Н, с), 3,82 (ЗН, с), 3,92 (ЗН, с), 5,14 (1Н, м), 5,32 (1Н, м), 7,15 (1Н, т), 7,47 (1Н,дд), 7,81(1 Н.дд). 8 1. д. Метил-3-[(1Н-имидазол-4-ил)метил]-2-метоксибензоат. Нагревают с обратным холодильником в течение 150 мин 45,5 г (189 ммоль) метил5 3-(2-хлор-2-пропенил)-2-метоксибензоата и 81,5 г (387 ммоль) м-хлорпербензойной кислоты в растворе сухого 300 мл хлороформа. Охлаждают до 0°С и удаляют полученный осадок фильтрованием. Последовательно 10 промывают фильтрат в водном насыщенном растворе тиосульфата натрия и в водном насыщенном растворе бикарбоната натрия. Раствор высушивают над сульфатом натрия, растворитель отгоняют при 15 пониженном давлении при температуре не выше 30°С. Суспензируют полученный остаток с 300 мл безводного метанола и смешивают с 111,3 г (1,32 моль) медквдиенерЫого бикар20 боната натрия. Сме.сь нагревают с обратным холодильником. Затем частями каждый час добавляют 137,6 г (1,32 моль) ацетата формамидина. Через 5,5 ч нагревания с обратным холодильником удаляют метанол 25 при пониженном давлении. Остаток разбавляют п 500 мл воды и экстрагируют этилацетатом. Органическую фазу высушивают над сульфатом натрия и выпаривают в вакууме. Остаток хроматографически очищают на 30 силикагеле (элюант : смесь дихлорметан : этанол : аммиак = 92,3:7:0,7 об.). Получают 13,8 г метил-3-[(1Н-имидазол-4-ил)метил]-2метоксибензоата. Выход. 30%. 35 Спектр ЯМР (ДМСО): дельта 3,73 (ЗН, с), 3,83 (ЗН, с), 3,89 (2Н, с), 6,78 (1Н, с), 7,11 (1Н, т), 7,24-7,73 (ЗН, м). Полученный продукт используется как он есть без дополнительной для приготовления соответствующей кисло40 ты (например, 2.В.1). Следующие соединения были получены методом, описанном ниже в В.1. 2. Метил-2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензоат. 45 Это соединение получают из метил-3-(2хлор-2-пропенил)-2-гидроксибензоата с выходом 35,4%, естественно, пропустив метилирование метилиодидом, которое описано в П.1.С. Температура плавления: 50 153-154°С. Полученный продукт идентичен продукту, полученному в примере 1.А.4. 3. Метил-3-[(1 Н-имидазол-4-ил)метил]-2н-пропоксибензоат. Выход: 20% (масло). Полученный продукт в неочищенном ви-55 де используется как есть для получения соответствующей кислоты (пример 2.В.2). С. Реакция Фриделя-Крафта. 1. Хлоргидрат метил-5-трет-бутил-2-гидрокси-3-[(1Н-имидазол-4-ил)метил]-бензоата. 9 12835 Проводят реакцию 50 г хлоргидрата 1Нимидазол-4-метанола с 60 г метил-5-трет-бутил-2-гидроксибензоатом в 150 мл концентрированной серной кислоты при 20°С в течение 21ч. Осторожно разлагают 5 реакционную смесь на льду. Твердый полученный продукт филыруют и хроматографически очищают, затем переводят его в хлоргидрат. Таким образом получают 1,1 г хлоргид- 10 рата метил-5-трет-бутил-2-гидрокси-3-[(1 Нимидазол-4-ил)метил]-бензоата. Выход: 2,8%. Температура плавления: 185-186°С. Спектр ЯМР (ДМСО): дельта 1,3 (9Н, с), 3,45 (ЗН, с), 4,1 (2Н, с), 7,3 (1Н, с),7,75 (2Н, м), 9,0 15 (Ж, с), 10,5-13,0 (ЗН). 2. Метил-2,6-дигидрокси-3-{(1 Н-имидазол-4-ил)метил]-бензоат. Смешивают 95,84 г (0,57 моль) метил-2,6дигидроксибензоата, 190 мл муравьи- 20 ной кислоты и 51,14 г (0,38 моль) хлоргидрата 1Н-имидазол-4-метанола. Нагревают смесь с обратным холодильником и в течение 15 мин отгоняют азеотроп муравьиная кислота-вода. Затем в течение 17 ч 25 нагревают смесь с обратным холодильником. Реакционную смесь выливают в воду. Экстрагируют избыток метил-2,6-дигмдроксибензоата толуолом, затем водную фазу при рН 7-8 нейтрализуют путем добавления 30 насыщенного водного раствора гидроксида натрия. Экстрагируют дихлорметаном, сушат органические фазы над сульфатом натрия и выпаривают растворитель при пониженном давлении. Остаток очищают 35 хроматографически на силикагеле (элюант:смесь дихлорметан-метанол-аммиак = 94:6:0,6 об.). Получают 14,8 г метил-2,6-дигидрокси-3-[(Ж-имидазол-4-ил)метил]бензоата, загрязненного следами остаточных 40 растворителей. Выход: 13%. Спектр ЯМР (ДМСО): дельта 3,72 (2Н, с), 3,81 (ЗН, с), 6,33 (1 Н,д), 6,86(1 Н, с), 7,06(1 Н, д), 7,73 (1Н, с), 9,5 до 10,2 (ЗН). Из-за нестабильности полученный про- 45 дукт используется как он есть без другой очистки для получения соответствующего бензамида (пример 4,9). Следующие соединения были получены по методике, описанной выше в п.С.2. 50 3. Метил-2,6-дигидрокси-3-[1-(1 Н-имидазол-4-ил)этил}-бензоат. Это соединение получают из хлоргидрата -метил-1Н-имидазол-4-метанола. Нагревают с обратным холодильником 55 проводят в течение 19 ч. Окончательно полученный остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметанметанол-аммиак = 95:4:0,5 об.). Выход: 43%. 10 Спектр ЯМР (СДСІз): дельта 1,52 (ЗН, д), 4,0 (ЗН, с), 4,48 (Ж, q), 6,40 (1Н, д), 6J9 (1Н, с), 7,14 (1Н, д), 7,47 (1Н, с). 10,0 (ЗН). Пилученный продукт используется как он есть без дополнительной очистки для получения соответствующего бензамида (пример 4.10). 4. Этил-6-гидрокси-3-[(1 Н-имидазол-4ил)метил]-2-метилбензоат и зтил-2-гидрокси-3-[(1Н-имидазол-4-ил)метил]-6-метилбензоат. Эти два соединения приготовляют одновременно из смеси 80 г (0,448 моль) этил2-гидрокси-6-метилбензоата, 225 мл муравьиной кислоты и 51,5 г (0,382 моль) хлоргидрата 1Н-имидазол-4-метанола, которую нагревают с обратным холодильником в течение 53 ч. Затем разделяют и очищают хроматографическими методами на силикагеле полученные продукты (элюент: смесь дихлорметан-метанол-аммиак = 94:6:0,5 об.). Получают 7,2 г этил-6-гидрокси-3-[(1Нимидазол-4-ил)-метил]-2-метилбензоата. Выход: 7,2%. Температура плавления: 4245°С. Спектр ЯМР (СДСІз). дельта 1,36 (ЗН, т), 2,34 (ЗН, с), 3,83 (2Н, с), 4,37 (2Н, q), 6,52 (1Н, с), 6,70 (1Н, д), 7,08 (1Н, д), 7,45 (Ж, с), 10,0 (2Н). Одновременно получают 2,8 г этил-2гидрокси-3-[(Ж-имидазол-4-ил)метил]-6-метилбензоата. Выход 2,8%. Температура плавления:101-103°С. Спектр ЯМР (СДСІз): дельта 1,40 )ЗН, т), 2,47 (ЗН, с), 3,88 (2Н, с), 4,39 (2Н, q), 6,62 (1Н, д), 6,76 (Ж, с), 7,12 (Ж, д), 7,43 (Ж, с), 10,0 (2Н). 5. Бромгидрат метил-2-гидрокси-3-[(Жимидазол-4-ил)метил]-4-метилбензоата. 5а. Метил-5-бром-2-гидрокси-3-[(1 Нимидазол-4-пл)метил]~4-метилбензоат. При комнатной температуре частями добавляют 95,6 г (0,71 моль) хлоргидрата Ж-имидазол-4-метанола к раствору 87 г (0,355 моль) метил-5-бром-2-гидрокси-4-метилбензоата в 900 мл концентрированной серной кислоты. Помешивают смесь в течение 234 ч. Затем осторожно выливают реакционную смесь на лед и доводят рН до 8 путем добавления водного насыщенного раствора гидроксида натрия. Проводят экстрагирование этилацетатом. Органическую фазу испаряют при пониженном давлении, остаток хроматографически очищают на 1,4 кг окиси кремния (элюент: смесь дихлорметан-метанол «= 9:1 об.). Полученный таким образом продукт хроматографически очищают второй раз на 400 г окиси кремния (элюент: смесь дихлорметанметанол =95:5:0,5 об.). Получают 2,4 г метил 11 12835 5-бром-2-гидрокси-3-[(1Н-имидазол-4-ил) метил ]-4-метил-бензоата, достаточно чистого, чтобы использовать как он есть на следующем этапе. Спектр ЯМР (ДМСО): дельта 2,43 (ЗН, с), 5 3.90 (ЗН, с), 3,96 (2Н, с), 6,62(1 Н, с), 7,51(1 Н, с), 7,87(1 Н, с). . 5.6. Бромгидрат метил-2-гидрокси-З[(1Н-имидазол-4-ил)метил]~4-метилбензоата. 10 Производят гидрогенолиз 2,38 г (7,2 ммоль) метил-5-бром-2-гидрокг,и-3-[(1 Н-имидазол-4-ил)метил]-4-метилбе»;.юата, растворенного в 70 мл метаноля. при давлении водорода 4,2 бар, при комнатной температу- 15 рэ и в присутствии 0.7 г палладия на угле (10%го). Катализатор отфильтровывают и растворитель выпаривают при пониженном давлении. Получают 1,83 г бромгидрата ме-тил2-гидрокси-3-[(1 Н-имидазол-4-ил)метил] 20 -4метиябензоата. Выход: 76%. Спектр ЯМР (ДМСО): дельта 2,34 (ЗН, с), 3.91 (ЗН. с), 4,02 (2Н, с), 6,87(1 Н, с), 7,13 (1Н, с), 7,68 (1Н, д), 8,00(1 И, с). Полученный таким образом продукт ис- 25 пользуется как он есть без дополнительной очистки для получения соответствующего бензамида (см. пример 4.14). П р и м е р 2. Получение исходных 1(1Н-имидазол~4-ил}алкил-бензойных кис- 30 лот формулы (IV). А. Путем окисления соответствующих 1(1 Н-имидазол-4-ил)~ал кил-бензол метанолов. 