Комбінація нуклеозидного інгібітора полімерази та макроциклічного інгібітора протеази та її застосування для лікування гепатиту с, фіброзу печінки та порушеної функції печінки
Номер патенту: 104754
Опубліковано: 11.03.2014
Автори: Портер Стівен Б., Саймондс Вілльям Т., Сміт Патрік Ф., Йєтцер Еллен С., де ла Роса Абель, Роджерс Майкл Д., Бредфорд Вілльямсон Ціглер



















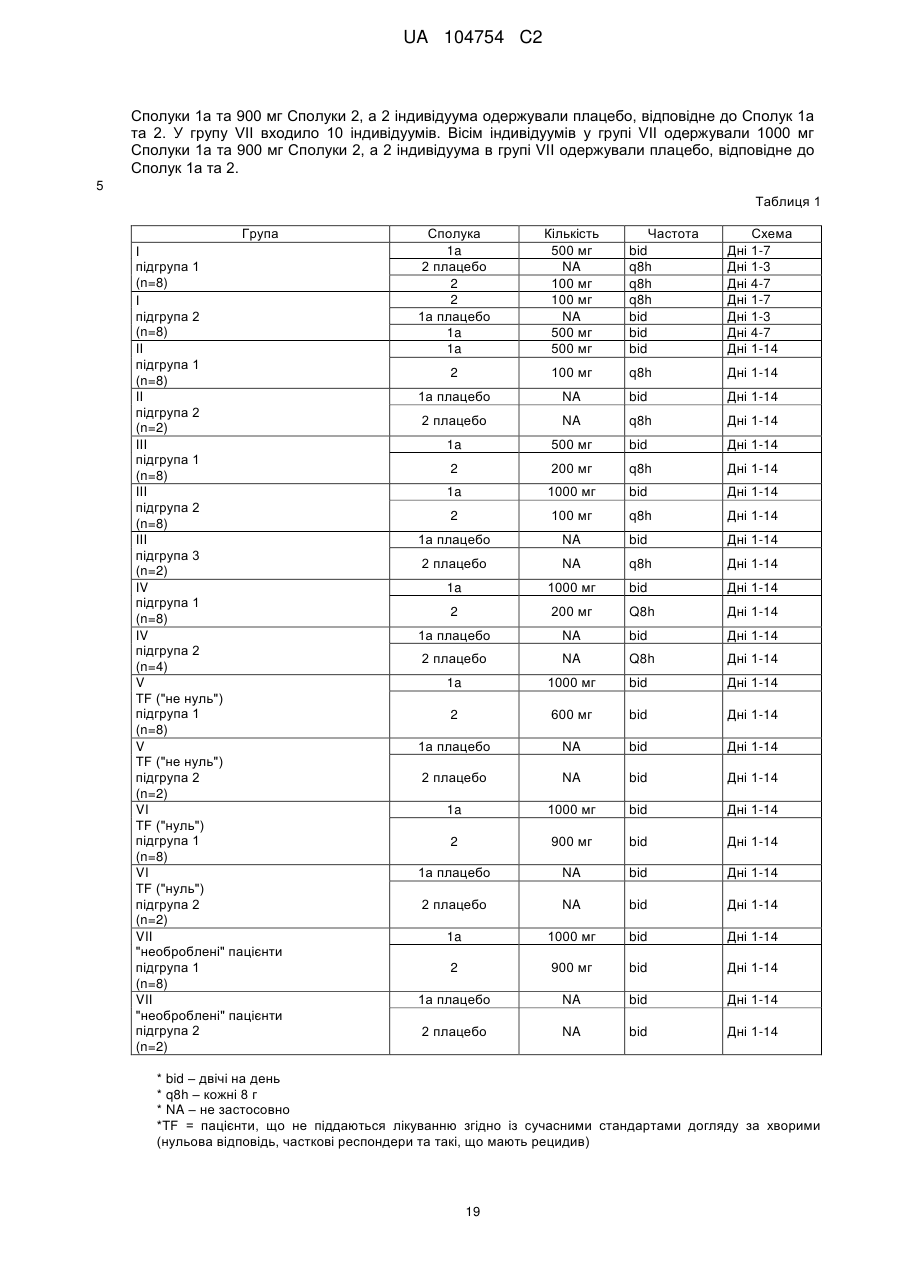


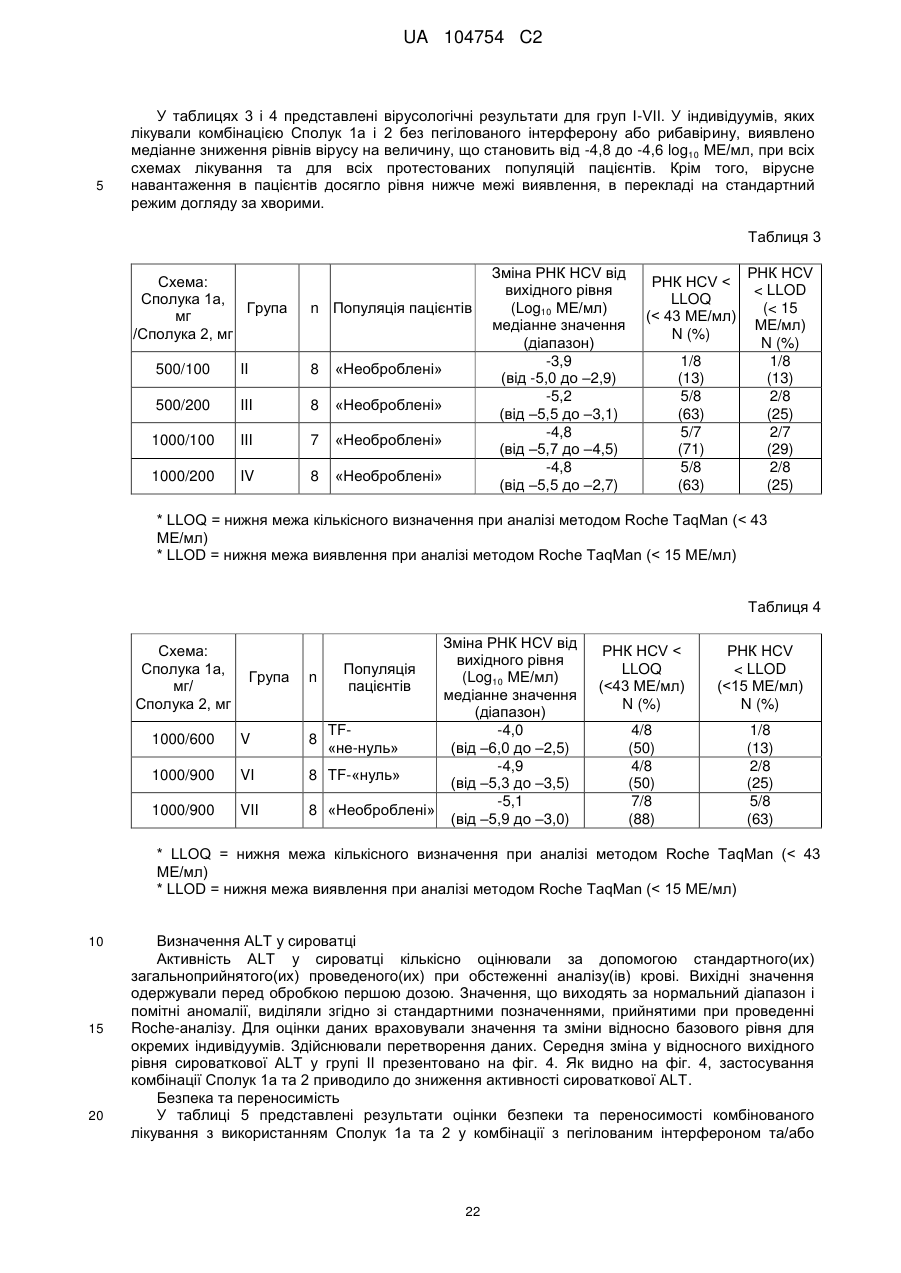
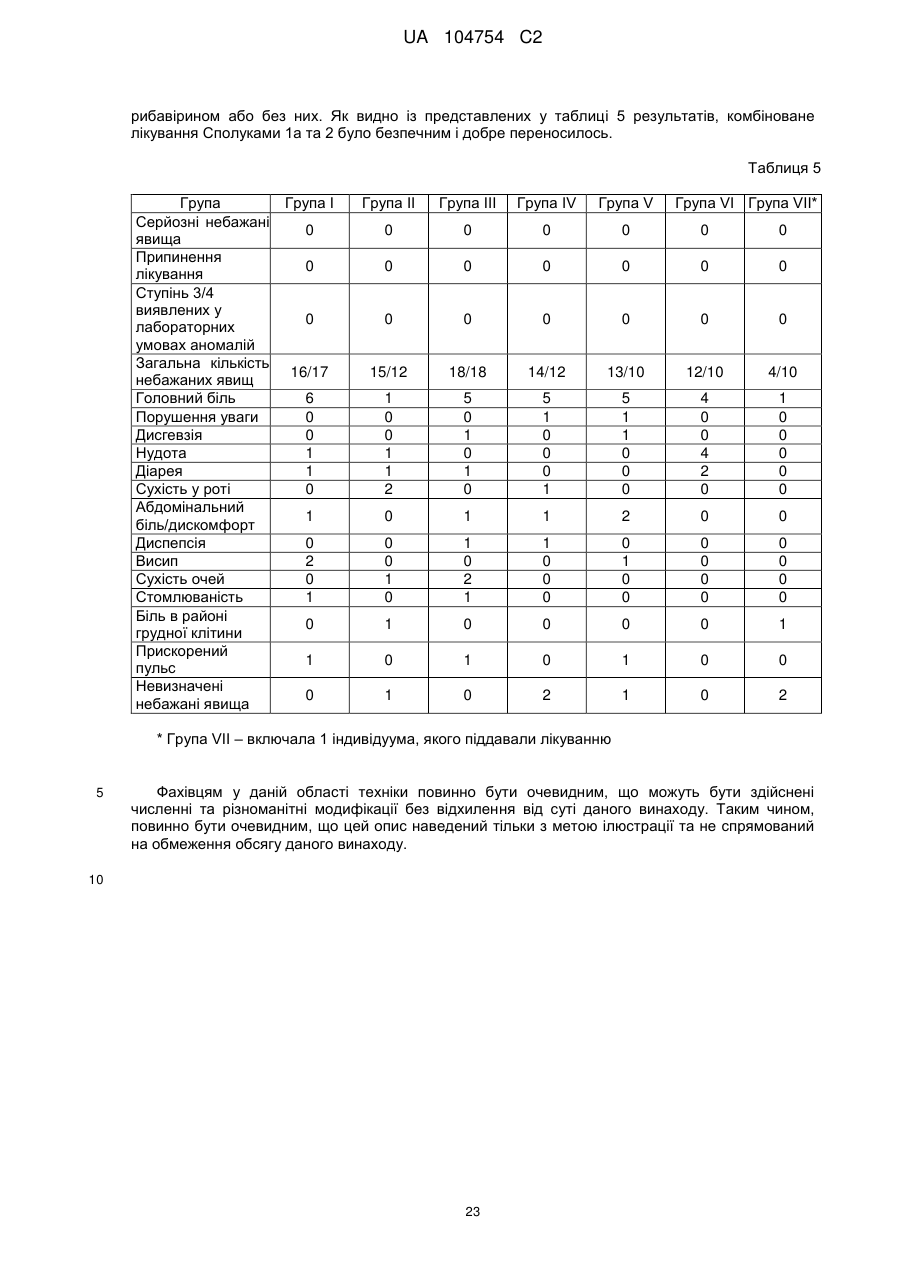



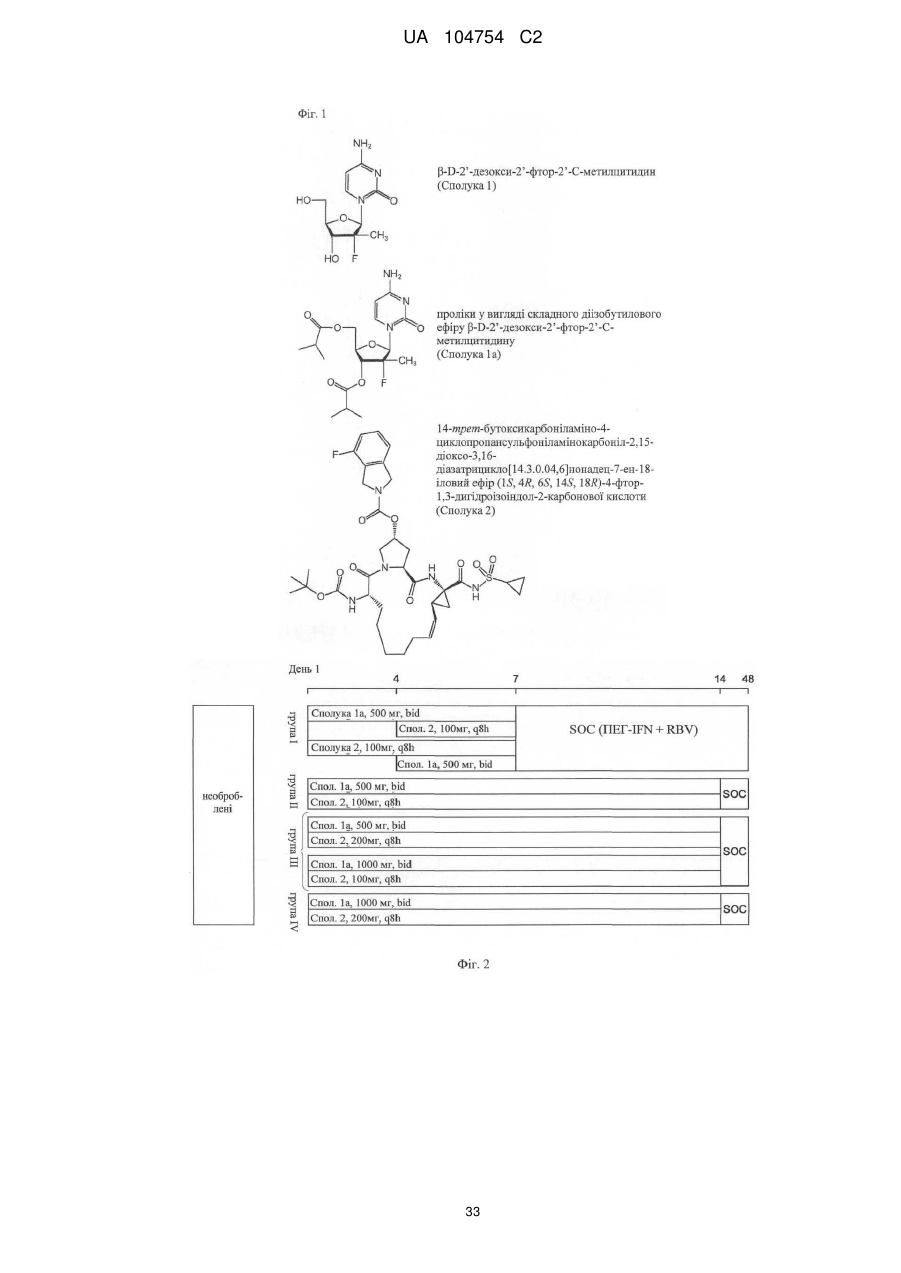

Формула / Реферат
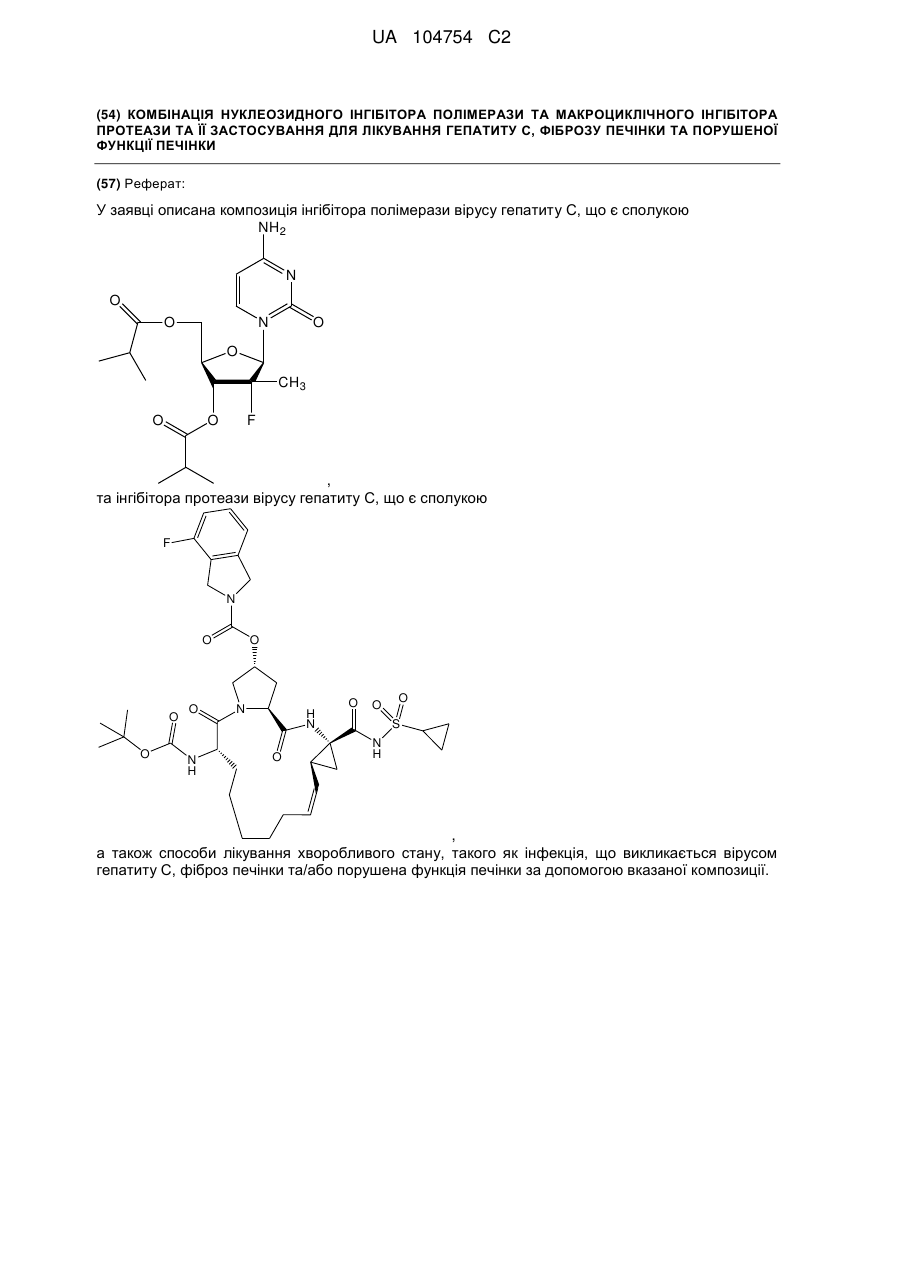
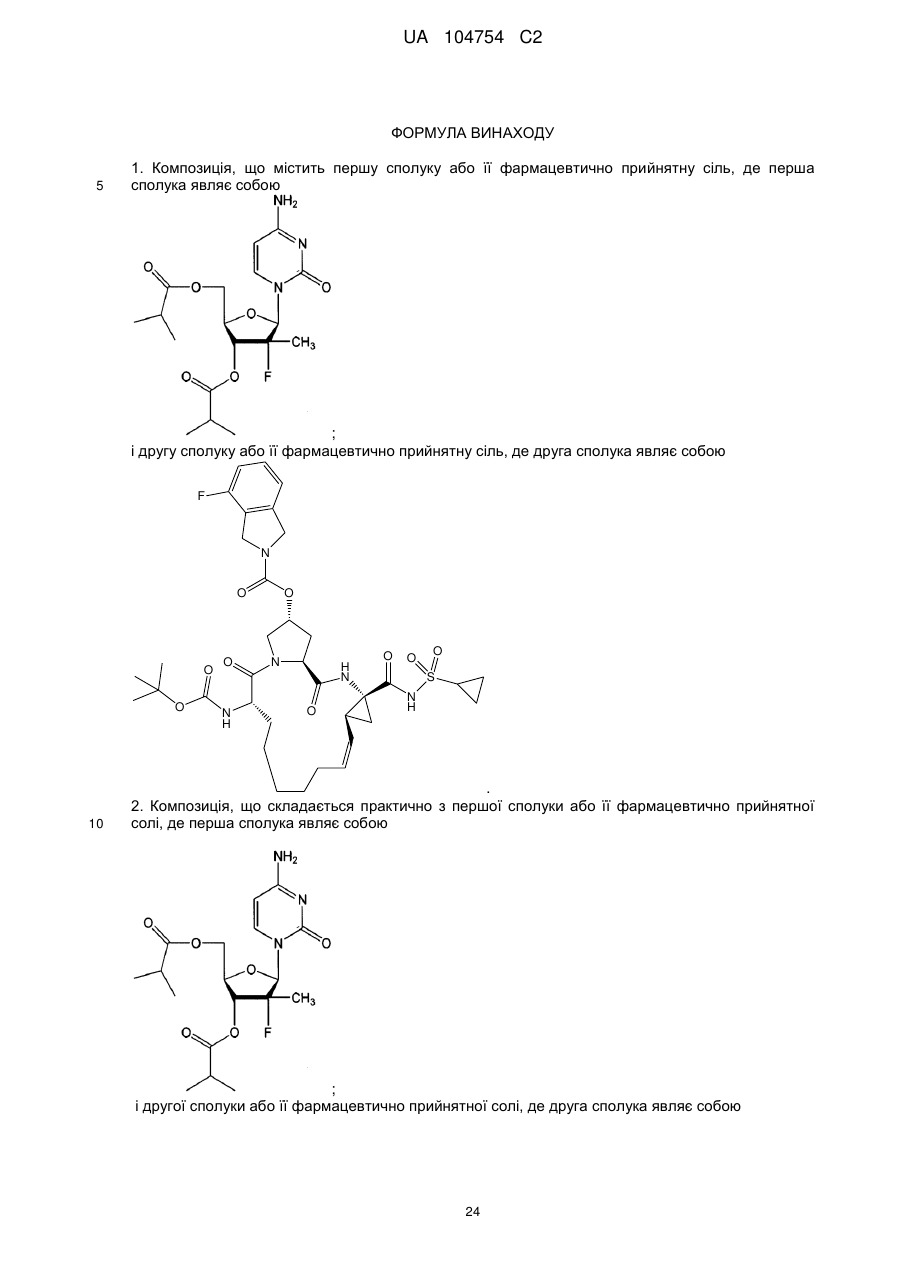
1. Композиція, що містить першу сполуку або її фармацевтично прийнятну сіль, де перша сполука являє собою
![]()
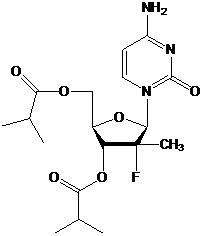 ;
;
і другу сполуку або її фармацевтично прийнятну сіль, де друга сполука являє собою
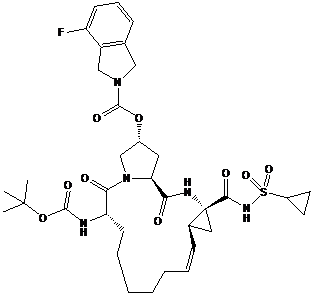 .
