Інгібітор регулюючої апоптотичні сигнали кінази







Формула / Реферат
1. Сполука формули (І):
 , (І)
, (І)
а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-N-(6-(4-ізопропіл-4Н-1,2,4-триазол-3-іл)піридин-2-іл)-2-фтор-4-метилбензамід, або її фармацевтично прийнятна сіль.
2. Сполука формули (І):
, (І)
а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-2-фтор-N-(6-(4-ізопропіл-4Н-1,2,4-триазол-3-іл)піридин-2-іл)-4-метилбензамід.
3. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки або солі за п. 1 та фармацевтично прийнятний носій.
4. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки за п. 2 та фармацевтично прийнятний носій.
5. Спосіб лікування хронічної ниркової недостатності, що включає введення терапевтично ефективної кількості сполуки за п. 2 або її фармацевтично прийнятної солі пацієнту, який цього потребує.
6. Спосіб лікування діабетичної нефропатії, що включає введення терапевтично ефективної кількості сполуки за п. 2 або її фармацевтично прийнятної солі пацієнту, який цього потребує.
7. Спосіб лікування фіброзу нирок, фіброзу печінки або фіброзу легенів, що включає введення терапевтично ефективної кількості сполуки або солі за п. 1 пацієнту, який цього потребує.
8. Спосіб лікування діабетичного захворювання нирок, що включає введення терапевтично ефективної кількості сполуки або солі за п. 1 пацієнту, який цього потребує.
9. Застосування сполуки або солі за п. 1 для одержання лікарського засобу для лікування хронічної ниркової недостатності.
10. Застосування сполуки або солі за п. 1 у терапії.
11. Проміжна сполука формули:
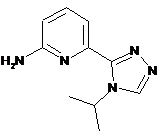 ,
,
а саме 2-аміно, 5-(4-ізопропіл-4Н-1,2,4-триазол-3-іл)піридин або його сіль, або захищена форма.
12. Проміжна сполука формули:
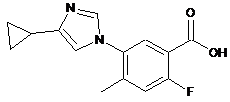 ,
,
а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-2-фтор-4-метилбензойна кислота, її сіль або захищена форма.
Текст
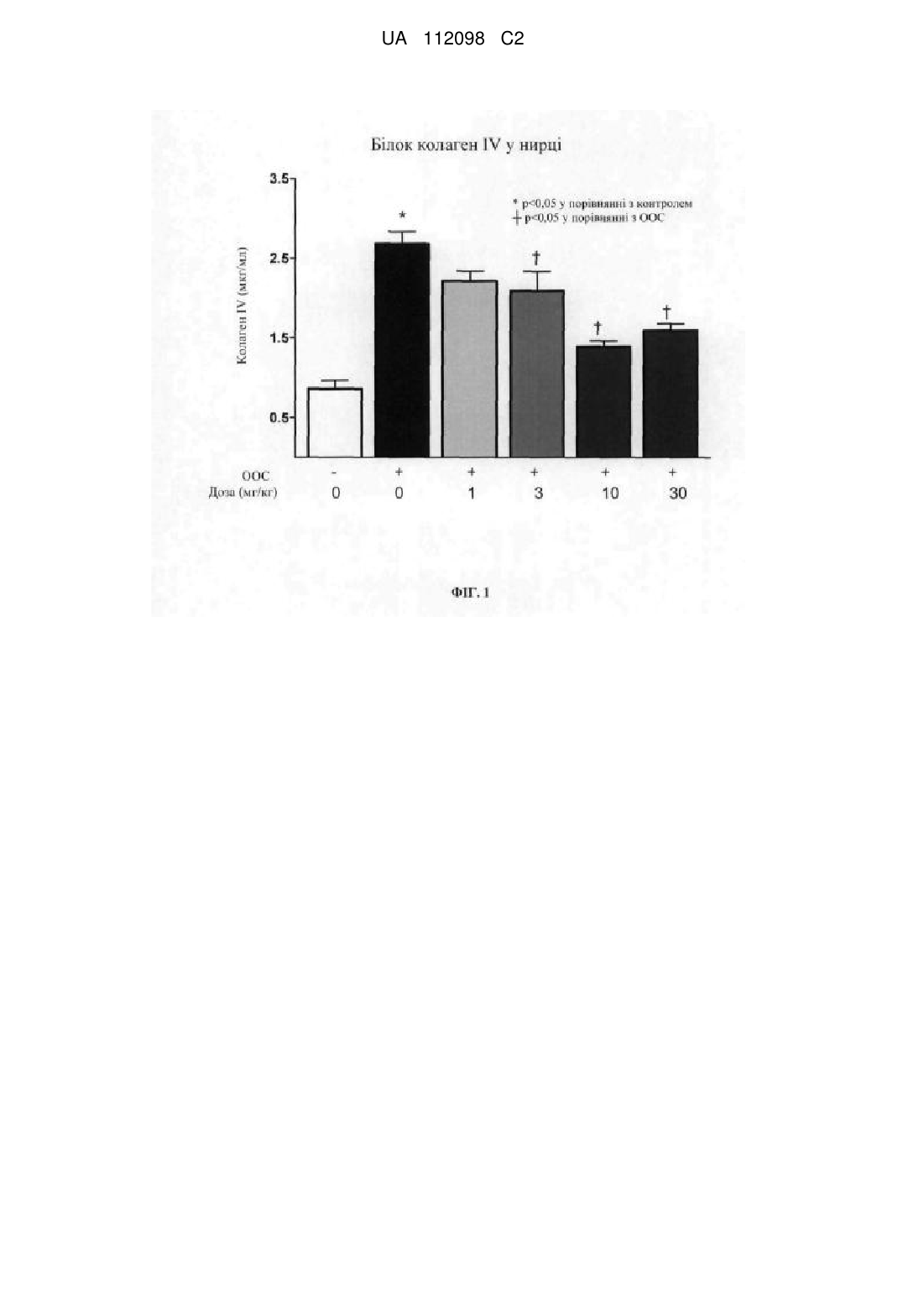
Реферат: Винахід стосується сполуки формули (І): N O N N H F N N N N , (І) яка має інгібуючу активність відносно регулюючої апоптотичні сигнали кінази і підходить у застосуванні для лікування захворювань, таких як захворювання нирок, діабетична нефропатія та фіброз нирок. UA 112098 C2 (12) UA 112098 C2 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 ГАЛУЗЬ ТЕХНІКИ Даний винахід відноситься до нової сполуки для застосування для лікування ASK1опосередкованих захворювань. Винахід також відноситься до проміжних сполук для одержання зазначеної нової сполуки та фармацевтичних композицій, що містять їх. РІВЕНЬ ТЕХНІКИ Регулююча апоптотичні сигнали кіназа 1 (ASK1) є членом сімейства кіназ кіназ мітогенактивуємих протеїнкіназ ("MAP3K"), які активують c-Jun N-термінальну протеїнкіназу ("JNK") та p38 MAP-кіназу (Ichijo, H., Nishida, E., Irie, K., Dijke, P. T., Saitoh, M., Moriguchi, T., Matsumoto, K., Miyazono, K., Gotoh, Y. (1997) Science, 275, 90-94). ASK1 активуються різними подразниками, включаючи окисний стрес, активні форми кисню (АФК), ЛПС, ФНО-α, FasL, ЕР-стрес та підвищені концентрації внутрішньоклітинного кальцію (Hattori, K., Naguro, I., Runchel, C., Ichijo, H. (2009) Cell Comm. Signal. 7:1-10; Takeda, K., Noguchi, T., Naguro, I., Ichijo, H. (2007) Annu. Rev. Pharmacol. Toxicol. 48: 1-8, 27; Nagai, H., Noguchi, T., Takeda, K., Ichijo, I. (2007) J. Biochem. Mol. Biol. 40:1-6). Фосфорилювання білку ASK1 може привести до апоптозу або інших клітинних відповідей залежно від типу клітин. Було показано, що активація та сигнальний шлях ASK1 відіграють важливу роль у широкому спектрі захворювань, включаючи нейродегенеративні, серцево-судинні, запальні, аутоімунні та метаболічні розлади. Крім того, було показано, що ASK1 бере участь у опосередкуванні ушкодження органів після ішемії та реперфузії серця, мозку та нирки (Watanabe et al. (2005) BBRC 333, 562-567; Zhang et al., (2003) Life Sci 74-37-43; Terada et al. (2007) BBRC 364: 1043-49). Показано, що АФК пов'язані з підвищеним виробленням запальних цитокінів, фіброзом, апоптозом та некрозом у нирці. (Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol 2011 Mar; 7(3): 176-184; Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001 Dec 13; 414 (6865): 813-820; Mimura I, Nangaku M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 2010 Nov; 6(11):667-678). Більше того, окисний стрес сприяє формуванню кінцевих продуктів посиленого глікозилювання (AGE), які викликають подальше ушкодження нирки та вироблення АФК. (Hung KY, et al. N-acetylcysteine-mediated antioxidation prevents hyperglycemia-induced apoptosis and collagen synthesis in rat mesangial cells. Am J Nephrol 2009; 29(3): 192-202). Тубулоінтерстиціальний фіброз у нирці є сильним прогностичним фактором прогресування захворювання до ниркової недостатності у пацієнтів, що страждають на хронічні захворювання нирок (Schainuck LI, et al. Structural-functional correlations in renal disease. Part II: The correlations. Hum Pathol 1970; 1: 631-641.). Однобічна обструкція сечоводу (ООС) у щурів є широко використовуваною моделлю тубулоінтерстиціального фіброзу. ООС викликає тубулоінтерстиціальне запалення, підвищену експресію трансформуючого фактору росту бета (ТФР-β) та накопичення міофібробластів, які секретують матриксні білки, такі як колаген та фібронектин. Модель ООС може використовуватися для дослідження потенційної здатності лікарського засобу лікувати хронічну ниркову недостатність шляхом пригнічення фіброзу нирок (Chevalier et al., Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy, Kidney International (2009) 75, 1145-1152). Таким чином, терапевтичні агенти, що діють як інгібітори сигналінгу ASK1, мають потенціал відносно лікування або поліпшення якості життя пацієнтів, що потребують лікування захворювань або станів, таких як нейродегенеративні, серцево-судинні, запальні, аутоімунні та метаболічні розлади. Зокрема, інгібітори ASK1 мають потенціал для лікування кардіоренальних захворювань, включаючи захворювання нирок, діабетичне захворювання нирок, хронічну ниркову недостатність, фіброзні захворювання (включаючи фіброз легенів та нирок), респіраторні захворювання (включаючи хронічне обструктивне захворювання легенів (ХОЗЛ) та гостре ушкодження легенів), гострі та хронічні ниркові недостатності. У публікації заявки на патент США №2007/0276050 описані способи ідентифікації інгібіторів ASK1, що підходять для запобігання та/або лікування серцево-судинного захворювання, та способи запобігання та/або лікування зазначеного серцево-судинного захворювання у тварини. У WO2009027283 описані тіазолопіридинові сполуки, способи їх одержання та способи лікування аутоімунних розладів, запальних захворювань, серцево-судинних захворювань та нейродегенеративних захворювань. У публікації заявки на патент США №2001/00095410A1, опублікованої 13 січня 2011 р., описані сполуки, що підходять для застосування як інгібітори ASK-1. Публікація заявки на патент США № 2001/00095410A1 відноситься до сполук формули (I): 1 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 (I) де: 1 R являє собою алкіл, алкеніл, алкініл, циклоалкіл, арил, гетероарил або гетероцикліл, кожен з яких необов'язково містить 1, 2, або 3 замісники, вибрані з галогену, оксо групи, алкілу, 6 6 6 6 циклоалкілу, гетероциклілу, арилу, арилокси групи, -NO2, R , -C(O)-R , -OC(O)-R -C(O)-O-R , 6 7 6 7 6 6 6 6 7 6 C(O)-N(R )(R ), -OC(O)-N(R )(R ), -S-R , -S(=O)-R , -S(=O)2R , -S(=O)2-N(R )(R ), -S(=O)2-O-R , 6 7 6 7 6 7 6 6 7 6 6 N(R )(R ), -N(R )-C(O)-R , -N(R )-C(O)-O-R , -N(R )-C(O)-N(R )(R ), -N(R )-S(=O)2-R , -CN та –O6 R, де зазначені алкіл, циклоалкіл, гетероцикліл, феніл та фенокси група необов'язково містять 1, 2 або 3 замісники, вибрані з алкілу, циклоалкілу, алкокси групи, гідроксилу та галогену; 6 7 де R та R незалежно вибрані з групи, що складається з водню, C 1-C15 алкілу, циклоалкілу, гетероциклілу, арилу та гетероарилу, кожен з яких необов'язково містить 1-3 замісники, вибрані з галогену, алкілу, моно- або диалкіламіно групи, аміду, алкілу або арилу або гетероарилу, -CN, нижчої алкокси групи, -CF3, арилу та гетероарилу; або 6 7 R та R разом з атомом азоту, до якого вони приєднані, утворюють гетероцикл; 2 R являє собою водень, галоген, ціано групу, алкокси групу або алкіл, що необов'язково містить як замісники галоген; 3 R являє собою арил, гетероарил або гетероцикліл, кожен з яких необов'язково містить один або більше замісників, вибраних з алкілу, алкокси групи, циклоалкілу, циклоалкілалкілу, арилу, арилалкілу, гетероарилу, гетероарилалкілу, гетероциклілу, гетероциклілалкілу, галогену, оксо 6 6 6 7 групи, -NO2, галогеналкілу, галогеналкокси групи, -CN, -O-R , -O-C(O)-R , -O-C(O)-N(R )(R ), -S6 6 7 6 6 6 7 6 6 7 6 R , -N(R )(R ), -S(=O)-R , -S(=O)2R , -S(=O)2-N(R )(R ), -S(=O)2-O-R , -N(R )-C(O)-R , -N(R )-C(O)7 6 6 7 6 6 6 7 6 7 O-R , -N(R )-C(O)-N(R )(R ), -C(O)-R , -C(O)-O-R , -C(O)-N(R )(R ) та -N(R )-S(=O)2-R , де зазначений алкіл, алкокси група, циклоалкіл, арил, гетероарил або гетероцикліл також необов'язково містить один або більше замісників, вибраних з галогену, оксо групи, -NO2, алкілу, 6 7 6 6 6 7 галогеналкілу, галогеналкокси групи, -N(R )(R ), -C(O)-R , -C(O)-O-R , -C(O)-N(R )(R ), -CN, –O6 R , циклоалкілу, арилу, гетероарилу та гетероциклілу; за умови, що гетероарильний або гетероциклільний фрагмент містить щонайменше один атом азоту у кільці; 1 2 3 4 5 6 7 8 4 4 X , X , X , X , X , X , X та X незалежно являють собою C(R ) або N, де кожен з R незалежно являє собою водень, алкіл, алкокси групу, циклоалкіл, арил, гетероарил, 6 6 6 7 гетероцикліл, галоген, -NO2, галогеналкіл, галогеналкокси групу, -CN, -O-R , -S-R , -N(R )(R ), 6 6 6 7 6 6 7 6 7 6 S(=O)-R , -S(=O)2R , -S(=O)2-N(R )(R ), -S(=O)2-O-R , -N(R )-C(O)-R , -N(R )-C(O)-O-R , -N(R )6 7 6 6 6 7 6 7 C(O)-N(R )(R ), -C(O)-R , -C(O)-O-R , -C(O)-N(R )(R ) або -N(R )-S(=O)2-R , де зазначений алкіл, циклоалкіл, арил, гетероарил та гетероцикліл також необов'язково містить один або більше 6 7 6 7 замісників, вибраних з галогену, оксо групи, -NO2, -CF3, -O-CF3, -N(R )(R ), -C(O)-R , -C(O)-O-R , 6 7 6 C(O)-N(R )(R ), -CN, –O-R ; або 5 6 6 7 X та X або X