Спосіб одержання похідних 5-галоідтієноізотіазол-3(2н)-oн-1,1-діоксидів




Формула / Реферат
Способ получения производных 5-гало-идтиеноизотиазол-3 (2Н)-ОН-1,1-диоксидов общей формулы
где R1 - галоид,
или их фармацевтически переносимых кислых аддитивных солей, отличающийся тем, что соединение общей формулы
где R1 – имеет указанное значение
X - галоид,
подвергают взаимодействию с 1-{2-пирими-динил)пиперазином в среде абсолютного диметилформамида при 60°С в течение 45мин. с последующим выделением целевого продукта в свободном состоянии или в виде фармацевтически переносимой кислой аддитивной соли.
Текст
Способ получения производных 5-галоидтиенои зотиазол-3(2 Н)-0 Н-1,1 -диоксидов общей формулы О О ..s где Ri - галоид, или их фармацевтически переносимых кислых аддитивных солей, о т л и ч а ю щ и й с я тем, что соединение общей формулы о о \ Rf f о где Ri - имеет указанное значение. X - галоид, подвергают взаимодействию с 1-{2-пиримидинил)пиперазином в среде абсолютного диметилформамида при 60°С в течение 45 мин с последующим выделением целевого продукта в свободном состоянии или в виде фармацевтически переносимой кислой ад дитивной соли. С > со 00 Изобретение относится к способу получения новых производных 5-галоидтиеноизотиазол-3(2Н)-ОН-1,1-диоксидов общей формулы о о о где Ri - галоид, или их фармацевтически переносимых кислых аддитивных солей, обладающих анксиолитической активностью. Известны 4-(1-пиперазинил)-бутилтиено- или бензоизотйазол-3(2Н)-оны, незамещенные 5-галоидом на тиофеновом кольце и также обладающие анксиолитическим действием. Цель изобретения - получение новых производных тиеноизотиазол-3(2Н)~она, обладающих большей активностью, чем известные структурные аналоги. Соединение общей формулы О 13481 О О У О -(ВДгх If где Ri имеет указанное значение; X - галоид, подвергают взаимодействию в 1-(2-пирими~ 10 динил)пиперазином в среде абсолютного диметилформамида при 60°С в течение 4 мин с последующим выделением целевого продукта всвободном состоянии или в виде фармацевтически переносимой кислой ад- 15 дитивной соли. П р и м е р 1. 5-Хлор-2{4-[2-пиримидинил)-1-пиперазинил]бутил}тиено(2,3-сО- изотиазол-3(2 Н)-он-1,1 -двуокись. 5,5 г (13,6 ммоль) 2-(4-йодбутил}-5-хлор- 20 тиено(2,3-й)изотиазол-3(2Н)-он-1, 1-двуоки-си смешивают с 25 мл абсолютного диметилформамида смешивают с 25 мл абсолютного диметилформамида и нагревают раствор до 40°С. Затем растворяют 2,23 г (13,6 ммоль) 25 1-(2-пиримидинил)пиперазина в абсолютном диметилформамиде при 60°С и закапы-в а ю т в т е ч е н и е 1 ми н. Ч ер е з 4 5 ми н выпаривают при 60°С растворитель и масля* нистый оранжевый остаток поглощают в 25 30 мл хлористого метилена. Фазу хлористого метилена экстрагируют путем встряхивания еще 2 раза, каждый раз с 20 мл воды, и затем 8 раз всего с 130 мл 2 и, соляной кислоты. Кислую, водную фазу нейтрализуют твер- 35 дым бикарбонатом натрия (рН 7,5) и затем 4 раза экстрагируют путем встряхивания, каждый раз с 25 мл хлористого метилена. Соединенные органические фазы сушат над сульфатом натрия, фильтруют и выпарива- 40 ют. Полученный сырой продукт (4,0 г, 57% от теории) растворяют в 45 мл изопропанола при температуре кипения и отфильтровывают в горячем состоянии немного нерастворимого побочного продукта (70 мг). 45 Маточный раствор выкристаллизовывают в низкотемпературном холодильнике при многократном растирании, затем желтый продукт фильтруют на нутче и трижды дигерируют с холодным, как лед, изопропано- 50 лом. Полученный сырой продукт (3,3 г, 7,67 ммоль) растворяют в 35 мл ацетона при температуре кипения, фильтруют и после охлаждения смешивают при перемешивании с 0,93 г (7,67 ммоль) 29,2%-ного раствора со- 55 л я ной кислоты в метаноле. Гидрохлорид выкристаллизовывают в низкотемпературном холодильнике при многократном растирании, отсасывают и дигерируют трижды с небольшим количеством холодного, как