Похідні амінодигідротіазину як інгібітори васе для лікування хвороби альцгеймера
Номер патенту: 102251
Опубліковано: 25.06.2013
Автори: ОДІА Джеймс Едмунд, Уотсон Брайан Морган, Мерготт Дастін Джеймс, Шіхан Скотт Мартін

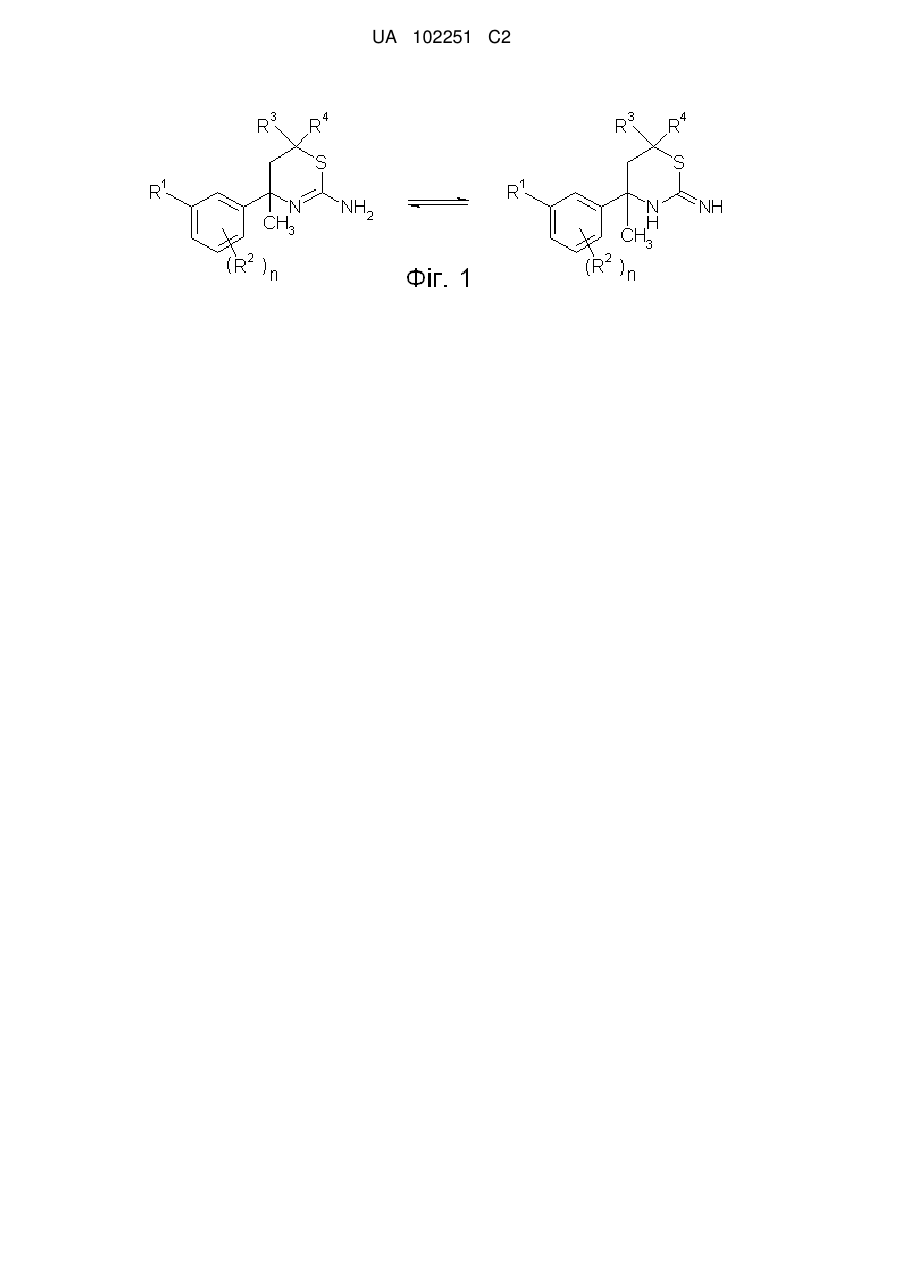






Формула / Реферат
1. Сполука Формули І:
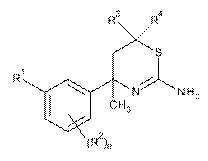 , I
, I
де:
n - 0, 1 або 2;
R1 - піримідиніл, піразиніл, факультативно заміщений хлором або фтором, або піридиніл, факультативно заміщений одним або двома замісниками, вибраними незалежно один від одного з групи, яку складають хлор, фтор та С1-С3-алкоксигрупа;
R2 у будь-якому випадку незалежно вибраний з групи, яку складають хлор та фтор;
R3 - водень або С1-С4-алкіл, факультативно заміщений гідроксилом; та
R4 - водень або С1-С3-алкіл;
або фармацевтично прийнятна сіль такої сполуки.
2. Сполука за п. 1, де R1 - піримідиніл, піридиніл, факультативно заміщений одним або двома замісниками, вибраними в кожному випадку незалежно один від одного з групи, яку складають хлор, фтор та метоксигрупа, або піразиніл, факультативно заміщений фтором; R2 - хлор або фтор; R3 - водень, метил, метил, заміщений гідроксилом, або ізопропіл, заміщений гідроксилом; R4 - водень; та n - 0, 1 або 2;
або фармацевтично прийнятна сіль такої сполуки.
3. Сполука за будь-яким із пп. 1-2, де R1 - піримідиніл, піридиніл, факультативно заміщений фтором, або піразиніл, факультативно заміщений фтором; R2 - фтор; R3 - водень або метил; R4 - водень; та n - 1 або 2;
або фармацевтично прийнятна сіль такої сполуки.
4. Сполука за будь-яким із пп. 1-3, де R1 - піримідиніл, R2 - фтор, R3 -водень, R4 - водень, та n - 2,
або фармацевтично прийнятна сіль такої сполуки.
5. Сполука за будь-яким із пп. 1-4, де конфігурація хірального центра, прилеглого до атома азоту амінотіазину, є (S),
або фармацевтично прийнятна сіль такої сполуки.
6. Сполука, яка являє собою 4-(2,4-дифтор-5-(піримідин-5-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-амін:
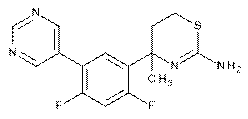 ,
,
або фармацевтично прийнятна сіль такої сполуки.
7. Сполука за п. 6, де сполука являє собою (S)-4-(2,4-дифтор-5-(піримідин-5-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-амін:
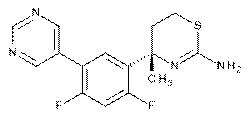 ,
,
або фармацевтично прийнятна сіль такої сполуки.
8. Фармацевтична композиція, яка містить сполуку за будь-яким із пп. 1-7 або фармацевтично прийнятну сіль такої сполуки у комбінації з фармацевтично прийнятним носієм, розріджувачем або наповнювачем.
9. Спосіб лікування хвороби Альцгеймера шляхом введення ефективної кількості сполуки за будь-яким із пп. 1-7 або фармацевтично прийнятної солі такої сполуки.
10. Сполука за будь-яким із пп. 1-7 для застосування у терапії.
11. Сполука за будь-яким із пп. 1-7 для застосування у лікуванні хвороби Альцгеймера.
12. Застосування сполуки за будь-яким із пп. 1-7 або фармацевтично прийнятної солі такої сполуки для виготовлення лікарського засобу для лікування хвороби Альцгеймера.
Текст
Реферат: Цей винахід пропонує інгібітори ВАСЕ Формули І: ,I їх застосування, проміжні сполуки та методики їх отримання. ДЛЯ ЛІКУВАННЯ ХВОРОБИ UA 102251 C2 (12) UA 102251 C2 UA 102251 C2 5 10 15 20 Цей винахід стосується лікування хвороби Альцгеймера та інших захворювань та розладів, пов’язаних із пептидом амілоїдом- (A) – нейротоксичним пептидним сегментом протеїнупопередника амілоїду (APP), який має високу схильність до агрегації. Зокрема, запропоновані ефективні інгібітори -секретази або -сайта ферменту, який розщеплює протеїн-попередник амілоїду (BACE). Показано, що повне або часткове інгібування BACE чинить значний вплив на пов’язані з тромбоцитами та залежні від тромбоцитів патологічні стани у піддослідних мишей; це дозволяє припустити, що навіть незначне зниження рівнів A може спричинити довготривале значне зниження тромбоцитного навантаження та синаптичної недостатності, забезпечуючи, таким чином, значний терапевтичний ефект. Описані на даний час інгібітори BACE являють собою пептидоміметичні аналоги перехідного стану, які у типових випадках містять гідроксіетильну групу. Численні сполуки такого типу є ефективними інгібіторами BACE, однак їх високі молекулярні маси та низька проникність через мембрани зумовлюють їх низьку придатність як потенціальних лікарських засобів. Має місце перехід від великорозмірних пептидоподібних молекул до менших молекул, таких як різноманітні похідні гідроксіетиламіну, а також до структур, які містять гетероцикли. Дивись, наприклад, Дерхем та Шепгерд (Durham and Shepherd, Current Opinion in Drug Discovery & Development, 9(6), 776-791 (2006)). Деякі похідні амінотіазину описані як інгібітори BACE у WO 2007/049532. Існує потреба в більш сильнодіючих та більш ефективних інгібіторах BACE для забезпечення лікування розладів, опосередкованих A-пептидом, таких як хвороба Альцгеймера. Цей винахід пропонує нові сильнодіючі та ефективні інгібітори BACE. Цей винахід пропонує сполуки Формули I: 3 R 4 R S 1 R N CH3 NH2 2 (R )n , 25 30 35 40 45 50 I де: n – 0, 1 або 2; 1 R – піримідиніл, піразиніл, факультативно заміщений хлором або фтором, або піридиніл, факультативно заміщений одним або двома замісниками, вибраними незалежно один від одного з групи, ÿêó ñêëàäàþòü хлор, фтор та C1-C3-алкоксигрупа; 2 R у будь-якому випадку незалежно вибраний з групи, ÿêó ñêëàäàþòü хлор та фтор; 3 R – водень або C1-C4-алкіл, факультативно заміщений гідроксилом; та 4 R – водень або C1-C3-алкіл; або фармацевтично прийнятну сіль такої сполуки. Цей винахід пропонує також спосіб лікування хвороби Альцгеймера у пацієнта, який включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. Цей винахід пропонує також спосіб запобігання розвитку у пацієнта помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера, який включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. Цей винахід пропонує також спосіб запобігання у пацієнта ризику розвитку хвороби Альцгеймера, який включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. Цей винахід пропонує також спосіб інгібування BACE у пацієнта, що включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. Цей винахід пропонує також спосіб інгібування опосередкованого -секретазою розщеплення протеїну-попередника амілоїду, який включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. Крім того, цей винахід пропонує спосіб інгібування продукування A-пептиду, який включає введення в організм пацієнта, який потребує такого лікування, ефективної кількості сполуки Формули I. 1 UA 102251 C2 5 10 15 20 25 30 35 40 45 50 55 Цей винахід пропонує також фармацевтичну композицію, яка містить сполуку Формули I у комбінації з фармацевтично прийнятним носієм, розріджувачем або наповнювачем. Крім того, цей винахід пропонує сполуку Формули I для застосування у терапії, зокрема, для лікування хвороби Альцгеймера або для запобігання розвитку помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера. Цей винахід пропонує також застосування сполуки Формули I для виготовлення лікарського засобу для лікування хвороби Альцгеймера. Цей винахід пропонує також застосування сполуки Формули I для виготовлення лікарського засобу для запобігання розвитку помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера. Крім того, цей винахід пропонуєзастосування сполуки Формули I для виготовлення лікарського засобу для інгібування BACE. Крім того, цей винахід пропонує застосування сполуки Формули I для виготовлення лікарського засобу для інгібування продукування A-пептиду. Крім того, цей винахід пропонує фармацевтичну композицію, придатну для лікування хвороби Альцгеймера. Крім того, цей винахід пропонує фармацевтичну композицію, придатну для запобігання розвитку помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера. Цей винахід пропонує також фармацевтичну композицію, придатну для інгібування BACE. Крім того, цей винахід пропонує фармацевтичну композицію, придатну для інгібування опосередкованого -секретазою розщеплення протеїну-попередника амілоїду. Крім того, цей винахід пропонує фармацевтичну композицію, придатну для лікування станів, які виникають внаслідок надлишкових рівнів A-пептиду, яка включає сполуку Формули I або її фармацевтично прийнятну сіль у комбінації з одним або кількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами. Загальновідомі хімічні терміни у вищенаведених формулюваннях вживаються у їхніх звичайних значеннях. Наприклад, термін "C 1-C3-алкіл" означає метил, етил, пропіл та ізопропіл. Термін "C1-C4-алкіл" означає метил, етил, пропіл, ізопропіл, бутил, ізобутил, втор-бутил та третбутил. Термін "C1-C4-алкіл, факультативно заміщений гідроксилом" означає C1-C4-алкіл, де один з атомів водню замінений гідроксильною групою. Термін "C1-C3-алкокси(група) означає C1-C3-алкіл, приєднаний до атома кисню, та стосується метокси-, етокси-, пропокси- та ізопропоксигрупи. Термін "група захисту азоту" вживається для позначення групи, яка є стабільною у вибраних умовах реакції, але може бути селективно відщеплена із застосуванням реагентів та умов реакції, сумісних із регенерованою аміногрупою. Такі групи добре відомі фахівцям у галузі та описані в літературі. Дивись, наприклад, Greene and Wuts (Гріні та Вутс), Protective Groups in Organic Synthesis, Third Edition, Chapter 7, John Wiley and Sons Inc., (1999). Термін "інгібування продукування A-пептиду" вживається для позначення зниження рівнів A-пептиду in vivo в організмі пацієнта до нормальних значень (якщо вони є надлишковими) або субнормальних значень (в разі потреби). Термін "ефективна кількість сполуки Формули I" вживається для позначення дози або доз сполуки Формули I, необхідних