1,2-гидрокси-3-[(1 Н-имидазол~4-ил)мет 35 ил]-бензойная кислота (хлоргидрат). В течение 2,5 ч нагревают при 180°С при хорошем перемешивании 1 г 2-гидрокси-З[(1Н-имидазол~4-пл)метил}-бензол метанола (получается по методике, описанной в приме- 40 ре 1 з-ки на европейский патент № 269.599 от имени фирмы заявительницы) в присутствии 7,5 г іидроксида калия. Затем реакционную смесь охлаждают и ее растворяют в 10 мл воды. Водный раствор подкисляют до рН 3-4 45 путем добавления концентрированной соляной кислоты. Образующийся осадок фильтрую т , с у ш а т и э к с т ра г и р у ю т в ки п я щ е м изопропиловом спирте. Затем изопропило-вый спирт удаляют при пониженном давле- 50 нии, полученный кристаллический осадок перекристаллизовывают в 5 мл 1 н.водного раствора соляной кислоты. Получают 0,74 г хлоргидрата 2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензойной кислоты. Выход: 58%. 55 Температура плавления: 257°С (разл). Спектр ЯМР (ДМСО): дельта 4,08 (2Н, с), 6,09 (1Н, т), 7,37 (1Н, д), 7,52 (1Н, дд), 7,79 (1Н, дд), 9,05 (1Н, д). Анализ для C11H10N2O3 12 Рассчитано: С 51,87, Н 4,32, N U,0. Получено: 51,68, 4,03, 10,61. Это соединение используется как исходное в примере 1.А.4. Точно таким же способом получают следующее соединение: 2. 2-гидрокси-3-[1-(1 Н-имидазол-4ил)зтил]-бензойная кислота. Это соеди нение используется как исходное в при мере. 1.А.2. получено из 2-гидрокси-3-[(1Нимидазол-4-ил)этил]-бензолметанола с выходом 50%. Свободную кислоту получают путем нейтрализации хлоргидрата и перекристаллизации в воде. Температура плавления: 278-280°С. Анализ C12H12N2O3 (в %). Рассчитано: С 62,06, Н 5,17, N 12,07. Получено: 62,05. 5,37, 11,72. Используемый в качестве исходного продукта 2-гидрокси-3-[1-(1 Н-имидазол-4ил)этил]-бензолметанол был получен по методике, описанной в примере 6,5 заявки на патент европейский № 269599 на имя фирмы заявительницы. 3. 2-гидрокси-3-[1-(1 Н-имидазол-4ил)пентил]бензойная кислота (хлоргид рат). В течение 5 ч нагревают при 170°С 3,86 г (! 4,8 ммоль) 2-гидрокси-3-[1-(1 Н-имидазол4-ил)пентил]5ензолметанола (получается по методике, описанной в примере 6,6 заявки на европейский патент № 269599 от имени фирмы-заявительницы) в присутствии 22 г гидроксида калия. Затем реакционную смесь охлаждают и растворяют в 100 мл воды. Нерастворимый осадок отфильтровывают, фильтрат подкисляют до рН 10 путем добавления концентрированной соляной кислоты. Образующиеся соли фильтруют, а фильтрат окончательно подкисляют до рН 1. Хлоргидрат 2-гидрокси-3-[1-(1 Н-имидазол4-ил)пентил]бензойной кислоты выпадает в осадок. Его перекристаллизовывают из 100 мл изопропилового спирта. Получают 1,44 г продукта. Температура плавления: 23925ГС. Спектр ЯМР (ДМСО): дельта 0,83 (ЗН, т), 1,17-1,32 (4Н, м), 1,94-2,07 (2Н, м), 4,43 (1Н, т), 6,79 (1Н, т), 7,33 (1Н, д), 7,47 (1н, с), 7,69 (1Н,д), 8,84(1 Н, с). Это соединение используется как исходное в примере 1.А.З. В. Путем гидролиза соответствующих эфиров, полученных превращением Клайзена. 1. 3-[(1 Н-имидазол-4-ил)метил]-2-метоксибензойная кислота. В течение 3 ч с обратным холодильником нагревают 4,3 г (17,5 ммоль) метил-3[(1Н-имидазол-4-ил)метил]-2-метоксибензо 13 12835 ата (как получено в примере 1.В.1), растворенного в 20 мл метанола и 21 мл водного раствора (1 н.) гидроксида натрия. Метанол испаряют при пониженном давлении, водный раствор подкисляют до рН 5 путем до- 5 бавляют 21 мл водного-раствора (1 н) соляной кислоты. Осадок отфильтровывают, фильтрат концентрируют наполовину его объема и фильтруют новый осадок, который образуется. Два остатка промывают гекса- 10 ном и высушивают в вакууме. Получают 3,16 г 3-[(1 Н-имидазол-4-ил)метил]-2-метоксибензойной кислоты, практически чистой. Выход: 78%. Спектр ЯМР (ДМСО+СРзСООН): дельта 15 3,78(ЗН, с), 4,07 (2Н, с), 6,96-7,82 (4Н, м), 8,91 Полученный продукт используется как он есть для получения соответствующего бензоамида (пример 5.1). 20 Точно таким же способом получают следующее соединение. 2. 3-[(1 Н-имидазол-4-ил)метил]-2-н-пропоксибензойная кислота. Она получается из метил-3-{(1 Н-имида- 25 зол-4-ил)метил]-2-н-пропоксибензола, пол ученного в примере 1.В.З. Полученный продукт используется как он есть для пол учения соответствующего бензамида (при мер 5.2). 30 С. Путем гидролиза соответствующих эфиров, полученных по реакции ФриделяКрафта. 2-гидрокси-3-[1Н-имидазол-4-ил)метил]бензоймая кислота и 2-гидрокси-5-{(1 Н-ими- 35 дазол-4-ил)мегил]-бензойная кислота (хлоргидрат). Частями добавляют 181 г (1,35 моль) хлоргидрата 1Н-имидазол-4-метанола к смеси 156 мл (1,2 моль) 2-гидроксибензоата метила и 675 г полифосфорной кислоты, на- 40 гретой до 80°С. При этой температуре при хорошем перемешивании смесь выдерживают в течение 288 ч. Затем смесь разлагают на льду и экстрагируют 2 раза толуолом. Жидкую фазу подщелачивают до рН 9,5 45 путем добавления 790 мл водного насыщенного раствора гидроксида натрия. Осаждаемые минеральные соли отделяют фильтрованием и промывают метанолом. Этот метаноловый промывной раствор сое- 50 диняют с водной фазой и смесь концентрируют путем частичного удаления метанола. Затем раствор подщелачивают до рН 10,3 путем добавления Юн. водного раствора гидроксида натрия, далее производят на- 55 гревание при 100°С в течение 1,5 ч для омыления эфиров. Водный раствор нейтрализуют до рН 7,5 путем добавления 10 н. соляной кислоты, фильтруют на норите 14 и фильтрат испаряют при пониженном давлении. Остаток трижды разбавляют последовательно в смеси толуолэтанол и сушат азеотропной дистилляцией. Затем его частично растворяют в горячем метаноле, нерастворимые минеральные соли отделяют фильтрованием. Фильтрат испаряют при пониженном давлении, остаток вновь растворяют в минимальном количестве воды и производят очистку путем пропускания через колонку с Амберлитом 1R 93 (высота колонки 60 см, диаметр 8 см. эквивалент 2,64 моль). Избыток 1 Н-имидазол-4-метанола, а также еги полимеры элюируют водой (рН элюата меняется от 11,2 до 7,3). Затем элюирование продолжают водным раствором соляной кислоты (4%). Кислый элюат (9 л) доводят до рН 7,7 путем добавления водного насыщенного раствора гидроксида натрия, затем его выпаривают при пониженном давлении. Полученный остаток вновь высушивают путем азеотропной дистилляции и смесью толуолэтанол, затем его разбавляют в 1,6 л ацетонитрила. После этого фильтруют. Остаток на фильтре (129 г) хроматографируют на окиси кремния (800 г, 15 мкм), после предварительного нанесения на 300 г окиси кремния (0,2-0,5 мм) [элюант: смесь этилацетат-этанол = 75:25 об.]. Таким образом получают 5,99 г 2-гидрокси-3-[(1Н-имидазол-4-ил)метил]-бензойной кислоты. Температура плавления: 245-252°С (вода). Анализ СцНю№Оз(в %): Рассчитано: С 60,56, Н 4,59, N 12,04. Получено: 60,32, 4,69, 12,41. Одновременно получают 31 г 2-гидрокси-5-[(1Н-имидазол-4-ил)метил]-бензойной кислоты. Его хлоргидрат используется как исходный продукт в примере 1.А.1. он плавится при 254-258°С (метанол-этиловый эфир). Анализ C11H10N2O3 * НСІ (в %): Рассчитано: С 51,87, Н 4,32, N 11,0, С! 13,40. Получено: 51,65, 4,24, 10,45, 13,73. П р и м е р 3. Получение исходных 2-гидрокси-3-[(1Н-имидазол-4-ил)-алкил]-бензойнитрилов формулы (V). 1. 2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензонитрил. 1а. Этил-4-бензилокси 2-[(1Н-имидазоп4-ил)метил]-3~оксобутаноат. При 10°С добавляют за один раз 182 г (0,77 моль) этил-4-бензилокси-З-оксо-бутаноата к раствору 16,9 г (0,735 моль) натрия в 590 мл абсолютного этанола. 15 12835 При комнатной температуре смесь перемешивают в течение 45 мин, затем охлаждают до -45°С и добавляют туда за один раз раствор 53,6 г (0,35 моль) хлоргидрата 4хлорметил-Ж-имидазола в 300 мл абсолютного этанола. Смесь доводят до комнатной температуры, а затем перемешивают в течение 1 ч. После этого суспензию выпаривают досуха. Далее остаток разбавляют раствором 35 мл концентрированной соляной кислоты в 900 мл воды и производят несколько раз экстрагирование диэтиловым эфиром. Водную фазу нейтрализуют раствором 18 г гидроксида натрия в 200 мл воды и осуществляют многократное экстрагирование этилацетатом. Органические фазы промывают водой и водным насыщенным раствором хлорида натрия. Затем производят сушку над сульфатом натрия и выпаривают при пониженном давлении. Получают 107 г этил4-бензилокси-2-[(1Н-имидазол-4-ил)метил]3-оксо-буганоата практически чистого. Выход: 97%. Спектр ЯМР (ДМСО): дельта 1,11 (ЗН, т), 2,98 (2Н, м), 4,05 (1Н, q), 4,08(1 Н,м), 4,25 (2Н, дд), 4,47 (2Н, с), 6,75 (1Н, с), 7,25-7,39 (5Н, м), 7,47 (1Н,д). 