Біополярні солі транс-каротиноїдів та їх використання























Формула / Реферат
1. Сполука – біполярна транс-каротиноїдна сіль, що має структуру:
YZ-TCRO-ZY,
де:
Y - катіон, яким є одновалентний іон металу, вибраний з групи, яку складають Na+, K+ та Li+, або органічний катіон, вибраний з групи, яку складають R4N+, R3S+, де R - H або CnH2n+1, де n – 1-10;
Z – полярна група, асоційована з катіоном, причому Z вибрана з групи, яку складають карбоксильна група (СOO-), сульфатна група (OSO3-) або монофосфатна група (ОРО3-), (ОР(ОН)O2-), дифосфатна група, трифосфат або їхні комбінації;
TCRO – транс-каротиноїдний скелет, який має бічні групи, де бічна група Х, яка може бути однаковою або різною, є лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю, або галогеном, і
де TCRO є
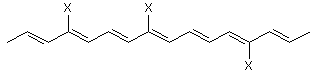 ,
,
де група X, яка може бути однаковою або різною, є
і) лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або
іі) галогеном, або де TCRO є
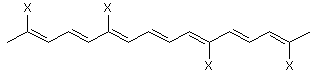 ,
,
де група X, яка може бути однаковою або різною, є
і) лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або
іі) галогеном, або де TCRO є
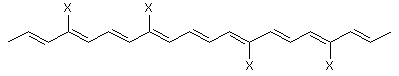 ,
,
де група X, яка може бути однаковою або різною, є
і) лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або
іі) галогеном, або де TCRO є
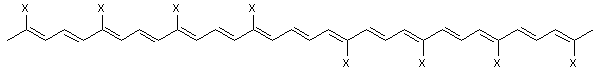 ,
,
де група X, яка може бути однаковою або різною, є
і) лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або
іі) галогеном;
причому дана сполука - біполярна транс-каротиноїдна сіль - не є ТКН (транс-кроцетинатом натрію), або сіллю норбіксину.
2. Сполука за п. 1, де TCRO є
,
де група X, яка може бути однаковою або різною, є Н, лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або галогеном.
3. Сполука за п. 1, де TCRO є
,
де група X, яка може бути однаковою або різною, є Н, лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або галогеном.
4. Сполука за п. 1, де TCRO є
,
де група X, яка може бути однаковою або різною, є Н, лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або галогеном.
5. Сполука за п. 1, де TCRO є
,
де група X, яка може бути однаковою або різною, є Н, лінійною або розгалуженою групою, що має 10 або менше атомів вуглецю і необов’язково включає галоген, або галогеном.
6. Спосіб підвищення коефіцієнта дифузії кисню у ссавця, який включає введення ссавцю терапевтично ефективної кількості сполуки за п. 1.
7. Спосіб за п. 6, який відрізняється тим, що введення здійснюють шляхом інгаляції.
8. Спосіб лікування хвороб дихальних органів, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
9. Спосіб лікування емфіземи, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
10. Спосіб лікування геморагічного шоку, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
11. Спосіб лікування серцево-судинного захворювання, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
12. Спосіб лікування атеросклерозу, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
13. Спосіб лікування астми, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
14. Спосіб лікування травм спинного мозку, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
15. Спосіб лікування набряку головного мозку, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
16. Спосіб лікування папілом, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
17. Спосіб лікування гіпоксії, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
18. Спосіб синтезу сполуки за п. 1, який включає такі стадії:
а) сполучення симетричного діальдегіду, який містить спряжені вуглець-вуглецеві подвійні зв’язки та являє собою 2,7-диметилокта-2,4,6-трієн-1,8-діал, з трифенілфосфораном;
b) омилення продукту стадії а).
19. Спосіб за п. 18, який відрізняється тим, що сполучення здійснюють із застосуванням [3-карбометокси-2-бутен-1-іліден]трифенілфосфорану.
20. Спосіб за п. 18, який відрізняється тим, що продукт стадії а) омилюють із застосуванням розчину NaOH і метанолу.
21. Спосіб за п. 18, який відрізняється тим, що після стадії а) виконують стадію виділення необхідного продукту реакції сполучення.
22. Спосіб омилення симетричного складного діефіру, що має спряжені вуглець-вуглецеві подвійні зв'язки, з одержанням сполуки за п. 1, що включає такі стадії:
a) розчинення симетричного складного диефіру, що має спряжені вуглець-вуглецеві подвійні зв'язки, зі сполукою, вибраною з групи, до складу якої входять метанол, етанол, пропанол і ізопропанол, і
b) змішування розчину стадії а) з основою, яка вибрана з групи, яку складають NaOH, KOH та LiOH.
23. Спосіб за п. 22, який відрізняється тим, що складний діефір омилюють із застосуванням метанолу і NaOH.
24. Сполука - біполярна транс-каротиноїдна сіль, синтезована за п. 18.
25. Спосіб лікування ішемії, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
26. Спосіб лікування травматичного пошкодження головного мозку, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
27. Спосіб підвищення життєвої активності ссавця, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
28. Спосіб лікування діабетичних ускладнень, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
29. Спосіб лікування хвороби Альцгеймера, що включає введення ссавцю, який потребує такого лікування, терапевтично ефективної кількості сполуки за п. 1.
Текст
Цей винахід має відношення до сполук - біполярних транс-каротиноїдних солей, способів їх розчинення, способів їх одержання та способів їх застосування. Ці сполуки - біполярні транс-каротиноїдні солі (БТКС) - є придатними для поліпшення коефіцієнта дифузії кисню між еритроцитами та тканинами тіла у ссавців, у тому числі людей. Каротиноїди являють собою клас вуглеводнів, які складаються з ізопренощних одиниць, з'єднаних таким чином, що в центрі молекули розташування бічних замісників відносно основного ланцюга змінюється на протилежне. Остов (скелет) молекули складається із спряжених вуглець-вуглецевих подвійних та простих зв'язків і може мати також бічні групи. Хоча раніше вважали, що скелет каротиноїду включає 40 атомів вуглецю, вже давно є визнаним, що каротиноїди можуть також мати вуглецеві скелети, що включають менш ніж 40 атомів вуглецю. Усі 4 прості зв'язки, що оточують вуглець-вуглецевий подвійний зв'язок, лежать у одній площині. У разі, якщо бічні групи знаходяться на одному боці вуглець-вуглецевого зв'язку, згадані групи позначаються як цис-; якщо ці групи знаходяться на протилежному боці вуглець-вуглецевого зв'язку, вони позначаються як транс-. Завдяки великій кількості подвійних зв'язків існують великі можливості геометричної (цис/транс) ізомерії каротиноїдів, і ізомеризація легко відбувається у розчині. Нещодавно видана серія книжок є надзвичайно добрим довідником відносно багатьох властивостей тощо каротиноїдів (["Carotenoids", під редакцією Г. Бріттон (G. Britton), С. Ліен-Йєнсен (S. Liaaen-Jensen) та Г. Пфандер (Η. Pfander), Birkhauser Verlag, Basel, 1995], які включені до цього опису у повному обсязі шляхом посилання). Багато каротиноїдів є неполярними і, таким чином, нерозчинними у воді. Ці сполуки є надзвичайно гідрофобними, що ускладнює одержання їх лікарських форм для біологічного застосування, оскільки для їх розчинення необхідно вдаватись до застосування не водного, але органічного розчинника. Інші каротиноїди є монополярними, і мають характеристики поверхнево-активних речовин (гідрофобна частина і гідрофільна полярна група). Як такі, ці сполуки не розчиняються у загальному об'ємі рідини, а притягаються до поверхні водного розчину. Існує незначна кількість природних біполярних каротиноїдних сполук, і ці сполуки включають центральний гідрофобний фрагмент і дві полярні групи, по одній на кожному кінці молекули. Повідомлялось ["Carotenoids", том 1А, стор.283] про те, що сульфати каротиноїдів мають "значну розчинність у воді до 0,4мг/мл". Інші каротиноїди, які могли б вважатись біполярними, також є не дуже розчинними у воді. До них належать діальдегіди та дикетони. Повідомлялось також про дипіридинову сіль кроцетину, але її розчинність у воді при кімнатній температурі є нижчою за 1мг/мл. Іншими прикладами біполярних каротиноїдів є кроцетин та кроцин (обидва знаходяться у пряно-смаковій речовині шафрані). Кроцетин, однак, є лише помірно розчинним у воді. Фактично, з усіх біполярних каротиноїдів лише кроцин демонструє значну розчинність у воді. [Патенти США №№4,176,179; 4,070,460; 4,046,880; 4,038,144; 4,009,270; 3,975,519; 3,965,261; 3,853,933 та 3,788,468] мають відношення до різних варіантів застосування кроцетину. [Патент США №5,107,030] має відношення до способу одержання 2,7-диметил-2,4,6-октатриєндіалу та його похідних. [Патент США №6,060,511] має відношення до транс-кроцетинату натрію (ТКН) та варіантів його застосування. ТКН одержують шляхом проведення реакції природного шафрану із гідроксидом натрію з подальшими екстрагуваннями. У роботі [Рой (Roy) та ін., Shock 10, 213-7 (1998)], повідомлялось про те, що пацюкам із кровотечею (55% об'єму циркулюючої крові) через 10хв. вводили транс-кроцетинат натрію (ТКН) у формі болюса і фізіологічний розчин ще через 30хв. Усі тварини, що піддавались обробці ТКН, залишились живими, у той час як усі контрольні тварини вмерли. Загальне споживання кисню у групі тварин, які одержувала ТКН, зросло, і через приблизно 15хв. досягало 75% від нормального значення у стані покою. Робота [Лейдіг (Laidig) та ін., J. Am. Chem. Soc. 120, 9394-9395 (1998)], має відношення до чисельного моделювання ТКН. Модельовану молекулу ТКН "гідратували" шляхом оточення її молекулами води. Гідрофобна упорядкованість води поблизу від ТКН полегшила дифузію молекул кисню через систему. Чисельне зростання коефіцієнта дифузії на приблизно 30% відповідало результатам, одержаним in vitro та під час експериментів із тваринами. У роботі Сінгер (Singer) та інші, Crit Care Med. 28, 1968-72 (2000), повідомлялось про те, що ТКН поліпшував гемодинамічний статус і подовжував термін доживання пацюків при моделюванні гострої гіпоксії на пацюках. Гіпоксію індукували за допомогою повітряної суміші з низькою концентрацією кисню (10%); через 10хв. тварини одержували фізіологічний розчин або ТКН. Гіпоксемія спричинювала зниження кровотоку і посилення основного розладу. У контрольній групі вижили лише 2 тварини з 6 тварин. У групі, яка одержувала ТКН, усі тварини вижили з доброю гемодинамічною стабільністю впродовж 2год. із повільним подальшим зниженням. Цей винахід має відношення до сполук - біполярних транс-каротиноїдних солей (БТКС) та синтезу таких сполук, які мають структуру: YZ-TCRO-ZY, де: Υ - катіон, Ζ - полярна група, асоційована з катіоном, і TCRO - транс-каротиноїдний скелет. Цей винахід має також відношення до окремих композицій сполук БТКС (у тому числі композиції ТКН), де оптична густина максимального піка (водного розчину композиції БТКС), що реєструється у видимому діапазоні довжини хвиль спектра, поділена на оптичну густину піка, що реєструється у УФ-діапазоні довжини хвиль, демонструє певні характеристичні значення. Цей винахід має також відношення до способу лікування різноманітних захворювань, що включає введення ссавцю, що потребує такого лікування, терапевтично ефективної кількості сполуки, що має формулу: YZ-TCRO-ZY. Цей винахід включає також декілька способів розчинення та синтезування сполук, що мають формулу: YZ-TCRO-ZY. Цей винахід має також відношення до інгалятора для доставки сполук за цим винаходом. Відкрито новий клас каротиноїдів та каротиноїд-споріднених сполук. Ці сполуки називають "біполярними трянс-каротиноїдними солями" (БТКС). Сполуки за цим винаходом Цей винахід має відношення до класу сполук, біполярних транс-каротиноїдних солей, що забезпечують можливість розчинення гідрофобного каротиноїдного або каротиноїд-спорідненого скелета у водному розчині, та способів їх одержання. Катіонами цих солей може бути ряд різновидів, однак за варіантом, якому віддають перевагу, натрій або калій (вони знаходяться у більшості біологічних систем). Патент США №6,060,511, що знаходиться у спільній власності і який включено до цього опису у повному обсязі шляхом посилання, описує спосіб екстрагування для одержання трянс-кроцетинату натрію, ТКН (БТКС), з шафрану. Біполярна трдис-каротиноїдна сіль має таку загальну структуру: YZ-TCRO-ZY, де: Υ (який може бути однаковим або різним на двох кінцях) - катіон, за варіантом, якому віддають перевагу, Na+, K+ або Li+. За варіантом, якому віддають перевагу, Υ є одновалентним іоном металу. Υ може також бути органічним катіоном, наприклад, R4N+, R3S+, де R - Η або СnН2n+1 де n - 1-10, за варіантом, якому віддають перевагу, 1-6. Наприклад, R може бути метилом, етилом, пропілом або бутилом. Ζ (яка може бути однаковою або різною на двох кінцях) - полярна група, асоційована з катіоном. У разі факультативного включення кінцевого атома вуглецю до складу каротиноїду (або каротиноїд-спорідненої сполуки) цією групою може бути карбоксильна (СОО) група або CO група. Цією групою може також бути сульфатна група (ОSО3-), монофосфатна група (ОРО3-), (ОР(ОН)О2-), дифосфатна група, трифосфат або їхні комбінації. TCRO - трйнс-каротиноїдний або каротиноїд-споріднений скелет (за варіантом, якому віддають перевагу, менше ніж 100 атомів вуглецю), який є нерозгалуженим, має бічні групи (визначені нижче), і, як правило, включає "спряжені" або переміжні вуглець-вуглецеві подвійні та прості зв'язки (за одним із варіантів здійснення, TCRO є не повністю спряженим, як у лікопені). Бічними групами є, як правило, метильні групи, однак це можуть бути і інші групи, як обговорюється нижче. За варіантом, якому віддають перевагу, структурні одиниці скелета з'єднані таким чином, що в центрі молекули розташування бічних замісників відносно основного ланцюга змінюється на протилежне. Усі 4 прості зв'язки, що оточують вуглець-вуглецевий подвійний зв'язок, лежать у одній площині. У разі, якщо бічні групи знаходяться на одному боці вуглецьвуглецевого зв'язку, згадані групи позначаються як цис-; якщо ці групи знаходяться на протилежному боці вуглець-вуглецевого зв'язку, вони позначаються як транс-. Сполуки цього винаходу мають транс-конфігурацію. Цис-ізомер, як правило, є шкідливою домішкою, що перешкоджає збільшенню коефіцієнта дифузії. За одним із варіантів здійснення може застосовуватись транс-ізомер, скелет якого залишається лінійним. Прикладами трянс-каротиноїдних або каротиноїд-споріднених скелетів є: де бічними групами X (які можуть бути однаковими або різними) є атоми водню (Н) або група з нерозгалуженим чи розгалуженим ланцюгом, що має 10 або менше атомів вуглецю, за варіантом, якому віддають перевагу, 4 або менше атомів (факультативно включає галоген), або галоген. Прикладами X є метальна група (СН3), етильна група (С2Н5 ), галогенвмісна алкільна група (С1-С10), наприклад, СН2СІ, або галоген, наприклад, СІ або Вr. Бічні групи X можуть бути однаковими або різними, однак групи X, що застосовуються, повинні зберігати лінійну форму скелета. Незважаючи на існування у природі багатьох каротиноїдів, каротиноїдних солей не існує. [Патент США №6,060,511] має відношення до транс-кроцетинату натрію (ТКН). ТКН одержали шляхом проведення реакції природного шафрану з гідроксидом натрію з подальшими екстрагуваннями, якими відбирали, головним чином, транс-ізомер. Присутність цис- та транс-ізомерів БТКС може визначатись шляхом дослідження у УФ-діапазоні спектра зразка каротиноїду, розчиненого у водному розчині. Приймаючи до уваги спектр, значення оптичної густини максимального піка, що реєструється у видимому діапазоні довжини хвиль спектра 416-423нм (значення залежить від застосованого розчинника), поділене