та X з'єднані з одержанням необов'язково заміщеного конденсованого арилу або необов'язково заміщеного конденсованого гетероарилу; та 2 3 4 4 за умови, що щонайменше один з X , X та X являє собою C(R ); 5 6 7 8 4 щонайменше два з X , X , X та X являють собою C(R ); та 2 3 4 5 6 7 8 щонайменше один з X , X , X , X , X , X та X являє собою N. Незважаючи на описані вище винаходи, існує необхідність у сполуках, які є високоактивними та проявляють поліпшені фармакокінетичні та/або фармакодинамічні властивості для лікування захворювань, пов'язаних з активацією ASK1. Заявники неочікувано виявили у межах патентної публікації США US2011/0009410A нову сполуку, що проявляє гарну активність та у цілому поліпшені фармакокінетичні та/або фармакодинамічні властивості у порівнянні із сполуками, запропонованими у зазначеній публікації. КОРОТКИЙ ОПИС ВИНАХОДУ Даний винахід відноситься до сполуки формули: 2 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 (I) або її фармацевтично прийнятної солі. Згідно з одним варіантом реалізації даний винахід відноситься до застосування сполуки формули (I) для лікування захворювання у пацієнта, який потребує зазначеного лікування, за допомогою інгібітору ASK1. Згідно з іншим варіантом реалізації даний винахід відноситься до фармацевтичної композиції, що містить сполуку формули (I) або її фармацевтично прийнятну сіль та один або більше фармацевтично прийнятних носіїв. Згідно з іншим варіантом реалізації даний винахід являє собою спосіб лікування діабетичної нефропатії або ускладнень діабету, що включає введення терапевтично ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі пацієнту, який цього потребує. Згідно з іншим варіантом реалізації даний винахід відноситься до способу лікування захворювання нирок або діабетичного захворювання нирок, що включає введення терапевтично ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі пацієнту, який цього потребує. Згідно з іншим варіантом реалізації даний винахід відноситься до способу лікування фіброзу нирок, фіброзу легенів або ідіопатичного легеневого фіброзу (ІЛФ), що включає введення терапевтично ефективної кількості сполуки формули (I) або її фармацевтично прийнятної солі пацієнту, який цього потребує. Згідно з іншим варіантом реалізації даний винахід відноситься до способу лікування діабетичного ниркової недостатності, діабетичної нефропатії, фіброзу нирок, фіброзу печінки або фіброзу легенів, що включає введення терапевтично ефективної кількості сполуки або солі формули (I) пацієнту, який цього потребує. Згідно з іншим варіантом реалізації даний винахід відноситься до проміжних сполук, що підходять для застосування для синтезу сполук формули (I). Згідно з іншим варіантом реалізації даний винахід відноситься до застосування сполуки формули (I) або її фармацевтично прийнятної солі для лікування хронічної ниркової недостатності. Згідно з іншим варіантом реалізації даний винахід відноситься до застосування сполуки формули (I) або її фармацевтично прийнятної солі для лікування діабетичної ниркової недостатності. Згідно з іншим варіантом реалізації даний винахід відноситься до застосування сполуки формули (I) або її фармацевтично прийнятної солі для виготовлення медикаменту для лікування хронічної ниркової недостатності. Згідно з іншим варіантом реалізації даний винахід відноситься до сполуки формули (I) для застосування у терапії. ДЕТАЛЬНИЙ ОПИС ВИНАХОДУ Фігури Фігура 1 являє собою гістограму, що показує рівні колагену IV у корковій речовині нирки щурів, яких піддали семиденній однобічній обструкції сечоводу та що одержували лікування носієм або сполукою формули (I) у концентрації 1, 3, 10 або 30 мг/кг два рази на день. На Фігурі 2 показані типові зображення зрізів коркової речовини нирки щурів, яких піддали семиденній однобічній обструкції сечоводу та що одержували лікування носієм або сполукою формули (I) у дозі 1, 3, 10 або 30 мг/кг два рази на день, пофарбованих на альфагладком'язовий актин (маркер активованих міофібробластів). Визначення та загальні характеристики Передбачається, що при використанні у даний заявці наведені далі слова та фрази мають значення, зазначені нижче, якщо інше не випливає з контексту, у якому вони використовуються. Якщо не приводиться позначення або визначення слова або фрази, тоді мається на увазі загальноприйняте значення зазначеного слова або фрази, яке можна виявити у відповідному словнику або повсюдному використанні, відомому фахівцеві у даній галузі техніки. Термін "хронічна ниркова недостатність" при використанні у даній заявці відноситься до прогресуючої втрати ниркової функції протягом тривалого періоду, як правило, декількох місяців або навіть років. Хронічну ниркову недостатність (ХНН) діагностує компетентний суб'єкт, що здійснює догляд, з використанням відповідної інформації, тестів або маркерів, відомих фахівцеві у даній галузі техніки. Хронічна ниркова недостатність має на увазі ниркову 3 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 недостатність. Термін "діабетична ниркова недостатність" при використанні у даній заявці відноситься до ниркової недостатності, викликаної діабетом, що збільшується діабетом або присутньої разом з діабетом. Діабетична ниркова недостатність являє собою форму хронічної ниркової недостатності, яка виникає приблизно у 30 % пацієнтів, що страждають на діабет. Вона визначається як діабет із супутньою альбумінурією та/або порушеною нирковою функцією (тобто зниженою швидкістю клубочкової фільтрації (Див. de B, I, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011 Jun 22; 305(24):2532-2539). Термін "фармацевтично прийнятна сіль" відноситься до солей фармацевтичних сполук, наприклад, сполуки формули (I), які зберігають біологічну ефективність та властивості вихідної сполуки та які не є небажаними з біологічної або іншої точки зору. Зазначені солі являють собою солі приєднання кислот та солі приєднання основ. Фармацевтично прийнятні солі приєднання кислот можуть бути отримані з неорганічних та органічних кислот. Кислоти та основи, застосовні для реакції з вихідною сполукою з утворенням фармацевтично прийнятних солей (солей приєднання кислоти або основи, відповідно), відомі фахівцеві у даній галузі техніки. Подібним чином, способи одержання фармацевтично прийнятних солей з вихідної сполуки (згідно з винаходом) відомі фахівцеві у даній галузі техніки та описані, наприклад, у джерелі Berge, at al. Journal of Pharmaceutical Science, Jan. 1977 vol. 66, No.1, та іншихджерелах. Солі, отримані з неорганічних кислот, включають, але не обмежуються зазначеними, солі соляної кислоти, бромисто-водневої кислоти, сірчаної кислоти, азотної кислоти, фосфорної кислоти тощо. Солі, отримані з органічних кислот, включають, але не обмежуються зазначеними, малеїнову кислоту, фумарову кислоту, винну кислоту, птолуолсульфонову кислоту тощо. Основи, застосовні для одержання солей приєднання основ, відомі фахівцеві у даній галузі техніки. Приклад фармацевтично прийнятної солі сполуки формули (I) являє собою гідрохлорид сполуки формули (I). При використанні у даний заявці "фармацевтично прийнятний носій" включає допоміжні речовини або агенти, такі як розчинники, розріджувачі, диспергуючі агенти, покриваючі агенти, антибактеріальні та протигрибкові агенти, ізотонічні та уповільнюючі абсорбцію агенти та т.п., які не виявляють несприятливого впливу на сполуку згідно з винаходом або її застосування. Застосування зазначених носіїв та агентів для одержання композицій фармацевтично активних речовин добре відомо у даній галузі техніки (див., наприклад, Remington's Pharmaceutical Sciences, Mace Publishing Co., Philadelphia, PA 17th Ed. (1985); та Modern Pharmaceutics, Marcel Dekker, Inc. 3rd Ed. (G.S. Banker & C.T. Rhodes, Eds.) Термін "кардіоренальні захворювання" при використанні у даний заявці відноситься до захворювань, пов'язаних з нирковою функцією, які викликані або збільшуються серцевосудинними порушеннями, такими як, наприклад, підвищений кровоносний тиск або гіпертензія. Вважається, що гіпертензія вносить важливий вклад у ниркову недостатність. Термін "респіраторні захворювання" при використанні у даний заявці відноситься до захворювань, що включають хронічне обструктивне захворювання легенів (ХОЗЛ) та ідіопатичний легеневий фіброз (ІЛФ). Термін "терапевтично ефективна кількість" відноситься до кількості сполуки формули (I), яка є достатньою для здійснення лікування відповідно до наведеного нижче визначення при введенні пацієнтові (зокрема, людині), що потребує зазначеного лікування, у вигляді однієї або більше доз. Терапевтично ефективна кількість варіюється залежно від пацієнта, захворювання, яке передбачається лікувати, маси тіла та/або віку пацієнта, маси захворювання або способу введення та визначається кваліфікованим медичним працівником або особою, що здійснюють догляд. Термін "лікування" або "проведення лікування" означає введення сполуки або фармацевтично прийнятної солі формули (I) для: (i) затримки виникнення захворювання, тобто перешкоджання розвитку клінічних симптомів захворювання або затримки їх розвитку; (ii) пригнічення захворювання, тобто стримування розвитку клінічних симптомів; та/або (iii) полегшення захворювання, тобто забезпечення регресії клінічних симптомів або їх важкості. Згідно із кращим варіантом реалізації винахід відноситься до застосування сполуки формули (I) для лікування хронічної ниркової недостатності, що включає введення терапевтично ефективної кількості пацієнту, який цього потребує. Відповідно до іншого кращого варіанту реалізації винахід відноситься до застосування сполуки формули (I) для лікування діабетичної ниркової недостатності, що включає введення терапевтично ефективної кількості пацієнту, який цього потребує. 