.
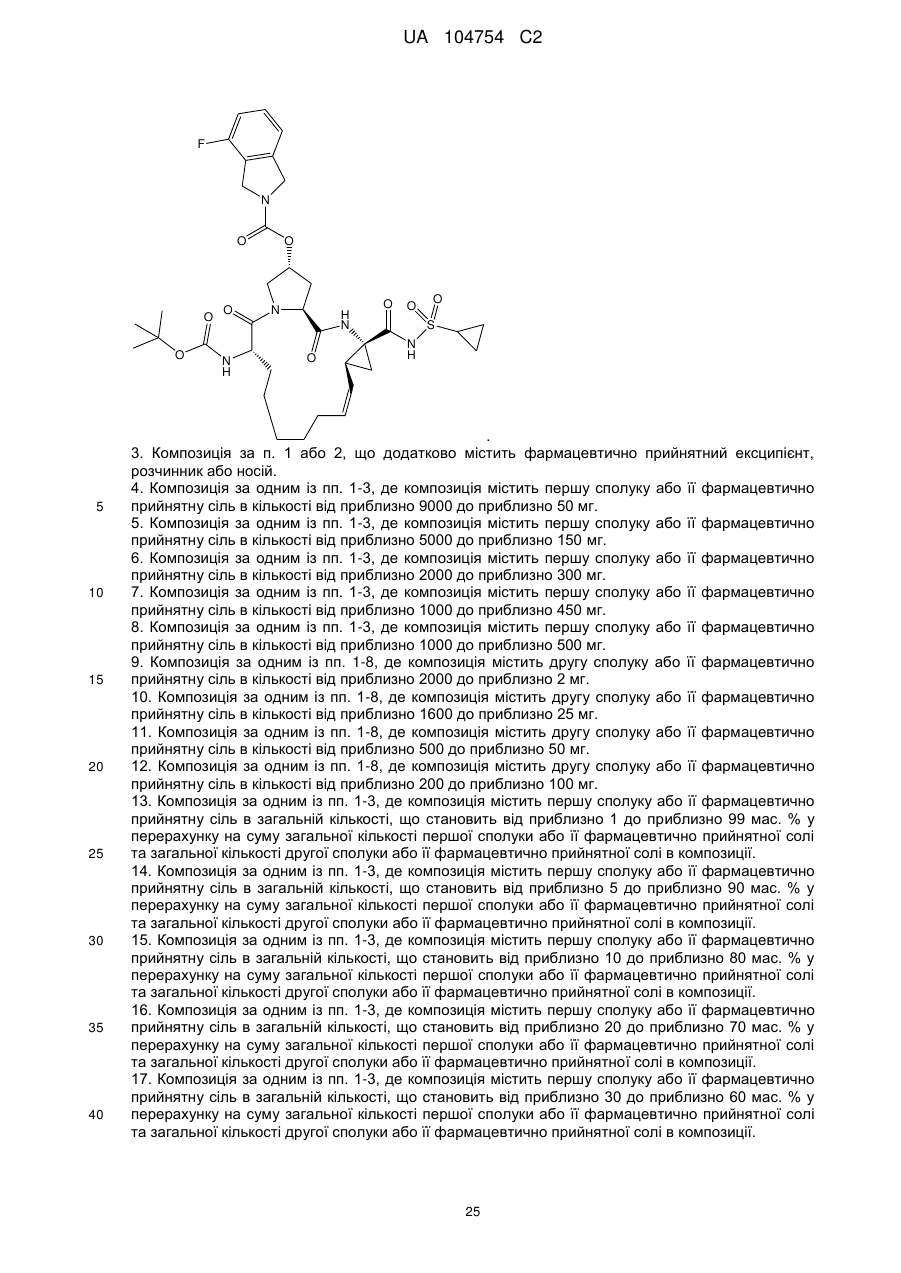
2. Композиція, що складається практично з першої сполуки або її фармацевтично прийнятної солі, де перша сполука являє собою
![]() ;
;
і другої сполуки або її фармацевтично прийнятної солі, де друга сполука являє собою
.
3. Композиція за п. 1 або 2, що додатково містить фармацевтично прийнятний ексципієнт, розчинник або носій.
4. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 9000 до приблизно 50 мг.
5. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 5000 до приблизно 150 мг.
6. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 2000 до приблизно 300 мг.
7. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 1000 до приблизно 450 мг.
8. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 1000 до приблизно 500 мг.
9. Композиція за одним із пп. 1-8, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 2000 до приблизно 2 мг.
10. Композиція за одним із пп. 1-8, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 1600 до приблизно 25 мг.
11. Композиція за одним із пп. 1-8, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 500 до приблизно 50 мг.
12. Композиція за одним із пп. 1-8, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в кількості від приблизно 200 до приблизно 100 мг.
13. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 1 до приблизно 99 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
14. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 5 до приблизно 90 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
15. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 10 до приблизно 80 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
16. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 20 до приблизно 70 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
17. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 30 до приблизно 60 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
18. Композиція за одним із пп. 1-3, де композиція містить першу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 40 до приблизно 50 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
19. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 1 до приблизно 99 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
20. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 5 до приблизно 90 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
21. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 10 до приблизно 80 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
22. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 20 до приблизно 70 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
23. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 30 до приблизно 60 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
24. Композиція за одним із пп. 13-18, де композиція містить другу сполуку або її фармацевтично прийнятну сіль в загальній кількості, що становить від приблизно 40 до приблизно 50 мас. % у перерахунку на суму загальної кількості першої сполуки або її фармацевтично прийнятної солі та загальної кількості другої сполуки або її фармацевтично прийнятної солі в композиції.
25. Композиція за одним із пп. 1-24, де кількість першої сполуки або її фармацевтично прийнятної солі в композиції є меншою кількості першої сполуки або її фармацевтично прийнятної солі, необхідної для досягнення практично такого ж зниження вірусного навантаження у випадку, коли першу сполуку або її фармацевтично прийнятну сіль застосовують як монотерапію.
26. Композиція за одним із пп. 1-25, де кількість другої сполуки або її фармацевтично прийнятної солі в композиції є меншою кількості другої сполуки або її фармацевтично прийнятної солі, необхідної для досягнення практично такого ж зниження вірусного навантаження у випадку, коли другу сполуку або її фармацевтично прийнятну сіль застосовують як монотерапію.
27. Композиція за одним із пп. 1-26, де композиція містить також додаткові терапевтичні засоби.
28. Композиція за п. 27, у якій один або декілька додаткових терапевтичних засобів вибрані із групи, що включає пірфенідон, інгібітор РНК-залежної РНК-полімерази NS5B, антагоніст фактора некрозу пухлини, тимозин-a, інтерферон-гамма (IFN-g), інтерферон-альфа (IFN-a), нуклеозидний аналог, що являє собою 3'-азидотимідин, 2',3'-дидезоксиінозин, 2',3'-дидезоксицитидин, 2,3-дидегідро-2',3'-дидезокситимідин, комбівір, абакавір, адефовіру дипівоксил, цидофовір, рибавірин, левовірин, вірамідин, L-нуклеозид або ізаторибин, ритонавір, інгібітор інозинмонофосфатдегідрогенази, інтерферон, додатковий інгібітор NS3-протеази, інгібітор NS5B-полімерази та інгібітор NS3-гелікази.
29. Композиція за п. 28, у якій нуклеозидний аналог вибраний із групи, що включає рибавірин, левовірин, вірамідин, L-нуклеозид та ізаторибин.
30. Композиція за п. 28, у якій антагоніст фактора некрозу пухлини вибраний із групи, що включає етанерцепт, іфліксимаб та адалімумаб.
31. Композиція за п. 28, у якій тимозин-a присутній у кількості, що становить від приблизно 1,0 до приблизно 1,6 мг.
32. Композиція за п. 28, у якій IFN-g присутній у кількості, що становить від приблизно 10 до приблизно 300 мкг.
33. Композиція за п. 28, у якій IFN-a являє собою монопегильований (30 кДа, лінійний) консенсусний IFN-a.
34. Композиція за п. 28, у якій IFN-a вибраний із групи, що включає кон'югат, що має молекулярну масу 40 кДа розгалуженого монометокси-ПЕГ та інтерферону a-2b та кон'югат, що має молекулярну масу 12 кДа монометокси-ПЕГ та інтерферону a-2b.
35. Композиція за п. 28, у якій IFN-a являє собою консенсусний IFN-α, INFERGEN.
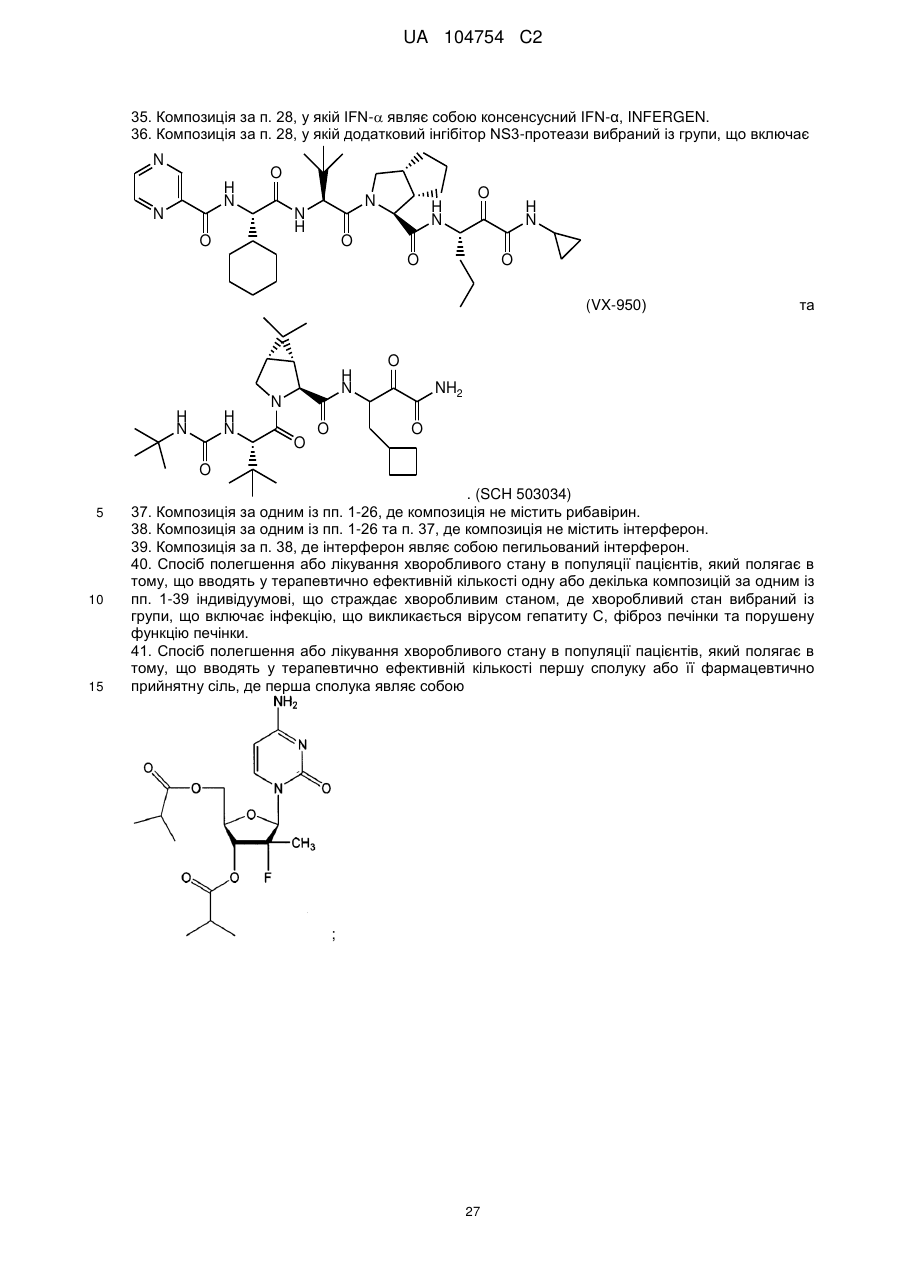
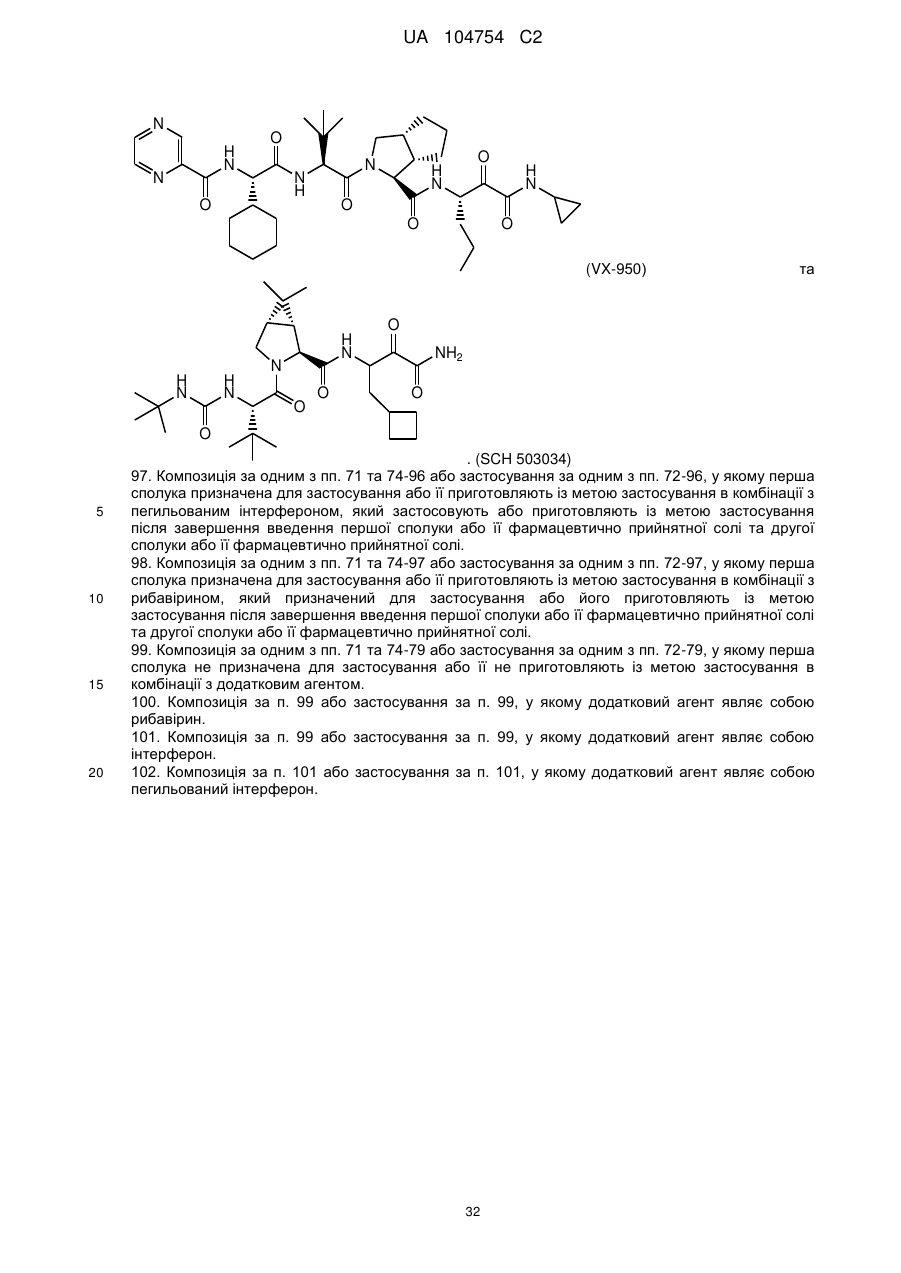
36. Композиція за п. 28, у якій додатковий інгібітор NS3-протеази вибраний із групи, що включає
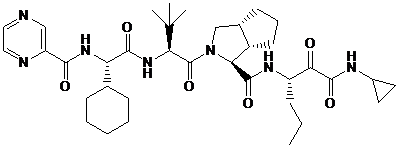 (VX-950) та
(VX-950) та 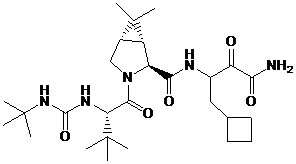 . (SCH 503034)
. (SCH 503034)
37. Композиція за одним із пп. 1-26, де композиція не містить рибавірин.
38. Композиція за одним із пп. 1-26 та п. 37, де композиція не містить інтерферон.
39. Композиція за п. 38, де інтерферон являє собою пегильований інтерферон.
40. Спосіб полегшення або лікування хворобливого стану в популяції пацієнтів, який полягає в тому, що вводять у терапевтично ефективній кількості одну або декілька композицій за одним із пп. 1-39 індивідуумові, що страждає хворобливим станом, де хворобливий стан вибраний із групи, що включає інфекцію, що викликається вірусом гепатиту C, фіброз печінки та порушену функцію печінки.