на оптичну густину піка, що реєструється у УФ-діапазоні довжини хвиль 250-256нм, може застосовуватись для визначення ступеня чистоти транс-ізомеру. У разі розчинення БТКС у воді, максимальний пік у видимому діапазоні довжини хвиль спектра буде припадати на приблизно 421нм, а пік у УФ-діапазоні довжини хвиль буде припадати на приблизно 254нм. За повідомленням [М. Кроу (М. Craw) та К. Ламберт (С. Lambert), Photochemistry and Photobiology, том 38 (2), 241-243 (1983)], роботу яких включено до цього опису у повному обсязі шляхом посилання, результат обчислень (у цьому разі аналізували кроцетин) становив 3,1, який збільшився до 6,6 після очищення. Нещодавно було встановлено, що розчинність ТКН у воді перевищує 10мг/мл при кімнатній температурі, що є дуже добрим показником для молекули, яка включає таку довгу гідрофобну частину. Було встановлено також, що ТКН підвищує коефіцієнт дифузії кисню через рідини. [Патент США №6,060,511] описує спосіб екстрагування для одержання ТКН із шафрану; однак інші біполярні каротиноїдні солі одержати за тією самою методикою не можна, оскільки застосування шафрану дозволяє включення до солі лише одного каротиноїдного скелета. Винахід, який розкривається у цьому описі, надає можливість синтезування цілого класу сполук, біполярних транс-каротиноїдних солей, які включають різні каротиноїдні або каротиноїд-споріднені скелети. Такі сполуки є розчинними у водних розчинах і мають варіанти корисного біологічного застосування, наприклад, забезпечення підвищення споживання кисню. Вважають, що це підвищення є результатом здатності гідрофобної частини (скелета) біполярної транс-каротиноїдної солі впливати на зв'язування сусідніх молекул води. Це фактично забезпечує можливість швидшої дифузії молекул кисню на цій ділянці. Розчинення сполук і композицій за цим винаходом Цей винахід надає можливість розчинення транс-каротиноїдних або каротиноїд-споріднених скелетних молекул у водних розчинах. Нижче наведені нові способи розчинення. Ці способи є придатними для будь-якої біполярної транс-каротиноїдної солі та її композицій. БТКС-вмісні фізіологічні інфузійні розчини При лікуванні геморагічного шоку вливають значні об'єми (які втричі перевищують визначену втрату крові) ізотонічного фізіологічного розчину (який називають також фізіологічним розчином хлориду натрію). Ізотонічний фізіологічний розчин містить 9г NaCl на літр води для того, щоб не порушити іонної сили плазми після вливання цього розчину до організму. Було показано, що наслідком додання ТКН до фізіологічного розчину є одержання надзвичайно доброї інфузійної рідини. Однак для одержання такого розчину порошкоподібний ТКН не можна просто змішати з фізіологічним розчином. У фізіологічному розчині хлориду натрію розчиняється приблизно 50% ТКН, незалежно від того, скільки ТКН додається (до декількох міліграм намл). Це означає, що присутніми все ще залишаються нерозчинені частинки ТКН. Для запобігання цього, концентрований розчин можна одержати шляхом додання подвійної необхідної кількості ТКН з подальшим відокремленням частинок, які не розчинились, шляхом центрифугування. Фактичний склад концентрованого розчину може перевірятись за допомогою УФ-спектроскопії. Цей концентрований розчин може додаватись до фізіологічного розчину хлориду натрію і ТКН залишається розчиненим. Цей спосіб може застосовуватись для розчинення БТКС у розчинах хлориду натрію інших типів, а також у розчинах інших солей, наприклад, KСl, Na2SO4, лактату тощо. До розчину таким чином може переходити декілька, наприклад, 1 - 3мг/мл. Розбавлений розчин карбонату натрію розчиняє БТКС БТКС, наприклад, ТКН, розчиняється у дуже розбавлених розчинах карбонату натрію. Розбавлений, наприклад, 0,00001 - 0,001М, розчин карбонату натрію може краплями додаватись до деіонізованої води, доки рН не досягне рівня 8,0 (рН деіонізованої води дорівнює, як правило, 5 - 6). Для цього вистачить усього декілька крапель дуже розбавленого розчину карбонату натрію на, скажімо, 50мл деіонізованої води. Цей розчин карбонату натрію-деіонізованої води є здатним до повного розчинення великої кількості ТКН (більше за 10мг/мл), що є дуже хорошим результатом, приймаючи до уваги гідрофобність каротиноїдної частини БТКС. БТКС може постачатись у вигляді порошку разом із флаконом натрійкарбонатної води. Цей концентрований розчин у подальшому може вводитись безпосередньо (можуть вводитись дуже малі об'єми розчинів, що мають іонну силу, меншу за іонну силу плазми) або ж концентрований розчин може додаватись до фізіологічного розчину хлориду натрію з подальшим введенням. Якщо ТКН розчиняється у розчиннику, який включає карбонат натрію-воду, і після цього додається ще більше того самого розчинника, ТКН залишається у розчині. За іншим варіантом здійснення, замість карбонату натрію застосовують бікарбонат натрію. Можуть застосовуватись також інші солі, які забезпечують одержання деіонізованої води, що має основний рН. За цією процедурою можна одержати концентрації каротиноїдних скелетів 5 - 10мг/мл. Вода розчиняє БТКС Незважаючи на те, що ТКН розчиняється у воді (водопровідній, дистильованій, деіонізованій), ці розчини залишаються стабільними лише у тому разі, якщо рН регулюється таким чином, що розчин залишається основним. ТКН, порівняно з нормальною водою, є більш розчинним у деіонізованій воді (присутня дуже невелика кількість іонів Na+). БТКС, наприклад, ТКН, буде розчинятись лише у щойно деіонізованій воді; якщо до цього розчину додається проста деіонізована вода, ТКН буде випадати до осаду. БТКС буде розчинятись лише у щойно деіонізованій воді, однак додаткова кількість деіонізованої води може викликати осадження БТКС, якщо рН не регулюється таким чином, щоб реакція цієї води була злегка основною. Інші способи розчинення БТКС БТКС може вводитись до складу системи доставки, що посилює доставку. Дивись розділ "Лікарські форми сполук за цим винаходом" нижче. Синтез сполук за цим винаходом Біполярні транс-каротиноїдні солі Нижче наведені нові способи синтезу, які можуть застосовуватись для синтезування біполярних транскаротиноїдних солей. Різні стадії синтезу можуть піддаватись змінам, які є очевидними для фахівця у цій галузі. А. Синтез ТКН Транс-кроцетинат натрію (ТКН) може синтезуватись шляхом сполучення симетричного С 10-діальдегіду, що включає спряжені вуглець-вуглецеві подвійні зв'язки, (2,7-диметилокта-2,4,6-триєн-1,8-діал), з [3карбометокси-2-бутен-1-іліден]трифенілфосфораном. Наслідком цього є одержання транс-диметилового складного ефіру кроцетину. Після цього цей диметиловий складний ефір перетворюють на кінцевий продукт ТКН шляхом омилення. За типовим варіантом, омилення здійснюють шляхом обробки складного ефіру водним розчином гідроксиду натрію або гідроксидом натрію, розчиненим у тетрагідрофурані; у цьому разі, однак, ці способи не забезпечували найкращих результатів. Омилення у цьому разі може дуже добре здійснюватись шляхом проведення реакції складного ефіру з розчином NaOH/метанолу. Після омилення ТКН виділяють шляхом висушування під вакуумом. С10-діальдегід і трифенілфосфоран, тобто реагенти, застосовані у цьому синтезі, можуть одержуватись різними шляхами. Наприклад, С10-діальдегід одержували за допомогою реакції Віттіга, використовуючи етилбромацетат та фуран як вихідні сполуки. Тиглінова кислота була вихідним матеріалом для одержання необхідного фосфорану. Каротиноїдні скелети різної довжини можна одержати шляхом з'єднання реагентів різної довжини (наприклад, С14-діальдегіду та трифенілфосфорану). Ця методика надає можливість одержання різних біполярних транс-каротиноїдних солей. Зміни можуть також вноситись таким чином, щоб одержати різні бічні групи (бічними групами ТКН є метильні групи). Одержаний таким чином ТКН є розчинним у воді (рН доводять до 8,0 за допомогою дуже розбавленого розчину карбонату натрію) на рівні >10мг/мл при кімнатній температурі. Інші біполярні трянс-каротиноїдні солі є розчинними при кімнатній температурі у воді, що має рН, який є нейтральним або перевищує його. Термін "розчинний", який уживають у цьому описі, означає, що у 1мл води при кімнатній температурі будуть розчинятись кількості, більші за 5мл (як зазначалось вище, у довідковій літературі з каротиноїдів вказується, що 0,4мг/мл є "високозначущою розчинністю", однак ця величина є нижчою за визначення розчинності, що наводиться у цьому описі). В. Загальний синтез Каротиноїдні або каротиноїд-споріднені структури можна одержати таким чином: (3-карбометокси-2-бутен-1-іліден)трифенілфосфоран (або споріднена сполука, у разі, коли X не є метальною групою) є ключовим попередником для додання ізопрено'щних одиниць (або ізопреноїдспоріднених одиниць) до обох кінців симетричного каротиноїду (або каротиноїд-спорідненої сполуки). Цей процес може повторюватись нескінченно. Наприклад, транс-диметилкроцетинат може відновлюватись до відповідного симетричного діальдегіду за допомогою реакцій, опис яких було наведено вище. Цей діальдегід може вступати до реакції із надлишком (3-карбометокси-2-бутен-1-іліден)трифенілфосфорану з одержанням відповідного складного діефіру. Ця послідовність реакцій синтезу може повторюватись знову і знову. Поліпшений синтез 2,7-диметил-2,4,6-октатриєндіал є ключовою проміжною хімічною сполукою у синтезі ТКН. Цей ключовий попередник має три подвійні зв'язки, завдяки чому можливими є декілька ізомерів. Для ТКН необхідним є ізомер, що має повністю тря«с-конфігурацію (Ε,Ε,Ε-ізомер). Загальний шлях синтезу являє собою 11стадійний процес із відносно низькими виходами і низьким рівнем вибірності на декількох стадіях (дивись Приклад 1). Як наслідок, для очищення декількох проміжних хімічних сполук під час здійснення синтезу необхідною є хроматографія на колонках. Поліпшений шлях синтезу є набагато простішим (дивись наведену нижче схему реакції). 3-стадійний процес, описаний [у патенті США 5,107,030], який включено до цього опису у повному обсязі шляхом посилання, забезпечує можливість одержання суміші геометричних ізомерів діальдегіду ([патент США 5,107,030] не вказує цієї суміші). Спосіб за цим винаходом, опис якого наведено у Прикладі 1, забезпечує одержання 96 - 97% необхідного ізомеру (ізомер, що має повністю транс-конфігурацію або Ε,Ε,Ε-ізомер) шляхом декількох перекристалізацій з метанолу або етилацетату з 59% виходом. Поліпшений спосіб синтезу за цим винаходом включає перетворення залишкової ізомерної суміші діальдегідів на необхідний транс-альдегід (Ε,Ε,Ε) шляхом ізомеризації із сульфіновою кислотою (RSO2H, де R – С1-С10-алкільна група з нерозгалуженим або розгалуженим ланцюгом або арильна група (заміщена фенільна група)), наприклад, паратолуолсульфоновою кислотою, у відповідному розчиннику, наприклад, 1,4-діоксані, тетрагідрофурані або діалкіловому простому ефірі, де алкільною групою є одна або дві С 1-С10-алкільні групи з нерозгалуженим або розгалуженим ланцюгом. Додатковий 8% вихід чистого необхідного діальдегіду одержують шляхом підвищення загального виходу останньої стадії з 59% до 67%. Таке підвищення виходу є важливим. Ця стадія ізомеризації може включатись до третьої стадії способу [патенту США 5,107,030] для одержання кращого виходу. Поліпшений шлях синтезу Два небажані ізомери: Ізомеризація небажаного до бажаного діальдегіду: Омилення може здійснюватись шляхом розчинення складного діефіру у метанолі з подальшим доданням основи, наприклад, NaOH (Υ БТКС у такому разі є Na+). За альтернативним варіантом складний діефір може розчинятись у метанолі, який вже включає основу. NaOH, як правило, застосовується у водному розчині (20 60% (мас.)), але може бути і твердою речовиною. Альтернативами метанолу для розчинення складного діефіру є етанол, пропанол і ізопропанол. Омилення може здійснюватись різними способами промисловим шляхом. Може застосовуватись одно- або двофазна система (одна органічна і одна водна фаза). Трас-кроцетин може також синтезуватись за вищеописаними способами. Окрім того, як повідомлялось для ТКН, такі сполуки БТКС підвищують коефіцієнт дифузії кисню через воду (це буде також залежати від природи гідрофобної частини, що входить до складу кінцевого продукту, наприклад, довжини вуглецевого ланцюга), оскільки вважають, що наслідком гідрофобних взаємодій каротиноїдного скелета з водою є підвищений коефіцієнт дифузії. Лікарські форми сполук за цим винаходом Концентрований розчин біполярної транс-каротиноїдної солі можна одержати, як описувалось вище, шляхом розчинення її у дуже розбавленому розчині карбонату натрію. Одержану суміш після цього можна застосовувати згаданим чином або її можна додатково розбавляти фізіологічним розчином хлориду натрію або іншими водними розчинниками. Окрім того, розчини біполярної транс-каротиноїдної солі можна одержати шляхом розчинення біполярної тррнс-каротиноїдної солі безпосередньо у фізіологічному розчині з подальшим позбавленням від будь-якого матеріалу, який не розчиняється. Біполярні транс-каротиноїдні солі є стійкими у сухій формі при кімнатній температурі і можуть зберігатись впродовж тривалих періодів часу. Перевага полягає у тому, що лікарська форма таких солей, у разі введення її через рот, абсорбується у травному каналі, а не у шлунку. Хоча сполуки за цим винаходом можуть вводитись самостійно, вони можуть вводитись також як частина фармацевтичної лікарської форми. Такі лікарські форми можуть включати фармацевтично прийнятні носії, відомі фахівцям у цій галузі, а також інші терапевтичні агенти (дивись нижче). Перевага полягає у тому, що лікарська форма не включає сполуки, яка пригнічує здатність сполук за цим винаходом до поліпшення коефіцієнта дифузії кисню. Відповідні дози сполук та композицій за цим винаходом будуть залежати від тяжкості стану, який піддається лікуванню. Для того, щоб доза була "терапевтично ефективною", вона повинна мати бажаний ефект, тобто підвищення коефіцієнта дифузії кисню. Це, у свою чергу, примусить повернутись до нормальних значень параметри, пов'язані з киснем. Введення може здійснюватись будь-яким прийнятним шляхом, у тому числі через рот, ніс, місцево, парентерально (у тому числі підшкірно, внутрішньом'язово, внутрішньовенно, внутрішньошкірно та внутрішньокістково), вагінально або ректально. Шлях введення, якому віддадуть перевагу, буде залежати від обставин. Інгаляційному шляху віддається перевага для лікування у невідкладних ситуаціях, коли БТКС повинні дуже швидко потрапити до кровотоку. Лікарські форми, таким чином, включають ті, що є придатними для введення такими шляхами (рідина або порошок, призначений для розпилювання). Слід розуміти, що шлях, якому віддають перевагу, може змінюватись, наприклад, у залежності від стану та віку пацієнта. Лікарські форми можуть бути зручно представлені у дозованій формі, наприклад, таблеток та капсул пролонгованої дії, і можуть одержуватись та вводитись способами, відомими у галузі фармації. Лікарська форма може бути лікарською формою негайного або повільного чи пролонгованого вивільнення БТКС. Дивись, наприклад, лікарську форму пролонгованої дії [у заявці WO 99/15150], яку включено до цього опису у повному обсязі шляхом посилання. Лікарські форми за цим винаходом, придатні для введення через рот, можуть бути представлені як дискретні одиниці, наприклад, драже, капсули, крохмальні облатки або таблетки, як порошок або гранули або як розчин, суспензія чи емульсія. До лікарських форм, придатних для введення через рот, додатково належать льодяники, пастилки та інгаляційні аерозолі, які вводяться з придатною основою або рідким носієм. Лікарські форми для місцевого нанесення на шкіру можуть бути представлені у вигляді мазей, кремів, гелів та паст, до складу яких входить активний