4 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 Відповідно до іншого кращого варіанту реалізації винахід відноситься до застосування сполуки формули (I) для лікування фіброзу легенів або фіброзу нирок, що включає введення терапевтично ефективної кількості пацієнту, який цього потребує. Концентрація напівмаксимального інгібування (IC50) терапевтичного агенту являє собою концентрацію зазначеного терапевтичного агенту, необхідну для забезпечення 50 % максимального інгібування відносно ферменту-мішені. Поставленим завданням є забезпечення терапевтичного агенту, наприклад, сполуки, яка інгібує регулюючу апоптотичні сигнали кіназу (ASK1) при низькій IC50. Таким чином, небажані побічні дії мінімізуються завдяки можливості застосування більш низької дози терапевтичного агенту для пригнічення ферменту ASK1. Подібним чином, поставленим завданням є забезпечення терапевтичного агенту, що має низьку константу дисоціації (Kd). Kd використовується для опису спорідненості між лігандом (таким як терапевтичний агент) та відповідною кіназою або рецептором; тобто міри того, наскільки сильно терапевтичний агент зв'язується з конкретною кіназою, наприклад, регулюючою апоптотичні сигнали кіназою ASK1. Таким чином, більш низькі значення K d у цілому є кращими для розробки лікарського засобу. Подібним чином, поставленим завданням є забезпечення сполуки, що має низьку EC50. EC50 являє собою концентрацію лікарського засобу, яка приводить до досягнення 50 % максимальної ефективності у клітині. Значення EC50 виражається як концентрація сполуки у аналітичному середовищі, необхідна для досягнення 50 % максимальної ефективності. Таким чином, більш низька EC50 у цілому є кращою для розробки лікарського засобу. Застосовна одиниця вимірювання, пов'язана з EC50, являє собою скоректовану з урахуванням зв'язування з білком EC50 (PBadj.EC50 при використанні у даній заявці). Зазначене значення описує кількість лікарського засобу, наприклад, сполуки формули (I), співвіднесену із фракцією лікарського засобу, незв'язаної з білком, яка забезпечує 50 % максимальної ефективності. Зазначене значення показує ефективність лікарського засобу, корельовану або скоректовану з урахуванням кількості лікарського засобу, доступного у місці дії мішені. Іншою бажаною властивістю сполуки є низьке відношення відтоку через клітинну мембрану, що визначається за допомогою досліджень проникності на клітинах лінії CACO. Відношення відтоку ((B/A) / (A/B)), що становить менше 3,0, є кращим. Очікується, що сполука, відношення відтоку якої становить більше 3, активно та швидко виходить із клітини та може не виявляти досить тривалого впливу у клітині для досягнення максимальної ефективності. Іншим поставленим завданням є забезпечення лікарського засобу, який проявляє мінімальне нецільове інгібування, тобто лікарський засіб, який мінімально інгібує ферменти Cyp450 (цитохром p450). Більш конкретно, бажаним є лікарський засіб, який є слабким інгібітором cyp3A4 - найбільш важливого з ферментів P450. Слабкий інгібітор являє собою сполуку, яке викликає щонайменше 1,25-разове, але менш ніж 2-разове підвищення значень ППК у плазмі крові або 20-50 % зниження кліренсу (сайт wikipedia.org/wiki/cyp3A4, відвіданий 11/12/11). У цілому, сполука, IC 50 якої у відношенні Cyp3A4 становить більше 10 мкМ, вважається слабким інгібітором. Вимірювання, застосовне для порівняння інгібування Сyp3A4 між кандидатами лікарських засобів, являє собою відношення інгібування Cyp3A4 та скоректованої з урахуванням зв'язування з білком EC50. Зазначене значення забезпечує показник відносної потенційної здатності інгібувати Cyp, з поправкою на EC50, скоректовану з урахуванням зв'язування з білком, специфічну для кожного лікарського засобу. Більш високе відношення для зазначеного вимірювання є кращим як показник більш низької потенційної здатності інгібувати cyp3A4. Неочікувано та вдало заявники виявили сполуку (у даний заявці сполуку формули (I)) у межах загального обсягу патентної публікації США № 2001/00095410A1, яка забезпечує переваги у порівнянні зі структурно близькими сполуками (які у даній заявці позначаються як сполука A та B), описаними у патентній публікації США № 2001/00095410A1: , Сполука A , Сполука B. Таким чином, завдання даного винаходу включає, але не обмежується зазначеними, забезпечення сполуки формули (I) або її фармацевтично прийнятної солі та способи 5 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 застосування зазначеної сполуки формули (I) для лікування ниркової недостатності, хронічної ниркової недостатності, діабетичної ниркової недостатності, діабетичної нефропатії, фіброзу нирок або фіброзу легенів. Комбінована терапія Пацієнти, що проходять лікування кардіоренальних захворювань, таких як хронічна ниркова недостатність, можуть мати позитивний результат від комбінованого лікарського лікування. Наприклад, сполуку згідно із даним винаходом можна комбінувати з одним або більше із інгібіторів ангіотензинперетворюючих ферментів (ACE), такими як еналаприл, каптоприл, раміприл, лізиноприл та хінаприл; або блокаторів рецептору II (ARB), таких як лазартан, олмесартан та ірбесартан; або гіпотензивними агентами, такими як амлодипін, ніфедипін та фелодипін. Позитивним результатом комбінування може бути підвищена ефективність та/або зменшення побічних ефектів компоненту, тому що доза зазначеного компонента може бути знижена для зменшення його побічних ефектів, тоді як позитивний результат від його ефективності підвищує ефективність сполуки формули (I) та/або іншого активного компоненту (компонентів). Пацієнти, що страждають на хронічну ниркову недостатність, що піддається лікуванню за допомогою інгібіторів ASKI, таких як сполука формули (I), також можуть страждати на стани, на лікування яких сприятливо впливає спільне введення терапевтичного агенту або агентів (за вказівкою кваліфікованої особи, що здійснює догляд), які являють собою антибіотики, анальгетики, антидепресанти та/або заспокійливі засоби у комбінації із сполукою формули (I). Комбіновані лікарські засоби можна вводити одночасно або послідовно з годинними інтервалами за вказівкою кваліфікованої особи, що здійснює догляд, або за допомогою лікарської форми з фіксованими дозами двох або більше активних агентів (усі активні інгредієнти об'єднані у одну форму дозування, наприклад, таблетку). Фармацевтичні композиції та введення Сполуки згідно із даним винаходом можна вводити у формі фармацевтичної композиції. У даному винаході, таким чином, запропоновані фармацевтичні композиції, які містять як активний інгредієнт сполуку формули (I) або її фармацевтично прийнятну сіль та одну або більше фармацевтично прийнятних допоміжних речовин та/або носіїв, включаючи інертні тверді розріджувачі та наповнювачі, розріджувачі, включаючи стерильний водний розчин та різні органічні розчинники, підсилювачі проникнення, солюбілізатори та ад'юванти. Фармацевтичні композиції можна вводити окремо або у комбінації з іншими терапевтичними агентами. Композиції для доставки можуть бути отримані у вигляді твердих таблеток, капсул, каплетів, мазей, трасдермальних пластирів, форм із уповільненим вивільненням, таблеток для розсмоктування, лікарських форм для інгаляції тощо. Типові фармацевтичні композиції одержують та/або вводять із використанням способів та/або процесів, добре відомих у галузі фармацевтики (див., наприклад, Remington's Pharmaceutical Sciences, Mace Publishing Co., Philadelphia, PA 17th Ed. (1985); and Modern Pharmaceutics, Marcel Dekker, Inc. 3rd Ed. (G.S. Banker & C.T. Rhodes, Eds). Лікарські форми для комбінованої терапії, що містять сполуку формули (I), можуть бути представлені у вигляді лікарських форм із фіксованою дозою, наприклад, таблеток, еліксирів, рідин, мазей, лікарських форм для інгаляції, гелів тощо, за допомогою методів, відомих фахівцеві у даній галузі техніки. Фармацевтичні композиції сполуки формули (I) можна вводити або у одній, або у декількох дозах за допомогою способів, включаючи, наприклад, ректальний, букальний, інтраназальний та трансдермальний спосіб введення; шляхом внутрішньоартеріальної ін'єкції, внутрішньовенно, внутрішньочеревинно, парентерально, внутрішньом'язово, підшкірно, перорально, місцево, за допомогою інгалятору або через просочене або покрите сполукою обладнання, таке як, наприклад, стент або вживлюваний у артерію циліндричний полімерний матеріал. Найбільш кращі способи введення включають пероральне, парентеральне та внутрішньовенне введення. Сполуку формули (I) можна вводити у фармацевтично ефективній кількості. Кожна одиниця дозування для перорального введення переважно містить від 1 мг до 500 мг сполуки формули (I). Більш переважно, доза сполуки формули (I) становить від 1 мг до 250 мг. Особливо переважно, доза сполуки формули (I) варіюється від приблизно 20 мг два рази на день до приблизно 50 мг два рази на день. Однак необхідно розуміти, що реальна кількість сполуки, що вводиться, як правило, визначається лікарем з урахуванням відповідних факторів, включаючи стан, який передбачається лікувати, вибраний спосіб введення, спільне введення сполуки, якщо застосовне, віку, маси тіла та відповіді на лікування конкретного пацієнта, важкості симптомів пацієнта тощо. 6 UA 112098 C2 Номенклатура Сполука згідно з даним винаходом має назву, отриману за допомогою програми ChemBioDraw Ultra 11: 5 10 15 20 25 30 35 40 5-(4-циклопропіл-1H-імідазол-1-іл)-N-(6-(4-ізопропіл-4H-1,2,4-триазол-3-іл)піридин-2-іл)-2фтор-4-метилбензамід, також відому як 5-((4-циклопропіл-1H-імідазол-1-іл)-2-фтор-N-(6-(4ізопропіл-4H-1,2,4-триазол-3-іл)піридин-2-іл)-4-метилбензамід. Синтез сполуки формули (I) Сполука згідно з винаходом може бути отримана з використанням способів, описаних у даний заявці, або їх модифікацій, які будуть очевидні з урахуванням описаного у даний заявці винаходу. Синтез сполуки згідно з винаходом можна здійснювати, як описано у наступному прикладі. Реагенти можуть бути отримані комерційним шляхом, якщо це доступно, наприклад, з компанії Sigma Aldrich або від інших виробників хімічних речовин. Альтернативно, реагенти можуть бути отримані з використанням схем реакцій та способів, відомих фахівцеві у даній галузі техніки. Параметри реакцій синтезу Терміни "розчинник", "інертний органічний розчинник" або "інертний розчинник" відносяться до розчиннику, інертного в умовах реакції, пов'язаних з ним, (включаючи, наприклад, бензол, толуол, ацетонітрил, тетрагідрофуран (ТГФ), диметилформамід (ДМФА), хлороформ, метиленхлорид (або дихлорметан), діетиловий ефір, петролейний ефір (ПЕ), метанол, піридин, етилацетат (ЕА) тощо. Якщо інше не зазначене, розчинники, використовувані у реакціях згідно із даним винаходом, являють собою інертні органічні розчинники, та реакції здійснюють в умовах інертного газу, переважно, азоту. Один зі способів одержання сполук формули (I) показаний на схемах реакцій 1 та 2 нижче. Схема 1 ДМФА-ДМА Одержання сполуки A До розчину метил 6-амінопіколінату (432 г, 2,84 моль) у MeOH (5 л) додавали NH2NH2.H2O (284 г, 5,68 моль, 2,0 екв.). Реакційну суміш нагрівали при температурі зворотної конденсації впродовж 3 годин та потім охолоджували до кімнатної температури. Утворений у суміші осад збирали шляхом фільтрування, промивали ЕА (2 л×2) та потім сушили у вакуумі з одержанням сполуки A (405 г, вихід 94 %) у вигляді твердої речовини білого кольору. Одержання сполуки B Суміш сполуки A (405 г, 2,66 моль) у диметилформаміді-диметилацеталі (ДМФА-ДМА) (3,54 л) нагрівали при температурі зворотної конденсації впродовж 18 годин, охолоджували до кімнатної температури та потім концентрували при зниженому тиску. Осад відбирали у ЕА (700 мл) та нагрівали при 50 °C впродовж 20 хвил. Після охолодження до кімнатної температури тверду речовину збирали шляхом фільтрування та сушили у вакуумі з одержанням сполуки B (572 г, вихід 82 %) у вигляді твердої речовини білого кольору. Одержання сполуки C До розчину сполуки B (572 г, 2,18 моль) у суміші CH3CN-AcOH (3,6 л, 4:1) додавали пропан2-амін (646 г, 5,0 екв.). Реакційну суміш нагрівали при температурі зворотної конденсації впродовж 24 годин та потім охолоджували до кімнатної температури та розчинник видаляли при 7 UA 112098 C2 5 10 15 20 25 30 зниженому тиску. Осад розчиняли у воді (2,8 л) та додавали 1 н водний розчин NaOH до досягнення pH 8,0. Осад збирали шляхом фільтрування, та фільтрат екстрагували ЕА (500 мл×3). Об'єднані органічні шари сушили над безводним Na 2SO4 та потім концентрували до досягнення об'єму 150 мл. До зазначеної суміші при 0 °C повільно додавали ПЕ (400 мл), та отриману суспензію фільтрували. Об'єднану тверду речовину перекристалізовували з ЕА-ПЕ з одержанням сполуки C (253 г, вихід 57 %) у вигляді твердої речовини не зовсім білого кольору. 1 H-ЯМР (400 МГц, CDCl3): δ 8,24 (s, 1 H), 7,52 (m, 2 H), 6,51 (dd, J=1,6, 7,2 Гц, 1 H), 5,55 (m, 1 + H), 4,46 (bs, 2 H), 1,45 (d, J=6,8 Гц, 6 H). МС (ІЕР+) m/z: 204 (M+1) . Сполука C є основною проміжною сполукою для синтезу сполуки формули (I). Таким чином, задачею даного винаходу також є одержання проміжної сполуки C: , її солей або захищених форм для одержання сполуки формули (I). Приклад солі сполуки C являє собою сіль приєднання HCl. Приклад захищеної форми сполуки C являє собою карбаматну сполуку, таку як сполука, отримана за допомогою Cbz-Cl. Захисні групи, способи їх одержання та застосування описані у джерелі Peter G.M. Wuts, Theodora W. Greene, Protective nd Groups in Organic Chemistry, 2 edition, 1991, Wiley and Sons, Publishers. Схема 2 Одержання Сполуки формули (I) продовжували наступним чином: Формула (I) Сполука 6 являє собою основну проміжну сполуку для синтезу сполуки формули (I). Таким чином, задачею даного винаходу також є одержання проміжної сполуки 6: , її солей або захищених форм, для одержання сполуки формули (I). Приклад солі сполуки 6 являє собою сіль приєднання HCl. Приклад захищеної форми сполуки 6 являє собою складний ефір (наприклад, метилові, етилові або бензилові складні ефіри) або карбаматну сполуку, таку як сполука, отримана за допомогою Cbz-Cl. Захисні групи, способи їх одержання та застосування описані у джерелі Peter G.M. Wuts and Theodora W. Greene, Protective Groups in nd Organic Chemistry, 2 edition, 1991, Wiley and Sons, Publishers. Етап 1 - Одержання 5-аміно-2-фтор-4-метилбензонітрилу - Сполуки (2) Вихідну сполуку 5-бром-4-фтор-2-метиланілін (1) (20 г, 98 ммоль) розчиняли у безводному 1метилпіролідоні (100 мл) та додавали ціанід міді (I) (17,6 г, 196 ммоль). Реакційну суміш 8 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 нагрівали до 180ºC впродовж 3 годин, охолоджували до кімнатної температури та додавали воду (300 мл) та концентрований розчин гідроксиду амонію (300 мл). Суміш перемішували впродовж 30 хвилин та екстрагували ЕА (3 × 200 мл). Об'єднані екстракти сушили над сульфатом магнію та розчинник видаляли при зниженому тиску. Маслянистий осад промивали гексаном (2 × 100 мл) та тверду речовину розчиняли у дихлорметані та наносили на колонку, заповнену силікагелем. У результаті елюювання у градієнті від 0 до 25 % ЕА у гексані +1 отримували 5-аміно-2-фтор-4-метилбензонітрил (10,06 г, 67,1 ммоль). РХ/МС (m/z:151 M ). Етап 2 - Одержання 5-(2-циклопропіл-2-оксоетиламіно)-2-фтор-4-метилбензонітрилу Сполуки (3) 5-Аміно-2-фтор-4-метилбензонітрил (12 г, 80 ммоль) розчиняли у безводному N, Nдиметилформаміді (160 мл) у атмосфері азоту та додавали при перемішуванні карбонат калію (13,27 г, 96 ммоль) та йодид калію (14,61 г, 88 ммоль) у вигляді твердої речовини. Реакційну суміш перемішували впродовж5 хвилин при кімнатній температурі та потім додавали бромметилциклопропілкетон (20,24 мл, 180 ммоль). Реакційну суміш нагрівали до 60ºC впродовж 3 годин, та потім розчинники видаляли при зниженому тиску. Осад розчиняли у ЕА (400 мл) та промивали 400 мл води. Органічний шар сушили над сульфатом магнію, та розчинник видаляли при зниженому тиску. Осад повторно розчиняли у мінімальній кількості ЕА та додавали гексан для досягнення відношення гексан:ЕА у розчині 3:1 по об'єму. Продукт осаджували з розчину та збирали шляхом фільтрування з одержанням 5-(2-циклопропіл-2+1 оксоетиламіно)-2-фтор-4-метил-бензонітрилу (14,19 г, 61,2 ммоль). РХ/МС (m/z: 233, M ). Етап 3 - Одержання 5-(4-циклопропіл-2-меркапто-1H-імідазол-1-іл)-2-фтор-4-метилбензонітрилу - Сполуки (4) 5-(2-Циклопропіл-2-оксоетиламіно)-2-фтор-4-метилбензонітрил (14,19 г, 61,2 ммоль) розчиняли у льодяній оцтовій кислоті (300 мл). Додавали при перемішуванні тіоціанат натрію (11,9 г, 122,4 ммоль) у вигляді твердої речовини. Реакційну суміш нагрівали до 110ºC впродовж 4 годин, потім розчинник видаляли при зниженому тиску. Осад відбирали у дихлорметані (200 мл) та промивали 200 мл води. Водний екстракт екстрагували додатковою порцією дихлорметану (2 × 200 мл), органічні екстракти об'єднували та сушили над сульфатом магнію. Розчинник видаляли при зниженому тиску, та отриманий маслянистий осад повторно розчиняли у ЕА (50 мл) та додавали 150 мл гексанів. Утворювався темний шар, та у колбу поміщали магнітну мішалку. Інтенсивне перемішування приводило до осадження продукту у вигляді твердої речовини персикового кольору. Продукт збирали шляхом фільтрування з одержанням 5-(4-циклопропіл-2-меркапто-1H-імідазол-1-іл)-2-фтор-4-метилбензонітрилу (14,26 г, 52,23 +1 ммоль). Аналітична РХ/МС (m/z: 274, M ). Етап 4 - Одержання 5-(4-циклопропіл-1H-імідазол-1-іл)-2-фтор-4-метилбензонітрилу Сполуки (5) У тригорлу круглодонну колбу на 500 мл наливали оцтову кислоту (96 мл), воду (19 мл) та перекис водню (30 %, 7,47 мл, 65,88 ммоль). Суміш нагрівали до 45ºC при перемішуванні у атмосфері азоту та слідкували за внутрішньою температурою. Потім додавали невеликими порціями 5-(4-циклопропіл-2-меркапто-1H-імідазол-1-іл)-2-фтор-4-метилбензонітрил (6,00 г, 21,96 ммоль) у вигляді твердої речовини впродовж 30 хвилин при підтриманні внутрішньої температури нижче 55ºC. Після завершення додавання тіоімідазолу реакційну суміш º перемішували протягом 30 хвилин при температурі 45 C та потім прохолоджували до кімнатної температури та повільно додавали 20 % (по масі) розчин сульфату натрію у воді (6 мл). Суміш перемішували протягом 30 хвилин та розчинники видаляли при зниженому тиску. Осад суспендували у 250 мл води та додавали 4 н водний розчин гідроксиду амонію для доведення pН до ~10. Суміш екстрагували дихлорметаном (3 × 200 мл), органічні шари об'єднували, сушили над сульфатом магнію та видаляли розчинник при зниженому тиску. Осад розчиняли у 20 мл ЕА та додавали 80 мл гексанів при перемішуванні. Розчинники зливали, залишаючи маслянистий осад. Зазначений процес повторювали та одержували продукт, 5-(4-циклопропіл1H-імідазол-1-іл)-2-фтор-4-метилбензонітрил, у вигляді в'язкого масла (5,14 г, 21,33 ммоль). +1 Аналітична РХ/МС (m/z: 242, M ). Етап 5 Одержання гідрохлориду 5-(4-циклопропіл-1H-імідазол-1-іл)-2-фтор-4метилбензойної кислоти (6) 5-(4-Циклопропіл-1H-імідазол-1-іл)-2-фтор-4-метилбензонітрил (11,21 г, 46,50 ммоль) поміщали у круглодонну колбу, оснащену зворотним конденсатором, та суспендували у 38 % розчині соляної кислоти (200 мл). Суміш нагрівали до 100ºC впродовж 4,5 годин та потім охолоджували до кімнатної температури. Розчинник видаляли при зниженому тиску з одержанням твердої речовини рожевого кольору, до якого додавали 100 мл ЕА. Твердий продукт збирали шляхом фільтрування та промивали 3 × 100 мл ЕА. До отриманого твердого 9 UA 112098 C2 5 10 15 20 25 продукту додавали 100 мл 10 % метанолу у дихлорметані, суміш перемішували та збирали фільтрат. Зазначену процедуру повторювали з використанням 2 додаткових порцій по 200 мл 10 % розчину метанолу у дихлорметані. Фільтрати об'єднували, та розчинник видаляли при зниженому тиску з одержанням неочищеного гідрохлориду 5-(4-циклопропіл-1H-імідазол-1-іл)-2фтор-4-метилбензойної кислоти. Додаткове очищення не проводили (11,13 г, 37,54 ммоль). +1 Аналітична РХ/МС (m/z: 261, M ). Етап 6 - Одержання 5-(4-циклопропіл-1H-імідазол-1-іл)-2-фтор-N-(6-(4-ізопропіл-4H-1,2,4триазол-3-іл)піридин-2-іл)-4-метилбензаміду - формули (I) Гідрохлорид 5-(4-циклопропіл-1H-імідазол-1-іл)-2-фтор-4-метилбензойної кислоти (1,5 г, 5,07ммоль) суспендували у безводному 1,2-дихлорметані (25 мл) при кімнатній температурі. Додавали оксалілхлорид (0,575 мл, 6,59 ммоль) при перемішуванні у атмосфері азоту з наступним додаванням N, N-диметилформаміду (0,044 мл, 0,507 ммоль). Суміш перемішували впродовж 4 годин при кімнатній температурі та потім розчинник видаляли при зниженому тиску. Осад розчиняли у 25 мл безводного дихлорметану. Швидко додавали 6-(4-ізопропіл-4H-1,2,4триазол-3-іл)піридин-2-амін (1,13 г, 5,58 ммоль) (сполука C) та 4-диметиламінопіридин (0,62 г, 5,07 ммоль) при перемішуванні у атмосфері азоту. Реакційну суміш перемішували впродовж 2 годин при кімнатній температурі та додавали водний насичений розчин NaHCO3 (15 мл). Суміш перемішували впродовж 10 хвилин та шари розділяли та водний шар промивали 1 × 20 мл дихлорметану. Об'єднані органічні шари сушили (MgSO 4), фільтрували та концентрували. Осад розчиняли у мінімальній кількості CH3CN та повільно додавали воду до тих пір, поки тверда речовина не осаджувалась з суміші Зазначену тверду речовину збирали шляхом фільтрування та сушили з одержанням 5-(4-циклопропіл-1H-імідазол-1-іл)-2-фтор-N-(6-(4-ізопропіл-4H-1,2,4триазол-3-іл)піридин-2-іл)-4-метилбензаміду з чистотою ~96 % (1,28 г, 2,88 ммоль). Аналітична +1 РХ/МС (m/z: 446, M ). Матеріал додатково очищували за допомогою ОФ-ВЕРХ (оберненофазової ВЕРХ) з одержанням зразку у вигляді солі HCl, чистого для аналізу. 