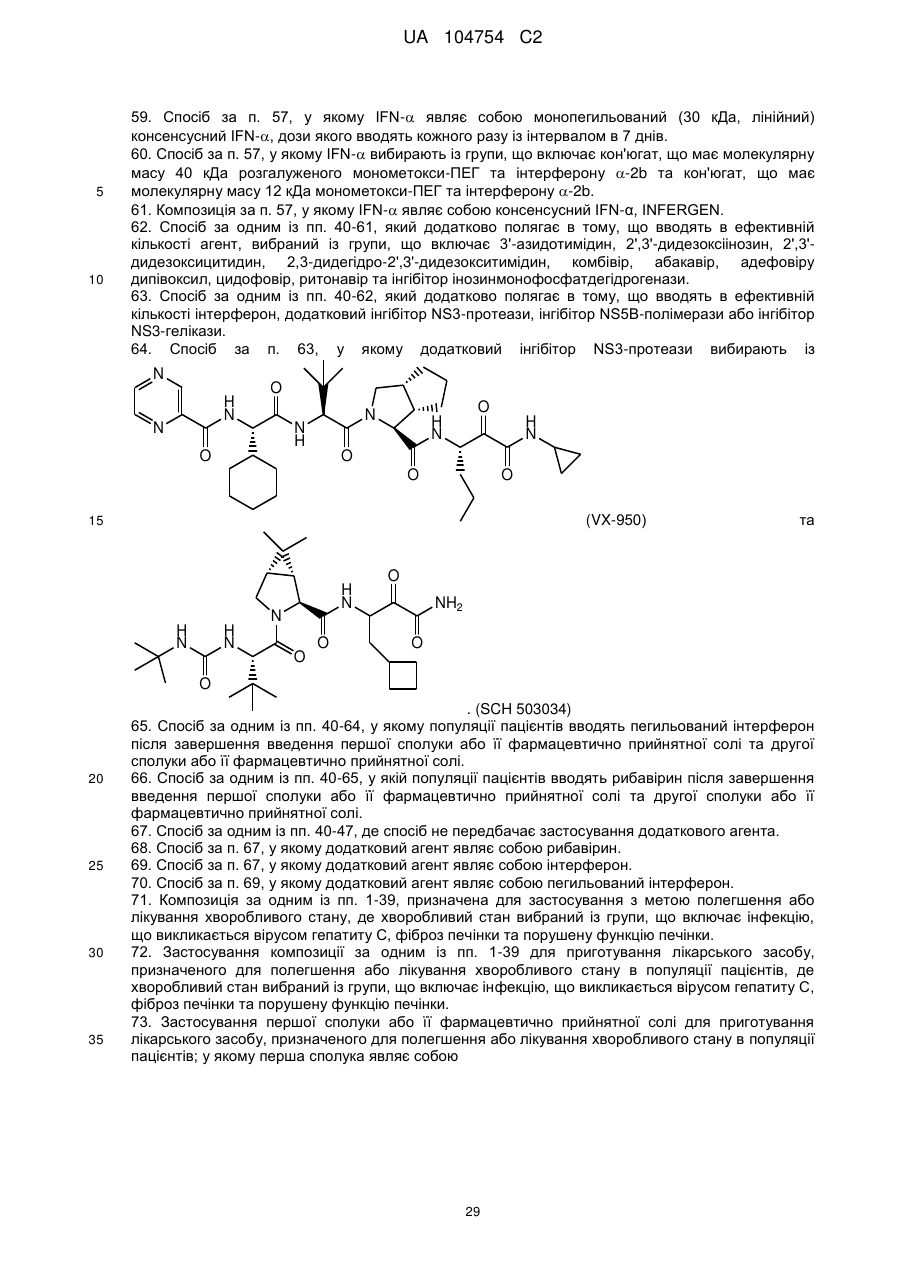
41. Спосіб полегшення або лікування хворобливого стану в популяції пацієнтів, який полягає в тому, що вводять у терапевтично ефективній кількості першу сполуку або її фармацевтично прийнятну сіль, де перша сполука являє собою
![]() ;
;
та в терапевтично ефективній кількості другу сполуку або її фармацевтично прийнятну сіль, де друга сполука являє собою ,
індивідуумові, що страждає хворобливим станом, де хворобливий стан вибраний із групи, що включає інфекцію, що викликається вірусом гепатиту C, фіброз печінки та порушену функцію печінки.
42. Спосіб за п. 41, у якому першу сполуку або її фармацевтично прийнятну сіль вводять до введення другої сполуки або її фармацевтично прийнятної солі.
43. Спосіб за п. 41, у якому першу сполуку або її фармацевтично прийнятну сіль вводять після введення другої сполуки або її фармацевтично прийнятної солі.
44. Спосіб за п. 41, у якому першу сполуку або її фармацевтично прийнятну сіль вводять практично одночасно із введенням другої сполуки або її фармацевтично прийнятної солі.
45. Спосіб за п. 41, у якому перша сполука або її фармацевтично прийнятна сіль та друга сполука або її фармацевтично прийнятна сіль разом входять до складу однієї лікарської форми.
46. Спосіб за одним із пп. 41-44, у якому перша сполука або її фармацевтично прийнятна сіль та друга сполука або її фармацевтично прийнятна сіль входятьдо складу різних лікарських форм.
47. Спосіб за одним із пп. 40-46, у якому хворобливий стан являє собою інфекцію, що викликається вірусом гепатиту C.
48. Спосіб за одним із пп. 40-47, який додатково полягає в тому, що вводять в ефективній кількості додатковий нуклеозидний аналог.
49. Спосіб за п. 48, у якому додатковий нуклеозидний аналог вибирають із групи, що включає рибавірин, левовірин, вірамідин, L-нуклеозид та ізаторибин.
50. Спосіб за одним із пп. 40-49, який додатково полягає в тому, що вводять в ефективній кількості пірфенідон.
51. Спосіб за одним із пп. 40-50, який додатково полягає в тому, що вводять в ефективній кількості інгібітор РНК-залежної РНК-полімерази NS5B.
52. Спосіб за одним із пп. 40-51, який полягає також у тому, що вводять в ефективній кількості антагоніст фактора некрозу пухлини, вибраний із групи, що включає етанерцепт, іфліксимаб та адалімумаб.
53. Спосіб за одним із пп. 40-52, який додатково полягає в тому, що вводять в ефективній кількості тимозин-a.
54. Спосіб за п. 53, у якому тимозин-a застосовують у кількості, що становить від приблизно 1,0 до приблизно 1,6 мг.
55. Спосіб за одним із пп. 40-54, який додатково полягає в тому, що вводять в ефективній кількості інтерферон-гамма (IFN-g).
56. Спосіб за п. 55, у якому IFN-g вводять підшкірно в кількості, що становить від приблизно 10 до приблизно 300 мкг.
57. Спосіб за одним із пп. 40-56, який додатково полягає в тому, що вводять в ефективній кількості інтерферон-альфа (IFN-a).
58. Спосіб за п. 57, у якому IFN-a являє собою монопегильований (30 кДа, лінійний) консенсусний IFN-a, для якого інтервал між введеннями доз становить кожного разу від 8 днів до 14 днів.
59. Спосіб за п. 57, у якому IFN-a являє собою монопегильований (30 кДа, лінійний) консенсусний IFN-a, дози якого вводять кожного разу із інтервалом в 7 днів.
60. Спосіб за п. 57, у якому IFN-a вибирають із групи, що включає кон'югат, що має молекулярну масу 40 кДа розгалуженого монометокси-ПЕГ та інтерферону a-2b та кон'югат, що має молекулярну масу 12 кДа монометокси-ПЕГ та інтерферону a-2b.
61. Композиція за п. 57, у якому IFN-a являє собою консенсусний IFN-α, INFERGEN.
62. Спосіб за одним із пп. 40-61, який додатково полягає в тому, що вводять в ефективній кількості агент, вибраний із групи, що включає 3'-азидотимідин, 2',3'-дидезоксиінозин, 2',3'-дидезоксицитидин, 2,3-дидегідро-2',3'-дидезокситимідин, комбівір, абакавір, адефовіру дипівоксил, цидофовір, ритонавір та інгібітор інозинмонофосфатдегідрогенази.
63. Спосіб за одним із пп. 40-62, який додатково полягає в тому, що вводять в ефективній кількості інтерферон, додатковий інгібітор NS3-протеази, інгібітор NS5B-полімерази або інгібітор NS3-гелікази.
64. Спосіб за п. 63, у якому додатковий інгібітор NS3-протеази вибирають із (VX-950) та . (SCH 503034)
65. Спосіб за одним із пп. 40-64, у якому популяції пацієнтів вводять пегильований інтерферон після завершення введення першої сполуки або її фармацевтично прийнятної солі та другої сполуки або її фармацевтично прийнятної солі.
66. Спосіб за одним із пп. 40-65, у якій популяції пацієнтів вводять рибавірин після завершення введення першої сполуки або її фармацевтично прийнятної солі та другої сполуки або її фармацевтично прийнятної солі.
67. Спосіб за одним із пп. 40-47, де спосіб не передбачає застосування додаткового агента.
68. Спосіб за п. 67, у якому додатковий агент являє собою рибавірин.
69. Спосіб за п. 67, у якому додатковий агент являє собою інтерферон.
70. Спосіб за п. 69, у якому додатковий агент являє собою пегильований інтерферон.
71. Композиція за одним із пп. 1-39, призначена для застосування з метою полегшення або лікування хворобливого стану, де хворобливий стан вибраний із групи, що включає інфекцію, що викликається вірусом гепатиту C, фіброз печінки та порушену функцію печінки.
72. Застосування композиції за одним із пп. 1-39 для приготування лікарського засобу, призначеного для полегшення або лікування хворобливого стану в популяції пацієнтів, де хворобливий стан вибраний із групи, що включає інфекцію, що викликається вірусом гепатиту C, фіброз печінки та порушену функцію печінки.
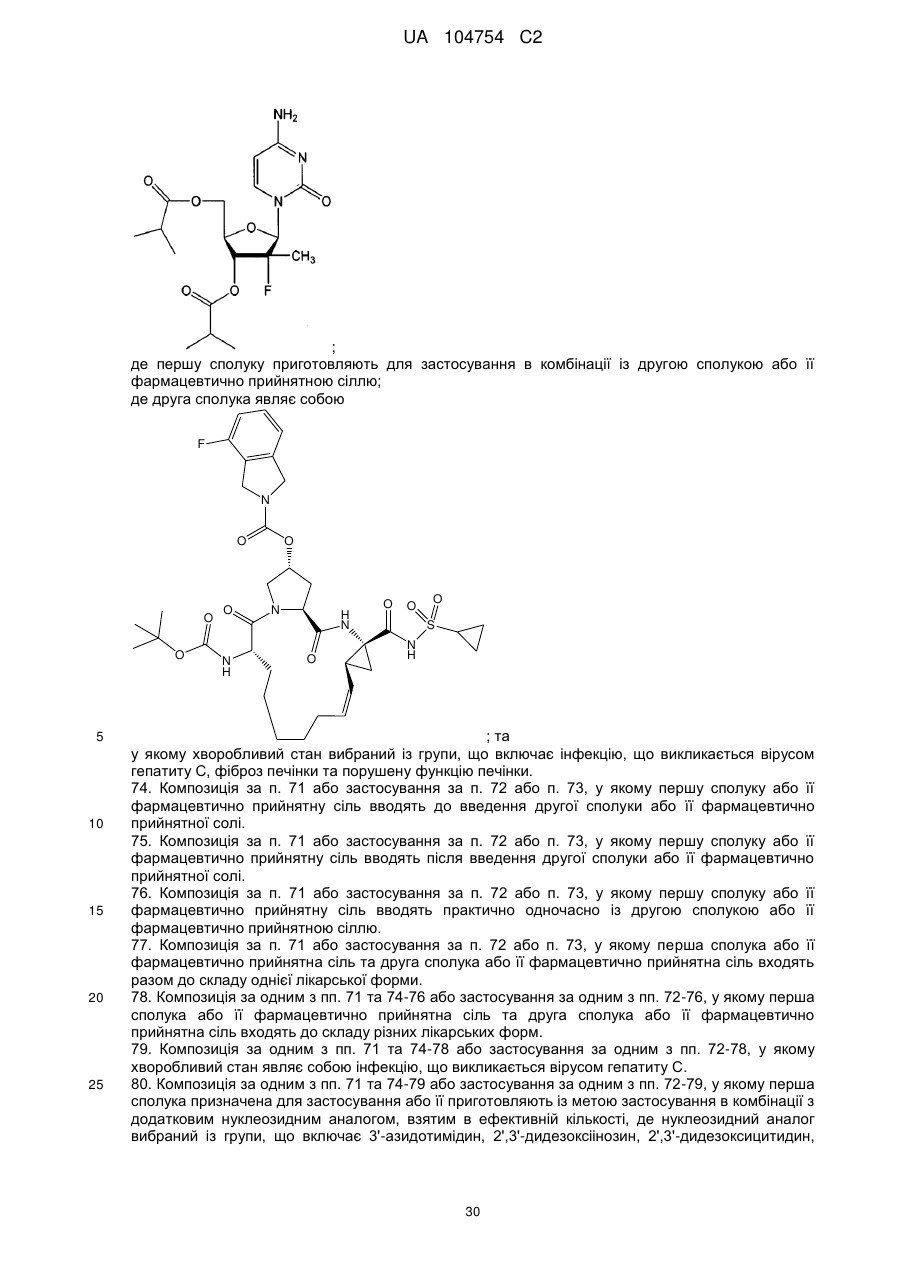
73. Застосування першої сполуки або її фармацевтично прийнятної солі для приготування лікарського засобу, призначеного для полегшення або лікування хворобливого стану в популяції пацієнтів; у якому перша сполука являє собою
![]() ;
;
де першу сполуку приготовляють для застосування в комбінації із другою сполукою або її фармацевтично прийнятною сіллю;
де друга сполука являє собою
; та
у якому хворобливий стан вибраний із групи, що включає інфекцію, що викликається вірусом гепатиту C, фіброз печінки та порушену функцію печінки.
74. Композиція за п. 71 або застосування за п. 72 або п. 73, у якому першу сполуку або її фармацевтично прийнятну сіль вводять до введення другої сполуки або її фармацевтично прийнятної солі.
75. Композиція за п. 71 або застосування за п. 72 або п. 73, у якому першу сполуку або її фармацевтично прийнятну сіль вводять після введення другої сполуки або її фармацевтично прийнятної солі.
76. Композиція за п. 71 або застосування за п. 72 або п. 73, у якому першу сполуку або її фармацевтично прийнятну сіль вводять практично одночасно із другою сполукою або її фармацевтично прийнятною сіллю .
77. Композиція за п. 71 або застосування за п. 72 або п. 73, у якому перша сполука або її фармацевтично прийнятна сіль та друга сполука або її фармацевтично прийнятна сіль входять разом до складу однієї лікарської форми.
78. Композиція за одним з пп. 71 та 74-76 або застосування за одним з пп. 72-76, у якому перша сполука або її фармацевтично прийнятна сіль та друга сполука або її фармацевтично прийнятна сіль входять до складу різних лікарських форм.
79. Композиція за одним з пп. 71 та 74-78 або застосування за одним з пп. 72-78, у якому хворобливий стан являє собою інфекцію, що викликається вірусом гепатиту C.
80. Композиція за одним з пп. 71 та 74-79 або застосування за одним з пп. 72-79, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з додатковим нуклеозидним аналогом, взятому в ефективній кількості, де нуклеозидний аналог вибраний із групи, що включає 3'-азидотимідин, 2',3'-дидезоксиінозин, 2',3'-дидезоксицитидин, 2,3-дидегідро-2',3'-дидезокситимідин, комбівір, абакавір, адефовіру дипівоксил, цидофовір, рибавірин, левовірин, вірамідин, L-нуклеозид та ізаторибин.
81. Композиція за п. 80 або застосування за п. 80, у якому додатковий нуклеозидний аналог вибраний із групи, що включає рибавірин, левовірин, вірамідин, L-нуклеозид та ізаторибин.
82. Композиція за одним з пп. 71 та 74-81 або застосування за одним з пп. 72-81, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з пірфенідоном, взятим в ефективній кількості.
83. Композиція за одним з пп. 71 та 74-82 або застосування за одним з пп. 72-82, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з інгібітором РНК-залежної РНК-полімерази NS5B, взятим в ефективній кількості.
84. Композиція за одним з пп. 71 та 74-83 або застосування за одним з пп. 72-83, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з антагоністом фактора некрозу пухлини, вибраним із групи, що включає етанерцепт, іфліксимаб та адалімумаб.
85. Композиція за одним з пп. 71 та 74-84 або застосування за одним з пп. 72-84, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з тимозином-a, взятому в ефективній кількості.
86. Композиція за п. 85 або застосування за п. 85, у якому тимозин-a призначений для застосування або його приготовляють із метою застосування у кількості, що становить від приблизно 1,0 до приблизно 1,6 мг.
87. Композиція за одним з пп. 71 та 74-86 або застосування за одним з пп. 72-86, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з інтерфероном-гамма (IFN-g), взятим в ефективній кількості.
88. Композиція за п. 87 або застосування за п. 87, у якому IFN-g призначений для застосування або його приготовляють із метою застосування шляхом підшкірного введення в кількості, що становить від приблизно 10 до приблизно 300 мкг.
89. Композиція за одним з пп. 71 та 74-86 або застосування за одним з пп. 72-86, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з інтерфероном-альфа (IFN-a), взятим в ефективній кількості.
90. Композиція за п. 89 або застосування за п. 89, у якому IFN-a являє собою монопегильований (30 кДа, лінійний) консенсусний IFN-a, призначений для застосування або його приготовляють із метою застосування шляхом введення доз із інтервалом, що становить кожного разу від 8 днів до 14 днів.
91. Композиція за п. 89 або застосування за п. 89, у якому IFN-a являє собою монопегильований (30 кДа, лінійний) консенсусний IFN-a, призначений для застосування або приготовлений з метою застосування шляхом введення доз із інтервалом, що становить кожного разу 7 днів.
92. Композиція за п. 89 або застосування за п. 89, у якому IFN-a вибраний із групи, що включає кон'югат, що має молекулярну масу 40 кДа розгалуженого монометокси-ПЕГ та інтерферону a-2b та кон'югат, що має молекулярну масу 12 кДа монометокси-ПЕГ та інтерферону a-2b.
93. Композиція за п. 89 або застосування за п. 89, у якому IFN-a являє собою консенсусний IFN-α, INFERGEN.
94. Композиція за одним з пп. 71 та 74-93 або застосування за одним з пп. 72-93, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з агентом, вибраним із групи, що включає 3'-азидотимідин, 2',3'-дидезоксиінозин, 2',3'-дидезоксицитидин, 2-,3-дидегідро-2',3'-дидезокситимідин, комбівір, абакавір, адефовіру дипівоксил, цидофовір, ритонавір та інгібітор інозинмонофосфатдегідрогенази, взятим в ефективній кількості.
95. Композиція за одним з пп. 71 та 74-94 або застосування за одним з пп. 72-94, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з інтерфероном, додатковим інгібітором NS3-протеази, інгібітором NS5B-полімерази або інгібітором NS3-гелікази, взятим в ефективній кількості.
96. Композиція за п. 95 або застосування за п. 95, у якому додатковий інгібітор NS3-протеази вибраний з
(VX-950) та . (SCH 503034)
97. Композиція за одним з пп. 71 та 74-96 або застосування за одним з пп. 72-96, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з пегильованим інтерфероном, який застосовують або приготовляють із метою застосування після завершення введення першої сполуки або її фармацевтично прийнятної солі та другої сполуки або її фармацевтично прийнятної солі.
98. Композиція за одним з пп. 71 та 74-97 або застосування за одним з пп. 72-97, у якому перша сполука призначена для застосування або її приготовляють із метою застосування в комбінації з рибавірином, який призначений для застосування або його приготовляють із метою застосування після завершення введення першої сполуки або її фармацевтично прийнятної солі та другої сполуки або її фармацевтично прийнятної солі.
99. Композиція за одним з пп. 71 та 74-79 або застосування за одним з пп. 72-79, у якому перша сполука не призначена для застосування або її не приготовляють із метою застосування в комбінації з додатковим агентом.
100. Композиція за п. 99 або застосування за п. 99, у якому додатковий агент являє собою рибавірин.
101. Композиція за п. 99 або застосування за п. 99, у якому додатковий агент являє собою інтерферон.
102. Композиція за п. 101 або застосування за п. 101, у якому додатковий агент являє собою пегильований інтерферон.