агент і фармацевтично прийнятний носій, або у вигляді черезшкірного пластиру. Лікарські форми, придатні для введення через ніс, де носієм є тверда речовина, включають дисперсні порошки, які можуть вводитись шляхом швидкого вдихання через носовий хід. Відповідні лікарські форми, де носієм є рідина, можуть вводитись, наприклад, як носовий спрей або краплі. Лікарські форми, придатні для парентерального введення, включають водні та неводні стерильні ін'єкційні розчини, до складу яких можуть входити антиоксиданти, буферні розчини, бактеріостатичні фактори та розчинені речовини, які надають лікарській формі ізотонічні властивості відносно крові передбачуваного реципієнта, та водні і неводні стерильні суспензії, до складу яких можуть входити суспендувальні агенти та загусники. Лікарські форми можуть бути представлені у однодозових або багатодозових контейнерах, наприклад, герметизованих ампулах і флаконах, і можуть бути ліофілізованими, потребуючи лише додання стерильного рідкого носія, наприклад, води для ін'єкцій, безпосередньо перед застосуванням. Ін'єкційні розчини та суспензії можна приготувати зі стерильних порошків, гранул і таблеток. Застосування сполук та композицій за цим винаходом Найрізноманітніші стани контролюються або опосередковуються шляхом доставки кисню до тканин тіла. Сполуки та композиції за цим винаходом можуть застосовуватись за такими самими варіантами фармацевтичного застосування, які були описані для кроцетину, і у таких самих ефективних кількостях; дивись [патенти США №№4,176,179; 4,070,460; 4,046,880; 4,038,144; 4,009,270; 3,975,519; 3,965,261; 3,853,933 та 3,788,468], кожен з яких включено до цього опису у повному обсязі шляхом посилання. Було показано, що ТКН підвищує коефіцієнт дифузії кисню через водні розчини на приблизно 30%. Таким чином, сполуки за цим винаходом є придатними для лікування захворювань/станів, які характеризуються низьким вмістом кисню (гіпоксія), наприклад, хвороб дихальних органів, геморагічного шоку та серцевосудинних захворювань, атеросклерозу, емфіземи, астми, гіпертензії, набряку головного мозку, папілом, пошкоджень спинного мозку тощо. Інші біполярні транс-каротиноїдні солі мають подібні властивості. Такі сполуки можуть також застосовуватись у поєднанні з іншими способами, які, зазвичай, пропонуються для підвищення споживання кисню у організмі, наприклад, оксигенотерапією та застосуванням гемоглобінів або фторвутлеців. За одним із варіантів здійснення винаходу, БТКС вводять пацієнту з одночасним введенням кисню. За альтернативним варіантом, гемоглобіни або фторвуглеці і БТКС можуть вводитись разом. У цих випадках реалізується адитивний ефект. Мінімальною дозою будь-якої з цих солей, необхідною для лікування, є доза, при якій підвищується коефіцієнт дифузії кисню. Ефективна доза сполук за цим винаходом буде залежати від стану, який підлягає лікуванню, тяжкості стану, стадії та індивідуальних характеристик кожного пацієнта-ссавця, який піддається лікуванню. Доза буде змінюватись, однак, від приблизно 0,001мг активної сполуки на кг маси тіла до приблизно 500мг/кг, а за варіантом, якому віддають перевагу, приблизно 0,01 - 30мг/кг маси тіла. Внутрішньовенне введення є таким шляхом, якому віддають перевагу, однак можуть використовуватись також інші шляхи ін'єкції, наприклад, внутрішньом'язовий, підшкірний або інгаляційний. Може застосовуватись також як введення через рот, так і черезшкірна доставка або внутрішньокісткова доставка. Хвороби дихальних органів Біполярні транс-каротиноїдні солі можуть застосовуватись для лікування хвороб дихальних органів. Ці хвороби описують як стан, при якому парціальний тиск кисню у артеріальній крові є зниженим до значення 60 70мм рт.ст. (8 - 9,3кПа), порівняно з нормальним значенням 90 - 100мм рт.ст. (12 - 13,3кПа). Такими хворобами є емфізема, гострий респіраторний дистрес-синдром або хронічне обструктивне захворювання легень. ТКН підвищує значення парціального тиску кисню у крові, коли останній є низьким (це симптом емфіземи, гострого респіраторного дистрес-синдрому і хронічного обструктивного захворювання легень). Підвищення парціального тиску кисню у крові ослаблює багато симптомів емфіземи, гострого респіраторного дистрессиндрому і хронічного обструктивного захворювання легень. ТКН не виліковує причину хвороби, але ослаблює окиснювальний дистрес і пошкодження, яке є наслідком першопричини. Геморагічний шок Геморагічний шок характеризується зниженням поглинання кисню. Біполярні транс-каротиноїдні солі підвищують поглинання кисню організмом шляхом посилення ступеня дифузії кисню з еритроцитів до тканин. Було показано, що ТКН підвищує поглинання кисню пацюками, що знаходяться у стані геморагічного шоку, а також компенсує інші симптоми шоку. Сполуки за цим винаходом підвищують низький тиск крові, зменшують частоту серцевих скорочень і ліквідують підвищений рівень кислот (ацидоз) у крові, що виникає під час шоку. Сполуки за цим винаходом зменшують також пошкодження органів унаслідок геморагічного шоку. Сполуки за цим винаходом можуть застосовуватись для лікування геморагічного шоку шляхом їх введення інгаляцією, ін'єкцією або доданням до стандартної реанімаційної рідини (лактатний розчин Рінгера-Локка або фізіологічний розчин хлориду натрію). Серцево-судинні захворювання Головною причиною смерті у західній культурі є ішемічна хвороба серця. Смерть може бути наслідком повільного погіршення здатності серця до скорочення або, частіше, раптової його зупинки. Раптова смерть унаслідок зупинки серця охоплює період часу, який розпочинається через 60с після появи симптомів, і триває впродовж подальших 24год. Такі смерті є, як правило, наслідком гострої коронарної оклюзії (закупорки) або фібриляції шлуночків (яка може виникати унаслідок оклюзії). Ішемія серцевого м'яза існує у разі недостатнього постачання кисню до серцевого м'яза. Коли коронарний кровотік є надзвичайно низьким, серцевий м'яз не може функціонувати і гине. У цьому разі кажуть, що ділянка серцевого м'яза відмирає унаслідок порушення її кровопостачання. Зменшення коронарного кровопостачання найчастіше викликається атеросклерозом, якому піддаються коронарні артерії. Наслідком ішемії є погіршення механічної та електричної активності і пошкодження м'язових клітин, що може призвести до летальної аритмії, яку називають фібриляцією шлуночків. У разі фібриляції електрична активність шлуночків серця є хаотичною, унаслідок чого одержують електрокардіограму з нестійким ритмом і картинами електричної активності серця, що не піддаються розпізнаванню. Фібриляція шлуночків часто відбувається разом з ішемією серцевого м'яза та інфарктом, і майже завжди є причиною раптової смерті унаслідок зупинки серця. Біполярні транскаротиноїдні солі є благотворними при лікуванні ішемії серцевого м'яза. Атеросклероз, який часто є попередником інфаркту серцевого м'яза, також може лікуватись цими солями. Ішемія Біполярні транс-каротиноїдні солі є також благотворними при лікуванні інших форм ішемії (недостатнього току крові до тканин або органів), наприклад, ішемії нирок, печінки, спинного і головного мозку, у тому числі інсульту. Гіпертензія Гіпертензія (або високий кров'яний тиск) часто пов'язується із серцево-судинними захворюваннями. Сполуки за цим винаходом можуть застосовуватись для зниження тиску крові. Підсилення життєвої активності БТКС підсилюють аеробний метаболізм шляхом підвищення рівнів поглинання кисню під час ходи, бігу, піднімання тощо. Підвищується також витривалість. Черепно-мозкова травма Наслідком гіпоксії після черепно-мозкової травми є посилення пошкодження головного мозку. БТКС підвищують рівні кисню у тканинах головного мозку після динамічної травми (вогнищева або дифузна травма). Прикладами динамічної травми є травми унаслідок дорожньо-транспортних аварій (автомобільні/мотоциклетні) та падінь. БТКС також збільшують кількість кисню, що досягає нормальних тканин головного мозку у разі застосування гіпербарооксигенотерапії. Хвороба Альцгеймера У разі хвороби Альцгеймера, БТКС підвищують рівні поглинання кисню головним мозком і тим самим полегшують її симптоми. Кровотік і поглинання кисню є на приблизно 30% нижчими за рівні, що спостерігаються у літніх людей, які не страждають на слабоумство [Уертман (Wurtman), Scientific American, том 252, 1985]. Підвищені рівні поглинання кисню головним мозком, що забезпечуються БТКС, зменшують також втрату пам'яті. Діабет БТКС є придатними для лікування діабетичних ускладнень, наприклад, виразок, гангрени та діабетичної ретинопатії. Краще загоюються діабетичні виразки ступнів у разі гіпербарооксигенотерапії [М. Калані (М. Kalani) та інші, Journal of Diabetes & Its Complications, том 16, №2, 153-158, 2002]. БТКС також допомагають при лікуванні ускладнень діабетичної ретинопатії, які пов'язуються з низьким тиском кисню [Деннінгхофф (Denninghofi) та інші, Diabetes Technology & Therapeutics, том 2, №1, 111-113, 2000]. Інші застосування Біполярні транс-каротиноїдні солі можуть також застосовуватись для лікування травми спинного мозку, набряку головного мозку та шкірних папілом. В усіх випадках вони полегшують стан, роблять його менш тяжким. Гадають, що це є наслідком підвищення поглинання кисню, яке є результатом застосування біполярних транс-каротиноїдних солей. БТКС також видаляють кисеньпохідні вільні радикали. Наведені нижче Приклади є ілюстративними і не обмежують композицій та способів за цим винаходом. Інші придатні модифікації та адаптації різноманітних станів та параметрів, з якими, як правило, стикаються і які є очевидними для фахівців у цій галузі, входять до суті та обсягу цього винаходу. Приклади Приклад 1 Синтез транс-кроцетинату натрію Трас-кроцетинат натрію синтезують шляхом сполучення симетричного С 10-діальдегіду, що включає спряжені вуглець-вуглецеві подвійні зв'язки, з [3-карбометокси-2-бутен-1-іліден]трифенілфосфораном. Після цього цей продукт омилюють за допомогою розчину NaOH/метанолу. Трифенілфосфін, розчинений у етилацетаті (з концентрацією приблизно 2моль/л), повільно додають до етилбромацетату. Після виділення та обробки основою, продукт обробляють спочатку метилйодидом, потім каустичною содою з одержанням фосфорану. Основну сполуку для одержання каротиноїдного скелета можна одержати, використовуючи як вихідний матеріал циклічну сполуку, наприклад, фуран у цьому випадку. Фуран реагує з бромом та метанолом із подальшою стадією селективного депротонування з одержанням моноальдегіду, який у подальшому сполучають із фосфораном. Кислотні умови забезпечують деблокування іншої диметилацетальної групи з одержанням вільного альдегіду. Після цього ця сполука знову реагує зі згаданим фосфораном з одержанням діетилового складного діефіру. Складноефірні групи відновлюють до спиртів, і наслідком подальшого окиснення (наприклад, за допомогою МnО2) є одержання С10-скелета у діальдегідній формі, який пізніше реагує з фосфораном, одержаним із тиглінової кислоти. Тиглінову кислоту естерифікують метанолом за кислотних умов з одержанням метилового складного ефіру з подальшою стадією бромування. Одержані таким чином ізомери алілброміду розділяють шляхом кристалізації. Подальша обробка необхідного броміду гідроксидом натрію забезпечує одержання необхідного фосфорану. Після цього цей фосфоран та Сю-діальдегід розчиняють у розчиннику, наприклад, толуолі або бензолі, і нагрівають до кипіння зі зворотним холодильником. Одержаний продукт виділяють у вигляді порошку, після чого омилюють 40% сумішшю NaOH/метанолу з одержанням ТКН після видалення розчинника. Транс-кроцетинат натрію 1 (ТКН) одержали шляхом 17-стадійної послідовності реакцій синтезу із загальним виходом 1,5%. У разі застосування етилбромацетату, фурану та тиглінової кислоти як вихідних матеріалів, у цілому одержали 4,1г ТКН. Транс-кроцетинат натрію (ТКН) синтезували шляхом омилення диметилкроцетинату, який одержали на основі загального синтезу, про який повідомлялось у роботі Бухта (Buchta) та Андре (Andree)1. Основою стратегії синтезу для одержання диметилкроцетинату є сполучення симетричного С 10-діальдегіду, (2,7диметилокта-2,4,6-триєн-1,8-діал), з [3-карбометокси-2-бутен-1-іліден]трифенілфосфораном. Незважаючи на те, що оригінальна стаття1 Бухта (Buchta) та Андре (Andree) мала назву "The Total Synthesis of trans-2,2-Bisdimethyl-crocetin-dimethyl ester and trans-Crocetin-dimethyl ester", експериментальні подробиці та виходу у ній не повідомлялись. Методики різних стадій, наслідком яких було одержання С10діальдегіду та фосфорану, були визначені після екстенсивного вивчення літератури. Зрештою, ТКН одержали шляхом 17-стадійної послідовності із загальним виходом 1,5% та застосуванням етилбромацетату, фурану та тиглінової кислоти як вихідних матеріалів. Симетричний С10-діальдегід одержали з етилбромацетату2 і фурану3 за допомогою реакції Віттіга. Етилбромацетат обробляли трифенілфосфіном і метилйодидом з одержанням фосфорану 6: а. трифенілфосфін, EtOAc, 92%; b. 1-н. NaOH, CH2Cl2; с CH3I, CH2Cl2; d. 1-н. NaOH, CH2Cl2. Вихід першої стадії був цілком задовільним і становив 92%. Кількісне визначення подальших стадій цієї послідовності ускладнювалось природою фосфорану 4 та солі фосфонію 5. Обидві ці сполуки являли собою надзвичайно в'язкі сиропи, які інтенсивно пінились під час концентрування на роторному випарнику. З обома сполуками можна зручно працювати у вигляді розчину у метиленхлориді і загальний вихід фосфорану 6 видається прийнятним з кількісної точки зору (за приблизними оцінками, більше за 75%). Кільце фурану розривали бромом з одержанням фумаральдегід-біс(диметилацеталю) 8.3 e. Вr2, МеОН; Na2SO4, 77%; f. Амберліст 15, Н2О, ацетон, 72%. Шляхом деблокування однієї функціональної групи біс(диметилацеталю) 8 за кислотних умов4 одержали альдегід 9, який у подальшому сполучали з фосфораном 6 з одержанням 10 з 45% виходом. Кислотні умови застосовувались для деблокування диметилацеталю 10. Шляхом обробки 11 фосфораном 6 одержали складний діефір 12. Складноефірні групи відновлювали до спиртів за допомогою DIBAL-H (діізобутиладипінат літію-Н) і шляхом подальшого окиснення за допомогою МnО2 одержали С10-діальдегід 14. Транс-стереохімію 14 визначали за даними ЯМР. Зокрема, С2 симетрія сполуки забезпечила очікувані 5 резонансів у 13С ЯМРспектрі і 1Н ЯМР-спектр продемонстрував сигнали при δ 9,54 (1Н), 7,07 (2Н) і 1,95 (3Н). g. CH2Cl2, 45%; h. Амберліст 15, H2O, ацетон, 42 - 65%; і. 6, СН2Сl2, 50-81%; j. DIBAL-H, гексан, 75 - 81%; k. МnО2, ацетон, 26 - 58%. Діапазон виходів на стадіях h - k відображає поліпшення виділення з початкових пілотних досліджень до реакцій у збільшених масштабах. Тиглінову кислоту 15 перетворювали на фосфоран 20 шляхом 4-стадійної послідовності. Шляхом проведення реакції Фішера з естерифікацією 15 одержали метиловий складний ефір 16. Унаслідок реакції з NBS (N-бромсукцинімід) одержали суміш 59% метил γ-бромтиглату, 26% метил α-бромтиглату з матеріальним балансом, який забезпечувався вихідним матеріалом, що не вступив до реакції. Одержання регіоізомерів очікували, виходячи з літературних повідомлень.5 На наступній стадії α/γ суміш солей фосфонію перекристалізовували з одержанням необхідного γ-фосфонію броміду 19.6 Шляхом подальшої обробки гідроксидом натрію одержали фосфоран 20. l. H2SO4, MeOh, 42%; m. N-бромсукцинімід, бензоїлу пероксид, 59%; n. трифенілфосфін, С6Н6, 40%; о. NaOH, H2O, 81%. Фосфоран 20 і С10-діальдегід 14 сполучали шляхом нагрівання зі зворотним холодильником у бензолі.6 Диметилкроцетинат 21 виділяли у вигляді порошку червоного кольору. Омилення метилового складного ефіру виявилось труднішою операцією, аніж очікувалась. Обробка складного ефіру 21 2екв. NaOH