1 30 35 40 45 50 55 C24H24FN7O-HCl. 446,2 (M+1). H-ЯМР (ДМСО): δ 11,12 (s, 1H), 9,41 (s, 1H), 9,32 (s, 1H), 8,20 (d, J=8,4 Гц, 1H), 8,07 (t, J=8,4 Гц, 1H), 7,95 (d, J=6,4 Гц, 1H), 7,92 (d, J=7,6 Гц, 1H), 7,79 (s, 1H), 7,59 (d, J=10,4 Гц, 1H), 5,72 (sept, J=6,8 Гц, 1H), 2,29 (s, 3H), 2,00-2,05 (m, 1H), 1,44 (d, J=6,8 Гц, 6H), 1,01-1,06 (m, 2H), 0,85-0,89 (m, 2H). Біологічні аналізи Аналіз кіназної активності ASK1 (регулюючої апоптотичні сигнали кінази 1) за допомогою TR-FRET (біохімічна IC50) Здатність сполук інгібувати кіназну активність ASK1 визначали з використанням методу резонансного переносу енергії флуоресценції з розділенням у часі [TR-FRET], у якому як білковий субстрат використовується біотинільований основний мієліновий білок [біотин-MBP]. Обладнання регулювання подачі рідини Beckman Biomek FX використовували для нанесення у невеликого об'єму 384-лункові поліпропіленові планшети [Nunc, #267460] сполуки у концентрації 2 мкл/лунку у 2,44 % водному розчині ДМСО з одержанням кінцевої концентрації сполуки, що становить між 100 мкМ та 0,5 нМ, для аналізу кіназної активності. Прилад Equator (Deerac Fluidics) використовували для нанесення 0,667 нг/мкл у об'ємі 3 мкл/лунку [Upstate Biotechnologies, #14-606, або еквівалентний білок власного виробництва] та 0,1665 нг/мл біотинMBP [Upstate Biotechnologies, #13-111] у буфері (85 мМ MOPS, pН 7,0, 8,5 мМ Mg-ацетат, 5 % гліцерин, 0,085 % NP-40, 1,7 мМ ДТТ та 1,7 мг/мл БСА) у планшети, що містять нанесені сполуки. Фермент попередньо інкубували із сполукою протягом 20 хвилин до ініціації кіназної реакції шляхом додавання 5 мкл/лунку 300 мкМ АТФ у буфері (50 мМ MOPS, pН 7,0, 5 мМ Mg-ацетат, 1 мМ ДТТ, 5 % ДМСО) за допомогою приладу Equator (Deerac Fluidics). Кіназним реакціям дозволяли протікати протягом 20 хвилин при кімнатній температурі та потім зупиняли шляхом додавання 5 мкл/лунку 25 мМ ЕДТА з використанням приладу Equator (Deerac Fluidics). Потім використовували обладнання Biomek FX для переносу кожної завершеної кіназної реакції у об'ємі 1 мкл/лунку у лунки білого полістиролового планшету OptiPlate-1536 [PerkinElmer, #6004299], який містив реагенти для виявлення, по 5 мкл/лунку (1,11 нМ міченого Eu-W1024 антитіла проти фосфотреоніну [PerkinElmer, #CR130-100] та 55,56 нМ алофікоціаніну стрептавідину [PerkinElmer, #CR130-100] у 1x буфері для виявлення LANCE [PerkinElmer, #CR97-100]). Сигнал TR-FRET потім зчитували на планшетному рідері Perkin Elmer Envision 10 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 після інкубації планшетів при кімнатній температурі протягом 2 годин. Лунки, що містять позитивний контроль, що відповідає 100 % інгібуванню, були отримані шляхом зміни порядку додавання розчинів ЕДТА та АТФ, описаних вище. Зазначені лунки та лунки, що відповідають 0 % інгібуванню, що містять краплі 2,44 % ДМСО на початку аналізу, використовували для розрахунків % інгібування для досліджуваних сполук. Результат IC50 сполуки формули (I) для інгібування ASK1 становила 3,0 нМ. Зазначені дані дають підстави вважати, що сполука формули (I) є активним інгібітором ASK1 у присутності конкурентного ліганду АТФ. В удосконаленому варіанті зазначеного вище аналізу інгібуючу активність сполуку згідно з винаходом у відношенні ASK1 оцінювали з використанням аналізу ASK1 за допомогою методу TR-FRET, що визначає кількість фосфату, перенесеного на пептидний субстрат від АТФ. Матеріали та способи Реагенти Дефосфорильовану рекомбінантну людську кіназу ASK1 одержували з компанії Gilead Sciences. Низькомолекулярний інгібітор кінази стауроспорин (каталожний # S6942) та дитіотреїтол (ДТТ, каталожний # 43815-5G) були отримані з компанії Sigma Chemicals (СентЛуїс, Міссурі). АТФ (каталожний # 7724) був отриманий з компанії Affymetrix (Санта-Клара, Каліфорнія), та сполука формули (I) була отримана з компанії Gilead Sciences. Набір для TM аналізу методом гомогенної розділеної у часі флуоресценції KinEASE -STK S3 одержували з компанії Cisbio (Бедфорд, Масочусетс). Усі інші реагенти являли собою комерційно доступні реагенти вищої якості. Аналізи Аналіз дозволяє вимірювати рівень фосфорилювання біотинільованого пептидного субстрату кіназою ASK1 шляхом виявлення за допомогою аналізу методом гомогенної розділеної у часі флуоресценції, HTRF (6,1). Зазначений аналіз являє собою конкурентний імунологічний аналіз резонансного переносу енергії флуоресценції з розділенням у часі (TRFRET), оснований на настанові для аналізу HTRF® KinEASE™-STK від Cisbio (6,1). Досліджувану сполуку, 1 мкМ пептидного субстрату STK3, 4 нМ кінази ASK1 інкубували з 10 мМ MOP буфером, pН 7,0, що містить 10 мМ Mg-ацетат, 0,025 % NP-40, 1 мМ ДТТ, 0,05 % БСА та 1,5 % гліцерину протягом 30 хвилин, потім додавали 100 мкМ АТФ для початку кіназної реакції 3+ та інкубували протягом 3 годин. Додавали пептидне антитіло, мічене 1X Eu криптатним буфером, що містить 10 мМ ЕДТА та 125 нМ стрептавідин XL665, для зупинки реакції та фосфорильований пептидний субстрат виявляли з використанням рідера для декількох міток Envision 2103 від компанії PerkinElmer. Флуоресценцію вимірювали при 615 нм (криптат) та 665 нм (XL665) та відношення 665 нм/615 нм розраховували для кожної лунки. Отриманий у результаті рівень TR-FRET (відношення 665 нм/615 нм) пропорційний ступеню фосфорилювання. У зазначених умовах аналізу ступінь фосфорилювання пептидного субстрату лінійно змінювалася залежно від часу та концентрації ферменту. За допомогою аналітичної системи були отримані узгоджувані результати у відношенні Km та специфічної активності для ферменту. У випадку експериментів щодо інгібування (значення IC 50) значення активності були представлені для постійних концентрацій АТФ, пептиду та декількох фіксованих концентрацій інгібітору. Стауроспорин, неселективний кіназний інгібітор, використовували як позитивний контроль. Усі дані щодо ферментативної активності показані як середнє чотирьох повторів вимірювань. Аналіз даних Значення IC50 розраховували за допомогою наступного рівняння: s y = межа /{1 + (x / IC50) } + вихідне значення, де x та y являють собою концентрацію інгібітору та ферментативну активність, відповідно. Ферментативну активність виражали як кількість фосфату, включеного у пептидний субстрат з АТФ. Межа являє собою максимальне значення y (відсутність інгібітору, контроль ДМСО) та s являє собою фактор нахилу кривої (6,2). Результати IC50 сполуки формули (I) становила 3,2 нМ при зазначених умовах проведення аналізу. Дані показали, що сполука формули (I) є активним інгібітором рецептору ASK-1 1. Клітинний аналіз ASK1 (регулюючої апоптотичні сигнали кінази 1) на клітинах лінії 293 (клітинна EC50) Клітинну активність сполук оцінювали на клітинах, що стабільно експресують AP1:репортерну конструкцію люциферази (клітини 293/AP1-Luc, Panomics Inc., 6519 Dumbarton Circle, Фрімонт, Каліфорнія). Клітини інфікували аденовірусом, що експресує активну кіназу 11 UA 112098 C2 ASK1 (631-1381 кДНК ASK1 щура), яка активує транскрипційний фактор AP-1 та підвищує експресію люциферази. Інгібітори ASK1 знижують ферментативну активність ASK1 і, таким чином, знижують активність транскрипційного фактору AP-1 та експресію люциферази. 1. МАТЕРІАЛИ, НЕОБХІДНІ ДЛЯ ЗДІЙСНЕННЯ ЗАЗНАЧЕНОГО ПРОТОКОЛУ 5 Середовища та реагенти Стабільна клітинна лінія 293, що експресує репортерний AP1 DMEM (з високим вмістом глюкози, без L-глутаміну, з піруватом, з HEPES) DMEM (з високим вмістом глюкози, без L-глутаміну, без пірувату, без HEPES, без фенолового червоного) HEPES, 1 М Піруват натрію, 100 мМ Ембріональна бичача сироватка, "ЕБС" Пеніцилін-стрептоміцин-глутамін, "ПСГ" Гігроміцин B ФСБ (фосфатно-сольовий буфер) Дульбекко (стерильний) Трипсин-ЕДТА (0,25 %) Система аналізу люциферази Steady-Glo Лабораторний посуд Флакони (покриті полі-D-лізином, 2 150 см , з вентильованою кришкою) Планшети (покриті полі-D-лізином, 384-лункові, білі/прозорі, стерильні, TCT) Біла фонова стрічка Клітинні фільтри (40 мкм, нейлонові, з синім кільцем, підходять для конічних пробірок на 50 мл) 10 15 20 25 Компанія - постачальник Каталожний номер Panomics Невідомий MediaTech 15-018-CM Invitrogen 31053-028 Invitrogen Invitrogen 15630-080 11360-070 Hyclone SH30088,03 Invitrogen 10378-016 Calbiochem 400052 MediaTech 21-030-CM Invitrogen 25200-056 Promega E2550 Джерело Каталожний № BD Biosciences 356538 Greiner (через VWR Scientific) 781944 (82051-354) PerkinElmer 6005199 VWR Scientific 21008-949 2. ВИХІДНІ МАТЕРІАЛИ Листок-вкладиш для стабільної клітинної лінії 293/AP1-Luc, Panomics. Листок-вкладиш для системи аналізу люциферази Steady-Glo, Promega. 3. НЕОБХІДНІ СЕРЕДОВИЩА Повне поживне середовище "ППС" DMEM (MediaTech) 10 % ЕБС 1 % ПСГ 100 мкг/мл гіграміцину B Аналітичне середовище, "АС" DMEM (Invitrogen) 25 мМ HEPES 1 мМ піруват натрію 1 % ПСГ 4. СПОСОБИ Підтримування: 293/AP1-Luc. Клітини лінії 293/AP1 підтримували відповідно до інструкцій виробника; при ~80 % моношару клітини збирали у флакони T150, як слідує далі: Відбирали середовище, обережно промивали ~12 мл стерильного D-ФСБ, рідину відбирали. 12 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 Додавали 5 мл розчину трипсин-ЕДТА, обережно нахиляли флакон, щоб покрити його поверхню, та інкубували протягом ~5 хвил. при 37 °C. Флакон не закривали кришкою; додавали 5 мл ППС, промивали флакон 4X суспензією клітин, переносили у конічну пробірку на 50 мл, центрифугували протягом 5 хвил. при 1200 об./хвил. Відбирали середовище від клітинного осаду, додавали 20-30 мл ППС, ресуспендували осад 6 разів шляхом піпетування, пропускали через клітинний фільтр, щоб розбити кластери (при необхідності), та рахували клітини за допомогою гемоцитометру. 