Текст
Реферат: У заявці описана композиція інгібітора полімерази вірусу гепатиту С, що є сполукою NH2 N O O N O O CH3 O O F , та інгібітора протеази вірусу гепатиту С, що є сполукою F N O O O O N H O N H N O O O O S N H , а також способи лікування хворобливого стану, такого як інфекція, що викликається вірусом гепатиту С, фіброз печінки та/або порушена функція печінки за допомогою вказаної композиції. UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 Область техніки, до якої відноситься винахід Даний винахід відноситься до композицій та способів лікування хворобливого стану, такого як інфекція, що викликається вірусом гепатиту C, фіброз печінки та порушена функція печінки. Опис винаходу Інфекція, що викликається вірусом гепатиту C (HCV), представляє собою найбільше широко розповсюджену в Сполучених Штатах хронічну, пов'язану із присутністю вірусу в крові, інфекцію. Хоча кількість нових випадків зараження знизилась, рівень хронічної інфекції є значним, за даними Центрів за контролем захворюваності в Сполучених Штатах налічується 3,9 мільйона (1,8 %) заражених людей. Хронічне захворювання печінки перебуває на десятому місці серед основних причин смерті дорослих людей у Сполучених Штатах, воно щорічно приводить до смерті приблизно 25000 людей, що відповідає приблизно 1 % від усіх випадків смерті. Дослідження показали, що 40 % випадків хронічного захворювання печінки пов'язане з HCV, що викликає 8000-10000 випадків смерті щороку. Асоційована з HCV кінцева стадія хвороби печінки є найбільш частим показанням для трансплантації печінки серед дорослих. Противірусна терапія хронічного гепатиту C швидко розвивалась протягом останнього десятиліття, що дозволило суттєво поліпшити ефективність лікування. Проте, навіть при використанні відповідної до стандарту догляду за хворими (SOC) комбінованої терапії на основі пегілованого IFN- плюс рибавірину (RBV), від 40 % до 50 % пацієнтів не піддаються терапії, тобто вони відносяться до нереспондерів або мають рецидив. Для цих пацієнтів на теперішній час відсутня ефективна альтернативна терапія. Зокрема, пацієнти із прогресуючим фіброзом або цирозом за даними біопсії печінки мають високий ризик розвитку ускладнень, пов'язаних із прогресуючим захворюванням печінки, включаючи асцити, жовтяницю, кровотечу з варикозно розширених вен, енцефалопатію та прогресуючу печінкову недостатність, а також у значній мірі підвищений ризик виникнення гепатоклітинної карциноми. Широке поширення хронічної HCV-інфекції обумовлює її важливе значення для суспільної охорони здоров'я, оскільки воно обумовлює в майбутньому високий рівень хронічних хвороб печінки в Сполучених Штатах. Дані третього національного дослідження (стану) здоров'я та харчування (NHANES III) свідчать про значне зростання рівня нових HCV-інфекцій, який спостерігався з кінця 1960-х до початку 1980-х років, насамперед у людей віком від 20 до 40 років. Оцінки показують, що кількість людей із тривалою HCV-інфекцією на протязі 20 або більш років може зрости більш ніж в 4 рази протягом періоду часу з 1990 р. по 2015 р., а саме, з 750000 до більш ніж 3 мільйонів. Пропорційне збільшення кількості інфікованих людей у віці 30 або 40 років може бути ще більш значним. Оскільки ризик асоційованого з HCV хронічного захворювання печінки пов'язаний із тривалістю інфекції, причому ризик розвитку цирозу прогресивно зростає в людей, інфікованих протягом більше 20 років, то найбільша імовірність значного підвищення пов'язаної із цирозом захворюваності та смертності існує для пацієнтів, заражених у період 1965-1985 років. HCV представляє собою вірус сімейства Flaviviridae, що має оболонку, геном якого представлений одноланцюговою РНК позитивної полярності. Одноланцюгова геномна РНК HCV складається приблизно з 9500 нуклеотидів і має одну відкриту рамку зчитування (ОРС), що кодує один великий поліпротеїн, який складається приблизно з 3000 амінокислот. У заражених клітинах цей поліпротеїн розщеплюється в декількох сайтах клітинними та вірусними протеазами з утворенням структурних і неструктурних (NS) білків вірусу (NS2, NS3, NS4, NS4A, NS4B, NS5A і NS5B). Короткий виклад суті винаходу Деякі варіанти здійснення винаходу, представлені в даному описі, відносяться до композиції, яка може включати першу сполуку або її фармацевтично прийнятну сіль або проліки, де перша сполука представляє собою β-D-2’-дезокси-2’-фтор-2’-C-метилцитидин (Сполука 1); і другу сполуку або її фармацевтично прийнятну сіль або проліки, де друга сполука представляє собою 14-трет-бутоксикарбоніламіно-4-циклопропансульфоніламінокарбоніл-2,15-діоксо-3,16діазатрицикло[14.3.0.04,6]нонадец-7-ен-18-іловий ефір (1S, 4R, 6S, 14S, 18R)-4-фтор-1,3дигідроізоіндол-2-карбонової кислоти (Сполука 2). Інші варіанти здійснення винаходу, представлені в даному описі, відносяться до композиції, яка практично складається з першої сполуки або його фармацевтично прийнятної солі або проліки, де перша сполука представляє собою Сполуку 1; і другої сполуки або її фармацевтично прийнятної солі або проліки, де друга сполука представляє собою Сполуку 2. Варіанти здійснення винаходу, представлені в даному описі, відносяться до застосування зазначених композицій для полегшення або лікування хворобливого стану в популяції пацієнтів та/або для приготування лікарського засобу, призначеного для полегшення або лікування зазначеного хворобливого стану. Хворобливий стан можна вибирати, наприклад, з інфекції, що 1 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 викликається вірусом гепатиту C, фіброзу печінки та порушеної функції печінки. В одному з варіантів здійснення винаходу проліки першої сполуки можуть представляти собою проліки у вигляді складного діізобутилового ефіру β-D-2’-дезокси-2’-фтор-2’-C-метилцитидину (Сполука 1a). У деяких варіантах здійснення винаходу друга сполука може представляти собою натрієву сіль 14-трет-бутоксикарбоніламіно-4-циклопропансульфоніламінокарбоніл-2,15-діоксо-3,16діазатрицикло[14.3.0.04,6]нонадец-7-ен-18-ілового ефіру (1S, 4R, 6S, 14S, 18R)-4-фтор-1,3дигідроізоіндол-2-карбонової кислоти (натрієва сіль Сполуки 2). Інші варіанти здійснення винаходу, представлені в даному описі, відносяться до застосування Сполуки 1 або її фармацевтично прийнятної солі або проліки для полегшення або лікування хворобливого стану в популяції пацієнтів та/або для приготування лікарського засобу, призначеного для полегшення або лікування зазначеного хворобливого стану, де Сполука 1 або її фармацевтично прийнятна сіль або проліки призначені для застосування та/або для приготування з метою застосування в комбінації із Сполукою 2 або її фармацевтично прийнятною сіллю або проліками. Інший варіант здійснення винаходу, представлений у даному описі, відноситься до застосування Сполуки 2 або її фармацевтично прийнятної солі або проліки для полегшення або лікування популяції пацієнтів та/або для приготування лікарського засобу, призначеного для полегшення або лікування зазначеного хворобливого стану, де Сполука 2 або її фармацевтично прийнятна сіль або проліки призначені для застосування та/або для приготування з метою застосування в комбінації із Сполукою 1 або її фармацевтично прийнятною сіллю або проліками. Наприклад, хворобливий стан можна вибирати з інфекції, що викликається вірусом гепатиту C, фіброзу печінки та порушеної функції печінки Інші варіанти здійснення винаходу, представлені в даному описі, відносяться до способів полегшення або лікування хворобливого стану в популяції пацієнтів, які можуть передбачати введення в терапевтично ефективній кількості першої сполуки або її фармацевтично прийнятної солі або проліки, де перша сполука представляє собою Сполуку 1; і в терапевтично ефективній кількості другої сполуки або її фармацевтично прийнятної солі або проліки, де друга сполука представляє собою Сполуку 2, індивідуумові, що страждає хворобливим станом. У деяких варіантах здійснення винаходу хворобливий стан можна вибирати з інфекції, що викликається вірусом гепатиту C, фіброзу печінки та порушеної функції печінки. В одному з варіантів здійснення винаходу проліки першої сполуки можуть представляти собою проліки у вигляді складного діізобутилового ефіру β-D-2’-дезокси-2’-фтор-2’-C-метилцитидину (Сполука 1a). У деяких варіантах здійснення винаходу друга сполука може представляти собою натрієву сіль 14-трет-бутоксикарбоніламіно-4-циклопропансульфоніламінокарбоніл-2,15-діоксо-3,16діазатрицикло[14.3.0.04,6]нонадец-7-ен-18-ілового ефіру (1S, 4R, 6S, 14S, 18R)-4-фтор-1,3дигідроізоіндол-2-карбонової кислоти (натрієва сіль Сполуки 2). Ці та інші варіанти здійснення винаходи описані більш докладно нижче. Короткий опис креслень На кресленнях показано: на фіг. 1 - структури β-D-2’-дезокси-2’-фтор-2’-C-метилцитидину (Сполука 1), проліків, що представляють собою складний діізобутиловий ефір β-D-2’-дезокси-2’-фтор-2’-C-метилцитидину (Сполука 1a), і 14-трет-бутоксикарбоніламіно-4-циклопропансульфоніламінокарбоніл-2,15діоксо-3,16-діазатрицикло[14.3.0.04,6]нонадец-7-ен-18-ілового ефіру (1S, 4R, 6S, 14S, 18R)-4фтор-1,3-дигідроізоіндол-2-карбонової кислоти (Сполука 2); на фіг. 2 і 3 - схема, що ілюструє сім режимів лікування з використанням сполук, представлених на фіг. 1; на фіг. 4 - графік, що демонструє зміни в рівні сироваткової аланінамінотрансферази (ALT) після 14 днів лікування Сполукою 1a і Сполукою 2. Докладний опис винаходу Варіантами здійснення винаходи є (але не обмежуючись тільки ними) терапевтичні композиції та їх застосування для лікування та/або полегшення хворобливого стану. У деяких варіантах здійснення винаходу хворобливий стан можна вибирати з інфекції, що викликається вірусом гепатиту C, фіброзу печінки та порушеної функції печінки. Якщо не зазначено інше, то всі технічні та наукові поняття, що використовуються в даному описі, мають загальноприйняті значення, відомі звичайному фахівцеві в області, до якої відносяться варіанти здійснення винаходу. Усі згадані в даному описі публікації включені в нього в якості посилання з метою розкриття та опису методів та/або матеріалів, стосовно до яких публікації процитовані. У контексті даного опису та у прикладеній формулі винаходу, згадування в однині має на увазі також згадування в множині, якщо з контексту ясно не випливає інше. Так, наприклад, 2 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 посилання на "метод" відноситься до безлічі таких методів, а посилання на "дозу" включає посилання на одну або декілька доз та їх еквіваленти, відомі фахівцям у даній області, і т.д. Коли зазначений діапазон значень, то слід розуміти, що будь-яке значення, що перебуває між нижньою та верхньою границею цього діапазону з точністю до однієї десятої від величини нижньої границі, якщо з контексту ясно не випливає інше, і будь-яке інше зазначене або що входить в зазначений діапазон значення, підпадає під обсяг варіантів винаходу. Верхні та нижні границі таких менших діапазонів, які незалежно можна включати в менші діапазони, також охоплюють згідно з винаходом діапазон з будь-який спеціально виключеною границею. Якщо зазначений діапазон включає одну або обидві границі, то діапазони, у яких виключена будь-яка одна або виключено обидві із зазначених включених границь, також підпадають під обсяг варіантів здійснення винаходу. Поняття "фармацевтично прийнятна сіль" відноситься до солі сполуки, яка не викликає вираженого подразнення в організмі, у який її вводять, і не усуває біологічну активність і властивості сполуки. У деяких варіантах здійснення винаходу сіль представляє собою кислотноадитивну сіль сполуки. Фармацевтичні солі можна одержувати взаємодією сполуки з неорганічними кислотами, такими як галогенводнева кислота (наприклад, соляна кислота або бромистоводнева кислота), сірчана кислота, азотна кислота, фосфорна кислота та т.і. Фармацевтичні солі можна одержувати також взаємодією сполуки з органічною кислотою, такими як аліфатичні або ароматичні карбонові або сульфонові кислоти, наприклад, оцтова, бурштинова, молочна, яблучна, винна, лимонна, аскорбінова, нікотинова, метансульфонова, етансульфонова, пара-толуолсульфонова, саліцилова або нафталінсульфонова кислота. Фармацевтичні солі можна одержувати також взаємодією сполуки з основою з одержанням такої солі, як амонієва сіль, сіль лужного металу, така як натрієва або калієва сіль, сіль лужноземельного металу, така як кальцієва або магнієва сіль, сіль органічних основ, таких як дициклогексиламін, N-метил-D-глюкамін, тріс(гідроксиметил)метиламін, C1-C7алкіламін, циклогексиламін, триетаноламін, етилендіамін, і солі з амінокислотами, такими як аргінін, лізин і т.і. Натрієва сіль Сполуки 2 представляє собою приклад (але не обмежуючись тільки ним) фармацевтично прийнятної солі. Поняття "проліки" відноситься до агента, який перетворюється в основний лікарський засіб in vivo. Проліки часто є цінними, оскільки в деяких ситуаціях їх простіше вводити, ніж материнський (основний) лікарський засіб. Вони можуть володіти, наприклад, біодоступністю при пероральному введенні, на відміну від основного лікарського засобу. Проліки можуть мати також підвищену розчинність у фармацевтичних композиціях, у порівнянні з основним лікарським засобом. Наприклад (але не обмежуючись тільки зазначеним), проліки можуть представляти собою сполуку, яку вводять у вигляді складного ефіру ("проліки") для полегшення проникнення через клітинну мембрану, коли розчинність у воді є несприятливим чинником, але яка потім метаболізується у результаті гідролізу з утворенням карбонової кислоти, тобто активної субстанції, після попадання в середину клітини, де розчинність у воді є корисною властивістю. Іншим прикладом проліків може бути короткий пептид (поліамінокислота), зв'язаний з кислотною групою, при цьому пептид метаболізується, "відкриваючи" активний фрагмент. Сполука 1a представляє собою приклад (але не обмежуючись тільки ним), проліків (у цьому випадку проліки Сполуки 1). Загальноприйняті процедури відбору та одержання прийнятних похідних, що представляють собою проліки, описані, наприклад, в Design of Prodrugs, під ред. H. Bundgaard, вид-во Elsevier, 1985, публікація включена в даний опис як посилання з метою опису процедур і шляхів одержання прийнятних похідних, що представляють собою проліки. Поняття "ефективна кількість" відноситься до кількості діючої речовини або фармацевтичного агента, яка викликає необхідну біологічну або медичну відповідь. Наприклад, ефективна кількість сполуки може представляти собою кількість, необхідну для попередження, ослаблення або полегшення симптомів хвороби, або пролонгування часу життя індивідуума, що піддається лікуванню. При лікуванні ця відповідь може мати місце в тканині, системі, тварині або людині та викликати ослаблення симптомів хвороби. Визначення ефективної кількості перебуває в компетенції фахівців у даній області, насамперед у світлі представленого докладного опису. Ефективну кількість сполук, представлених у даному описі, виражають у вигляді дози, яка повинна залежати від способу введення, типу тварини, включаючи людину, що піддається лікуванню, та фізичних характеристик конкретної розглянутої тварини. Дозу можна спеціально пристосовувати (цілеспрямовано змінювати) для досягнення необхідної дії, але вона повинна залежати від таких факторів, як вага, дієта медикаментозна, супроводжуюче лікування та інші фактори, які повинні бути очевидні фахівцям-медикам. У цілому, ефективна кількість композицій, представлених у даному описі, і необов'язково одного або декількох додаткових 3 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 противірусних агентів, представляє собою кількість, ефективну у відношенні зниження вірусного навантаження або для досягнення тривалої вірусної відповіді на терапію. У контексті даного опису поняття "лікування", "лікувати" і т.і. відносяться до одержання необхідної фармакологічної та/або фізіологічної дії. Дія може бути профілактичною у контексті повного або часткового попередження хвороби або її симптомів та/або може бути терапевтичною у контексті часткового або повного лікування хвороби та/або небажаних явищ, пов'язаних із хворобою. Поняття "лікування" у контексті даного опису відноситься до будь-якого лікування хвороби у ссавця, зокрема людини, і включає: (a) попередження виникнення хвороби в індивідуума, який може бути схильний до хвороби, але в якого вона ще не діагностована; (б) інгібування хвороби, тобто припинення її розвитку; та (в) ослаблення хвороби, тобто регрес хвороби. Поняття "індивідуум", "хазяїн", "суб'єкт" і "пацієнт" у контексті даного опису використовують взаємозамінно, та вони відносяться до ссавця, включаючи (але не обмежуючись тільки ними), представників мишачих, мавп, людину, сільськогосподарських тварин із класу ссавців, тварин із класу ссавців, призначених для спорту, свійських тварина із класу ссавців. У контексті даного опису поняття "гепатичний фіброз", яке застосовують взаємозамінно із поняттям "фіброз печінки", відноситься до росту рубцевої тканини в печінці, який може мати місце при хронічній інфекції печінки. У контексті даного опису поняття "функція печінки" відноситься до нормальної функції печінки, включаючи (але не обмежуючись тільки ними) функцію синтезу, включаючи (але не обмежуючись тільки ними) синтез білків, таких як сироваткові білки (наприклад, альбумін, фактори згортання, лужна фосфатаза, амінотрансферази (наприклад, аланінтрансаміназа, аспартаттрансаміназа), 5’-нуклеозидаза, γ-глутамінілтранспептидаза та т.д.), синтез біліруміну, синтез холестерину та синтез жовчних кислот; метаболічну функцію печінки, включаючи (але не обмежуючись тільки ними) метаболізм вуглеводів, амінокислот і метаболізм аміаку, метаболізм гормонів і метаболізм ліпідів; детоксикацію екзогенних лікарських засобів; гемодинамічну функцію, включаючи вісцеральну та портальну гемодинаміку; і т.і. Поняття "тривала вірусна відповідь" (SVR; який позначають також як "тривала відповідь" або "довготривала відповідь") у контексті даного опису відноситься до відповіді індивідуума на режим лікування HCV-інфекції в поняттях титру HCV у сироватці. Наприклад, "тривала вірусна відповідь" відноситься до відсутності РНК HCV (наприклад, менш ніж приблизно 500, менш ніж приблизно 200 або менш ніж приблизно 100 геномних копій на мілілітр сироватки), що виявляється у сироватці пацієнта протягом періоду часу, що становить принаймні приблизно 1 місяць, принаймні приблизно 2 місяця, принаймні приблизно 3 місяця, принаймні приблизно 4 місяця, принаймні приблизно 5 місяців та/або принаймні приблизно 6 місяців, після припинення лікування. Продемонстровано, що сполука β-D-2’-дезокси-2’-фтор-2’-C-метилцитидин (далі Сполука 1) ефективно інгібує реплікацію HCV. Хоча даний винахід не обмежений будь-якою конкретною теорією, імовірно, що Сполука 1 інгібує реплікацію HCV шляхом інгібування РНК-полімерази HCV, ферменту, який бере участь у реплікації вірусу гепатиту C. Сполуку 1 можна одержувати за допомогою методів, відомих фахівцям у даній області, наприклад, методів, які описані в US 7419572, який повністю включений у даний опис як посилання. У композиціях, представлених у даному описі, можна застосовувати фармацевтично прийнятні солі та проліки Сполуки 1. Наприклад, встановлено, що проліки у вигляді складного діізобутилового ефіру β-D-2’-дезокси2’-фтор-2’-C-метилцитидину, представленого на фіг. 1, мають підвищену здатність до проникнення, що приводить до підвищеної експозиції в плазмі та в результаті до покращеної противірусної ефективності. Встановлено, що сполука 14-трет-бутоксикарбоніламіно-4циклопропансульфоніламінокарбоніл-2,15-діоксо-3,16-діазатрицикло[14.3.0.04,6]нонадец-7-ен18-іловий ефір (1S, 4R, 6S, 14S, 18R)-4-фтор-1,3-дигідроізоіндол-2-карбонової кислоти (далі Сполука 2) ефективно інгібує реплікацію HCV. Вищевказану сполуку можна одержувати за допомогою методів, відомих фахівцям у даній області, наприклад, методів, які описані в US 7491794, який повністю включений у даний опис як посилання. Хоча даний винахід не обмежений будь-якою конкретною теорією, імовірно, що Сполука 2 інгібує протеазу HCV, зокрема NS3/4A-протеазу. У композиціях, представлених у даному описі, можна застосовувати фармацевтично прийнятні солі та проліки Сполуки 2. Наприклад, у композиції, що представлені в даному описі, можна включати натрієву сіль Сполуки 2. Структура та методи одержання натрієвої солі описані в публікації US № 2007-0054842, зареєстрованої 21 липня, 2006 р., яка повністю включена в даний опис в якості посилання. 