у суміші тетрагідрофурану з Н2О при кімнатній температурі і нагрівання зі зворотним холодильником залишили матеріал без змін. Розчинність виявилась суттєвою проблемою, тому додали піридин. Незважаючи на те, що це призвело до розчинення більшості твердих матеріалів, при нагріванні суміші піридину та 2,5-н. розчину NaOH зі зворотним холодильником продукту не одержали. Стандартні умови омилення (суміш тетрагідрофурану/2,5-н. розчину NaOH) також жодного впливу на складний ефір не виявили. Успіху, зрештою, досягли при нагріванні до кипіння 40% суміші NaOH/метанолу впродовж ночі. Завдяки цій операції ТКН 1 одержали у вигляді твердої речовини оранжевого кольору. p. C6H6, нагрівання зі зворотним холодильником; q. MeOH, 40% водний розчин NaOH, 58 - 65%. Робились спроби розчинення ТКН з метою одержання 1H ЯМР-спектра. Однак ТКН був практично нерозчинним у більшості звичайних органічних розчинників (хлороформ, диметилсульфоксид, піридин, метанол, ацетон і льодяна оцтова кислота). Характеристики ТКН, який одержали унаслідок здійснення цього проекту, визначали засобами ІЧ- та УФ-спектроскопії, високоефективної рідинної хроматографії та елементного аналізу. ІЧ-спектроскопія показала характеристичну оптичну густину при 1544см1 та 1402см-1 (що відповідає спряженим карбоксилатам). Дані УФ-спектроскопії та високоефективної рідинної хроматографії відповідали автентичному ТКН.7 Елементні аналізи дали задовільні результати. Загальний вихід послідовності реакцій становив 1,5% (виходячи з фурану). Синтез докладно описано нижче: Усі реактиви та хімічні продукти були закуплені від фірм Aldrich або Sigma і застосовувались у тому вигляді, у якому були одержані, якщо не вказувалось інше. Розчинники були закуплені від фірми Fisher Scientific як ACS реактиви (реактиви, що відповідають вимогам Американської хімічної спілки) або реактиви хроматографічної чистоти, і застосовувались без додаткового очищення. Безводні розчинники були закуплені від фірми Aldrich у бутлях Sure/Seal™, і застосовувались безпосередньо без додаткового очищення. Деіонізовану воду одержали з власної системи водопідготовки Culligan. Температуру плавлення визначали на Mel-Temp II без коригування. Інфрачервоні спектри визначали за допомогою спектрофотометра Perkin- Elmer 1600 FTIR. Ядерні магнітні спектри визначали за допомогою спектрометра JEOL FX90Q із застосуванням 5мм багатоядерного зонда з внутрішнім або зовнішнім дейтерієвим затвором, у залежності від природи зразка. Хімічні зсуви протонного та вуглецевого ЯМР визначали відносно тетраметилсилану або дейтерованого розчинника, відповідно. Спектр ЯМР фосфору визначали, як правило, у режимі протонної розв'язки із співвісною вставною пробіркою з 5% водним розчином фосфорної кислоти, як зовнішній стандарт. Поточні газохроматографічні аналізи для визначення розвитку реакції або складу продукту здійснювали на газовому хроматографі Varian 3700, спорядженому полум'яно-іонізаційним детектором та інтегратором Hewlett Packard 3394A. 1мл розчину вносили до 15м колонки DB5 (внутрішній діаметр 0,53мм, товщина плівки 1,5мкм) із газом-носієм гелієм за температурною програмою від 50°С до 250°С при 20°С/хв і 10хв. витримуванням при температурі 250°С. Температуру дозатора і детектора встановлювали, як правило, на рівні 250°С. Хроматографування в тонкому шарі проводили на силікагелевих пластинках Baker-flex 2,5см´7,5см з/без флуоресцентного індикатора (1В2 або 1B2-F) у залежності від способу виявлення. Складові на проявлених пластинках виявляли за допомогою УФ випромінювання. Елементні аналізи були здійснені фірмою Quantitative Technologies, Inc., Whitehouse, штат Нью-Джерсі. [(Етоксикарбоніл)метилен]трифенілфосфоран (4)2 (ACL-G29-1) Трифенілфосфін (235,6г, 0,90моль) розчиняли у EtOAc (540мл). Для розчинення усіх твердих речовин було необхідно приблизно 30хв. Процес був ендотермічним (розчин охолоджували до температури 13°С, коли температура навколишнього середовища дорівнювала 20°С). Впродовж 1,5год. краплями додавали розчин етилбромацетату (100мл, 0,90моль) у EtOAc (400мл). Під час додання утворився осад білого кольору. Перемішували впродовж ночі (20год.) при температурі навколишнього середовища (18°С). Тверді речовини: відфільтровували під вакуумом з промиванням великою кількістю Et2O. Сушили впродовж ночі у вакуумі при температурі 45°С з одержанням 3 у вигляді твердої речовини білого кольору (356,3г, 92,6% вихід, 0,83моль). 1H ЯМР відповідав літературним значенням. Тверду речовину розчиняли у метиленхлориді (3л) і обробляли 1Μ розчином NaOH (3,6л) у 12л колбі з енергійним перемішуванням впродовж 45 хв. Органічний шар відділяли, водну фазу екстрагували додатковою кількістю метиленхлориду (2´1л). Органічні шари сушили (MgSO4) і концентрували до одержання приблизно 1л об'єму. Невелику кількість матеріалу видаляли, і визначали 1H ЯМР; було встановлено, що 1Н ЯМР відповідав літературним значенням. [1-(етоксикарбоніл)етиліден]трифенілфосфонію йодид (5)2 (ACL-G29-2) Матеріал з ACL-G29-1 обробляли йодометаном (64,0мл, 1,03моль) з охолодженням реакційної колби на льодяній бані. Реакційну суміш перевіряли засобами хроматографування в тонкому шарі (силікагель, 10% МеОН/СНСІ 3) після завершення додання (1год.). Хроматографія в тонкому шарі показала залишок значної кількості вихідного матеріалу. Льодяну баню видаляли, і реакційну суміш перевіряли засобами хроматографування в тонкому шарі через 1,5год. Реакція виглядала завершеною, виходячи з ущільнення головної смуги (невелика кількість домішок). Реакційну суміш концентрували на роторному випарнику, після того як більшу частину розчинника було видалено; продукт починав пінитись і повільно підійматись по паропроводу. Сіль фосфонію 5 виглядала як надзвичайно в'язкий сироп, який утримували у формі розчину у метиленхлориді для полегшення маніпулювання. Приймаючи до уваги природу 5, матеріал не піддавали кількісному визначенню. [1-(етоксикарбоніл)етиліден]трифенілфосфоран (6)2 (ACL-G29-2A) Частину 5 розчиняли у СН2Сl2 (350мл) і енергійно перемішували з 1Μ розчином NaOH (500мл) впродовж 45хв. Органічний шар відділяли, і водний шар екстрагували СН2СІ 2 (2´100мл). Об'єднані органічні шари сушили (MgSO4) і концентрували з одержанням 6 у вигляді твердої речовини жовтого кольору (8,0г). Спектр 1Н ЯМР відповідав літературним значенням. Фумаральдегід-біс(диметилацеталь) (8)3 (ACL-G29-3) Розчин фурану (88,0г, 1,29моль) у безводному МеОН (650мл) охолоджували до температури -45°С у атмосфері N2. Розчин брому (68,0мл, 1,32моль) краплями додавали впродовж 2,5год. зі швидкістю, яка забезпечували утримування температури на рівні £-45°С. Розчин червоного кольору нагрівали до температури -10°С впродовж 2,5год. і додатково витримували впродовж 2год. Реакційна суміш мала блідо-бурштиновий колір. Додання 5г Nа2СО3 викликало виділення значної кількості газу і проходження екзотермічної реакції (4°С). Реакційну суміш охолоджували твердою вуглекислотою, і залишкову кількість Na2CO3 (у цілому 210г) додавали впродовж 50хв. Після витримування при температурі -10°С впродовж ночі (11год.) охолоджувальну баню видаляли, реакційну суміш витримували з нагріванням до кімнатної температури і перемішували впродовж 20год. Солі відфільтровували під вакуумом, фільтрат переганяли під вакуумом на колонці Вітре до одержання приблизно 150мл. Додаткова кількість солі випадала до осаду і викликала сильне кипіння перегінного кубу. Після фільтрування переганяли ще 150мл, і з розчину одержували додаткову кількість солі. Проблему, знову таки, створювало сильне кипіння. Перегінний куб охолоджували, вміст фільтрували, фільтрат обробляли Et2O (400мл), осад відфільтровували під вакуумом. Було зібрано щонайменше 120г солі (початкові виходи відкидали без кількісного визначення). Більшу частину Et2O видаляли на роторному випарнику при температурі 25°С з водовідсмоктувальним пристроєм. Перегонку відновлювали на колонці Вітре; 8 збирали (175,2г) у вигляді прозорої безбарвної рідини (76,9% вихід), температура кипіння 86 - 92°С/9мм рт.ст. (1,2кПа) (літературні дані: 85 - 90°С/15мм рт.ст. (2,0кПа)). Спектр 1 Н ЯМР відповідав необхідному продукту. Газохроматографічний аналіз: 81,9% чистота. Фумаральдегід-моно(диметилацеталь) (9)4 (ACL-G29-4) Фумаральдегід-біс(диметилацеталь) 8 (5,29г, 0,03моль) розчиняли у ацетоні (120мл). Послідовно додавали Н2О (1,80мл) і смолу Амберліст 15 (Amberlyst 15) (1,20г). Суміш енергійно перемішували впродовж 5хв., після чого фільтрували для видалення смоли. За цей час безбарвний розчин набув жовтого кольору. Фільтрат концентрували на роторному випарнику при кімнатній температурі, і залишок світло-брунатного кольору відганяли на трубці з кульовим розширенням (температура 37°С/0,2мм рт.ст. (26,7Па)) з одержанням 9 у вигляді рідини жовтого кольору (2,80г, 71,8% вихід). Невелику кількість матеріалу було втрачено під час кипіння перегінного кубу на початку процесу. Спектр 1H ЯМР відповідав необхідному продукту, газохроматографічний аналіз показав 80% чистоту. (ACL-G29-7) Фумаральдегід-біс(диметилацеталь) 8 (72,1г, 0,41моль) розчиняли у ацетоні (1600мл). Додавали Н2О (25,0мл) і смолу Амберліст 15 (16,7г, попередньо промиту ацетоном). Суміш енергійно перемішували впродовж 5 хв, після чого фільтрували для видалення смоляної кислоти. Реакційна суміш мала дещо жовтуватий відтінок, набагато слабший, аніж препарат, який було попередньо одержано у великому масштабі. Газохроматографічний аналіз показав 34,5% продукту і 46,1% домішок. Обробку смолою здійснювали впродовж 5хв. Газохроматографічний аналіз показав 59,5% продукту і 21,7% домішок. Обробку смолою здійснювали ще впродовж 10хв. (загальний час 20хв.). Газохроматографічний аналіз показав 73,9% продукту і 2,0% домішок. Фільтрат концентрували на роторному випарнику при кімнатній температурі з одержанням масла брунатного кольору (54г). Шляхом вакуумної перегонки одержали масло жовто-зеленого кольору (34,48г). Газохроматографічний аналіз показав 64,7% чистоту (8,22хв.) з основною масою домішок 17,5% (9,00хв.) і 6,9% (9,14хв.). Сумарний виділений вихід становив 22,3г (0,17моль). Газохроматографічний аналіз першого погону показав надзвичайно брудний матеріал. (ACL-G29-13) Амберліст 15 (8,61г) перемішували у ацетоні (100мл) впродовж 30хв. і відфільтровували. Ацеталь 8 (35,0г, 0,16моль) розчиняли у ацетонітрилі (620мл), і додавали (з одночасним механічним перемішуванням) смоляну кислоту і деіонізовану Н2О (10,0мл, 0,55моль). Проходження реакції контролювали засобами хроматографії в тонкому шарі (гексан:Еt2О, 10:3). Через 15хв. відбулось перетворення більшості вихідного матеріалу. Через 20хв. виявляли лише слідові кількості диметилацеталю. Смолу відфільтровували, і фільтрат концентрували на роторному випарнику при температурі £-40°С. Неочищений продукт вносили до колонки Biotage (7,5см´9,0см) і елюювали 15% Et2O у гексані, одержуючи 19,8г. 65% вихід. 6,6-диметокси-2-метилгекса-2,4-дієноат (10)2 (ACL-G29-5) Ілід 6 (7,80г, 22ммоль) розчиняли у метиленхлориді (65мл). Додавали розчин фумаральдегідмоно(диметилацеталю) 9 (2,80г, 17ммоль), і розчин перемішували впродовж ночі. Розчинник видаляли під зниженим тиском на роторному випарнику. 1 Н ЯМР неочищеного матеріалу показав присутність необхідного продукту. Після відстоювання виросли кристали (ймовірно, трифенілфосфіну оксиду). Тверду речовину (14,1г після висушування фільтруванням під вакуумом) суспендували у петролейному простому ефірі і фільтрували. Фільтрат концентрували з одержанням масла жовтого кольору; тверді речовини, що випадали з масла до осаду, розчиняли у метиленхлориді (15мл) і хроматографували на колонці Biotage (4см´7,5см) з елююванням метиленхлоридом, одержуючи 10 у вигляді масла жовтого кольору (1,8г, 50% вихід). Спектр 1Н ЯМР масла жовтого кольору відповідав літературним значенням, однак, оскільки залишались слідові кількості метиленхлориду (0,75екв.), матеріал вносили до роторного випарника на 45 хв. Масу зменшили до 1,5г, 40,6% вихід, і резонанс метиленхлориду зник. Газохроматографічний аналіз головного піка на 12,6хв., 87,5% (50°С, 5хв. витримування, 20°С/хв. до кінцевої температури 250°С). (ACL-G29-6) Розчин іліду 6 (59,2г, 0,16моль) у метиленхлориді (650мл) охолоджували на льодяній бані і додавали розчин 9 (25,7г, 0,19моль). Розчин перемішували впродовж ночі до розтоплення льодяної бані. Хроматографія в тонкому шарі (гексан:Еt2O, 10:3) показала щонайменше 3 інші сполуки, які йшли дуже близько до продукту. Газохроматографічний аналіз альдегіду показав 50,0% чистоту. Розчинник видаляли з одержанням суміші твердої речовини/масла. (ACL-G29-8) Ілід 6 (59,2г, 0,16моль) і ацеталь 9 (0,19моль) об'єднували у метиленхлориді (1,1л) і обробляли, як описувалось вище, з одержанням масла жовто-зеленого кольору (80г). Частину неочищеної реакційної суміші (4,13г від початкових 80г) вносили до трубки з кульовим розширенням і переганяли при температурі 50°С/0,250мм рт.ст. (33,3Па). Конденсували безбарвне масло (2,28г). 1Н ЯМР показав, що це був початковий альдегід, у той час як продукт 10 залишався у перегінному кубі (1,85г). Леткі складові видаляли із загального об'єму неочищеного продукту шляхом перегонки у трубці з кульовим розширенням при температурі 50°С/0,2мм рт.ст. (26,7Па) (сумарний вихід 35г). Етил-2-метил-6-оксо-гекса-2,4-дієноат (11)2 (ACL-G29-9) Ацеталь 10 з пілотного перегінного кубу (ACL-G29-8, 1,85г, 9ммоль) розчиняли у ацетоні (33мл). Додавали деіонізовану Н2О (0,50мл) і смолу Амберліст 15 (0,35г, попередньо промиту ацетоном). Суміш перемішували впродовж 20хв., фільтрували і концентрували на роторному випарнику з одержанням масла жовто-зеленого кольору (1,53г). Хроматографували на колонці Biotage (4,5см´7см) з елююванням 15% Εt2Ο у гексані. Ця система забезпечили неповне розділення, однак 0,32г головної складової виділили і піддали аналізу; спектр 1 H ЯМР відповідав літературним даним і дані ІЧ-спектроскопії (1711см-1, 1682см-1) відповідали необхідному продукту. Газохроматографічний аналіз: 95,6%. Додатково виділили 0,35г продукту, хоча він і був забруднений як менш, так і більш полярним матеріалами. Спектр 1 H ЯМР показав доволі чистий матеріал. Газохроматографічний аналіз: 90,6%. Вихід: 42%. Діетил-2,7-диметилокта-2,4,6-триєн-1,8-діоат (12)2 (ACL-G29-10) Альдегід 11 (0,65г, 3,5ммоль) з G29-9 розчиняли і за допомогою магнітної мішалки перемішували у метиленхлориді. Додавали ілід (1,59г, 4,4ммоль). Через декілька хвилин розчин світлого жовто-зеленого кольору набирав темнішого жовтого відтінку. Хроматографія в тонкому шарі через 10хв. вказала майже повне вичерпання вихідного матеріалу. Після перемішування впродовж 20год. реакційну суміш (розчин брунатного кольору) фільтрували через піпетку, частково заповнену силікагелем. Фільтрат концентрували з одержанням твердої речовини брунатного кольору. Тверду речовину розчиняли у 5% Εt2O3 у гексані з невеликою кількістю СНСІ 3 і хроматографували на колонці Biotage (4см´7,5см) з елююванням 5% Et2O у гексані. Головний продукт виділяли у вигляді твердої кристалічної речовини білого кольору (0,45г, 50% вихід). Спектр 1H ЯМР відповідав літературним даним. (ACL-G29-14) Додаткову кількість 12 одержали, як описано вище, з одержанням 21,8г, 81,6% після хроматографічного очищення. Спектр 1H ЯМР відповідав необхідному продукту. 