1 день аналізу: Збирали клітини, як описано вище, за винятком ресуспендування клітинного осаду. 5 Клітини рахували та розводили до концентрації 1,5 × 10 клітин на мл; додавали аденовірус для досягнення співвідношення 5 інфекційних одиниць на клітину. Відбирали (20-30 мл) та поміщали клітини на покриті полі-D-лізином 384-лункові планшети 4 Greiner у концентрації 1,2 × 10 клітин на лунку за допомогою приладу uFill, BioTek (80 мкл на лунку). Негайно у планшети вводили серії доз сполуки по 0,4 мкл (у 100 % ДМСО) та інкубували 24 години у інкубаторі із зволоженням (37 °C, 5 % CO2). 2 день аналізу: Планшети обробляли (у відповідності з інструкціями виробника) як слідує далі: Планшети поміщали у ламінарний бокс та залишали без кришки на 30 хвилин при кімнатній температурі для охолодження. Видаляли 60 мкл АС з аналізованих лунок. Додавали по 20 мкл на лунку субстрату Steady-Glo, Firefly, залишали на 10-20 хвилин при кімнатній температурі. Дно аналітичного планшету покривали білою фоновою стрічкою. Дані отримували за допомогою флуоресцентного планшетного рідеру. Лунки з позитивним контролем, що відповідали 100 % інгібуванню, отримували шляхом інфікування клітин аденовірусом, що експресує мутант ASK1 з інактивованою каталітичною активністю, що містить заміну лізину на аргінін у положенні залишку 709. Результат EC50 сполуки формули (I) становила 2,0 нМ. Визначення Kd Аналізи кіназної активності Штами фага T7 з кіназною міткою одержували у хазяїні E. coli, отриманому зі штаму BL21. E. coli ростили до фази логарифмічного росту та інфікували фагом T7 та інкубували при перемішуванні при температурі 32 °C до лізування. Лізати центрифугували та фільтрували для видалення клітинних залишків. Інші кінази продукували у клітинах лінії HEK-293 та потім мітили ДНК для виявлення за допомогою кількісної ПЛР. Покриті стрептавідином магнітні кульки обробляли біотинільованими низькомолекулярними лігандами протягом 30 хвилин при кімнатній температурі для одержання смол афінності для аналізу кіназної активності. Покриті лігандом гранули блокували за допомогою надлишку біотину та промивали блокуючим буфером (SeaBlock, Pierce), 1 % БСА (бичачий сироватковий альбумін), 0,05 % твін 20, 1 мМ ДТТ (дитіотреїтол) для видалення незв'язаного ліганду та зниження неспецифічного зв'язування. Реакції зв'язування проводили шляхом об'єднання кіназ, пов'язаних з лігандом гранул для аналізу афінності та досліджуваних сполук у 1x зв'язуючому буфері (20 % SeaBlock, 0,17x ФСБ, 0,05 % Твін 20, 6 мМ ДТТ). Усі реакції проводили у полістиролових 96-лункових планшетах у кінцевому об'ємі 0,135 мл. Аналітичні планшети інкубували при кімнатній температурі при перемішуванні протягом 1 години та гранули для аналізу афінності промивали промивним буфером (1x ФСБ, 0,05 % Твін 20). Потім гранули ресуспендували у елююючому буфері (1x ФСБ, 0,05 % твін 20, 0,5 мкМ небіотинільований ліганд для аналізу афінності) та інкубували при кімнатній температурі при перемішуванні протягом 30 хвилин. Концентрацію кінази в елюатах вимірювали за допомогою кількісної ПЛР. Константи зв'язування (Kd) розраховували за допомогою стандартної кривої залежності ефекту від дози з використанням рівняння Хіла. Результат Kd для сполуки формули (I) становила 0,24 нМ. Зазначені дані дають підстави вважати, що сполука формули (I) активно зв'язується з рецептором ASK1 за відсутності АТФ. Визначення відсоткової кількості сполуки, пов'язаної із плазмою крові Схема експерименту: У зазначених експериментах використовували 1 мл тефлонових діалізних лунок від компанії 13 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 Harvard Apparatus (Холістон, Масачусетс, США). До початку проведення дослідження діалізну мембрану змочували протягом приблизно двох годин на 0,133 M фосфатному буфері, pН 7,4. Сполуку у невеликій концентрації 2 мкМ вводили у 1 мл плазми або 1 мл клітинного поживного середовища. Загальний об'єм рідини з кожної сторони лунки становив 1 мл. Через 3 години інкубації у рівноважному стані на водній бані при 37 °C аліквоти зразків з кожної сторони лунки поміщали у підходящі пробірки, що містять або 1 мл людської плазми (клітинне поживне) середовище, або буфер. Пробірки зі зразком зважували та дані фіксували. Відбирали аліквоти по 100 мкл та додавали до 400 мкл розчину, що зупиняє реакцію, (50 % метанол, 25 % ацетонітрил, 25 % вода та внутрішній стандарт). Зразки перемішували на вортексі та центрифугували протягом 15 хвилин при 12000 G. 200 мкл супернатанту відбирали та поміщали у новий 96-лунковий планшет. Додавали додаткові 200 мкл суміші ACN:вода, 1:1. Потім вміст планшету перемішували на вортексі та піддавали РХ-МС аналізу. Відсоток незв'язаної аналізованої сполуки у плазмі розраховували з використанням наступного рівняння: % Незв'язаної сполуки = 100(Cf /Ct) де Cf та Ct являють собою концентрації у буфері та плазмі після діалізу, відповідно. Результати Відсоток незв'язаної сполуки формули (I), виміряної у плазмі крові людини, становив 11,94 %. Визначення відношення відтоку для клітин CACO-2 Експериментальна частина: Клітини лінії Caco-2 підтримували у модифікованому Дульбекко середовищі Ігла (DMEM) з піруватом натрію, Glutamax з додаванням 1 % розчину пеніцилін/стрептоміцин, 1 % NEAA та 10 % ембріональної бичачої сироватки у інкубаторі, для якого були встановлені температура 37ºC, вологість 90 % та вміст CO2 5 %. Клітини Caco-2 на 62-72 пасажі висівали у концентрації 2100 клітин/лунку та ростили до моношару протягом щонайменше 21 дня на 24-лункових PET (поліетилен - терефталатних) планшетах (BD Biosciences). Акцепторна лунка містила HBSSбуфер (10 мМ HEPES, 15 мМ глюкоза з pН, доведеним до pН 6,5) з додаванням 1 % БСА, pН, доведений до pН 7,4. Після попереднього зрівноважування буфером переносу зчитували значення TEER для дослідження цілісності досліджуваної мембрани. Додавали буфери, що містять досліджувані сполука, та відбирали 100 мкл розчину через 1 та 2 години з акцепторних камер. Видалений буфер заміщували на свіжий буфер, та всі розрахунки коректували з урахуванням видаленого матеріалу. Експеримент проводили у двох повторах. Усі зразки відразу збирали у 400 мкл 100 % ацетонітрилової кислоти для осадження білку та стабілізації досліджуваних сполук. Дозу вводили до клітин з апікального або базолатерального боку для визначення прямої (від A до B) та зворотної (від B до A) проникності. Проникність через безклітковий трансвел також визначали як міру клітинної проникності через мембрану та неспецифічного зв'язування. Для дослідження неспецифічного зв'язування та нестабільності сполуки визначали відсоткове відновлення. Зразки аналізували за допомогою РХ/МС/МС. Очевидну проникність, Papp, та % відновлення розраховували, як слідує далі: Papp = (dR/dt) x Vr/(A x D0) % Відновлення = 100 x ((Vr x R120) + (Vd x D120))/ (Vd x D0) де: dR/dt являє собою зниження сумарної концентрації у акцепторній камері в залежності від часу, мкМ/с, визначене на основі концентрацій акцептору, виміряних через 60 та 120 хвилин. 3 Vr та Vd являють собою об'єм у акцепторній та донорній камері у см , відповідно. 2 A являє собою площу клітинного моношару (0,33 см ). D0 та D120 являють собою виміряну концентрацію у донорній камері на початку та кінці експерименту, відповідно. R120 являє собою концентрацію у акцепторній камері у кінці експерименту (120 хвилин). Класифікація абсорбції та відтоку: -6 Papp (A до B) ≥ 1,0 × 10 см/с -6 Висока -6 1,0 × 10 см/с > Papp (A до B) ≥ 0,5 × 10 см/с Середня -6 Papp (від A до B) < 0,5 × 10 см/с Papp (від B до A)/ Papp (від A до B) ≥ 3 Низька Значний відтік % відновлення < 20 % Може впливати на виміряну проникність Papp за відсутності клітин < 15 Може впливати на виміряну проникність 14 UA 112098 C2 5 10 15 20 25 30 35 40 45 Результат Спостерігали, що значення A→B сполуки формули (I) для клітин CACO становило 27; та значення B→A для клітин CACO становило 35, що приводило до значення відношенні відтоку (B→A)/(A→B) 1,3. Визначення метаболічної стабільності у мікросомальній фракції печінки: Експериментальна частина: Метаболічну стабільність оцінювали з використанням кофакторів для окисного метаболізму (NADPH) та кон'югації (UDP-глюкуронова кислота (UDPGA)). По дві аліквоти сполуки формули (I) (3 мкл 0,5 мМ вихідного розчину ДМСО) або метаболічно стабільних стандартів (буспірон) додавали до вихідного розчину мікросом, розведеного калій-фосфатним буфером, pН 7,4, для одержання концентрації білку 1,0 мг/мл, та що містять аламетицин як пермеабілізуючий агент. Метаболічні реакції ініціювали шляхом додавання відтворюючої системи NADPH та кофактору UDPGA. Кінцевий склад кожної реакції був наступним: 3 мкМ досліджуваної сполука, 1 мг мікросомального білку/мл, 5 мМ UDPGA, 23,4 мкг/мл аламетицину, 1,25 мМ NADP, 3,3 мМ глюкозо-6-фосфату, 0,4 од/ мл глюкозо-6-фосфатдегідрогенази а та 3,3 мМ MgСl2 у 50 мМ калій-фосфатному буфері, pН 7,4. Через 0, 2, 5, 10, 15, 30, 45 та 60 хвил. аліквоти реакційної суміші по 25 мкл переносили у планшети, що містять 250 мкл IS/Q (зупиняючого розчину, що містить внутрішній стандарт). Після зупинки реакцій планшети центрифугували при 3000 g протягом 30 хвилин та аліквоти по 10 мкл супернатанту аналізували з використанням РХ/МС для одержання відношення площі піку сполуки, що аналізують, /внутрішнього стандарту. Метаболічну стабільність у мікросомальних фракціях печінки визначали шляхом вимірювання швидкості зникнення сполуки формули (I). Дані (% збереження вихідної сполуки) наносили на напівлогарифмічну шкалу та приводили за допомогою експонентного наближення: Ct C0 eK t та T1/ 2 ln 2 / K , де Ct % вихідної речовини, що залишилася на момент часу t C0 % вихідної речовини, що залишилася на момент часу 0 t час (годин) -1 K Константа швидкості виведення першого порядку (години ) T½ Час напіввиведення in vitro (години) Прогнозований печінковий кліренс розраховували, як слідує