4 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 У сполуках, представлених у даному описі, кожний стереогенний атом вуглецю може перебувати в R- або S-конфігурації. Хоча в даному описі конкретні, наведені в якості прикладів сполуки, можуть бути представлені в конкретній конфігурації, однак, якщо спеціально не зазначено інше, то під обсяг винаходу підпадають також сполуки, що мають будь-яку протилежну стереохімію на будь-якому конкретному хіральному центрі, або їх суміш. Коли хіральні центри виявлені в солях або проліках сполук, то слід розуміти, що, якщо спеціально не зазначено інше, ці сполуки включають усі можливі стереоізомери. Крім того, мається на увазі, що в будь-якій сполуці, представленій в даному описі, що має один або декілька подвійний(их) зв'язок(ів), утворюються геометричні ізомери, які можна позначати як E- або Z-ізомери, кожний подвійний зв'язок може незалежно один від одного перебувати в E- або Z-орієнтації або представляти собою їх суміш. Під обсяг винаходу підпадають також усі таутомерні форми. Деякі варіанти здійснення винаходу, представлені в даному описі, відносяться до композиції, яка може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки; та Сполуку 2 або її фармацевтично прийнятну сіль або проліки. В одному з варіантів здійснення винаходу проліки Сполуки 1 можуть представляти собою Сполуку 1a. У деяких варіантах здійснення винаходу сіль Сполуки 2 може представляти собою натрієву сіль. Один з варіантів здійснення винаходу, представлений у даному описі, відносяться до композиції, яка практично складається із Сполуки 1 або її фармацевтично прийнятної солі або проліки; та Сполуки 2 або її фармацевтично прийнятної солі або проліки. У деяких варіантах здійснення винаходу проліки Сполуки 1 може представляти собою Сполуку 1a. У деяких варіантах здійснення винаходу сіль Сполуки 2 може представляти собою натрієву сіль. У деяких варіантах здійснення винаходу композиція може включати також фармацевтично прийнятний ексципієнт, розчинник та/або носій, які зазначені в даному описі. Сполука 1 або її фармацевтично прийнятна сіль або проліки можуть входити у композиції, що представлені в даному описі, в різних кількостях. У деяких варіантах здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 9000 до приблизно 50 мг. В інших варіантах здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 5000 до приблизно 150 мг. В інших варіантах здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 2000 до приблизно 350 мг. У наступних варіантах здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 1000 до приблизно 450 мг. В одному з варіантів здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 1000 до приблизно 500 мг. Аналогічно цьому Сполука 2 або її фармацевтично прийнятна сіль або проліки можуть входити в композиції в різних кількостях. У деяких варіантах здійснення винаходу композиція може включати Сполуку 2 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 2000 до приблизно 2 мг. В інших варіантах здійснення винаходу композиція може включати Сполуку 2 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 1600 до приблизно 25 мг. В інших варіантах здійснення винаходу композиція може включати Сполуку 2 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 500 до приблизно 50 мг. В одному з варіантів здійснення винаходу композиція може включати Сполуку 2 або її фармацевтично прийнятну сіль або проліки в кількості, що становить від приблизно 200 до приблизно 100 мг. Потенційна перевага застосування комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліки може полягати в зниженні необхідної кількості однієї або декількох сполук, які є ефективними у відношенні лікування хворобливого стану, зазначеного в даному описі (наприклад, що викликається HCV), у порівнянні з лікуванням у вигляді монотерапії, порівнянної за іншими параметрами популяції пацієнтів з використанням окремо кожної із Сполук 1 або 2 або їх фармацевтично прийнятних солей або проліки. У деяких варіантах здійснення винаходу кількість Сполуки 1 або її фармацевтично прийнятної солі або проліки в композиції може бути нижчою, в порівнянні з кількістю Сполуки 1 або її фармацевтично прийнятної солі або проліки, необхідної для досягнення такого ж зниження вірусного навантаження при застосуванні у вигляді монотерапії. У деяких варіантах здійснення винаходу кількість Сполуки 2 або її фармацевтично прийнятної солі або проліки в композиції є нижчою, в порівнянні з кількістю Сполуки 2 або її фармацевтично прийнятної солі або проліки, необхідною для досягнення такого ж зниження вірусного навантаження при застосуванні у вигляді монотерапії. В одному з варіантів здійснення винаходу сума кількості Сполуки 1 або її фармацевтично прийнятної солі 5 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 або проліки та кількості Сполуки 2 або її фармацевтично прийнятної солі або проліки є нижчою, ніж є очікуваним або прогнозованим на підставі адитивної дії комбінації Сполуки 1 або її фармацевтично прийнятної солі або проліки при окремому застосуванні та Сполуки 2 або її фармацевтично прийнятної солі або проліки при окремому застосуванні, у відношенні лікування хворобливого стану, такого як такий, що викликається HCV, стан. Додатковими перевагами застосування комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліки можуть бути невисокий рівень або відсутність перехресної стійкості при застосуванні Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліки; різні шляхи елімінації Сполук 1 і 2 або фармацевтично прийнятних солей або проліки; невисокий рівень або відсутність токсичності, що перекривається, між Сполуками 1 і 2 або їх фармацевтично прийнятними солями або проліками; невисокий рівень або відсутність вираженого впливу на цитохром P450; та/або невисокий рівень або відсутність фармакокінетичних взаємодій між Сполуками 1 і 2 або їх фармацевтично прийнятними солями або проліками. Можна також варіювати процентний вміст Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліки у композиції. Наприклад, у деяких варіантах здійснення винаходу композиція може включати Сполуку 1 або її фармацевтично прийнятну сіль або проліки в кількості від приблизно 1 до приблизно 99 мас. % у перерахуванні на суму кількості Сполуки 1 або її фармацевтично прийнятної солі або проліки та кількості Сполуки 2 або її фармацевтично прийнятної солі або проліки в композиції. У додаткових варіантах здійснення винаходу кількість Сполуки 1 або її фармацевтично прийнятної солі або проліки може становити (але не обмежуючись тільки зазначеним) від приблизно 5 до приблизно 90, від приблизно 10 до приблизно 80, від приблизно 20 до приблизно 70, від приблизно 30 до приблизно 60 і від приблизно 40 до приблизно 50 мас. % у перерахуванні на суму кількості Сполуки 1 або її фармацевтично прийнятної солі або проліки та кількості Сполуки 2 або її фармацевтично прийнятної солі або проліки в композиції. Відносно Сполуки 2 в одному з варіантів здійснення винаходу композиція може включати Сполуку 2 або її фармацевтично прийнятну сіль або проліки в кількості від приблизно 1 до приблизно 99 мас. % у перерахуванні на суму кількості Сполуки 1 або її фармацевтично прийнятної солі або проліки та кількості Сполуки 2 або її фармацевтично прийнятної солі або проліки в композиції. У додаткових варіантах здійснення винаходу кількість Сполуки 2 або її фармацевтично прийнятної солі або проліки може становити, наприклад, від приблизно 5 до приблизно 90, від приблизно 10 до приблизно 80, від приблизно 20 до приблизно 70, від приблизно 30 до приблизно 60 і від приблизно 40 до приблизно 50 мас. % у перерахуванні на суму кількості Сполуки 1 або її фармацевтично прийнятної солі або проліки та кількості Сполуки 2 або її фармацевтично прийнятної солі або проліки в композиції. У композицію, що містить Сполуку 1 і 2 або їх фармацевтично прийнятні солі або проліки, можуть входити додаткові терапевтичні засоби. У деяких варіантах здійснення винаходу додатковий терапевтичний засіб може представляти собою противірусний засіб. В одному з варіантів здійснення винаходу противірусний засіб може представляти собою противірусний засіб проти HCV. Прикладами прийнятних терапевтичних засобів є (але не обмежуючись тільки ними) нуклеотиди та нуклеозидні аналоги (такі як азидотимідин (AZT) (зидовудин) та їх аналоги та похідні; 2’,3’-дидезоксиінозин (DDI) (диданозин) та його аналоги та похідні; 2’,3’дидезоксицитидин (DDC) (дидезоксицитидин) та його аналоги та похідні; 2’,3’-дидегідро-2’,3’дидезокситимідин (D4T) (ставудин) та його аналоги та похідні; комбівір; абакавір; адефовіра дипівоксил; цидофовір; рибавірин; аналоги рибавірину; левовірин, вірамідин, ізаторибин і т.і., пірфенідон або аналоги пірфенідону, інгібітори РНК-залежної РНК-полімерази NS5B, антагоністи факторів некрозу пухлини (такі як етанерцепт, іфліксімаб та адалімумаб), тимозинα- (Zadaxin™) , агоніст(и) рецептора інтерферону, інгібітори α-глюкозидази, антагоністи TNF-α, інгібітори NS3-геликаз, інгібітори NS5B-полімерази, інгібітори NS3-протеази (наприклад, (VX950) та (SCH 503034)), рітонавір ([5S-(5R*,8R*,10R*,11R*)]-5-тіазолілметиловий ефір 10гідрокси-2-метил-5-(1-метилетил)-1-[2-(1-метилетил)-4-тіазоліл]-3,6-діоксо-8,11-біс(фенілметил)2,4,7,12-тетраазатриденкан-13-онової кислоти, фірми Abbott Laboratories) і рибозими, такі як TM Heptazyme , та фосфоротіоатні олігонуклеотиди, комплементарні білковим послідовностям HCV, які інгібують експресію корових білків вірусів. Одним із обмежень раніше застосовуваної терапії на основі інтерферону (IFN) був швидкий кліренс білка із крові. Хімічна дериватизація IFN за допомогою поліетиленгліколю (ПЕГ) дозволила одержати білки із суттєво поліпшеними фармакокінетичними властивостями. ® PEGASYS представляє собою кон'югат α-2a, що має молекулярну масу 40 кДа розгалуженого ® монометокси-ПЕГ, і PEG-INTRON представляє собою кон'югат α-2b, що має молекулярну масу 6 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 12 кДа монометокси-ПЕГ (B.A. Luxon та ін., Clin. Therapy, 24(9), 2002, сс. 13631-1383; та A. Kozlowski і J.M Harris, J. Control. Release, 72, 2001, сс. 217-224). Однак деякі пацієнти не могли або не прагли піддаватись інтерфероновій терапії з однієї або декількох причин, таким, наприклад, як необхідність здійснювати самоін'єкції та/або наявність однієї або декількох побічних дій, пов'язаних з інтерфероновою терапією. Композиції, що містять Сполуку 1 і 2 або їх фармацевтично прийнятні солі або проліки, можуть додатково включати агоніст рецептора інтерферону, такий як агоніст рецептора інтерферону типу I та/або агоніст рецептора інтерферону типу II. В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу II може представляти собою інтерферон-γ (IFN-γ). В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу I може представляти собою інтерферон-α (IFN-α), наприклад, монопегілований (30 кДа, лінійний) консенсусний IFN-α, консенсусний IFN-α, INFERGEN, кон'югат, що має молекулярну масу 40 кДа розгалуженого монометокси-ПЕГ та інтерферону α-2b та/або кон'югат, що має молекулярну масу 12 кДа монометокси-ПЕГ та інтерферону α-2b. У деяких варіантах здійснення в композицію, що містить Сполуку 1 і 2 або їх фармацевтично прийнятні солі або проліки, може входити також рибавірин. В інших варіантах здійснення в композицію, що містить Сполуку 1 і 2 або їх фармацевтично прийнятні солі або проліки, не входить агоніст рецептора інтерферону. Наприклад, агоніст рецептора інтерферону може представляти собою агоніст рецептора інтерферону типу I. В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу I представляє собою пегілований агоніст рецептора інтерферону типу I, наприклад, представлений у даному описі. У наступних варіантах здійснення винаходу в композицію, що містить Сполуку 1 і 2 або їх фармацевтично прийнятні солі або проліки, не входить рибавірин. У контексті даного опису поняття "агоніст рецептора інтерферону" відноситься до будь-якого типу агоністів рецептора інтерферону типу I, агоністів рецептора інтерферону типу II або агоністів рецептора інтерферону типу III. У контексті даного опису поняття "агоніст рецептора інтерферону типу I" відноситься до будь-якого, що зустрічається в природних умовах або, що не зустрічається в природних умовах ліганду рецептора людського інтерферону типу I, який зв'язується з рецептором і викликає трансдукцію сигналу через рецептор. Агоністи рецептора інтерферону типу I включають інтерферони, у тому числі інтерферони, що зустрічаються в природних умовах, модифіковані інтерферони, синтетичні інтерферони, пегіловані інтерферони, злиті білки, що включають інтерферон, і гетерологічний білок, перестановлений інтерферонами; антитіло, специфічне відносно рецептора інтерферону; непептидні хімічні агоністи та т.і. У контексті даного опису поняття "агоніст рецептора інтерферону типу II" відноситься до будьякого, що зустрічається в природних умовах або, що не зустрічається в природних умовах ліганду рецептора людського інтерферону типу II, який зв'язується з рецептором і викликає трансдукцію сигналу через рецептор. Агоністи рецептора інтерферону типу II включають нативний людський інтерферон-γ, рекомбінантні види IFN-α, глікозовані види IFN-γ, пегіловані види IFN-γ, модифіковані види або варіанти IFN-γ, злиті білки, що включають IFN-γ, агоністи антитіл, специфічних відносно рецептора, непептидні агоністи та т.і. У контексті даного опису поняття "агоніст рецептора інтерферону типу III" відноситься до будь-якого, що зустрічається в природних умовах або, що не зустрічається в природних умовах ліганду людського рецептора α IL-28 ("IL-28R"), амінокислотна послідовність якого описана в Sheppard та ін., вище, який зв'язується з рецептором і викликає трансдукцію сигналу через рецептор. Прийнятними інгібіторами α-глюкозидаз є будь-які описані вище іміноцукри, включаючи похідні іміноцукрів з довгим алкільним ланцюгом, які описані в публікації патенту США № 2004/0110795; інгібітори асоційованих з ендоплазматичним ретикулом α-глюкозидаз; інгібітори ® пов'язаної з мембраною α-глюкозидази; міглітол (Glyset ) та його активні похідні та аналоги; та ® акарбоза (Precose ) та її активні похідні та аналоги. Композиції, представлені в даному описі, можна вводити хворій людині per se або у вигляді композицій, у яких вони змішані з іншими діючими речовинами, як у випадку комбінованої терапії, або з носіями, розчинниками, екціпієнтами або їх комбінаціями, і на їхній основі можна приготовляти препарати у твердій, напівтвердій, рідкій або газоподібній формі, такі як таблетки, капсули, порошки, гранули, мазі, розчини, супозиторії, форми, що вводяться за допомогою ін'єкції, інгалянти та аерозолі. Вимога до препаративної форми визначається вибраним шляхом введення. Методики приготування препаративних форм і введення композицій, представлених у даному описі, відомі фахівцям у даній області. Фармацевтично прийнятні ексціпієнти відомі фахівцям у даній області та описані в різних публікаціях, включаючи, наприклад, A. Gennaro, "Remington: The Science and Practice of Pharmacy", 20-е вид., вид-во Lippincott, Williams, & Wilkins, 2000; Pharmaceutical Dosage Forms and Drug Delivery Systems, під ред. H.C. Ansel та ін., 7 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 7-е вид., вид-во Lippincott, Williams, & Wilkins, 1999; та Handbook of Pharmaceutical Excipients, під ред. A.H. Kibbe та ін., 3-е вид., вид-во Amer. Pharmaceutical Assoc, 2000. Композиції, представлені в даному описі, можна приготовляти добре відомим методом, наприклад, за допомогою загальноприйнятих методів змішування, розчинення, грануляції, виготовлення драже, розтирання в порошок, емульгування, капсулювання, включення або таблетування. Крім того, діючі речовини входять у кількості, ефективній для досягнення наміченої мети. Багато із сполук, застосовуваниху композиціях, представлених у даному описі, можна приготовляти у вигляді солей з фармацевтично прийнятними протиіонами. У деяких варіантах здійснення винаходу сполуки або їх фармацевтично прийнятні солі або проліки (наприклад, Сполука 1, 1a і 2) приготовляють у водному буфері. Прийнятними водними буферами є (але не обмежуючись тільки ними) ацетатний, сукцинатний, цитратний та фосфатний буфери різної концентрації, що становить від приблизно 5 до приблизно 100 мМ. У деяких варіантах здійснення винаходу водний буфер включає реагенти, що забезпечують одержання ізотонічного розчину. Такі реагенти включають (але не обмежуючись тільки ними) хлорид натрію; та цукри, наприклад, манніт, декстрозу, сахарозу та т.і. У деяких варіантах здійснення винаходу водний буфер додатково включає неіоногенну поверхнево-активну речовину, таку як полісорбат 20 або 80. Необов'язково препаративні форми можуть включати також консервант. Прийнятними консервантами є (але не обмежуючись тільки ними) бензиловий спирт, фенол, хлорбутанол, бензалконійхлорид і т.і. У багатьох випадках препаративну форму зберігають при температурі приблизно 4 ºC. Препаративні форми можна також ліофілізувати, у цьому випадку вони, як правило, включають кріопротектори, такі як сахароза, трегалоза, лактоза, мальтоза, манніт і т.і. Ліофілізовані препаративні форми можна зберігати протягом тривалих періодів часу навіть при температурі навколишнього середовища. Прийнятні шляхи введення можуть представляти собою, наприклад, пероральний, ректальний, місцевий, через слизувату оболонку або кишковий шлях введення; парентеральне введення, включаючи внутрішньом'язові, підшкірні, внутрішньовенні, інтрамедулярні ін'єкції, а також внутрішньооболонкові, безпосередньо в шлуночок, внутрішньочеревні, внутрішньоносові, внутрішньоочні ін'єкції або застосування у вигляді аерозольної інгаляції. Композиції повинні бути, як правило, призначені для конкретного очікуваного шляху введення. В одному з варіантів даного опису композиції, представлені в даному описі, можна застосовувати перорально. Підшкірне введення можна здійснювати з використанням стандартних методів і обладнань, наприклад, голки та шприца, системи введення на основі ін'єкційного підшкірного порту та т.