2,7-диметилокта-2,4,6-триєн-1,8-діол (13)2 (ACL-G29-11) Складний діефір 12 (0,45г, 1,8ммоль) розчиняли у безводному гексані (15,0мл). Видавалось, що деяка частина матеріалу розчинилась, але суміш залишалась зовсім каламутною. Видавалось, що більше матеріалу виділилось із розчину, коли суміш охолодили на бані з температурою -78°С. Чистий DIBAL-H (2,50мл) розчиняли у безводному гексані (загальний об'єм 10,0мл), і частину (приблизно 2,0мл) через недогляд сифонували у реакційну суміш, коли складний діефір охолоджували на бані з твердою вуглекислотою. Додаткову кількість розчину DIBAL-H додавали доти, доки загальний об'єм доданого розчину не досягнув 5,0мл (6,7ммоль). СО2-баню витримували до нагрівання. Після перемішування впродовж 2год. 50хв. хроматографія в тонкому шарі вказала повне вичерпання складного діефіру. Температуру бані доводили до 20°С з подальшим нагріванням до 0°С впродовж 20хв. Впродовж 30хв. здійснювали обробку сумішшю Н2О/силікагелю (2мл/7г). Додавали K2СО3 і MgSO4. Фільтрували для видалення твердих речовин і ретельно промивали метиленхлоридом. Концентрували з одержанням твердої речовини білого кольору (0,14г, 50% вихід). Примітка: (хроматографія в тонкому шарі) Rf=0,21 (5% МеОН/СНСІ 3) є повністю полярним. Промивання метиленхлоридом могло бути недостатнім для виділення усього продукту. Спектр 1H ЯМР відповідав літературним значенням. (ACL-G29-15) Складний діефір (5,4г, 21ммоль) розчиняли у безводному гексані (175мл, слабка розчинність), охолоджували на бані з температурою -78°С і впродовж 35хв. обробляли розчином DIBAL-H (14,5мл у 50мл безводного гексану). Під час додання спостерігалось енергійне виділення газу. Забарвлення суспензії спочатку змінилось з білого на темно-жовте, після чого світлішало по мірі додання додаткової кількості DIBALH. Суміш витримували з нагріванням до температури -40°С впродовж 2год., після чого на ніч перенесли на баню з температурою -28°С. Реакційну суміш обробляли однорідною сумішшю Н2О/силікагелю (4мл/14,4г) впродовж 30хв. Додавали (MgSO4) (7,5г) і K2СО3 (5,1г), і реакційну суміш виймали з охолоджувальної бані. Перемішували впродовж 20хв., після чого фільтрували на лійці зі спеченим склом. Тверді речовини промивали метиленхлоридом. Унаслідок цього утворювалась значна кількість осаду. Тверді речовини, що випали до осаду, розчинялись при нагріванні на роторному випарнику. Тверді речовини, які залишились на лійці зі спеченим склом, промивали EtOAc (4´75мл), і фільтрат концентрували. Після промивання СН2Сl2 одержали тверду речовину блідо-жовтого кольору (1,7г). Спектр 1H ЯМР відповідав літературним значенням; після промивання EtOAc одержали тверду речовину жовтуватого кольору (1,0г). Спектр 1H ЯМР відповідав літературним значенням; загальне виділення 2,7г, 75% вихід. (ACL-G29-17) Складний діефір (16,4г, 6,5ммоль) перемішували у безводному гексані (500мл) у атмосфері N2 і охолоджували до температури -78°С. Впродовж 1год. додавали розчин DIBAL-H (45мл, 253ммоль) у гексані (150мл). Витримували з нагріванням до температури -30°С і перемішували впродовж ночі (загальний час 17,5год.). Додавали однорідну суміш Н2О/силікагелю (12,3г/43,7г) і вручну перемішували впродовж 45хв. Додавали K2СО3 (15,5г) і MgSO4 (23,5г). Додатково перемішували впродовж 30хв. Фільтрували на лійці зі спеченим склом, промивали метиленхлоридом (утворювався осад, причиною чого, ймовірно, було випарне охолодження), і фільтрат концентрували. Тверді речовини декілька разів промивали EtOAc (приблизно 100мл порціями, загальний об'єм 2л) і об'єднували з вихідним фільтратом. Унаслідок концентрації одержали тверду речовину жовтого кольору, 8,9г, 81% вихід у неочищеному стані. Спектр 1Н ЯМР відповідав необхідному продукту. 2,7-диметилокта-2,4,6-триєн-1,8-діал (14)2 (ACL-G29-12) Суспензію МnО2 (7,80г, 90ммоль) охолоджували на льодяній бані у атмосфері N2. Розчин діолу 13 (0,14г, 0,8ммоль) додавали за допомогою піпетки у вигляді ацетонового розчину (5,0мл). Для промивання колби і завершення перенесення додатково застосовували 2,0мл ацетону. Льодяну баню витримували до розтоплення з перемішуванням реакційної суміші впродовж ночі. Тверді речовини відфільтровували через шар Hyflo і концентрували з одержанням твердої речовини жовтого кольору. Матеріал розчиняли у 10% Et2O/гексану з мінімальною кількістю СНСІ з і вносили до силікагелевої колонки (30мм´190мм) з елююванням 10% Еt2О/гексану. Продукт може відслідковуватись як смуга жовтого кольору по мірі елюювання, 14 виділяли у вигляді твердої речовини світло-жовтого кольору, 37мг, 26% вихід. Спектр 1H ЯМР відповідав літературним значенням. (ACL-G29-16) Розчин діолу 13 (2,70г, 16ммоль) у ацетоні (500мл) охолоджували на льодяній бані у атмосфері N2. Порціями впродовж 20хв. додавали МnО2 (60,0г, 0,69моль). Льодяну баню витримували до розтоплення з перемішуванням реакційної суміші впродовж ночі. Реакційну суміш фільтрували через шар Hyflo, і фільтрат концентрували з одержанням твердої речовини жовтого кольору, 1,6г, 61% вихід у неочищеному стані. Спектр 1 Н ЯМР відповідав літературним значенням. Неочищену тверду речовину жовтого кольору розчиняли у метиленхлориді (додавали разом з невеликою кількістю 10% розчину Et2O у гексані) і вносили до силікагелевої колонки Biotage (4см´7,5см). Елюювання розпочинали 10% розчином простого ефіру у гексані (1л), після чого підвищували полярність до 15% Et2O (1л) і 20% Et2O (0,5л). Виділили тверду речовину жовтого кольору, 1,0г, 38% вихід. Спектр 1 H ЯМР відповідав необхідному продукту. (ACL-G29-21) Розчин діолу (9,31г, 60ммоль) у ацетоні (500мл) охолоджували на льодяній бані у атмосфері N2. Додавали МnО2 (100г, 1,15моль), і суміш перемішували із розтопленням льодяної бані впродовж ночі. Перевіркою засобами ІЧ-спектроскопії було встановлено утворення значної кількості продукту, однак присутньою все ще залишалась значна доля спирту. Додатково додали 50 г окисника, і перемішування здійснювали впродовж ще однієї ночі. Частину реакційної суміші фільтрували і перевіряли на 1 H ЯМР; реакція виглядала завершеною, виходячи із споживання вихідного матеріалу. Залишок реакційної суміші фільтрували через шар Hyflo і ретельно промивали ацетоном. Шляхом концентрування одержали тверду речовину темно-жовтого кольору. Азеотропну суміш піддали одноразовому кип'ятінню з 40мл бензолу, після чого сушили у вакуумі при температурі 40°С впродовж 5год., потім при кімнатній температурі впродовж ночі. Виділили 5,28г, 58% вихід. Спектр 1Н ЯМР та ІЧ-спектр відповідали необхідному продукту. Метилтиглат (16) У 2л 3-горлій колбі, спорядженій навісною мішалкою, конденсатором та термометром, розчин тиглінової кислоти 15 (89,8г; 0,9моль) та 5мл концентрованої сірчаної кислоти (0,09моль) у 900мл метанолу нагрівали зі зворотним холодильником впродовж 20год. Розчин охолоджували до температури 25°С, і надлишок метанолу відганяли при температурі 30°С під вакуумом (27 дюймів рт.ст. (685,8мм рт.ст.=91,4кПа)) на роторному випарнику. Дані газорідинного хроматографічного аналізу виділеного метанолового дистиляту показали присутність продукту у верхніх погонах. Одержаний двофазний концентрат світло-брунатного кольору розчиняли у 500мл етилового простого ефіру і послідовно промивали 250мл води, 250мл 10% водного розчину бікарбонату натрію і 250мл насиченого розсолу. Ефірний розчин сушили над безводним карбонатом калію, фільтрували і упарювали на роторному випарнику при температурі 25°С під вакуумом (17 дюймів рт.ст. (431,8мм рт.ст.=57,5кПа)) з одержанням неочищеного метилтиглату у вигляді майже безбарвного масла; 43,6г (42% вихід). Дані газорідинного хроматографічного аналізу показали один головний леткий продукт із часом утримування 2,7хв., порівняно з 3,8хв. для вихідної тиглінової кислоти. Протонний ЯМР у CDCI3 показав очікувані сигнали з деяким забрудненням слідовими кількостями етилового простого ефіру: 1,79млн-1 (d, 3Н), 1,83 (s, 3Н), 3,73 (s, 3Н), 6,86 (q, 6,6Гц). ІЧ (чистий на KВr): карбоніл складного ефіру при 1718см-1. Це масло застосовували як є на наступній стадії. Метил-γ-бромтиглат (17)5 У 1л 4-горлій колбі, спорядженій навісною мішалкою, термометром та конденсатором, перемішувану суміш неочищеного метилтиглату (43,6г; 0,38моль), N-бромсукциніміду (68г; 0,38моль) та 70% бензоїлу пероксиду (5,34г; 0,015моль) у 500мл чотирихлористого вуглецю нагрівали зі зворотним холодильником впродовж 2 год. Після охолодження до температури 20°С нерозчинний сукцинімід (38,1г, 100% виділення) видаляли шляхом вакуум-фільтрування. Фільтрат тричі промивали 250мл води, сушили над MgSO4, після чого упарювали на роторному випарнику при температурі 25°С під вакуумом (26 дюймів рт.ст. (660,4мм рт.ст.=88,0кПа)) з одержанням масла жовтого кольору; 78,8г. Протонний ЯМР цього масла у CDCl3 дав складний спектр. Протони метилену необхідного γ-бромзаміщеного складного ефіру були віднесені до дублету, центрованого при 4,04млн-1 (8,6Гц), тоді як ті самі протони α-бромзаміщеного ізомеру були віднесені до синглету при 4,24млн-1. Протонне інтегрування цих сигналів та метального мультиплету у межах від 1,6млн1 до 2,0млн-1 дозволило припустити таку композицію (% (моль)): γ-бромзаміщений складний ефір: 59% α-бромзаміщений складний ефір: 26% вихідний матеріал: 15% Це неочищене масло застосовували на наступній стадії без будь-якого додаткового очищення. Цю реакцію здійснювали також у масштабі 0,05моль із застосуванням лише 0,87екв. N-бромсукциніміду за інших ідентичних умов. Склад цього неочищеного масла визначали, виходячи з його спектра протонного ЯМР, як 52% γ-бромзаміщеного складного ефіру, 24% α-бромзаміщеного складного ефіру і 23% метилтиглату, що не вступав до реакції. Дані газорідинного хроматографічного аналізу цього масла були дещо складнішими і показували інші другорядні складові. Трифенілфосфонієва сіль метил-γ-бромтиглату (19)6 У 2л 4-горлій колбі, спорядженій термометром, 100мл краплинною лійкою постійного тиску та конденсатором, підключеним до статичної азотної системи, перемішуваний розчин неочищеного метил-γбромтиглату (78,8г) у 350мл бензолу обробляли шляхом додання краплями розчину трифенілфосфіну (95г; 0,36моль) у 350мл бензолу впродовж 1,75год. Температура суміші (екзотермічна реакція) дещо зростала від 24°С до 27°С за умов навколишнього середовища. Після додання реакційну суміш енергійно перемішували впродовж ночі з одержанням суспензії твердої речовини білого кольору, до складу якої входила смола жовтуватого кольору, яка прилипала до стінок колби. Тверду речовину білого кольору відфільтровували під вакуумом на лійці зі спеченим склом, не зачіпаючи згаданої смоли жовтуватого кольору. Колбу двічі промивали 100мл бензолу, і суміш виливали на фільтр. Осад на фільтрі промивали 50мл бензолу, потім двічі 50мл гексану. Вологий осад сушили у вакуумній печі при температурі навколишнього середовища впродовж 5,5год. Сухий порошок білого кольору [93г; температура плавлення=125°С (розклад)] розчиняли у 150мл ацетонітрилу з нагріванням з одержанням прозорого розчину жовтого кольору. До цього гарячого розчину додавали етилацетат (300мл), і продукт починав кристалізуватися після додання приблизно 100мл етилацетату. Колбу зберігали у холодильнику впродовж ночі. Продукт піддавали вакуум-фільтруванню і промивали мінімальною кількістю ацетонітрилу та етилацетату (1:2); 45,0г, температура плавлення=187-190°С (розклад), температура плавлення за літературними джерелами=183°С (розклад). Смолисті тверді речовини у реакційній колбі перекристалізовували з 10мл ацетонітрилу та 20мл етилацетату. Окрім того, з бензольного маточного розчину впродовж ночі до осаду випала додаткова кількість твердих речовин. Ці тверді речовини відфільтровували і перекристалізовували таким самим чином. Обидва зразки охолоджували впродовж 2год. і піддавали вакуум-фільтруванню з одержанням додаткового продукту; 13,3г. Бензольний фільтрат упарювали на роторному випарнику, масло жовтого кольору розчиняли у 10мл ацетонітрилу і осаджували 20мл етилацетату. Суспензію зберігали у холодильнику впродовж ночі з одержанням додаткового продукту у вигляді твердої речовини білого кольору; 4,6г, температура плавлення 185 - 187°С (розклад). Загальний вихід необхідної фосфонієвої солі у вигляді твердої речовини білого кольору становив 62,9г або 36,2% вихід, виходячи з неочищеного метилтиглату. Протонний ЯМР (CDCI3, тетраметилсилан) млн-1, 1,55 (d, 4Гц, 3Н), 3,57 (s, 3Н), 4,9 (dd, 15,8Гц та 7,9Гц, 2Н), 6,55 (широкий q, 6,6-7,9Гц, 1H), 7,4-7,9 (m, 15H). ЯМР фосфору з протонною розв'язкою (CDCI 3, 5% водний розчин Н3РО4 як співвісний зовнішній стандарт), 22,08млн-1. Частковий вуглецевий ЯМР (CDCl3): СО2СН3, (166,6млн-1, d, JCP=3Гц), олефіновий СН (117,5млн-1, d, JCP=86,1Гц), СО3СН3, (52,0млн-1), Ph3PCH2 (25,4млн-1, d, JCP=50,6Гц) та СН3 (13,4млн-1,d, JCP=2,4Гц). Частковий ІЧ-спектр (таблетка KВr): карбоніл складного ефіру при 1711см-1. (3-карбометокси-2-бутен-1-іліден)трифенілфосфоран (20)6 У 5л 5-горлій колбі, спорядженій навісною мішалкою, краплинною лійкою та термометром, розчин гідроксиду натрію (5,12г; 0,128моль) у 250мл води краплями додавали до енергійно перемішуваного розчину трифенілфосфонієвої солі метил-γ-бромтиглату (58,3г; 0,128моль) у 2500мл води впродовж 41хв. при температурі 25°С. Суспензію жовтого кольору перемішували впродовж 10хв. при кімнатній температурі, після чого піддавали вакуум-фільтруванню. Осад на фільтрі промивали 1800мл води, після чого ретельно сушили на фільтрі у атмосфері азоту. Тверду речовину жовтого кольору після цього сушили впродовж ночі у вакуумному ексикаторі над Р2О5 при кімнатній температурі під вакуумом (27 дюймів рт.ст. (685,8мм рт.ст.=91,4кПа)); 35,3г (73,7% вихід). Температура плавлення=145 - 150°С. Температура плавлення за літературними джерелами=145 - 165°С. ЯМР фосфору з протонною розв'язкою у CDCI 3 показав два піки при 17,1млн-1 та 21,1млн-1 у співвідношенні 93:7. Протонний ЯМР (CDCI3, тетраметилсилан) млн.