1.6. Этил-4-бензилокси-3-гидрокси-2{(1Н-имидазол-4-ил)метил]-бутаноат. К раствору 101,2 г (0,32 моль) этил-4бензилокси-2-[(1Н-имидазол-4-ил)метил]-3 оксо-бутаноата в 600 мл этанола, охлажденному до -20°С, добавляют за один раз ледяной раствор 6,03 г (0,16 моль) боргидрида натрия в 25 мл воды. Смесь доводят до комнатной температуры и перемешивают в течение 1 ч. Затем добавляют 25 мл ацетона. Раствор выпаривают досуха и остаток разбавляют в 500 мл воды. Производят многократное экстрагирование этилацетатом. Органические фазы промывают водой и насыщенным водным раствором хлорида натрия. Высушивают над сульфатом натрия и выпаривают растворитель при пониженном давлении. Остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метанол-аммиак = 93,5:6:0,5 об.). Получают 95,8 г этил-4-бензилокси-З-гидрокси-2-[(1Н-имидазол-4-ил)метил]-бутаноата (смесь диастереоизомеров). Выход: 94%. Спектр ЯМР (СДСІ)з: дельта 1,15 и 1,16 (ЗН, 2т), 2,90-3,05 (ЗН,0 м), 3,51-3,58 (2Н, м), 3,96-4,11 (ЗН, 2q+iM), 4,51 и 4,53 (2Н, 2с), 6,73 и 6,75 (1Н, 2с), 7,25-7,36 (5Н, м). 1. с. Хлоргидрат 4-гидрокси-3-{(1 Н-имидазол-4-ил)метил}-дигидро-2-{3 Н)-фуран она. Производят гидрогенолиз 93,9 г (0,295 моль) этил-4-бензилокси-3-гидрокси-2-[(1 Нимидазол-4-ил)метил]-бутаноата, раство 16 ренного в 500 мл абсолютного этанола и 65 мл 6,8 н.раствора соляной кислоты в этаноле, в присутствии 5 г палладия на угле (10%-го), при давлении водорода 3,5 бар. 5 Затем катализатор отфильтровывают, а растворитель удаляют при 65°С при пониженном давлении. Получают 67,1 г хлоргидрата 4-гирокси-3-[(1 Н-имидазол-4ил)метил]-дигидро-2(ЗН)-фуранона (смесь 10 диастереоизомеров). Выход практически количественный. Полученный продукт используется как он есть на следующем этапе. 1. д. 3-[(1Н-имидазол-4-ил)метил]-2(5Н)15 фуранон. В течение 75 мин нагревают при 160°С при давлении 0,0013 мбар67,1 г (0,295 моль) хлоргидрата 4-гидрокси-3-{(1 Н-имидазол-4-ил) метил]-дигидро-2(ЗН)-фуранона. Охлаждают и 20 разбавляют 125 мл абсолютного этанола. Производят нейтрализацию путем добавления 70 мл 5 н.раствора аммиака в этаноле. Суспензию отфильтровывают и удаляют растворитель при пониженном давлении. Оста25 ток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метаноламмиак = 91,5:8:0,5 об.). После перекристаллизации из ацетонитрила получают 27,5 г 3[(1Н-имидазол-4-ил)метил}-2(5Н)-фуранона. 30 Выход: 53% (рассчитан на этапах 1.с и I.g вместе). Температура плавления: 123°С. Спектр ЯМР (СДСІз): дельта 3,63 (2Н, q), 4.79 (2Н. q), 6,90 (1Н, д), 7,25 (1Н, квинтип35 лет), 7,52(1 Н,д). 1.е. 2-гидрокси-3-[(1 Н-имидазол-4ил)метил]-бензонитрил. К суспензии 24,6 г (0,15 моль) 3-[(1Нимидазол-4-ил)метил]-2(5Н)-фуранона в 225 40 мл безводного акрилонитрила добавляют последовательно 63 мл (0,45 моль) безводного триэтиламина и 57 мл (0,45 моль) триметилхлорсилана. Смесь нагревают с обратным холодильником (72-74°С) в тече45 ние 4 ч. Затем испаряют при пониженном давлении. Остаток обрабатывают за один раз, 75 мл концентрированной бромистоводородной кислоты и выдерживают при 80°С в течение 2 мин. Затем раствор выливают на 50 лед, разбавляют путем добавления 300 мл этилацетата и 300 мл воды, потом нейтрализуется твердым бикарбонатом натрия. Затем фильтруют на целите и фильтрат многократно экстрагируют этилацетатом. Органи55 ческие фазы промывают водой, водным насыщенным раствором хлорида натрия, потом высушивают над сульфатом натрия и выпаривают при пониженном давлении. Полученный остаток растирается в порошок вдиэтиловом эфире. Получают22,8 г2-гидро 17 кси-3-[( 1 Н-имидазол-4-ил)метил]-бензонитрила. Выход: 76%. Спектр ЯМР (ДМСО): дельта 3,90 (2Н, с), 6,88 (1Н,т), 7,06(1 Н, с), 7,41 (1Н,дд), 7,47(1 Н, дд), 7,99 (1Н, д). Его хлоргидрат плавится при 245°С. 2. 2-гидрокси-3-[1-(1 Н-имидазол-4ил)этил]-бензонитрил. 2.а. Этил-4-бензилокси-2-[1 -(1 Н-имидазол-4-ил)этил]-3-оксо~бутаноат. Это соединение получают, как указано выше о п.1, а из этил-4-бензилокси-З-оксобутаноата и 4-(1-хлорэтил)-1Н-имидаола. Выход: 68% (смесь диастереоизомеров). Спектр ЯМР (СДСІз): дельта 1,10 и 1,22 (ЗН, 2т), 1,33 и 1,36 (ЗН, 2д), 3,60-3,72 (1Н, м), 3,93-4,20 (ЗН, м), 4,47 и 4,55 (1Н, 2с), 6,70 и 6,74 (1Н, с), 7,26-7,35 (5Н, м). 7,41 и 7,45 (1Н, с+д). 2.6. Этил-4-бензилокси-3-гидрокси-2-[1(1Н-имидазол-4-ил)этил]-бутаноат. Это соединение получается, как указано выше в п. 1.6, путем восстановления этил-4бензилокси-2-[1-(1Н-имидазол-4-ил)этил]-3оксо-бутаноата. Выход: 91% (смесь диастереоизомеров). Масс-спектр: 332 (М+), 314, 287, 211, 181, 135,95,91. 2.с. Хлоргидрат 4-гидрокси-3-[1-(1Нимидазол-4-ил)этил]-дигидро-2(ЗН)-фуранона. Это соединение получается, как указано выше в п.1.с, путем гидрогенолиза этил-4беизилокси-3-гидрокси-2-[1-(1Н-имидазол4-ил)этил]-бутаноата. Выход практически количественный. Получают смесь диастереоизомеров, которая используется как она есть на следующем этапе. 2.д. 3-[1-(1 Н-имидазол-4-ил)этил]-2(5Н)фуранон. В течение часа при 170°С при давлении 13,3 м бар нагревают 75,1 г, (0,32 моль) хлоргидрата 4-гидрокси-3-[1-(1 Н-имидазол-4ил)этил]-дигидро-2(ЗН)-фуранона в 30 мл этиленгликоля. Затем растворитель удаляют при давлении 0,0013 мбар. Остаток разбавляют в 300 мл абсолютного этанола и нейтрализуют путем добавления 63.2 мл 5 н.раствора аммиака в этаноле. Суспензию фильтруют, растворитель испаряют при пониженном давлении. Остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метанол-аммиак = 91,5:8:0,5 объем). После перекристаллизации из ацетонитрила получают 41,9 г 3-[1(1Н-имидазол-4-ил)этил}-2(5Н)-фуранона. Выход: 74% (рассчитан совместно на этапах 2.с и 2.д). 12835 18 Температура плавления: 127-129°С. Спектр ЯМР (ДМСО): дельта 1,40 (ЗН, д), 3,72 (1Н, q), 4,84 (2Н, т). 6,80 (1Н, т), 7,35 (1Н, q), 7,51 (1Н, с). 5 2.е. 2-гидрокси-3-[1-(1 Н-имидазол-4ил)этил}-бензонитрил. К суспензии 17,8 г (0,1 моль) 3-[1-{1Нимидазол-4-ил)этил]-2(5Н)-фуранона в 150 мл безводного акрилонитрила добавляют 10 последовательно 56 мл (0.4моль) безводного триэтиламина и 50,7 мл (0,4 моль) триметилхлорсилана. Смесь нагревают с обратным холодильником в течение 3,5 ч. Затем реакционную смесь испаряют при по-15 ниженном давлении. Остаток обрабатывают за один раз 50 мл концентрированной соляной кислоты и выдерживают при 80°С в течение 2 мин. Потом раствор выливают на лед, нейтрализуют водным насыщенным 20 раствором бикарбоната натрия и многократно экстрагируют этилацетатом. Органические фазы промывают водой и водным насыщенным раствором хлорида натрия, затем высушивают над сульфатом натрия и 25 испаряют при пониженном давлении. Остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-мета-ноламмиак - 93,5:6:0,5 об.). Получают 17,6 г 2гидрокси-3-[1-(1Н-имидазол-4-ил)этил]-бен 30 зонитрила, практически чистого. Выход: 83%. Температура плавления: 172°С. Спектр ЯМР (ДМСО): дельта 1,52 (ЗН, д), 4,21 (1Н, q), 6,86 (1Н, т), 7,02 (1Н, с), 7,41 (1Н, 35 дд), 7,44 (1Н,дд), 7,85(1 Н, с). П р и м е р 4. Получение 1-(1Н-имидазол4-ил)-алкил-бензамидов формулы (путем ре акции эфиров формулы (II) с азотосодержащим соединением формулы (!Н). 40 1. Хлоргидрат 2-гидрокси-3~[(1 Н-имидазол-4-ил)метил]-бензамида (А). Газообразный аммиак, осушенный над гидроксидом калия, пропускают через раствор 18.1 г (78 ммоль) метил-2-гидрокси-З-45 [(1 Н-имидазол-4-ил)метил]бензоата, полученного как в примерах 1.В.2 и 1.А.4, в 400 мл безводного метанола в течение ночи. Затем в течение 2 ч нагревают с обратным холодильником. Реакционную смесь испа-50 ряют при пониженном давлении и остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метаноламмиак - 89,5:10:0,5 об.). Получают 16,6 г 2гидрокси-3-[(1Н-имидазол-4-ил)метил]-бен-55 замида. Выход: 98%. Температура плавления: 197,6°С. Анализ C11H11N3O2 (%): Рассчитано: С 60,83, Н 5.07. N 19,35. Получено: 60,91, 5.06. 