далі {посилання 1}: CL int K V YP P або CL int K V YH H CL h (CL int Qh ) /(CL int Qh ) , де CLh Прогнозований печінковий кліренс (л/год./кг маси тіла) CLint Внутрішній печінковий кліренс (л/год./кг маси тіла) V Інкубаційний об'єм (л) YP Вихід мікросомального білку (мг білку/кг маси тіла) YH Вихід гепатоцитів (мільйонів клітин/кг маси тіла) P Маса булку у інкубаційному об'ємі (мг) H Кількість гепатоцитів в інкубаційному об'ємі (млн) Qh Печінковий кровоток (л/год./кг маси тіла) Потім розраховували прогнозоване печінкове виведення шляхом порівняння прогнозованого печіночного кліренсу з печінковим кровотоком. Сполуку вважали стабільною, якщо зниження концентрації субстрату було 395 хвил. у мікросомальних фракціях та > 39,5 годин у гепатоцитах). Значення, використовувані для розрахунків прогнозованого печінкового кліренсу, показані у таблицях нижче: 15 UA 112098 C2 Таблиця 1 Значення, отримані з аналізу мікросомальної стабільності, використовувані для розрахунку прогнозованого печінкового кліренсу Мікросоми печінки Вид Щур Яванський макак Макак-резус Собака Людина V (л) 0,001 0,001 0,001 0,001 0,001 P (мг) 1,0 1,0 1,0 1,0 1,0 Y (мг/кг) 1520 684 1170 1216 977 Qh (л/кг) 4,2 1,6 2,3 1,8 1,3 Результат: Прогнозований печінковий кліренс у людини, визначений на основі експериментів на мікросомальних фракціях in vitro, становив 0,1 л/год./кг. 5 10 15 20 25 30 35 40 Визначення CL та Vss у щурів для досліджуваної сполуки Фармакокінетика досліджуваних сполук після ВВ інфузії дози 1 мг/кг та пероральної дози 5,0 мг/кг у щурів Досліджуваний виріб та лікарська форма Для ВВ введення досліджувану сполуку готували у суміші 60:40 PEG 400:вода з 1 еквівалентом HСl у концентрації 0,5 мг/ мл. Лікарська форма являла собою розчин. Для ПО (перорального) введення досліджувану сполуку готували у суміші 5/75/10/10 етанол/ПГ/солютол/вода у концентрації 2,5 мг/ мл. Лікарська форма являла собою розчин. Використовувані тварини Кожна із груп внутрішньовенного (ВВ) та перорального (ПО) введення дози складалася з 3 самців щурів лінії SD. На момент введення дози маса тварин, у цілому, становила між 0,317 та 0,355 кг. Тварин не годували протягом ночі до введення дози та протягом до 4 годин після введення дози. Дозування Для групи ВВ інфузії досліджувану сполуку вводили шляхом інфузії протягом 30 хвилин. Швидкість інфузії регулювали відповідно до маси тіла кожної тварини для доставки дози 1 мг/кг у об'ємі 2 мл/кг. Для групи перорального введення дози досліджуваний виріб вводили шляхом примусового годування у об'ємі 2 мл/кг для дози 5,0 мг/кг. Збір зразку Серії зразків венозної крові (приблизно по 0,4 мл кожен) відбирали у кожної тварини у певні M точки часу після введення дози. Зразки крові збирали у ампули Vacutainert (Becton-Disckinson Corp, Нью-Джерсі, США), що містять ЕДТА як антикоагулянт, та швидко піддавали центрифугуванню на льоді для одержання плазми. Визначення концентрації сполуки формули (I) у плазмі крові Для вимірювання концентрації досліджуваної сполуки у плазмі крові використовували метод РХ/МС/МС. Розрахунки Некомпартментний фармакокінетичний аналіз проводили на даних концентрація у плазмі час. Результати Для сполуки формули (I) CL становила 0,09 л/год./кг; пероральна біодоступність становила 75 %; t1/2 становив 5,07 годин та Vss становив 0,55 л/кг у щурів. Аналіз інгібування Cyp Мета: Оцінити потенційну здатність досліджуваної сполуки інгібувати основні ізоформи цитохрому P450, CYP1A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 та CYP3A4 (2 субстрати). Визначення IC50 для інгібування цитохрому P450 (8 ізоформ, 9 субстратів) Короткий опис протоколу 16 UA 112098 C2 5 10 15 20 25 30 35 40 45 50 55 60 Досліджувану сполуку (0,1 мкМ - 25 мкМ) інкубували з мікросомами печінки людини та NADPH у присутності маркерного субстрату, специфічного для ізоформи цитохрому P450. У випадку реакцій, специфічних для CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 та CYP3A4, за метаболітами слідкували за допомогою мас-спектрометрії. За активністю CYP1A слідкували шляхом вимірювання утворюваного флуоресцентного метаболіту. Зниження утворення метаболіту у порівнянні з носієм як контроль використовували для розрахунку значення IC 50 (концентрація досліджуваної сполуки, яка забезпечує 50 % інгібування). Вимоги до проведення аналізу 500 мкл 10 мМ розчину досліджуваної сполуки у ДМСО. Хід експерименту Інгібування CYP1A Шість концентрацій досліджуваної сполуки (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,3 %) інкубували з мікросомами печінки людини (0,25 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату етоксирезоруфину (0,5 мкМ) впродовж 5 хвил. при 37 °C. Селективний інгібітор CYP1A, альфа-нафтофлавон, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP2B6 Шість концентрацій досліджуваної сполуки (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,3 %) інкубували з мікросомами печінки людини (0,1 мг/мл) та NADPH (1 мМ) у присутності маркерного субстрату бупропіону (110 мкМ) впродовж 5 хвил. при 37 °C. Селективний інгібітор CYP2B6, тиклопідин, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP2C8 Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,3 %) інкубували з мікросомами печінки людини (0,25 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату паклітакселу (7,5 мкМ) впродовж 30 хвил. при 37 °C. Селективний інгібітор CYP2C8, монтелукаст, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP2C9 Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,25 %) інкубували з мікросомами печінки людини (1 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату толбутаміду (120 мкМ) впродовж 60 хвил. при 37 °C. Селективний інгібітор CYP2C9, сульфафеназол, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP2C19 Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,25 %) інкубували з мікросомами печінки людини (0,5 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату мефенітоїну (25 мкМ) впродовж 60 хвил. при 37 °C. Селективний інгібітор CYP2C19, транілципромін, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP2D6 Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,25 %) інкубували з мікросомами печінки людини (0,5 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату декстрометорфаном (5 мкМ) впродовж 5 хвил. при 37 °C. Селективний інгібітор CYP2D6, хінідин, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP3A4 (мідазолам) Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,26 %) інкубували з мікросомами печінки людини (0,1 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату мідазоламу (2,5 мкМ) впродовж 5 хвил. при 37 °C. Селективний інгібітор CYP3A4, кетоконазол, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. Інгібування CYP3A4 (тестостерон) Досліджувану сполуку у шести концентраціях (0,1, 0,25, 1, 2,5, 10, 25 мкМ у ДМСО; кінцева концентрація ДМСО = 0,275 %) інкубували з мікросомами печінки людини (0,5 мг/ мл) та NADPH (1 мМ) у присутності маркерного субстрату тестостерону (50 мкМ) впродовж 5 хвил. при 37 °C. Селективний CYP3A4 інгібітор, кетоконазол, використовували у скринінговому аналізі разом з досліджуваними сполуками як позитивним контролем. У випадку інкубації з CYP1A реакції зупиняли метанолом і за утворенням метаболіту, резоруфіну, стежили за допомогою флуоресценції (довжина хвилі порушення = 535 нм, довжина 17 UA 112098 C2 5 10 хвилі випромінювання = 595 нм). У випадку інкубації з CYP2B6, CYP2C9, CYP2C19, CYP2D6 та CYP3A4 реакції зупиняли за допомогою метанолу. Зразки потім центрифугувалита супернатанти об'єднували для одночасного аналізу 4-гідрокситолбутаміду, 4гідроксимефенітоїну, декстрарфану та 1-гідроксімідазоламу за допомогою РХ-МС/МС. Гідроксибупропіон, 6-гідроксипаклітаксел та 6-гідрокситестостерон аналізували окремо за допомогою РХ-МС/МС. До кінцевого зразку до початку проведення аналізу додавали мурашину кислоту у деіонізованій воді (кінцева концентрація = 0,1 %), що містить внутрішній стандарт. Зниження утворення метаболітів у порівнянні з носієм як контроль використовували для розрахунків значення IC50 (концентрації досліджуваної сполуки, яка забезпечує 50 % інгібування). Результати Ізоформа CYP450 1A 1A2 2B6 2C8 2C9 2C19 2D6 3A4 3A4 15 20 25 30 35 40 45 Субстрат Етоксирезоруфін Фенацетин Бупропіон Паклітаксел Толбутамід S-мефенітоїн Декстрометорфан Мідазолам Тестостерон Метаболіт Резоруфін Ацетамінофен Гідроксибупропіон 6-Гідроксипаклитаксел 4-Гідрокситолбутамід 4-Гідроксимефенітоїн Декстрорфан Гідроксімідазолам 6 -Гідрокситестостерон Розрахована IC50 (мкМ) > 25 мкМ > 25 мкМ 19,2 мкМ 21,6 мкМ >25 мкМ > 25 мкМ 17,7 мкМ 2,7 мкМ 10,5 мкМ Загальна схема дослідження для моделі фіброзу нирок, викликаного однобічною обструкцією сечоводу (ООС), у щурів. Самців щурів лінії Sprague-Dawley забезпечували нормальним кормом, утримували у стандартних умовах та дозволяли акліматизуватися протягом щонайменше 7 днів до проведення операції. На початку дослідження щурів ділили на групи на основі маси тіла та вводили (2 мл/кг перорально два рази на день) носій, сполуку у одній із чотирьох доз (1, 3, 10 або 30 мг/кг) шляхом примусової годівлі. Щурів піддавали анестезії за допомогою ізофлурану на носовий конус та проводили лапаротомію. Щурів піддавали повній обструкції правого сечоводу (ООС) за допомогою стерилізованих нагріванням інструментів та асептичного хірургічного методу. Щурам вводили 50 мкл пеніциліну G (внутрішньом'язово) безпосередньо після операції. Щурам дозволяли відновлюватися у чистій клітці з підігрівом до повернення у нормальні умови віварію. Щурам вводили сполуку у дозі, описаній вище, два рази на день (з 12-годинними інтервалами) протягом наступних один за одним 7 днів. На 7 день після операції щурів піддавали анестезії за допомогою ізофлурану та збирали сироватку, плазму та сечу. Потім тварин присипляли, вилучали нирки та брали біопсії коркової речовини нирки для морфологічного, гістологічного та біохімічного аналізу. Усі тканини для біохімічного аналізу були швидкозамороженими у рідкому азоті та зберігалися при -80 °C, тканини для гістологічного аналізу фіксували у 10 % нейтральному забуференому формаліні. Фіброз нирки оцінювали шляхом вимірювання