Похідні заміщеного азепіну як модулятори рецептора серотоніну
Номер патенту: 88786
Опубліковано: 25.11.2009
Автори: Робердж Майкл Дж., Тамі Лоуренс, Беннані Юссеф Л., Варга Норберт, Бом Девід К.








Формула / Реферат
1. Сполука формули І
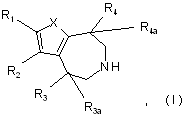
у якій
X являє собою S, О або NR5;
R1 та R2 незалежно вибрані з групи, яка включає Н, галоген, С1-8алкіл, С1-8алкіларил, С1-8алкілгетероарил, С1-8алкеніл, пергалоалкіл, CN, OR5, SR5, N(R5)2, CON(R5)2, NR5COR5, NR5CO2R5, SO2N(R5)2, NR5SO2R5, арил та гетероарил, де вказані арил або гетероарил можуть бути необов'язково заміщені за допомогою до трьох замісників, вибраних з алкілу, галогену та алкоксигрупи;
R3 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -СН2-О-С1-8 алкілу, -СН2ОН, -СОО-С1-8алкілу, -CON(R5)2 та арилу;
R3a являє собою Н або R3 та R3a, взяті разом, являють собою СН2СН2- або
R2 та R3 утворюють 5- або 6-членне кільце;
R4 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8 алкіларилу, С1-8алкілгетероарилу, OR5, -СН2-О-С1-8алкілу, -СН2ОН, -СОО- С1-8алкілу, -CON(R5)2 та арилу;
R4a являє собою Н або R4 та R4a, взяті разом, являють собою -СН2СН2-, за умови, що принаймні один з R1, R2, R3, R3a, R4 та R4a повинен приймати значення, відмінне від водню;
та додатково за умови, що, якщо R3 являє собою ОН, тоді принаймні один з R1, R2, R4 та R4a повинен приймати значення, відмінне від водню;
якщо R4 являє собою арил та R2 являє собою Н, CN або Вr, тоді принаймні один з R1, R3 та R3a повинен приймати значення, відмінне від водню; та
якщо X являє собою О, тоді принаймні один з R3, R3a, R4 та R4a повинен приймати значення, відмінне від водню; та
R5 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, арилу, гетероарилу та пергалоалкілу;
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, у якій: X являє собою S;
R1 вибраний з групи, яка включає галоген, С1-8алкіл, OR5, SO2N(R5)2 тa пергалоалкіл;
R2 вибраний з групи, яка включає водень, галоген, С1-8алкіл та OR5, або разом з R3 утворює 5-членне кільце;
R3 являє собою водень або С1-8алкіл;
R3а являє собою водень;
R4 являє собою водень або С1-8алкіл;
R4а являє собою водень; і
R5 являє собою водень або С1-8алкіл.
3. Сполука за п. 1, яка вибрана з групи, що включає:
2-бром-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2,3-дибром-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-хлор-4-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-хлор-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2,3-дихлор-4-метил-5,6,7,8-тетрагідро-тієно[2,3-d]азепін;
2-(4-фторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(2,5-дифторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(3-хлор-4-фторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(2,5-дихлорфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(5-фтор-2-метоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(3,4,5-триметоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(4-етоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(4-етилфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(3-метоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-феніл-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-нафтален-1-іл-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-нафтален-2-іл-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(2,6-дифторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
3-(2,6-дифторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(2-хлор-6-фторбензил)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
3-бром-2-(2-хлор-6-фторбензил)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-8-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-2-сульфокислоти;
диметиламід 3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-2-сульфокислоти;
2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-аза-циклопента[сd]азулен;
2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-аза-циклопента[сd]азулен;
2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; та
2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
або її фармацевтично прийнятна сіль.
4. Сполука за п. 1, яка вибрана з групи, що включає:
2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-бром-8-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-2-сульфокислоти;
диметиламід 3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-2-сульфокислоти;
2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-аза-циклопента[сd]азулен;
2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-аза-циклопента[сd]азулен;
2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; та
2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
або її фармацевтично прийнятна сіль.
5. Сполука за п. 1, яка вибрана з групи, що включає:
2-(4-трифторметоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
2-(2-трифторметилфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; та
2-(піролідин-1-сульфоніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін;
або її фармацевтично прийнятна сіль.
6. Фармацевтична композиція, яка містить принаймні одну сполуку за п. 1 та фармацевтично прийнятний носій.
7. Спосіб лікування захворювання, розладу та/або стану у пацієнта, де бажаною є модуляція функції 5-НТ2с, який включає введення пацієнту, який потребує такого лікування, ефективної кількості принаймні однієї сполуки за п. 1.
Текст
1. Сполука формули І 2 (19) 1 3 88786 4 2-(4-фторфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-(2,5-дифторфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(3-хлор-4-фторфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2,5-дихлорфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(5-фтор-2-метоксифеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(3,4,5-триметоксифеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(4-етоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-(4-етилфеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-(3-метоксифеніл)-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-феніл-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; 2-нафтален-1-іл-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-нафтален-2-іл-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-(2,6-дифторфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 3-(2,6-дифторфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2-хлор-6-фторбензил)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 3-бром-2-(2-хлор-6-фторбензил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; 2-бром-3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-8-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-сульфокислоти; диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти; 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; та 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; або її фармацевтично прийнятна сіль. 4. Сполука за п.1, яка вибрана з групи, що включає: 2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; 2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-метокси-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-8-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-сульфокислоти; диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти; 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; та 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; або її фармацевтично прийнятна сіль. 5. Сполука за п.1, яка вибрана з групи, що включає: 2-(4-трифторметоксифеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2-трифторметилфеніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; та 2-(піролідин-1-сульфоніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; або її фармацевтично прийнятна сіль. 6. Фармацевтична композиція, яка містить принаймні одну сполуку за п.1 та фармацевтично прийнятний носій. 7. Спосіб лікування захворювання, розладу та/або стану у пацієнта, де бажаною є модуляція функції 5-НТ2с, який включає введення пацієнту, який потребує такого лікування, ефективної кількості принаймні однієї сполуки за п.1. Даний винахід стосується ряду сполук, фармацевтичних композицій, що містять ці сполуки, та застосування цих сполук і композицій як фармацевтичних агентів. Більш конкретно сполуки даного винаходу являють собою сполуки гексагідротієноазепіну та октагідротієноазепіну. Ці сполуки є лігандами рецептора серотоніну (5-НТ) і вони є корисними для лікування захворювань, розладів та станів, де бажаною є модуляція рецепторів (5-НТ) серотоніну (наприклад, наркоманія, стан тривоги, депресія та ожиріння). Встановлено, що серотонін є задіяний у ряді захворювань, розладів та станів, які беруть початок з центральної нервової системи, включаючи захворювання, розлади та стани, які відносяться до, наприклад, сну, прийому їжі, болю, який відчувається, контролю температури тіла, контролю кров'яного тиску, депресії, стану тривоги, наркоманії та шизофренії. Серотонін також відіграє важли 5 88786 ву роль в периферичних системах, таких як шлунково-кишкова система, де, як було виявлено, він опосередковує ряд скорочувальних, секреторних та електрофізіологічних ефектів. Внаслідок широкого розподілення серотоніну в організмі, є потреба в ліках, які впливають на серотонергічні системи. Зокрема агоністи, часткові агоністи та антагоністи серотонергічних систем є цікавими для лікування широкого ряду розладів, включаючи стан тривоги, депресію, гіпертензію, мігрень, ожиріння, розлади, пов'язані з нав'язливістю, шизофренію, аутизм, нейродегенеративні розлади (напр. хворобу Альцгеймера, паркінсонізм та хорею Хантінгтона) і нудоту, викликану хіміотерапією. Головні класи рецепторів серотоніну (5-НТ1-7) включають від одного до семи окремих рецепторів, які були формально класифіковані. Див Glennon et al., Neuroscince and Behavioral Reviews, 1990, 14, 35; та D. Hoyer, et al., Pharmacol. Rev. 1994, 46, 157-203. Наприклад, сімейство 5-НТ2 рецепторів включає підтипи 5-НТ2а, 5-НТ2b та 5-НТ2c, які були сгруповані на основі первинної структури, вторинної месенджерної системи та операційного профілю. Інші три підтипи 5-НТ2 являють собою зв'язані Gпротеїном, активовані фосфоліпазою С як основним механізмом трансдукції, і включають семитрансмембранну доменну структуру. Є чіткі розділення у розподіленні трьох підтипів 5-НТ2 у ссавців. Рецептори 5-НТ2b та 5-НТ2a широко розподілені у периферичній нервовій системі, причому 5НТ2a також знаходять у мозку. Рецептори 5-НТ2c знаходять лише у центральній нервовій системі і він є високо експресованим у багатьох зонах людського мозку. Див. G Baxter, et al., Trends in Pharmacol Sci. 1995,16, 105-110. Підтип 5-НТ2а асоціюється з ефектами, включаючи вазоконстрикцію, агрегацію бляшок та бронхоконстрикцію а також і деякі ефекти ЦНС, в той час як підтип 5-НТ2c асоціюється із захворюваннями, які включають депресію, стан тривоги, розлад, пов'язаний зі станом нав'язливого неврозу, наркоманію, панічні розлади, фобії, психіатричні синдроми та ожиріння. Дуже мало відомо про фармакологічну роль рецептора 5-НТ2b. Див. F. Jenck, et al., Exp Opin Invest. Drugs, 1998, 1587-1599; M. Bos, et al., J. Med. Chem., 1997. 40, 2762-2769; J.R. Martin at al., The Journal of Pharmacology and Experimental Therapeutics, 1998, 286, 913-924; S.M. Bromidge, et al., J. Med. Chem., 1998, 41, 15981612; G.A. Kenneth, Drugs, 1998, 1. 4, 456-470; та A. Dekeyne, et al., Neuropharmacology, 1999, 38, 415-423. WO 93/13105 описує похідні тіофену; Патент US 4414225 описує похідні тіофену, фурану та піролу та WO 96/12201 описує похідні фурану. Даний винахід стосується сполук формули (I) 6 де X являє собою S, О або NR5; R1 та R2 незалежно вибрані з групи, яка включає Н, галоген, С1-8алкіл, С1-8алкіларил, С1-8алкілгетероарил, С1-8алкеніл, пергалоалкіл, CN, OR5, SR5, N(R5)2, CON(R5)2, NR5COR5, NR5CO2R5, SO2N(R5)2, NR5SO2R5, арил та гетероарил, де вказані арил та гетероарил можуть бути необов'язково заміщені від одного до трьох замісниками, вибраними з алкілу, галогену та алкокси, або R1 та R2 ,взяті разом, утворюють 5-або 6членне кільце; R3 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -CH2-O-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R3a являє собою Η або R3 та R3а, взяті разом, являють собою -СН2СН2-, або R2 тa R3 утворюють 5- або 6-членне кільце; R4 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -СН2-О-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R4a являє собою Н або R4 та R4a, взяті разом, являють собою –СН2СН2-; і R5 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, арилу, гетероарилу та пергалоалкілу або разом з атомом, до якого він приєднаний, R5 утворює гетероарильне кільце. Інший варіант втілення даного винаходу пропонує фармацевтичну композицію, яка містить сполуку формули (І) або її фармацевтично прийнятну сіль та фармацевтично прийнятний носій. Ще один варіант втілення даного винаходу пропонує спосіб лікування або попередження захворювань, розладів і/або станів центральної нервової системи. Спосіб включає введення терапевтично ефективної кількості сполуки формулі (І) або її фармацевтично прийнятної солі ссавцю. Конкретні захворювання, розлади і/або стани включають ожиріння, депресію, шизофренію, тривогу, розлад, пов'язаний зі станом нав'язливого неврозу, наркоманію, панічні розлади, розлади сну, мігрень, діабети II типу, епілепсію, фобіє та психіатричні синдроми. Використані наступні визначення, якщо не вказано інше. Як використано тут, термін "алкіл" включає прямі ланцюгові та розгалужені вуглеводневі групи, які містять вказане число атомів вуглецю, зазвичай метил, етил та прямі ланцюгові та розгалужені пропільні та бутильні групи. Термін "алкіл" також охоплює циклоалкіл, тобто циклічну С3-С8 вуглеводневу групу, таку як циклопропіл, циклобутан, циклопентил та циклогексил. Посилання на індивідуальну групу чи складову, таку як "пропіл" охоплює лише групу чи складову з прямим ланцюгом. Розгалужений ланцюговий ізомер, такий як "ізопропіл" вказується конкретно. Як використано тут, термін "алкеніл", один або у комбінації, стосується заміщеного або незаміщеного з прямим ланцюгом або заміщеного або незаміщеного з розгалуженим ланцюгом алкенільного радикала, який містить від 2 до 10 атомів 7 вуглецю. Приклади таких радикалів включають, але без обмеження, етеніл, Е- та Z-пентеніл, деценіл та ін. Як використано тут, термін "алкокси", один або у комбінації, стосується радикалу алкілетеру, де термін "алкіл" визначено вище. Приклади придатних радикалів алкілетеру включають, але без обмеження, метокси, етокси, n-пропокси, ізопропокси, n-бутокси, ізо-бутокси, сек-бутокси, третбутокси та ін. Термін "гало", як використано тут, включає фтор, хлор, бром або йод. Подібним чином термін "галоген" означає тут фтор, хлор, бром та йод. Термін "аміно", один або у комбінації, включає групу -NH2 або NRaRb, де Ra та Rb незалежно являють собою алкіл, алкіларил або арил. Термін "арил", один або у комбінації, означає тут моно циклічну або біциклічну ароматичну групу (напр. феніл чи нафтил), яка може бути незаміщеною або заміщеною, наприклад, одним або більше, зокрема від одного до трьох наступними замісниками, вибраними з групи, яка складається з Н, гало; CN, NO2, CF3, N3, С1-6алкілу, ОН, NRaRb, ОС1a a b a b 6алкілу, OR , С(=О) NR R , C(=S) NR R , тетраозоілу, тріазолілу, амідинілу, гуанідинілу, тіогуанідинілу, ціаногуанідинілу та арилу. Загалом "арил" означає фенільну групу, або орто-злиту біциклічну карбоциклічну групу, яка містить від дев'яти до десяти атомів кільця, де принаймні одне кільце є ароматичним (напр. нафтил чи тетрагідронафтил). Термін "арил" також є скороченим у багатьох хімічних структурах до "Аr". Термін "гетероарил", як використано тут, включає моноциклічну, біциклічну чи трициклічну кільцеву систему, яка містить один, два чи три ароматичних кільця та включає принаймні один атом кисню або сірки у ароматичному кільці, я яка може бути незаміщеною або заміщеною, наприклад, одним чи більше, зокрема від одного до трьох замісниками, такими як гало, алкіл, гідрокси, гідроксиалкіл, алкокси, алкоксиалкіл, галоалкіл, нітро, аміно, алкіламіно, ациламіно, алкілтіо, алкілсульфоніл та алкілсульфоніл. Приклади гетероарильних груп включають, але без обмеження, 2Нпіроліл, 3Н-індоліл, 4Н-хінолізиніл, 4nНкарбазоліл, акридиніл, бензо[b]тієніл, бензотіазоліл, 13-карболініл, карбазоліл, хроменіл, цінаолініл, дибензо[b,d]фураніл, фуразиніл, фурил, імідазоліл, імідізоліл, індазоліл, індолізиніл, індоліл, ізобензофураніл, ізоіндоліл, ізохіноліл, ізотіазоліл, ізоксазоліл, нафтіридиніл, нафто[2,3-b], оксазоліл, перимідиніл, фенантридиніл, фенантролініл, фенарсалізиніл, феназиніл, фенотіазиніл, феноксатіініл, феноксазиніл, фталазиніл, птеридиніл, пууриніл, піраніл, піразиніл, піразоліл, піридазиніл, піриділ, піримідиніл, піримідиніл, піроліл, хіназолініл, хіноліл, хіноксалініл, тіадіазоліл, тіантреніл, таізоліл, тієніл, тіазоліл та ксантеніл. В одному варіанті втілення термін "гетероарил" позначає моноциклічне ароматичне кільце, яке містить п'яти чи шість атомів кільця, які містять вуглець, та 1, 2, 3 або 4 гетероатомів, незалежно вибраних з групи, яка включає не-пероксидний кисень, сірку та N(Z), 88786 8 де Ζ відсутній або являє собою Н, О, С1-4алкіл, феніл чи бензил. В іншому варіанті втілення позначає орто-злиту біциклічне гетерокільце з від одного до десяти атомами кільця, які походять від нього, зокрема бенз-похідну чи похідну, одержану шляхом прикріплення пропіленового чи тетраметиленового дирадикалу. Термін "Het" загалом означає гетероциклічну групу, насичену або частково ненасичену, яка містить принаймні один гетероатом, вибраний з групи, яка складається з кисню, азоту та сірки, і необов'язково заміщеною С1-6алкілом чи C(=O)OR6. Зазвичай "Het" являє собою мооциклічну, біциклічну або трициклічну групу, яка містить один або більше гетероатомів, вибраних з групи, яка складається з кисню, азоту та сірки. Група "Het" також може включати оксо-групу (=O), приєднану до кільця. Приклади, яку не обмежують, включають 1,3-дигідробензофуран, 1,3-діоксан, 1,4-діоксан, 1,4-дитіан, 2Н-піран, 2-піразолін, 4Н-піран, хроманіл, імідазолідиніл, імідазолініл, індолініл, ізохроманіл, ізоіндолініл, морфолін, піперазиніл, піперидин, піпериділ, піразолідин, піразолідиніл, піразолініл, піролідин, піролін, хінуклідин та тіоморфолін. Кращі варіанти втілення даного винаходу включають: Варіант 1: де R1 та R2 незалежно вибрані з групи, яка включає Н, галоген, С1-8алкіл, С1-8алкіларил, С1-8алкілгетероарил, С1-8алкеніл, пергалоалкіл, CN, OR5, SR5, N(R5)2, CON(R5)2, NR5COR5, NR5CO2R5, SO2N(R5)2, NR5SO2R5, арил та гетероарил, де вказані арил та гетероарил можуть бути необов'язково заміщені від одного до трьох замісниками, вибраними з алкілу, галогену та алкокси або R1 та R2 взяті разом, утворюють 5-або 6членне кільце; R3 вибраний з групи, яка складається з Н, С1-8алкілу, OR5, -СН2-O-С1-8алкілу, -СН2ОН, -СООС1-8алкілу, -CON(R5)2 та арилу; R3a являє собою Η або R3 та R3a, взяті разом, являють собою -СН2СН2- або R2 та R3 утворюють 5- або 6-членне кільце; R4 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -CH2-O-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R4a являє собою Н або R4 та R4a, взяті разом, являють собою -СН2СН2-; і R5 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, арилу, гетероарилу та пергалоалкілу або разом з атомом, до якого він приєднаний, R5 утворює гетероарильне кільце. 