-1, 1,89 (s, 3Н), 3,58 (s, 3Н), 7,3-7,8 (m, 17H). У цьому спектрі був явно виражений невеликий, але такий, що піддавався виявленню, синглет при 1,74млн-1, який відносили на рахунок домішок. Цю тверду речовину застосовували без додаткового очищення на наступній стадії. Диметилкроцетинат (21)6 (ACL-G29-18) Діальдегід 14 (0,48г, 2,9ммоль) вносили до 100мл круглодонної колби. Додавали бензол (20мл), і тверді речовини розчиняли з перемішуванням за допомогою магнітної мішалки. Додавали ілід; додатково застосовували 10мл бензолу для промивання сполуки у колбі. Нагрівали з енергійним кипінням впродовж 6 год. Реакційну суміш витримували з охолодженням впродовж ночі. У протилежність літературним повідомленням, одержали дуже невелику кількість твердої речовини. Реакційну суміш концентрували, осад розчиняли у МеОН (30мл) і кип'ятили впродовж 30хв. Після охолодження до температури навколишнього середовища тверді речовини збирали шляхом вакуум-фільтрування. Зразок для визначення ЯМР одержали шляхом розчинення 20мг у 0,5мл CDCl3. Деякий подив викликало те, що для повного розчинення необхідним виявилось нагрівання за допомогою нагрівача. Реєстрували спектр 1Н ЯМР і встановили його відповідність необхідному продукту. Залишковий матеріал розчиняли у гарячому бензолі, фільтрували, фільтрат концентрували, розчиняли у МеОН, охолоджували на льодяній бані і збирали тверді речовини червоного кольору, 334мг, 33% вихід. Цей матеріал виявився ніскільки не більш розчинним, аніж початково виділений матеріал. (ACL-G29-18A) Діальдегід 14 (5,78г, 35ммоль) розчиняли у бензолі (300мл) у атмосфері N2. Додавали ілід 20 (35,3г, 94ммоль), і суміш нагрівали зі зворотним холодильником впродовж 6год. з одержанням розчину темночервоного кольору. Після витримування реакційної суміші з охолодженням впродовж ночі, тверді речовини червоного кольору збирали шляхом вакуум-фільтрування і промивали метанолом. Переносили до 500мл круглодонної колби і нагрівали зі зворотним холодильником з приблизно 65мл метанолу впродовж 30хв. Охолоджували і збирали тверду речовину червоного кольору. Промивали холодним метанолом і сушили у вакуумі з одержанням 21 у вигляді твердої речовини червоного кольору, 3,00г. Спектр 1Н ЯМР та ІЧ-спектр відповідали необхідному продукту. Вихідний фільтрат (з реакційної суміші) концентрували на роторному випарнику, і залишок темного кольору розчиняли у 100мл метанолу і нагрівали зі зворотним холодильником впродовж 40хв. Охолоджували на льодяній бані і шляхом вакуум-фільтрування збирали тверду речовину червоного кольору. Промивали холодним метанолом і сушили у вакуумі з одержанням 21 у вигляді твердої речовини червоного кольору, 1,31г. Спектр 1Н ЯМР відповідав необхідному продукту. Фільтрати об'єднували, концентрували, розчиняли у 75мл метанолу і витримували з осадженням впродовж ночі при кімнатній температурі. Тверду речовину червоного кольору виділяли шляхом вакуумфільтрування: 0,38 г. Спектр 1Н ЯМР відповідав необхідному продукту. У фільтраті утворилось більше твердих речовин. Виділяли шляхом вакуум-фільтрування з одержанням твердої речовини червоного кольору, 0,127г. ІЧ-спектр відповідав вищезгаданому. Загальне виділення: 4,89г, 39% вихід. Спроба омилення за допомогою суміші тетрагідрофурану/NaOH (ACL-G29-19) Додавали перемішувану суспензію складного діефіру 21 (100мг, 0,28ммоль) у тетрагідрофурані (2мл) і 1-н. розчин NaOH (0,56мл, 2екв). Перемішували при кімнатній температурі впродовж ночі. Хроматографія в тонкому шарі показала лише вихідний матеріал. Нагрівали зі зворотним холодильником; жодних змін через декілька годин. Додали тетрагідрофуран (6мл) у спробі розчинити більшість твердих речовин, однак це не вияло жодного ефекту. Продовжували нагрівати до кипіння впродовж ночі. Додали ще тетрагідрофурану (приблизно 6мл, хроматографія в тонкому шарі показала лише вихідний матеріал) і нагрівали зі зворотним холодильником впродовж ще однієї ночі. Концентрували і перевірили на 1Н ЯМР. Виявили лише вихідний матеріал (на основі інтегрування метилу та метилового складного ефіру). Розчиняли у піридині (10мл) з одночасним нагріванням за допомогою нагрівальної оболонки. Додали 2,5-н. розчин NaOH (1,0мл). Через декілька хвилин розчин темно-оранжевого кольору набув темно-червоного забарвлення. Нагрівальну оболонку видалили, почалось утворення твердих речовин. Через 30хв. нагрівальну оболонку встановили знову, після чого перемішування продовжували при кімнатній температурі впродовж ночі. Концентрували під високим вакуумом. Залишок не розчинявся у хлороформі, диметилсульфоксиді, піридині і помірно розчинявся у Н2О. ІЧ-спектр (паста з вазелінового масла) показав оптичну густину С=О , характерну для вихідного матеріалу. Омилення 2,5-н. розчином NaOH і тетрагідрофураном (ACL-G29-20) Складний діефір 21 (37мг, 0,10ммоль) відважували до колби і перемішували у діетиловому простому ефірі (4мл). Розчинник набув оранжевого забарвлення, однак тверді речовини все ще були присутніми. Додали 1мл 2,5-н. розчину NaOH, і нагрівали зі зворотним холодильником. Через півгодини більшість простого ефіру випарилась. Простий ефір замінили на тетрагідрофуран (3мл), і нагрівання до кипіння продовжували на протязі декількох годин. Тверду речовину збирали шляхом вакуум-фільтрування, промивали деіонізованою водою, після чого сушили у вакуумній печі. ІЧ-спектр показав лише вихідний матеріал. Омилення 40% розчином NaOH (1) (ACL-G29-22) Складний діефір 21 (32мг, 8,9ммоль) відважували до колби і перемішували у метанолі (1,5мл). Розчинник набув оранжевого/червоного забарвлення, однак тверді речовини все ще були присутніми. Додали 1,5мл 40% розчину NaOH і нагрівали зі зворотним холодильником впродовж 17год. Після охолодження до кімнатної температури тверді речовини оранжевого кольору збирали шляхом вакуум-фільтрування і промивали деіонізованою водою. Сушили у вакуумі при температурі 40°С з одержанням 1 у вигляді порошку оранжевого кольору (21мг, 59%). ІЧ (таблетка KВr) 3412см-1, 1544см-1, 1402см-1, сполука, ймовірно, є гігроскопічною, зсув карбонілу у бік сильного поля відповідає спряженню. (ACL-G29-22A) Повторили з 35мг складного діефіру 1 із нагріванням до кипіння впродовж 15год. Реакційну суміш охолоджували на льодяній бані, збирали шляхом вакуум-фільтрування і промивали холодною деіонізованою водою. Сушили у вакуумі при температурі 40°С. Виділили 1 у вигляді твердої речовини оранжевого кольору (25,5мг, 65%). (ACL-G29-23) Складний діефір 21 (0,48г, 1,3ммоль) розчиняли у метанолі (15,0мл) і 40% розчині гідроксиду натрію (15,0мл) і нагрівали зі зворотним холодильником. Однорідна суміш червоного кольору через приблизно 2год. перетворилась на оранжеву. Нагрівання припинили через 6 год, і суміш витримували з охолодженням впродовж ночі. Тверду речовину оранжевого кольору збирали шляхом вакуум-фільтрування і промивали холодною деіонізованою водою. Шляхом висушування у вакуумі одержали крихку тверду речовину оранжевого кольору (0,36г, 68% вихід). (ACL-G29-24) Складний діефір 21 (1,10г, 3,1ммоль) вносили до 100мл відновної колби і нагрівали зі зворотним холодильником у метанолі (20мл) і 40% розчині NaOH (20мл) впродовж 12год. Після охолодження на льодяній бані тверду речовину оранжевого кольору збирали шляхом вакуум-фільтрування і промивали деіонізованою водою. Після висушування у вакуумі одержали 1,4г, 100%. Аналітично розраховано для C20H22O4Na2-0,4H2O: С, 63,29; Η, 6,05; Na, 12,11;H2O, 1,90. Встановлено: С, 63,41; Η, 6,26; Na, 11,75; Η2Ο, 1,93. (ACL-G29-25) Складний діефір 21 (3,00г, 8,4ммоль) нагрівали зі зворотним холодильником у метанолі (80мл) і 40% розчині NaOH (60мл) впродовж 12 год. Продукт виділяли у вигляді твердої речовини оранжевого кольору, як описувалось вище (2,7г, 880%). Аналітично розраховано для C20H22O4Na2-0,4H2O: С, 63,29; Η, 6,05; Na, 12,11; H2O, 1,90. Встановлено: С, 63,20; Η, 6,00; Na, 11,93; Η2Ο, 1,81. Зразки ACL-G29-23, -24 і -25 подрібнювали у агатовій ступці і об'єднували, як ACL-G29-A. Посилання I.E. Buchta and F. Andree Naturwiss. 1959, 46, 74. 2. F.J.H.M. Jansen, M. Kwestro, D. Schmitt, J. Lugtenburg Reel. Trav. Chim. Pays-Bas 1994, 113, 552. 3. R. Gree, H. Tourbah, R. Carrie, Tetrahedron Letters 1986, 27, 4983. 4. G.M. Coppola, Syn. Commun. 1984, 1021. 5. D.S. Letham and H. Young, Phytochemistry 1971, 10, 2077. 6. E. Buchta and F. Andree, Chem. Ber. 1960, 93, 1349. Приклад 2 Синтез транс-норбіксинату калію Транс-корбїксттат калію синтезують шляхом сполучення симетричного С 20-діальдегіду, що включає спряжені вуглець-вуглецеві подвійні зв'язки, з [1-(етоксикарбоніл)метиліден]трифенілфосфораном. Одержання цієї сполуки є подібним до одержання, яке описували вище для транс-кроцетинату натрію, за винятком того, що вихідний матеріал, фуран, замінюють на відповідну циклічну структуру. Цей продукт у подальшому омилюють за допомогою розчину КОН/метанолу. Приклад 3 Синтез довших БТКС Вищезгадану сполуку синтезують шляхом додання симетричного С10-діальдегіду, що включає спряжені вуглець-вуглецеві подвійні зв'язки, до надлишку [3-карбометокси-2-бутен-1-іліден]трифенілфосфорану. Одержання цієї сполуки є подібним до одержання, яке описували вище для транс-кроцетинату натрію, за винятком того, що вихідний матеріал, фуран, замінюють на відповідну циклічну структуру. Потім одержаний продукт із 40 атомів вуглецю, що має трянс-конфігурацію, виділяють за допомогою, наприклад, такої процедури, як хроматографування. Цей продукт у подальшому омилюють за допомогою розчину NaOH/метанолу. Приклад 4 Введення ТКН шляхом інгаляції ТКН вводили пацюкам інгаляційним шляхом. Десятьом пацюкам ТКН вводили безпосередньо до легень. Це робили шляхом введення трубки до трахеї і розпилювання 0,2мл розчину ТКН (ТКН розчиняли у розбавленому розчині карбонату натрію) з приблизно 3 - 6мл повітря. При усіх досліджених дозах (0,5 - 2мг/кг) приблизно 20% лікарського засобу були присутні у кровотоці через 1хв. після введення. У разі доз 0,8 1,6мг/кг, лікарський засіб був присутнім у кровотоці впродовж щонайменше 2год. Приклад 5 Поліпшений спосіб синтезу Одержання тетраетил-2-бутеніл-1,4-біфосфонату 250мл 3-горла колба була споряджена вкритою тефлоном термопарою, 60мл краплинною лійкою постійного тиску і простою дистиляційною насадкою. У атмосфері азоту чистий триетилфосфіт (59мл; 0,344моль) нагрівали за допомогою нагрівальної оболонки, температура якої контролювалась за допомогою регулятора Jkem, при температурі 140°С. Впродовж 93хв. при температурі 134 - 144°С краплями додавали розчин транс-1,4-дихлор-2-бутену (26,9г; 0,215моль) і триетилфосфіту (35мл; 0,204моль). Прозорий розчин після цього витримували при температурі 140°С у атмосфері азоту. Через 37хв. газова хроматографія аліквоти (1 крапля) у 1мл етилацетату показала необхідний продукт, проміжний продукт і два вихідні матеріали. Через 15,5год. при температурі 140°С газова хроматографія аліквоти (1 крапля у 0,5мл EtOAc) показала необхідний продукт без виявлення вихідного дихлориду або проміжного продукту. Через 16год. розчин блідожовтого кольору охолоджували до кімнатної температури у атмосфері азоту. Масло блідо-жовтого кольору переганяли у трубці з кульовим розширенням із двоколбовим приймачем і додатковою колбою, яку охолоджували на бані із твердою вуглекислотою-ацетоном при температурі 25 - 100°С і під тиском 0,1 - 0,2мм рт.ст. (13,3 - 26,6Па) з одержанням безбарвного масла (14,8г) у першому погоні фракції. Газова хроматографія показала лише продукт у перегінному кубі трубки з кульовим розширенням. Це масло світло-бурштинового кольору переганяли у трубці з кульовим розширенням при температурі 140°С і під тиском 0,1 - 0,15мм рт.ст. (13,3 - 20,0Па) з одержанням дистиляту у вигляді безбарвного масла; 66,45г (94,1% вихід). Газова хроматографія показала лише одну летку складову. Газохроматографічний-мас-спектроскопічний аналіз показав, що цією складовою був необхідний продукт, який давав невеликий молекулярний іон при 328 m/z і основний іон при 191 m/z (втрата РО3Еt2). Протонний ЯМР відповідав необхідному продукту. Вуглецевий ЯМР також відповідав необхідному біс(фосфонатному складному діефіру), показуючи лише далекодіючий (слабкий) та нормальний зв'язок вуглецю-фосфору з вуглецем алілу. Залишок у перегінному кубі - масло світло-жовтого кольору - 0,8г. Одержання 1,1,8,8-тетраметилокси-2,7-диметил-2,4,6-октатриєну У атмосфері азоту суміш тетраетил-транс-2-бутеніл-1,4-бісфосфонату (3,3г; 10,0ммоль), піровиноградного альдегіду диметилацеталю (2,6мл; 21,5ммоль) у 10мл толуолу та 10мл циклогексану, перемішувану за допомогою магнітної мішалки, послідовно обробляли безводним карбонатом калію (10,2г; 73,8ммоль) і порошкоподібним гідроксидом натрію (1,25г; 31,2ммоль). Розчин негайно набув жовтого забарвлення. Одержану суспензію перемішували при температурі навколишнього середовища у атмосфері азоту. Реакційна суміш повільно нагрівалась із досягненням максимальної температури 38°С через приблизно 25хв. Утворився також смолистий осад, який заважав роботі магнітної мішалки. Через 2,5год. газова хроматографія аліквоти розчину жовто-оранжевого кольору (1 крапля у 0,5мл толуолу) показала два вихідні матеріали та 3 інші нові складові. Через 16,75год. при температурі навколишнього середовища газова хроматографія аліквоти розчину оранжевого кольору (1 крапля у 0,5мл толуолу) показала лише невелику кількість вихідного складного біс(фосфонатного складного діефіру). Одержану суміш оранжевого кольору зі смолистою масою (яка заважала роботі магнітної мішалки) охолоджували на льодяній бані і різко охолоджували 100мл холодного 10% водного розчину NaCl. Тверді речовини розчиняли у цьому водному розчині за допомогою шпателя. Суміш після цього екстрагували 200мл суміші простого ефіру:гексану (1:1). Органічний шар промивали спочатку 10% водним розчином NaCl (200мл), потім насиченим розсолом (100мл). Безбарвний органічний шар сушили над Na2SO4. Газова хроматографія показала три головні складові без виявлення вихідного складного біс(фосфонатного складного діефіру). Хроматограмма в тонкому шарі показала дві головні плями і одну другорядну пляму. Na2SO4 видаляли шляхом вакуум-фільтрування і промивали простим ефіром. Фільтрат концентрували на роторному випарнику при температурі 35°С з одержанням безбарвного масла; 1,8г. Газохроматографічний-мас-спектроскопічний аналіз показав, що трьома головними леткими складовими були ізомерні продукти, що давали молекулярні іони при 256 m/z і основні іони при 75 m/z [(MeO)2CH+]. Протонний ЯМР також відповідав суміші ізомерних продуктів разом з іншими невизначеними домішками. Вихід неочищеного продукту=70,3%. Одержання 1,1,8,8-тетраметилокси-2,7-диметил-2,4,6-октатриєну Механічно перемішувану суміш тетраетил-транс-2-бутеніл-1,4-бісфосфонату (63,2г; 0,19моль), піровиноградного альдегіду диметилацеталю (50мл; 0,41моль) у 200мл толуолу та 200мл циклогексану послідовно обробляли безводним карбонатом калію (196г; 1,42моль) і порошкоподібним гідроксидом натрію (24,0г; 0,60моль). Розчин негайно набув жовтого забарвлення. Одержану суспензію перемішували при температурі навколишнього середовища у атмосфері азоту. Реакційна суміш нагрівалась до температури 61°С через приблизно 11хв., і перемішувану суміш охолоджували на льодяній бані для зниження температури до 35°С. Через 4,7год. при температурі 29 - 35°С газова хроматографія аліквоти (3 краплі у 0,5мл толуолу) показала відсутність вихідного біс(фосфонату). Через 5год. суміш охолоджували на льодяній бані до температури 13°С і при підвищенні температури до 30°С додавали 10% водний розчин хлориду натрію (400мл). Додатково додавали 10% водний розчин хлориду натрію (1500мл), і суміш екстрагували 3000мл суміші простого ефіру:гексану (1:1). Органічний шар жовтого відтінку промивали спочатку 10% водним розчином хлориду натрію (2´1000мл), потім насиченим розсолом (1000мл). Органічний шар жовтого відтінку сушили над Na2SO4, фільтрували і концентрували на роторному випарнику при температурі 30°С з одержанням масла світло-жовтого кольору; 43,4г. Газова хроматографія показала три головні складові, що становили 89% суміші, без виявлення вихідного біс(фосфонату). Хроматографічний аналіз в тонкому шарі показав одну головну та 3 другорядні складові. Протонний ЯМР показав ізомерний продукт плюс толуол. Масло додатково випарювали на трубці з кульовим розширенням при температурі 50°С та під тиском 0,2мм рт.