лед, ацетона. Высушенные при 40°С и 20 мбар 2,93 г гидрохлорида суспендируют в 45 мл воды, доводят с помощью насыщенного раствора бикарбоната натрия до величины рН 7,5 и экстрагируют 4 раза, каждый раз с 30 мл хлористого метилена. Соединенные органические фазы сушат над сульфатом натрия, смешивают с активным углем, отфильтровывают и выпаривают (2,75 г). Окончательную очистку производят хроматографией на колонке (KG 60, 50:1, растворитель - простой диэтиловый эфир). Продукт снова перекристаллизовывают на 25 мл изопропанола. Выход 2,25 г слабо-желтых кристаллов (38% от теории). Т.пл. 134-135,5°С (изопропанол). Вычислено, %: С 46,20; Н 4,56; N 15,85. Ci7H2oN5C!03S2 (мол.м. 441,96). Найдено, %: С 46,04; Н 4,62; N 15,70. Н-ЯМР (ядерный магнитный резонанс), С СІз, б ррт: 8,29 (d, J - 4,9 Гц, 2Н, Руг-Н4 и Не (7,28) S, 1Н, Т1-Нб); 6,47 (t, J - 4,9 Гц, 1Н, Руг-Нб); 3,90-3,70(т, 6Н, Р1р-Н3и Н5.Tl-CH2); 2,56-2,36(т, 6Н, РІр-Нг и Не, -СНг-РІр); 2,001.50 (т, 4Н, ТІ-С-СН2- и -СНг-С-Рір). Получение исходного продукта 2(4бромбутил)-5-хлортиено(2,3-d) изотиазол3(2 Н)-он-1,1 -двуокиси. 15 г (67,1 ммоль) 5-хлортиено(2,3-гі)изотиазол-3(2Ну-он-1,1 -двуокиси растворяют в 100 мл абсолютного диметилформамида. Затем 2,82 г (70,5 ммоль) 60%-ной суспензии гидрида натрия промывают 4 раза абсолютным бензолом и медленно добавляют при охлаждении льдом и сильном магнитном перемешивании в раствор диметилформамида таким образом, чтобы температура не поднималась выше 15°С. Реакционную смесь после 15-минутнОго перемешивания при комнатной температуре нагревают до 60°С и смешивают в течение 30 мин с 43,5 г (202 ммоль) 1,4-дибромбутана. После 3 ч при 60°С раствор выпаривают при 70°С и 1,5 мбар. Остающееся желтое масло суспендируют в 40 мл насыщенного раствора бикарбоната натрия и экстрагируют трижды, каждый раз с 50 мл хлористого метилена. Затем соединенные органические фазы экстрагируют путем встряхивания дважды, каждый раз с 50 мл насыщенного раствора бикарбоната натрия, и дважды всего с 110 мл воды. Органическую фазу сушат над сульфатом натрия, смешивают с активным углем, фильтруют и выпаривают. Сырой продукт растворяют в 100 мл простого диэтилового эфира при температуре кипения и отсасывают 50 мг бесцветного побочного продукта. После выпаривания растворителя остаются 19,74 г твердого сырого продукта, который можно применять без дальнейшей очистки в 6 13481 следующей стадии реакции. 0,7 г подвергают очистке методом хроматографии на колонке (KG 60,40:1, растворитель - хлористый метилен, выход 0,62 г). Выход 17,48 г бесцветных кристаллов 5 (73% от теории). Т.пл. 75-76°С (простой диэтиловый эфир). 2,3 г (15,3 ммоль) йодистого натрия растворяют в 70 мл абсолютного ацетона и сразу добавляют 5,5 г (15,3 ммоль) 10 2-(4бромбутил)-5-хлортиено(2,3-гі)изотиа-эол3(2Н)-он-1,1 -двуокиси. При сильном магнитном перемешивании нагревают 90 мин при флегме, причем образуется бесцветный рыхлый осадок. Реакционную смесь выпари- 15 вают, поглощают в 40 мл хлористого метилена и дважды экстрагируют, каждый раз с 60 мл насыщенного раствора бисульфита натрия. Фазу хлористого метилена экстрагируют встряхиванием с 25 мл воды. 20 Органическую фазу сушат над сульфатом натрия, фильтруют и выпаривают. Сырой продукт можно непосредственно применять в следующей стадии. 1 г сырого продукта очищают хроматографией на колонке (KG-60, 25 30:1, растворитель - хлористый метилен'.петролейный эфир 2:1, выход 0,94 г). Выход 5,57 г слабо-желтых кристаллов (90% от теории). Т.пл. 86-87°С (хлористый метилен). 30 Пример 2 (сравнительный). Аналогично примеру 1 получали 2{4-{4-пиримидинил)-1 пиперазинил]бутил}тиено(2,3-0)-изотиазол-3(2Н)-он-1,1 -двуокись (известное соединение). Т.пл. 117-118°С (изопропанол). 