9 Варіант 2: де R1 та R2 незалежно вибрані з групи, яка включає Н, галоген, С1-8алкіл, С1-8алкіларил, С1-8алкілгетероарил, С1-8алкеніл, пергалоалкіл, CN, OR5, SR5, N(R5)2, CON(R5)2, NR5COR5, NR5CO2R5, SO2N(R5)2, NR5SO2R5, арил та гетероарил, де вказані арил та гетероарил можуть бути необов'язково заміщені від одного до трьох замісниками, вибраними з алкілу, галогену та алкокси або R1 та R2 взяті разом, утворюють 5-або 6членне кільце; R3 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіл арилу, С1-8алкілгетероарилу, OR5, -СН2-О-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R3a являє собою Η або R3 та R3a, взяті разом, являють собою -СН2СН2- або R2 та R3 утворюють 5- або 6-членне кільце; R4 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -СН2-О-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R4a являє собою Н або R4 та R4a, взяті разом, являють собою -СН2СН2-; і R5 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, арилу, С18алкілгетероарилу, гетероарилу та пергалоалкілу або разом з атомом, до якого він приєднаний, R5 утворює гетероарильне кільце. Варіант 3 де R1 та R2 незалежно вибрані з групи, яка включає Н, галоген, С1-8алкіл, С1-8алкіларил, С1-8алкілгетероарил, С1-8алкеніл, пергалоалкіл, CN, OR5, SR5, N(R5)2, CON(R5)2, NR5COR5, NR5CO2R5, SO2N(R5)2, NR5SO2R5, арил та гетероарил, де вказані арил та гетероарил можуть бути необов'язково заміщені від одного до трьох замісниками, вибраними з алкілу, галогену та алкокси або R1 та R2 взяті разом, утворюють 5-або 6членне кільце; R3 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -CH2-O-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; R3a являє собою Η або R3 та R3a, взяті разом, являють собою -СН2СН2- або R2 та R3 утворюють 5- або 6-членне кільце; R4 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкіларилу, С1-8алкілгетероарилу, OR5, -CH2-O-С1-8алкілу, -СН2ОН, -СОО-С1-8алкілу, CON(R5)2 та арилу; 88786 10 R4a являє собою Н або R4 та R4a, взяті разом, являють собою -СН2СН2-; і R5 вибраний з групи, яка складається з Н, С1-8алкілу, С1-8алкілгетероарилу, С1-8алкіларилу, арилу, гетероарилу та пергалоалкілу або разом з атомом, до якого він приєднаний, R5 утворює гетероарильне кільце. Переважно X являє собою S; R1 вибраний з групи, яка включає галоген, С1-8алкіл, OR5, SO2N(R5)2 та пергалоалкіл. R2 вибраний з групи, яка включає водень, галоген, С1-8алкіл та OR5 або разом з R3 утворює 5членне кільце; R3 являє собою водень або С1-8алкіл; R3 являє собою водень; R4 являє собою водень або С1-8алкіл; R4 являє собою водень; і R5 являє собою водень або С1-8алкіл, або разом з атомом, до якого він приєднаний, утворює гетероарильне кільце. На даний час кращими сполуками є: 5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; 2-бром-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; 2,3-дибром-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; (R,S) 5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін4-ол; 2-хлор-4-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-хлор-4-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2,3-дихлор-4-метил-5,6,7,8-тетрагідротієно[2,3-d]азепін; 2-(4-трифторметокси-феніл)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; 2-(3-трифторметил-феніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 2-(2-трифторметил-феніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 2-(4-фтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2,5-дифтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(3-хлор-4-фтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2,5-дихлор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(5-фтор-2-метокси-феніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 2-(3,4,5-триметокси-феніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 2-(4-етокси-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(4-етил-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(3-метокси-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-феніл-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 11 2-(3-фтор-біфеніл-4-іл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2-фтор-біфеніл-4-іл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-4-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-8-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(пірролідин-1-сульфоніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-сульфокислоти; диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти; 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; трифторметил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-нафтален-1-іл-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-нафтален-2-іл-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2,6-дифтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 3-(2,6-дифтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(2-хлор-6-фтор-бензил)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 3-бром-2-(2-хлор-6-фтор-бензил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; етиловий естер 2-аміно-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-3-карбонової кислоти; етиловий естер 2-аміно-5,6,7,8-тетрагідро-4Нтієно[2,3-с]азепін-3-карбонової кислоти; етиловий естер 5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-3-карбонової кислоти та етиловий естер 5,6,7,8-тетрагідро-4Нтієно[2,3-с]азепін-3-карбонової кислоти. Особливо кращі сполуки включають: 5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін; 2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; 2-бром-3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-4-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-бром-8-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 2-(пірролідин-1-сульфоніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; 88786 12 диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-сульфокислоти; диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти; 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; 2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен; трифторметил-5,6,7,8-тетрагідро-4Н-тієно [2,3d]азепін; 3-бром-2-трифторметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; та 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін. Деякі сполуки винаходу можуть існувати в різних ізомерних (напр., енантіомери та дистереізомери) формах. Винахід охоплює всі такі ізомери як у чистій формі, так і у суміші, включаючи рацемічні суміші. Енольні форми також включені. Сполуки винаходу можуть існувати як у несольватованих так і у сольватованих формах, включаючи гідратовані форми, напр., гемігідрат. Загалом, сольватовані форми з фармацевтично прийнятними розчинниками, такими як вода, етанол та ін. для вирішення задач винаходу еквівалентні несольватованим формам. Деякі сполуки винаходу можуть також утворювати фармацевтично прийнятні солі, напр., кислі адитивні солі. Наприклад, атоми азоту можуть утворювати солі з кислотами. Прикладами придатних кислот для утворення солі є хлористоводнева, сірчана, фосфорна, оцтова, лимонна, щавлева, малонова, саліцилова, яблучна, фумарола, бурштинова, аскорбінова, малеїнова, метансульфонова та інші мінеральні карбонові кислоти, які добре відомі у галузі. Солі одержують контактуванням вільної основної форми з достатньою кількістю бажаної кислоти для одержання солі для одержання солі у звичайний спосіб. Вільні кислотні форми можуть бути регенеровані шляхом обробки солі придатним розбавленим водним основним розчином, таким як розбавлений водний гідроксидний карбонат калію, аміак та бікарбонат натрію. Вільні основні форми відрізняються від їх відповідних сольових форм деякими фізичними властивостями, такими як розчинність у полярних розчинниках, але кислі солі еквівалентні їх відповідним основним формам для вирішення задач винаходу. (Див, напр., S. Μ. Berge et al., "Pharmaceutical Salts," J. Pharm Sci, 66: 1-19 (1977), де вказане джерело включено шляхом посилання. Як використано тут, термін "композиція" охоплює продукт, який містить конкретні інгредієнти у конкретних кількостях, або будь-який продукт, який походить прямо чи непрямо, з комбінації конкретних інгредієнтів у конкретних кількостях. Сполуки даного винаходу можуть бути використані у формі фармацевтично прийнятних солей, які походять з неорганічних чи органічних кислот. Фраза "фармацевтично прийнятна кислота" позначає такі солі, які у межах озвученої медичної оцінки, придатні для використання в контакті з тканинами людей та нижчих тварин без небажаної токсичної, подразнювальної, алергічної відповіді та ін. і які відпові 13 дають прийнятному співвідношенню користь/ризик. Фармацевтично прийнятні солі добре відомі у галузі. Наприклад, S. М. Berge докладно описує фармацевтично прийнятні солі у J. Pharmaceuitical Sciences, 1977, 66: 1 et seq. Солі можна одержати in situ під час кінцевого виділення та очищення сполук винаходу, або роздільно шляхом реакції вільної основної функції з придатною органічною кислотою. Репрезентативні кислі адитивні солі включають, але без обмеження, ацетат, адипат, альгінат, цитрат, аспартат, бензоат, бензольсульфонат, бісульфат, бутират, камфорат, камфорсульфонат, диглюконат, гліцерофосфат, гемісульфат, гептаноат, гексоноат, фумарат, гідрохлорид, гідробромід, гідройодид, 2-гідроксиетансльфонат (ізотіонат), лактат, малеат, метансульфонат, нікотінат, 2-нафталенсульфонат, оксалат, пальмітоат, пектинат, персульфат, 3-фенілпропіонат, пікрат, півалат, пропіонат, сукцинат, тартрат, тіоціанат, фосфат, глутамат, бікарбонат, ртолулолсульфонат та ундеканоат. Також основні групи, що містять азот, можна кватернізувати з такими агентами як нижчі алкілгалоїди, такі як метил, етил, пропіл та бутилхлориди, броміди та йодиди; діалкілсульфати, такі як диметил, диетил, дибутил та діамілсульфати; галоїди з довгим ланцюгом, такі як децил, лаурил, міристил та стерарил хлориди, броміди та йодиди; арилалкілгалоїди, такі як бензил та фенетил броміди та ін.. Таким чином одержують розчинні чи здатні до диспергування у воді або олії продукти. Приклади кислот, які також можуть бути використані для одержання фармацевтично прийнятних кислих адитивних солей включають такі неорганічні кислоти як соляна кислота, бромистоводнева кислота, сірчана кислота та фосфорна кислота та такі органічні кислоти як щавлева кислота, малеїнова кислота, бурштинова кислота та лимонна кислота. Основні адитивні солі можна приготувати in situ під час кінцевого виділення та очищення сполук винаходу шляхом реакції групи, що містить карбонову кислоту, з придатною основою, такою як гідроксид, карбонат чи бікарбонат фармацевтично прийнятного катіону металу або з аміаком, або з органічним первинним, вторинним або третинним аміном. Фармацевтично прийнятні солі включають, але без обмеження, катіони на основі лужних металів або лужноземельних металів, таких як солі літію, натрію, калію, кальцію, магнію та алюмінію та ін. нетоксичний четвертинний аміак та катіони аміну, включаючи амоній, тетраметиламоній, тетраетиламоній, метиламоній, диметиламоній, триметиламоній, триетиламоній, диетиламоній та етил амоній серед інших. Інші репрезентативні органічні аміни, придатні для утворення основних адитивних солей включають етилендіамін, етаноламін, диетаноламін, піперидин, піперазин та ін.. Дозовані форми для місцевого застосування сполуки даного винаходу включають порошки, спреї, мазі та продукти для інгаляції. Активну сполуку змішують в стерильних умовах з фармацевтично прийнятним носієм та будь-якими необхідними консервантами, буферами або витискувачами, які можуть бути потрібні. Офтальмологічні композиції, очні мазі, порошки та розчини 88786 14 також слід розглядати як такі, що знаходяться у межах даного винаходу. Фактичні рівні доз активних інгредієнтів у фармацевтичних композиціях даного винаходу можуть варіювати для одержання кількості активної(их) сполуки(к), яка є ефективною для одержання бажаної терапевтичної відповіді для конкретного пацієнта, композицій та шляху введення. Вибраний рівень дози буде залежати від активності конкретної сполуки, шляху введення, серйозності стану, який лікують, та попередньої історії хвороби пацієнта, якого лікують. Однак, у галузі прийнято, що треба починати з доз, які є меншими, ніж ті, які потрібні для досягнення бажаного терапевтичного ефекту, і поступово збільшувати дозування доки не буде досягнуто бажаного ефекту. Коли використовують у вищевказаних або інших способах лікування, терапевтично ефективну кількість однієї зі сполук даного винаходу можна використовувати у чистій формі, або там, де такі форми існують, у формі фармацевтично прийнятної солі, естеру або проліків. Альтернативно, сполуку можна вводити як фармацевтичну композицію, що містить сполуку, яка представляє інтерес, у комбінації з одним або більше фармацевтично прийнятними ексципієнтами. Фраза "фармацевтично прийнятна кількість" сполуки винаходу означає, що достатню кількість сполуки для лікування розладів з розумним співвідношенням вигода/ризик, яке є прийнятне для будь-якого медичного лікування. Цілком зрозуміло, що загальна денна норма сполук та композицій даного винаходу буде встановлюватись лікаря-куратора у межах озвученого медичного висновку. Конкретний рівень терапевтично ефективної дози для будь-якого конкретного пацієнта залежати від різноманітних факторів, включаючи розлад, який лікують, та серйозність розладу; активності конкретної сполуки, яку застосовують; конкретної композиції, яку застосовують; віку, маси тіла, загального стану здоров'я; статі та дієти пацієнта; часу введення, шляху введення та швидкості екскреції конкретної сполуки, яку застосовують; тривалості лікування; ліків, які застосовують у комбінації або випадково з конкретною сполукою яку використовують та інших факторів, які добре відомі у медичній галузі. Наприклад, у галузі добре відомо, що треба починати з доз, які є меншими, ніж ті, які потрібні для досягнення бажаного терапевтичного ефекту, і поступово збільшувати дозування доки не буде досягнуто бажаного ефекту. Загальна денна доза сполук винаходу, які вводять людині чи нижчій тварині може знаходитись у межах від приблизно 0,0001 до приблизно 1000мг/кг/день. Для цілей орального введення кращі дози можуть знаходитись у межах від приблизно 0,001 до приблизно 5мг/кг/день. При потребі ефективну денну дозу можна розділити на мультик ратні дози для цілей введення; отже, однократні дозовані композиції