19.32. 1 19 12835 Амид, обработанный в этаноле с 1,2 эквивалентом соляной кислоты образуют хлоргидрат с выходом 73%. Температура плавления: 287,8°С. Анализ C11H11N3O2 HCI (в %): Рассчитано: С 52,07, Н 4,73, N 16,57, CI 14,00. Получено: 52,04. 4,76, 16.54, 13,94. 2. 2-гидрокси-5-[( 1 Н-имидазол-4-ил)метил]-бензамид (G). Это соединение получают, как описано выше в п. 1, из хлоргидрата этил-2-гидрокси5-[(1 Н-имидазол-4-ил)метил]-бензоата (полученного, как в примере 1.А.1). Реакционную смесь перемешивают в течение 3 суток при температуре окружающей среды. Продукт реакции очищают хроматографически на силикат еле (элюант: дихлорметан-метанол = 85:15 об.). Температура плавления: 180-185°С (изопропиловый спирт). Анализ: СііНцМзОг(в %): Рассчитано: С 60,83, Н 5,07, N 19,35. Получено: 60.71, 5,25, 19,01, 3. Хлоргидрат 2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-Ы-метилбензамида (с). В автоклаве при 75°С в течение 3 ч нагревают 6,96 (30 ммоль) метил-2-гидрокси-3-[(1 Н-имидазол-3 -ил)метил]-бензоата (полученного в примере 1.В.2) и 60 мл метиламина в растворе в 350 мл этанола. Смесь выпаривают при пониженном давлении. Остаток разбавляют водой и проводят экстрагирование 3 раза этилацетатом. Органическую фазу сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метанол-аммиак = 91,5:8:0,5 об.). После испарения растворителей полученный продукт кристаллизуют в этилацетате. Получают 4,95 г 2-гидрокси-3[(1Н-имидазол-4-ил)метил]-М-метилбензамида. Выход: 71%. Амид, обработанный в смеси этанолпростой эфир, с 1,2 эквивалентными соляной кислоты дает 4,7 г хлоргидрата. Выход: 59%. Температура плавления: 233,9°С. Анализ C12H13N3O2 • HCI (в %): Рассчитано: С 53,83, Н 5,23, N 15,70. Получено: 53,74, 5,17, 15,55. 4. 2-гидрокси-3-[(1 Н-имидазол-4-ил)метилрбенаогидразид (D)_. В течение 13 ч с обратным холодильником нагревают 10 г (43,1 ммоль) метил2-гидрокси-3-[(1Н-имидазол-4-ил)метил]бензоата (получен, как в примере 1.В.2) и 4,31 г (86,2 ммоль) гидрата гидразина, 20 растворенного в 100 мл метанола. Добавляют 2,15 г (43,1 ммоль) гидрата гидразина, нагревание продолжают в течение 24 ч с обратным холодильником. Затем смесь вы5 паривают при пониженном давлении. Остаток разбавляют в 100 мл воды (рН 8). Этот раствор насыщают хлоридом натрия и проводят экстрагирование этилацетатом. Органическую фазу высушивают над 10 сульфатом натрия и растворитель выпаривают при пониженном давлении. Остаток кристаллизуют в этаноле. Получают 6,1 г 2-гидрокси-3-[(1Н-имидазол-4-ил)метил]бензогидразида, который рекристаллизуют 15 из метанола. Выход: 61%. Температура плавления: 189,7°С. Анализ C11H12N4O2 в %: Рассчитано: С 56,88, Н 5,21, N 24,13. 20 Получено: 56,91, 5,24, 24,00. 5. Хлоргидрат 2-гидрокси-3-[1-(1Н-имидазол-4-ил)этил]-бензамида (В). В течение 90 ч перемешивают при комнатной температуре и в присутствии катали25 тического количества 20 мг метилата натрия, раствор 2 г этил-2-гидрокси-3-[1(1 Н-имидазол-4-ил)этил]-бензоата (полученного, как в примере 1.А.2) в 50 мл метанола, через который пропускают ток газообразно30 го аммиака. Затем метанол испаряют, а остаток очищают хроматографически на силикагеле (элюант: дихлорметан-метаноламмиак = 8,5:1:0,5 об.). Полученный продукт переводят в хлор35 гидрат в этанольном растворе хлороводородной кислоты, в присутствии диэтилового простого эфира. Получают 1,2 г хлоргидрат 2-гидрокси-3-[1 -(1 Н-имидазол-4-ил)этил]бензамида. 40 Выход: 68%. Температура плавления: 240-243°С. Анализ C12H13N3O2 • HCI (в %). Рассчитано: С 53,83, Н 4,86, N 15,70, CI 13,27. 45 Получено: 54,0, 4,88, 15,73, 13,17. После хроматографической очистки на силикагеле полученный амид разделяют на два энантиомера при помощи хроматографии на хиральной фазе альфа-гликопроте50 ина (элюант: изопропиловый спирт буферный фосфатный раствор 0,02М, рН 7, 1: 99 об.). Затем каждый из энантиомеров амида превращают в соответствующий хлоргидрат указанным способом. Таким 55 образом получают почти равные количества: а) хлоргидрата гидратированного (+)-2гидрокси-3-[-(1Н-имидазол-4-ил)этил]-бензамида (р). Температура плавления: 107,8°С (вода). 21 [ a ]&5 * +82,04° (С » 1, метанол). Спектр ЯМР (ДМСО): дельта 1,57 (ЗН, д), 3,30(5Н,м), 4,56 (1H,q), 6,83 (Ж, т), 7,23 (Ж, дд),7,40(1Н,с),7,82(1Н,дд),7,92(1Н,м),8,51 (Ж,м), 8,98 (Ж, д), 13,5-14,5 (Ж, м); б) хлоргидрат гидратированного (-)-2гидрокси-3-[1 -(1 Н-имидазол-4-ил)этил]-бензамида. Температура плавления: 107,4°С (вода). [ а 3&5 -79,13° (С = 1, метанол). Спектр ЯМР, идентичен указанному. 6. 2-гидрокси-5-[(1 Н-имидазол-4-ил)метил]-г\1-метилбензамид. Это соединение получают по методике, описанной в п.З, из хлоргидрата этил2-гидрокси-5[(1Н-имидазол-4-ил)-метил}-бензоата (получен по примеру 1 .А. 1) и метиламина. Предварительно выделяют основание из хлоргидрата небольшим избытком (1,2 эквивалента) метилата натрия. Полученный продукт после выпаривания растворителя хроматографически очищают на силикагеле (элюант: смесь дихлорметан-этанол-аммиак = 97,5:12:0,5 объем). Выход: 67% (после перекристаллизации в этаноле). Температура плавления: 219,5°С. Анализ C12H13N3O2 (в %): Рассчитано: С 62,32, Н 5,67, N 18,17. Получено: 62,23, 5,65, 18,06. Спектр ЯМР (ДМСО): дельта 2,80 (ЗН, д), 3,27 (Ж, с), 3,76 (2Н, с), 6,67 (Ж, с), 6,80(Ж, д), 7,23 (Ж, дд), 7,49 (Ж, с), 7,87(Ж, д), 8,80 (Ж, с), 11,8 (Ж, с). 7. 1-[2-гидрокси-5-[(1 Н-имидазол-4ил)метил]-бензоил]-пиролидин. В течение 30 мин нагревают с обратным холодильником 300 мг хлоргидрата этил-2гидрокси-5-[(1Н-имидазол-4-ил)метил]-бензоата (полученного по примеру 1.А.1), растворенного в 5 мл пиролидина. Избыток амина удаляют при пониженном давлении, остаток хроматографически очищают на окиси кремния (элюант: смесь дихлорметанэтанол-аммиак = 88,5:11:0,5 об.). Получают 120 мг 1-[2-гидрокси-5~[(1Н-имидазол-4ил)метил}-бензоил]-пиролидина. Температура плавления: 96-98°С. Выход: 36%. Спектр ЯМР (ДМСО): дельта 1,81 (4Н, с), 3,35 (Ж, с), 3,74 (2Н, с), 6,7 (Ж, с), 6,78 (1Н, д), 7,04 (Ж, д), 7,08 (Ж, дд), 7,50 (Ж, с), 9,85 (Ж, с), 11,8 (Ж, с). 8. 5-трет-бутил-2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-бензамид (J). Это соединение получают, как указано в а.2, из хлоргидрата метил-5-трет-бутил-2гидрокси-3-[(1Н-имидазол-4-ил)метил]-бензоата (как получено в примере 1.С.1). 12835 22 Выход: 22,7%. іемпература плавления: 209-211 °С (тетрагидрофуран). Анализ C15H19N3O2 (в %): Рассчитано: С 65,91, Н 7,00, N 15.37. 5 Получено: 65,42, 7,06, 15,14. 9; Хлоргидрат 2,6-дигидрокси-3-[(1Нимидазол-4-ил)метил]-бензамид (I). Это соединение получают, как описано в п.1, из метил-2,6-дигидрокси-3-[(1Н-ими-10 дазол4-ил)метил]-бензоата (который получается, как в примере 1.С.2). Газообразный аммиак пропускают в течение 3 ч через раствор, находящийся при комнатной температуре, затем раствор испаряют, остаток 15 перекристаллизовывают в диоксане. Выход: 81%. Амид, обработанный в этаноле с 1,2 эквивалентами соляной кислоты, образует хлоргидрат с выходом 73%. Температура 20 плавления 290,3°С (разл.). Анализ C11H11N3O3 • НСІ (в %): Рассчитано: С 48.99, Н 4.49, N 15,58, СГ 13,15. Получено: 48,79, 4,43, 15,44. 13,16. 25 10. Хлоргидрат 2,6-дигидрокси-3-[1-(Жимидазол-4-ил(этил]-бензамид. Это соединение получают, как описано в п.9. из метил-2,6-дигидрокси-3-[1-(1Н-ими-дазол-4ил)этил]-бензоата (получается по 30 примеру 1.С.З). Остаток перекристаллизовывают в толуоле. Выход: 85%. Амид обрабатывается в смеси этанолпростой эфир с 1.1 эквивалентом соляной 35 кислоты и образуют хлоргидрат. Выход: 77% (после перекристаллизации в воде). Температура плавления: 288,8°С (разл.). Анализ C12H13N3O3 • НСІ (в %): Рассчитано: С 50,80, Н 4,97, N 14,81, СГ 40 12,50. Получено: 50,68, 4,92, 14,67, 12.09. 11. Хлоргидрат 6-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-2-метилбензамида. В автоклаве в течение 72 ч при 60°С 45 нагревают раствор 6,1 г (23,4 ммоль) этил-6гидрокси-3-[(Ж-имидазол-4-ил)метил]-2-метилбензоата (получается в примере 1.С.4) в 400 мл жидкого аммиака. Затем смесь выпаривают при пониженном давлении, пол-50 ученный остаток хроматографически очищают на силикагеле (элюант: смесь зтилацетат-этанол-аммиак = 80:20:0,5 об.). Получают 3,3 г 6-гидрокси-3-[(1Н-имидазол-4ил)метил]-2-метилбензамида. 