ст. (26,6Па) впродовж 30хв.; 31,9г. Протонний ЯМР показав ізомерний продукт, біс(ацеталь), без виявлення толуолу. Вихід=65,5%. Одержання 2,7-диметил-2,4,6-октатриєндіалу при більшому корисному навантаженні У атмосфері азоту розчин неочищених ізомерів 1,1,8,8-тетраметилокси-2,7-диметил-2,4,6-октатриєну (31,9г; 124,4ммоль) у тетрагідрофурані (160мл), воді (80мл) та льодяній оцтовій кислоті (320мл), перемішуваний за допомогою магнітної мішалки, нагрівали при температурі 45°С за допомогою нагрівальної оболонки, температура якої контролювалась за допомогою регулятора Jkem через покриту тефлоном термопару (9:03 ранку). Реакційна суміш нагрівалась із досягненням максимальної температури 54°С через 30хв., після чого її температура повернулась до заданих 45°С. Газова хроматографія аліквоти (3 краплі у 0,5мл тетрагідрофурану) через 3год. показала деяку залишкову частину вихідного матеріалу, два головні та один другорядний продукт. Реакційний розчин жовтого кольору охолоджували на льодяній бані до температури 21°С, після чого розбавляли сумішшю простого ефіру:дихлорметану (4:1, 2000мл). Після цього цей розчин послідовно промивали 20% водним розчином NaCl (2000мл´2), сумішшю 20% водного розчину NaCl:1Μ водного розчину NaOH (4:1, 2000мл´3) (Перші два змиви, очевидно, видалили оцтову кислоту, про що свідчить нейтральний рН. Третій змив набув червоного забарвлення і все ще залишався основним, що дозволяє припустити видалення побічного продукту) та 20% водним розчином NaCl (1000мл´2). Органічний шар жовтого кольору сушили над MgSO4, фільтрували і концентрували на роторному випарнику з одержанням твердої речовини жовтого кольору; 18,9г. Газова хроматографія показала одну головну і одну другорядну складову, вихідний біс(ацеталь). Хроматографічний аналіз в тонкому шарі показав одну головну пляму і декілька другорядних, більш полярних домішок. Цю тверду речовину розчиняли у 250мл нагрітого до кипіння метанолу, охолоджували до кімнатної температури, потім, впродовж 1год., на льодяній бані. Суспензію піддавали вакуум-фільтруванню з одержанням пухнастих голчастих кристалів жовтого кольору; 14,15г. Газова хроматографія показала суміш ізомерних діальдегідів (95:5). Цю тверду речовину знову перекристалізовували з 200мл нагрітого до кипіння метанолу, охолоджували до кімнатної температури, після чого охолоджували у холодильнику впродовж ночі. Суспензію піддавали вакуум-фільтруванню і промивали охолодженим у морозильній камері метанолом з одержанням голчастих кристалів жовтого кольору; 11,2г. Газова хроматографія показала суміш ізомерних діальдегідів (97:3). Хроматографічний аналіз в тонкому шарі показав одну пляму. Голчасті кристали сушили у вакуумній печі при температурі 45°С впродовж 160хв. до набуття ними постійної маси; 10,75г. Некоригована температура плавлення=154 - 156°С. Температура плавлення за літературними джерелами [Dictionary of Organic Compounds. Version 10:2, Sept, 2002] =161 - 162°C. Протонний і вуглецевий ЯМР відповідали необхідному симетричному діальдегіду. Два метанолові фільтрати перекристалізацій об'єднували. Хроматограмма в тонкому шарі показала продукт плюс інші домішки. Фільтрати концентрували, і різні виходи збирали, як показано нижче: Вихід 2 3 4 Вигляд Кількість (г) Ізомерний склад 1,4 80:20 2,6 75:25 4,45 46:30 Порошок жовтого кольору Порошок жовтого кольору Тверда речовина жовтого кольору Вихід 2 і 3: Ці об'єднані виходи розчиняли у 20мл нагрітого до кипіння етилацетату, охолоджували до кімнатної температури, після чого, впродовж 1год., охолоджували у морозильній камері. Суспензію піддавали вакуум-фільтруванню і промивали охолодженим у морозильній камері етилацетатом з одержанням голчастих кристалів жовтого кольору; 1,95г. Газова хроматографія показала суміш ізомерів 86:14. Цю тверду речовину знову перекристалізовували у етилацетаті (10мл) з одержанням голчастих кристалів жовтого кольору; 1,55г. Газова хроматографія показала ізомерний склад 92:8. Після третьої перекристалізації з етилацетату (10мл) одержали голчасті кристали жовтого кольору; 1,25г. Температура плавлення =152 - 154°С. Газова хроматографія показала ізомерний склад 94:6. Протонний ЯМР підтвердив, що ця речовина є необхідним діальдегідом. Газохроматографічний-мас-спектроскопічний аналіз відповідав необхідному діальдегіду і показав чітко визначений М+ іон при 164 m/z і основний іон при 91 m/z. Етилацетатний фільтрат об'єднували з твердою речовиною жовтого кольору з метанолового фільтрату (вихід 4) і концентрували на роторному випарнику з одержанням твердої речовини жовтого кольору; 6,0г. Газова хроматографія показала суміш (53:34) двох ізомерів разом з іншими домішками. Тверду речовину розчиняли у 100мл дихлорметану і додавали силікагель (33,5г) класу Davisil. Суміш упарювали на роторному випарнику при температурі 35°С. Після цього силікагель з адсорбованим матеріалом вносили до модуля введення проби до системи Biotage, яка вже містила шар скловати і шар піску. Після цього силікагель накривали фільтрувальним папером. Колонку Biotage 75S попередньо зволожували сумішшю розчинників із радіальним стисненням 35 фунт/дюйм2 (241,15 кПа) і тиском розчинників 20фунт/дюйм2 (137,80кПа). Колонку елюювали сумішшю гексану:етилацетату (85:15, 6000мл). Вільний об'єм із включенням стадії попереднього зволоження приймали рівним 1000мл. На основі хроматографічного аналізу в тонкому шарі, збирали і об'єднували 250мл фракції. Ці фракції концентрували на роторному випарнику при температурі 35°С, як показано нижче. Фракція 1 2-3 4 5-10 11-18 19-20 Вміст Контроль А Слідова кількість А В Слідова кількість В або слідова кількість С Слідова кількість В або С та D Вигляд Тверда речовина жовтого кольору Кількість (г) 3,9 Пояснювальна примітка Фракція продукту Жодних ознак елюйованої у безпосередній близькості домішки Фракції 5-10: Тверду речовину жовтого кольору суспендували у гексані і піддавали вакуум-фільтруванню з одержанням твердої речовини світло-жовтого кольору; 2,5г. Газова хроматографія показала суміш ізомерів діальдегіду зі співвідношенням 67:33. Загальний вихід 96 - 97% Е,Е,Е-Діальдегіду =10,75+1,25=12,0г (58,8% вихід). Ізомеризація 2,7-диметил-2,4,6-октатриєндіалу паратолуолсульфіновою кислотою У атмосфері азоту ізомерну суміш (2:1) 2,7-диметил-2,4,6-октатриєндіалу та його другорядного ізомеру (2,5г; 15,2ммоль), 4-толуолсульфінову кислоту (0,35г; 2,2ммоль) і 50мл безводного 1,4-діоксану нагрівали зі зворотним холодильником впродовж 15хв. Аліквоту (7 крапель) розбавляли у 0,5мл суміші простого ефіру:дихлорметану (4:1) і сушили над K2СО3. Газова хроматографія показала суміш (91:9) необхідного та другорядного ізомерів. Після охолодження впродовж ночі при кімнатній температурі одержану суспензію розчиняли у 100мл суміші простого ефіру:дихлорметану (4:1) і послідовно промивали водою (50мл´3), 0,2Μ водним розчином NaOH (50мл), водою (50мл´2) і насиченим розсолом (50мл´3). Після розділення шарів залишковий осадовий шар розчиняли у дихлорметані. Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували на роторному випарнику при температурі 40°С з одержанням твердої речовини оранжевого кольору; 2,2г. Газова хроматографія показала 93:7 співвідношення необхідного діальдегіду та другорядного ізомеру. Цю тверду речовину суспендували у гексані і піддавали вакуум-фільтруванню з одержанням твердої речовини оранжевого кольору; 2,15г. Цю тверду речовину перекристалізовували з 20мл нагрітого до кипіння етилацетату шляхом охолодження до температури 30 - 40°С з подальшим охолодженням у морозильній камері впродовж 1 год. Суспензію піддавали вакуум-фільтруванню і промивали охолодженим у морозильній камері етилацетатом з одержанням голчастих кристалів жовто-оранжевого кольору; 1,65г. Температура плавлення =158 - 160°С. Температура плавлення за літературними джерелами =161 - 162°С. Газова хроматографія показала 96:4 співвідношення необхідного діальдегіду та другорядного ізомеру. Протонний та вуглецевий ЯМР відповідали необхідному ізомеру діальдегіду. Вихід=66%. Одержання метилтиглату з розчином тіонілхлориду у метанолі у збільшеному об'ємі Механічно перемішуваний розчин тиглінової кислоти (397,35г; 3,97моль) у 3000мл метанолу обробляли (доданням краплями) чистим тіонілхлоридом (397мл; 5,44моль) впродовж 130хв. при зростанні температури від 14°С до максимального рівня у 50°С через 80хв. без зовнішнього охолодження. Газова хроматографія аліквоти показала повне перетворення на складний ефір без виявлення тиглінової кислоти. Після перемішування при температурі навколишнього середовища впродовж 1 год, розчин переганяли під атмосферним тиском на посрібленій колонці Вітре з вакуумною оболонкою (400мм´20мм). Конденсат із температурою, переважно, 57 - 61°С збирали при температурі перегінного кубу 58 - 63°С; 630мл за 2год. Газова хроматографія показала значну кількість метилового складного ефіру у дистиляті. Колонку Вігре замінили на колонку меншої ефективності (30см´2см, з меншою кількістю заглиблень) для прискорення швидкості перегонки. При температурі перегінного кубу 69 - 71°С, дистилят збирали з температурою головного погону 65 - 59°С; 1300мл впродовж 2,25год. Газова хроматографія показала значну кількість метилового складного ефіру в дистиляті. Атмосферну перегонку продовжували, доки температура кубу не досягла 87°С; дистилят впродовж цього періоду збирали з температурою головного погону 69 - 83°С; 975мл впродовж 2год. Газова хроматографія показала значно більшу кількість метилового складного ефіру у дистиляті, аніж у попередніх фракціях. Двофазну суміш жовтого кольору у перегінному кубі екстрагували простим ефіром (300мл та 200мл), сушили над K2СО3, фільтрували і концентрували на роторному випарнику при температурі 25°С з одержанням масла оранжевого кольору; 132,6г (29,3% вихід). Газова хроматографія показала продукт. Протонний та вуглецевий ЯМР відповідали необхідному продукту зі слідовими кількостями етилового простого ефіру. Газова хроматографія ефірного конденсату показала деяку кількість метилового складного ефіру у верхніх погонах. Дистилят 3: Третій метаноловий дистилят (975мл) концентрували на роторному випарнику при температурі 25°С з одержанням двофазної суміші (100 - 150мл). Цю суміш екстрагували простим ефіром (100мл та 50мл), сушили над K2СО3. Дистилят 2: Другий метаноловий дистилят (1300мл) концентрували на роторному випарнику при температурі 25°С з одержанням двофазної суміші (30 - 50мл). Цю суміш екстрагували простим ефіром (2´50мл), сушили над K2СО3. Концентровані ефірні екстракти дистиляту 2 та дистиляту 3 об'єднували, піддавали вакуум-фільтруванню і концентрували на роторному випарнику при температурі 25°С з одержанням безбарвного масла; 77,3г. Протонний та вуглецевий спектри відповідали попереднім спектрам необхідного метилового складного ефіру. Загальний вихід =132,6+77,3=209,9г (46,3%) За альтернативним варіантом, 1) метилтиглат є комерційно доступним від фірм Alfa, Lancaster або Acros і 2) можна провести пілотні експерименти з одержання солі фосфонію за методикою, що наводиться у JOC [(Journal of Organic Chemistry), 64, 8051-8053 (1999)]. Бромування метилтиглату Механічно перемішувану суспензію метилтиглату (209,9г; 1,84моль) та N-бромсукциніміду (327,5г; 1,84моль), 70% розчину бензоїлпероксиду (3,2г; 0,009моль) у 2000мл чотирихлористого вуглецю нагрівали зі зворотним холодильником (78 - 81°С) у 1л сполучній трубці з кульовим розширенням між 5л реакційною колбою та парціальним конденсатором гарячого зрошення. Через 2год. нагрівання до кипіння припиняли, нагрівальну оболонку видаляли, мішалку відключали. Усі тверді речовини плавали у розчині ССl4, що дозволяє припустити присутність сукциніміду з незначною кількістю N-бромсукциніміду. Суспензію охолоджували на льодяній бані до температури 20°С і піддавали вакуум-фільтруванню з одержанням твердої речовини жовтуватого кольору; 180,7г. Не промивали. Фільтрат жовтого кольору промивали водою (1л´3), сушили над MgSO4. Газова хроматографія показала вихідний метилтиглат і два моноброміди у співвідношенні 1:2:1, а також інші другорядні складові. Після відфільтровування MgSO4, фільтрат світло-жовтого кольору концентрували на роторному випарнику при температурі 35°С з одержанням масла світло-жовтого кольору; 327,1г. Протонний ЯМР і газова хроматографія дозволяють припустити такий склад: Складова ЯМР Газова (моль %) хроматографія (площина %) γ-бромзаміщений складний ефір α-бромзаміщений складний ефір α,γ-дибромзаміщений складний ефір (?) Метилтиглат Інше 50% 49% 26% 21% 7% 4% 6% 11% 10% Вихід необхідного продукту приведено до 50% кількісного аналізу =46,0%. Це масло застосовують як є на наступній стадії. Реакція метил-γ-бромтиглату з трифенілфосфіном у ацетонітрилі у збільшеному об'ємі з дещо більшим корисним навантаженням Неочищену суміш метил-γ-бромтиглату (322,6г; 85% алілбромід; 1,42моль) у 1300мл безводного ацетонітрилу піддавали механічному перемішуванню у 5л 4-горлій колбі у атмосфері азоту. Краплями впродовж 4год. додавали розчин трифенілфосфіну (387,0г; 1,48моль) у 2000мл етилацетату. Під час додання температура зросла з 22°С до максимального рівня у 30°С після додання приблизно 40% впродовж перших 75хв. Після додання 60% розчину трифенілфосфіну впродовж 120хв., розчин став каламутним, і впродовж подальшого додання до осаду продовжували випадати тверді речовини. Після завершення додання лійку промивали етилацетатом (600мл) і додавали цей об'єм до реакційної суміші. Суспензію кремового кольору перемішували при температурі навколишнього середовища з суботи до понеділка. Суспензію білого кольору піддавали вакуум-фільтруванню, і осад на фільтрі промивали сумішшю етилацетату:ацетонітрилу (2:1, 150мл´3). Тверду речовину білого кольору (352,55г) сушили у вакуумній печі при температурі 40°С впродовж 4год. (постійна маса через 2год.); 322,55г. Температура плавлення =187 188°С (розклад), температура плавлення за літературними джерелами=183°С (розклад). Протонний та вуглецевий ЯМР відповідали попереднім спектрам необхідної солі фосфонію. Газохроматографічний-масспектроскопічний аналіз показав одну головну складову, мас-спектр з електророзпиленням якої у позитивному режимі відповідав необхідній солі фосфонію з виділенням молекулярного іону при 375 m/z. ЯМР фосфору показав один сигнал фосфору при 22,0млн-1. Вихід на основі вихідного метилтиглату= =100х322,55/(455,32´1,84´322,6/327,1)=39,0%. Одержання (3-карбометокси-2-Z-бутен-1-іліден)трифенілфосфорану Механічно перемішувану рідку суспензію (3-карбометокси-2-Е-бутен-1-іліден)трифенілфосфонію броміду (154,8г; 0,34моль) у 3400мл деіонізованої води обробляли (доданням краплями) розчином гідроксиду натрію (13,6г; 0,34моль) у 500мл води при температурі 23°С впродовж 32хв. без явного підвищення температури, однак з негайним випаденням до осаду твердої речовини світло-жовтого кольору. Після перемішування впродовж 15хв. суспензію світло-жовтого кольору піддавали вакуум-фільтруванню, промивали водою (1500мл) і сушили під вакуумом з одержанням твердої речовини світло-жовтого кольору; 151,7г. Цю тверду речовину сушили у вакуумній печі при температурі 35 - 45°С (3:50 вечора) впродовж ночі. Після сушіння у вакуумній печі при температурі 35 - 45°С впродовж 22,5год. одержали постійну масу; 107,8г. Температура плавлення=144 - 160°С, температура плавлення за літературними джерелами =145 165°С. Протонний ЯМР був подібним до попереднього спектра необхідного іліду, приймаючи до уваги різниці напруженості поля ЯМР. Вуглецевий ЯМР показав вуглець метилу при 50,2млн-1 і 11,8млн-1 зі складною ароматичною ділянкою і без явних сигналів атомів вуглецю олефіну і атома вуглецю іліду. Вихід=84,7%. Пілотне одержання диметилкроцетинату Суміш (3-карбометокси-2-Z-бутен-1-іліден)трифенілфосфорану (12,8г; 34,2ммоль) і 2,7-диметил-2,4,6октатриєндіалу (2,1г; 12,8ммоль) у бензолі (128мл), перемішувану за допомогою магнітної мішалки, нагрівали зі зворотним холодильником у атмосфері азоту впродовж 6год. із застосуванням таймера. Одержану суспензію охолоджували на льодяній бані впродовж 40хв., піддавали вакуум-фільтруванню, промивали бензолом і сушили під вакуумом для розтоплення замерзлого бензолу з одержанням твердої речовини червоного кольору; 2,1г. Хроматографічний аналіз в тонкому шарі показав одну пляму жовтого кольору. Цю тверду речовину сушили у вакуумній печі при температурі 40 - 45°С впродовж 70хв.; 1,85г (40,5% вихід). Некоригована температура плавлення =210 - 213°С, температура плавлення за літературними джерелами [Е. Бухта (Е. Buchta), Φ. Андре (F. Andree), Chem Ber, 93, 1349 (1960)] =214 - 216°С. Протонний ЯМР був подібним попередньому спектру диметилкроцетинату на 90МГц приладі. Вуглецевий ЯМР показав усі 11 специфічних вуглецевих сигналів із коригованим хімічним зсувом для необхідного диметилового складного ефіру з одним сигналом другорядної домішки, якою може бути залишковий бензол. Мас-спектр з елекгророзпилювання дозволяє припустити розклад і рекомбінацію фрагментів. Хроматографічний аналіз в тонкому шарі показав, що фільтрат червоного кольору включав додатковий продукт, трифенілфосфіну