можуть містити такі кількості або їх суб-мультикратні кількості, які складають денну дозу. Даний винахід також пропонує фармацевтичні композиції, які містять сполуки даного винаходу разом з одним чи більше нетоксичними фармаце 15 втично прийнятними носіями. Фармацевтичні композиції можна спеціально одержувати для орального прийому у твердій чи рідкій формі, для парентеральних ін'єкцій, чи для ректального введення. Фармацевтичні композиції даного винаходу можна вводити людям та іншим ссавцяморально, ректально, парентерально, інтравагінально, інтраперітоніально, місцево (у формі порошків, мазей чи крапель), букально або як оральний чи назальний спрей. Термін «парентерально», як його використано тут, стосується способів введення, які включають внутрішньовенні, внутрішньом'язові, інтраперітоніальні, інтрастернальні, підшкірні та інтраартикулярні ін'єкції та інфузії. В іншому аспекті даний винахід пропонує фармацевтичну композицію, яка містить компонент данного винаходу та фізіологічно прийнятний розчинник. Даний винахід включає одну або більше описаних вище сполук, сформовані у композицію разом з одним або більше нетоксичними фізіологічно прийнятними або придатними розріджувачами, носіями, ад'ювантами або наповнювачами, які разом охоплюються терміном розріджувачі, для парентеральної ін'єкції, для інтраназальної доставки, для орального введення у твердій чи рідкій формі, для ректального чи міцевого введення серед інших. Композиції також можна доставляти за допомогою катерера для місцевої доставки у місцемішень, за допомогою інтракоронарного стенту (трубкоподібного пристрою, який складається з тонкого дроту), або за допомогою полімеру, який піддається біодеградації. Сполуки також можуть утворювати комплекси з лігандами, такими як антитіла, для цільової доставки. Композиції, придатні для парентеральної ін'єкції, можуть містити фізіологічно прийнятні стерильні водні чи неводні розчини, дисперсії, суспензії чи емульсії та стерильні порошки для перетворення у придатні для ін'єкцій розчини або дисперсії. Приклади придатних водних та неводних носіїв, розріджувачів, розчинників чи наповнювачів включають воду, етанол, поліоли (пропіленгліколь, поліетиленгліколь, гліцерин та ін.), рослинні олії (такі як оливкова олія), придатні для ін'єкцій органічні естери, такі як етилолеат та їх придатні суміші. Ці композиції можуть також містити ад'юванти, такі як консервуючі, зволожувальні, емульгуючі та диспергуючі агенти. Попередження дії мікроорганізмів можна досягти за допомогою різних антибактеріальних та протигрибкових агентів, наприклад, парабенів, хлорбутанолу, фенолу, сорбінової кислоти та iн. Також може бути бажаним включити ізотонічні агенти, наприклад, цукри, хлорид натрію та ін. Пролонговане поглинання придатної для ін'єкції фармацевтичної форми можна досягти завдяки використанню агентів, які уповільнюють поглинання, наприклад, моностеарату алюмінію та желатину. Суспензії на додаток до активних сполук можуть містити суспендуючі агенти, як, наприклад, етоксильовані ізостеарильні спирти, естери поліоксиетильованого сорбіту та сорбітану, мікрокристалічну целюлозу, метагідроксид алюмінію, бентоніт, агар-агар та трагакант або суміші цих матеріалів та ін. 88786 16 У деяких випадках для пролонгації ефекту ліків бажаним є уповільнити поглинання ліків, які надійшли з підшкірною чи внутрішньом'язовою ін'єкцією. Цього можна досягти шляхом застосування рідкої суспензії кристалічного чи аморфного матеріалу з низькою розчинністю у воді. Швидкість поглинання ліків тоді буде залежати від швидкості його розчинення, яка в свою чергу буде залежати від розміру кристалів та кристалічної форми. Альтернативно, уповільнене поглинання введеної парентерально лікарської форми досягається шляхом розчинення чи суспендування ліків в олійному носії. Придатні для ін'єкцій "депо-форми" виготовляють шляхом формування мікрокапсульованих матриць ліків у здатних до біологічної деградації полімерах, таких як поліактид-полігліколід. В залежності від співвідношення ліків до полімеру та природи конкретного полімеру, який використовують, можна контролювати швидкість вивільнення ліків. Приклади таких здатних до біологічної деградації полімерів включають полі(ортоестери) та полі(ангідриди). Здатні до ін'єкцій композиції у формі "депо" також можна приготувати шляхом введення ліків у ліпосоми або мікроемульсії, які є сумісними з тканинами тіла. Здатні до ін'єкцій композиції можна стерилізувати, наприклад, шляхом фільтрування крізь фільтр, який затримує бактерії, або шляхом введення стерилізуючих агентів у формі стерильних твердих композицій, які можуть розчинятись чи диспергуватись у стерильній воді чи іншому здатному до введення шляхом ін'єкції середовищі безпосередньо перед застосуванням. Тверді дозовані форми для орального застосування включають капсули, таблетки, пігулки, порошки та гранули. В таких твердих дозованих формах активна сполука може бути змішана з принаймні одним інертним, фармацевтично прийнятним ексципієнтом чи носієм, таким як цитрат натрію або фосфорнокислий кальцій і/або а) наповнювачами чи заповнювачами, такими як крохмалі, лактоза, цукроза, глюкода, маніт та саліцилова кислота; b) зв'язувальні агенти, такі як карбоксиметилцелюлоза, альгінати, желатин, полівінілпіролідон, цукроза та гуміарабік; с) зволожувальні агенти, такі як гліцерин; d) дезінтегруючі агенти, такі як агар-агар, карбонат кальцію, крохмаль з картоплі чи тапіоки; e) агенти, які уповільнюють розчинення, такі як парафін; g) агенти, які прискорюють поглинання, такі як четвертинні амонієві сполуки; h) абсорбенти, такі як каолін та бентонітова глина та і) змащувачі, такі як тальк, стеарат кальцію, стеарат магнію, тверді поліетиленгліколі, лаурилсульфат натрію та їх суміші. У випадку капсул, таблеток та пігулок дозована форма може також включати буферні агенти. Тверді композиції схожого типу можуть також бути включеними фк наповнювачі у м'які та тверді наповнені желатинові капсул из використанням таких ексципієнтів лактоза чи молочний цукор а також поліетиленгліколів з великою молекулярною массою та ін. Тверді дозовані форми таблеток, драже, капсул, пігулок та гранул можуть бути приготовані з покриттями та оболонками, такими як 17 ентеросолюбільне покриття та інші покриття, які є добре відомими у галузі фармацевтичних композицій. Вони можуть необов'язково містити агенти для надання непрозорості і вони можуть включати композицію такого типу, яка виділяє активний інгредієнт(и) лише, або переважно у певній частині кишкового тракту, необов'язково, уповільненим способом. Приклади композицій, які можуть бути використані, охоплюють полімерні матеріали та воски. Активні сполуки можуть бути також при потребі у мікрокапсульованій формі з одним чи більше вищевказаними ексципієнтами. Рідкі дозовані форми для орального введення включають фармацевтично прийнятні емульсії, розчини, суспензії, сиропи та еліксири. На додаток до активних сполук рідкі дозовані форми можуть містити інертні розчинники, які зазвичай використовуються у галузі, такі як, наприклад, воду чи інші розчинники, агенти, що сприяють розчиненню, та емульгатори, такі як етиловий спирт, ізопропіловий спирт, етилкарбонат, етилацетат, бензиловий спирт, бензилбензоат, пропіленгліколь, 1,3бутиленгліколь, диметилформамід, олії (зокрема, олії бавовника, земляного горіха, кукурудзи, зачатків, оливи, касторову та сезамову олії), гліцерин, тетрагідрофурфуриловий спирт, поліетиленгліколі та естери жирної кислоти сорбітану та їх суміші. Окрім інертних розчинників оральні композиції можуть також включати ад'юванти, такі як зволожуючі агенти, емульгуючі та суспендуючі агенти, підсолоджувачі, ароматизатори та агенти для надання запахів. Композиції для ректального чи вагінального введення переважно являють собою суппозиторії, які можна приготувати шляхом змішування сполук винаходу з придатними неподразнюючими ексципієнтами або носіями, такими як кокосове масло, поліетиленгліколь або суппозиторний віск, які є твердими при кімнатній температурі, Але стають рідкими при температурі тіла і, таким чином, розплавляються в прямій кишці або вагінальній порожнині та виділяють активну сполуку. Сполуки данного винаходу також можна вводити у формі ліпосом. Як відомо у галузі, ліпосоми загалом походять від фосфоліпідів чи інших ліпідних субстанцій. Ліпосоми утворюються моно- чи мультиламелярними гідратованими рідкими кристаллами, які можуть бути диспергованими у водному середовищі. Може бути використаний будьякий нетоксичний, фізіологічно прийнятний та здатний до метаболізму ліпід, який може утворювати ліпосоми. Дані композиції у формі ліпосом на додаток до сполуки данного винаходу можуть містити стабілізатори, консерванти, ексципієнти та ін. Кращими ліпідами є природні та синтетичні фос 88786 18 фоліпіди та фосфатиділхоліни (лецитини), які використовують окремо чи разом. Способи одержання ліпосом добре відомі у галузі. Див., наприклад, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. Y. (1976), p.33 et seq. Термін "фармацевтично прийнятні проліки", як його використано тут, представляє такі проліки сполук даного винаходу, які у межах озвученого медичного висновку, є придатними для використання у контакті з тканинами людей та нижчих тварин без небажаних токсичності, подразнення, алергічної відповіді та ін., і відповідають прийнятному співвідношенню вигода/ризик та є ефективними для призначеного застосування, а також цвіттер-іонної форми, там, де це потрібно, сполук винаходу. Проліки даного винаходу можуть бути швидко перетворені in vivo на вихідну сполуку вказаної формули, наприклад, шляхом гідролізу у крові. Докладне обговорення наводиться в Т. Higuchi та V. Stella, Pro-drugs as Novel Delivery Systems, V.14, A.C.S. Symposium Series та в Edward В. Roche, ed. Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press (1987), які включені шляхом посилання. Сполуки даного винаходу можуть бути одержані за допомогою методик, як представлені на Схемах. Загальні вказані аналітичні умови використовувались у всіх Прикладах. Загальні аналітичні умови: Аналіз HPLC та очищення проводили з використанням бінарного градієнтного насосу Waters 2525, пристрою для управління зразками Waters 2767, детектора Waters 2487 UV (220 та 254пМ) та мас-спектрального детектора Waters Micromass ZQ з електророзпиленням. Micromass ZQ був відрегульований як на позитивну, так і на негативну іонізацію (конусна напруга =25 та 50 відповідно). Аналітичний HPLC аналіз виконували наступним чином: Колонка Waters XTerra MS С 18 50´4,6мм 3,5мкм Рухома фаза: 10мМ буфер ацетату амонію при рН5,75 та ацетонітрил Ацетонітрил: 10-75% - 3,5хв., 75-99% - 3,9хв., 99% утримування до 4,2хв., 99-10% - 4,5хв, повторне урівноважування. Препаративну HPLC виконували наступним чином: Рухома Фаза: 10 мМ буфер ацетату амонію при 5,75 рН та ацетонітрил Ацетонітрил: 10-99% - 8хв., 99% час утримування до 9хв., 99-10% - 9,5хв., повторне урівноважування. ЯМР аналіз проводили на Bruker BioSpin UltraShield NMR (300МГц). 19 88786 Схема 1a: Синтез трифторметил та моногалогенованих тієнілазепінів Схема 1b: Синтез дігалогенованих та 3-бром-2-трифторметил-тієнілазепінів Схема 2: Синтез біарилтієнілазепінів. 20 21 88786 22 Схема 3: Синтез аналогів алкіл- та бензилтієнілазепінів. Схема 4: Синтез 3-бром-3-метил-тієнілазепіну та 2,3-Дихлортієнілазепіну та споріднених аналогів. Схема 5: Синтез 3-метокситієнілазепінів. 23 88786 Схема 6: Синтез галогенованих 4-метил-тієнілазепінів та споріднених сполук * позначає, що енантіомерно чистий матеріал було ізольовано Схема 7: Синтез галогенованих 8-метил-тієнілазепінів та споріднених сполук * позначає, що енантіомерно чистий матеріал було ізольовано Схема 8: Синтез 2-сульфонамідо- та 2-амідо-тієнілазепінів 24 25 88786 26 Схема 9: Синтез 3-алкілтієнілазепінів та споріднених сполук * позначає, що енантіомерно чистий матеріал було ізольовано Схема 10: Синтез похідних трициклічного тіофеназепіну * позначає, що енантіомерно чистий матеріал було ізольовано Схема 11: Синтез фуранілазепінів Приклад 1. 2-бром-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 1а) а.) етиловий естер[етоксикарбоніл-(2-тіофен-2іл-етил)аміно]-оцтової кислоти; 27 2-тіофен-2-іл-етиламін (21г, 165ммоль) розмішували в 1 літрі DCM. Додавали етилгліоксилат (165ммоль, 50% у толуолі), а потім - 50мкл НОАс. Реакційну суміш перемішували протягом 10 хвилин, після чого повільно додавали NaBH(OAc)3 (214ммоль, 45г). Через 15 хвилин додавали НОАс (214ммоль) і реакційну суміш перемішували протягом 20 хвилин. Реакційну суміш концентрували і сировинний матеріал розчиняли в 500мл THF та води. Додавали NаНСО3 (42г, 500ммоль), після чого додавали етилхлорформіат (21мл, 214ммоль). До реакційної суміші додавали насичений NaHCO3, доки виділення газу не стало мінімальним. Після перемішування протягом ночі реакційну суміш розбавляли ЕtOАс (400мл). Продукт екстрагували 2´ЕtOАс, висушували над MgSO4 і концентрували для одержання продукту, вказаного у підзаголовку, у формі темної олії, який використовували далі без додаткового очищення. b.) [етоксикарбоніл-(2-тіофен-2-іл-етил)-аміно]оцтова кислота; Сировинний матеріал зі стадії а) (~165ммоль) розчиняли в ЕtOН (700мл) і обробляли 600мл 1М NaOH. Після перемішування протягом ночі реакційну суміщ підкислювали концентрованою НСI до рН ~1. Сировинну реакційну суміш розбавляли ЕtOАс (400мл) і промивали водою. Вода була зворотно екстрагована за допомогою EtOAc. Комбіновані органічні екстракти промивали водою (2´) і висушували над MgSO4. Після концентрування та випаровування від толуолу (2´) одержали продукт, вказаний у підзаголовку, у вигляді твердого тіла, який використовували далі без додаткового очищення. с.) етиловий естер 4-оксо-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт зі стадії b.) (~165ммоль) розчиняли в 1л DCM. Додавали DMF (100мкл), а потім повільно додавали оксалілхлорид (21,7мл, 247ммоль). Після 1 години реакційну суміш концентрували до сухого стану і сировинний матеріал знову розчиняли в DCE (1л). Обережно додавали АІСl3 (55г, 410ммоль) і реакційну суміш перемішували при кімнатній температурі протягом ½ години. Сировинну реакційну суміш різко охолоджували льодом, промивали ЕtOН (300мл), промивали водою (3´) і висушували над MgSO4. Продукт, вказаний у заголовку, очищали хроматографією на силікагелі (30% ЕtOАс в гексані) для одержання 10,5г продукту, вказаного у підзаголовку, не зовсім білого кольору. MS:ESI (позитивний): 240 (М+Н). d.) етиловий естер 4,5,7,8-тетрагідро-тієно[2,3d]азепін-6-карбонової кислоти; АІСl3 (3,95г, 29,7ммоль) додавали до 50мл DCM при 0°С. Додавали комплекс боран-tбутиламіну (5,2г, 59,5ммоль), після чого - продукт реакції стадії с) (2,37г, 9,9ммоль), розчинений в DCM (50мл). Реакційну суміш перемішували протягом 2 годин при кімнатній температурі, після чого додавали інші 3,95г АІСl3 (29,7ммоль). Після перемішування протягом 10 хвилин реакційну суміш обережно гасили 0,1Μ НСI (~50мл). Після концентрації органічного розчинника сировинну реакційну суміш розділяли між 1М НСI та EtOAc (70 кожний). Водний шар піддавали зворотній екстра 88786 28 кції 1´ЕtOАс. Комбіновані органічні шари висушували над MgSO4 і концентрували. Після очищення шляхом хроматографії на силікагелі (ЕtOАс/градієнт-гексан) (одержали продукт, вказаний у підзаголовку (1,45г). 1Н ЯМР (300МГц, CDCI3) 6,96 (d, J=5Гц, 1H), 6,76 (d, J=5Гц, 1Н), 4,18 (d, J=1Гц, 2H), 3,52-3,78 (m, 4H), 2,78-3,08 (m, 4H), 1,28 (t, J=7Гц, 3Н). MS:ESI (позитивний): 226 (Μ+Η). е.) 5,6,7,8-тетрагідро-4Н-тіено[2,3,-d]азепін; Продукт стадії d) (220мг, 0,89ммоль) розчиняли в 15мл СНСІ3 та піддавали обробці TMSI (4,5ммоль, 600мкл). Після нагрівання протягом ночі при 70°С реакційну суміш обережно гасили МеОН (10мл) та 1Μ NaOH (20мл). Продукт, вказаний у підзаголовку, екстрагували в DCM (3´20мл). Екстракти висушували над MgSO4 і концентрували для одержання 178мг продукту, вказаного у підзаголовку. 1H ЯМР (300МГц, DMSO) 7,20 (d, J=5Гц, 1Н), 6,85 (d, J=5Гц, 1H), 3,42-3,61 (m, 4H), 2,71-3,03 (m, 4H). MS:ESI (позитивний): 154 (Μ+Η). f.) трет-бутиловий естер 4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії e) (296мг, 1,93ммоль), розчинений в 50:50 суміші ацетону/води (8мл), піддавали обробці NaHCO3 (340,5мг, 4,03ммоль) при 0°С і перемішували протягом 30 хвилин. До одержаного розчину додавали ді-трет-бутил дикарбонат (463мг, 2,12ммоль). Реакційну суміш перемішували при кімнатній температурі протягом 14 годин. Реакційну суміш вливали у воду (50мл) та екстрагували EtOAc (3´50мл). Комбіновані органічні фази промивали розсолом (75мл), висушували (MgSO4) і концентрували у вакуумі для одержання сировинного продукту у вигляді олії. Очищення шляхом хроматографії на силікагелі (ЕtOАс/градієнтгексан) дало в результаті вказаний у підзаголовку продукт у вигляді чистої олії; вихід (93%). 