для інгібування BACE у мірі, достатній для зниження рівнів Aпептиду in vivo в організмі пацієнта до нормальних або субнормальних значень. Термін "лікування" охоплює уповільнення або припинення розвитку захворювання у пацієнта. Помірне погіршення пізнавальної функції визначається як потенціальна продромальна фаза деменції, пов’язаної із хворобою Альцгеймера, діагностована на основі клінічних ознак та їх розвитку у пацієнтів, які з часом виявляють схильність до помірного погіршення пізнавальної функції як ознаки деменції, зумовленої хворобою Альцгеймера (Морріс та ін. – Morris, et al., Arch. Neurol., 58, 397-405 (2001); Петерсен та ін. – Petersen, et al., Arch. Neurol., 56, 303-308 (1999)). Термін "запобігання розвитку помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера" означає уповільнення, припинення або повернення до попереднього рівня розвитку у пацієнта помірного погіршення пізнавальної функції, зумовленого хворобою Альцгеймера. Для фахівця у галузі зрозуміло, що сполуки Формули I можуть існувати у таутомерних формах, як показано на Фіг. (1). В разі будь-якого посилання у цій заявці на один із конкретних таутомерів сполуки Формули I мається на увазі охоплення обох таутомерних форм та усіх сумішей таких форм. 2 UA 102251 C2 Для фахівця у галузі зрозуміло, що сполуки Формули I включають молекулярну структуру, яка містить щонайменше один хіральний центр: 5 10 15 20 25 30 Цей винахід охоплює усі індивідуальні енантіомери, а також суміші енантіомерів згаданих сполук; однак перевага віддається сполукам Формули I, які мають при атомі, позначеному 1, абсолютну конфігурацію, показану на Фіг. (2). Крім того, за умови відповідного заміщення, перевага віддається сполукам Формули I, які мають при атомі, позначеному 2, абсолютну конфігурацію, показану на Фіг. (3). Крім того, для фахівця у галузі зрозуміло, що у сполуках за цим винаходом можуть бути утворені додаткові хіральні центри шляхом вибору певних змінних груп. Цей винахід охоплює усі індивідуальні енантіомери або діастереомери, а також суміші енантіомерів та діастереомерів згаданих сполук, в тому числі рацемати. Для фахівця у галузі також зрозуміло, що позначення усіх хіральних центрів як (R) або (S) за Каном-Інгольдом-Прелогом (Cahn-Ingold-Prelog) змінюється залежно від сукупності замісників у конкретній сполуці. Індивідуальні енантіомери або діастереомери можна одержати із застосуванням хіральних вихідних реагентів або стереоселективних чи стереоспецифічних способів синтезу. В альтернативних варіантах індивідуальні енантіомери або діастереомери можна виділити із сумішей, застосовуючи стандартні способи хіральної хроматографії або кристалізації, на будь-якій зручній для цього стадії синтезу сполук за цим винаходом. Варіантом здійснення винаходу, якому віддається перевага, є індивідуальні енантіомери або діастереомери сполук за цим винаходом. Сполуки за цим винаходом є амінами і, відповідно, здатні реагувати з будь-якою із численних неорганічних та органічних кислот, з утворенням фармацевтично прийнятних солей з кислотами. Фармацевтично прийнятні солі та загальна методологія їх одержання добре відомі в галузі. Дивись, наприклад, P. Stahl, et al. (Шталь та ін.) Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., (Берж та ін.) 3 UA 102251 C2 5 10 15 20 25 30 35 40 45 50 55 60 "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Перевага серед фармацевтично прийнятних солей віддається солям із хлористоводневою кислотою. Усі сполуки Формули I є корисними інгібіторами BACE, однак перевага віддається певним класам сполук. Нижче подано перелік таких класів, яким віддається перевага: 1 a) R - піримідиніл; 1 b) R - піразиніл, факультативно заміщений фтором; 1 c) R - піридиніл, факультативно заміщений одним або двома замісниками, вибраними в кожному випадку незалежно один від одного з групи, яку складають хлор, фтор та метоксигрупа; 1 d) R - піридиніл, факультативно заміщений фтором або метоксигрупою; 1 є) R - піридиніл, факультативно заміщений фтором; 2 f) R - фтор; 2 g) R - хлор; h) n - 0; і) n - 1; j) n - 2; 3 k) R - водень; 3 1) R - метил, заміщений гідроксилом; 3 m) R - метил; 3 n) R - ізопропіл, заміщений гідроксилом; 4 о) R - водень; 4 р) R - метил; q) Сполука Формули І має абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; r) Сполука Формули І є вільною основою; s) Сполука Формули І є фармацевтично прийнятною сіллю; t) Сполука Формули І є гідрохлоридом; u) Сполука Формули І є дигідрохлоридом. Варіант здійснення цього винаходу, якому віддається перевага, стосується сполук 1 Формули I, де R – піримідиніл, піридиніл, факультативно заміщений одним або двома замісниками, вибраними в кожному випадку незалежно один від одного з групи, яку складають 2 хлор, фтор та метоксигрупа, або піразиніл, факультативно заміщений фтором; R – хлор або 3 фтор; R – водень, метил, заміщений гідроксилом метил або заміщений гідроксилом ізопропіл; 4 R – водень або метил; та n – 0, 1 або 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. Інший варіант здійснення цього винаходу, якому віддається перевага, стосується сполук 1 Формули I, де R – піримідиніл, піридиніл, факультативно заміщений одним або двома замісниками, вибраними в кожному випадку незалежно один від одного з групи, яку складають 2 хлор, фтор та метоксигрупа, або піразиніл, факультативно заміщений фтором; R – хлор або 3 фтор; R – водень, метил, заміщений гідроксилом метил або заміщений гідроксилом ізопропіл; 4 R – водень; та n – 0, 1 або 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. Варіант здійснення цього винаходу, якому віддається більша перевага, стосується сполук 1 Формули I, де R – піримідиніл, піридиніл, факультативно заміщений одним або двома замісниками, вибраними в кожному випадку незалежно один від одного з групи, яку складають 2 3 хлор та фтор, або піразиніл, факультативно заміщений фтором; R – хлор або фтор; R – 4 водень, метил; R – водень; та n – 0, 1 або 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. 1 Другий варіант здійснення цього винаходу стосується сполук Формули I, де R – піримідиніл, піридиніл, факультативно заміщений фтором або метоксигрупою, або піразиніл, факультативно 2 3 4 заміщений фтором; R – фтор; R – водень або метил; R – водень; та n – 1 або 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. 4 UA 102251 C2 5 10 15 Варіант здійснення цього винаходу, якому віддається більша перевага, стосується сполук 1 Формули I, де R – піримідиніл, піридиніл, факультативно заміщений фтором, або піразиніл, 2 3 4 факультативно заміщений фтором; R – фтор; R – водень або метил; R – водень; та n – 1 або 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. Варіант здійснення цього винаходу, якому віддається особлива перевага, стосується сполук 1 2 3 4 Формули I, де R – піримідиніл; R – фтор; R – водень; R – водень; та n – 2; або фармацевтично прийнятних солей таких сполук. У зазначеному варіанті здійснення цього винаходу перевага віддається сполукам, які мають абсолютну конфігурацію (S) у хіральному центрі, прилеглому до атома азоту амінотіазинового циклу; або фармацевтично прийнятним солям таких сполук. Подальшим варіантом здійснення цього винаходу, який стосується сполук Формули I та якому віддається особлива перевага, є сполука N S N N CH3 F 20 F або її фармацевтично прийнятна сіль. Ще одним варіантом здійснення цього винаходу, який стосується сполук Формули I та якому віддається особлива перевага, є сполука N S N N CH3 F 25 30 35 40 45 NH2 NH2 F або її фармацевтично прийнятна сіль. Сполуки Формули I є інгібіторами BACE. Відповідно, цей спосіб пропонує також спосіб інгібування BACE в організмі пацієнта, що потребує згаданого лікування, який включає введення в організм пацієнта достатньої для інгібування BACE кількості сполуки Формули I. Серед пацієнтів, які підлягають лікуванню шляхом введення в організм сполук Формули I, перевага віддається людині. Як інгібітори BACE, сполуки за цим винаходом є корисними для пригнічення продукування A-пептиду і, отже, для лікування розладів, які є наслідками надлишкових рівнів A-пептиду, пов’язаних із надлишковим продукуванням та/або знижим кліренсом A-пептиду. Подальшим варіантом здійснення цього винаходу є застосування сполук Формули I для виготовлення лікарських засобів для лікування захворювань або хворобливих станів, які можна полегшити або яким можна запобігти шляхом інгібування BACE. Тому вважається, що сполуки Формули I є корисними при лікуванні або запобіганні хвороби Альцгеймера, помірного погіршення пізнавальної функції, синдрому Дауна, спадкових мозкових крововиливів з амілоїдозом датського типу, мозкової амілоїдної ангіопатії, інших дегенеративних деменцій, таких як деменції змішаного судинного та дегенеративного походження, деменція, пов’язана з хворобою Паркінсона, деменція, пов’язана з прогресивним супрануклеарним паралічем, деменція, пов’язана з кортикальною базальною дегенерацією, та хвороби Альцгеймера типу дифузного тільця Леві. Сполуки за цим винаходом можуть бути одержані за різноманітними методиками; деякі з них ілюстровано поданими нижче схемами. Для фахівця у галузі зрозуміло, що окремі стадії у поданих нижче схемах можна змінювати, маючи на увазі одержання сполук Формули I. Конкретний порядок виконання стадій, необхідний для одержання сполук Формули I, залежить від конкретної сполуки, яку синтезують, вихідної сполуки та відносної лабільності групзамісників. Продукти, одержувані на кожній стадії поданих нижче схем, можна виділити 5 UA 102251 C2 5 10 15 звичайними способами, в тому числі екстрагуванням, випарюванням, осадженням, хроматографією, фільтруванням, розтиранням та кристалізацією. У поданих нижче схемах деякі стереохімічні центри залишені непозначеними, та деякі замісники не вказані з міркувань ясності, причому ці особливості схем не призначені для будьякого обмеження обсягу інформації, поданої на них. Крім того, індивідуальні ізомери, енантіомери або діастереомери можуть бути виділені на будь-якому зручному для цього етапі синтезу сполук Формули I відповідними способами, такими як хіральна хроматографія. Крім того, проміжні сполуки, вказані на поданих нижче схемах, містять певну кількість груп захисту азоту. Змінні групи захисту можуть бути в кожному випадку однаковими або різними залежно від конкретних умов реакції та конкретних перетворень, які слід виконувати. Умови приєднання та відщеплення груп захисту добре відомі фахівцям у галузі та описані в літературі. Дивись, наприклад, вищезгадану монографію Greene and Wuts, Protective Groups in Organic Synthesis. У поданих нижче схемах усі замісними відповідають поданим вище визначенням, якщо не зазначено інше. Мається на увазі, що сполуки формул (1a) – (1e), (2) та (3) можуть бути без утруднень виготовлені за способами, добре відомими та стандартними для галузі, в тому числі за способами та методиками, аналогічними розкритим у цьому описі. Необхідні вихідні матеріали або є наявними на ринку, або можуть бути виготовлені з наявних на ринку матеріалів способами, добре відомими фахівцям у цій галузі. 