55 Выход: 61%. Амид, обработанный в этаноле с 1,1 эквивалентом соляной кислоты, образует хлоргидрат. Выход: 66% (после перекристаллизации в воде). Температура плавления: 262,8°С. 23 12835 Анализ C12H13N3O2 * НС! (в %): Рассчитано: С 53,84, Н 5,27. N 15.70, СГ 13.24. Получено: 54,22, 5,29, 15,74, 13,12. 12. Хлоргидрат 2-гидрокси-3-[(1Н-имидазол-4-ил)метил]-6-метилбензамида (К). Это соединение получают, как описано в п. 11, из 2 г (7,68 ммоль) этил-2-гидрокси-З[(1Н-имидазол-4-ил)метил]-6-метилбензоата (получается по примеру 1.С.4) и 12 мл жидкого аммиака. Затем нагревают при 60°С в течение 24 ч. далее смесь выпаривают при пониженном давлении. Сырой амид обрабатывают без предварительной очистки 1,1 эквивалентом соляной кисл оты в смеси этанол-диэтиловый эфир. Хлоргидрат перекристаллизовывают в тетрагидрофуране. Выход: 49%. Температура плавления: 1С4,8°С (смесь аморфного и кристаллического продуктов). Анализ C12H13N3O2 * HCI (в %): Рассчитано: С 53,84, Н 5,27, N 15,70, СГ 13,24. Получено: 52,55, 5,30, 15,35, 13,92. 13. Хлоргидрат-моногидрат Ы,2-дигидрокси-3-[(1Н-имидазол-4-ил)метил]-бензамида (м). К смеси 4,1 г (25 ммоль) сульфата гидроксиламина и 25 г кускового льда добавляют каплями раствор 5 г (125 ммоль) гидроксида натрия в 15 мл воды. Когда температуру доведут до 0°С, добавляют 0,5 г твердого сульфита натрия, затем 6,15 г (25 ммоль) этил-2-гидрокси-3-[(1Н-имидазол-4-ил)мет ил]-бензоата, после полного растворения реакционную смесь нейтрализуют 20,8 мл водного 6 н.раствора соляной кислоты. Полученный белый осадок фильтруют, промывают водой и хроматографически очищают на силикагеле (элюант: смесь дихлорметан-метанол-уксусная кислота-вода = 76:20:2:2 об.). Полученный ацетат после выпаривания растворителей разбавляют в 30 мл воды, к которой добавляют 5 мл концентрированной соляной кислоты. Хлоргидрат Ы,2-дигидрокси-3-[(1Н-имидазол-4-ил)метил]-беизамида осаждается в виде моногидрата. Получают 4,8 г чистого продукта. Выход: 55%. Температура плавления: 240°С (разлож.). Анализ C11H11N3O3 • НС! • НгО (в %): Рассчитано: С 45,92, Н 4,90, N 14,61, СГ 12,32. Получено: 46,33, 4,63, 14,69, 12,57. Этил-2-гидрокси-3-[(1Н-имидазол-4-ил) метил]-бензоат, используемый как исходное вещество, получается по методу, описанному в примере 7 заявки на европейский патент № 269599 от имени фирмы-заявительницы. 24 14.2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-4-метилбензамид (L). Это соединение получают, как описано в п.2, из бромгидрата метил-2-гидрокси-З5 [(1 Н-имидазол-4-ил)метил]-4-бромбензоата (полученного по примеру 1.С.5). Продукт ре акции очищают хроматографически на сили кагеле (элюант: см есь дихлорметан-метанол-аммония = 95:5:0,5 об.). 10 Выход: 44%. Температура плавления: 172-178°С (диэтиловый эфир). Анализ C12H13N3O2 (в %): Рассчитано: С 62,33, Н 5,62, N 18,18. 15 Получено: 62,42, 5,61, 18,08. 15. Хлоргидрат 2-гидрокси-3-[(1Н-имидазол-4-ил)пентил]-бензамида. Это соединение получают, как описано в п.5, из этил~2-гидрокси-3-[1-(1Н-имидазол-20 4ил)пентил]-бензоата (полученного по примеру 1.А.З). Полученный остаток после испарения растворителя растворяется в 2,5 н.растворе соляной кислоты в этаноле, хлоргидрат 2гидрокси-3-[1-(1 Н-имидазол-4-ил)пен-25 тил]бензамида осаждается при добавлении диэтилового эфира. Выход: 50%. Спектр ЯМР (ДМСО): дельта 0,83 (ЗН, т), 1,08-1,34 (4Н, м), 3,3-3,5 (2Н, м), 4,44 (1Н, т), 6,82 (1Н, т), 7,37-7,40 (2Н, с+д), 7,82 (1Н, д), 30 8,97 (1Н, с). Точно таким же способом получают следующие соединения. 16.2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-ІЧ,М-диметилбензамид. 35 Температура плавления: 208°С. Анализ C13H16N3O2 (в %): Рассчитано: С 63,67, Н 6,12, N 17,14. Получено: 63,58, 6,06, 17,09. 17. Хлоргидрат 2-гидрокси-М-(2-гидро-40 ксиэтил)-3-[(1 Н-имидазол-4-ил)метил]-бензамида (Е). Температура плавления: 200,6°С. Анализ для C13H15N3O3 • НС! (в %): Рассчитано: С 52,44, Н 5,38, N 14,12. 45 Получено: 52.49, 5,36, 13,98. П р и м е р 5. Получение 1-(1Н-имидазол-4-ил)алкил-бензамидов формулы (I) путем реакции кислот формулы (IV) с азотосодержащим соединением формулы (III). 50 1. Хлоргидрат 3-[(1 Н-имидазол-4-ил)метил]-2-метоксибензамида (F). Охлаждают при 0°С суспензию 3,1 г (13,4 ммоль) 3-[(1 Н-имидазол-4-ил)метил]-2метоксибензойной кислоты (полученной по 55 примеру 2.В.1) и 4,06 г (40,2 ммоль) триэтиламина в 30 мл сухого дихлорметана. Добавляют 4,36 г (40,2 ммоль) этилхлорформиата и растворенного в 10 мл сухого дихлорметана. После добавления перемешивают при 25 12835 0°С еще в течение получаса, а затем - в течение получаса при комнатной температуре. После этого через реакционную смесь в течение ночи пропускают газообразный аммиак, просушенный над гидроксидом калия. 5 Затем смесь нагревают в течение получаса с обратным холодильником, испаряют растворитель при пониженном давлении и остаток хроматографически очищают на силикагеле (элюант: смесь дихлорметан-ме- 10 танол-аммония = 89:10:1 об.). Получают 2,9 г 3[(1 Н-имидазол-4-ил)метил]-2-метоксибен замида, который кристаллизуется в ацетонитриле. Выход: 95%. 15 Амид, обработанный с 1,2 эквивалента соляной кислоты в этаноле, образует хлоргидрат. Температура плавления: 160,5°С (изолропиловый спирт). Анализ для C12H13N3O2 * HCI (в %): 20 Рассчитано: С 53,84, Н 5,27, N 15,73. Получено: 53,94, 5,30, 15,81. Точно таким же способом получают следующее соединение. 2. 3-[(1Н-имидазол-4-ил)метил]-2-н-про- 25 поксибензамид. Это соединение получают из 3-[(1Н-имидазол-4-ил)метил]-2-н-пропоксибензойной кислоты (полученной по примеру 2.В.2) с выходом 52%. Температура плавления: 161 °С. 30 Анализ для C14H17N3O2 (в %): Рассчитано: С 64,86, Н 6,56, N 16,22. Получено: 64,93, 6,60, 16,14. П р и м е р 6. Получение 1-{1Н-имидазол-4-ил)алкил-бензамидов формулы (1) пу- 35 тем гидролиза 2-гидрокси-3-[1-(1Н-имидазол-4-ил)алкил]-бензонитрилов формулы (V). 1. 2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-бензамид. Перемешивают до полного растворения 40 13,1 г (66 ммоль) 2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-бензонитрила (получен по примеру 3.1) в 50 мл водного раствора серной кислоты (80 об.%). Затем нагревают в течение 3 ч при 65°С. Реакционную смесь 45 выливают на лед и нейтрализуют бикарбонатом натрия. Фильтруют и многократно экстрагируют фильтрат этилацетатом. Органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над 50 сульфатом натрия и выпаривают при пониженном давлении. Остаток очищают хроматографически на силикагеле (элюант: смесь дихлорметан-метанол-аммиак = 84:15:1 об.). Получают 9,8 г 2-гидрокси-3-[(1 Н-имидазол- 55 4-ил)метил]-бензамида, идентичного продукта, полученному в примере 4.1. Выход: 68%. 2. 2-гидрокси-3-[1-(1 Н-имидазол-4ил)этил]-бензамид. 26 В течение 3 ч при 65°С перемешивают суспензию 1.07 г (5 ммоль) 2-гидрокси-3-[1(1 Н-имидазол-4-ил)этил]~бензонитрила (получен по примеру 3,2) в 4 мл водного раствора серной кислоты (80 об.%). Реакционную смесь затем выливают на лед и нейтрализуют бикарбонатом натрия. Проводят многократное экстрагирование этилацетатом. Органические фазы сушатся над сульфатом натрия, растворитель выпаривают при пониженном давлении. Остаток разбавляют в 50 мл 6 н.водного раствора соляной кислоты, а затем нейтрализуют при помощи 1 н.водного раствора гидроксида натрия. Образуемый осадок фильтруют промывают водой и диэтиловым эфиром и сушат в вакууме. Получают 0,7 г 2-гидрокси-3-[1-(1 Н-имидазол-4-ил)этил]-бензамида, практически чистого. Выход 70%. Хлоргидрат этого продукта идентичен продукту, полученному в примере 4.5. 