1H ЯМР (300МГц, CDCI3) δ 6,96 (d, J=5Гц, 1H), 6,76 (d, J=5Гц, 1H), 3,44-3,68 (m, 4H), 2,76-3,06 (m, 4H), 1,50 (s, 9H). MS:ESI (позитивний): 254 (Μ+Η). g.) трет-бутиловий естер 2-бром-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії f) (10мг, 0,039ммоль), розчинений в 50:50 суміші хлороформу/оцтової кислоти (1мл), піддавали обробці N-бромсукциімідом (7мг, 0,041ммоль). Реакційну суміш перемішували протягом 30 хвилин при кімнатній температурі. Реакційну суміш вливали у воду (5мл) та екстрагували СНСІ3 (3´5мл). Комбіновані органічні фази промивали 10% розчином КОН (5мл), розсолом (5мл), висушували (MgSO4) і концентрували для одержання сировинного продукту у вигляді олії. Очищення методом HPLC дало в результаті продукт, вказаний у підзаголовку, у формі олії. MS:ESI (позитивний): 332, 334 (М+Н). h.) 2-бром-5,6,7,8-тетрагідро-4Н-тієно [2,3-d] азепін; Продукт стадії g) (0,39ммоль), розчинений в етері (1мл), піддавали обробці 4Μ НСІ/діоксаном (1мл). Реакційну суміш перемішували протягом 19 годин при кімнатній температурі. Одержаний осад фільтрували і промивали безводним діетиловим етером для одержання продукту, вказаного у заголовку, як його НСI солі. 1H ЯМР (300МГц, CDCI3) δ 29 9,40 (s, 2H), 7,05 (s, 1H), 3,14-3,33 (m, 4H), 2,943,23 (m, 2H), 2,76-3,06 (m, 2H). MS:ESI (позитивний): 232, 234 (Μ+Η). Приклад 2 2-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін (Схема 1а) а.) трет-бутиловий естер 2-хлор-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 1, стадія f) (10мг, 0,039ммоль, Приклад 1) в СНСІ3 (1мл) та НОАс (1мл) піддавали обробці N-хлорсукцинімідом (6мг, 0,041ммоль). Реакційну суміш перемішували протягом 12 годин при кімнатній температурі. Реакційну суміш вливали у воду (5мл) та екстрагували СНСl3 (3´5мл). Комбіновані органічні фази промивали 10% розчином КОН (5мл), розсолом (5мл), висушували (MgSO4) і концентрували для одержання сировинного продукту у вигляді олії. Очищення методом HPLC дало в результаті продукт, вказаний у підзаголовку, у формі олії. MS:ESI (позитивний): 288 (М+Н). b.) 2-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; Сполуку, вказану у заголовку, одержали за способом Прикладу 1, стадія h), використовуючи продукт зі стадії а). 1H ЯМР (300МГц, DMSO) δ 6,72 (s, 2H), 2,78-2,85 (m, 2H), 2,72-2,79 (m, 4Н), 2,51-2,70 (m, 2Н). MS:ESI (позитивний): 188 (Μ+Η). Приклад 3 2,3-дибром-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 1b) а.) етиловий естер 23-дибром-4,5,7,8тетрагідро-4Н-тієно[2,3-d]азелін-6-карбоної кислоти; Продукт Прикладу 1, стадія d) (95мг, 0,42ммоль) розчиняли в СНСl3 (1мл) та НОАс (1мл) та піддавали обробці NBS (66,7мг, 0,42ммоль). Реакційну суміш перемішували протягом 20 хвилин при кімнатній температурі. До цієї суміші додавали ацетат натрію (138мг, 1,68ммоль) та додатково NBS (133,4мг, 0,84ммоль). Реакційну суміш перемішували при 60°С, доки реакцій не закінчилась, як було визначено LC/MS. Реакційну суміш охолоджували до кімнатної температури, розбавляли насиченим бікарбонатом натрію (2мл) і екстрагували СНСІ3 (3´2мл). Комбіновані органічні фази промивали розсолом (10мл), висушували (MgSO4) і концентрували у вакуумі для одержання сировинного продукту у вигляді олії. В результаті очищення флеш-хроматографією одержали продукт, вказаний у підзаголовку, у формі олії. MS:ESI (позитивний): 384 (М+Н). b.) 2,3-дибром-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; Продукт стадії b) (0,42ммоль) розчиняли в DCM (2мл) та піддавали обробці йодтриметилсиланом (0,46ммоль). Реакційну суміш перемішували при кип'ятінні зі зворотним холодильником протягом 24 годин). Реакційну суміш вливали в насичений бікарбонат натрію (10мл) та екстрагу 88786 30 вали дихлорметаном (3´5мл). Комбіновані органічні фази промивали розсолом (10мл), висушували (MgSO4) і концентрували у вакуумі для одержання сировинного продукту у вигляді олії. В результаті очищення препаративною HPLC одержали продукт, вказаний у заголовку. 1H ЯМР (300МГц, CDCI3) δ 2,94-3,05 (m, 4H), 2,85-2,92 (m, 4H), 1,92 (s, 1H). MS:ESI (позитивний): 312 (Μ+Η). Приклад 4 2,3-дихлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 4) а.) етиловий естер (4,5-дихлор-тіофен-2-іл)оксо-оцтової кислоти; При 5-10°С етиловий естер хлор-оксо-оцтової кислоти (5,43мл, 48,7ммоль) додавали до 2,3дихлортіофену (5г, 32,6ммоль). Розчин АІСІ3 (6,49г, 48,7ммоль), розчинений у дихлорметані (13мл), додавали по краплинах, таким чином, щоб внутрішня температура реакції не піднімалась вище 10°С. Через 1 годину реакційну суміш вливали в льодяну воду і екстрагували СН2СІ2 (2´100мл). Органічний шар промивали 10% NaHCO3 (2´50мл), водою (1´50мл) та розсолом (1´50мл). В результаті висушування (Na2SO4) та концентрування одержали світло-оранжеве тверде тіло, яке після очищення хроматографією на силікагелі' (EtOAc/градієнт-гексан) дало в результаті 6,8г (82%) продукту, вказаного у підзаголовку. b.) етиловий естер (4,5-дихлор-тіофен-2-іл)гідрокси-оцтової кислоти; Розчин продукту зі стадії а) (23,0г, 90,9ммоль) в THF (500мл) піддавали обробці NaBH(OAc)3 (23,1г, 109ммоль) та АсОН (250мкл) при 60°С протягом 1 години. Реакційну суміш гасили АсОН (8мл) та концентрували до ~250мл. Вміст розбавляли Н2О (400мл) та екстрагували СН2СІ2 (1´400мл; 1´400мл). Органічний шар висушували (MgSO4) і концентрували, що дало в результаті 23г сполуки, вказаної у підзаголовку. 1H ЯМР (300МГц, CDCI3) δ 6,91 (s, 2H); 5,25 (dd, J1=6Гц, J2=1Гц, Η); 4,22-4,40 (m, 2H); 3,52-3,60 (br, m, 1Η); 1,33 (t, J=7Гц, 3Н). с.) етиловий естер (4,5-дихлор-3метоксикарбонілметил-тіофен-2-іл)-оцтової кислоти; Розчин продукту зі стадії b) (12,2г, 48,0ммоль) у декаліні (145мл) піддавали обробці триметилортоацетатом (34,5мл, 192ммоль) та гексанової кислоти (0,61мл). Колбу обладнали колонкою Вігре і нагрівали до 180°С. Періодично протягом 6 годин додавали додаткову кількість гексанової кислоти і реакційну суміш нагрівали протягом ночі. Реакційну суміш концентрували на роторному випаровувачі, а залишок екстрагували за допомогою МеОН (100мл´2). Екстракти МеОН концентрували і очищали за допомогою хроматографії на силікагелі (EtOAc/градієнт-гексан), що дало в результаті 4,36г (29%) продукту, вказаного у підзаголовку. MS:ESI (позитивний): 311, 313 (М+Н). d.) (3-карбоксиметил-4,5-дихлор-тіофен-2-іл)оцтова кислота; 31 Розчин продукту зі стадії с) (1,14г, 3,66ммоль) в МеОН (7мл) при 0°С піддавали обробці по краплинах 2М NaOH (3,8мл). Реакційну суміш нагрівали до 22°С і перемішували протягом ночі. Розчинник випаровували і залишок розчиняли в 2Μ NaOH (50мл) і екстрагували етером (2м´50мл). Основний шар охолоджували до 0°С та підкислювали до рН1 за допомогою 6Μ НСI. Кислий шар піддавали зворотній екстракції ЕtOАс (4´100мл), а органічний шар висушували (MgSO4) і концентрували. Сировинний твердий продукт розтирали у порошок з гексаном та фільтрували, що дало в результаті 2,75г (73%) продукту, вказаного у підзаголовку. MS:ESI (позитивний): 267, 269 (М+Н). е.) 2-[4,5-дихлор-3-(2-гідрокси-етил)-тіофен-2іл]-етанол; Розчин продукту зі стадії d) (2,5г, 9,33ммоль) в THF (85мл) охолоджували до 0°С і по краплинах протягом 10 хвилин додавали 1М розчин BH3-THF (46,6мл, 46,6ммоль) та перемішували протягом наступних 20 хвилин після закінчення додавання. Реакційну суміш нагрівали до 22°С та перемішували протягом 2 годин. Реакційну суміш вливали в льодяний, холодний насичений NaHCO3 (150мл) та екстрагували за допомогою EtOAc. Сировинний матеріал пропускали крізь набивку з силікагелю та промивали EtOAc. Концентрація елюента забезпечила одержання 1,99г (88%) сполуки, вказаної у підзаголовку. f.) етиловий естер 2-[4,5-дихлор-2-(2метансульфонілокси-етил)-тіофен-3-іл]метансульфокислоти; Розчин продукту за стадії e) (1,99г, 8,25ммоль) в СН2СІ2 (41мл) охолоджували до 0°С та піддавали обробці триетиламіном (3,4мл, 24,7ммоль), після чого по краплинах протягом 10 хвилин додавали метансульфонілхлорид (1,4мл, 18,1ммоль). Через 45 хвилин сировинну реакційну суміш розбавили СН2Сl2 (100мл) та промили льодяною водою (25мл), 10% лимонною кислотою (2´25мл), насиченим NaHCO3 (2´25мл) та розсолом (1´25мл). Органічний шар висушували (MgSO4), концентрували до 29мл і розбавляли безводним діоксаном (76мл). Суміш концентрували для вилучення залишкового СН2СІ2 і розчин діоксану, що залишився, перенесли у наступну реакцію. g.) 6-бeнзил-2,3-диxлop-5,6,7,8-тeтpaгiдpo-4Hтiєнo[2,3-d]aзeпiн; Розчин бісмезилату діоксану, одержаний на стадії f), перевели у реакційну колбу з 3-ма шийками, обладнану зливною воронкою та конденсатором. Додавали безводний карбонат калію (4,93г, 35,7ммоль) та вміст нагрівали зі зворотним холодильником. Потім по краплинах на протязі 45 хвилин додавали розчин бензиламіну (2,71г, 25,3ммоль) у безводному діоксані (27мл) та продовжували нагрівання протягом 16 годин. Солі відфітровували і розчинник концентрували. Сировинний продукт піддавали очищенню хроматографією на силікагелі (ЕtOАс/градієнт-гексан), що дало в результаті 1,43г (62%) продукту, вказаного у підзаголовку. 1H ЯМР (300МГц, CDCl3) δ 7,207,40 (m, 5H); 3,73 (s, 2H); 2,68-2,89 (m, 8H); MS:ESI (позитивний): 312, 314 (Μ+Η). 88786 32 h.) гідрохлорид 2,3-дихлор-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепіну; Розчин продукту зі стадії g) (727г, 2,33ммоль) в безводному діхлоретані охолоджували до 0°С, піддавали обробці 1-хлоретилхлорформіатом (1,27мл, 11,65ммоль) і реакційну суміш нагрівали до 22°С протягом 1 години. Реакційну суміш розбавляли СH2Сl2 (50мл) і промивали насиченим NаНСО3 (25мл). Насичений NаНСО3 піддавали зворотній ексктрації з СН2Сl2 і комбіновані органічні шари промивали розсолом (25мл), висушували (MgSO4) і концентрували, в результаті чого одержали маслоподібний залишок, який перевели у безводний МеОН (75мл) і нагрівали зі зворотним холодильником протягом 1 години. МеОН випаровували і сировинний залишок розтирали в порошок і фільтрували, в результаті чого одержали 323г (54%) сполуки, вказаної у підзаголовку. 1H ЯМР (300МГц, DMSO) δ 9,60 (br s, 2H), 3,14-3,28 (m, 6H); 2,97-3,50 (m, 6H); 2,97-3,50 (m, 2H) MS:ESI (позитивний): 222, 224 (Μ+Η). Приклад 5 2-бром-3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 1b) а.) трет-бутиловий естер 2-бром-3-хлор4,5Л,8-тетрагідро-4Н-тієно[2,3-d]азепін-6-карбоної кислоти; Продукт Прикладу 1, стадії f) (54мг, 0,21ммоль) розчиняли в СНСl3 (1мл) та НОАс (1мл) та піддавали обробці гідрохіноном (2мг, 0,02ммоль) та N-бромсукциімідом (38мг, 0,21ммоль). Після перемішування при кімнатній температурі протягом 20 хвилин додавали Nхлорсукциімід (28мг, 0,21моля) та продовжували перемішування при кімнатній температурі протягом 48 годин. Реакційну суміш вливали в насичений бікарбонат натрію (10мл) та екстрагували хлороформом (3´5мл). Комбіновані органічні фази промивали розсолом (10мл), висушували (MgSO4) і концентрували, в результаті чого одержали сировинну олію, яку використовували без подальшого очищення. b.) гідрохлорид 2-бром-3-хлор-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепіну; Вказану в заголовку сполуку одержали за способом Прикладу 1, стадія h), використовуючи продукт стадії а). 1H ЯМР (300МГц, DMSO) δ 9,12 (bs, 2H); 3,22-3,31 (m, 4H); 3,13-3,20 (m, 2H); 2,98-3,05 (m, 2H) MS:ESI (позитивний): 266, 268 (Μ+Η). Приклад 6 2-(4-трифторметокси-феніл)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін (Схема 2) a) трет-бутиловий естер 2-(4-трифторметоксифеніл)-4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт Прикладу 1, стадія g) (20мг, 0,06ммоль) розчиняли в DMF (5мл) та піддавали обробці ацетатом паладію (1мг, 0,004ммоль), трифенілфосфіном (4,7мг, 0,018ммоль) та 1Μ кар 33 бонатом натрію (Nа2СО3; 0,45мл). Суміш перемішували при кімнатній температурі протягом 5 хвилин і потім піддавали обробці 4-трифторметокси фенілборною кислотою (28,4мг, 0,138ммоль). Після нагрівання протягом 5 годин при 85°С додавали додаткову 4-трифторметокси фенілборну кислоту (6,2мг, 0,03ммоль) і перемішування продовжували ще 12 годин. Реакційну суміш охолоджували до кімнатної температури, пропускали крізь набивку з сухого целіту та фільтрували крізь набивку з кремнезему з елююванням DCM (5мл) та EtOAc (5мл). Фільтрат випаровували для одержання продукту, вказаного у підзаголовку, у вигляді сировинної олії, яку використовували без подальшого очищення. b.) 2-(4-трифторметокси-феніл)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; Продукт, вказаний у заголовку, одержували за способом Прикладу 1, стадія h), використовуючи продукт стадії а). Реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання продукту, вказаного у заголовку. 1H ЯМР (300МГц, DMSO) δ 7,76 (d, J=8Гц, 2H); 7,48 (d, J=8Гц, 2Н); 7,36 (s, 1H); 2,77-3,06 (m, 8H), 2,0 (s, 1H) MS:ESI (позитивний): 314 (Μ+Η). Приклад 7 2-(4-трифторметил-феніл)-5,6,7.8-тетpагідро4Н-тієно[2,3-d]азепін (Схема 2) а) трет-бутиловий естер 2-(4-трифторметилфеніл)-4,5,7.8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи 2трифторметил-фенілборну кислоту. b) 2-(4-трифторметил-феніл)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а). Реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 298 (М+Н). Приклад 8 2-(4-фтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 2) а.) трет-бутиловий естер 2-(4-фтор-феніл)4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи 4фтор-феніл борну кислоту. b). 2-(4-фтор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а). Реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 248 (М+Н). Приклад 9 88786 34 2-(3-хлор-4-фтор-феніл)-5,6,7,8-тетрагідро-4Нтіено[2,3-d]азепін (Схема 2) а.) трет-бутиловий естер 2-(3-хлор-4-фторфеніл)-4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи 3хлор-4-фтор-фенілборну кислоту. b). 2-(3-хлор-4-фтор-феніл)-4,5,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а). Реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 282 (М+Н). Приклад 10 2-(2,5-дихлор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 2) а.) трет-бутиловий естер 2-(2,5-дихлор-феніл)4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи 2,4-дихлорфеніл борну кислоту. b.) 2-(2,5-дихлор-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а). Реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 298 (М+Н). Приклад 11 2-(4-етил-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-діазепін (Схема 2) а.) трет-бутиловий естер 2-(4-етил-феніл)4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи 4етилфеніл борну кислоту. b.) 2-(4-етил-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а), за винятком того, що реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS-ESI (позитивний): 258 (М+Н). Приклад 12 2-(2-метокси-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 2) 35 а.) трет-бутиловий естер 2-(3-метокси-фетл)4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а.), використовуючи 3-метоксифеніл борну кислоту. b.) 2-(3-метокси-феніл)-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а), за винятком того, що реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 260 (М+Н). Приклад 13 2-феніл-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 2) а.) трет-бутиловий естер 2-феніл-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом прикладу 6, стадія а), використовуючи фенілборну кислоту. b.) 2-феніл-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; Продукт, вказаний у підзаголовку, одержали за способом прикладу 1, стадія h), використовуючи продукт зі стадії а), за винятком того, що реакційну суміш концентрували у вакуумі та очищали препаративною HPLC для одержання сполуки, вказаної у заголовку. MS.ESI (позитивний): 230 (М+Н). Приклад 14 2-(2-хлор-6-фтор-бензил)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін (Схема 3) а.) етиловий естер 2-бром-4,5,7,8-тетрагідро4Н-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 1, стадія d) (300мг, 1,33ммоль) розчиняли у хлороформі (3мл) та оцтовій кислоті (3мл) і піддавали обробці Nбромсукцинімідом (248мг, 1,40ммоль) та перемішували при кімнатній температурі протягом 30 хвилин. Реакційну суміш обережно гасили розбавленням насиченим бікарбонатом натрію (20мл) та екстрагували хлороформом (3´20мл). Комбіновані органічні фази промивали розсолом (50мл), висушували (MgSO4) і випаровували у вакуумі для одержання сировинної олії. В результаті очищення препаративною TLC (гексан/етилацетат) одержали продукт, вказаний у підзаголовку у вигляді олії. MS:ESI (негативний): 302 (М-Н). b.) етиловий естер 2-(2-хлор-6-фтор-бензил)4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт стадії а) (144мг, 0,48ммоль) в 2мл сухого діетилового етеру піддавали обробці NiCl2 88786 36 (dppp) (3,5ммоль), після чого по протягом 30 хвилин при кімнатній температурі, краплинах додавали 2-хлор-6-фторбензил магнійбромід (0,25Μ розчин, 1,2ммоль, 4,8мл). Реакційну суміш нагрівали до температури кипіння зі зворотним холодильником протягом 12 годин. Реакційну суміш охолоджували до кімнатної температури, гасили 1Μ НСI (10мл) та екстрагували діетиловим ефіром (3´10мл). Комбіновані екстракти етеру промивали водою (30мл), висушували (MgSO4) та випаровували для одержання сировинної олії. В результаті очищенні за допомогою HPLC одержали сполуку, вказану у підзаголовку. MS, розраховане для C18H19CIFNO2S+H 369, показала значення 369. с.) 2-(2-хлор-6-фтор-бензил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; Сполуку, вказану у заголовку, одержали за способом Прикладу 3, стадія b), використовуючи продукт стадії b). 