20 або 5 25 30 35 40 На Схемі I показана реакція відповідної сполуки будь-якої з формул (1a) – (1e), де R – група захисту азоту, така як ацетил, бензоїл або трет-бутоксикарбоніл, з відповідною сполукою формули (2) або формули (3) з одержанням сполуки Формули I після відщеплення групи захисту від проміжної сполуки (4). Проводять реакцію сполучення за Судзукі між сполукою будь-якої з формул (1a) – (1e) та сполукою формули (2) із застосуванням відповідного паладієвого реагента, такого як хлорид біс(трифенілфосфін)паладію(II), тетракістрифенілфосфінпаладію, PdCl 2 або ацетат паладію(II), у присутності прийнятної основи, такої як карбонат цезію, карбонат натрію або карбонат калію. Такі реакції проводять у прийнятному розчиннику, такому як 1,2-диметоксіетан, вода, етанол, ацетонітрил, диметилформамід або діоксан, або в їх сумішах. За альтернативним варіантом проводять реакцію сполучення за Штілле між сполукою будьякої із формул (1a) – (1e) та сполукою формули (3) із застосуванням прийнятного паладієвого реагента, такого як хлорид біс(трифенілфосфін)паладію(II), PdCl 2 або тетракістрифенілфосфінпаладій, у присутності відповідної допоміжної сполуки, такої як хлорид літію або фторид цезію. Такі реакції проводять у прийнятному розчиннику, такому як толуол або ДМФ, або в їх сумішах. На факультативній стадії може бути одержана фармацевтично прийнятна сіль сполуки Формули I шляхом проведення реакції відповідної вільної основи сполуки Формули I з відповідною фармацевтично прийнятною кислотою у відповідному розчиннику в стандартних 6 UA 102251 C2 5 10 15 20 25 умовах. Крім того, одержання таких солей можна виконувати одночасно з відщепленням групи захисту азоту. Одержання таких солей добре відоме та є звичайним для галузі. Сполуки формули (1a) можна одержати двома способами. На Cхемі II та Cхемі III показані етапи синтезу з використанням відповідної вихідної сполуки формули (i) для одержання сполуки 6 5 формули (1a), де R – метил або етил, та R – група захисту азоту, така як ацетил, бензоїл або трет-бутоксикарбоніл. Як показано на Схемі II, проводять реакцію сполуки формули (i) з амідом 2-метилпропан-2сульфінової кислоти у присутності Ti(OEt)4 у прийнятному розчиннику, такому як THF, і одержують сполуку формули (ii). Сполуку формули (iii) одержують шляхом додання n-BuLi до діізопропіламіну у прийнятному розчиннику, такому як THF. Додають відповідний ацетат, а потім надлишок триізопропоксихлортитану. Сполуку формули (ii) додають до розчину сполуки формули (iii), і одержують сполуку формули (iv). Виконують відщеплення групи захисту аміногрупи способами, відомими у галузі, після чого відновлюють складний ефір до спирту способами, добре відомими у галузі, наприклад, із застосуванням алюмогідриду літію або боргідриду літію. До спирту (v) додають бензоїлізотіоціанат. Одержану проміжну сполуку обробляють HCl як для сприяння утворенню тіазену, так і для видалення бензоїльної групи, а потім вводять відповідну групу захисту азоту, і одержують сполуку формули (1a). Як показано на Схемі III, додають надлишок вінілмагнійброміду до сполуки формули (i) у прийнятному розчиннику, такому як THF, і одержують сполуку формули (vi). Спирт (vi) обробляють тіонілхлоридом або PBr3 у прийнятному розчиннику, такому як гексан або етанол, після чого додають тіосечовину, і одержують сполуку формули (vii). Сполуку формули (vii) 7 UA 102251 C2 обробляють кислотою при підвищеній температурі, і одержують рацемічний амінотіазин, до якого приєднують відповідну групу захисту азоту, і піддають одержану сполуку очищенню, наприклад, способом хіральної хроматографії або кристалізації, одержуючи сполуку формули (1a). 5 10 15 На Схемі IV показано етапи синтезу з використанням відповідної сполуки формули (ii) як вихідного матеріалу для одержання сполуки формули (1b). Надлишок 2метилалілмагнійхлориду додають до розчину сполуки формули (ii) у прийнятному розчиннику, такому як дихлорметан. Одержану проміжну сполуку обробляють розчином HCl у прийнятному розчиннику, такому як діоксан, і одержують сполуку формули (viii). Проводять реакцію аміну (viii) з бензоїлізотіоціанатом у прийнятному розчиннику, такому як THF, і одержують сполуку формули (ix). Шляхом оброблення сполуки формули (ix) надлишком йоду у прийнятному розчиннику, такому як дихлорметан, одержують сполуку формули (x). Нарешті, додаючи гідрид три-н-бутилолова та AIBN у прийнятному розчиннику, такому як толуол, одержують сполуку формули (1b). 20 25 На Схемі V показано етапи синтезу сполуки формули (1c) з використанням як вихідного 5 матеріалу відповідної сполуки формули (iv). X – бром або хлор. R – відповідна група захисту 6 азоту. R – метил або етил. Сполуку (xi) одержують шляхом проведення реакції N,Oдиметилгідроксиламіну з надлишком бутиллітію у прийнятному розчиннику, такому як THF. Додають сполуку формули (iv) до розчину сполуки формули (xi), і одержують сполуку формули (xii). Додають надлишок відповідного галогеніду магнію (xiii) до розчину сполуки формули (xii) у 8 UA 102251 C2 5 10 15 20 25 30 35 прийнятному розчиннику, такому як THF. Одержаний кетон (xiv) відновлюють до спирту (xv) із застосуванням умов, добре відомих та загальноприйнятих у галузі, наприклад, боргідридом натрію у прийнятному розчиннику, такому як метанол. Додають до спирту (xv) бензоїлізотіоціанат. Одержану проміжну сполуку обробляють HCl, після чого приєднують відповідну групу захисту азоту, і одержують сполуку формули (1c). На Схемі VI показано етапи синтезу сполуки формули (1d) та сполуки формули (1e) з 5 використанням як вихідного матеріалу відповідної сполуки формули (xvi), де R – відповідна група захисту азоту. Сполуку формули (xvii) одержують, проводячи спочатку реакцію броміду (метил)трифенілфосфонію з н-бутиллітієм у прийнятному розчиннику, такому як THF. До одержаного розчину повільно додають сполуку формули (xvi), наприклад, із застосуванням завантажувальної лійки або шприц-насоса. Сполуку формули (xviii) одержують шляхом додавання етилгліоксалату та трифторметилсульфонату ітербію до сполуки формули (xvii) у прийнятному розчиннику, такому як ацетонітрил. Сполуку формули (xix) одержують за тристадійним способом. Спочатку спиртову групу у сполуці формули (xviii) перетворюють у відщеплювану групу, наприклад, шляхом проведення реакції з ангідридом трифторметансульфонової кислоти у присутності прийнятної амінної основи, такої як 2,6-лутидин або діізопропілетиламін, у прийнятному розчиннику, такому як дихлорметан. Додають надлишок тіосечовини, після чого одержану проміжну сполуку додають до надлишку сірчаної кислоти. Одержану аміногрупу сполуки формули (xix) захищають відповідною групою захисту азоту способами, добре відомими або описаними у галузі, і одержують сполуку формули (xx). Реакцію з використанням сполуки формули (xx) можна виконувати у двох варіантах, одержуючи або сполуку формули (1d), або сполуку формули (1e). Сполуку формули (1d) можна одержати шляхом відновлення складного ефіру сполуки формули (xx) способами, добре відомими або описаними у галузі, наприклад, надлишком боргідриду літію у прийнятному розчиннику, такому як THF. Сполуку формули (1e) можна одержати шляхом проведення реакції сполуки формули (xx) з надлишком метилмагнійхлориду у прийнятному розчиннику, такому як THF. Підготовчі синтези та приклади Описані нижче підготовчі синтези та приклади додатково ілюструють цей винахід. ® Назви сполук за цим винаходом утворено із застосуванням програми ChemDraw Ultra, версія 10.0. Абревіатури, вжиті в цьому описі, відповідають визначенням, поданим у Aldrichimica Acta, Vol. 17, No. 1, 1984. Інші абревіатури мають такі значення: "SCX" – сильний катіонообмінник; "ca." – приблизно; "EtOAc" – етилацетат; "MeOH" – метанол; "DCM" – дихлорметан; "THF" – тетрагідрофуран; "Et2O" – діетиловий ефір; "(OEt)" – етилат; "екв." – еквіваленти; "FRET" 9 UA 102251 C2 5 10 15 резонансне перенесення енергії при флуоресценції; "RFU" – відносна одиниця інтенсивності флуоресценції; "DMEM" – середовище Ігла (Eagle), модифіковане за Дюльбекко (Dulbecco); "F12" – середовище Хема (Ham) F12; "FBS" – сироватка плода корови. Мас-спектрометричні дані, якщо не зазначено інакше, одержано методом рідинної хроматографії у сполученні з мас-спектроскопією. Колонка Xbridge