3. Хлоргидрат 2-тидрокси-3-[(1Н-имидазол-4-ил)метил]-бензамида. Добавляют 2 г 2-гидрокси-3-[1Н~имидазол-4-ил)метил]-бензонитрила (получают по примеру 3,1) и 4 мл воды в 40 мл метанола, который был предварительно насыщен газообразной соляной кислотой при -10°С. Смесь перемешивают при комнатной температуре в течение 24 ч. Затем концентрируют раствор при помощи пониженного давления и нагревают остаток при 75°С в течение 3 ч. Далее производят дважды перекристаллизацию остатка в воде. Получают 1,5 г хлоргидрата 2-гидрокси-3-[(1Н-имида~ зол-4-ил)метил]-бензамида, идентичного продукта, полученному в примере 4.1. Выход: 59%. Для фармакологических испытаний были взяты следующие соединения в соответствии с настоящим изобретением: хлоргидрат 2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-бензамида (соединение А), хлоргидрат 2-гидрокси-3-[1-(1Н-имидазол-4-ил)этил]-бензамида (соединение В), хлоргидрат 2-гидрокси-3-[(1 Н-имидазол-4-ил)метил]-Ы-метилбензамида (соединение С). 2-гидрокси-3-[(1Н-имидазол-4-ил)метил] -бензогидразид (соединение D), хлоргидрат 2-гидрокси-Г\1-(2-гидроксиэтил)-3-[(1Н-имидазол-4-ил)метил]-бензамида (соединение Е), хлоргидрат-3-[(1Н-имидазол-4-ил)метил}-2-метоксибензамида (соединение F), 2-гидрокси-5-[(1Н-имидазол-4-ил)метил] -бензамид (соединение G), хлоргидрат 2,6-дигидрокси-3-[(1 Нимидазол-4-ил)метил]-бензамида (соедине ние I), _ 27 12835 5-трет-бутил-2~гидрокси-3-{(1Н-имидазол-4-ил)метил]-бензамид (соединение J), хлоргидрат 2-гидрокси-3-[(1 Н-имидазол-4-ил)мет ил]-6-метил-бензамида (соеди нение К) 5 2-гидрокси-3-[(1Н-имидазол-4-ил)метил] -4-метилбензамид (соединение L), хлоргидрат-моногидрат N, 2-дигидрокси-3-[(1Н-имидазол-4-ил)метил]-бензамида (соединение М). 10 хлоргидрат (+)-2-гидрокси-3-[1-(1 Нимидазол-4-ил)этил]-бензамид (соединение Р). 1. Антиишемическая сердечная актив ность. 15 а). Модель острой сердечной недостаточности у живой собаки. У живой и подключенной к аппарату собаки (пневматический манжет вокруг отходящей нижней коронарной артерии, 20 внугрисердечные электроды) в течение 6 мин, при помощи пневматических манжет осуществлялась закупорка коронарной артерии Вследствие уменьшении поступления кислорода это закупоривание вызывает 25 миокардическую ишемию, которая выраже-нч на кардиограмме воспроизводимым и количественным увеличением сегмента ST. Антиишемическое действие исследуемого соединения зыражается в уменьшении ве- 30 личины сегмента ST. В табл.1 показана доза DE20 мкмоль/кг для исследованных соединений, которая при внутривенном введении группе из 10 животных вызывает уменьшение в среднем на 35 20% роста сегмента ST для всех животных. В качестве эталонного вещества использо-взлея 1-(изопропиламино)-3-(1-нафтилокси)-2пропанол (или пропанолол). Из эгой таблицы следует, что соеди- 40 нения в соответствии с изобретением имеют хорошую антиишемическую активность. б). Тест нагрузками на бегущей дорож ке. 45 В этих испытаниях бралась группа из не менее 4 собак, подключенных к аппаратуре (внутри сердечные электроды), у которых на уровне коронарной артерии имеется органический стеноз. Этот стеноз провоцирует 50 дисбаланс между потребностью в кислороде и его полу іением, когда животное получает нагрузку. Этот дисбаланс проявляется на электрокардиограмме возрастанием сегмента ST. При испытаниях собака бежит со 55 скоростью 12 км/ч по бегущей дорожке, которая имеет наклон 15°. Эта максимальная нагрузка длится в течение 1 мин. При испытаниях регистрируют возрастание сегмента ST и естественный рост частоты сердечных 28 сокращений. Опыт повторяют не менее 4 раз и среднее из полученных величин берут как репер (100%) для группы животных. Затем животных оставляют в покое в течение минимум 24 ч, после чего проводят исследование с нагрузкой при воздействии исследуемого соединения. Исследуемое соединение вводят медленно внутривенно (в течение 1 мин) за 5 мин перед новым исследованием с нагрузкой. В ходе исследований регистрируют изменения тех же параметров. В табл.2 приведены для указанной дозы (мкмоль/кг) среднее наблюдаемое уменьшение сегмента ST (в %) и частота сердечных сокращений (ударов/мин) по сравнению с величинами, полученными при начальных измерениях. Из этой таблицы следует, что соединения в соответствии с изобретением имеют хорошую антиишемическую активность, это видно по сильному уменьшению сегмента ST, то же наблюдалось и для пропранолола, но при более высокой дозе. Кроме того, в отличие от пропранолола, который одновременно вызывает сильное уменьшение сердечного ритма в ходе опыта и является нежелательным и даже вредным для выдерживания нагрузки, соединения в соответствии с изобретением не препятствуют естественному возрастанию частоты сердечных сокращений в ходе опыта. Следовательно, эти вещества способствуют правильный адаптации частоты сердечных сокращений к нагрузке и препятствуют ишемии. 2. Антиишемическая церебральная активность. а). Общая постоянная церебральная ишемия у крыс. Мужские особи крыс Вистора (200-250 г) анестезируют путем ингаляции (от 1 до 5%), находящегося в смеси N2O-O2 (70:30). Две общие сонные артерии одновременно перевязывают вблизи соединения внутренней сонной и наружной сонной артерий. Испытуемое соединение вводят интраперитонально, первый раз за 30 мин перед перевязыванием и затем от 30 до 270 мин после перевязывания. На следующий день и через день определяют невралгический дефицит выживших животных. В этих опытах принимают во внимание сенсорно-двигательные функции: спонтанная подвижность, рефлексы хватания, реакции на индивидуальную установку и при потере опоры, рефлексы на изгиб лап. рефлекс распрямления и тест на подвешивание за хвост. Максимальный возможный показатель для животного, не имеющего ишемии, равен 17. 29 12835 В табл.З даны результаты для соединения А, введенного интраперитонально при дозе 0,76 мкг/кг (3,2 наномоль) - средних значениях для неврологических показателей, определенных для оставшихся в живых 5 животных из контрольной группы и для об работанной группы; эти показатели регистрируют через 2 дня после перевязывания. Статистическое значение (Р) наблюдаемой разницы между этими величинами опреде- 10 ляют по месту Ман-Витнея. Из этой таблицы следует, что соединение А при очень малой дозе у обработанных животных значительно уменьшает неврологический дефицит, вызванный ишемией. 15 б). Односторонняя многоочаговая церебральная ишемия у крыс. У мужских крыс Спраха-Доулея SPF возрастом от 8 до 9 недель вызывают постоянную одностороннюю церебральную 20 ишемию (или эмболизацию) путем введения в правую сонную аретрию 2000 микросфер. Исследуемое соединение вводят в пер вый раз за 30 мин до и второй раз через 30 25 мин после эмболизации, а контрольной группе животных вводят только физиологи ческую сыворотку. Затем крыс оставляют в покое. Через б дней после восстановления измеряют у выживших животных: 30 1) остаточный неврологический дефи цит - при помощи теста на позу и на пере движение животных (тест А, максимальный показатель 4). Этот тест оценивают: а) ненормальное положение задних ног, 35 б) боковой наклон тела при движении, с) продольный изгиб тела, относящийся к той же стороне, д) ненормальная походка. 2) сенсорно-двигательные функции 40 (спонтанная подвижность, хватательный ре флекс, реакции на визуальную установку, потеря опоры). Тест Б, максимальный пока затель 10. 3) боковую сенсорно-двигательную ре- 45 акцию (относится к противоположной сто роне). Тест С, максимальный показатель 3. Ее определяют путем комбинирования измерений рефлекса визуальной установки, рефлекса ориентирования головы к боково- 50 му звуковому раздражителю и кожного подошвенного рефлекса. 4) Тактильное ослабление левой сторо ны (тест D, максимальный показатель 300). В противоположность другим указанным те- 55 стам для которых дефицит будет тем ярко выражен, чем выше показатель, в данном тесте дефицит будет тем больше проявляться, чем ближе показатель к 300. ЗО На 7 день восстановления наблюдается отек в различных, относящихся к той же стороне церебральных структурах. В табл.4 для соединения А, вводимого интраперитонально при дозе 0,76 мкг/кг (3,2 наномоль/кг), приводятся результаты для тестов А, В, С, D, т.