1H ЯМP (300МГц, C6D6) δ 6,94 (d, J=8Гц, 1H); 6,52-6,70 (m, 2H); 6,49 (s, 1H); 2,47-2,78 (m, 8H), 2,03 (s, 1H) MS:ESI (позитивний): 296 (Μ+Η). Приклад 15 3-бром-2-(2-хлор-6-фтор-бензил)-5,6,7,8тетрагїдро-4Н-тієно[2,3-d]азепін (Схема 3) а.) етиловий естер 3-бром-2-(2-хлор-6-фторбензил)-4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін-6карбонової кислоти; Продукт Прикладу 14, стадія b) (49мг, 0,13ммоль) розчиняли в суміші 1:1 СН3СІ/НОАс (1мл) та піддавали обробці ацетатом натрію (43мг, 0,52ммоль) та N-бросукциніміду (27мг, 0,15моля). Реакційну суміш нагрівали при 60°С та перемішували протягом 30 хвилин. Реакційну суміш охолоджували до кімнатної температури, розбавляли водою (5мл) та екстрагували хлороформом (3´5мл). Комбіновані органічні екстракти промивали 10% КОН, висушували (MgSO4) та випаровували у вакуумі для одержання продукту, вказаного у підзаголовку, у вигляді олії, яку використовували без подальшого очищення. b.) 3-бром-2-(2-хлор-6-фтор-бензил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін; Сполуку, вказану у заголовку, одержали за способом Прикладу 3, стадія b), використовуючи продукт стадії b). 1H ЯМР (300МГц, C6D6) δ 6,336,78 (m, 3Н), 4,09 (s, 2H), 2,18-3,20 (m, 8H), 1,62 (s, 1H). MS, розраховане для C15H14BrCIFNS+H 374, показала значення 374. Приклад 16 2-бром-3-метил-5,6,7,8-тетрагвдро-4Нтієно[2,3-d]азепін (Схема 4) а.) етиловий естер (5-бром-4-метил-тіофен-2іл)-гідрокси-оцтової кислоти; 2-бром-3-метил тіофен (13,2мл, 113ммоль) перемішували в 1 літрі DCM при 0°С. Додавали етил хлор оксалат (13,9мл, 124ммоль) , після чого 37 додавали АlСl3 (16,5г, 124ммоль). Після перемішування протягом 10 хвилин реакційну суміш обережно виливали над льодом (~500мл) та EtOH (~300мл). Після нагрівання до кімнатної температури, продукт екстрагували в DCM (2´) та висушували над MgSO4. В результаті концентрування одержали 37,5г жовтогарячого твердого тіла, яке розчинили в THF (1 літр) та піддавали обробці NaBH(OAc)3 (36г, 170ммоль). Після нагрівання до 60°С протягом 1 години реакційну суміш охолоджували та гасили НОАс (13,6мл, 226ммоль). Реакційну суміш концентрували та осад розділяли між DCM/EtOH (5:1) та водою. Водний шар екстрагували 1´DCM та комбіновані органічні екстракти висушували над MgSO4 та концентрували для одержання продукту, вказаного у підзаголовку (35г), який використовували без подальшого очищення. b.) етиловий естер (5-бром-3етоксикарбонілметил-4-метил-тіофен-2-іл)-оцтової кислоти; Продукт стадії а) (~113ммоль) піддавали обробці 300мл декаліну, 103мл триетилортоацетату (565ммоль) та гексановою кислотою (6,2мл, 50ммоль). Реакційну суміш нагрівали до 180°С протягом 10 хвилин, після чого додавали додаткові 50ммоль гексанової кислоти. Після нагрівання протягом 10 хвилин додавали додаткову гексанові кислоту (50ммоль) і реакційну суміш знову нагрівали протягом 10 хвилин. Реакційну суміш охолоджували та концентрували у вакуумі з нагріванням для одержання сполуки, вказаної у підзаголовку, у вигляді олії, яку використовували без подальшого очищення. с.) 2-[5-бром-3-(2-гідрокси-етил)-4-метилтіофен-2-іл]-етанол; Продукт стадії b) (113ммоль) розчиняли в ЕtOН (1 літр) та охолоджували до 0°С, а потім піддавали обробці 170мл 2М NaOH. Після перемішування протягом 24 годин одержаний осад піддавали фільтруванню та промивали етанолом для одержання 15г динатрієвої солі. Частину цього матеріалу (3г, 8,9ммоль) розчиняли в 90мл THF та піддавали обробці 4М НСI в діоксані (4мл, 16ммоль). Після інтенсивного перемішування протягом ½ години додавали ВН3-THF (44,4мл, 1М) і розчин перемішували при кімнатній температурі протягом 2 годин. Реакційну суміш гасили та обережно, насичували NaHCO3 та концентрували. Сировинний осад розділяли між ЕtOАс та водою (150мл кожний). Органічний шар висушували над MgSO4 та концентрували для одержання 1,94г сполуки, вказаної у підзаголовку. d.) 6-бензил-6-бром-3-метил-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін: Продукт стадії с) (1,94г, 7,3ммоль) розчиняли в 75мл DCM, охолоджували до 0°С та піддавали обробці Еt3N (29,3ммоль, 4,1мл), після чого додавали MsCl (22ммоль, 1,71мл). Після 1 години додавали додаткову кількість Et3N та MsCl (відповідно 2екв. та 3екв.) і реакційну суміш перемішували ще одну годину. Реакційну суміш виливали над 5% лимонною кислотою, екстрагували в DCM та промивали насиченим NaHCO3. Після висушування органічного розчину над MgSO4 та концентрування 88786 38 сировинний продукт розчиняли в діоксані (200мл) та піддавали обробці K2СО3 (36,6ммоль, 5,0г). Після нагрівання зі зворотним холодильником додавали BnNt2 (22ммоль, 2,4мл) і реакційну суміш нагрівали зі зворотним холодильником протягом ночі. Реакційну суміш охолоджували, фільтрували і концентрували. Сполуку, вказану у підзалоловку, очищали хроматографією на силікагелі (10% ЕtOАс в гексані) для одержання 419мг сполуки, вказаної у підзаголовку. e.) 2-бром-3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії d) (80мг, 0,24ммоль) розчиняли в 2мл DCE. Додавали 2-хлоретил хлор форміат (103мкл, 0,95ммоль) і реакційну суміш перемішували при кімнатній температурі протягом 15 хвилин. Реакційну суміш гасили 3мл МеОН і сировинну реакційну суміш швидко нагрівали до кипіння зі зворотним холодильником і потім концентрували до сухого стану. Сировинний залишок розчиняли в ½мл МеОН та розтирали в порошок з етером для одержання 47мг сполуки, вказаної у заголовку у вигляді білого твердого тіла. 1H ЯМР (300МГц, CD3OD) δ 3,36-3,30 (m, 1H), 3,12 (t, J=5,2Гц, 2H), 3,00 (t, J=5,1Гц, 2H), 2,11 (s, 3H); MS:ESI (позитивний): 246, 248 (Μ+Η). Приклад 17 2-метокси-2-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) а.) діетиловий естер 5-оксо-азепан-1,4дикарбоної кислоти; Етиловий естер 4-оксо-піперидин-1-карбонової кислоти (20г, 117ммоль) розчиняли в 120мл Et2O та охолоджували до -30°С. Одночасно протягом 30 хвилин додавали BF3-OEt2 (14,8мл) та етилдіазоацетат (16мл, 152ммоль) (кожен в 15мл Et2O), підтримуючи внутрішню температуру на рівні приблизно -20°С. Реакційну суміш охолоджували до кімнатної температури і перемішували протягом 3 годин, після чого реакційну суміш обережно гасили 30% K2СО3 (60мл). Органічний шар висушували над K2СО3 та концентрували для одержання 30,4г сполуки, вказаної у підзаголовку. b.) діетиловий естер 3-гідрокси-4,5,7,8тетрагідро-4Н-тієно[2,3-d]азепін-2,6-дикарбонової кислоти; Продукт стадії а) (20г, 77,8ммоль) розчиняли в 300мл EtOH. Розчин охолоджували до 0°С та в розчин протягом 10 хвилин пропускали бульбашки газу НСI. Додавали етилтіогліколат (7,8мл, 77,8 моля) і знову протягом 3 хвилин в розчин пропускали бульбашки газу НСI. Після перемішування протягом 4 днів при кімнатній температурі реакційну суміш концентрували, нейтралізували насиченим NаНСО3 та екстрагували в етер (200мл). Після висушування екстрактів над MgSO4 та концентрування залишок розчиняли в EtOH (100мл) та піддавали обробці NaOEt (100мл 21% р-ну NaOEt в EtOH). Після перемішування протягом ночі реакційну суміш розбавляли 500мл води і промивали 300мл DCM. DCM екстрагували 150мл 39 води і потім кілька разів екстрагували 5% КОН (~10´75мл). Комбіновані водні екстракти підкислювали концентрованою НСI та екстрагували в DCM (4´200мл). Екстракти DCM висушували над MgSO4 та концентрували для одержання 7,5г сполуки, вказаної у підзаголовку у вигляді олії. с.) діетиловий естер 3-метокси-4,5,7,8тетрагідро-4Н-тієно[2,3-d]азепін-2,6-дикарбонової кислоти; Продукт стадії b) (2,3г, 7,3ммоль) розчиняли в 60мл 1:1 МеОН:THF. Додавали діізопропілетиламін (1,9мл, 10,9ммоль), після чого додавали TMSCHN2 (10,9мл 2b). Реакційну суміш перемішували протягом ночі при кімнатній температурі, а потім обережно гасили 0,4мл НОАс. Після перемішування протягом ½ години реакційну суміш розділяли між DCM та 1М HCL (100мл кожен). Органічний шар висушували над MgSO4 та концентрували для одержання 2,75г сполуки, вказаної у підзаголовку. d.) діетиловий естер 2-гідроксиметил-3метокси-4,5,7,8-тетрагідро-4Н-тієно[2,3-d]азепін2,6-дикарбонової кислоти; Продукт стадії с) (2,5г, 7,8ммоль) розчиняли в 175мл сухого THF та піддавали обробці LiCL (0,65г, 15,3ммоль), після чого піддавали обробці LiBH4 (15,3мл, 2М). Після перемішування протягом 1 години реакційну суміш обережно гасили EtOH та НОАс до припинення виділення газу. Сировинну реакційну суміш розділяли між водою та DCM.Органічний екстракт висушували над MgSO4 та концентрували для одержання 2,3г чистої олії, яку очищали хроматографією на кремнеземі (30% EtOAc/Hex) для одержання 1,03г сполуки, вказаної у підзаголовку. e.) 2-метокси-2-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії d) (70мг, 0,25 моль) розчиняли в 10мл ЕtОАс та піддавали обробці 35мг 10% Pd/C (вологий, компанії Degusssa, тип Е101) та швидко розтирали в атмосфері Н2 протягом 3 годин. Реакційну суміш фільтрували та концентрували. Сировинний осад розчиняли в 1мл EtOH та піддавали обробці 1мл 40% КОН. Після нагрівання до 80°С протягом ночі реакційну суміш розбавляли водою та продукт екстрагували 2х в DCM. Продукт, вказаний у заголовку, одержували шляхом очищення препаративною HPLC-MS. 1H ЯМР (300МГц, CDCI3) δ 3,69 (s, 3Н), 3,21-3,14 (m, 4H), 3,00 (t, J=5,2Гц, 2Н), 2,88 (t, J=5,1Гц, 3Н), 2,29 (s, 3Н); MS:ESI (позитивний): 198 (Μ+Η). Приклад 18 2-бром-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) а.) 6-етиловий естер 3-метокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-2,6-дикарбонової кислоти; Продукт з Прикладу 17, стадія с) (1,0г, 3,19ммоль) перемішували в етанолі (20мл) з 1М NaOH (6,38мл, 6,38ммоль) при 80°С протягом ночі. Реакційну суміш охолоджували до кімнатної тем 88786 40 ператури, підкислювали 10% розчином НСI та екстрагували 5% EtOH в DCM (х2). Органічні екстракти комбінували та промивали водою. Органічний шар висушували над Na2SO4, фільтрували та концентрували для одержання 912мг сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. MS:ESI (позитивний): 300 (М+Н). b.) етиловий естер 2-третбутоксикарбоніламіно-3-метокси-4,5,7,8тетрагідро-тієно[2,3-d]aзепін-6-карбонової кислоти; Розчин продукту зі стадії а) (790мг, 3,01ммоль) перемішували з дифенілфосфорилазидом (647мкл, 3,01ммоль) та третидаміном (418мкл, 3,01ммоль) у трет-бутанолі (20мл) при 80°С протягом ночі. Реакційну суміш охолоджували до кімнатної температури та вливали в насичений водний розчин NaHСО3. Одержану суміш екстрагували етилацетатом. Органічний екстракт висушували над MgSO4 та очищали хроматографією на силікагелі (EtOAc/градієнт-гексан) для одержання 852мг сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 370 (М+Н). с.) етиловий естер 3-метокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт зі стадії b) (256мг, 0,69ммоль) перемішували в TFA (2мл) протягом 30 хвилин. Реакційну суміш концентрували у вакуумі та розчиняли в EtOH (5мл). До розчину додавали ізоамілнітрит (140мкл, 1,04ммоль) та Сu(ОАс)2 (188мг, 1,04ммоль) і реакційну суміш перемішували при кімнатній температурі протягом ночі. Реакційну суміш фільтрували крізь набивку із силікагелю і очищали хроматографією на силікагелі (EtOAc/градієнт-гексан) для одержання 17мг сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 271 (М+Н). d.) етиловий естер 2-бром-3-метокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії с) (17мг, 0,067ммоль) розчиняли в 1мл в суміші 1:1 оцтової кислоти: СНСІ3 та піддавали обробці N-бромсукциімідом (12мг, 0,068ммоль). Після 30 хвилин реакційний розчин по краплинах додавали до розчину насиченого бікарбонату натрію та екстрагували етилацетатом. Органічні екстракти концентрували для одержання сполуки, вказаної у підзаголовку,яку використовували без подальшого очищення. e.) 2-бром-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; 40% розчин КОН у воді (1мл) додавали до продукту зі стадії d) (~0,067ммоль) в 1мл етанолу. Після нагрівання до 80°С протягом ночі реакційну суміш охолоджували до кімнатної температури і розділяли між водою та DCM. Органічний екстракт концентрували і очищали за допомогою препаративної LCMS для одержання сполуки, вказаної у підзаголовку. 1Н ЯМР (300МГц, CD3OD) δ 3,83 (s, 3Н), 3,22-3,28 (m, 4H), 3,05 (t, J=5,1Гц, 2H), 2,93 (t, J=5,1Гц, 2Н); MS:ESI (позитивний): 262 (Μ+Η). Приклад 19 2-хлор-3-метокеи-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) 41 а.) етиловий естер 2-хлор-3-метокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Сполуку, вказану у підзаголовку, одержували за способом Прикладу 18, стадія d), використовуючи продукт з Прикладу 18, стадія с) та Nхлорсукциімід. b.) 2-хлор-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Сполуку, вказану у заголовку, одержували за способом Прикладу 18 , стадія e), використовуючи продукт зі стадії а). 1H ЯМР (300МГц, CD3OD) δ 3,86 (s, 3H), 3,19-3,26 (m, 4H), 3,01 (t, J=5,4Гц, 2Н), 2,88 (t, J=5,4Гц, 2Н); MS:ESI (позитивний): 218 (Μ+Η). Приклад 20 2-ізофеніл-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) а.) етиловий естер 2-ізофеніл-3-метокси4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 17, стадія с) (59мг, 0,19ммоль) розчиняли в 5мл THF і охолоджували до 0°С. Додавали MeMgBr (0,54мл, 1,4Μ) і реакційну суміш перемішували при кімнатній температурі протягом 1 години. Реакційну суміш гасили водою і НОАс (~3мл, 10:1) і екстрагували в DCM (2´5мл). Органічні екстракти концентрували для одержання продукту, вказаного у підзаголовку у вигляді олії, який використовували без подальшого очищення. b.) 2-ізофеніл-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії а) (~0,19ммоль) піддавали обробці 2мл, кожен з яких містив ЕtOН та 40% водний розчин КОН. Після нагрівання до 100°С протягом 16 годин реакційну суміш розбавляли водою і продукт екстрагували в DCM (2´4мл). Органічні екстракти концентрували і продукт, вказаний у заголовку, очищали препаративною HPLC-MS. 1H ЯМР (300МГц, CD3OD) δ 5,41 (s, 1H); 5,02 (t, J=1,6Гц, 1H), 3,14-3,08 (m, 4H), 2,96 (t, У=5,1Гц, 2H), 2,82 (t, J=5,1Гц, 2H), 2,09 (s, 3H); MS:ESI (позитивний): 224 (Μ+Η). Приклад 21 2-трет-бутил-3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 9) а.) етиловий естер 3-метил-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 16, стадія e) (475мг, 1,68ммоль) розчиняли в 50мл DCM і піддавали обробці Εt2Ν (585мкл, 4,2ммоль) та етилхлорформіатом (210мкл, 2,18ммоль). Після перемішування протя 88786 42 гом 3 днів при кімнатній температурі реакційну суміш концентрували над силікагелем і очищали хроматографією на силікагелі (ЕtOАс/градієнтгексан) для одержання 380мг етилкарбамату. Етилкарбамат (60мг, 0,19ммоль) розчиняли в 4мл EtOH і піддавали обробці ~10мг 10% Pd/C (вологий, компанії Degusssa, тип Ε101) та перемішували в атмосфері водню протягом 12 годин. Після фільтрування та концентрування одержали продукт, вказаний у підзаголовку. b.) етиловий естер 2-трет-бутил-3-метил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії а) (37мг, 0,16ммоль) перемішували в 1,5мл DCE. Додавали t-бутанол (19мкл, 0,20ммоль), після чого додавали BF3-OEt2 (20мкл, 0,16ммоль). Реакційну суміш нагрівали до 60°С протягом 1 години. В результаті концентрування одержали сполуку, вказану у підзаголовку, яку використовували без подальшого очищення. с.) 2-трет-бутил-3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії b) піддавали обробці 2мл, кожен з яких містив EtOH та 40% водний розчин КОН. Після нагрівання до 100°С протягом 12 годин реакційну суміш охолоджували, розбавляли водою та екстрагували 2х в DCM. Продукт, вказаний у заголовку, очищали за допомогою препаративної HPLC-MS. MS:ESI (позитивний): 224 (М+Н). Приклад 22 2-ізопропіл-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) а.) етиловий естер 3-ізопропіл-3-метокси4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 20, стадія а) (35мг, 0,13ммоль) розчиняли в 2мл EtOH і піддавали обробці 50мг 10% Pd/C (вологий, компанії Degusssa, тип Е101). Після перемішування протягом 3 годин при кімнатній температурі в атмосфері водню реакційну суміш фільтрували та концентрували для одержання сполуки, вказаної у підзаголовку, у вигляді олії. b.) 2-ізопропіл-3-метокси-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Продукт стадії а) піддавали депротекції за методикою, описаною в Прикладі 20, стадія b). В результаті очищення за допомогою препаративної HPLC-MS одержали сполуку, вказану у заголовку. 