C18 (2,1 мкм50 мкм3,5 мкм) при температурі 5010C, швидкість потоку 1 мл/хв. Елюент – градієнт від 5% до 100% ацетонітрилу у воді з домішкою 10 мМ бікарбонату амонію (pH 10) протягом 7,0 хв із подальшим витримуванням 100%ацетонітрилу протягом 1,0 хв у комбінації з іонізацією електророзпиленням (діапазон сканування 100-800 атомних одиниць маси (amu); крок сканування 0,2 amu; фрагментатор 80 В; підсилення 1,0; поріг 80). ® Деякі сполуки очищені методом РХВЕ, методика A: колонка Xterra RP18 (30300 мм) при температурі навколишнього середовища при швидкості потоку 40 мл/хв. Система елюювання являє собою або містить ізократичний градієнт 0:100 (ацетонітрил: (0,1% HCl в H 2O)) протягом 1-5 хв із подальшим лінійним градієнтом від 0:100 (ацетонітрил:(0,1% HCl в H 2O)) до 50:50 (ацетонітрил:(0,1% HCl в H2O)) протягом 20 хв. Інші умови РХВЕ вказано у конкретних описах. Підготовчий синтез 1 [1-(5-бром-2,4-дифторфеніл)етиліден]амід (R)-2-метилпропан-2-сульфінової кислоти O S N Br F F 20 25 30 До розчину 1-(5-бром-2,4-дифторфеніл)етанолу (19 г, 64,7 ммоль, 1 екв.) та аміду (R)-2метилпропан-2-сульфінової кислоти (10,2 г, 84,1 ммоль, 0,76 екв.) у THF (0,3 M, 215 мл) додають Ti(OEt)4 (29,5 г, 129 ммоль, 2,0 екв.) однією порцією при температурі навколишнього середовища. Реакційну суміш нагрівають до 70C, та витримують при перемішуванні 18 год. Реакційну суміш охолоджують до температури навколишнього середовища, та виливають у воду. Одержану суспензію фільтрують через шар діатомової землі, та промивають етилацетатом. Фільтрат збирають, та екстрагують етилацетатом. Органічні шари об’єднують, сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском до одержання залишку. Залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (3:1) протягом 20 хв, та одержують вказану в заголовку сполуку (вихід 81%): MS (m/z): 338, 340 (M+1). Сполуки, наведені нижче в Таблиці 1, одержують по суті як описано у підготовчому синтезі [1-(5-бром-2,4-дифторфеніл)етиліден]аміду (R)-2-метилпропан-2-сульфінової кислоти. 35 Підготовчий синтез 2 Метиловий складний ефір дифторфеніл)масляної кислоти (S)-3-((R)-2-метилпропан-2-сульфініламіно)-3-(5-бром-2,4 10 UA 102251 C2 O S NH O Br OMe F 5 10 15 20 F До охолодженого до -78C розчину діізопропіламіну (10,6 г, 105 ммоль, 2 екв.) у THF (262 мл) додають н-бутиллітій (41,9 мл, 105 ммоль, 2 екв.) (2,5 M розчин у гексані). Через 15 хв додають краплями метилацетат (7,7 г, 105 ммоль, 2 екв.), та реакційну суміш залишають при перемішуванні протягом 30 хв. До реакційної суміші додають краплями розчин триізопропоксихлортитану (31,6 г, 115 ммоль, 2,2 екв.) у THF (50 мл). Після перемішування протягом 60 хв при -78C додають краплями розчин [1-(3-бромфеніл)етиліден]аміду 2метилпропан-2-сульфінової кислоти (12,2 г, 40.4 ммоль, 1 екв.) у THF (50 мл). Реакційну суміш перемішують протягом 3 год при -78C. Реакційну суміш гасять насиченим розчином хлориду амонію (100 мл), нагрівають до температури навколишнього середовища, та розводять водою (100 мл). Одержану суспензію фільтрують через шар діатомової землі, та промивають етилацетатом. Фільтрат збирають, та екстрагують етилацетатом. Органічні шари об’єднують, сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском до одержання залишку. Залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від гексан:етилацетат (5:1) до гексан:етилацетат (10:7) протягом 20 хв, та одержують вказану в заголовку сполуку (вихід 72%): MS (m/z): 412, 414 (M+1). Сполуки, наведені нижче в Таблиці 2, одержують по суті як описано у підготовчому синтезі метилового складного ефіру (S)-3-((R)-2-метилпропан-2-сульфініламіно)-3-(5-бром-2,4дифторфеніл)масляної кислоти. Підготовчий синтез 3 Гідрохлорид (S)-метил-3-аміно-3-(2,4-дифторфеніл)бутаноату 25 ClH NH2 O Br F 30 35 OMe F До розчину метилового складного ефіру (S)-3-((R)-2-метилпропан-2-сульфініламіно)-3фенілмасляної кислоти (15,5 г; 37,6 ммоль; 1 екв.) та метанолу (100 мл) однією порцією додають хлористий водень (4М розчин у діоксані) (100 мл, 400 ммоль, 11 екв.). Реакційну суміш перемішують при кімнатній температурі протягом 1 год. Розчинник видаляють під зниженим тиском, та одержують вказану в заголовку сполуку, яку використовують без додаткового очищення (вихід >95%): MS (m/z): 306, 308 (M+1). Сполуки, наведені нижче в Таблиці 3, одержують по суті як описано у підготовчому синтезі гідрохлориду (S)-метил-3-аміно-3-(2,4-дифторфеніл)бутаноату. 11 UA 102251 C2 Підготовчий синтез 4 (S)-3-аміно-3-(5-бром-2,4-дифторфеніл)бутан-1-ол 5 NH2 Br F 10 15 OH F До охолодженого до 0°С розчину гідрохлориду (S)-етил-3-аміно-3-(2,4дифторфеніл)бутаноату (40,2 г, 90,5 ммоль, 1 екв.) у THF (180 мл) додають алюмогідрид літію (1 М розчин у THF) (118 мл, 118 ммоль) протягом 45 хв, підтримуючи при цьому внутрішню температуру реакційної суміші нижче 15°С. Реакційну суміш витримують для нагрівання до температури навколишнього середовища, та перемішують протягом 1,5 год. Реакційну суміш охолоджують до 0°С, та гасять, додаючи краплями воду (4,5 мл), 2 М розчин гідроксиду натрію (4,5 мл) та воду (13,6 мл). Одержану тверду речовину видаляють фільтруванням, та промивають етилацетатом. Фільтрат сушать над MgSO4, та фільтрують. Розчинник видаляють під зниженим тиском, та одержують вказану в заголовку сполуку, яку використовують без додаткового очищення (вихід 71%, чистота 73% за даними LCMS): MS (m/z): 280, 282. Підготовчий синтез 5 (S)-3-аміно-3-(3-бромфеніл)бутан-1-ол 20 NH2 Br 25 30 OH До охолодженого до 0°С розчину гідрохлориду (S)-метил-3-аміно-3-(4-фторфеніл)бутаноату (14 г, 38,6 ммоль, 1 екв.) у THF (200 мл) обережно додають боргідрид літію (1,67 г, 77,1 ммоль, 2 екв.). Через 5 хв реакційну суміш нагрівають до 50 С, та перемішують. Після завершення реакції суміш охолоджують на льодяній бані, та гасять, додаючи краплями воду. Реакційну суміш підкислюють 1-н. розчином HCl (100 мл). Після перемішування протягом 1 год одержаний розчин підлуговують 5-н. розчином NaOH, та екстрагують дихлорметаном. Органічні шари об’єднують, сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 94%): MS (m/z): 244,0 та 246,0 (M+1). Сполуки, наведені нижче в Таблиці 4, одержують по суті як описано у підготовчому синтезі (S)-3-аміно-3-(3-бромфеніл)бутан-1-олу. Таблиця 4 Підг. Синт. № 5a 5b 5c Хімічна назва (S)-3-аміно-3-(3-бром-4-фторфеніл)бутан-1-ол (S)-3-аміно-3-(5-бром-2-фторфеніл)бутан-1-ол (S)-3-аміно-3-(5-бром-2-хлорфеніл)бутан-1-ол 12 Фізичні дані MS (m/z) 262, 264 (M+1) 262, 264 (M+1) 278, 280 (M+1) UA 102251 C2 Підготовчий синтез 6 (S)-1-бензоїл-3-[1-(5-бром-2,4-дифторфеніл)-3-гідрокси-1-метилпропіл]тіосечовина O S N H HN Br F OH F 5 10 До розчину (S)-3-аміно-3-(5-бром-2,4дифторфеніл)бутан-1-олу (9,5 г, 34 ммоль, 1 екв.) у THF (50 мл) додають біс(триметилсиліл)трифторацетамід (8,7 г, 34 ммоль, 1 екв.). Через 2 год додають краплями бензоїлізотіоціанат (5,5 г, 34 ммоль, 1 екв.). Реакційну суміш перемішують 18 год, гасять водою, та екстрагують етилацетатом. Об’єднані органічні фази екстрагують 1-н. розчином HCl та насиченим водним розчином NaCl. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід >95%, чистота 90% за даними LCMS): MS (m/z): 443, 445 (M+1). Сполуки, наведені нижче в Таблиці 5, одержують по суті як описано у підготовчому синтезі (S)-1-бензоїл-3-[1-(5-бром-2,4-дифторфеніл)-3-гідрокси-1-метилпропіл]тіосечовини. 15 Підготовчий синтез 7 (S)-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4Н-1,3-тіазин-2-амін 20 S Br F 25 30 35 N NH2 F До розчину (S)-1-бензоїл-3-[1-(5-бром-2,4-дифторфеніл)-3-гідрокси-1метилпропіл]тіосечовини (41 г, 68 ммоль) у 1,4-діоксані (20 мл) додають водний розчин НСl (5-н. розчин, 407 мл, 2,0 моль, 30 екв.). Одержану суспензію витримують при 100C. Після перемішування протягом 20 год, реакційну суміш концентрують під зниженим тиском. Одержану суміш обробляють водним розчином HCl (5-н. розчин, 407 мл, 2,0 моль), та перемішують при 100C протягом 18 год. Одержану суспензію охолоджують до 10C, та pH доводять до pH 10 доданням 50% водного розчину NaOH. Одержаний водний розчин екстрагують етилацетатом. Органічні шари об’єднують, сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Залишок очищають флеш-хроматографією на колонці із силікагелем, елююючи ступінчастим градієнтом від гексан:ацетон (4:1) до гексан:ацетон (3:1), та одержують вказану в заголовку сполуку (вихід 57%, чистота 85% за даними LCMS): MS (m/z): 321, 323 (M+1). Сполуки, наведені нижче в Таблиці 6, одержують по суті як описано у підготовчому синтезі (S)-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-аміну. 