е. среднее из неврологических показателей, определенных для выживших животных из контрольной группы и обработанной группы, через 6 дней после восстановления. В таблице также указывается среднее изменение (в г) веса тела на 7 день после восстановления. Статическое значение (Р) разницы между средними расчетными величинами для контрольных животных и обработанных животных оценивают по тесту Ман-Витнея. Из таблицы следует, что соединение А значительно ослабляет неврологический и поведенческий дефицит, созданный ишемией, и улучшает вес обработанных животных. В табл.5 приведены данные по количеству воды (среднее в процентах), содержащейся в различн ых цереб ральных структурах, относящихся к одной стороне, у контрольных животных и обработанных животных, выживших на 7 дней после эмболизации. Полученные данные показывают, что лечение соединением А значительно уменьшает отек в различных церебральных структурах, относящихся к одной стороне. 3. Агонистические а2-адронергические свойства. а) Конкурентное связывание с радиоактивным лигандом. Опыты по соревновательному фиксированию имеют целью измерить средство, которое имеет соединение в соответствии с данным изобретением, по отношению к (Х2-адренергическим рецепторам. Эти классические эксперименты определяют соревновательности фиксирования на б?2-эдренергических рецепторах исслед уемого соед инения радиолиганда, которым в данном случае является (ЗН) клонидин, который известен как селективный «г-адренергический агонист. Кривые смещения фиксирования ( Н) клонидина определяют для 9 концентраций соединения А в пределах от 10" до 10* (моль/л) и для трех различных мембранных препаратов мозга крыс. Образцы инкубируют в течение 30 мин, затем фильтруют под вакуумом на фильтре Wharman GF/B. Фильтры трижды промывают 5 мл буферного раствора трис-HCI (рН 7,5 при 0°С), затем сушат в течение минуты. Радиоактивность измеряют в среде Econofluor (NEN Corp.) (3H) клонидин (25,5 С1/ммоль). 31 12835 32 доказывают, что соединения в соответствии Поставляется фирмой AMERSAAM. с изобретением воздействуют специфичеСродство соединения А к «г-адренергиским механизмом через «2-агонистическое ческим рецепторам, рассчитывают по кри3 действие. вой смещения ( Н) хлонидина. Оно вырас). Стимулирование тонкой кишки моржается в концентрациях (ICso в моль/л) сое- 5 ской свинки. динения А, которые необходимы, чтобы полФрагменты продольных мышц, приучить торможение 50% фиксирования крепленных к индикатору изометрической радиолиганда на рецепторе. Полученные силы, погружают в раствор Тирода и натягиданные показывают,'что соединение А имеет значительное сродство к О£-адренерги- 10 вают с силой 1 г. Электрическое стимулирование парасимпатических нервов, относяческим рецепторам: щихся к фрагментам тонкой кишки, вызыва9 ет сокращение мышцы. Это сокращение ICGO « 8,90 ± 0,72 10" моль/л. уменьшается в присутствии пресинаптиче0) Стимулирование изолированного 15 ского 2-агониста, уменьшение зависит от концентрации используемого агониста. предсердия морской свинки. Этому эффекту противодействует одновреВыделение норадреналина на уровне менное присутствие «2-антагонистическонервных окончаний регулируется механизго соединения, например альфаиохимбин. мам ретроконтроля пресинаптическими 0*220 Исследуемые соединения проверялись при адренергическими рецепторами. Этот возрастающих концентрациях от 10"10до 10" 3 механизм был доказан на предсердии мормоль/л. Определяют концентрацию (ICso в ской свинки. Электрическая стимуляция моль/л), которая уменьшает на 50% интенизолированного предсердия морской свинсивность сокращения мышцы. ки приводит к выделению норадреналина, что проявляется в возрастании частоты со- 25 В табл.7 приведены концентрации ICso (в моль/л) для соединений в соответствии с кращений сердца (тахикардия). Эта тахикаризобретением. Эти результаты показывают, дия тормозится «2-агонистом, например что эти соединения достаточно активны при клонидином, в пропорции, которая зависит очень малых концентрациях. от дозы используемого агониста. Действие 2-агониста может быть ограничено в присут- 30 В присутствии альфа-иохимбина при его концентрации 10 моль/л концентраствии 2-специфического антагониста, нация соединений, например А или В, котопример альфаиохимбина. Активность In рая требует для уменьшения интенсивvitro соединений в соответствии с данным ности сокращения мышцы на 50%, будет изобретением по отношению к пресиноптическим о*2-адренергическим рецепторам 35 более высокая и превышает 10 моль/л, что является дополнительным подтвержбыла исследована на изолированном преддением того, что соединения в соответстсердии морской свинки, электрически стивии с изобретением воздействуют на уровне мулированном по известной методике. пресинаптических ф-іздренергических преИсследуемое соединение тестируют при возрастающих концентрациях от 10"10 40 паратов. 4. Диуретическая активность. до 10"3 ммоль/л. Определяют концентраДиуретическая активность соединений в цию (Юзов моль/л, которая вызывает уменьсоответствии с изобретением определяшение на 30% максимальной тахикардии, лась у собак Бигля (6 мужских особей и 6 первоначально полученной при электростимуляции предсердия, когда отсутствовало 45 женских) при помощи исследования перекрестных перестановок, рандомизированисследуемое соединение. ных по 6 путям. В табл.6 приведены концентрации ІСзо Исследуемое соединение вводят внут(моль/л) для соединений в соответствии с ривенно при возрастающих дозах: 2; 6,5; 20; изобретением и для клонидина. Из этой таблицы следует, что соеди- 50 65; 200 мкг/кг. В течение первых трех часов вслед за введением измеряют объем выденения в соответствии с изобретением ляемой мочи. препятствуют тахикардии, вызванной В табл.8 приводится для соединения А электрической стимуляцией, при очень относительное увеличения (в об.%) выдемалых концентрациях, В присутствии альфа-иохимбина в кон- 55 ленной мочи по отношению к группе животных, которые не получали этого соединецентрации 10'6 моль/л концентрация сония. единения А, необходимая для Результаты показывают, что минимальуменьшения тахикардии на 30%, превыная доза, которая вызывает статистически шает 10*7 моль/л. Эти результаты хорошо 12835 33 значимое увеличение (Р « 0,05) выделения мочи, равна < 6,5 мкг/кг у мужских особей, а у женских особей эта доза составляет < 2 мкг/кг. 5. Токсичность. Токсичность соединений в соответствии с изобретением определялась у мышей (мужской особи) NMR1 при помощи теста Ирвина. 34 Возрастающие дозы исследуемого вещества вводились интраперитональным путем группам по три мыши до тех пор, пока не получалась смертельная доза (доза, вызывающая смерть двух животных из трех в течение 48 ч). В табл.9 приведены смертельные дозы соединений в соответствии с изобретением. Из этой таблицы следует, что эти соединения очень мало токсичные. Таблица 1 Соединение ДЕго, мкмоль/кг А 0,03 В 0,02 С 0,3 D 3 Е
ДивитисяДодаткова інформація
Автори англійськоюZhan Gober
Автори російськоюЖан Гобер
МПК / Мітки
Мітки: хлоргідрати, 1-(1н-імідазол-4-іл)алкіл-бензаміди, alрha2-адренергічних, протиішемічні, рецепторів, властивості, ізомери, мають, оптичні, антагоністичні
Код посилання
<a href="https://ua.patents.su/21-12835-1-1n-imidazol-4-ilalkil-benzamidi-kh-khlorgidrati-ta-optichni-izomeri-yaki-mayut-protiishemichni-ta-antagonistichni-vlastivosti-alrha2-adrenergichnikh-receptoriv.html" target="_blank" rel="follow" title="База патентів України">1-(1н-імідазол-4-іл)алкіл-бензаміди, їх хлоргідрати та оптичні ізомери, які мають протиішемічні та антагоністичні властивості alрha2-адренергічних рецепторів</a>
Хлоргідрати біс-основних ефірів 3,6-дигало-генфлуоренонів, які мають імуностимулюючу активність

Номер патенту: 1264
Опубліковано: 30.12.1993
Автори: Чижов Новомир Павлович, Уманський Юліан Олександрович, Богатський Олексій Всеволодович, Лемпарт Галина Василівна, Філіппова Тетяна Олеговна, Якименко Людмила Вікторівна, Литвинова Людмила Олександрівна, Мелехін Віктор Давидович, Балицький Костянтин Петрович, Андронаті Сергій Андрійович, Пінчук Вадим Григорович, Строганов Віталій Іванович, Синяченко Володимир Власович
МПК: A61K 31/085, A61P 37/04, C07C 217/14, C07C 49/80, C07C 13/00
Мітки: ефірів, імуностимулюючу, біс-основних, мають, активність, 3,6-дигало-генфлуоренонів, хлоргідрати
Формула / Реферат:
Хлоргидраты бис-основных эфиров 3,6-дига-логенфлуоренонов общей формулы:где R — хлор, бром, обладающие иммуностимулирующей активностью.обладающий антигипоксическим и актопротекторным действием.