1 H ЯМР (CD3OD) δ 3,69 (s, 3Н); 3,29-3,21 (m, 5Н), 3,03 (t, J=5,2Гц, 2Н), 2,88 (t, J=5,4Гц, 2Н), 1,24 (t, J=6,9Гц, 6Н); MS:ESI (позитивний): 226 (Μ+Η). Приклад 23 2-бром-4-метил-5,6,7,8-тетрагідро-4Н-тієно [2,3-d]азепін (Схема 6) а.) етиловий естер 4-метилен-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; 43 Метилтифенілфосфонійбромід (6,3г, 17,6ммоль) розчиняли в 150мл THF та охолоджували до 0°С. Порціями додавали KHMDS (3,2г, 16,2ммоль) і реакційну суміш перемішували протягом ½ години. Додавали продукт Прикладу 1, стадія с) (3,0г, 12,5ммоль) у формі розчину у 25мл THF. Реакційну суміш перемішували нагрівали до кімнатної температури та перемішували протягом 1 години. Суміш концентрували і продукт, вказаний у підзаголовку, за допомогою хроматографії на силікагелі (EtOAc/градієнт-гексан) для одержання 2,6г сполуки, вказаної у підзаголовку. b.) етиловий естер 4-метил-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії а) (2,6г, 10,8ммоль) розчиняли в 100мл EtOH та обробляли 0,5г 10% Pd/C (вологий, компанії Degusssa, тип Е101). Після швидкого перемішування протягом 14 годин в атмосфері водню реакційну суміш фільтрували крізь целіт та концентрували для одержання 2,3г сполуки, вказаної у підзаголовку у вигляді прозорої олії. MS:ESI (позитивний): 240 (М+Н). с.) етиловий естер 2,3-дибром-4-метил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії b) (5,6г, 23,4ммоль) розчиняли в 250мл циклогексану та піддавали обробці NаНСО3 (11, 8г, 140ммоль). Повільно додавали бром (3,6мл, 70,3ммоль) і реакційну суміш перемішували протягом ½ години при кімнатній температурі, після чого реакційну суміш гасили Na2SO3 (180мл, 5% розчин). Після швидкого перемішування протягом 15 хвилин додавали EtOH (~100мл) і органічний вилучали та висушували над MgSO4 для одержання 9,1г сполуки, вказаної у підзаголовку. Рацемічний матеріал відділяли, використовуючи колонку Chiralpak® AD-RH® 20´50мм від Chiral Technologies (рухома фаза 10мл/хв. МеОН) для одержання енантіомеру 1 (час утримування =9,8 хвилин) та енантіомеру 2 (час утримування =11,4 хвилини) сполуки, вказаної у підзаголовку. d.) етиловий естер 4-метил-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії с) (800г, 2,0ммоль) розчиняли в 150мл EtOH та піддавали обробці 800мг 10% Pd/C (вологий, компанії Degusssa, тип Е101). Після перемішування додавали інші 300мг Pd і перемішування продовжували ще 3 години. Реакційну суміш фільтрували крізь целіт, розбавляли DCM (300мл) і промивали водою (1´300мл). Органічний шар висушували над MgSO4 і концентрували для одержання 475мг сполуки, вказаної у підзаголовку. e.) етиловий естер 2-бром-4-метил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії d) (80мг, 0,35ммоль) розчиняли в 2мл 1:1 СНСН3/НОАс. Додавали nбромсукцинімід (62мг, 0,35ммоль) і реакційну суміш перемішували протягом 15 хвилин. В результаті концентрування та очищення хроматографією на силікагелі одержали сполуку, вказану у підзаголовку, у вигляді жовтої олії. f.) 2-бром-4-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії e) піддавали депротекції за методикою, описаною для Прикладу 20, стадія b) для одержання цієї сполуки. 1Н ЯМР (CD3OD) δ 3,97 (s, 88786 44 1H), 3,46-3,11 (m, 7H), 1,39 (t, J=7,2Гц, 3Н); MS:ESI (позитивний): 246, 248 (Μ+Η). Приклад 24 2-бром-8-метил-5,6,7,8-тетрагідро-4Н-тієно [2,3-d]азепін (Схема 7) a) 2-тіофен-3-іл-етиламін; Тіофен-3-іл-ацетонітрил (5,0г, 40,6ммоль) розчиняли в 50мл THF. Повільно додавали ВН3-THF (61мл, 1М в THF). Реакційну суміш нагрівали до 60°С протягом ночі, а потім оборежно гасили 45 водною НСI доки перестали спостерігати виділення бульбашок. Сировинну реакційну суміш потім розділили на частини між ЕtOАс та водою (300мл кожна). Водний шар підкислювали 30% NaOH до рН~12 і продукт екстрагували в DCM/EtOH (4:1, 3х). Органічні екстракти висушували над MgSO4 і концентрували для одержання 2,9г сполуки, вказаної у підзаголовку, у вигляді олії. b.) етиловий естер [етоксикарбоніл-(2-тіофен3-іл-етил)-аміно]-оцтової кислоти; Продукт, вказаний у підзаголовку, одержали з продукту зі стадії а), використовуючи методику, описану в Прикладі 1, стадія а). с) [етоксикарбоніл-(2-тіофен-3-іл-етил)-аміно]оцтова кислота; Продукт, вказаний у підзаголовку, одержали з продукту зі стадії b), використовуючи методику, описану в Прикладі 1, стадія b). d.) етиловий естер 8-оксо-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт, вказаний у підзаголовку, одержали з продукту зі стадії с), використовуючи методику, описану в Прикладі 1, стадія с). e.) етиловий естер 8-метилен-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; МеРРh3Вr (785мг, 2,2ммоль) розчиняли в 7мл THF та піддавали обробці KHDMS (40мг, 2.04ммоль). Після перемішування протягом 30 хвилин додавали продукт стадії d) (350мг, 1,5ммоль) у формі розчину в 3мл THF. Після перемішування протягом 1 години реакційну суміш розводили EtOAc та промивали водою. Органічний шар концентрували і продукт очищали хроматографією на силікагелі (ЕtOАс/градієнт-гексан) для одержання 188мг сполуки, вказаної у підзаголовку. f.) етиловий естер 8-метил-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Продукт зі стадії а) (59мг, 0,25ммоль) розчиняли в 5мл EtOH та піддавали обробці 75мг 10% Pd/C (вологий, компанії Degusssa, тип Ε101). Після перемішування протягом 1 години в атмосфері водню реакційну суміш фільтрували і концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. g.) 2-бром-8-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії f) розчиняли в 2мл 1:1 СНСІ3/НОАс та піддавали обробці NBS (62мг, 0,35ммоль). Після перемішування протягом 10 хвилин реакційну суміш концентрували до сухого 45 стану і фільтрували крізь набивку з сілікагелю, елюювали EtOAc. Елюент концентрували і осад піддавали обробці 2мл, кожен з яких містив EtOH до 40% водний КОН. Після нагрівання протягом 14 годин при 100°С реакційну суміш розбавляли водою та екстрагували 2х в DCM. Сполуку, вказану у підзагловку, очищали препаративною HPLC-MS. Два енантіомери розділяли, використовуючи колонку Chiralpak® AD-RH® 20´50мм від Chiral Technologies (рухома фаза 10мл/хв. МеОН) для одержання енантіомеру 1 (час утримування =11,6 хвилин) та енантіомеру 2 (час утримування =13,6 хвилини) сполуки, вказаної у заголовку. 1Н ЯМР (CD3OD) δ 6,92 (s, 1Н), 3,48-3,34 (m, 3H), 3,17-2,99 (m, 4H), 1,44 (t, J=7,2Гц, 3Н); MS.-ESI (позитивний): 246,248 (Μ+Η). Приклад 25. 2-бром-8-спіроциклопропіл-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін (Схема 7) а.) етиловий естер 8-спіроциклопропіл-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6 карбонової кислоти: Розчин ZnCl2 (2,49мл, 1М в гексагені) та СН2І2 (100uL, 1.24ммоль) змішували при 0°С в DCM протягом 30 хвилин. Продукт з Прикладу 24, стадія e) (59мг, 0.25ммоль) додавали як розчин в DCM (1мл) і реакційну суміш перемішували при кімнатній температурі протягом 48 годин. Реакційну суміш розбавляли водою і екстрагували в DCM. Органічний шар промивали насиченим NH4CI, водою та розсолом. В результаті концентрування органічного шару одержали продукт, вказаний у підзаголовку, який використовували без подальшого очищення. MS:ESI (позитивний): 252 (М+Н). b.) етиловий естер 2-бром-8-спіроциклопропіл4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт, вказаний у підзаголовку, одержали за способом, описаним у Прикладі 18, стадія d), використовуючи продукт стадії а) і використовували його у сировинній формі без подальшого очищення. с.) 2-бром-8-спіроциклопропіл-5,6,7,8тетрагідро-тієно[2,3-d]азепін: Продукт, вказаний у заголовку, одержали за способом, описаним у Прикладі 18, стадія e), використовуючи продукт зі стадії b). 1H ЯМР (CD3OD) δ 6,84 (s, 1H), 3,20 (t, J=5,1Гц, 2Н), 3,02 (t, J=5,1Гц, 2Н), 3,06 (s, 2H), 1,08 (d, J=9,6Гц, 2Н), 1,05 (d, J=9,6Гц, 2Н). MS:ESI (позитивний): 258 (Μ+Η). Приклад 26. 2-(піролідин-1-сульфоніл)-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін (Схема 8) а.) етиловий естер 2-хлоросульфоніл-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6 карбонової кислоти: Продукт з Прикладу 1, стадія d) (400мг, 1,8ммоль) розчиняли в 10мл СНСl3 і очищали хло 88786 46 рсульфоновою кислотою (355мкл, 1,8ммоль). Після 5 хвилин реакційну суміш гасили льодом та відразу екстрагували в DCM (2´10мл). Органічні екстракти висушували над MgSO4 та концентрували для одержання 200мг продукту, вказаного у підзаголовку, як твердого тіла, який використовували без подальшого очищення. b.) 2-(піролідин-1-сульфоніл)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін: Продукт стадії а) (45мг, 0.14ммоль) перемішували в 2мл СНСІ3 піддавали обробці піролідином (46мкл, 0,55ммоль). Після перемішування протягом 5 хвилин реакційну суміш концентрували до сухого стану, залишок розчиняли в 2мл, кожен з яких включав ЕtOН та 40% водного КОН. Реакційну суміш нагрівали у герметичній посудині при 100°С протягом ночі. Після охолодження реакційну суміш розбавляли водою і продукт екстрагували в DCM (4´3мл). Органічні екстракти концентрували і продукт, вказаний у заголовку, очищали препаративною HPLC-MS. 1Н ЯМР (CD3OD) δ 7,42 (s, 1H), 3,43-3,34 (m, 4H), 3,31-3,24 (m, 6H), 3,13 (t, J=5,2Гц, 2H), 1,81-1,76 (m, 4H); MS: ESI (позитивний): 287 (Μ+Η). Приклад 27. Диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-сульфокислоти (Схема 8) Продукт, вказаний у заголовку, одержували за методикою, описаною в Прикладі 26, стадія b) використовуючи диметиламінгідрохлорид та Еt3N. 1Н ЯМР (CD3OD) δ 7,38 (s, 1Н), 3,44-3,27 (m, 4H), 3,32-3,26 (m, 2H), 3,17-3,14 (m, 2H), 2,72 (s, 6H); MS: ESI (позитивний): 261 (Μ+H). Приклад 28. Диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти (Схема 8) а.) етиловий естер 2-хлорсульфоніл-3-метил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт, вказаний у підзаголовку, одержували за методикою, описаною у Прикладі 26, стадія а), використовуючи проміжний продукт з Прикладу 21, стадія а). Продукт було використано без подальшого очищення. b.) диметиламід 3-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-сульфокислоти; Продукт вказаний у заголовку було приготовлено згідно з процедурою описаною в прикладі 26, стадії b), використовуючи продукт з стадії а) та гідрохлорид диметиламіну і Et3N. 1H ЯМР (CD3OD) δ 3,41-3,35 (m, 4H), 3,24 (dd, J=5,1, 6,6Гц, 2H), 3,04 (t, J=5,2Гц, 2Н), 2,75 (s, 6Н), 2,41 (s, 3Н); MS:ESI (позитивний): 275 (Μ+Η). Приклад 29. Циклопентиламід 5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-карбонової кислоти (Схема 8) 47 а.) етиловий естер 2-циклопентилкарбоніл4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт з Прикладу 1, стадія d) (50мг, 0,222ммоль) був змішаний з ізоціанатоциклопентаном (25мкл, 0,222ммоль) та АІСЬ (35мг, 0,266ммоль) в дихлороетані (2мл) при кімнатній температурі протягом 4 годин. Додатково додавали ізоціанато-циклопентан (25uL, 0,222ммоль) та АІСl3 (35мг, 0.266ммоль) і реакційну суміш перемішували протягом ночі. Реакція була розподілена між водою та DCM. Органічний шар промивали водою та концентрували для отримання продукту вказаного у підзаголовку, який було використано без подальшого очищення. MS:ESI (позитивний): 337 (М+Н). b.) циклопентиламід 5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-карбоксильної кислоти; Продукт зі стадії b) (37мг, 0,111ммоль) очищали за допомогою TMSI (50uL, 0,333ммоль) в DCM (2мл) і перемішували при 50°С протягом ночі. Потім реакційну суміш піддавали обробці метанолом (1мл), концентрували до сухого стану і очищали препаративною LC-MS для одержання сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 265 (М+Н). Приклад 30. Диметиламід 5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін-2-карбонової кислоти (Схема 8) а.) етиловий естер 2-диметилкарбамоіл4,5,7,8-тетрагідро-тієно[2,3-d]азепін-2-карбонової кислоти; Продукт з Прикладу 1, стадії d) (50мг, 0.22ммоль) перемішували в дихлоретані з хлоридом просгенімінію (84мг, 0,518ммоль) та АІСl3 (35мг, 0,266ммоль) при 75°С протягом ночі. Реакційну суміш охолоджували до кімнатної температури, гасили водою (2мл) та екстрагували в DCM. Потім органічний шар пропускали крізь набизку з целіту і концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. MS:ESI (позитивний): 297 (М+Н). b.) Диметиламід 5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін-2-карбонової кислоти; Сполуку, вказану у заголовку, одержували за способом, описаним в Прикладі 29, стадія b), використовуючи продукт зі стадії a). MS:ESI (позитивний): 225 (М+Н). Приклад 31. (R,S)-2-хлор-8-метил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 7) 88786 48 Сполуку, вказану у заголовку, одержували з проміжного продукту, описаного в Прикладі 24, стадія f) та N-хлор-сукциніміду, використовуючи методику з Прикладу 24, стадія g). MS:ESI (позитивний): 202, 204 (М+Н). Приклад 32. 2,3-дибром-4метил-5,6,7,8-тетрагідро-4Н-тієно [2,3-d]азепін (Схема 6) Продукт Прикладу 23, стадія с) піддавали депротекції за методикою, описаною в Прикладі 20, стадія b) для одержання сполуки, вказаної у підзаголовку. 1Н ЯМР (CD3OD) δ 3,60-3,52 (m, 3Н), 3,353,11 (m, 3Н), 3,35-3,11 (m, 4H), 1,33 (d, J=6,9Гц, 3Н); MS:ESI (позитивний): 324,326,328 (М+Н). Приклад 33. (R,S)-2-бром-4-Метокси-5,6,7,8-тетрагідро-4Hтієно[2,3-d]азепін (Схема 6) а.) етиловий естер (R,S)-4-гідрокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Боргідрид натрію (154мг, 4,18ммоль) додавали до розчину продукту зі стадії 1, стадія с) (200мг, 0,837ммоль) в етанолі (2мл) та перемішували при кімнатній температурі протягом 30 хвилин. Реакційну суміш гасили оцтовою кислотою та розділяли між DCM і водою. Органічний шар промивали водою і концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. MS:ESI (позитивний): 264 (M+Na). b) етиловий естер (R,S)-4-метокси-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт зі стадії а) (160мг, 0,664ммоль) в безводному THF (2,0мл) охолоджували до -78°С. До розчину додавали розчин 1М LHMDS в THF (800мкл, 0,797ммоль) і реакційну суміш нагрівали до кімнатної температури. До реакційної суміші додавали МеІ (63мкл, 0,996ммоль) і перемішували при кімнатній температурі протягом 72 годин. Реакційну суміш розділяли між етилацетатом і водою і органічний шар концентрували для одержання сполуки, вказаної у підзаголовку, яку очищали хроматографією на силікагелі (EtOAc/градієнтгексан) перед використанням в наступних стадіях. MS:ESI (позитивний): 278 (M+Na). с.) етиловий естер (R,S)-2-бром-4-метокси4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Сполуку, вказану у підзаголовку, одержували за способом Прикладу 18, стадія d), використовуючи продукт зі стадії b). d.) (R,S)-2-бром-4-метокси-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Сполуку, вказану в підзаголовку, одержували за способом, описаним в Прикладі 18 , стадія e), використовуючи продукт зі стадії с). MS:ESI (позитивний): 262 (М+Н). Приклад 34. 49 2-бром-8,8-диметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 6) a.) етиловий естер 8,8-диметил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; DCM (5мл) охолоджували до -78°С та піддавали обробці ТіСl4 (274мкл, 2,5ммоль), після чого Me2Zn (1,3мл 2М розчину в толуолі). Після перемішування темно-червоного розчину при -78°С протягом 15 хвилин повільно додавали продукт Прикладу 24, стадія d) (100мг, 0,42ммоль) у формі розчину в 5мл DCM. Реакційну суміш нагрівали до 0°С та перемішували протягом 2 годин. Органічні екстракти висушували над MgSO4 та концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. b.) 2-бром-8,8-диметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії а) (45мг, 0,18ммоль) розчиняли в 4мл 1:1 НОАс/СНСl3 та піддавали обробці NBS (44мг, 0,25ммоль), Після перемішування протягом ½ години реакційну суміш концентрували до сухого стану та піддавали обробці 2мл, кожен з яких містив EtOH та 40% водний КОН. Суміш нагрівали до 100°С протягом ночі, охолоджували та розводили водою. Продукт екстрагували 2х в DCM (2´4мл), концентрували та очищали препаративною HPLC-MS. 1H ЯМР (CD3OD) δ 7,19 (s, 1H), 3,40-3,34 (m, 2H), 3,28 (m, 2H), 3,21-3,17 (m, 2H), 1,41 (s, 6H); MS:ESI (позитивний): 260, 262 (Μ+Η). Приклад 35. (R,S)-2-бром-4-етил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 9) а.) етиловий естер (E,Z)-4-етиліден-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; KHMDS (233мг, 1,17ммоль) додавали до розчину броміду (етил)трифенілфосфонію (466мг, 1,26ммоль) в THF (5мл), охолоджували до 0°С і одержаний розчин перемішували протягом 20 хвилин. До реакційної суміші додавали продукт Прикладу 1, стадія с) (200мг, 0,837ммоль) в THF (5мл) і реакційну суміш нагрівали до кімнатної температури протягом 1 години. Реакційну суміш гасили водою і розділяли між етилацетатом і водою. Органічний шар концентрували і сировинний продукт очищали хроматографією на силікагелі (ЕtOАс/градієнт-гексан) для одержання 151мг сполуки, вказаної у підзаголовку, як суміші Ε та Ζ ізомерів. MS:ESI (позитивний): 252 (М+Н). b.) етиловий естер (R,S)-4-етил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії а) (151мг, 0,602ммоль) перемішували з 10% Pd/C (50мг) в етанолі (3мл) в атмосфері Н2 (1атм.) протягом ночі. Реакційну суміш фільтрували над целітом і концентрували до сухого стану для одержання 131мг сполуки, вказаної у підзаголовку, як пурпурної олії, яку використовува 88786 50 ли без подальшого очищення. MS:ESI (позитивний): 254 (М+Н). с.) етиловий естер (R,S)-2-бром-4-етил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Сполуку, вказану у підзаголовку, одержували за способом Прикладу 18, стадія d), використовуючи продукт зі стадії b) і використовували у сировинній формі без подальшого очищення. d.) (R,S)-2-бром-4-етил-4,5,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Сполуку, вказану у підзаголовку, одержували за способом, описаним у Прикладі 18, стадія e), використовуючи продукт зі стадії с). 