і. (див., наприклад, US 3547119; 4755173; 4531937; 4311137 і 6017328). Комбінацію підшкірного ін'єкційного порту та обладнання для введення пацієнтові через порт фармацевтичної композиції, що пропонується у винаході, позначають у контексті даного опису як "система введення на основі ін'єкційного підшкірного порту". У багатьох варіантах здійснення винаходу підшкірну ін'єкцію здійснюють шляхом болюсного введення за допомогою голки або шприца. У препаратах, призначених для перорального введення, сполуку можна застосовувати окремо або в комбінації з добавками, придатними для одержання таблеток, порошків, гранул або капсул, наприклад, із загальноприйнятими добавками, такими як лактоза, манніт, кукурудзяний крохмаль або картопляний крохмаль; зі зв'язуючими агентами, такими як кристалічна целюлоза, похідні целюлози, аравійська камедь, кукурудзяний крохмаль або желатини; з розпушувачами, такими як кукурудзяний крохмаль, картопляний крохмаль або натрієва сіль карбоксиметилцелюлози; із замаслювачами, такими як тальк або стеарат магнію; і при необхідності з розчинниками, забуферовуючими агентами, зволожувачами, консервантами та коригентами. Сполуки можна приготовляти для включення в препарати для ін'єкції шляхом розчинення, суспендування або емульгування їх у водному або неводному розчиннику, такому як рослинна або інші аналогічні олії, синтетичні гліцериди аліфатичних кислот, ефіри вищих аліфатичних кислот або пропиленгліколю; і при необхідності із загальноприйнятими добавками, такими як солюбілізатори, агенти, що надають ізотонічність, суспендуючі агенти, емульгатори, стабілізатори та консерванти. Крім того, на основі сполук можна приготовляти супозиторії, шляхом змішування з різними основами, такими як основи що емульгуються, або водорозчинні основи. Згідно з варіантами здійснення винаходу сполуки можна вводити ректально за допомогою супозиторія. Супозиторій може включати наповнювачі, такі як масло какао, карнаубський віск і поліетиленгліколі, що плавляться при температурі тіла, які знаходяться в твердому стані при кімнатній температурі. Можна приготовляти стандартні дози лікарського засобу, призначені для введення пероральним або ректальним шляхом, наприклад, у формі сиропів, еліксирів і суспензій, де кожна стандартна одиниця, наприклад, чайна ложка, столова ложка, таблетка або супозиторій, 8 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 містить у заздалегідь визначеній кількості композицію, що включає один або декілька інгібіторів. Аналогічно цьому стандартна доза лікарського засобу, призначена для введення шляхом ін'єкції або внутрішньовенного введення, може містити інгібітор(и), що входять до складу композиції, що представляє собою розчин у стерильній воді, нормальному фізіологічному розчині або іншому фармацевтично прийнятному носії. Поняття "стандартна доза лікарського засобу" у контексті даного опису відноситься до фізично дискретних одиниць, придатних у якості унітарних доз для людини та тварин, кожна одиниця містить попередньо визначену кількість сполук, що є варіантами здійснення винаходу, яка розрахована як кількість, достатня для одержання необхідної дії, у комбінації з фармацевтично прийнятним розчинником, носієм або наповнювачем. Специфікації нових стандартних доз лікарського засобу, що є варіантами здійснення винаходу, залежать від конкретної застосовуваної сполуки та дії, яку очікують досягти, та фармакодинамічних характеристик, якими володіє кожна сполука в організмі-хазяїні. Композиції, представлені в даному описі, можна вводити перорально, парентерально або за допомогою імплантованого резервуара. В одному з варіантів здійснення винаходу композицію можна вводити перорально або вводити шляхом ін'єкції. Композицію можна застосовувати також для місцевої обробки, а не системно, наприклад, шляхом ін'єкції композиції безпосередньо в уражену область, часто у вигляді депо або у вигляді препаративної форми із пролонгованим вивільненням. Крім того, композицію можна застосовувати з використанням системи для спрямованого введення лікарського засобу, наприклад, з використанням ліпосоми, покритої специфічним для тканини антитілом. Ліпосоми повинні направлено доставлятись до органа та вибірково поглинатись органом. При необхідності композиція може знаходитись в упакуванні або диспенсерному пристрої, який може містити одну або декілька стандартних доз лікарського засобу, що включають діючу речовину. Упакування може містити, наприклад, металеву або пластикову фольгу, наприклад, у випадку блістерного упакування. Упакування або диспенсерний пристрій може бути постачене інструкціями із застосування. Упакування або диспенсер можуть також бути постачені повідомленням, встановленим урядовим агентством, яке регулює виробництво, застосування або продаж фармацевтичних засобів, де повідомлення сповіщає про те, що застосування форми лікарського засобу в медицині або ветеринарії схвалено агентством. Таке повідомлення може, наприклад, представляти собою етикетку, схвалену Управлінням з контролю над харчовими продуктами та лікарськими препаратами США для лікарських засобів, або схвалену листівку-вкладиш в упакуванні. Композиції, що містять сполуки, представлені в даному описі, приготовляють у вигляді форми, у якій може бути присутнім також сумісний фармацевтичний носій, поміщений у відповідний контейнер, і етикетка із вказівкою способу застосування для лікування відповідного стану. Деякі варіанти здійснення винаходу, представлені в даному описі, відносяться до способу полегшення або лікування хворобливого стану, які полягають у тому, що вводять у певній кількості Сполуку 1 або її фармацевтично прийнятну сіль або проліки та у певній кількості Сполуку 2 або її фармацевтично прийнятну сіль або проліки, де хворобливий стан може представляти собою інфекцію, викликувану вірусом гепатиту C, фіброз печінки та/або порушену функцію печінки. В одному з варіантів здійснення винаходу проліки Сполуки 1 можуть представляти собою Сполуку 1a. Для полегшення або лікування хворобливого стану можна використовувати різні лікарські форми Сполуки 1 або її фармацевтично прийнятної солі або проліки та/або Сполуки 2 або її фармацевтично прийнятної солі або проліки. У деяких випадках Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки можуть бути присутніми в одній і тій же лікарській формі, наприклад, у вигляді композицій, представлених у даному описі. В інших випадках Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки можна вводити у вигляді різних лікарських форм. Наприклад, Сполуку 1 або її фармацевтично прийнятну сіль або проліки можна застосовувати у вигляді першої таблетки, а Сполуку 2 або її фармацевтично прийнятну сіль або проліки можна застосовувати у вигляді другої таблетки. Коли Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки входять до складу різних лікарських форм, то лікарські форми можуть бути однаковими (наприклад, обидві представляти собою пігулки) або різними (наприклад, одна сполука може бути включена в препаративну форму, що представляє собою пігулку, а друга сполука може бути включена в препаративну форму, призначену для ін'єкцій). Порядок введення Сполуки 1 або її фармацевтично прийнятної солі або проліки та Сполуки 2 або її фармацевтично прийнятної солі або проліки можна варіювати. Коли Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки входять до складу різних лікарських форм, то 9 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 лікарські форми можна вводити одночасно або послідовно. У деяких варіантах здійснення винаходу лікарську форму, що містить Сполуку 1 або її фармацевтично прийнятну сіль або проліки, можна вводити перед лікарською формою, що містить Сполуку 2 або її фармацевтично прийнятну сіль або проліки. В інших варіантах здійснення винаходу лікарську форму, що містить Сполуку 1 або її фармацевтично прийнятну сіль або проліки, можна вводити після лікарської форми, що містить Сполуку 2 або її фармацевтично прийнятну сіль або проліки. У наступних варіантах здійснення винаходу лікарську форму, що містить Сполуку 1 або її фармацевтично прийнятну сіль або проліки, можна вводити приблизно одночасно з лікарською формою, що містить Сполуку 2 або її фармацевтично прийнятну сіль або проліки. У деяких варіантах здійснення винаходу Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки можна застосовувати спільно. У контексті даного опису поняття "спільно" означає, що обидві сполуки в ефективних концентраціях присутні в організмі індивідуума одночасно. При спільному застосуванні, Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки можна використовувати в одній і тій же лікарській формі або в різних лікарських формах. В інших варіантах здійснення винаходу Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки можна застосовувати послідовно. У контексті даного опису поняття "послідовно" означає, що спочатку на протязі першого періоду часу вводять одну сполуку, а потім на протязі другого періоду часу вводять другу сполуку, при цьому перший і другий період часу не перекриваються. Індивідуумові, що має хворобливий стан, можна вводити додаткові терапевтичні засоби. Не обмежуючий обсяг винаходу перелік додаткових терапевтичних засобів представлений вище. Коли застосовують один або декілька додатковий(их) терапевтичний(их) засіб(ів), то додатковий(і) засіб(и) можна вводити в такій же лікарській формі, що і лікарська форма, яка містить Сполуку 1 або її фармацевтично прийнятну сіль або проліки, та/або Сполуку 2 або її фармацевтично прийнятну сіль або проліки. Наприклад, додатковий(і) терапевтичний(і) засіб(и) можна включати в композицію, яка містить Сполуку 1 або її фармацевтично прийнятну сіль або проліки, але не містить Сполуку 2, або її фармацевтично прийнятну сіль або проліки; або в композицію, яка містить Сполука 2 або її фармацевтично прийнятну сіль або проліки, але не містить Сполуку 1, або її фармацевтично прийнятну сіль або проліки; або в композицію, представлену у даному описі (наприклад, композицію, яка містить Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки). В альтернативному варіанті додатковий(і) терапевтичний(і) засіб(и) можна використовувати у вигляді однієї або декількох окремих лікарських форм. Якщо здійснюють введення у вигляді однієї або декількох окремих лікарських форм, то кожна лікарська форма, що містить один або декілька додаткових лікарських засобів, може представляти собою таку ж лікарську форму, яка включає Сполуку 1 або її фармацевтично прийнятну сіль або проліки, та/або лікарську форму, яка включає Сполуку 2 або її фармацевтично прийнятну сіль або проліки, або форму, відмінну від лікарської форми, що включає Сполуку 1 або її фармацевтично прийнятну сіль або проліки, та/або відмінну від лікарської форми, що включає Сполуку 2 або її фармацевтично прийнятну сіль або проліки. Коли один або декілька додаткових терапевтичних засобів знаходяться в одній або декількох окремих лікарських формах, то лікарські форми, що містять один або декілька додатковий(их) терапевтичний(их) засіб(ів), можна вводить до, після, у проміжку, спільно або послідовно із введенням Сполуки 1 або її фармацевтично прийнятної солі або проліків та/або Сполуки 2 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу додатковий терапевтичний засіб може представляти собою агоніст рецептора інтерферону, такий як агоніст рецептора інтерферону типу I та/або агоніст рецептора інтерферону типу II. В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу II може представляти собою інтерферон-γ (IFN-γ). В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу I може представляти собою інтерферон-α (IFN-α). У деяких варіантах здійснення винаходу агоніст рецептора інтерферону типу I можна вибирати із групи, що включає монопегілований (30 кДа, лінійний) консенсусний IFN-α, консенсусний IFNα, а саме, INFERGEN, кон'югат, що має молекулярну масу 40 кДа розгалуженого монометоксиПЕГ та інтерферону α-2b і кон'югат, що має молекулярну масу 12 кДа монометокси-ПЕГ та інтерферону α-2b. В одному з варіантів здійснення винаходу додатковий терапевтичний засіб може представляти собою рибавірин. У деяких варіантах здійснення винаходу Сполуки 1 і 2 можна застосовувати без введення одного або декількох додаткових терапевтичних агентів, таких як агоніст рецептора інтерферону та/або рибавірин. В одному з варіантів здійснення винаходу агоніст рецептора інтерферону може представляти собою агоніст рецептора інтерферону типу I, такий як пегілований агоніст рецептора інтерферону типу I. Один або декілька додаткових терапевтичних засобів можна застосовувати після Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично 10 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 прийнятної солі або проліків. Наприклад, один або декілька додаткових терапевтичних засобів можна застосовувати після закінчення режиму лікування з використанням Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу додатковий терапевтичний засіб може представляти собою агоніст рецептора інтерферону, такий як агоніст рецептора інтерферону типу I. В одному з варіантів здійснення винаходу агоніст рецептора інтерферону типу I може представляти собою пегілований агоніст рецептора інтерферону типу I. У деяких варіантах здійснення винаходу додатковий терапевтичний засіб може представляти собою рибавірин. Чи має спосіб, що пропонується у винаході, ефективність у відношенні лікування інфекції, що викликається HCV, можна визначати різними шляхами, наприклад, за зниженням вірусного навантаження, за зниженням часу до сероконверсії (вірус неможливо виявити в сироватці пацієнта), за підвищенням рівня тривалої вірусної відповіді на терапію, за зниженням захворюваності або смертності при оцінці клінічного результату, або за іншими показниками реакції захворювання. Таким чином, питання про те, чи має спосіб, що пропонується у винаході, ефективність у відношенні лікування інфекції, що викликається HCV, можна вирішувати, оцінюючи вірусне навантаження, або оцінюючи параметр, асоційований з інфекцією, що викликається HCV, такими як (але не обмежуючись тільки ними) фіброз печінки, підвищення в сироватці рівнів трансаміназ та активність некрозапального процесу в печінці. В деяких варіантах здійснення винаходу введення та/або застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків у комбінації може знижувати вірусне навантаження більшою мірою, ніж зниження вірусного навантаження, що досягається при окремому застосуванні Сполуки 1 або її фармацевтично прийнятної солі або проліків, практично у такій же кількості. Наприклад, використання комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може знижувати вірусне навантаження, принаймні, приблизно на 10 %, принаймні, приблизно на 15 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 40 %, принаймні, приблизно на 50 %, принаймні, приблизно на 60 %, принаймні, приблизно на 70 %, принаймні, приблизно на 80 % або принаймні, приблизно на 90 % або більше, у порівнянні зі зниженням вірусного навантаження HCV, яка досягається при застосуванні Сполуки 1 або її фармацевтично прийнятної солі або проліків практично у такій же кількості в якості монотерапії. У деяких варіантах здійснення винаходу введення та/або застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків у комбінації може знижувати вірусне навантаження більшою мірою, ніж зниження вірусного навантаження, як досягається при окремому застосуванні Сполуки 2 або її фармацевтично прийнятної солі або проліків практично в такій же кількості. Наприклад, використання комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може знижувати вірусне навантаження, принаймні, приблизно на 10 %, принаймні, приблизно на 15 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 40 %, принаймні, приблизно на 50 %, принаймні, приблизно на 60 %, принаймні, приблизно на 70 %, принаймні, приблизно на 80 % або принаймні, приблизно на 90 % або більше, у порівнянні зі зниженням вірусного навантаження HCV, яке досягається при застосуванні Сполуки 2 або її фармацевтично прийнятної солі або проліків практично в такій же кількості в якості монотерапії. У деяких варіантах здійснення винаходу кількість Сполуки 1 або її фармацевтично прийнятної солі або проліків та кількість Сполуки 2 або її фармацевтично прийнятної солі або проліків представляє собою синергічну кількість. У контексті даного опису "синергічна комбінація" або "синергічна кількість" відноситься до об'єднаної дози, яка є більш ефективною у відношенні терапевтичного або профілактичного лікування інфекції, що викликається HCV, ніж інкрементальне поліпшення результату лікування, яке можна прогнозувати або очікувати для комбінації, що має тільки адитивну дію у відношенні (I) терапевтичного або профілактичного сприятливого впливу Сполуки 1 або її фармацевтично прийнятної солі або проліків при її введенні в такій же дозі в якості монотерапії, та (II) терапевтичного або профілактичного сприятливого впливу Сполуки 2 або її фармацевтично прийнятної солі або проліків при її введенні в такій же дозі в якості монотерапії. Поняття "синергічна комбінація" або "синергічна кількість" відноситься також до об'єднаної дози, яка є більш ефективною у відношенні терапевтичного або профілактичного лікування інфекції, що викликається HCV, ніж прогнозовано або очікувано для комбінації на основі правила сумішей у відношенні (I) терапевтичного або профілактичного сприятливого впливу Сполуки 1 або її фармацевтично прийнятної солі або проліків та (II) терапевтичного або профілактичного сприятливого впливу Сполуки 2 або її фармацевтично прийнятної солі або проліків. Таким чином, у деяких варіантах 11 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 здійснення винаходу використання комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може знижувати вірусне навантаження, принаймні, приблизно на 10 %, принаймні, приблизно на 15 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 40 %, принаймні, приблизно на 50 %, принаймні, приблизно на 60 %, принаймні, приблизно на 70 %, принаймні, приблизно на 80 % або, принаймні, приблизно на 90 % або більше, у порівнянні зі зниженням вірусного навантаження HCV, прогнозованого або очікуваного на основі правила сумішей або прогнозованого або очікуваного для адитивної комбінації у відношенні зниження вірусного навантаження при введенні Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу вищевказані рівні зниження вірусного навантаження є усередненими для популяції індивідуумів. Вірусне навантаження HCV і зниження вірусного навантаження можна визначати за допомогою методів, відомих у даній області техніки. Наприклад, вірусне навантаження HCV можна визначати, оцінюючи рівні РНК HCV за допомогою прийнятного аналізу, такого як ПЦРаналіз зі зворотною транскриптазою. В одному з варіантів здійснення винаходу аналіз ® ® представляє собою RUO-(тільки для дослідницьких цілей)-тест COBAS AmpilPrep/COBAS ® Taqman HCV. Комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може скорочувати період часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію. Наприклад, комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може скорочувати період часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію, у порівнянні з періодом часу, необхідного для досягнення у індивідуума тривалої вірусної відповіді, при застосуванні практично в такій же кількості Сполуки 1 або її фармацевтично прийнятної солі або проліків в якості монотерапії. В аналогічному або альтернативному варіанті комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може скорочувати період часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію, у порівнянні з періодом часу, необхідного для досягнення в індивідуума тривалої вірусної відповіді, при застосуванні практично в такій же кількості Сполуки 2 або її фармацевтично прийнятної солі або проліків в якості монотерапії. В одному з варіантів здійснення винаходу комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може скорочувати період часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію, принаймні, приблизно на 10 %, принаймні, приблизно на 15 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 40 %, принаймні, приблизно на 50 %, принаймні, приблизно на 60 %, принаймні, приблизно на 70 %, принаймні, приблизно на 80 % або принаймні, приблизно на 90 % або більше, у порівнянні з очікуваним на основі правила сумішей або прогнозованим або очікуваним для адитивної комбінації у відношенні періоду часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію, при застосуванні практично в такій же кількості Сполуки 1 або її фармацевтично прийнятної солі або проліків в якості монотерапії, та періоду часу, необхідного для забезпечення досягнення в індивідуума тривалої вірусної відповіді на терапію, при застосуванні практично в такій же кількості Сполуки 2 або її фармацевтично прийнятної солі або проліків в якості монотерапії. У деяких варіантах здійснення винаходу періоди часу, необхідні для забезпечення досягнення тривалої вірусної відповіді, є усередненими для популяції індивідуумів. Як відзначалось вище, чи є спосіб, що пропонується у винаході, ефективним у відношенні лікування інфекції, що викликається HCV, можна визначати, оцінюючи параметр, асоційований з інфекцією, що викликається HCV, такий як фіброз печінки. Методи визначення ступеню фіброзу печінки відомі фахівцям у даній області. У деяких варіантах здійснення винаходу рівень сироваткового маркера фіброзу печінки свідчить про ступінь фіброзу печінки. Наприклад (але не обмежуючись тільки цим), оцінюють рівні в сироватці аланінамінотрансферази (ALT) за допомогою стандартних аналізів. У цілому, рівень ALT є нижчим приблизно 45 міжнародних одиниць (МЕ), вважається нормальним. У деяких варіантах здійснення винаходу комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків знижує рівень у сироватці маркера фіброзу печінки, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше, у порівнянні з рівнем маркера в індивідуума, що 12 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 зазнав монотерапії (такий як індивідуальне застосування практично в такій же кількості Сполуки 1 або 2 або її фармацевтично прийнятної солі або проліків). В інших варіантах здійснення винаходу комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків знижує рівень у сироватці маркера фіброзу печінки, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше, у порівнянні з рівнем, очікуваним на основі правила сумішей або з очікуваним для адитивної комбінації у відношенні зниження рівнів у сироватці маркера фіброзу печінки при використанні практично в такій же кількості Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків. У деяких варіантах здійснення винаходу рівні в сироватці маркера фіброзу печінки є усередненими для популяції індивідуумів. В індивідуума, у якого лікують хворобливий стан, може бути виявлена стійкість до одного або декількох терапевтичних засобів (наприклад, до Сполуки 1 або її фармацевтично прийнятної солі або проліків та/або Сполуки 2 або її фармацевтично прийнятної солі або проліків). Поняття "стійкість" у контексті даного опису відноситься до пацієнта, для якого характерна відкладена, знижена відповідь та/або відсутність відповіді на терапевтичний(і) засіб(и). Наприклад, вірусне навантаження HCV у пацієнта, який надбав стійкість до противірусного засобу або їх комбінації, може знижуватись меншою мірою, в порівнянні з рівнем зниження вірусного навантаження, характерним для індивідуума до того, як він надбав стійкість до противірусного засобу або їх комбінації, та/або в порівнянні із установленим нормальним середнім зниженням