35 П р и м е р 3. Анксиолитическую активность соединения примера 1 исследовали в конфликтном тесте с крысами (passive avoidance test). На одну дозу испытывали группу по 40 меньшей мере из 10 животных (крысы мужского пола, рода Spraque Dawley. Charles River U.K. весом 230-290 г). Каждую группу делили на 2 подгруппы, которые испытывали в различные дни. В качестве контрольной 45 служила группа животных, которые получали только инъекцию растворителя. "Тренировка" животных. Каждое животное помещали в освещенную отдельную половину тест-бокса. После 50 времени приспосабливания 60 с открывали дверь для неосвещенной половины бокса и Упорядник Замовлення 4120 пускали секундомер. Записывали время, которое проходило до перехода крысы в темную половину (крысы предпочитают темные помещения). Сразу после перехода в темную половину крысы получали слабый электрический удар (0,45 мА, 3 с) через металлическую решетку основания. По окончании шока животных помещали обратно в клетки. Тест. Через день после тренировки животные получали испытуемое вещество или растворитель вн лрибрюшинно. Через 30 мин после инъекции животных снова помещали в освещенную часть тест-бокса и измеряли время до перехода в тем ,ую часть. При этом по снижению времени »с рехода подопытных животных по сравнению с контрольными животными судят об анксиолитической активности испытуемого вещества. Определили, что как 1 мг/кг веса тела, так и 5 мг/кг вещества примера 1 вызывали 50%-ное снижение времени перехода. Поэтому вещество примера 1 является сильным анксиолитическим средством. П р и м е р 4. Повторяли пример 3 с применением вещества примера 2. Однако в области концентраций 1-10 мг/кг веса тела не получали никакого статистически значительного сокращения времени перехода. Поэтому в исследованной области концентраций вещество примера 2 не имеет анксиолитической активности. П р и м е р 5. Для определения острой токсичности б мышей (род Charles River Wlga, Зульцефельд, ФРГ, вес тела 18-20 г, 3 мужского пола, 3 женского пола) подвергали тесту на токсичность. Мыши получали через рот однократную дозу вещества примера 1 как суспензию в 0,5% карбоксиметилцеллюлозы в количестве 2000 мг/кг веса тела. После приема вещества мышей исследовали в течение 2 недель на признаки отравления или смертельные случаи. Через 2 недели мышей умерщвляли и подвергали вскрытию. Смертельные случаи не имели места, признаков отравлейия не смоги установить. При вскрытии не было установлено никаких признаков патологического изменения на органах. Техред М.Моргентал Коректор М.Керецман Тираж Підписне Державне патентне відомство України, 254655, ГСП, КиТв-53, Львівська пл., 8 Відкрите акціонерне товариство "Патент", м. Ужгород, вул.Гагаріна, 101
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07D 275/00, C07D 295/00, A61K 31/425, C07D 233/00, C07D 239/00, C07D 513/04
Мітки: спосіб, 5-галоідтієноізотіазол-3(2н)-oн-1,1-діоксидів, одержання, похідних
Код посилання
<a href="https://ua.patents.su/4-13481-sposib-oderzhannya-pokhidnikh-5-galoidtiehnoizotiazol-32n-on-11-dioksidiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних 5-галоідтієноізотіазол-3(2н)-oн-1,1-діоксидів</a>
Спосіб одержання похідних 1,1,2-трифенілпропана або 1,1,2-трифенілпропена, або стереоізомерів, або суміші стереоізомерів, або їх гідрохлоридних солей

Номер патенту: 5145
Опубліковано: 28.12.1994
Автори: Лайош Толдь, Єва Кіш, Ілона Сенте, Янош Борвендег, Гізела Абрахам, Кальман Торі, Ендре Чаньі, Тібор Хорват
Мітки: спосіб, солей, суміші, 1,1,2-трифенілпропана, похідних, стереоізомерів, одержання, гідрохлоридних, 1,1,2-трифенілпропена
Формула / Реферат:
(57) Способ получения производных 1,1,2-трифенилпропана или 1,1,2-трифенилпропена общей формулыгде А и В - водород или они совместно образуют валентную связь;X и Y - одинаковые или различные и представляют собой незамещенный или замещен-ный в п —положении хлором фенил;R1 - C1 - С4 - алкил, метоксиметил, эпоксинизший алкил илигде R2 и R3 - С1 –С4 -алкил или вместе с атомом азота образуют морфолино-,...