1-(гідразинокарбоніл)алкил-1, 2-дигідро-3н-1, 4-бенздиазипін-2-они, що мають транквілізуючі та протисудорожні властивості
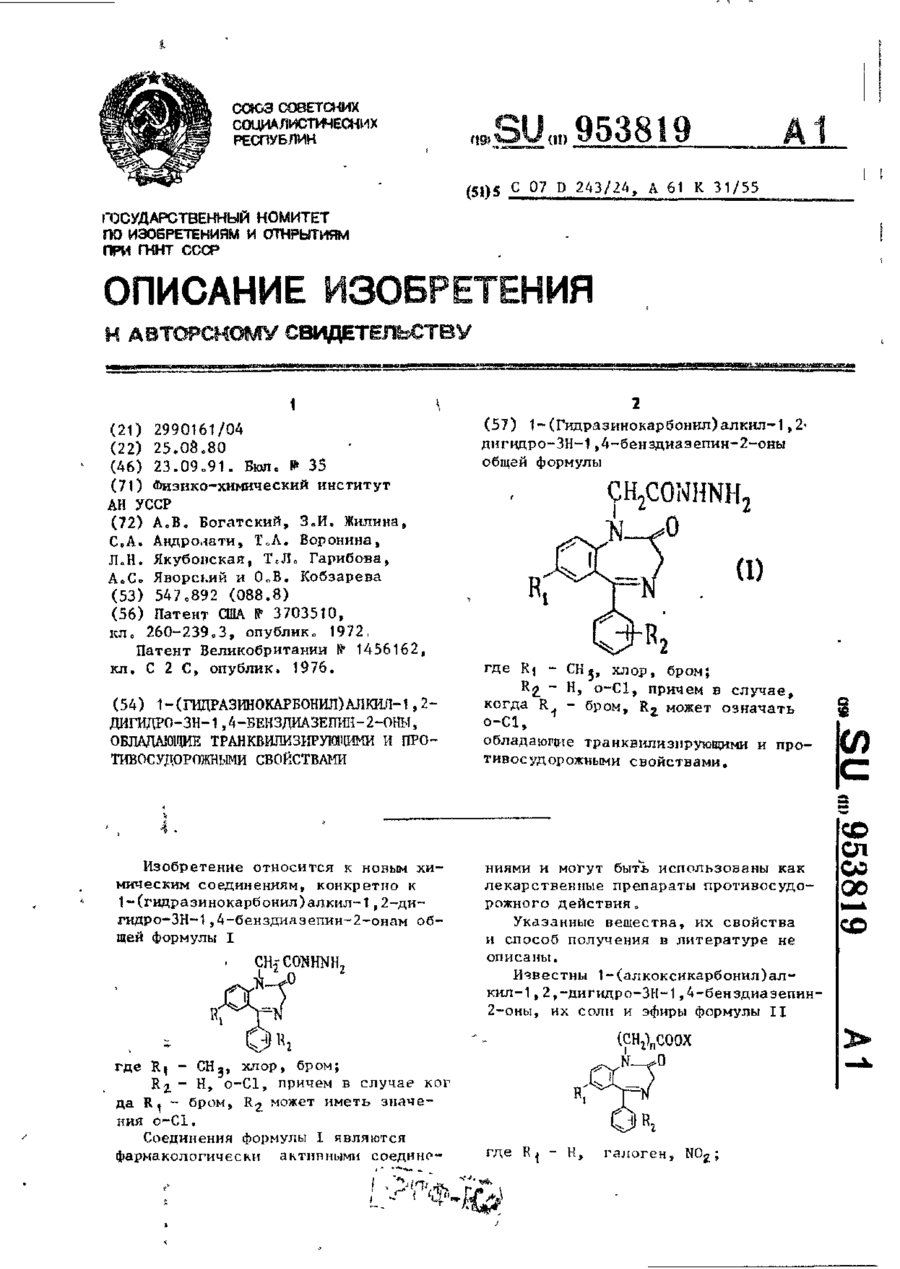
Номер патенту: 8184
Опубліковано: 29.03.1996
Автори: Богатський Олексій Всеволодович, Вороніна Тетяна Олександрівна, Гарібова Таісія Леонівна, Кобзарева Ольга Василівна, Андронаті Сергій Андрійович, Якубовська Лідія Миколаївна, Жиліна Зінаіда Іванівна, Яворський Олександр Степанович
МПК: A61P 25/22, A61K 31/5513, A61P 25/08, C07D 243/24
Мітки: 4-бенздиазипін-2-они, транквілізуючі, 1-(гідразинокарбоніл)алкил-1, мають, 2-дигідро-3н-1, протисудорожні, властивості
Формула / Реферат:
1 - (Гидразинокарбонил) алкил-1,2-дигидро-3Н-1,4-бенздиазепин-2-оны общей формулыгде R1 - СН3, хлор, бром;R2 - Н, о-Сl, причем в случае, когда R1 - бром, R2 может означать о-Сl, обладающие транквилизирующими и противосу-дорожными свойствами.
Beta-[beta’-(2,4-ді-трет-амілфенокси)-етокси] етилхлорид як напівпродукт для одержання речовин, які мають антимікробні властивості

Номер патенту: 10153
Опубліковано: 30.09.1996
Автори: Свертілова Ізабела Олександрівна, Ютілов Юрій Михайлович
МПК: C07C 39/00, A61K 31/02, A61P 31/04
Мітки: напівпродукт, мають, антимікробні, властивості, beta-[beta'-(2,4-ді-трет-амілфенокси)-етокси, етилхлорид, одержання, речовин
Формула / Реферат:
b-b'-(2,4-Ди-трет-амилфенокси)-этокси]-этилхлорид формулы в качестве полупродукта для получения веществ, обладающих антимикробными свойствами.
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Жерар Шарль Санз, Альфонс Герман Маргарета Реймакерс, Едді Жан Едгард Фрейн
МПК: A61K 31/4433, C07D 403/06, C07D 405/14, A61P 43/00, C07D 401/14, C07D 403/14, A61P 35/00, A61K 31/415, A61P 19/06, A61K 31/443, A61K 31/44, C07D 417/14, A61K 31/425, A61P 17/00, A61K 31/4427, C07D 409/14, C07D 521/00, A61P 5/00, A61K 31/47
Мітки: похідних, металів, бензімідазола, фармацевтично, 1н-імідазол-1-ілметіл)замішаного, кислоти, одержання, прийнятих, стереоізомерів, спосіб, солей
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Кристалічні мікропористі силікати лужних металів, що мають молекулярно-ситові властивості та спосіб їх одержання

Номер патенту: 3367
Опубліковано: 27.12.1994
Автори: Турутіна Наталія Вікторівна, Ільїн Володимир Георгійович
МПК: C01B 33/32
Мітки: силікати, молекулярно-ситові, лужних, одержання, металів, кристалічні, властивості, мають, спосіб, мікропористі
Формула / Реферат:
1. Кристаллические микропористые силикаты щелочных металлов с общей формулой Ме2О • nSіО2 • mН2О, где Ме-Lі, Nа, К, Rb, Сs, n-SiO2/Ме2О=1-40, m - Н2О/Ме2О=1-20, обладающие молекулярно-ситовыми свойствами, в качестве сорбентов.2. Способ получения кристаллических микропористых силикатов щелочных металлов по п. 1, отличающийся тем, что аморфный кремнезем подвергают взаимодействию с водным раствором щелочи при соотношении...
Попередній патент: Спосіб одержання нетканих голкопроколюваних матеріалів з термопластичних волокон
Наступний патент: Склад для отримання композиційних електролітичних покриттів на основі металів групи заліза
Випадковий патент: Спосіб змазування корінних та шатунних підшипників ковзання по крайній мірі для двоциліндрового поршньового аміачного компресора та система для його здійснення