вірусного навантаження. У деяких варіантах здійснення винаходу, рівень стійкості хворобливого стану до терапії може виявитись зниженим у порівнянні з рівнем стійкості, виявленим в індивідуума, якому вводять практично в такій же кількості Сполуки 1 або її фармацевтично прийнятної солі або проліків в якості монотерапії. У деяких варіантах здійснення винаходу рівень стійкості хворобливого стану до терапії може виявитись зниженим у порівнянні з рівнем стійкості, виявленим в індивідуума, якому вводять практично в такій же кількості Сполуки 2 або її фармацевтично прийнятної солі або проліків в якості монотерапії. В інших варіантах здійснення винаходу комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків знижує рівень стійкості хворобливого стану до терапії, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше, у порівнянні з рівнем, очікуваним на основі правила сумішей або очікуваним для адитивної комбінації у відношенні рівнів стійкості при застосуванні практично в такій же кількості Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків. У деяких варіантах здійснення винаходу рівні стійкості є усередненими для популяції індивідуумів. Для деяких індивідуумів, що стосовно яких проводили лікування із приводу HCV, у яких розвилась стійкість або які мали стійкість до одного або декількох терапевтичних засобів, може мати місце явище "віддачі" (по типу рикошету) вірусного навантаження. Поняття "явище віддачі" (по типу рикошету) вірусного навантаження" у контексті даного опису відноситься до істотного, що становить ≥0,5 log МЕ/мл, підвищенню вірусного навантаження відносно його найнижчого рівня, виявленого до закінчення лікування, де найнижчий рівень характеризується зниженням на ≥0,5 log МЕ/мл відносно вихідного рівня. У деяких варіантах здійснення винаходу спільне застосування Сполуки 1 або її фармацевтично прийнятних солей або проліків і Сполуки 2 або її фармацевтично прийнятних солей або проліків приводить до зменшення кількості індивідуумів, у яких виявлено явище "віддачі" вірусного навантаження, у порівнянні з відповідною кількістю індивідуумів, яких піддавали монотерапії з використанням Сполуки 1 або її фармацевтично прийнятної солі або проліків, або Сполуки 2 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу спільне застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків приводить до зниження, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше кількості індивідуумів, у яких виявлено явище "віддачі" вірусного навантаження, у порівнянні з відповідною кількістю індивідуумів, яких 13 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 піддавали монотерапії з використанням Сполуки 1 або її фармацевтично прийнятних солей або проліків, або Сполуки 2 або її фармацевтично прийнятних солей або проліків. У деяких варіантах здійснення винаходу спільне застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків приводить до зниження, принаймні, приблизно до 75 %, до зниження принаймні, приблизно до 50 %, до зниження, принаймні, приблизно до 40 %, до зниження, принаймні, приблизно до 30 %, до зниження, принаймні, приблизно до 20 %, до зниження, принаймні, приблизно до 10 % або до зниження, принаймні, приблизно до 5 % популяції пацієнтів, у яких виявлено явище "віддачі" вірусного навантаження. В інших варіантах здійснення винаходу застосування комбінації Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків знижує відсоток популяції пацієнтів, у яких виявлено явище "віддачі" вірусного навантаження, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше у порівнянні з відсотком, очікуваним на основі правила сумішей. Деякі індивідууми, яких лікували із приводу HCV, у яких розвилась стійкість або які мали стійкість до одного або декількох терапевтичних засобів, можуть бути або можуть ставати нереспондерами. Поняття "нереспондер" у контексті даного опису відноситься до пацієнта, у якого зниження вірусного навантаження в процесі лікування становить ≤ 0,5 log МЕ/мл. У деяких варіантах здійснення винаходу спільне застосування Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків приводить до зниження кількості пацієнтів, що є нереспондерами, у порівнянні з відповідною кількістю пацієнтів, яких піддавали монотерапії з використанням Сполуки 1 або її фармацевтично прийнятної солі або проліків, або Сполуки 1 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу, спільне застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків приводить до зниження, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 % або більше кількості пацієнтів, які є нереспондерами, у порівнянні з відповідною кількістю пацієнтів, яких піддавали монотерапії. У деяких варіантах здійснення винаходу, спільне застосування Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків приводить до зниження, принаймні, приблизно до 75 %, до зниження принаймні, приблизно до 50 %, до зниження принаймні, приблизно до 40 %, до зниження, принаймні, приблизно до 30 %, до зниження, принаймні, приблизно до 20 %, до зниження, принаймні, приблизно до 10 % або до зниження, принаймні, приблизно до 5 % популяції пацієнтів, що є нереспондерами. В інших варіантах здійснення винаходу, комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків знижує відсоток популяції пацієнтів, що є нереспондерами, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або, принаймні, приблизно на 80 % або більше, у порівнянні з відсотком, очікуваним на основі правила сумішей. У додаткових або альтернативних варіантах здійснення винаходу, початок прояву стійкості хворобливого стану до терапії можна затримувати в порівнянні з початком прояву стійкості в пацієнта, якому вводили практично в такій же кількості Сполуки 1 або її фармацевтично прийнятної солі або проліків в якості монотерапії. У деяких варіантах здійснення винаходу початок прояву стійкості хворобливого стану до терапії можна затримувати, в порівнянні з початком прояву стійкості в пацієнта, якому вводили практично в такій же кількості Сполуки 2 або її фармацевтично прийнятної солі або проліків в якості монотерапії. В одному з варіантів здійснення винаходу, Сполука 1 або її фармацевтично прийнятна сіль або проліки можуть затримувати початок прояву стійкості HCV до Сполуки 2 або її фармацевтично прийнятної солі або проліків. Наприклад, Сполука 1 або її фармацевтично прийнятна сіль або проліки можуть затримувати початок прояву стійкості HCV до Сполуки 2 або її фармацевтично прийнятної солі або проліків, що встановлюють за допомогою аналізу реплікону HCV. У контексті даного опису фраза "початок прояву стійкості" відноситься до моменту часу, коли в індивідуума виявляється стійкість до одного або декількох терапевтичних сполук. В одному з варіантів здійснення 14 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 винаходу хвороба може викликатись HCV. У деяких варіантах здійснення винаходу комбінація Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків може представляти собою синергічну комбінацію, при використанні якої початок прояву стійкості може бути затриманий, принаймні, приблизно на 10 %, принаймні, приблизно на 15 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 40 %, принаймні, приблизно на 50 %, принаймні, приблизно на 60 %, принаймні, приблизно на 70 %, принаймні, приблизно на 80 % або, принаймні, приблизно на 90 % або більше, у порівнянні з початком прояву стійкості, прогнозованим або очікуваним на основі правила сумішей, або для адитивної комбінації, що містить практично в такій же кількості Сполуки 1 і 2 або їх фармацевтично прийнятні солі або проліки. У деяких варіантах здійснення винаходу момент початку прояву стійкості представляє собою середнє значення для популяції індивідуумів. Часто при лікування терапевтичними засобами, такими як противірусні сполуки, в індивідуумів можуть бути виявлено одну або декілька побічних дій. У деяких випадках побічні дії можуть бути такого ступеню, що стає неможливо здійснювати або рекомендувати лікування з використанням конкретного засобу, у результаті чого таке лікування не може бути застосоване для деяких індивідуумів або лікування повинне бути припинено. Шляхом зменшення або зниження кількості та/або серйозності побічних дій можна поліпшувати дотримання індивідуумом режиму та схеми лікування (підвищувати "піддатливість" індивідуума лікуванню). У деяких варіантах здійснення винаходу, кількість побічних дій, асоційованих зі спільним застосуванням Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків можна знижувати, в порівнянні з кількістю побічних дій, виявлених в індивідуума, якому вводили практично в такій же кількості Сполуку 1 або її фармацевтично прийнятну сіль або проліки в якості єдиної діючої речовини. У деяких варіантах здійснення винаходу кількість побічних дій, асоційованих зі спільним застосуванням Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків можна знижувати, в порівнянні з кількістю побічних дій, виявлених в індивідуума, якому вводили практично в такій же кількості Сполуку 2 або її фармацевтично прийнятну сіль або проліки в якості єдиної діючої речовини. У деяких варіантах здійснення винаходу в індивідуума, якому вводять комбінацію Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків, може бути виявлена менша кількість побічних дій, ніж кількість побічних дій, прогнозованих або очікуваних на основі правила сумішей або для адитивної комбінації при введенні індивідуумові практично в такій же кількості Сполук 1 і 2 або їх фармацевтично прийнятних солей або проліків, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або, принаймні, приблизно на 80 %. У деяких варіантах здійснення винаходу кількість побічних дій представляє собою середнє значення для популяції індивідуумів. Як відзначалось раніше, "піддатливість" індивідуумів противірусному лікуванню можна підвищувати шляхом зниження серйозності однієї або декількох побічних дій, асоційованих з монотерапією з використанням діючих речовин. У деяких варіантах здійснення винаходу серйозність побічних дій, асоційованих із застосуванням комбінації Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків, знижують, у порівнянні із серйозністю побічних дій, виявлених в індивідуума при введенні йому Сполуки 1 або її фармацевтично прийнятної солі або проліків в якості монотерапії. В одному з варіантів здійснення винаходу серйозність побічних дій, асоційованих із застосуванням комбінації Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків, знижують у порівнянні із серйозністю побічних дій, виявлених в індивідуума при введенні йому Сполуки 2 або її фармацевтично прийнятної солі або проліків в якості монотерапії. У деяких варіантах здійснення винаходу серйозність побічних дій, асоційованих із застосуванням комбінації Сполуки 1 або її фармацевтично прийнятної солі або проліків та Сполуки 2 або її фармацевтично прийнятної солі або проліків, може бути знижена, принаймні, приблизно на 10 %, принаймні, приблизно на 20 %, принаймні, приблизно на 25 %, принаймні, приблизно на 30 %, принаймні, приблизно на 35 %, принаймні, приблизно на 40 %, принаймні, приблизно на 45 %, принаймні, приблизно на 50 %, принаймні, приблизно на 55 %, принаймні, приблизно на 60 %, принаймні, приблизно на 65 %, принаймні, приблизно на 70 %, принаймні, приблизно на 75 % або принаймні, приблизно на 80 %, у порівнянні із серйозністю побічних дій, прогнозованих або очікуваних на основі правила сумішей або для адитивної комбінації у відношенні 15 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 серйозності побічних дією, асоційованих із введенням практично в такій же кількості Сполуки 1 або її фармацевтично прийнятної солі або проліків та практично в такій же кількості Сполуки 2 або її фармацевтично прийнятної солі або проліків. У деяких варіантах здійснення винаходу серйозність побічної дії є усередненою для популяції індивідуумів. Як має бути очевидним фахівцеві в даній області техніки, придатна для введення in vivo доза та конкретний шлях введення повинні варіюватись залежно від віку, ваги, серйозності поразки та видів ссавців, що підлягають лікуванню, конкретних застосовуваних сполук і конкретної мети, для якої використовують зазначені сполуки (див., наприклад, в Fingl та ін., в: "The Pharmacological Basis of Therapeutics", 1975, публікація повністю включена в даний опис в якості посилання з конкретною вказівкою на главу 1, с. 1). Визначення ефективних рівнів доз, що представляють собою рівні доз, необхідні для досягнення необхідного результату, може здійснювати фахівець у даній області техніки на основі загальноприйнятих фармакологічних методів. Як правило, введення людині продуктів, рекомендованих для клінічного застосування, починають із більш низьких рівнів доз, потім рівень доз підвищують до досягнення необхідної дії. В альтернативному варіанті можна застосовувати досліди in vitro для визначення необхідних доз і шляхів введення композицій, зазначених у способах, що пропонуються у винаході, використовуючи прийняті фармакологічні методи. Хоча точну дозу можна визначати на основі вивчення взаємодії лікарського засобу з лікарським засобом (drug-by-drug), у більшості випадків можна зробити деякі узагальнення у відношенні дози. Доза може представляти собою однократну дозу або серії із двох або більшої кількості доз, які приймаються у вигляді курсу на протязі одного або декількох днів, залежно від потреб індивідуума. У деяких варіантах здійснення винаходу сполуку слід застосовувати в режимі тривалої терапії, наприклад, на протязі одного або декількох тижнів, або на протязі декількох місяців, або років. У випадку, коли дози сполук, призначені для людини, встановлені, принаймні, для якогось стану, можна застосовувати такі ж дози або дози, що становлять приблизно від 0,1 % до 500 %, більш переважно приблизно від 25 % до 250 % від встановленої для людини дози. Якщо доза для людини не встановлена, як буває у випадку вперше розроблених фармацевтичних композицій, то висновок про прийнятну для людини дозу можна робити на основі значень ED 50 або ID50 або інших придатних значень, встановлених у дослідах in vitro або in vivo, що визначаються у токсикологічних дослідження та у дослідах по оцінці ефективності на тваринах. У випадках застосування фармацевтично прийнятної солі, дози можна перераховувати на вільну основу. Фахівцям у даній області має бути очевидним, що в певних ситуаціях може виявитись необхідним вводити сполуки, що представлені в даному описі, у кількості, що перевищує, або навіть значно перевищує зазначений вище діапазон доз, для того щоб ефективно та енергійно лікувати найбільш агресивні захворювання або інфекції. Рівень дози та інтервал між введеннями доз можна регулювати окремо для забезпечення рівнів у плазмі активного фрагменту, достатніх для підтримки дій, що модулюють, або мінімальної ефективної концентрації (MEC). Величина MEC може варіюватись для кожної сполуки, але її можна визначати на основі даних, отриманих in vitro. Дози, необхідні для досягнення MEC, повинні залежати від індивідуальних характеристик і шляху введення. При цьому ЖХВР-аналізи або біологічні аналізи можна застосовувати для визначення концентрацій у плазмі. Інтервали між введенням доз можна визначати також, використовуючи значення MEC. Композиції слід застосовувати згідно зі схемою приймання, яка забезпечує підтримку рівнів, що перевищують MEC на протязі 10-90 % часу, переважно 30-90 % і найбільш переважно 50-90 %. У випадках місцевого застосування або вибіркового поглинання ефективна місцева концентрація лікарського засобу може не бути пов'язана з концентрацією в плазмі. Слід зазначити, що штатний лікар повинен знати, як і коли припиняти, переривати або регулювати введення через токсичність або дисфункції органів. Крім того, штатний лікар повинен знати також, як регулювати лікування, підвищуючи рівні доз, якщо клінічна відповідь не є адекватною (запобігання токсичності). Рівень дози, що вводиться при лікуванні порушення, що представляє інтерес, повинен варіюватись залежно від серйозності стану, що підлягає лікуванню, і шляхи введення. Серйозність стану можна, наприклад, частково визначати за допомогою стандартних прогностичних методів оцінки. Крім того, доза та можлива частота застосування доз повинна варіюватись також залежно від віку, ваги тіла та відповіді окремого пацієнта. Програму, порівнянну з обговореної вище, можна використовувати у ветеринарії. У дослідах на тваринах крім людини застосування перспективних продуктів починають, використовуючи більш високі рівні доз, зі зниженням дози доти, поки необхідна дія більше не досягається, або до припинення побічних дій. Дозу можна варіювати в широких межах залежно 16 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 від необхідних дій і терапевтичного показання. В іншому варіанті дози можуть бути зв'язані та розраховані з урахуванням площі поверхні тіла пацієнта, що повинно бути очевидним фахівцям у даній області техніки. У сполук, представлених у даному описі, можна оцінювати ефективність і токсичність, використовуючи відомі методи. Наприклад, токсикологічні дані для конкретної сполуки або набору сполук, що характеризуються певними хімічними залишками, можна одержувати, визначаючи токсичність in vitro у відношенні клітинної лінії, такої як клітинна лінія ссавців і переважно людини. Результати таких досліджень часто можуть дозволяти прогнозувати токсичність для тварин, таких як ссавці та більш конкретно людей. В альтернативному варіанті можна визначати токсичність конкретних сполук із використанням тваринних моделей, таких як миші, пацюки, кролики або мавпи, за допомогою відомих методів. Ефективність конкретної сполуки можна визначати, використовуючи декілька методів дослідження, таких як методи in vitro, тваринні моделі або клінічні дослідження за участю людей. Аналогічно цьому, прийнятні тваринні моделі можна застосовувати для визначення ефективності хімічних сполук у відношенні лікування зазначених станів. При виборі моделі для визначення ефективності фахівець у даній області може керуватись рівнем техніки в даній області для вибору відповідної моделі, дози та шляху введення та схеми введення лікарського засобу. Очевидно, що клінічні дослідження за участю людей можна застосовувати також для визначення ефективності сполуки або композицій для людей. Будь-які композиції та способи, представлені в даному описі, можна застосовувати для індивідуумів, у яких діагностована інфекція, що викликається HCV. Будь-які композиції та способи, представлені в даному описі, можна застосовувати для індивідуумів, які не піддавались попередньому лікуванню інфекції, що викликається HCV ("пацієнти, що не піддаються лікуванню (лікування яких є безуспішним)», включаючи як нереспондерів, так і пацієнтів, що мають рецидив). У багатьох варіантах здійснення винаходу найбільший інтерес представляють індивідууми, у яких клінічно діагностовано HCV. Індивідуумів, інфікованих HCV, ідентифікують за присутністю РНК HCV у крові та/або за присутністю антитіла до HCV у сироватці. Такі індивідууми включають індивідуумів, позитивних за антитілами до HCV за даними ELISA, та індивідуумів, позитивних за даними аналізу методом рекомбінантного імуноблоттингу (RIBA). У таких індивідуумів можуть бути також, але не обов'язково, підвищені рівні ALT у сироватці. Індивідууми, у яких клінічно діагностовано наявність заражень HCV, включають "необроблених" індивідуумів (наприклад, індивідуумів, яких раніше не лікували із приводу HCV, насамперед індивідуумів, яких раніше не піддавали терапії на основі IFN-α та/або рибавірину) та індивідуумів, які не піддавались попередньому лікуванню інфекції, що викликається HCV ("пацієнти, що не піддаються лікуванню"). До пацієнтів, що не піддаються лікуванню, відносяться нереспондери (тобто індивідууми, в організмі яких титр HCV несуттєво або недостатньо знижувався при раніше здійснюваному лікуванні інфекції, що викликається HCV, наприклад, при застосовуваній раніше монотерапії на основі IFN-α, застосовуваній раніше комбінованій терапії на основі IFN-α та рибавірину, або застосовуваній раніше комбінованій терапії на основі пегілованого IFN-α та рибавірину); та пацієнти, що мають рецидив (тобто індивідууми, яких раніше лікували з приводу HCV, наприклад, яких піддавали раніше монотерапії на основі IFN-α, піддавали раніше комбінованій терапії на основі IFN-α та рибавірину або піддавали раніше комбінованій терапії на основі пегілованого IFN-α та рибавірину, в організмі яких титр HCV спочатку знижувався, але потім відбувалось його підвищення). В одному з варіантів здійснення винаходу в HCV-позитивних індивідуумів титр HCV 5 5 становить, принаймні, приблизно 10 , принаймні, приблизно 5 × 10 або принаймні, приблизно 6 6 10 , або принаймні, приблизно 2 × 10 геномних копій HCV на мілілітр сироватки. Пацієнт може бути заражений HCV будь-якого генотипу (таким як генотип 1, включаючи 1a та 1b, 2, 3, 4, 6 і т.