Спосіб одержання похідних акрилової кислоти або їх ізомерів

Номер патенту: 6305
Опубліковано: 29.12.1994
Автори: Ян Фергусон, Майкл Гордон Хічінгс, Патрік Джелф Кроулі, Пол Дефрейн, Крістофер Річард Ейлз Годфрі, Джон Мартін Клаф, Вів'єнн Маргарет Ентоні
МПК: A01N 43/54, C07D 215/24, C07D 239/40, C07D 239/93, C07D 213/70
Мітки: ізомерів, спосіб, акрилової, одержання, кислоти, похідних
Формула / Реферат:
Способ получения производных акриловой кислоты общей формулыгде W - пирвдиинп или пиримидинил, возможно замещенные галогенами С1-С4-алкилом, который, в свою очередь, может быть замещен галогеном, фенвлом, С1-С4-алкоксигруппой, феноксигруппой, которая, в свою очередь, может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогругаюй, циано-, нитро-, амино-, формамидогруппой или N-оксидной группой, или...
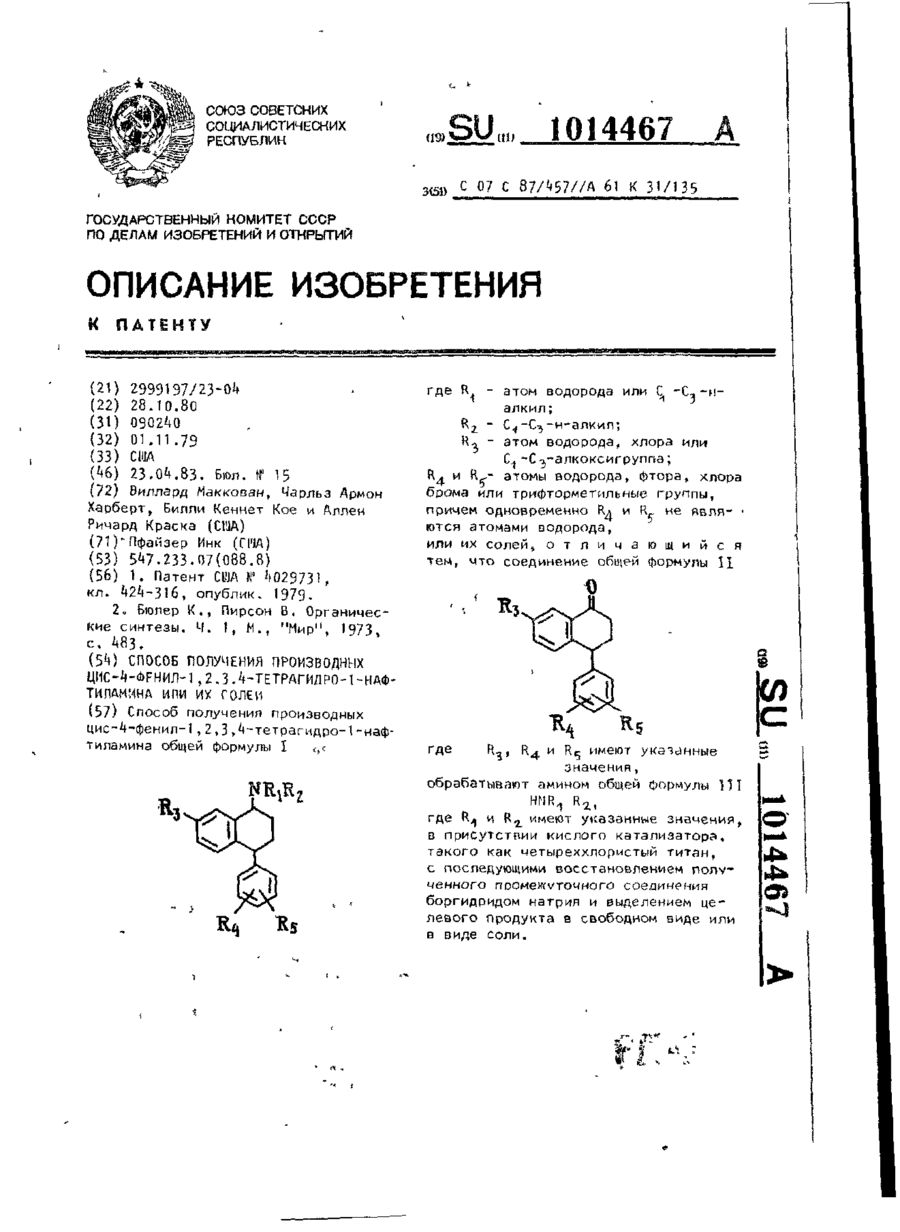
Спосіб одержання похідних ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна або його солей
Номер патенту: 6301
Опубліковано: 29.12.1994
Автори: Чарльз Армон Херберт, Віллард Маккован, Біллі Кеннет Кое, Аллен Річард Краска
МПК: C07C 45/46, C07C 45/00, A61K 31/135, A61P 25/24, C07C 211/41, C07C 255/58, A61K 31/13, C07C 217/74, A61K 31/137, C07C 211/42, C07C 43/21, A61P 25/26, C07C 45/28, C07C 67/00, C07C 209/00, C07C 213/00, A61K 31/165, C07C 49/697, C07C 57/00
Мітки: похідних, одержання, спосіб, солей, ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна
Формула / Реферат:
Способ получения производных цис-4-фенил-1,2,3,4-тетрагидро-1-нафтиламина общей формулы Ігде R1 - атом водорода или С1-С3-н-алкил; R2-С1-С3-н-алкил; R3 - атом водорода, хлора или С1-С3 - алкоксигруппа; R4 и R5 - атомы водорода, фтора, хлора, брома или трифторметильные группы, причем одновременно R4 и R5 не являются атомами водорода, или их солей, отличающийся тем, что соединение общей формулы IIгде R3, R4 и R5...