1Н ЯМР (CD3OD) δ 6,88 (s, 1Н), 3,19-3,28 (m, 3H), 2,92-3,15 (m, 4H), 1,61-1,84 (m, 2H), 0,98 (t, J=7,3Гц, 3Н); MS:ESI (позитивний): 260 (Μ+Η). Приклад 36. 2-бром-3,4-Диметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 9) а.) етиловий естер 3-бром-4-метил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт Прикладу 23, стадія с) (енантіомер 2, 0,75г, 1,9ммоль) та Zn (0,25г, 3,8ммоль) нагрівали до кипіння зі зворотним холодильником в 20мл, кожен з яких містив воду та НОАс. Після ½ години, реакційну суміш охолоджували, розбавляли EtOAc і промивали 2х водою. Органічний шар висушували над MgSO4 і концентрували для одержання 490мг продукту, вказаного у підзаголовку, у вигляді олії, який використовували без подальшого очищення. b.) етиловий естер 3,4-диметил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії а) (150мг, 0,47ммоль) розчиняли в 3мл діоксану та піддавали обробці Me2Zn (0,47мл 2М розчину в толуолі) та Pd(ddf)2Cl2 (11мг, 0,014ммоль). Після нагрівання до 100°С протягом 3 годин реакційну суміш гасили водою та фільтрували. Фільтрат розділяли між EtOAc та водою (7мл кожний). Органічний шар висушували над MgSO4 і концентрували для одержання 92мг сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. с.) етиловий естер 2-бром-3,4-диметил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Продукт стадії b) (92мг, 0,36ммоль) розчиняли в 4мл 1:1 НОАс/СНСI3 та піддавали обробці NBS (67мг, 0,38ммоль). Після перемішування протягом ½ години реакційну суміш розбавляли EtOAc (70мл), промивали водою (3´30мл) та 1М NaOH (2´30мл). Органічний шар висушували над MgSO4 і концентрували. Сировинний продукт очищали хроматографією на силікагелі (EtOAc/Hex-градієнт) для одержання 90мг сполуки, вказаної у підзаголовку. d.) 2-бром-3,4-диметил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Продукт стадії с) піддавали депротекції, як описано в Прикладі 20, стадія b). Сполуку, вказану у підзаголовку, одержали після очищення препаративною HPLC-MS. 1Н ЯМР (CD3OD) δ 3,95-3,08 51 (m, 7Н), 2,13 (s, 3Н), 1,31 (d, J=7,2Гц, 3H); MS:ESI (позитивний): 260, 262 (Μ+Η). * Енантіомер 1 можна одержати подібним способом. Приклад 37. 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6-азациклопента[сd]азулен (Схема 10) а.) етиловий естер (Е,Z)-4етоксикарбонілметилен-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбоновоі кислоти; До розчину триетилфосфоноацетату (4мл, 16,74ммоль) в безводному THF (100мл) додавали 1,6М розчин LHMDS в THF (15мл), суміш перемішували протягом 15 хвилин, після чого додавали продукт Прикладу 1, стадія с) (2,0г, 8,37ммоль). Реакційну суміш перемішували протягом ночі, а потім піддавали обробці додатковим розчином LHMDS (3,2мл, 1,6М) та триетилфосфоноацетату (800мл, 3,3ммоль). Після перемішування протягом 3 годин, реакційну суміш гасили водою та розбавляли DCM. Органічний шар висушували над MgSO4 і концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення MS:ESI (позитивний): 310 (М+Н). b.) етиловий естер (R,S)-4етоксикарбонілметил-4,5,7,8-тетрагідро-тієно[2,3d]азепін-6-карбонової кислоти; Продукт стадії а) (2,47г, 8ммоль) перемішували з 2,0г 10% 10% Pd/C (вологий, компанії Degusssa, тип Е101) у метанолі (8мл) в атмосфері Н2 (1атм.) протягом 72 годин. Реакційну суміш фільтрували над целітом і концентрували до сухого стану для одержання продукту, вказаного у підзаголовку, у вигляді олії, який використовували без подальшого очищення. MS:ESI (позитивний): 312 (М+Н). с.) етиловий естер (R,S)-4-карбоксиметил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти. Продукт стадії b) (2,47г, 8ммоль) перемішували в етанолі (60мл) з 1М NaOH (30мл) при кімнатній температурі протягом ночі. Реакційну суміш підкислювали 1М НСI і розділяли між DCM і водою. Органічний шар промивали водою, висушували над MgSO4 і концентрували до сухого стану для одержання 2,13г продукту, вказаного у підзаголовку, у вигляді жовтої олії, який використовували без подальшого очищення. MS:ESI (позитивний): 248 (М+Н). d.) етиловий естер (R,S)-3-оксо-3,4,4а,5,7,8гексагідро-1-тіа-6-аза-циклопентал[cd]азулен-6карбонової кислоти; Оксалілхлорид (3мл, 37,7ммоль) та каталітичну кількість DMF додавали до розчину продукту зі стадії с) (2,13г, 7,54ммоль) в DCM (40мл) і реакційну суміш перемішували при кімнатній температурі протягом 1 години. Реакційну суміш концентрували до сухого стану та знову розчиняли в дихлоретані (100мл). До розчину додавали АlСl3 (2,0г, 15,1ммоль) і реакційну суміш перемішували 88786 52 при кімнатній температурі протягом ночі. Реакційну суміш гасили льодом і розділяли між DCM і водою. Органічний шар концентрували для одержання сполуки, вказаної у підзаголовку, яку очищали хроматографією на силікагелі (ЕtOАс/градієнт-гексан, виділені 1,02г) до використання на наступних стадіях. MS:ESI (позитивний): 266 (М+Н). е.) етиловий естер (R,S)-3,4,4а,5,7,8гексагідро-1-тіа-6-аза-циклопентал[cd]азулен-6карбонової кислоти; До ΒΗ3tBuΝΗ2 (492г, 5,66ммоль) в DCM (2мл) додавали АlСl3 (627мг, 4,72ммоль) при 0°С. Розчин перемішували протягом 10 хвилин, а потім піддавали обробці продуктом зі стадії d) (250мг, 0,943ммоль) у формі розчину в DCM (1мл). Реакційну суміш розбавляли в 1М НСI і екстрагували в ЕtOАс. Органічний шар концентрували для одержання продукту, вказаного у підзаголовку, який очищали хроматографією на силікагелі (ЕtOАс/градієнт-гексан) до використання на наступних стадіях. MS:ESI (позитивний): 252 (М+Н). f.) етиловий естер (R,S)-2-бром-3,4,4а,5,7,8гексагідро-1-тіа-6-аза-циклопентал[сd]азулен-6карбонової кислоти; Сполуку, вказану у підзаголовку, одержували за способом Прикладу 18, стадія d), використовуючи продукт зі стадії e) і одержаний продукт використовували у сировинній формі без очищення. g.) 2-бром-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6аза-циклопента[c,d]азулен; Сполуку, вказану у заголовку, одержували за способом Прикладу 18, стадія e), використвуючи продукт зі стадії f) і очищали препаративною HPLC-MS. Два енантіомери розділяли, використовуючи колонку Chiralpak® AD-RH® 20´50мм від Chiral Technologies (рухома фаза 10мл/хв. МеОН) для одержання енантіомеру 1 (час утримування =8,6 хвилин) та енантіомеру 2 (час утримування =10,8 хвилини) сполуки, вказаної у заголовку. HPLC-MS. 1H ЯМР (CD3OD) δ 3,45-3,56 (m, 2H), 2,89-3,08 (m, 3Н), 2,45-2,65 (m, 4H), 1,99-2,04 (m, 2H). MS:ESI (позитивний): 260 (Μ+Η). Приклад 38. (R,S)-2-метил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа6-аза-циклопента[сd]азулен (Схема 10) a) етиловий естер (R,S)-2-метил-3,4,4а,5,7,8гексагідро-1-тіа-6-аза-циклопента[сd]азулен-6карбонової кислоти; Розчин 2М Μe2Ζn в толуолі (1,5мл) додавали до продукту Прикладу 37, стадія f) (50мг, 0,150ммоль) та Pd(dppf)2Cl2 (4мг, 0,0045ммоль) в діоксані (1мл). Після нагрівання до 100°С протягом 3 годин реакційну суміш гасили водою та екстрагували в етилацетаті. Органічні шари комбінували та концентрували для одержання сполуки, вказаної у підзаголовку, яку використовували у сировинній формі без очищення MS:ESI (позитивний): 266 (М+Н). b) (R,S)-2-метил-4,4а,5,6,7,8-гексагідро-3Н-1тіа-6-аза-циклопента[cd]азулен; 53 Сполуку, вказану у заголовку, одержували за способом, який описано в Прикладі 18, стадія e), використовуючи продукт стадії а), та очищаючи препаративною HPLC-MS. 1H ЯМР (CD3OD) δ 3,523,64 (m, 2Н), 3,25-3,30 (m, 2Н), 2,86-3,10 (m, 4Н), 2,46-2,73 (m, 4Н), 2,23 (s, 3Η). MS:ESI (позитивний): 194 (Μ+Η). Приклад 39. 2-трет-бутил-4,4а,5,6,7,8-гексагідро-3Н-1-тіа-6аза-циклопента[cd]азулен (Схема 10) a.) етиловий естер (R,S)-2-трет-бутил3,4,4а,5,7,8-гексагідро-1-тіа-6-азациклопента[cd]азулен-6-карбонової кислоти; Продукт з Прикладу 37, стадія e) (100мг, 0,398ммоль), ВF3Оеt (50мкл, 0,398ммоль) та третбутанол (56мкл, 0,597ммоль) нагрівали до 75°С протягом 2 годин у дихлоретані (1мл). Реакційну суміш гасили водою та екстрагували в DCM. Органічні шари комбінували та екстрагували протягом ночі для одержання сполуки, вказаної у підзаголовку, яку використовували у сировинній формі без подальшого очищення. MS:ESI (позитивний): 308 (М+Н). b.) 2-трет-бутил-4,4а,5,6,7,8-гексагідро-3Н-1тіа-6-аза-циклопента[cd]азулен; Сполуку, вказану у заголовку, одержували за способом Прикладу 18, стадія e), використовуючи продукт зі стадії а). Два енантіомери розділяли, використовуючи колонку Chiralpak® AD-RH® 20´50мм від Chiral Technologies (рухома фаза 10мл/хв. МеОН) для одержання енантіомеру 1 (час утримування =7,7 хвилин) та енантіомеру 2 (час утримування =10,2 хвилини) сполуки, вказаної у заголовку. 1Н ЯМР (CD3OD) δ 3,20-3,34 (m, 2Н), 2,91-3,01 (m, 1H), 2,56-2,91 (m, 5H), 2,27-2,39 (m, 2H), 1,74-1,88 (m, 1H), 1,30 (s, 9H). MS:ESI (позитивний): 236 (Μ+Η). Приклад 40. 2-трифтометил-5,6,7,8-тетрагідро-4Н-тієно [2,3-d]азепін (Схема 1а) а.) етиловий естер 2-йодо-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту з Прикладу 1, стадія d) (470мг, 2,09ммоль) в СНСl3 (5мл) та АсОН (5мл) піддавали обробці N-йодосукциімідом (493мг, 2,19ммоль) при 22°С. Після 1 години реакційну суміш розбавляли СН2СІ2 (20мл) і вливали в насичений NаНСО3 (20мл). Органічний шар промивали розсолом (1´20мл), висушували (MgSO4) і концентрували, в результаті чого одержали сировинний продукт, вказаний у підзаголовку у вигляді жовтої олії, який використовували без подальшого очищення. b.) етиловий естер 2-трифторметил-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії а) (75мг, 0,21ммоль) в DMF (0,5мл) та NMP (0,5мл) піддавали обробці біпиридилом (43мг, 0,27ммоль), Cul (44мг, 88786 54 0,23ммоль), KF (13мг, 0,23ммоль) та триметилсилілтрифторметаном (2,1мл, 1,05ммоль). Реакційну суміш нагрівали до 80°С протягом 3 днів. Реакційну суміш охолоджували і фільтрували крізь целіт, промиваючи за допомогою EtOAc (20мл). Елюент промивали розсолом (2´3мл) і висушували (MgSO4), в результаті чого одержали сполуку, вказану у підзаголовку у вигляді коричневої олії, яку використовували без подальшого очищення. с.) 2-трифтометил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін; Розчин сировинного продукті зі стадії b) (26мг, 0,089ммоль) в СН2Сl2 (0,5мл) піддавали обробці йодтриметилсиланом (19мкл, 0,134ммоль) при 50°С протягом 12 годин. Реакційну суміш гасили додаванням МеОН і розчинник випаровували. Залишок очищали препаративною LC/MS, в результаті чого одержали сполуку, вказану у заголовку. 1 H ЯМР (CD3OD) δ 7,18 (s, 1Н); 3,42-3,56 (m, 6H); 3,31-3,39 (m, 2H); 2,09 (s, 1H); MS:ESI (позитивний): 222 (Μ+Η). Приклад 41. 3-хлор-2-трифтометил-5,6,7,8-тетрагідро-4Нтієно [2,3-d]азепін (Схема 1b) a.) етиловий естер 2,3-дихлор-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту з Прикладу 4, стадія Η (323мг, 1,25ммоль) в СН2СІ2 (6,25мл) охолоджували до 0°С та додавали триетиламін (522мкл, 3,75ммоль), після чого додавали етилхлорформіат (144мкл, 1,5ммоль). Після 1,5 години реакційну суміш вливали у воду (25мл) та розбавляли ЕtOАс (50мл). Органічний шар висушували (MgSO4) і концентрували, в результаті чого одержали 341мг (93%) сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. MS:ESI (позитивний): 294,296 (М+Н). b) етиловий естер 3-хлор-2-йод-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії а) (129мг, 0,44ммоль)у безводному THF (2,2мл) охолоджували до -78°С та піддавали обробці 1,5екв. n-BuLi (412мкл, 0,66ммоль). Через 1 годину реакційну суміш гасили THF розчином I2 (167мг, 0,66ммоль). Реакційну суміш нагрівали до 22°С і розбавляли ЕtOАс (15мл). Органічний шар промивали насиченим NaSO3 (5мл), розсолом (5мл) та висушували (MgSO4), в результаті чого одержали 98мг (58%) сировинної сполуки, вказаної у підзаголовку, яку викорисовували без подальшого очищення. MS:ESI (позитивний): 386 (М+Н). с.) етиловий естер 3-хлор-2-трифторметил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії b) (140мг, 0,363ммоль) в NMP (0,9мл) та DMF (0,9мл) піддавали обробці Cul (76мг, 0,39ммоль), KF (46,4мг, 0,79ммоль), біпиридилом (74мг, 0,472ммоль), та TMSCF3 (267мкл, 1,81ммоль). Суміш нагрівали до 80°С протягом 12 годин. Сировинну реакційну суміш розбавляли ЕtОАс і фільтрували крізь целіт. 55 Органічну фазу промивали Н2О (2´1мл), висушували (MgSO4) і концентрували. Сировинний продукт очищали препаративною TLC, використовуючи 10:1 гексан/EtOAc, в результаті чого одержали 14,4мг (12,1%) сполуки, вказаної у підзаголовку. d.) 3-хлор-2-трифтометил-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Розчин продукту зі стадії с) (14,4мг, 0,044ммоль) і СН2Сl2 (220мкл) піддавали обробці йодтриметилсиланом (19мкл, 0,13ммоль) при 60°С протягом 12 годин. Залишок очищали препаративною LC/MS, в результаті чого одержали 2,3мг (18%) сполуки, вказаної у заголовку. MS:ESI (позитивний): 256 (М+Н). Приклад 42. 3-бром-2-трифтометил-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 1b) а.) етиловий естер 3-бром-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту з Прикладу 3, стадія а) (148мг, 0,39ммоль) в АсОН (80мкл) та Н2О (1,92мл) піддавали обробці Zn (76мг, 1,17ммоль) при 105°С протягом 4,5 години. Потім вміст охолоджували до 22°С, вливали в насичений NаНСО3 (25мл) та екстрагували за допомогою EtOAc (2´25мл). Органічний шар висушували (MgSO4) і концентрували, в результаті чого одержали 80мг (68%) сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. b.) етиловий естер 3-бром-2-йод-4,5,7,8тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії а) (124мг, 0,41ммоль) с СНСI3 (1,0мл) та АсОН (1,0мл) піддавали обробці N-йодсукцинімідом (97мг, 0,43ммоль) протягом 30 хвилин. Потім сировинну реакційну суміш вливали в насичений NаНСО3 (5мл) та екстрагували за допомогою СН2СІ2 (2´3мл). Органічний шар промивали насиченим NаНСО3 (3мл), висушували (колонка Extrelut), концентрували і очищали препаративною TLC (80% гексану:20% EtOAc), в результаті чого одержали 134мг (76%) сполуки, вказаної у підзаголовку. b.) етиловий естер 3-бром-2-трифторметил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії b) (134мг, 0,31ммоль) в NMP (1мл) та DMF (1мл) піддавали обробці KF (20мг, 0,34ммоль), Cul (65мг, 0,34ммоль) та біпиридилом (62мг, 0,4ммоль) при 50°С протягом 15 хвилин. Потім додавали 0,5Μ розчин триметилсилілтрифторметану (3,1мл, 1,55ммоль) і реакційну суміш перемішували протягом 16 годин при 80°С. Реакційну суміш охолоджували до 22°С, розбавляли СН2СІ2 (5мл) і промивали розсолом (10мл). Водний шар екстрагували за допомогою СН2Сl2 (3´5мл) і комбіновані органічні шари висушували (колонка Extrelut), пропускали крізь набивку з силікагелю, концентрували і очищали препаративною LC/MS, в результаті чого одержали 26мг (23%) сполуки, вказаної у підзаголовку. 88786 56 d.) 3-бром-2-трифтометил-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Розчин продукту зі стадії с) (51мг, 0,137ммоль) в СН2СІ2 (685мкл) піддавали обробці йодтриметилсиланом (59мкл при 50°С протягом 16 годин). Реакційну суміш гасили додаванням МеОН і розчинник випаровували. Залишок очищали препаративною LC/MS, в результаті чого одержали 16мг (39%) сполуки, вказаної у заголовку. 1H ЯМР (300МГц, CDCI3) δ 4,10-4,40 (br s, 1H); 2,90-3,60 (m, 8H); MS:ESI (позитивний): 300, 302 (Μ+Η). Приклад 43. 3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін (Схема 3) а.) етиловий естер 3-метил-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбоноваї кислоти; Розчин продукту з Прикладу 42, стадія а) (122мг, 0,40ммоль) в сухому THF (2мл) піддавали обробці NiCl2 (dppp) (2-3мг, 0,004ммоль), після чого по краплинах при 22°С додавали 1,4Μ розчин броміду метилмагнію (0,71мл, 1ммоль), а потім реакційну суміш нагрівали зі зворотним холодильником протягом 16 годин. Реакційну суміш охолоджували до 22°С, розбавляли етером (5мл) і гасили 1N HCl (2мл). Водний шар піддавали зворотній екстракції етером (3´5мл). Комбіновані органічні екстракти промивали водою (10мл) і висушували (MgSO4). Розчинник випаровували в результаті чого одержали сировинний продукт, вказаний у підзаголовку у вигляді жовтувато-коричневої олії, який використовували без подальшого очищення. b.) 3-метил-5,6,7,8-тетрагідро-4Н-тієно[2,3d]азепін; Розчин продукті зі стадії а) в СН2Сl2 (1мл) піддавали обробці йодтриметилсиланом при 50°С протягом 16 годин). Реакційну суміш гасили додаванням МеОН і розчинник випаровували. Залишок очищали препаративною LC/MS, в результаті чого одержали 1,6мг сполуки, вказаної у заголовку. 