13 UA 102251 C2 Таблиця 6 Підг. синт. № Хімічна назва гідрохлорид (S)-4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро4H-[1,3]тіазин-2-іламіну гідрохлорид (S)-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-аміну (S)-4-(5-бром-2-хлорфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин2-амін 7a 7b 7c 5 10 15 20 S N F 30 303, 305 (M+1) 285, 287 (M+1) 319, 321 (M+1) Підготовчий синтез 8 Трет-бутиловий складний ефір (S)-[4-(5-бром-2-фторфеніл)-4-метил-5,6-дигідро-4H[1,3]тіазин-2-іл]карбамінової кислоти Суміш (S)-1-бензоїл-3-[1-(5-бром-2-фторфеніл)-3-гідрокси-1-метилпропіл]тіосечовини (0,79 г, 1,8 ммоль) та водного розчину HCl (5-н. розчин, 25 мл, 71 ммоль) нагрівають до 100(C. Після перемішування протягом 6 год реакційну суміш охолоджують до температури навколишнього середовища, та залишають для вистоювання протягом ночі. Реакційну суміш концентрують під зниженим тиском до одержання неочищеного гідрохлориду (S)-4-(5-бром-2-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2аміну [MS (m/z): 303, 305 (M+1)]. До суміші розчину гідрохлориду (S)-4-(5-бром-2-фторфеніл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-аміну у THF (30 мл) та насиченого водного розчину бікарбонату натрію (15 мл) додають ди-трет-бутилдикарбонат (0,77 г, 3,5 ммоль). Через 4 год реакційну суміш розводять водою, екстрагують етилацетатом, сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Залишок очищають флеш-хроматографією на колонці із силікагелем, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (3:1), та одержують вказану в заголовку сполуку (вихід 69%): MS (m/z): 403, 405 (M+1). Підготовчий синтез 9 (S)-трет-бутил-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-іл-карбамат Br 25 Фізичні дані MS (m/z) O N H O F До розчину (S)-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-аміну (14,3 г, 39 ммоль 1 екв.) у 1,4-діоксані (190 мл) при температурі навколишнього середовища додають насичений водний розчин бікарбонату натрію (190 мл) та воду (30 мл). Одержану суспензію перемішують протягом 5 хв, після чого додають ди-трет-бутилдикарбонат (17 г, 78 ммоль, 2 екв.). Через 1 год реакційну суміш розводять водою, екстрагують етилацетатом, сушать над сульфатом магнію, фільтрують, та концентрують під зниженим тиском. Залишок очищають хроматографією на силікагелі, елююючи сумішшю гексан:етилацетат (4:1), та одержують вказану в заголовку сполуку (вихід 78%): MS (m/z): 421, 423 (M+1). Сполуки, наведені нижче в Таблиці 7, одержують по суті як описано у підготовчому синтезі (S)-трет-бутил-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамату. 35 Таблиця 7 Підг. синт. № 9a 9b 9c Фізичні дані MS (m/z) трет-бутиловий складний ефір (S)-[4-(3-бром-4-фторфеніл)-4-метил-5,6- 403, 405 дигідро-4H-[1,3]тіазин-2-іл]карбамінової кислоти (M+1) (S)-трет-бутил-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2385, 387 ілкарбамат (M+1) (S)-трет-бутил-4-(5-бром-2-хлорфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2- 419, 421 ілкарбамат (M+1) Хімічна назва 14 UA 102251 C2 Підготовчий синтез 10 2-бром-3-ілбут-3-ен-2-ол Br OH 5 10 15 До розчину 3-бромацетофенону (50 г, 250 ммоль; 1 екв.) у MTBE (375 мл) при 10(C додають краплями вінілмагнійбромід (0,7 M розчин у THF, 250 ммоль; 360 мл, 1 екв.). Реакційну суміш витримують при нагріванні зі зворотним холодильником протягом 16 год. Реакційну суміш охолоджують, та гасять насиченим водним розчином хлориду амонію. Одержану суміш розводять водою, екстрагують етилацетатом, сушать над сульфатом магнію, фільтрують, та концентрують під зниженим тиском. Залишок очищають хроматографією на силікагелі, елююючи ступінчастим градієнтом від гексан:етилацетат (9:1) до гексан:етилацетат (4:1), та 1 одержують вказану в заголовку сполуку (40 г, вихід 42%): H ЯМР (400 МГц, CDCl3): 1,64 (s, 3H), 5,18 (d, J=14 Гц, 1H), 5,30 (d, J=23 Гц, 1H), 6,13 (dd, J=14 Гц, 23 Гц, 1H), 7,21(t, J=11 Гц, 1H), 7,36-7,40 (m, 2H), 7,63 (s, 1H). Підготовчий синтез 11 Гідрохлорид 2-[3-(3-бромфеніл)-бут-2-еніл]ізотіосечовини NH S Br NH2 ClH 20 25 30 До охолодженого до 0C розчину 2-бром-3-ілбут-3-ен-2-олу (11 г, 29 ммоль, 1 екв.) та гексану (20 мл) додають тіонілхлорид (6,9 г, 58 ммоль, 2 екв.). Реакційну суміш витримують для нагрівання до температури навколишнього середовища, при цьому інтенсивно виділяється газ. Реакційну суміш перемішують при температурі навколишнього середовища до припинення виділення газу, та видаляють розчинник під зниженим тиском. Одержаний залишок розчиняють у ацетонітрилі (100 мл). Додають тіосечовину (2,2 г, 29 ммоль, 1 екв.), та реакційну суміш витримують при 50C. Через 2 год реакційну суміш охолоджують до температури навколишнього середовища. Одержаний осад збирають фільтруванням, промивають ацетонітрилом, та одержують вказану в заголовку сполуку (вихід 90%): MS (m/z): 285,0, 287,0 (M+1). Підготовчий синтез 12 4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-[1,3]тіазин-2-іламін S Br N NH2 35 40 45 Суспензію гідрохлориду 2-[3-(3-бромфеніл)бут-2-еніл]ізотіосечовини (17 г, 52 ммоль) у 12 M розчині HCl (53 мл, 640 ммоль, 12 екв.) витримують при 100(C. Через 24 год одержаний розчин охолоджують до температури навколишнього середовища. Доводять pH розчину до 10 доданням 2-н. водного розчину NaOH, та екстрагують етилацетатом. Органічні шари об’єднують, сушать над сульфатом магнію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 70%): MS (m/z): 285,0, 287,0 (M+1). Підготовчий синтез 13 N-(4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-іл)ацетамід 15 UA 102251 C2 S Br 5 10 15 20 N O N H До охолодженого до 0(C розчину 4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-[1,3]тіазин-2іламіну (10 г, 35 ммоль, 1 екв.) та триетиламіну (4,3 г, 42 ммоль, 1,2 екв.) у дихлорметані (70 мл) додають краплями ацетилхлорид (2,8 г, 35 ммоль, 1 екв.) протягом 5 хв. Реакційну суміш витримують для нагрівання до температури навколишнього середовища. Через 1 год реакційну суміш розводять дихлорметаном, та екстрагують водою. Органічні шари розділяють, сушать над сульфатом магнію, фільтрують, та концентрують під зниженим тиском. Одержаний залишок очищають хроматографією на силікагелі, елююючи сумішшю гексан:етилацетат (1:1), та одержують вказану в заголовку сполуку (вихід 87%): MS (m/z): 327, 329 (M+1). Підготовчий синтез 14 (S)-N-[4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-[1,3]тіазин-2-іл]ацетамід 4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-[1,3]тіазин-2-іламін (20 г, 61 ммоль) очищають розділенням методом хіральної рідинної хроматографії високої ефективності (РХВЕ) [колонка: 832 см, Chiralpak AD; елюент: 60:40:0,2 (ізопропіловий спирт:гептан:диметилетиламін); швидкість потоку: 350 мл/хв, УФ детектор, довжина хвилі 260 нм]. Ізомер, який елюювався другим, виділяють, одержуючи енантіомерно збагачену вказану в заголовку сполуку (вихід 35%): MS (m/z): 327, 329 (M+1). Підготовчий синтез 15 Трет-бутиловий складний ефір (S)-[4-(2,4-дифтор-5-піримідин-5-ілфеніл)-4-метил-5,6дигідро-4H-[1,3]тіазин-2-іл]карбамінової кислоти N S N N F 25 30 35 O N H O F До нагрітого до 100(C розчину (S)-4-(5-бром-2,4-дифторфеніл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-аміну (12,6 г, 29,9 ммоль, 1 екв.) у суміші 1,2-диметоксіетан:вода:етанол (15:7:5, 300 мл) додають піримідин-5-борну кислоту (25 г, 203 ммоль, 6,8 екв.), після чого карбонат цезію (58 г, 180 ммоль, 6 екв.) та хлорид біс(трифенілфосфін)паладію(II) (4,2 г, 6,0 моль, 0,2 екв.). Через 40 хв реакційну суміш охолоджують до температури навколишнього середовища, розводять водою, та екстрагують етилацетатом. Одержану органічну фазу сушать над сульфатом магнію, фільтрують, та концентрують під зниженим тиском. Залишок очищають хроматографією на силікагелі, елююючи ступінчастим градієнтом від гексан:етилацетат (7:3) до гексан:етилацетат (1:1), та одержують вказану в заголовку сполуку (вихід 67%): MS (m/z): 421 (M+1). Сполуки, наведені нижче в Таблиці 8, одержують по суті як описано у підготовчому синтезі трет-бутилового складного åô³ðó (S)-[4-(2,4-дифтор-5-піримідин-5-ілфеніл)-4-метил-5,6-дигідро4H-[1,3]тіазин-2-іл]карбамінової кислоти. Таблиця 8 Підг. синт. № 15a 15b 15c 15d 15e 15f Хімічна назва (S)-N-{4-[3-(5-метоксипіридин-3-іл)феніл]-4-метил-5,6-дигідро-4H[1,3]тіазин-2-іл}ацетамід (S)-N-(4-(3-(5-хлор-2-фторпіридин-3-іл)феніл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-іл)ацетамід трет-бутиловий складний ефір (S)-[4-(4-фтор-3-піримідин-5-ілфеніл)-4метил-5,6-дигідро-4H-[1,3]тіазин-2-іл]карбамінової кислоти трет-бутиловий складний ефір (S)-[4-(2-фтор-5-піримідин-5-ілфеніл)-4метил-5,6-дигідро-4H-[1,3]тіазин-2-іл]карбамінової кислоти (S)-N-[4-метил-4-(3-піримідин-5-ілфеніл)-5,6-дигідро-4H-[1,3]тіазин-2іл]ацетамід трет-бутиловий складний ефір (S)-{4-[4-фтор-3-(2-фторпіридин-3іл)феніл]-4-метил-5,6-дигідро-4H-[1,3]тіазин-2-іл}карбамінової кислоти 16 Фізичні дані MS (m/z) 356 (M+1) 378 (M+1) 403 (M+1) 403 (M+1) 327 (M+1) 420 (M+1) UA 102251 C2 Продовження таблиці 8 15g 15h 15i 15j 15k 1 трет-бутиловий складний ефір (S)-{4-[2,4-дифтор-5-(2-фторпіридин-3іл)феніл]-4-метил-5,6-дигідро-4H-[1,3]тіазин-2-іл}карбамінової кислоти (S)-трет-бутил-4-(2,4-дифтор-5-(5-фторпіридин-3-іл)феніл)-4-метил-5,6дигідро-4H-1,3-тіазин-2-ілкарбамат (S)-4-(2-хлор-5-(5-хлор-2-фторпіридин-3-іл)феніл)-4-метил-5,6-дигідро-4H1 1,3-тіазин-2-амін N-{4-[3-(5-метокси-піридин-3-іл)феніл]-4-метил-5,6-дигідро-4H-[1,3]тіазин2-іл}ацетамід (S)-N-(4-(3-(2-фторпіридин-3-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин2-іл)ацетамід 438 (M+1) 438 (M+1) 370, 372 (M+1) 356 (M+1) 344 (M+1) Трет-бутоксикарбонільна група була відщеплена в умовах реакції. Підготовчий синтез 16 (S)-4-(3-(2-фторпіридин-3-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-амін N F S N NH2 5 10 15 До розчину (S)-N-(4-(3-(2-фторпіридин-3-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2іл)ацетаміду (450 мг, 1,3 ммоль) у метанолі (40 мл) додають розчин K2CO3 (210 мг, 1,5 ммоль) у суміші метанол:вода (2:1, 15 мл). Реакційну суміш перемішують при кімнатній температурі протягом 6 год. Розчинник видаляють під зниженим тиском, та залишок розчиняють в етилацетаті. Етилацетатний шар промивають водою та насиченим водним розчином NaCl, сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском до одержання залишку. Залишок очищають, застосовуючи хроматографію на колонці із SCX, та одержують вказану в заголовку сполуку (вихід 65%): MS (m/z): 302 (M+1). Підготовчий синтез 17 (S)-4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-амін S Br N NH2 F 20 25 30 35 До розчину (S)-трет-бутил-4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2ілкарбамату (1,1 г, 2,7 ммоль) у метанолі (10 мл) додають трифтороцтову кислоту (10 мл). Реакційну