Спосіб одержання похідних фенілгуанідину та похідні сульфокислоти як проміжні продукти в синтезі похідних фенілгуанідину

Номер патенту: 13323
Опубліковано: 28.02.1997
Автори: Петер Шаркезі, Ференц Шпербет, Атілла Немет, Чаба Хусар, Єва Шомфаї, Лайошне Палі
МПК: C07C 279/00, C07C 323/43, C07C 277/00, C07C 309/00, C07C 317/42, C07C 319/00
Мітки: одержання, фенілгуанідину, синтезі, продукти, похідні, спосіб, похідних, проміжні, сульфокислоти
Формула / Реферат:
(57) 1. Способ получения производных фенилгуанидина общей формулы I:где R - алкил с 1-4 атомами углерода; М - водород, натрий, калий или кальций; А - группа -S-, -SO- или -SO2-,отличающийся тем, что производное тиокарбамида общей формулы II:где R и М имеют вышеуказанные значения, подвергают окислению с последующей обработкой полученного при этом производного сульфокислоты общей формулы...
Спосіб одержання похідних циклопропана

Номер патенту: 7046
Опубліковано: 31.03.1995
Автори: Пол Ентоні Вортінгтон, Крістофер Джон Урч, Патрік Джелф Кроулі
МПК: C07C 67/00, C07C 33/00, C07C 27/00, C07C 29/44, C07C 43/23, C07C 29/56, C07C 29/38, C07C 43/295, C07C 41/00, B01J 23/06, C07D 249/08, C07B 61/00
Мітки: циклопропана, спосіб, похідних, одержання
Формула / Реферат:
Формула изобретения1. Способ получения производных циклопропана общей формулыгде Y - хлор;R1-Сі-С4-алкил.отличающийся тем, что соединение общей формулыгде Y и R1 имеют указанные значения, обрабатывают в присутствии металлического цинка соединением общей формулыгде Z1 и Z2, одинаковые или различные, галогены.2. Способ по п.1, отличающийся тем, что процесс ведут в...
Попередній патент: Спосіб одержання похідних тієно (3′,4′: 4,5) імідазо (2,1- b) тіазолу
Наступний патент: Вітродвигун
Випадковий патент: Спосіб профілактики спайкоутворення у ранньому післяопераційному періоді при гострій абдомінальній патології