оксид, і складову оранжевого кольору з Rf, дещо нижчим за відповідний показник виділеної твердої речовини. Фільтрат червоного кольору концентрували на роторному випарнику при температурі 35°С з одержанням твердих речовин червоного кольору; 13,2г. Цю тверду речовину нагрівали зі зворотним холодильником у метанолі (25мл). Після цього одержану суспензію охолоджували на льодяній бані, через 60хв. піддавали вакуум-фільтруванню і промивали метанолом з одержанням твердої речовини червоного кольору; 0,6г. Цю тверду речовину сушили у вакуумній печі при температурі 45°С впродовж 135хв.; 0,5г. Температура плавлення =203 - 208°С. Протонний ЯМР показав необхідний складний діефір із залишковими домішками. Вуглецевий ЯМР показав лише сигнали необхідного продукту. Хроматографічний аналіз в тонкому шарі показав смугасту пляму продукту. Фільтрат концентрували і зберігали. Другий спосіб одержання диметилового складного ефіру кроцетину 2,7-диметил-2,4,6-октатриєндіал (11,95г; 12,8ммоль) однією порцією додавали у атмосфері азоту до механічно перемішуваної суспензії (3-карбометокси-2-7-бутен-1-іліден)трифенілфосфорану (73,0г; 195,0ммоль) у 400мл бензолу, після чого суміш розбавляли 330мл бензолу. Одержану суспензію брунатного кольору нагрівали зі зворотним холодильником впродовж 6год. із застосуванням таймера і охолоджували до кімнатної температури впродовж ночі у атмосфері азоту. Одержану суспензію охолоджували на льодяній бані до температури 6 - 10°С, піддавали вакуумфільтруванню і промивали бензолом (50мл´2) з одержанням твердої речовини червоного кольору; 10,05г. Хроматографічний аналіз в тонкому шарі показав одну пляму жовтого кольору. Цю тверду речовину сушили у вакуумній печі при температурі 40°С (9:00 ранку) впродовж 3,5год. без втрати маси; 10,05г (38,7% вихід). Температура плавлення =211 - 214°С, температура плавлення за літературними джерелами =214 - 216°С. Протонний та вуглецевий ЯМР відповідали попереднім спектрам необхідного диметилового складного ефіру кроцетину. Фільтрат червоного кольору концентрували на роторному випарнику при температурі 40°С з одержанням твердої речовини червоного кольору; 84,4г. Хроматографічний аналіз в тонкому шарі був подібним до пілотного експерименту. Цю тверду речовину суспендували у 165мл метанолу з нагріванням із зворотним холодильником і перемішуванням за допомогою магнітної мішалки. Після цього одержану суспензію охолоджували на льодяній бані впродовж 2,5год., піддавали вакуум-фільтруванню і промивали мінімальною кількістю метанолу з одержанням пасти оранжевого кольору; 10,5г. Хроматографічний аналіз в тонкому шарі показав одну пляму жовтого кольору. Цю пасту сушили у вакуумній печі при температурі 45°С впродовж 190хв.; 5,6г. Температура плавлення =201 - 208°С. ЯМР показав необхідний складний діефір із невідомими ароматичними домішками. Цю неочищену тверду речовину і дві інші подібні тверді речовини з попередніх серій експериментів (загальною масою 6,5г) розчиняли у киплячому хлороформі (75мл), розбавляли метанолом і охолоджували у холодильнику впродовж ночі. Суспензію піддавали вакуум-фільтруванню і промивали мінімальною кількістю метанолу з одержанням кристалічної твердої речовини червоного кольору: 6,1г. Цю тверду речовину сушили у вакуумній печі при температурі 45°С впродовж 3год. до одержання постійної маси; 4,25г. Температура плавлення =211 - 213°С. Протонний і вуглецевий ЯМР показали інші олефінові або ароматичні домішки. Цю тверду речовину розчиняли у киплячому толуолі (150мл) і, зрештою, охолоджували у холодильнику впродовж 130хв. Суспензію піддавали вакуум-фільтруванню і промивали толуолом з одержанням твердої речовини червоного кольору; 2,05г. Цю тверду речовину сушили у вакуумній печі при температурі 45°С впродовж 50хв. без зміни маси; 2,05г. Температура плавлення =214 - 216°С. Протонний ЯМР показав необхідний диметилкроцетин з деякою залишковою кількістю толуолу і незначними домішками другорядного ізомеру. Вуглецевий ЯМР показав необхідний диметилкроцетин без виявлення домішок другорядного ізомеру і 2 - 3 нові залишкові сигнали, які відповідали толуолу. Вихід=45,5%. Одержання двонатрієвої солі кроцетину Механічно перемішувану суміш диметилкроцетину (13,95г; 39,1ммоль), водного розчину (40% (мас.)) гідроксиду натрію (273мл; 3,915моль) і метанолу (391мл) нагрівали зі зворотним холодильником при температурі 74°С впродовж 12год. із застосуванням таймера. Одержану суспензію оранжевого кольору піддавали вакуум-фільтруванню на лійці Бюхнера з фільтрувальним папером і лійкою зі спеченим склом. Повільне фільтрування (фільтрування відбувалось швидше через спечене скло, доки фільтр не забився після висушування. Закупорення фільтра, однак, вдалось ліквідувати промиванням водою). Суспензію з лійки зі спеченим склом додавали до твердих речовин у лійці Бюхнера. Пасту оранжевого кольору промивали спочатку водою (100мл´3), потім метанолом (50мл´3). Пасту оранжевого кольору сушили у вакуумній печі при температурі 45 - 50°С. Через 21год. маса грудок оранжевого кольору становила 24,25г. Матеріал подрібнювали шпателем і сушили у вакуумній печі при температурі 45 - 50°С. Після висушування впродовж у цілому 65,5 год, кількість порошку оранжевого кольору досягла 23,1г. ІЧспектр показав додаткові смуги, порівняно з повідомленим ІЧ-спектром ТКН, з особливо широкими смугами при 3424см-1 та 1444см-1. Протонний ЯМР не мав ознак метилових складних ефірів. Об'єднання олефінової та метальної ділянок було відсутнім, можливо, унаслідок проблем фазування. Приймаючи, що надлишкова маса була обумовлена гідроксидом натрію, тверду речовину оранжевого кольору перемішували за допомогою магнітної мішалки у 400мл деіонізованої води впродовж 35хв. Суспензію піддавали вакуум-фільтруванню, і осад на фільтрі промивали деіонізованою водою (50мл´2) з одержанням пасти оранжевого кольору. Цей матеріал сушили у вакуумній печі при температурі 45 - 50°С до одержання постійної маси. Через приблизно 7год. тверду речовину подрібнили, розпилили і додатково висушили у вакуумній печі при температурі 45°С впродовж ночі. Після 21год. висушування при температурі 45°С кількість твердої речовини становила 13,25г. Після додаткового розпилювання та сушіння у вакуумній печі при температурі 45°С кількість твердої речовини становила 13,15г. Інфрачервоний спектр відповідав повідомленому ІЧ-спектру. Протонний ЯМР відповідав двонатрієвій солі. Високоефективний рідинний хроматографічний аналіз показав одну головну складову з, можливо, однією другорядною домішкою. Спектр з електророзпилюванням маси негативних іонів головної складової відповідав необхідній двонатрієвій солі кроцетину. Вуглецевий ЯМР показав усі десять специфічних вуглецевих сигналів для двонатрієвої солі кроцетину, що підтверджувало симетрію молекули. З вихідного фільтрату води, гідроксиду натрію та метанолу до осаду під час промивання водою випала додаткова кількість твердих речовин. Цю суспензію піддавали вакуум-фільтруванню, промивали водою з одержанням пасти оранжевого кольору. Цю пасту сушили у вакуумній печі при температурі 45°С впродовж 18,5год. з одержанням твердої речовини оранжевого кольору; 0,65г. Спектральні дані відповідали необхідній двонатрієвій солі кроцетину. Цю тверду речовину об'єднували з першим виходом. Вихід=13,15+0,65=13,8г (94,8%). Елементний аналіз першого виходу показав неприйнятні значення для необхідного продукту, що дозволяє припустити забруднення двонатрієвої солі кроцетину гідроксидом натрію. Промивання двонатрієвої солі кроцетину водою Двонатрієву сіль кроцетину (13,6г) суспендували у 150мл деіонізованої води і перемішували за допомогою магнітної мішалки при кімнатній температурі впродовж 1 год. Суспензію піддавали вакуум-фільтруванню на лійці Бюхнера. Пасту оранжевого кольору після цього промивали водою з контролюванням рН фільтрату оранжевого кольору. Пасту оранжевого кольору сушили під вакуумом на фільтрі з гумовою перегородкою. Цю пасту сушили під вакуумом при температурі 25 - 55°С впродовж 5,5год. з одержанням крихкої твердої речовини оранжевого кольору; 11,2г. Цю тверду речовину розпилювали, переносили до флакона і сушили у вакуумній печі при температурі 45°С впродовж ночі. Кількість=11,1г. Виділення=81,6%. ІЧ-спектр та спектр протонного ЯМР відповідали попереднім ІЧспектрам та спектрам протонного ЯМР необхідної двонатрієвої солі кроцетину. Високоефективний рідинний хроматографічний аналіз показав одну складову при 420нм, мас-спектр з електророзпилюванням якої у режими негативних іонів відповідав кроцетину. Вуглецевий ЯМР показав усі десять специфічних сигналів вуглецю з коригованими хімічними зсувами для необхідної двонатрієвої солі кроцетину. Елементний аналіз дав прийнятні дані для необхідного продукту. Посилання 1. Tetrahedron Letters, 27, 4983-4986 (1986). 2. F.J.H.M. Jansen, M. Kwestro, D. Schmitt & J. Lugtenburg, Reel. Trav. Chem. Pays-Bas, 113, 552-562 (1994) та посилання, цитовані в цій роботі. 3. J.H. Babler, Патент США 4,107,030, 12 квітня, 1992. 4. T.W. Gibson & P. Strassburger, J. Org. Chem., 41, 791 (1976) & J.M. Snyder & C.R. Scholfield, J. Am. Oil Chem. Soc, 59, 469 (1982). Приклад 6 Визначення чистоти ТКН, який одержали за поліпшеним способом синтезу Для ТКН, синтезованого за способом Прикладу 5, відношення оптичної густини при 421нм до оптичної густини при 254нм становило 11,1, що визначалось за допомогою УФ-спектрофотометра. Приклад 7 Пероральне введення ТКН На пацюках було показано, що ТКН, у разі введення через рот (за допомогою шлункового зонда), абсорбується до кровотоку. На двох пацюках було встановлено, що до кровотоку, через 15 - 30хв. після введення, попадає від 1% до 2% дози. Абсорбування максимальної кількості через рот відбувалось фактично раніше цього періоду часу. Фахівцям у цій галузі є легко зрозумілою можливість внесення численних модифікацій та доповнень до цих сполук та композицій, а також споріднених способів без відхилення від розкритого винаходу.
ДивитисяДодаткова інформація
Назва патенту англійськоюTrans carotenoid bipolar salts and uses thereof
Автори англійськоюGainer John L., Grabiak Raymond C.
Назва патенту російськоюБиполярные соли транс-каратиноидов и их использование
Автори російськоюГейнер Джон Л., Гребьяк Раймонд К.
МПК / Мітки
МПК: C07C 305/00, A61P 9/00, A61P 25/28, A61P 11/06, C07F 9/113, A61P 25/00, C07C 69/602, A61P 9/10, A61P 11/00, A61P 15/14, A61K 31/202, C07C 57/00
Мітки: солі, використання, біополярні, транс-каротиноїдів
Код посилання
<a href="https://ua.patents.su/23-83341-biopolyarni-soli-trans-karotinodiv-ta-kh-vikoristannya.html" target="_blank" rel="follow" title="База патентів України">Біополярні солі транс-каротиноїдів та їх використання</a>
Похідні піридилу, їх енантіомери, цис- або транс-ізомери і їх солі

Номер патенту: 26976
Опубліковано: 28.02.2000
Автори: СОЙКА Райнер, ВЕЙСЕНВЕРГЕР Йоханес, МЮЛЛЕР Томас
МПК: C07D 213/55, C07D 401/06, C07F 7/08, C07D 401/12
Мітки: піридилу, транс-ізомери, цис, похідні, енантіомери, солі
Формула / Реферат:
1. Производные пиридила общей формулы (І):где А - углерод-азотная связь или неразветвленная алкиленовая группа с 1 - 4 атомами углерода,X - нитрометиленовая группа, цианометиленовая группа, замещенная цианогруппой, аминокарбонилом или алкоксикарбонилом с 1 - 3 атомами углерода в алкоксильной группе, мли группа формулы =N-R7, где R7 означает цианогруппу, алкансульфонил с 1 - 3 атомами углерода, фенилсульфонил,...
Солі ізотіазол-4-карбоксаміду та їх використання як антигіперпроліферативних агентів

Номер патенту: 74221
Опубліковано: 15.11.2005
Автори: Уільямс Гленн Роберт, Гант Томас Дж.
МПК: A61P 17/06, A61P 37/06, A61K 31/425, A61P 29/00, A61P 13/08, A61P 9/10, A61P 13/12, A61P 27/02, A61P 9/00, A61P 17/00, C07D 275/00, A61P 35/02, A61P 3/10, A61P 35/00, A61P 19/02
Мітки: агентів, ізотіазол-4-карбоксаміду, використання, солі, антигіперпроліферативних
Формула / Реферат:
1. Гідробромідна сіль 3-(4-бром-2,6-дифторбензилокси)-5-[3-(4-піролідин-1-ілбутил)уреїдо]ізотіазол-4-карбоксаміду.2. Сіль згідно з п. 1, при цьому вказана сіль має спектр порошкової рентгенографії, такий самий, що й спектр порошкової рентгенографії, показаний на Фіг. 2.3. Геміцитратна сіль 3-(4-бром-2,6-дифторбензилокси)-5-[3-(4-піролідин-1-ілбутил)уреїдо]ізотіазол-4-карбоксаміду.4. Сіль згідно з п. 3, при цьому вказана...
Фракція гіалуронової кислоти або її солі для застосування в офтальмології, спосіб одержання фракції натрієвої солі гіалуронової кислоти та фармацевтичний препарат для використання в офтальмології

Номер патенту: 27229
Опубліковано: 15.08.2000
Автори: Ауреліо Ромео, Сільвана Лоренці
МПК: A61K 9/08, C08B 37/08, A61P 27/02, A61K 31/715
Мітки: одержання, натрієвої, фармацевтичний, фракція, препарат, офтальмології, спосіб, гіалуронової, використання, кислоти, солі, фракції, застосування
Текст:
...Получаемую в результате пасту помещают в стеклянный сосуд с 10 объемами безводного ацетона или этанола, содержащими 0,1% весового объема специфического хелатообразующего агента, способного образовывать комплекс с железом Примерами использования хелатообразующих агентов являются 1,10 - фенантролин (С A R N 66-71-7) или его диметиловые производные Стеклянные емкости и специфические хелатообразователи используются для того, чтобы избежать...
Нові солі врс-пептидів з органопроективною активністю, спосіб їх отримання і їх використання в терапії

Номер патенту: 61955
Опубліковано: 15.12.2003
Автори: Ротквич Іво, Петек Маріян, Зайверт Свен, Туркович Бранко, Сікірич Предраг, Дувняк Марко, Грабаревич Желько, Удовічич Іван, Місе Степан
МПК: A61K 38/00, C07K 7/06, C07K 7/08, A61P 43/00, C07K 14/47, A61P 39/00
Мітки: спосіб, використання, солі, терапії, врс-пептидів, отримання, органопроективною, нові, активністю
Формула / Реферат:
1. Сіль пептидної сполуки для захисту організму (ВРС), що містить 8 амінокислотних залишків, де аніон солі є негативно заряджений пептид, що має загальну формулу [Zaa Pro Pro Pro Xaa Yaa Pro Ala] (-)або(2-),деXaa є нейтральний аліфатичний амінокислотний залишок,Yaa є основний амінокислотний залишок, іZaa є кислий амінокислотний залишок,і де катіон солі є катіоном неорганічної або органічної...
Солі e-2-метокси-n-(3-{4-[3-метил-4-(6-метилпіридин-3-ілокси)феніламіно]хіназолін-6-іл}аліл)ацетаміду, спосіб їх одержання і використання для лікування раку

Номер патенту: 75482
Опубліковано: 17.04.2006
Автори: Ріхтер Деніел Тайлер, Кат Джон Чарльз
МПК: A61P 17/00, A61P 13/12, A61P 7/00, A61P 1/00, A61P 5/00, A61P 25/00, A61P 35/00, A61P 13/02, A61K 39/395, A61P 43/00, A61P 1/18, A61K 31/675, A61K 31/475, A61K 45/00, A61K 31/4745, C07D 401/12, A61P 11/00, A61P 15/00, A61P 27/02, A61P 13/08, A61P 35/02, A61K 33/24, A61K 31/138, A61K 31/573, A61K 31/7072, A61P 19/00, A61K 31/704, A61K 31/337, A61K 31/282, A61K 31/517, A61K 38/43, A61P 13/10, A61K 31/513
Мітки: солі, лікування, раку, e-2-метокси-n-(3-{4-[3-метил-4-(6-метилпіридин-3-ілокси)феніламіно]хіназолін-6-іл}аліл)ацетаміду, одержання, використання, спосіб
Формула / Реферат:
1. Сіль Е-2-метокси-N-(3-{4-[3-метил-4-(6-метилпіридин-3-ілокси)феніламіно]хіназолін-6-іл}аліл)ацетаміду у вигляді сукцинату.2. Сполука за п. 1, де сукцинатною сіллю Е-2-метокси-N-(3-{4-[3-метил-4-(6-метилпіридин-3-ілокси)феніламіно]хіназолін-6-іл}аліл)ацетаміду є сесквісукцинат.3. Спосіб лікування аномального росту клітин у ссавця, при якому вводять зазначеному ссавцеві сіль...
Попередній патент: Автозаправна установка
Наступний патент: Спосіб квазічастотного пуску асинхронного двигуна та пристрій для його реалізації
Випадковий патент: Стенд для визначення рідинних та газових фільтраційних характеристик зразків гірських порід