кількості колагену IV у нирці за допомогою методу ІФА та шляхом оцінки накопичення позитивно пофарбованих на альфа-гладком'язовий актин міофібробластів у нирці за допомогою імуногістохімії. Для проведення першого аналізу невелику частину замороженої коркової речовини нирки переносили та гомогенізували у RIPAбуфері, потім центрифугували при 14000 x g протягом 10 хвилин при 4 °C. Супернатант збирали у попередньо охолоджені ампули та визначали концентрацію білку. Еквівалентна кількість загального білку піддавали ІФА-аналізу на колаген IV (Exocell) відповідно до інструкцій виробника. Фіксовану формаліном та залиту парафіном тканину нирки забарвлювали на альфагладком'язовий актин, як описано раніше (Stambe et al., The Role of p38 Mitogen-Activated Protein Kinase Activation in Renal Fibrosis J Am Soc Nephrol 15: 370-379, 2004). Результати: Було виявлено, що сполука формули (I) у дозах від 3 до 30 мг/кг значно знижує індукцію колагену IV у нирці (фігура 1) та накопичення позитивне пофарбованих на альфагладком'язовий актин міофібробластів (фігура 2). Порівняльні дані для сполуки формули (I) та сполук порівняння У наступній таблиці представлені порівняльні результати для сполуки формули (I) та сполук порівняння A та B, описаних у патентній публікації США № 2001/00095410A1, опублікованій 13 18 UA 112098 C2 січня 2011 р. Необхідно відзначити, що експерименти, порівняння результатів яких наведене нижче, проводили у подібних умовах, але необов'язково одночасно. Таблиця Сполука формули (I) 3 2 17 27/ 35 Сполука A 5 3,4 71 (4X) 3,1 /18,5 Сполука B 6,5 18 (9X) 563 (33X) 0,26 / 4 1,3 12 6 4,8 15,4 3,2 11 1,1 (10X) 4 (2,8X) 647 0,55 0,11 75 5,07 IC50 (нМ) EC50 (нМ) PBadj EC50 (нМ) CACO (A/B, B/A) Відношення відтоку (B/A)/(A/B) fu IC50 тестостерону (TST) для Cyp3A4) (мкМ) IC50 /PBAdj.EC50 для Cyp3A4 Vss (л/кг) у щурів CL (л/год./кг) у щурів %F у щурів t ½ у щурів (год.) 15 (43X) 0,17 (3,2X) 0,30 (2,7X) 11 (6,8X) 0,59 (8,6X) 7 (92X) 0,54 0,39 (3,6X) 50 (1,5X) 1,3 (3,9X) () значення у круглих дужках показують, у скільки разів сполука формули (I) має перевагу у порівнянні із зазначеною сполукою для зазначеного параметру. 5 10 15 20 З описаних вище порівняльних даних можна вивести наступне: EC50 сполуки формули (I) порівнянна із EC50 сполуки A. Функціональна EC50 сполуки формули (I) порівнянна з EC50 сполук A та B. Скоректована з урахуванням зв'язування з білком EC50 сполуки формули (I) у 4 рази нижче у порівнянні із сполукою A та у 33 рази нижче у порівнянні із сполукою B. Сполука формули (I) є більш слабким інгібітором Cyp3A4 у порівнянні із сполуками A та B. Значення IC50 CYP3A4/ Pbadj.EC50 сполуки формули (I) у 43 рази вище у порівнянні із сполукою формули A та у 92 рази вище у порівнянні із сполукою формули B. Значення CL сполуки формули (I) у щурів у 2,7 рази нижче у порівнянні із сполукою формули A та у 3,6 рази нижче у порівнянні із сполукою формули B. Процентна біодоступність сполуки формули (I) у щурів у 6,8 разів вище у порівнянні із сполукою A та у 1,5 рази вище у порівнянні із сполукою B. Період напіввиведення сполуки формули (I) у щурів у 8,6 разів більше у порівнянні із сполукою A та у 3,9 разів більше у порівнянні із сполукою B. Наведені вище дані дають достатні підстави вважати, що сполука формули (I) має несподівані та кращі властивості у порівнянні із сполуками формули A та B; і що зазначена сполука формули (I), імовірно, є кращим кандидатом для подальшої розробки засобів для лікування хронічної ниркової недостатності, фіброзу легенів та/або нирок та/або кардіоренальних захворювань. ФОРМУЛА ВИНАХОДУ 25 1. Сполука формули (І): N O N N H F N N N N , (І) 30 19 UA 112098 C2 а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-N-(6-(4-ізопропіл-4Н-1,2,4-триазол-3-іл)піридин-2-іл)2-фтор-4-метилбензамід, або її фармацевтично прийнятна сіль. 2. Сполука формули (І): N O N N H N N N N F , (І) 5 10 15 20 25 а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-2-фтор-N-(6-(4-ізопропіл-4Н-1,2,4-триазол-3іл)піридин-2-іл)-4-метилбензамід. 3. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки або солі за п. 1 та фармацевтично прийнятний носій. 4. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки за п. 2 та фармацевтично прийнятний носій. 5. Спосіб лікування хронічної ниркової недостатності, що включає введення терапевтично ефективної кількості сполуки за п. 2 або її фармацевтично прийнятної солі пацієнту, який цього потребує. 6. Спосіб лікування діабетичної нефропатії, що включає введення терапевтично ефективної кількості сполуки за п. 2 або її фармацевтично прийнятної солі пацієнту, який цього потребує. 7. Спосіб лікування фіброзу нирок, фіброзу печінки або фіброзу легенів, що включає введення терапевтично ефективної кількості сполуки або солі за п. 1 пацієнту, який цього потребує. 8. Спосіб лікування діабетичного захворювання нирок, що включає введення терапевтично ефективної кількості сполуки або солі за п. 1 пацієнту, який цього потребує. 9. Застосування сполуки або солі за п. 1 для одержання лікарського засобу для лікування хронічної ниркової недостатності. 10. Застосування сполуки або солі за п. 1 у терапії. 11. Проміжна сполука формули: H2N N N N N , 30 а саме 2-аміно, 5-(4-ізопропіл-4Н-1,2,4-триазол-3-іл)піридин або його сіль, або захищена форма. 12. Проміжна сполука формули: N O N OH F 35 , а саме 5-(4-циклопропіл-1Н-імідазол-1-іл)-2-фтор-4-метилбензойна захищена форма. 20 кислота, її сіль або UA 112098 C2 Комп’ютерна верстка Л. Бурлак Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 21
ДивитисяДодаткова інформація
Назва патенту англійськоюApoptosis signal-regulating kinase inhibitor
Автори англійськоюNotte, Gregory
Автори російськоюНоттэ Грэгори
МПК / Мітки
МПК: A61K 31/4164, A61P 13/12, C07D 401/14, C07D 233/56, C07D 401/04
Мітки: апоптотичні, сигнали, кінази, інгібітор, регулюючої
Код посилання
<a href="https://ua.patents.su/23-112098-ingibitor-regulyuyucho-apoptotichni-signali-kinazi.html" target="_blank" rel="follow" title="База патентів України">Інгібітор регулюючої апоптотичні сигнали кінази</a>
Інгібітор кінази

Номер патенту: 94622
Опубліковано: 25.05.2011
Автори: Зілберстейн Ешер, Аллен Елізабет М., Гіллеспі Тімоті А., Ейнотт Пол, Ю Кін Т.
МПК: C07D 487/04, A61P 19/02, A61K 31/4985
Формула / Реферат:
1. Сполука формули (І): (І)або відповідний N-оксид, або проліки, або фармацевтично прийнятна сіль, або сольват такої сполуки і N-оксид і проліки солі і сольвату. 2. Фармацевтична композиція, що містить сполуку за п. 1 разом із щонайменше одним фармацевтично прийнятним носієм або наповнювачем. 3. Спосіб лікування...
v-карбоксіарил заміщена дифенілсечовина як інгібітор raf-кінази

Номер патенту: 73731
Опубліковано: 15.09.2005
Автори: Вуд Джілл Е., Дюма Жак, Сміт Роджер А., Натеро Рейна, Монахан Мері-Кетрін, Скотт Уільям Дж., Ренік Джоел, Кхайр Удей, Сіблі Роберт Н., Ловінгер Тімоті Б., Рідл Бернд
МПК: C07C 275/36, C07C 275/28, C07C 275/30, C07C 311/29, C07D 213/81, C07D 213/75, C07D 295/192, C07D 209/48, C07C 275/32, C07D 295/135, C07C 317/22, C07C 275/40, C07D 295/13
Мітки: дифенілсечовина, raf-кінази, v-карбоксіарил, заміщена, інгібітор
Формула / Реферат:
1. Сполука Формули І:A-D-B (І)або її фармацевтично прийнятна сіль, де D — це -NH-C(O)-NH-, A — це заміщена складова, що включає до 40 атомів вуглецю формули -L-(M-L1)q, де L — це 5- або 6-членна циклічна структура, з'єднана безпосередньо з D, L1 містить заміщену циклічну складову, що має щонайменше 5 членів, М — зв'язуюча група, що має щонайменше один атом, q — це число від 1 до 3, і кожна циклічна структура L і L1 містить 0—4...
Інгібітор p38 map-кінази

Номер патенту: 105014
Опубліковано: 10.04.2014
Автори: Чаррон Кетрін Елізабет, Мюррей Пітер Джон, Уільямс Джонатан Гарет, Іто Казухіро, Кінг-Андервуд Джон, Рейппорт Уільям Гарт, Оніонс Стюарт Томас, Стронг Пітер
МПК: A61K 31/415, A61P 29/00, C07D 401/12, A61P 11/00
Мітки: map-кінази, інгібітор
Формула / Реферат:
1. Сполука формули (І) (І)або її фармацевтично прийнятна сіль або сольват, включаючи всі її таутомери.2. Фармацевтична композиція, яка містить сполуку за п. 1 в комбінації з одним або більше фармацевтично прийнятними розріджувачами або носіями.3. Спосіб лікування стану, вибраного з:COPD (включаючи хронічний бронхіт і емфізему), астми,...
2-[(5-хлор-2-{[3-метил-1-(1-метилетил)-1н-піразол-5-іл]аміно}-4-піридиніл)аміно]-n-метилбензамід як інгібітор кінази фокальної адгезії (fak)

Номер патенту: 103208
Опубліковано: 25.09.2013
Автори: Каспарец Іржі, Адамс Джеррі Лерой, Джонсон Нейл В., Пенг Сінь, Фейтг Томас Х., Лін Хонг, Се Рен, Меллінджер Марк
МПК: A61K 31/535, A61P 35/00
Мітки: кінази, адгезії, фокальної, fak, інгібітор, 2-[(5-хлор-2-{[3-метил-1-(1-метилетил)-1н-піразол-5-іл]аміно}-4-піридиніл)аміно]-n-метилбензамід
Формула / Реферат:
1. Сполука, яка являє собою 2-[(5-хлор-2-{[3-метил-1-(1-метилетил)-1Н-піразол-5-іл]аміно}-4-піридиніл)аміно]-N-метилбензамід та має структурну формулу ,або її фармацевтично прийнятна сіль.2. Фармацевтична композиція, що містить сполуку за п. 1 і фармацевтично прийнятний носій.3. Фармацевтична композиція за п. 2, яка відрізняється тим, що має...
Фармацевтична композиція, що містить інгібітор альдегідредуктази та інгібітор ангіотензинперетворюючого ферменту

Номер патенту: 64761
Опубліковано: 15.03.2004
Автори: Коттер Мері Енн, Кері Френк, Таффін Девід Патрік, Кемерон Норман Юджін
МПК: A61K 31/165, A61K 38/55, A61K 45/06
Мітки: інгібітор, альдегідредуктази, фармацевтична, ангіотензинперетворюючого, композиція, містить, ферменту
Формула / Реферат:
1. Фармацевтична композиція, яка відрізняється тим, що включає інгібітор альдегідредуктази та інгібітор ферменту перетворення ангіотензину (АСЕ) разом з фармацевтично прийнятним носієм і/або розріджувачем.2. Фармацевтична композиція за п. 1, яка відрізняється тим, що інгібітор альдегідредуктази вибирають з епалрестату, толрестату, понолрестату, зополрестату, AD-5467, SNK-860, ADN-138, AS-3201, зенарестату, сорбінілу, метосорбінілу,...
Попередній патент: Тепловий акумулятор
Наступний патент: Фармацевтична композиція з гемостатичною дією
Випадковий патент: Сполучений пристрій для викиду газів в атмосферу