1H ЯМР (300МГц, CDCI3) δ 6,69 (d, J=1Гц, 1H); 4,8-5,2 (br s, 1H); 3,20-3,40 (m, 6H); 3,00-3,1 (m, 2H); 2,14 (d, J=1Гц, 3Н); MS:ESI (позитивний): 168 (Μ+Η). Приклад 44. 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін (Схема 5) а.) етиловий естер 2-трет-бутил-3-метил4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; До перемішаного розчину продукту Прикладу 18, стадія а) (20мг, 0,078ммоль) в 0,5мл DCE додавали t-бутанол (25мкл, 0,26ммоль), після чого додавали BF3-Ot2 (10мкл, 0,078ммоль). Реакційну суміш нагрівали до 75°С протягом 2 годин. Реакційну суміш охолоджували до кімнатної температури, концентрували до сухого стану і залишок використовували без подальшого очищення. MS:ESI (позитивний): 312 (М+Н). 57 b.) 2-трет-бутил-3-метокси-5,6,7,8-тетрагідро4Н-тієно[2,3-d]азепін; Продукт стадії а) піддавали обробці 0,5мл EtOH та 40% водним КОН. Після нагрівання при 100°С протягом 18 годин реакційну суміш охолоджували, розбавляли водою та екстрагували в DCM (2´10мл). Органічний шар концентрували до сухого стану і залишок очищали препаративною HPLC-MS для одержання сполуки, вказаної у заголовку. 1H ЯМР (300МГц, CD3OD) δ 3,78 (s, 3Н); 3,30-3,38 (m, 4H); 3,06-3,15 (m, 2H); 2,94-3,03 (m, 2H); 1,45 (s, 9H); MS:ESI (позитивний): 240 (Μ+Η). Приклад 45. 2-трет-бутил-5,6,7,8-тетрагідро-4Н-тієно [2,3d]азепін (Схема 9) До перемішаного розчину продукту з Прикладу 1, стадія d) (66мг, 0,29ммоль) в 2,5мл DCE додавали t-бутанол (36мкл, 0,38ммоль) та BF3-Oet2 (36мкл, 0,29ммоль). Реакційну суміш нагрівали до 60°С протягом 2 годин. Суміш нагрівали до 100°С протягом ночі, охолоджували та розбавляли водою. Реакційну суміш концентрували до сухого стану і піддавали обробці 2мл, кожен з яких містив EtOH та 40% КОН. Продукт екстрагували в DCM (2´10мл), концентрували і очищали препаративною HPLC-MS для одержання сполуки, вказаної у заголовку. MS:ESI (позитивний): 210 (М+Н). Приклад 46. 2-трет-бутил-3-хлор-5,6,7,8-тетрагідро-4Нтієно [2,3-d]азепін (Схема 4) Приклад 47. 3-хлор-2-(1,1,3,3-тетраметил-бутил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін (Схема 4) Приклад 48. 3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін (Схема 4) а.) етиловий естер 3-хлор-4,5,7,8-тетрагідротієно[2,3-d]азепін-6-карбонової кислоти; Розчин продукту з Прикладу 41, стадія а.) (382мг, 1,3ммоль) в THF (6,5мл) охолоджували до -78°С та піддавали обробці 1,6Μ розчином n-BuLi (2,26мл, 1,36ммоль). Через 1 годину реакційну суміш гасили водою та екстрагували за допомогою EtOAc (2´30мл). Органічний шар промивали розсолом (1´15мл), висушували (MgSO4) і концентрували. Сировинну жовту олію очищали хроматографією на силікагелі (EtOAc/градієнт-гексан), в результаті чого одержали 156мг (46%) сполуки, вказаної у підзаголовку. 1Н ЯМР (300МГц, CDCL3) δ 6,88 (s, 3Н); 4,19 (q, J=7Гц, 2H); 3,47-3,72 (m, 4H); 2,92-3,03 (m, 2H); 2,81-2,92 (m, 2H); 1,29 (q, J=7Гц, 3Н). b.) етиловий естер 2-трет-бутил-3-хлор4,5,7,8-тетрагідро-тієно[2,3-d]азепін-6-карбонової кислоти; 88786 58 Розчин продукту зі стадії а) (156мг, 0,6ммоль) в дихлоретані (3мл) піддавали обробці t-BuOH (172мкл, 1,8ммоль) та BF3-OEt2 (113мкл, 0,9ммоль). Реакційну суміш нагрівали при 85°С протягом 6 годин, після чого після випаровування одержали сировинну сполуку, вказану у підзаголовку. с.) 2-трет-бутил-3-хлор-5,6,7,8-тетрагідро-4Нтієно[2,3-d]азепін, Приклад 44; 3-хлор-2-(1,1,3,3-тетраметил-бутил)-5,6,7,8тетрагідро-4Н-тієно[2,3-d]азепін, Приклад 45; 3-хлор-5,6,7,8-тетрагідро-4Н-тієно[2,3-d]азепін, Приклад 46; Розчин сировинного продукту зі стадії b.) в EtOH (4мл) та 40% водному КОН (4мл) піддавали обробці бромідом тетра-бутиламонію (20мг) та нагрівали при 95°С протягом 2 днів. Потім реакційну суміш охолоджували до 22°С та екстрагували за допомогою СН2СІ2 (3´25мл). Органічний шар промивали розсолом (1´10мл), висушували (MgSO4) і концентрували до сухого стану. Залишок очищали препаративною LC/MS, в результаті чого одержали сполуки, вказані у підзаголовку. (Α) 1H ЯМP (300МГц, CDCL3) δ 6,50-6,80 (br s, 1H); 3,053,14 (m, 4H); 2,87-2,99 (m, 4H); 1,43 (s, 9H); MS:ESI (позитивний): 244 (Μ+Η); (Β) 1H ЯМР (300МГц, CDCL3) δ 3,58-3,70 (br s, 1H); 2,98-3,09 (m, 4H); 2,88-2,94 (m, 2H); 2,82-2,87 (m, 2H); 1,91 (s, 2H); 1,46 (s, 6H); 0,81 (s, 9H); MS:ESI (позитивний): 300 (Μ+Η); (С) 1H ЯМР (300МГц, CDCL3) δ 6,75 (s, 1H); 2,92-3,11 (m, 6H); 2,85-2,90 (m, 2H); 2,72-2,82 (br, s 1H); MS:ESI (позитивний): 188 (Μ+Η). Приклад 49. 2-бром-5,6,7,8-тетрагідро-4Н-фуро [2,3d]азепін (Схема 11) а.) етиловий естер фуран-2-іл-оксо-оцтової кислоти; Фуран-2-іл-оксо-оцтову кислоту (15г, 107ммоль), розчинену в СНСІ3 (420мл), піддавали обробці EtOH (9,6мл, 165ммоль) та H2SO4 (1мл) та нагрівали до 63°С протягом 12 годин. Потім реакційну суміш переводили в ділильну лійку та промивали насиченим NaHCO3 (100мл). Органічний шар промивали розсолом (100мл), висушували (MgSO4) і концентрували, в результаті чого одержали 16,9г (66%) сполуки, вказаної у підзаголовку, яку використовували без подальшого очищення. b.) фуран-2-іл-гідрокси-оцтова кислота; Розчин продукту зі стадії а) (25г, 135ммоль) в EtOH (250мл) охолоджували до 0°С та піддавали обробці розчином NaBH4 (2,5г, 66ммоль) в Н2О (27мл) протягом 5 хвилин. Потім реакційну суміш гасили АсОН (17мл) та Н2О (271мл) та концентрували до сухого стану. Сировинну олію розчиняли в СН2Сl2 (300мл), промивали розсолом (2´100мл), висушували (MgSO4) і концентрували, в результаті чого одержали 17,5г (70%) вказаної у підзаголовку сполуки, яку використовували без подальшого очищення. с.) (3-карбоксиметил-фуран-2-іл)-оцтова кислота; 59 Розчин продукту зі стадії b.) (10,9г, 64ммоль) у декаліні (193мл) піддавали обробці триметлортоацетатом (48,2мл, 384ммоль) та гексановою кислотою (2,0мл). Потім реакційну суміш введи у колонку Vigreaux та нагрівали до 180°С протягом 18 годин. Додаткові гексанову кислоту (3´1,5мл) аліквоти гексанової кислоти додавали кожні 2 години для перших 6 годин часу реакції. Потім реакційну суміш охолоджували до 22°С і екстрагували МеОН, в результаті чого одержали 27г сировинної суміші диестеру та декаліну. Цю суміш розчиняли в МеОН (250мл), охолоджували до 0°С і піддавали обробці 2М NaOH (150мл). Після 12 годин розчинник випаровували і осад вводили в 2N NaOH (100мл) і промивали етером (2´150мл). Основний шар підкислювали 4М НСI до рН1 і знову екстрагували ЕtOАс (4´100мл). Органічний шар промивали розсолом, висушували (MgSO4) і концентрували, в результаті чого одержали 6,4г (54%) сполуки, вказаної у підзаголовку. d.) 2-[3-(гідрокси-етил)-фуран-2-іл]-етанол; Розчин продукту зі стадії с.) (6,4г, 35ммоль) в сухому THF (400мл) охолоджували до 0°С та по краплинах через 10 хвилин додавали 1,0Μ розчин ВН3 в THF (174мл, 174ммоль). Після завершення додавання суміш перемішували додаткові 20 хвилин при 0°С, а потім нагрівали до 22°С протягом 2 годин. Потім суміш вливали в охолоджений до льоду насичений NaHCO3 (300мл) і екстрагували за допомогою ЕtOАс (2´200мл). Органічний шар висушували (MgSO4) і концентрували, в результаті чого одержали 3,58г (65%) сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 157 (М+Н). e.) етиловий естер 2-[3-(2метансульфонілокси-етил)-фуран-2-іл]метансульфокислоти; Розчин продукту зі стадії d.) (3,58г, 22,9ммоль) в СН2Сl2 (114мл) охолоджували до 0°С та піддавали обробці триетиламіном (9,56мл, 68,7ммоль), після чого по краплинах додавали хлорид метансульфонілу (3,88мл, 50,4ммоль) протягом 10 хвилин. Після 1 години реакційну суміш переводили у ділильну лійку та екстрагували за допомогою льодяної води (1´50мл). 10% лимонною кислотою (2´50мл), насиченим NaHCO3 (2´50мл) та розсолом (1´50мл). Органічний шар висушували (MgSO4), концентрували до 20мл, розбавляли сухим діоксаном (42мл) і потім концентрували для вилучення СН2СІ2, що залишився. Одержаний діоксановий розчин бісмезилату негайно переводили на стадію f). f.) 6-бензил-5,6,7,8-тетрагідро-4Н-фуро[2,3d]азепін; Діоксановий розчин бісмезилату, одержаний на стадії e.) розводили сухим діоксаном (168мл) та переводили в реакційну посудини з 3-ма шийками, обладнану ділильною лійкою та конденсатором. Додавали безводний K2СО3 (46,5г, 337ммоль) і суміш нагрівали до 102°С. Потім по краплинах через 45 хвилин додавали розчин бензиламіну (7,5г, 70,1ммоль) у діоксані (74,4мл) і реакційну суміш нагрівали зі зворотним холодильником протягом 18 годин. Суміш охолоджували до 22°С, солі відфільтровували і розчинник випаровували. Сировинну олію очищали за допомогою хроматографією на силікагелі (ЕtOАс/гексан-градієнт), в 88786 60 результаті чого одержали 2,56г (49%) (комбінований вихід стадій e та d) сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 228 (М+Н). g.) гідрохлорид 5,6,7,8-тетрагідро-4Н-фуро[2,3d]азепіну; Розчин продукту зі стадії f.) (2,56мг, 11,3ммоль) у безводному дихлоретані (56мл) охолоджували до 0°С, піддавали обробці 1хлоретилхлорформіатом (6,11мл, 56,4ммоль) і реакційну масу нагрівали до 22°С протягом 1 години. Реакційну масу розбавляли СН2СІ2 (10мл) та промивали насиченим NаНСО3 (50мл). Насичений NаНСО3 піддавали зворотній екстракції за допомогою СН2СІ2 і комбіновані органічні шари промивали розсолом (50мл), висушували (MgSO4) і концентрували, в результаті чого одержали маслоподібний залишок, який ввели у безводний МеОН (150мл) та нагрівали зі зворотним холодильником. МеОН випаровували, а сировинний продукт розтирали в порошок з етером та фільтрували, в результаті чого одержали 1,71г (87%) сполуки, вказаної у підзаголовку. 1H ЯМР (300МГц, DMSO) δ 9,56 (br s, 2H); 7,43 (d, J=2Гц,1Н); 6,34 (d, J=2Гц,1Н); 3,18-3,30 (br m, 4H); 3,03-3,10 (br m, 2H); MS:ESI (позитивний): 138 (Μ+Η). h.) трет-бутиловий естер 4,5,7,8-тетрагідрофуро[2,3-d]азепін-6-карбонової кислоти; Розчин продукту зі стадії g) (500мг, 2,88ммоль) в ацетоні (7,2мл) та воді (7,2мл) піддавали обробці NaHCO3 (484мг, 5,76ммоль) та ди-третбутилкарбонатом (691мг, 3,17ммоль) при інтенсивному перемішуванні протягом 1 години. Вміст розбавляли за допомогою H2О (10мл) та екстрагували за допомогою EtOAc (2-50мл), органічний шар висушували (MgSO4), концентрували та очищали хроматографією (EtOAc/гексан-градієнт), в результаті чого одержали 643мг (94%) сполуки, вказаної у підзаголовку. MS:ESI (позитивний): 238 (М+Н). і.) 2-бром-5,6,7,8-тетрагідро-4Н-фуро[2,3d]азепін; Розчин продукту зі стадії h.) (50мг, 0,21ммоль) в СНСl3 (527мкл) та АсОН (527мкл) піддавали обробці N-бросукциімідом (38,1мг, 0,21ммоль) при 22°С. Після 1 години вміст переводили в насичений NaHCO3 та екстрагували за допомогою ЕtOАс (2´5мл). Органічний шар промивали розсолом (1´5мл), висушували (MgSO4) та очищали препаративною TLC (80% гексану: 20% EtOAc), в результаті чого одержали трет-бутиловий естер 2бром-4,5,7,8-тетрагідро-4Н-фуро[2,3-d]азепін-6карбонової кислоти, який піддавали прямій обробці 4М НСI у діоксані (2мл). Діоксан випаровували, а залишок розчиняли в МеОН та очищали препаративною LC/MS, в результаті чого одержали 1,3мг сполуки, вказаної у заголовку. 1Н ЯМР (300МГц, CDCL3) δ 6,08 (s, 1H); 3,02-3,07 (m, 4H); 2,89-2,92 (m, 2H); 2,57-2,70 (m, 3H); MS:ESI (позитивний): 216, 218 (Μ+Η). Для оцінки репрезентативних сполук даного винаходу як агоністів рецептора 5НТ2c використовували нижченаведену методику. Результати цього досліду наведені в Таблиці 1. Культура клітин
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted azepine derivatives as serotonin receptor modulators
Автори англійськоюBennani Youssef L., Robarge Michael J., Bom David C., Varga Norbert, Tumey Lawrence N.
Назва патенту російськоюПроизводные замещенного азепина как модуляторы рецептора серотонина
Автори російськоюБеннани Юссеф Л., Робердж Майкл Дж., Бом Девид К., Варга Норберт, Тами Лоуренс
МПК / Мітки
МПК: C07D 487/02, A61K 31/55
Мітки: азепіну, серотоніну, рецептора, модулятори, заміщеного, похідні
Код посилання
<a href="https://ua.patents.su/33-88786-pokhidni-zamishhenogo-azepinu-yak-modulyatori-receptora-serotoninu.html" target="_blank" rel="follow" title="База патентів України">Похідні заміщеного азепіну як модулятори рецептора серотоніну</a>
Діарильні і арилгетероарильні похідні сечовини як модулятори 5-нт2а-рецептора серотоніну, придатні для профілактики і лікування пов’язаних з ним захворювань

Номер патенту: 86774
Опубліковано: 25.05.2009
Автори: Страх-Плейнет Соня, Йаякумар Хоннаппа, Доса Петер Ян, Тігарден Бредлі, Лі Хунмей
МПК: C07D 231/16, A61K 31/415
Мітки: модулятори, придатні, похідні, 5-нт2а-рецептора, захворювань, діарильні, пов'язаних, арилгетероарильні, серотоніну, ним, профілактики, сечовини, лікування
Похідні піперидину як модулятори активності рецептора хемокіну

Номер патенту: 76148
Опубліковано: 17.07.2006
Автори: Еванз Річард, Перрі Мет'ю, Спрінґторп Брайан
МПК: C07D 417/14, C07D 401/14, C07D 211/06, A61P 31/16, A61P 37/00, C07D 409/14, A61P 11/06, C07D 401/04, A61K 31/445
Мітки: модулятори, похідні, активності, рецептора, хемокіну, піперидину
Формула / Реферат:
1. Сполука формули (I):, (I)де:T - C(О) або S(O)2;W - C(O) або S(O)2;X - CH2, O або NH;Y - CR5 або N;R1 - як варіант, заміщений арил або, як варіант, заміщений гетероцикліл;R2 - гідроген або C1-6алкіл;R3 - гідроген або, як варіант, заміщений C1-6алкіл;R4 - алкіл, як варіант, заміщений арил, як варіант,...
Похідні сульфонаміду як модулятори рецептора хемокіну

Номер патенту: 80283
Опубліковано: 10.09.2007
Автори: Бенніон Колін, Боннерт Роджер Віктор, Меґані Премджі, Кук Ентоні Роналд, Ебден Марк Річард
МПК: A61P 37/06, A61P 19/08, A61P 29/00, A61P 11/00, A61P 37/04, C07D 413/14, A61P 25/10, C07D 401/12, A61P 15/06, A61P 25/28, A61P 35/00, A61K 31/5377, A61P 15/08, A61P 21/04, A61P 19/10, C07D 409/12, A61P 17/06, A61P 25/06, A61P 37/08, A61P 17/02, A61P 11/06, C07D 403/12, A61P 43/00, A61P 1/04, C07D 405/12, A61P 17/08, A61P 3/10, A61P 35/04, A61P 17/00, C07D 239/48, A61K 31/506, A61P 5/14, A61P 9/10, A61P 37/02, C07D 417/12, C07D 239/69, A61P 13/08, A61P 31/04, A61P 13/12, A61P 27/02, A61P 19/02, A61P 11/02, A61K 31/505, A61P 17/04, A61P 15/00, A61P 7/04, A61P 25/14, A61P 17/14, A61P 25/00, A61P 31/18, A61P 1/12, C07D 413/12, A61P 21/00
Мітки: хемокіну, похідні, сульфонаміду, модулятори, рецептора
Формула / Реферат:
1. Сполука формули (1), її фармацевтично прийнятна сіль, сольват або здатний до гідролізу in vivo естер:, (1)де R1 вибрано з груп: С3-7карбоцикліл, С1-8алкіл, С2-6алкеніл та С2-6алкініл; де групу, як варіант, заміщено 1, 2 або 3 замісниками, незалежно вибраними з флуору, нітрилу, -OR4, -NR5R6, -CONR5R6, -COOR7, -NR8COR9, -SR10, -SО2R10, -SO2NR5R6, -NR8SO2R9, фенілу або гетероарилу; де феніл та гетероарил, як варіант, заміщено...
Похідні піперидину як модулятори рецептора ccr5 хемокіну, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 88002
Опубліковано: 10.09.2009
Автори: Фолл Алан, Такер Говард
МПК: C07D 401/14, A61K 31/4545, A61P 19/00, A61P 1/00, C07D 403/06, A61P 17/00, A61K 31/496, C07D 401/06, A61P 11/00, C07D 417/14
Мітки: модулятори, фармацевтична, основі, піперидину, похідні, одержання, спосіб, композиція, рецептора, хемокіну
Формула / Реферат:
1. Сполука формули (І):,де:А відсутня або є (СН2)2;L є СН або N;M є NR1, O, S, S(O) aбo S(O)2;R1 є гідрогеном, С1-6алкілом [як варіант, заміщеним фенілом {заміщеним галогеном, С1-4алкілом, С1-4алкокси, ціано, нітро, CF3, OCF3, (С1-4алкіл)С(О)NН, S(O)2NH2, С1-4алкілтіо, S(O)(C1-4алкіл) або S(O)2(C1-4алкіл)} або гетероарилом...
Фторовані похідні 4-азастероїду як модулятори рецептора андрогену

Номер патенту: 78029
Опубліковано: 15.02.2007
Автори: Мейсснер Роберт С., Перкінс Джеймс Дж.
МПК: A61K 31/40, A61K 31/22, A61P 7/06, A61K 31/675, A61K 31/663, A61P 19/00, A61P 21/00, A61K 38/23, A61P 15/10, A61P 29/00, A61K 31/4418, A61K 45/00, A61K 31/565, A61K 31/505, A61K 31/473, A61P 15/12, A61P 19/02, A61K 31/366, A61K 31/47, A61P 17/00, A61K 33/16, A61K 38/00, A61P 19/10, A61K 31/138, A61K 38/27, A61P 3/04, A61K 31/4535, A61K 31/404, A61P 3/06, A61P 1/02, A61P 9/10, A61K 31/661, A61P 13/08
Мітки: модулятори, фторовані, похідні, андрогену, рецептора, 4-азастероїду
Формула / Реферат:
1. Сполука структурної формули І: (I)або її фармацевтично прийнятна сіль, або її енантіомер; де n дорівнює 0, 1 або 2;a-b означає CF=CH, CHFCH2 або CF2CH2;R1 являє собою водень, гідроксиметил або С1-3алкіл, де алкіл є незаміщеним або заміщений одним-сімома атомами фтору;R2 являє собою водень;R3 вибраний з...
Попередній патент: Похідні n-гідроксіаміду та їх застосування
Наступний патент: Іграшка (варіанти) і музичний механізм для цієї іграшки
Випадковий патент: Горілка особлива "аристократ"