д., та підтипи (наприклад, 2a, 2b, 3a і т.д.)), найбільше важко лікувати інфекцію, що викликається таким генотипом, як генотип 1 HCV та конкретні підтипи та квазівиди HCV. У деяких варіантах здійснення винаходу HCV-позитивні індивідууми (описані вище) включають індивідуумів, що страждають серйозним фіброзом або раннім цирозом (недекомпенсований, класу А або нижче за шкалою Чайлда-П'ю), або більш запущеним цирозом (декомпенсований, класу B або C за шкалою Чайлда-П'ю), обумовленим хронічним зараженням HCV, та індивідуумів, які мають віремію незважаючи на попереднє противірусне лікування за допомогою заснованих на застосуванні IFN-α терапій, або індивідуумів, які не можуть переносити засновані на застосуванні IFN-α терапії, або індивідуумів, які мають протипоказання до зазначених терапій. В одному з варіантів здійснення винаходу HCV 17 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 позитивних індивідуумів, що мають 3 або 4 стадії фіброзу печінки за шкалою METAVIR, можна лікувати за допомогою композицій та способів, представлених у даному описі. В інших варіантах здійснення винаходу індивідууми, яких можна лікувати за допомогою композицій та способів, що пропонуються у даному описі, представляють собою пацієнтів з декомпенсованим цирозом, що мають клінічні прояви, включаючи пацієнтів з давно запущеним цирозом печінки, включаючи таких, що потребують трансплантації печінки. У наступних варіантах здійснення винаходу індивідууми, яких можна лікувати за допомогою композицій та способів, що пропонуються у даному описі, представляють собою пацієнтів з більш слабким ступенем фіброзу, у тому числі з раннім фіброзом (1 та 2 стадії за шкалою METAVIR та за шкалами Людвіга та Шейєра; або 1, 2 або 3 стадії за шкалою Ісхака). Приклади Варіанти здійснення винаходу описані більш детально в наступних прикладах, які жодним чином не спрямовані на обмеження обсягу формули винаходу. Приклад 1 Протоколи лікування Проводили рандомізоване, подвійно-сліпе, плацебо-контрольоване, з використанням різних доз, дослідження Сполуки 1a та Сполука 2 на дорослих пацієнтах, що страждають хронічним гепатитом C генотипу 1. У дослідження було включено приблизно 54 "необроблених" чоловіків і жінок віком з 18 до 65 років (включно), заражених генотипом 1 HCV, яких раніше не лікували інтерфероном або досліджуваним терапевтичним засобом проти HCV. У дослідження було включено додатково приблизно 20 пацієнтів, що раніше лікувались, які не піддавались лікуванню, проведеному згідно із сучасними стандартами догляду за хворими (нульова відповідь, часткові респондери та такі, що мають рецидив). В індивідуумів брали біопсію печінки або їх піддавали неінвазивній (наприклад, за допомогою обладнання Фіброскан) процедурі в період часу, що становить 24 календарних місяця до введення першої дози, за допомогою яких підтверджений діагноз хронічного гепатиту С з цирозом. Приймали також дані біопсії печінки, отримані на протязі 5 років захворювання, що демонструють фазу захворювання F0 або F1. Із цього конкретного протоколу виключені пацієнти із цирозом печінки або з неповним/ цирозом, що має перехідну форму, або іншими формами хвороби печінки, анемією, ВІЛ- або HBVінфекцією, гепатоклітинною карциномою, хворобою серця або хворобою нирок, а також вагітні жінки та жінки в період лактації, жінки потенційно дітородного віку та чоловіки, що є партнерами вагітних жінок або жінок у період лактації. Жінок потенційно недітородного віку визначали як таких, що перебувають на стадії постменопаузи (відсутність спонтанних менструацій на протязі, принаймні, року, що підтверджено результатами лабораторної оцінки FSH та LH); стерилізованих хірургічним шляхом (статус після гістеректомії або перев'язки труб, принаймні, за 6 місяців); та/або таких, що мають природню стерильність (аменорея на протязі принаймні, 1 року). Дослідження проводили в 7 групах (I, II, III, IV, V, VI і VII) індивідуумів. Зразки крові відбирали у відповідні моменти часу для визначення фармакокінетичних параметрів, включаючи Tmax, Cmax, Kel, T1/2, AUC, CL/F, Vd/F та рівень накопичення. Конкретні дані у відношенні груп I, II, III, IV, V, VI і VII представлені нижче в таблиці 1 та проілюстровані на фіг. 2 і 3. Група два складалась із двох підгруп, при цьому в підгрупу 1 входило 8 індивідуумів і в підгрупу 2 входило 8 індивідуумів. У підгрупі 1 Сполуку 1a вводили на протязі усіх семи днів, а Сполуку 2 вводили в останній половині семиденного періоду. У підгрупі 2 Сполуку 2 вводили на протязі усіх семи днів, а Сполуку 1a вводили в останній половині семиденного періоду. У групу II входило в цілому 10 індивідуумів. Індивідууми одержували або Сполуку 1a та 2, або плацебо на протязі усіх чотирнадцяти днів. Дослідження групи I та групи II завершували послідовно. У групах III і IV індивідуумам вводили Сполуку 1a та 2 або плацебо на протязі усіх чотирнадцяти днів. У групу III входило 18 індивідуумів, яким вводили або 500 мг Сполуки 1a і 200 мг Сполуки 2, або 1000 мг Сполуки 1a та 100 мг Сполуки 2, або плацебо, відповідне Сполукам 1a та 2. У групу IV входило 12 індивідуумів. 8 індивідуумів у групі IV одержували 1000 мг Сполуки 1a та 200 мг Сполуки 2, а 4 індивідуума одержували плацебо. Дослідження груп III і IV завершували послідовно після одержання даних про безпеку для груп I і II. У групах V, VI і VII індивідууми одержували Сполуку 1a та 2 або плацебо на протязі усіх чотирнадцяти днів. У групу V входило 10 індивідуумів, класифікованих як "такі, що не повністю не піддаються лікуванню (не нульова відповідь)». Вісім індивідуумів у групі V одержували 1000 мг Сполуки 1a та 600 мг Сполуки 2, а 2 індивідуума в групі V одержували плацебо, відповідне до Сполук 1a та 2. У групу VI входило 10 індивідуумів, класифікованих як "такі, що повністю не піддаються лікуванню" (нульова відповідь)». Вісім індивідуумів у групі VI одержували 1000 мг 18 UA 104754 C2 Сполуки 1a та 900 мг Сполуки 2, а 2 індивідуума одержували плацебо, відповідне до Сполук 1a та 2. У групу VII входило 10 індивідуумів. Вісім індивідуумів у групі VII одержували 1000 мг Сполуки 1a та 900 мг Сполуки 2, а 2 індивідуума в групі VII одержували плацебо, відповідне до Сполук 1a та 2. 5 Таблиця 1 Група I підгрупа 1 (n=8) I підгрупа 2 (n=8) II підгрупа 1 (n=8) II підгрупа 2 (n=2) III підгрупа 1 (n=8) III підгрупа 2 (n=8) III підгрупа 3 (n=2) IV підгрупа 1 (n=8) IV підгрупа 2 (n=4) V TF ("не нуль") підгрупа 1 (n=8) V TF ("не нуль") підгрупа 2 (n=2) VI TF ("нуль") підгрупа 1 (n=8) VI TF ("нуль") підгрупа 2 (n=2) VII "необроблені" пацієнти підгрупа 1 (n=8) VII "необроблені" пацієнти підгрупа 2 (n=2) Сполука 1a 2 плацебо 2 2 1a плацебо 1a 1a Кількість 500 мг NA 100 мг 100 мг NA 500 мг 500 мг Частота bid q8h q8h q8h bid bid bid Схема Дні 1-7 Дні 1-3 Дні 4-7 Дні 1-7 Дні 1-3 Дні 4-7 Дні 1-14 2 100 мг q8h Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA q8h Дні 1-14 1a 500 мг bid Дні 1-14 2 200 мг q8h Дні 1-14 1a 1000 мг bid Дні 1-14 2 100 мг q8h Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA q8h Дні 1-14 1a 1000 мг bid Дні 1-14 2 200 мг Q8h Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA Q8h Дні 1-14 1a 1000 мг bid Дні 1-14 2 600 мг bid Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA bid Дні 1-14 1a 1000 мг bid Дні 1-14 2 900 мг bid Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA bid Дні 1-14 1a 1000 мг bid Дні 1-14 2 900 мг bid Дні 1-14 1a плацебо NA bid Дні 1-14 2 плацебо NA bid Дні 1-14 * bid – двічі на день * q8h – кожні 8 г * NA – не застосовно *TF = пацієнти, що не піддаються лікуванню згідно із сучасними стандартами догляду за хворими (нульова відповідь, часткові респондери та такі, що мають рецидив) 19 UA 104754 C2 5 10 15 20 25 30 35 40 45 50 55 60 Оцінки фармакокінетичних параметрів Зразки крові обсягом по 4 мл одержували у такий спосіб: Група I Сполука 1a - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 1a о 12 г - День 3: в "переддозовий" період і через 0,5, 1, 2, 3, 4, 8 і 12 г після введення дози - День 4: перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 2, 3, 4, 8 і 12 г після введення дози Сполука 2 - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 2 о 8 г - День 3: в "переддозовий" період і через 0,5, 1, 1,5, 2, 3, 4 і 8 г після введення дози - День 4: перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 1,5, 2, 3, 4 і 8 г після введення дози Групи II, III і IV Сполука 1a - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 1a о 12 г - День 4: перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 2, 3, 4, 8 і 12 г після введення дози - День 14: в "переддозовий" період Сполука 2 - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 2 о 8 г - День 4: перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 1,5, 2, 3, 4 і 8 г після введення дози - День 14: в "переддозовий" період Групи V, VI і VII Сполука 1a - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 1a о 12 г - День 4: перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 2, 3, 4, 8 і 12 г після введення дози - День 10: в "переддозовий" період - День 14: в "переддозовий" період Сполука 2 - День 1: в "переддозовий" період перед введенням першої дози досліджуваних лікарських засобів і безпосередньо перед дозуванням Сполуки 1a о 12 г - День: 4 перед ранковим введенням дози - День 7: в "переддозовий" період і через 0,5, 1, 2, 3, 4, 8 і 12 г після введення дози - День 10: в "переддозовий" період - День 14: в "переддозовий" період Концентрації в плазмі Сполуки 1 (і при необхідності її метаболітів) і Сполуки 2 вимірювали валідованими методами рідинної хроматографії/тандемної мас-спектрометрії (РХ-МС/МС). Фармакокінетичні параметри кожної сполуки визначали за допомогою стандартних некомпартментальних методів з використанням програми WinNonlin (версія 5.2, фірма Pharsight Co.). Визначення вірусного навантаження РНК HCV та оцінка стійкості вірусу Зразки крові для оцінки РНК HCV (противірусна активність ± стійкість) збирали під час лікування та у наступний період у такий спосіб: Група I - Скринінг - День 1: у межах 1 г до ранкового введення доз і через 4 і 12 г після ранкового введення доз - Дні 5, 6, і 7: у межах 1 г до ранкового введення доз - Дні 8, 14, 35 і 91 Групи II, III і IV - Скринінг - День 1: у межах 1 г до ранкового введення доз і через 4 і 12 г після ранкового введення доз 20 UA 104754 C2 - Дні 2, 3, і 4: у межах 1 г до ранкового введення доз і через 12 г після ранкового введення доз - Дні 5, 6, 7, 10, 13 і 14: у межах 1 г до ранкового введення доз - Дні 15, 21, 42 і 98 Групи V, VI і VII - Скринінг - День 1: у межах 1 г до ранкового введення доз і через 4, 12 і 16 г після ранкового введення 5 доз - Дні 2 і 14: у межах 1 г до ранкового введення доз і через 12 г після ранкового введення доз - Дні 3 і 4: у межах 1 г до ранкового введення доз і через 12 і 16 г після ранкового введення 10 доз 15 20 25 30 35 40 - Дні 5, 6, 7, 10 і 13: у межах 1 г до ранкового введення доз - Дні 21, 42 і 98. Отримані в "переддозовий" період зразки відбирали в межах 1 г до дозування. Приблизно 10 мл крові застосовували як для визначення вірусного навантаження РНК HCV, так і для оцінки ® ® стійкості вірусу. Рівні РНК HCV визначали за допомогою RUO-тесту COBAS AmpilPrep/COBAS ® Taqman HCV. Він представляв собою ПЦР у реальному часі. Оцінку HCV і РНК здійснювали в певні моменти часу. Для кожної підгрупи в кожній групі одержували середні та індивідуальні дані про вірусне навантаження (абсолютні та зміна від вихідного рівня). Визначали перелік індивідуальних змін від вихідного рівня. Для підгрупи, яку піддавали лікуванню, представлено узагальнення даних про рівень РНК HCV у кожний номінальний момент часу. Певні зразки крові, які збирали для оцінки вірусного навантаження, використовували для фенотипічного аналізу та аналізу послідовностей для моніторингу розвитку стійкості до Сполуки 1 та Сполуки 2 в індивідуумів, у яких було виявлено або явище "віддачі" вірусного навантаження, або відсутність відповіді при лікуванні Сполукою 1 та/або Сполукою 2. Популяційне секвенування повної кодуючої послідовності NS5B-полімерази та/або NS3/4A HCV усіх отриманих у вихідній момент часу зразків здійснювали за допомогою стандартної технології секвенування. Для індивідуумів, у яких було виявлено явище "віддачі" вірусного навантаження, були спроби визначити кодуючу послідовність популяції NS5B (а) у вихідній момент часу та (б) у першому зразку, узятому після виявлення явища "віддачі" вірусного навантаження. Визначали амінокислотні заміни в зразках після явища "віддачі" вірусного навантаження в порівнянні з відповідною послідовністю у вихідній момент часу для кожного відібраного індивідуума. Вторинний аналіз включав секвенування повного геному HCV, секвенування зразків, отриманих від індивідуумів, які мали вірусологічну відповідь, та визначення послідовностей другорядних квазівидів. Здійснювали досліди з оцінки фенотипу для моніторингу стійкості до Сполуки 1 та Сполуки 2 зразків, описаних у підпунктах (a) та (б), і вони включали аналіз зразків від індивідуумів, що мають вірусологічну відповідь. Оцінку перехресної стійкості до інших інгібіторів HCV та аналізи послідовностей здійснювали на відібраних зразках, і для цієї мети може бути необхідним проведення ампліфікації та субклонування послідовностей з геному HCV. У таблиці 2 представлені результати оцінки кінетики вірусів для групи I, підгрупи 1 і 2 для варіанта, у якому Сполуку 1a або Сполуку 2 використовували в якості монотерапії, після 7 днів лікування. Результати представлені у вигляді середньої зміни (в log) рівня РНК-полімерази HCV (МЕ/мл) від вихідного рівня. 45 Таблиця 2 Підгрупа 1 Підгрупа 2 Сполука 2, монотерапія, 100 мг, q8h Сполука 1a, монотерапія, 750 мг, bid -2,9 -3,2 -1,7 -1,3 Як видно із результатів, представлених у таблиці 2, в індивідуумів, яких лікували комбінацією Сполук 1a і 2, виявлено зниження вірусного навантаження, в порівнянні з індивідуумами, яких лікували Сполукою 1a або Сполукою 2 у якості монотерапії. 21 UA 104754 C2 5 У таблицях 3 і 4 представлені вірусологічні результати для груп I-VII. У індивідуумів, яких лікували комбінацією Сполук 1a і 2 без пегілованого інтерферону або рибавірину, виявлено медіанне зниження рівнів вірусу на величину, що становить від -4,8 до -4,6 log10 МЕ/мл, при всіх схемах лікування та для всіх протестованих популяцій пацієнтів. Крім того, вірусне навантаження в пацієнтів досягло рівня нижче межі виявлення, в перекладі на стандартний режим догляду за хворими. Таблиця 3 Схема: Сполука 1a, Група мг /Сполука 2, мг n Популяція пацієнтів 500/100 II 8 «Необроблені» 500/200 III 8 «Необроблені» 1000/100 III 7 «Необроблені» 1000/200 IV 8 «Необроблені» Зміна РНК НCV від вихідного рівня (Log10 МЕ/мл) медіанне значення (діапазон) -3,9 (від -5,0 до –2,9) -5,2 (від –5,5 до –3,1) -4,8 (від –5,7 до –4,5) -4,8 (від –5,5 до –2,7) РНК HCV < LLOQ (< 43 МЕ/мл) N (%) 1/8 (13) 5/8 (63) 5/7 (71) 5/8 (63) РНК HCV < LLOD (< 15 МЕ/мл) N (%) 1/8 (13) 2/8 (25) 2/7 (29) 2/8 (25) * LLOQ = нижня межа кількісного визначення при аналізі методом Roche TaqMan (< 43 МЕ/мл) * LLOD = нижня межа виявлення при аналізі методом Roche TaqMan (< 15 МЕ/мл) Таблиця 4 Схема: Сполука 1a, мг/ Сполука 2, мг Група n 1000/600 V 8 1000/900 VI 8 1000/900 VII 8 Зміна РНК НCV від вихідного рівня Популяція (Log10 МЕ/мл) пацієнтів медіанне значення (діапазон) TF-4,0 «не-нуль» (від –6,0 до –2,5) -4,9 TF-«нуль» (від –5,3 до –3,5) -5,1 «Необроблені» (від –5,9 до –3,0) РНК HCV < LLOQ (
ДивитисяДодаткова інформація
Назва патенту англійськоюCombination of a nucleoside polymerase inhibitor with a macrocyclic protease inhibitor and use thereof in the treatment of hepatitis c, liver fibrosis and impaired liver function
Автори англійськоюPorter, Steven, B., Bradford, Williamson, Ziegler, Smith, Patrick, F., Yetzer, Ellen, S., De La Rosa, Abel, Rogers, Michael, D., Symonds, William, T.
Автори російськоюПотер Стивен Б., Бредфорд Вылльямсон Циглер, Смит Патрик Ф., Йетцер Еллен С., Де Ла Роса Абель, Роджерс Майкл Д., Саймондс Вилльям Т.
МПК / Мітки
МПК: A61P 1/16, A61P 31/14, A61K 31/7068, A61K 31/4709
Мітки: протеази, макроциклічного, полімерази, функції, лікування, фіброзу, комбінація, застосування, нуклеозидного, печінки, порушеної, інгібітора, гепатиту
Код посилання
<a href="https://ua.patents.su/36-104754-kombinaciya-nukleozidnogo-ingibitora-polimerazi-ta-makrociklichnogo-ingibitora-proteazi-ta-zastosuvannya-dlya-likuvannya-gepatitu-s-fibrozu-pechinki-ta-porusheno-funkci-pechinki.html" target="_blank" rel="follow" title="База патентів України">Комбінація нуклеозидного інгібітора полімерази та макроциклічного інгібітора протеази та її застосування для лікування гепатиту с, фіброзу печінки та порушеної функції печінки</a>
Фармацевтична композиція інгібітора протеази вірусу гепатиту c

Номер патенту: 104195
Опубліковано: 10.01.2014
Автори: Чень Фен-Цзін, Швабе Роберт Дж.
МПК: A61K 9/107
Мітки: інгібітора, композиція, протеази, гепатиту, вірусу, фармацевтична
Формула / Реферат:
1. Рідка фармацевтична композиція, яка містить(а) сполуку формули (1): (1)або її фармацевтично прийнятну сіль;(б) один або декілька фармацевтично прийнятних ліпідів; і(в) одну або декілька фармацевтично прийнятних гідрофільних поверхнево-активних речовин.2. Фармацевтична композиція за п. 1, в якій сполука формули (1) присутня у...
Комбінація інгібітора протеази ns3 hcv з інтерфероном і рибавірином

Номер патенту: 103496
Опубліковано: 25.10.2013
Автори: Хуан Девід, Стерн Джеррі О., Штайнманн Герхард Густав
МПК: A61K 38/21, A61K 31/4709, A61K 31/7056, A61P 31/12
Мітки: комбінація, інгібітора, протеази, інтерфероном, рибавірином
Формула / Реферат:
1. Застосування комбінації, що включає:(а) сполуку наступної формули (1) або її фармацевтично прийнятну сіль:(1);(б) пегільований інтерферон альфа; і(в) рибавірин,для готування лікарського засобу, призначеного для лікування інфекції, викликаної вірусом гепатиту С (НCV), або полегшення одного або декількох її симптомів.2....
Спосіб підвищення біодоступності інгібітора протеази ns3/4а вірусу гепатиту с

Номер патенту: 103801
Опубліковано: 25.11.2013
Автор: Тран Джонатан К.
МПК: A61P 31/12, A61K 31/404, A61K 31/426
Мітки: гепатиту, протеази, вірусу, біодоступності, інгібітора, спосіб, підвищення
Формула / Реферат:
1. Спосіб підвищення біодоступності інгібітора протеази NS3/4A вірусу гепатиту С формули І у пацієнта, який включає спільне введення пацієнту сполуки формули І(I),та інгібітора монооксигенази цитохрому Р450, де інгібітор монооксигенази присутній в кількості, достатній для підвищення рівнів в крові І, де інгібітор монооксигенази цитохрому Р450 являє собою...
Комбінація інгібітора нmg-cоa-редуктази й інгібітора фосфодіестерази 4, призначена для лікування запальних захворювань легенів

Номер патенту: 98466
Опубліковано: 25.05.2012
Автори: Воллін Штефан-Лутц, Маркс Дегенхард, Вольсен Андреа, Браун Клеменс
МПК: A61K 31/502, A61K 45/06, A61K 31/40, A61P 11/00
Мітки: нmg-coa-редуктази, лікування, фосфодіестерази, призначена, запальних, інгібітора, легенів, комбінація, захворювань
Формула / Реферат:
1. Фармацевтична композиція, що являє собою фармацевтичну лікарську форму, що містить у певній кількості інгібітор PDЕ4, у певній кількості інгібітор HMG-CoAредуктази і щонайменше одну фармацевтично прийнятну допоміжну речовину, де інгібітор PDE4 являє собою 2-{4-[(4аS,8аR)-4-(3,4-диметоксифеніл)-1-оксо-4а,5,8,8а-тетрагідро-1Н-фталазин-2-іл]піперидин-1-іл}ацетамід або його фармацевтично прийнятну сіль, інгібітор НМG-СоА-редуктази являє...
Застосування антагоністів рецептора св1 для виробництва композиції для лікування фіброзу печінки

Номер патенту: 94025
Опубліковано: 11.04.2011
Автори: Жюльєн Боріс, Тран ван Нхьє Жанна, Гренар Паскаль, Лотерштайн Софі, Малла Аріан
МПК: A61K 31/415, A61P 1/16, A61K 31/454
Мітки: антагоністів, рецептора, композиції, лікування, застосування, фіброзу, св1, виробництва, печінки
Формула / Реферат:
1. Застосування антагоніста рецептора СВ1 у виробництві композиції для лікування фіброзу печінки.2. Застосування за п. 1, яке відрізняється тим, що антагоніст рецептора СВ1 являє собою специфічний антагоніст рецептора СВ1.3. Застосування за п. 1 або 2, яке відрізняється тим, що антагоніст являє собою сполуку формули II або одну з її фармацевтично прийнятних солей, де g2, g3, g4, g5 і g6 та w2, w3, w4, w5 і w6 є однаковими або...
Попередній патент: Поліморф 6-оксо-6,7,8,9,10,11-гексагідроциклогепта[с]хромен-3-ілсульфамату
Наступний патент: Бетононасос
Випадковий патент: Лінія для виробництва агломераційних недогарків