суміш витримують при 60C. Через 15 год розчинник видаляють під зниженим тиском. До одержаного залишку додають воду, та одержану суміш підлуговують насиченим бікарбонатом натрію. Лужну водну фазу екстрагують дихлорметаном. Одержану органічну фазу відділяють, сушать над сульфатом натрію, фільтрують, розчинник видаляють під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 62%): MS (m/z): 303, 305 (M+1). Підготовчий синтез 18 (S)-N-(4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-іл)ацетамід До розчину (S)-4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-аміну (550 мг, 1,8 ммоль, 1,0 екв.) у тетрагідрофурані (20 мл) додають піридин (720 мг, 9,0 ммоль, 5 екв.) та ангідрид оцтової кислоти (220 мг, 2,2 ммоль, 1,2 екв.). Через 10 хв реакційну суміш виливають у воду, та одержану водну суміш екстрагують дихлорметаном. Органічну фазу відділяють, та промивають 1-н. розчином HCl та насиченим водним розчином NaCl, сушать над сульфатом магнію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 83%): MS (m/z) 345, 347 (M+1). Підготовчий синтез 19 (S)-трет-бутил-4-(4-фтор-3-(піразин-2-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат 17 UA 102251 C2 N S N N O N H O F 5 10 15 20 Розчин трет-бутилового складного ефіру (S)-[4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро4H-[1,3]тіазин-2-іл]карбамінової кислоти (100 мг, 250 мкмоль, 1 екв.), тетракіс(трифенілфосфін)паладію (14 мг, 12,40 мкмоль, 0,05 екв.) та 2-трибутилстанілпіразину (96 мг, 250 мкмоль, 1 екв.) у діоксані (3 мл) опромінюють у лабораторній мікрохвильовій печі до досягнення температури 130(C, та витримують протягом 20 хв. Розчинник видаляють під зниженим тиском, та залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (1:4) за 20 хв, та одержують вказану в заголовку сполуку (вихід 14%, чистота 90% за даними LCMS): MS (m/z): 403 (M+1). Підготовчий синтез 20 (S)-N-(4-(4-фтор-3-(3-фторпіразин-2-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2іл)ацетамід До розчину (S)-N-(4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-іл)ацетаміду (500 мг, 1,5 ммоль, 1 екв.) та 2-фтор-3-(трибутилстаніл)піразину (1,7 г, 4,3 ммоль, 3,0 екв.) у толуолі (15 мл) додають хлорид біс(трифенілфосфін)паладію(II) (51 мг, 72 мкмоль, 0,05 екв.) та хлорид літію (92 мг, 2,2 ммоль, 1,5 екв.). Реакційну суміш опромінюють у лабораторній мікрохвильовій печі до досягнення температури 130C, та витримують протягом 3 год. Розчинник видаляють під зниженим тиском, та залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (1:1) за 20 хв, та одержують вказану в заголовку сполуку (вихід 31%): MS (m/z): 363 (M+1). Сполуку, вказану нижче в Таблиці 9, одержують по суті як описано у підготовчому синтезі (S)-N-(4-(4-фтор-3-(3-фторпіразин-2-іл)феніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-іл)ацетаміду. 25 Таблиця 9 Підг. синт. № Хімічна назва Фізичні дані MS (m/z) 20a (S)-N-(4-(2,4-дифтор-5-(3-фторпіразин-2-іл)феніл)-4-метил-5,6-дигідро-4H1,3-тіазин-2-іл)ацетамід 381 (M+1) Підготовчий синтез 21 (R)-N-((S)-2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2-іл)-2-метилпропан-2-сульфінамід KG2-E01905-021-2 30 O S NH Br F 35 40 До охолодженого до 0(C розчину (R,Z)-N-(1-(3-бром-4-фторфеніл)етиліден)-2-метилпропан2-сульфінаміду (10 г, 31 ммоль, 1,0 екв.) у дихлорметані (100 мл) повільно додають 2метилалілмагнійхлорид (0,5 M розчин у THF, 250 мл, 125,92 ммоль, 4 екв.). Через 2 год реакційну суміш гасять насиченим розчином хлориду амонію, та екстрагують етилацетатом. Розчинник видаляють під зниженим тиском, та залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від дихлорметану до суміші 10% дихлорметан:етилацетат, та одержують вказану в заголовку сполуку (вихід 35%): MS (m/z): 376, 378 (M+1). Підготовчий синтез 22 (S)-2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2-амін NH2 Br F 18 UA 102251 C2 5 10 До розчину (R)-N-((S)-2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2-іл)-2-метилпропан-2сульфінаміду (1,8 г, 5,1 ммоль, 1 екв.) у 1,4-діоксані (6 мл) додають хлористий водень (4,0 M розчин у 1,4-діоксані, 15 мл). Реакційну суміш перемішують протягом 5 хв, та розчинник видаляють під зниженим тиском. До залишку додають насичений водний розчин бікарбонату натрію, та одержану суміш екстрагують етилацетатом. Об’єднану органічну фазу сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в 1 заголовку сполуку (вихід 97%): H ЯМР(CDCl3, 400 МГц) 7,66 (dd, 1H, J=6,40 Гц, J=2,80 Гц), 7,37-7,33 (m, 1H), 7,02 (t, 1H, J=8,40 Гц), 4,83 (s, 1H), 4,62 (s, 1H), 2,47 (d, 1H, J=13,2 Гц), 2,35 (d, 1H, J=13,2 Гц), 1,44 (s, 3H), 1,36 (s, 3H). Підготовчий синтез 23 (S)-N-(2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2-ілкарбамотіоїл)бензамід S Br N H O N H F 15 20 До розчину (S)-2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2-аміну (1,3 г, 4,6 ммоль, 1,0 екв.) у THF (5 мл) додають бензоїлізотіоціанат (0,63 мл, 4,6 ммоль, 1 екв.). Реакційну суміш перемішують при кімнатній температурі протягом 3 год. Розчинник видаляють під зниженим тиском, та залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від 20% дихлорметан:гексан до 50% дихлорметан:гексан, та одержують вказану в заголовку сполуку (вихід 84%): MS (m/z): 457, 459 (M+23). Підготовчий синтез 24 N-((4S)-4-(3-бром-4-фторфеніл)-6-(йодметил)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2іл)бензамід 25 I S Br N O N H F 30 35 До охолодженого до 0(C розчину (S)-N-(2-(3-бром-4-фторфеніл)-4-метилпент-4-ен-2ілкарбамотіоїл)бензаміду (1,3 г, 3,0 ммоль, 1,0 екв.) у дихлорметані (40 мл) додають йод (1,5 г, 5,9 ммоль, 2,0 екв.). Реакційну суміш перемішують при 0C протягом 1 год, та поступово нагрівають до кімнатної температури. Реакційну суміш гасять насиченим водним розчином тіосульфату натрію. Водний шар екстрагують дихлорметаном. Об’єднану органічну фазу сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 82%): MS (m/z): 561, 563 (M+1). Підготовчий синтез 25 (S)-N-(4-(3-бром-4-фторфеніл)-4,6,6-триметил-5,6-дигідро-4H-1,3-тіазин-2-іл)бензамід S Br N O N H F 40 45 До розчину N-((4S)-4-(3-бром-4-фторфеніл)-6-(йодметил)-4,6-диметил-5,6-дигідро-4H-1,3тіазин-2-іл)бензаміду (0,13 г, 0,23 ммоль, 1,0 екв.) у толуолі (1,5 мл) додають 2-2’-азо-бісізобутиронітрил (0,006 г, 0,03 ммоль, 0,15 екв.) та гідрид три-н-бутилолова. Реакційну суміш перемішують при кімнатній температурі протягом 3 год, та концентрують під зниженим тиском. Розчинник видаляють під зниженим тиском, залишок очищають хроматографією на силікагелі, елююючи лінійним градієнтом від 5% етилацетат:гексан до 20% етилацетат:гексан, та одержують вказану в заголовку сполуку (вихід 25%): MS (m/z): 435, 437 (M+1). Підготовчий синтез 26 (S)-N-(4-(4-фтор-3-(піримідин-5-іл)феніл)-4,6,6-триметил-5,6-дигідро-4H-1,3-тіазин-2іл)бензамід 19 UA 102251 C2 N S N N O N H F 5 10 15 До нагрітого до 97(C розчину (S)-N-(4-(3-бром-4-фторфеніл)-4,6,6-триметил-5,6-дигідро-4H1,3-тіазин-2-іл)бензаміду (0,067 г, 0,15 ммоль, 1,0 екв.) у 1,2-диметоксіетані (1,5 мл), етанолі (0,7 мл) та воді (1,0 мл) додають піримідин-5-борну кислоту (0,095 г, 0,77 ммоль, 5,0 екв.), карбонат цезію (0,301 г, 0,92 ммоль, 6,1 екв.) та хлорид біс(трифенілфосфін)паладію(II) (0,022 г, 0,03 ммоль, 0,2 екв.). Реакційну суміш перемішують при 97C протягом 20 хв. Після охолодження до кімнатної температури реакційну суміш виливають у воду, та екстрагують етилацетатом. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Розчинник видаляють під зниженим тиском, залишок очищають флеш-хроматографією на колонці із силікагелем, елююючи лінійним градієнтом від дихлорметану до суміші 15% етилацетат:дихлорметан, та одержують вказану в заголовку сполуку (вихід 46%): MS (m/z): 435 (M+1). Підготовчий синтез 27 (S)-3-(3-бромфеніл)-N-метокси-N-метил-3-(R)-(2-метилпропан-2-сульфініламіно)бутирамід O S NH O Br 20 25 30 N O До охолодженого до -78(C розчину гідрохлориду N,O-диметилгідроксиламіну (12 г, 130 ммоль, 5,0 екв.) у THF (200 мл) додають н-бутиллітій (100 мл, 250 ммоль, 10 екв.) (2,5 M розчин у гексані) через канюлю. Реакційну суміш перемішують протягом 15 хв, та додають краплями розчин метилового складного ефіру (S)-3-((R)-2-метилпропан-2-сульфініламіно)-3фенілмасляної кислоти (9,5 г, 25 ммоль, 1,0 екв.) у THF (50 мл). Реакційну суміш нагрівають до 60C, та витримують при цій температурі протягом 1 год. Реакційну суміш гасять насиченим водним розчином хлориду амонію, розводять водою, та екстрагують етилацетатом. Одержану органічну фазу екстрагують водою, насиченим водним розчином NaCl, сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід 60%): MS (m/z): 405, 407 (M+1). Сполуки, наведені нижче в Таблиці 10, одержують по суті як описано у підготовчому синтезі (S)-3-(3-бромфеніл)-N-метокси-N-метил-3-(R)-(2-метилпропан-2-сульфініламіно)бутираміду. Таблиця 10 Підг. синт. № 27a 27b Хімічна назва Фізичні дані MS (m/z) (S)-3-(3-бром-4-фторфеніл)-3-((R)-1,1-диметилетилсульфінамідо)-N-метокси- 423, 425 N-метилбутанамід (M+1) (S)-3-(5-бром-2,4-дифторфеніл)-3-((R)-1,1-диметилетилсульфінамідо)-N441, 443 метокси-N-метилбутанамід (M+1) Підготовчий синтез 28 [(S)-1-(3-бромфеніл)-1-метил-3-оксобутил]амід (R)-2-метилпропан-2-сульфінової кислоти 35 O S NH O Br 20 UA 102251 C2 5 10 До охолодженого до -78(C розчину (S)-3-(3-бромфеніл)-N-метокси-N-метил-3-(R)-(2метилпропан-2-сульфініламіно)бутираміду (1,5 г, 3,7 ммоль; 1,0 екв.) у THF (53 мл) додають метилмагнійбромід (6,2 мл, 18,5 ммоль, 5,0 екв.), та реакційну суміш залишають для нагрівання до температури навколишнього середовища. Через 1 год реакційну суміш охолоджують до 78C, та гасять насиченим водним розчином хлориду амонію. Одержану суміш розводять водою, та екстрагують етилацетатом. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку (вихід >95%): MS (m/z) 360, 362 (M+1). Сполуки, наведені нижче в Таблиці 11, одержують по суті як описано у підготовчому синтезі [(S)-1-(3-бромфеніл)-1-метил-3-оксобутил]аміду (R)-2-метилпропан-2-сульфінової кислоти. Таблиця 11 Підг. синт. № Хімічна назва (R)-N-((S)-2-(3-бром-4-фторфеніл)-4-оксопентан-2-іл)-2метилпропан-2-сульфінамід (R)-N-((S)-2-(5-бром-2,4-дифторфеніл)-4-оксопентан-2-іл)-2метилпропан-2-сульфінамід 28a 28b Фізичні дані MS (m/z) 378, 380 (M+1) 396, 398 (M+1) Підготовчий синтез 29 (R)-N-((2S)-2-(3-бромфеніл)-4-гідроксипентан-2-іл)-2-метилпропан-2-сульфінамід PG6-E01647-028 15 O S NH OH Br 20 25 30 До розчину [(S)-1-(3-бромфеніл)-1-метил-3-оксобутил]аміду (R)-2-метилпропан-2сульфінової кислоти (1,34 г, 3,5 ммоль, 1,0 екв.) у метанолі (20 мл) додають тетрагідроборат натрію (1,4 г, 35 ммоль, 10,0 екв.). Реакційну суміш перемішують при температурі навколишнього середовища протягом ночі. Реакційну суміш обережно гасять водою, та екстрагують етилацетатом. Одержану органічну фазу сушать над сульфатом натрію, та розчинник видаляють під зниженим тиском, одержуючи суміш діастереомерів. Діастереомери розділяють хроматографією на силікагелі (120 г), елююючи градієнтом від (50:50) етилацетат:гексан до (100:0) етилацетат:гексан. Ізомер, який елюювався другим, виділяють, розчинник видаляють під зниженим тиском, та одержують вказану в заголовку сполуку як єдиний діастереомер (вихід 45%): MS (m/z): 362, 364 (M+1). Сполуки, наведені нижче в Таблиці 12, одержують по суті як описано у підготовчому синтезі (R)-N-((2S)-2-(3-бромфеніл)-4-гідроксипентан-2-іл)-2-метилпропан-2-сульфінаміду. Таблиця 12 Підг. синт. № 29a 29b 2 3 35 Хімічна назва (R)-N-((2S)-2-(3-бром-4-фторфеніл)-4-гідроксипентан-2-іл)-22 метилпропан-2-сульфінамід (R)-N-((2S)-2-(5-бром-2,4-дифторфеніл)-4-гідроксипентан-2-іл)-23 метилпропан-2-сульфінамід Фізичні дані MS (m/z) 380, 382 (M+1) 398, 400 (M+1) Вказану сполуку виділяли та використовували у формі суміші діастереомерів. Вказану сполуку виділяли та використовували у формі суміші діастереомерів (65:35). Підготовчий синтез 30 21 UA 102251 C2 (S)-4-аміно-4-(3-бромфеніл)пентан-2-ол HCl NH2 OH Br 5 10 Розчин хлористого водню (5 мл; 13 екв.; 20 ммоль) (4 M розчин у діоксані) та [(S)-1-(3бромфеніл)-3-гідрокси-1-метилбутил]аміду (R)-2-метилпропан-2-сульфінової кислоти (570 мг, 1,6 ммоль, 1,0 екв.) як єдиного діастереомера перемішують протягом 5 хв. Розчинник видаляють під зниженим тиском, та залишок підлуговують насиченим водним розчином бікарбонату натрію. Водну фазу екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, концентрують під зниженим тиском, та одержують вказану в заголовку сполуку: MS (m/z): 358, 360 (M+1). Сполуку, наведену нижче в Таблиці 13, одержують по суті як описано у підготовчому синтезі (S)-4-аміно-4-(3-бромфеніл)-пентан-2-олу. Таблиця 13 Підг. Хімічна назва синт. № 4 30a (4S)-4-аміно-4-(5-бром-2,4-дифторфеніл)пентан-2-ол 4 Фізичні дані MS (m/z) 380, 382 (M+1) Вказану в заголовку сполуку виділяли у формі суміші діастереомерів 15 Підготовчий синтез 31 Трет-бутил-(4S,6R)-4-(3-бромфеніл)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат S Br N O N H O 20 25 30 35 40 45 До розчину (S)-4-аміно-4-(3-бромфеніл)пентан-2-олу (410 мг, 794 мкмоль) як єдиного діастереомера у THF (10 мл) додають біс(триметилсиліл)трифторацетамід (204 мг, 0,79 ммоль). Через 1 год додають краплями бензоїлізотіоціанат (259 мг, 1,7 ммоль). Реакційну суміш перемішують протягом 1 год. Реакційну суміш концентрують під зниженим тиском. До одержаного залишку додають 5-н. розчин хлористого водню (25 мл, 125 ммоль), та реакційну суміш витримують при 100C. Через 48 год розчинник видаляють під зниженим тиском, та залишок розподіляють між THF (20 мл) та насиченим розчином бікарбонату натрію (10 мл). До одержаної суміші додають ди-трет-бутилдикарбонат (347 мг, 1,6 ммоль), та реакційну суміш перемішують протягом 48 год. Реакційну суміш розводять водою, та екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Одержаний продукт очищають силікагелем, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (5:2) протягом 20 хв, та одержують вказану в заголовку сполуку (вихід 52%, чистота 70% за даними LCMS): MS (m/z): 399, 401 (M+1). Підготовчий синтез 32 Трет-бутил-(4S,6R)-4-(3-бром-4-фторфеніл)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат Розчин (R)-N-((2S)-2-(3-бром-4-фторфеніл)-4-гідроксипентан-2-іл)-2-метилпропан-2сульфінаміду у вигляді суміші діастереомерів (1:5, 2 г, 1,0 екв.) у діоксані (5 мл) додають краплями до розчину 4-н. розчину хлористого водню у діоксані (20 мл, 80 ммоль) при 0C. Реакційну суміш нагрівають до кімнатної температури, та перемішують протягом 5 хв. Розчинник видаляють під зниженим тиском, та залишок підлуговують насиченим водним розчином бікарбонату натрію. Водну фазу екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Одержаний залишок розчиняють у THF (50 мл), та охолоджують до 0C. Додають краплями бензоїлізотіоціанат (1,7 г, 10,5 ммоль). Реакційну суміш перемішують протягом 1 год. Потім 22 UA 102251 C2 5 10 15 реакційну суміш концентрують під зниженим тиском. Залишок розчиняють у діоксані (5 мл), та переносять у товстостінну скляну реакційну посудину. До одержаного розчину додають 5-н. розчин хлористого водню (75 мл, 375 ммоль). Реакційну посудину закривають кришкою, та витримують при 100C. Через 24 год розчинник видаляють під зниженим тиском, та залишок розподіляють між THF (20 мл) та насиченим водним розчином бікарбонату натрію (10 мл). До одержаної суміші додають ди-трет-бутилдикарбонат (1,7 г, 7,9 ммоль), та реакційну суміш перемішують протягом 2 год. Реакційну суміш розводять водою, та екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Залишок очищають хроматографією на колонці із силікагелем (340 г), елююючи градієнтом від (0:100) етилацетат:гексан до (50:50) етилацетат:гексан протягом 25 хв. Діастереомер, який елююється другим, збирають, та розчинник видаляють під зниженим тиском, одержуючи 595 мг суміші: вказаної в заголовку сполуки, трет-бутил-(4S,6R)-4-(3-бром-4-фторфеніл)-4,6-диметил-5,6дигідро-4H-1,3-тіазин-2-ілкарбамату: MS (m/z): 417, 419 (M+1) (Підготовчий синтез 32); та N-((4S,6R)-4-(3-бром-4-фторфеніл)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2-іл)бензаміду: MS (m/z) 421, 423 (M+1) (Підготовчий синтез 32a). Підготовчий синтез 33 (4S)-4-(5-бром-2,4-дифторфеніл)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2-амін S Br 20 25 30 35 40 F N NH2 F До охолодженого до 0(C розчину (4S)-4-аміно-4-(5-бром-2,4-дифторфеніл)пентан-2-олу (1,3 г, 4,4 ммоль) у вигляді суміші діастереомерів у THF (50 мл) додають краплями бензоїлізотіоціанат (1,4 г, 1,2 ммоль), та реакційну суміш перемішують протягом 1 год. Реакційну суміш концентрують під зниженим тиском. Залишок розчиняють у діоксані (5 мл), та переносять у товстостінну скляну реакційну посудину. До одержаної суміші додають 5-н. розчин хлористого водню (75 мл, 375 ммоль), та реакційну суміш витримують при 100C. Через 36 год розчинник видаляють під зниженим тиском, та залишок розчиняють у воді. Водну суміш екстрагують етилацетатом. Водну фазу підлуговують 5-н. розчином NaOH, та екстрагують сумішшю 3:1 хлороформ:IPA. Одержану фазу хлороформ:IPA сушать над сульфатом натрію, фільтрують, розчинник видаляють під зниженим тиском, та одержують вказану в заголовку сполуку. Етилацетатний екстракт сушать над сульфатом натрію, фільтрують, та розчинник видаляють під зниженим тиском. Одержаний залишок пропускають через врівноважену MeOH колонку із SCX, промивають метанолом, після чого елююють 2-н. розчином NH3 у MeOH (50 мл). Промивний елюат (2-н. розчин NH3 у MeOH) збирають, та розчинник видаляють під зниженим тиском. Залишок об’єднують із вказаною в заголовку сполукою, одержаною шляхом екстрагування сумішшю хлороформ:IPA, та одержують вказану в заголовку сполуку у вигляді суміші (6R та 6S) діастереомерів (4S)-4-(5-бром-2,4-дифторфеніл)-4,6-диметил-5,6-дигідро-4H1,3-тіазин-2-аміну. (вихід 64%, чистота 80% за даними LCMS): MS (m/z): 335, 337 (M+1). Підготовчий синтез 34 Трет-бутил-(4S,6R)-4-(5-бром-2,4-дифторфеніл)-4,6-диметил-5,6-дигідро-4H-1,3-тіазин-2ілкарбамат S Br 45 50 F N O N H O F До суміші (6R та 6S) діастереомерів (4S)-4-(5-бром-2,4-дифторфеніл)-4,6-диметил-5,6дигідро-4H-1,3-тіазин-2-аміну у THF (20 мл) та насиченого водного розчину бікарбонату натрію (10 мл) додають ди-трет-бутилдикарбонат (900 мг, 4,1 ммоль). Через 2 год реакційну суміш розводять водою, та екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Одержаний продукт очищають силікагелем, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (4:1) протягом 5 хв. Діастереомер, який елююється другим, збирають, та розчинник видаляють 23 UA 102251 C2 5 під зниженим тиском, одержуючи вказану в заголовку сполуку (вихід 43%): MS (m/z): 435, 437 (M+1). Підготовчий синтез 35 Трет-бутиловий складний ефір [(4S,6R)-4,6-диметил-4-(3-піримідин-5-ілфеніл)-5,6-дигідро4H-[1,3]тіазин-2-іл]карбамінової кислоти N N 10 15 20 S N O N H O До нагрітого до 100(C розчину трет-бутил-(4S,6R)-4-(3-бромфеніл)-4,6-диметил-5,6-дигідро4H-1,3-тіазин-2-ілкарбамату у суміші 1,2-диметоксіетан:вода:етанол [(3:1,5:1), 12 мл] однією порцією додають піримідин-5-борну кислоту (128 мг, 5,9 ммоль, 2,5 екв.), хлорид біс(трифенілфосфін)паладію(II) (29 мг, 41 мкмоль, 0,1 екв.) та карбонат цезію (1,24 г, 1,24 ммоль, 3 екв.). Через 20 хв реакційну суміш охолоджують до температури навколишнього середовища, розводять водою, та екстрагують дихлорметаном. Одержану органічну фазу сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Залишок очищають флеш-хроматографією на колонці із силікагелем, елююючи лінійним градієнтом від гексану до суміші гексан:етилацетат (5:2) протягом 20 хв, та одержують вказану в заголовку сполуку (вихід 39%): MS (m/z): 399 (M+1). Сполуки, наведені нижче в Таблиці 14, одержують по суті як описано у підготовчому синтезі трет-бутилового складного åô³ðó [(4S,6R)-4,6-диметил-4-(3-піримідин-5-ілфеніл)-5,6-дигідро-4H[1,3]тіазин-2-іл]-карбамінової кислоти. Таблиця 14 Підг. синт. № 35a 35b Хімічна назва трет-бутил-(4S,6R)-4-(4-фтор-3-(піримідин-5-іл)феніл)-4,6-диметил5 5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат трет-бутил-(4S,6R)-4-(2,4-дифтор-5-(піримідин-5-іл)феніл)-4,6диметил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат Фізичні дані MS (m/z) 417 (M+1) 435 (M+1) 5 Виділяють у вигляді суміші вказаної сполуки та N-((4S,6R)-4-(3-бром-4-фторфеніл)-4,6диметил-5,6-дигідро-4H-1,3-тіазин-2-іл)бензаміду. MS (m/z): 421 (M+1). Підготовчий синтез 36 1-бром-3-(проп-1-ен-2-іл)бензол 25 Br 30 35 40 Метилтрифенілфосфонійбромід (35,7 г, 97,9 ммоль, 1,3 екв.) суспендують у тетрагідрофурані (100 мл), та охолоджують до 0(C. До одержаної суміші через крапельну лійку повільно додають N-бутиллітій (2,5 M розчин у гексані, 27,0 г, 97,7 ммоль, 39,2 мл, 1,3 екв.). Одержаний розчин перемішують протягом 1 год при 0C. Додають повільно через крапельну лійку розчин 3-бромацетофенону (15,0 г, 75,3 ммоль, 10,0 мл, 1,0 екв.) у тетрагідрофурані (50 мл). Одержану суміш нагрівають до кімнатної температури, та перемішують протягом 3 год. Реакційну суміш охолоджують до 0C, та гасять насиченим водним розчином хлориду амонію. Шари розділяють у ділильній лійці, та водну фазу екстрагують гексаном. Об’єднану органічну фазу сушать над безводним сульфатом натрію, фільтрують, та залишають для вистоювання протягом ночі при кімнатній температурі. Одержану органічну фазу декантують з осаду, та концентрують під зниженим тиском. Одержану тверду речовину розводять гексаном, та фільтрують. Одержаний осад промивають гексаном. Фільтрат концентрують, та одержану суміш очищають флеш 24 UA 102251 C2 5 хроматографією на колонці із силікагелем (гексан), та одержують вказану в заголовку сполуку 1 (вихід 83%): H ЯМР (CDCl3, 400M Гц) 7,59 (t, 1H, J=1,72 Гц), 7,40-7,36 (m, 2H), 7,18 (t, 1H, J=8 Гц), 5,36 (s, 1H), 5,12-5,10 (m, 1H), 2,13-3,11 (m, 3H). Сполуку, наведену нижче в Таблиці 15, одержують по суті як описано у підготовчому синтезі 1-бром-3-(проп-1-ен-2-іл)бензолу. Таблиця 15 Підг. синт. № 36a Хімічна назва 2-бром-1-фтор-4-(проп-1-ен-2-іл)бензол Фізичні дані 6 Дивись нижче 6 1 H ЯМР(CDCl3, 400M Гц): ( 7,61 (dd, 1H, J=6,8 Гц, 2,2 Гц), 7,36-7,32 (m, 1H), 7,05 (t, 1H, J=8,4 Гц), 5,29 (s, 1H), 5,09-5,07 (m, 1H), 2,10-2,09 (m, 3H). Підготовчий синтез 37 Етил-4-(3-бромфеніл)-2-гідроксипент-4-еноат 10 OH O Br O 15 20 25 До розчину 2-бром-1-фтор-4-(проп-1-ен-2-іл)бензолу (12,3 г, 62,2 ммоль, 1,0 екв.) у ацетонітрилі (124 мл, 0,5 M) додають етилгліоксалат (38,1 г, 37 мл, 187 ммоль, 3 екв.) та гідрат трифторметансульфонату ітербію (7,72 г, 12,4 ммоль, 0,2 екв.). Одержану суміш перемішують протягом ночі при кімнатній температурі. Потім суміш концентрують під зниженим тиском, та розводять діетиловим ефіром. Одержаний розчин промивають двічі водою. Органічний шар сушать над безводним сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Залишок очищають флеш-хроматографією на колонці із силікагелем (330 г) двома порціями, застосовуючи градієнт 0-100% етилацетат/гексан, та одержують вказану в заголовку сполуку 1 (вихід 87%): H ЯМР (CDCl3, 400 МГц): 7,53 (t, 1H, J=1,9 Гц), 7,41-7,37 (dt, 1H), 7,33-7,30 (dt, 1H), 7,18 (t, 1H, J=7,6 Гц), 5,37 (s, 1H), 5,22 (s, 1H), 4,26-4,21 (m, 1H), 4,17-3,99 (m, 2H), 2,99 (dd, J=14,8 Гц, 4,8 Гц), 2,83-2,72 (m, 2H), 1,23 (t, 3H, J=7,6 Гц). Сполуку, наведену нижче в Таблиці 16, одержують по суті як описано у підготовчому синтезі етил-4-(3-бромфеніл)-2-гідроксипент-4-еноату. Таблиця 16 Підг. Хімічна назва синт. № 37a етил-4-(3-бром-4-фторфеніл)-2-гідроксипент-4-еноат Фізичні дані 7 Дивись нижче 7 1 30 H ЯМР (CDCl3, 400M Гц): ( 7,59-7,55(m, 1H), 7,32-7,27 (m, 1H), 7,08-7,03 (m, 1H), 5,33 (s, 1H), 5,20 (s, 1H), 4,26-4,20 (m, 1H), 4,19-4,02 (m, 2H), 2,96 (dd, J=14,8 Гц, 4,8 Гц), 2,80-2,73 (m, 2H), 1,24 (t, 3H, J=7,6 Гц). Підготовчий синтез 38 Етил-2-аміно-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-6-карбоксилат O O S Br N NH2 35 До розчину етил-4-(3-бромфеніл)-2-гідроксипент-4-еноату (5,1 г, 17 ммоль) у ацетонітрилі (68 мл) додають 2,6-літидин (2,19 г, 20,4 ммоль, 1,2 екв.). Реакційну суміш охолоджують до 0C, 25 UA 102251 C2 5 10 15 та додають краплями трифторметансульфоновий ангідрид (3,30 мл, 19,6 ммоль, 1,15 екв.) протягом приблизно 5 хв. Одержану суміш перемішують при 0C протягом 20 хв. Додають тіосечовину (2,59 г, 34,0 ммоль, 2 екв.), та нагрівають реакційну суміш до кімнатної температури. Через 45 хв одержану суміш концентрують під зниженим тиском. Одержане густе оранжеве масло потім додають за допомогою великої піпетки до перемішуваної сірчаної кислоти (17,8 M розчин, 8 мл) при кімнатній температурі. Через 20 хв одержану суміш додають краплями при інтенсивному перемішуванні до охолодженого до 0°C розчину K 2CO3 (приблизно 50 г у 50 мл H2O). Додають додаткову кількість води для уможливлення перемішування при утворенні твердої речовини протягом гасіння. Одержану жовтувато-коричневу/оранжеву тверду речовину відділяють фільтруванням. Цю тверду речовину залишають для висихання на фільтрувальному папері під струменем повітря протягом 1 год, та одержують вказану в заголовку сполуку у вигляді суміші діастереомерів, яку використовують без додаткового очищення: MS (m/z): 357, 359 (M+1). Сполуку, наведену нижче в Таблиці 17, одержують по суті як описано у підготовчому синтезі етил-2-аміно-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-6-карбоксилату. Таблиця 17 Підг. синт. № Фізичні дані MS (m/z) етил-2-аміно-4-(3-бром-4-фторфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин375, 377 8 6-карбоксилат (M+1) Хімічна назва 38a 8 20 Рацемічна суміш діастереомерів Підготовчий синтез 39 Етил-4-(3-бромфеніл)-2-(трет-бутоксикарбоніламіно)-4-метил-5,6-дигідро-4H-1,3-тіазин-6карбоксилат O O S Br 25 30 N O N H O До суспензії етил-2-аміно-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3-тіазин-6-карбоксилату (6,1 г, 17 ммоль) у 1,4-діоксані (35 мл), воді (18 мл) та насиченому водному розчині бікарбонату натрію (18 мл) додають ди-трет-бутилдикарбонат (7,42 г, 34,0 ммоль, 2 екв.). Одержану суміш перемішують протягом приблизно 60 год. Реакційну суміш розводять водою, та екстрагують тричі CH2Cl2. Органічний шар сушать над Na2SO4, фільтрують, та концентрують під зниженим тиском до одержання коричневого масла. Це масло очищають флеш-хроматографією на колонці із силікагелем (150 г), елююючи градієнтом 0-100% етилацетат/гексан, та одержують вказану в заголовку сполуку як суміш діастереомерів (вихід 64%): MS (m/z): 457, 459 (M+1). Сполуку, ìàâåäåну нижче в Таблиці 18, одержують по суті як описано у підготовчому синтезі етил-4-(3-бромфеніл)-2-(трет-бутоксикарбоніламіно)-4-метил-5,6-дигідро-4H-1,3-тіазин-6карбоксилату. 35 Таблиця 18 Підг. синт. № 39a 9 Хімічна назва етил-4-(3-бром-4-фторфеніл)-2-(трет-бутоксикарбоніламіно)-4-метил-5,69 дигідро-4H-1,3-тіазин-6-карбоксилат Рацемічна суміш діастереомерів 26 Фізичні дані MS (m/z) 475, 477 (M+1) UA 102251 C2 Підготовчий синтез 40 (4S,6S)-етил-4-(3-бромфеніл)-2-(трет-бутоксикарбоніламіно)-4-метил-5,6-дигідро-4H-1,3тіазин-6-карбоксилат O O S Br N O N H O 5 10 15 Етил-4-(3-бромфеніл)-2-(трет-бутоксикарбоніламіно)-4-метил-5,6-дигідро-4H-1,3-тіазин-6карбоксилат (3,5 г, 7,7 ммоль) очищають хіральною РХВЕ у дві стадії: (колонка: Chiralcel OJ 8(32 см; елюент: 1:3 (спирт 3A:гептан); швидкість потоку: 400 мл/хв, УФ детектування на довжині хвилі 240 нм), одержуючи фракцію 1, яка містить піки 1 та 2 із загальної кількості 3; після чого піки 1 та 2 знову очищають додатковою хіральною хроматографією (колонка: Chiralcel OD 832 см; елюент 1:9 (ізопропіловий спирт:гептан); швидкість потоку: 400 мл/хв, УФ детектування на довжині хвилі 240 нм). Виділяють ізомер, який елюювався другим, та одержують вказану в заголовку сполуку після концентрування фракцій під зниженим тиском (вихід 14%). Підготовчий синтез 41 (+/-)-Трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(гідроксиметил)-4-метил-5,6-дигідро-4H-1,3тіазин-2-ілкарбамат OH S Br N O O N H 20 25 30 35 40 До охолодженого до 0(C розчину етил-4-(3-бромфеніл)-2-(трет-бутоксикарбоніламіно)-4метил-5,6-дигідро-4H-1,3-тіазин-6-карбоксилату (2,0 г, 4,4 ммоль) у тетрагідрофурані (87 мл) та етанолі (25 мл) додають боргідрид літію (289 мг, 13,1 ммоль, 3 екв.). Реакційну суміш нагрівають до кімнатної температури, та перемішують протягом 4 год. Реакційну суміш гасять насиченим розчином NH4Cl. Шари розділяють, та водний шар екстрагують етилацетатом. Об’єднані органічні шари промивають насиченим водним розчином NaCl, та сушать над Na 2SO4. Одержану суміш фільтрують, концентрують, та одержують світло-жовте масло. Це масло очищають хроматографією на колонці із силікагелем (120 г), застосовуючи градієнт 0-100% етилацетат/гексан, та одержують вказану в заголовку сполуку (вихід 29%): MS (m/z): 415, 417 (M+1). Підготовчий синтез 42 Трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(гідроксиметил)-4-метил-5,6-дигідро-4H-1,3-тіазин-2ілкарбамат (+/-) Трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(гідроксиметил)-4-метил-5,6-дигідро-4H-1,3тіазин-2-ілкарбамат (870 мг, 2,1 ммоль) очищають хіральним розділенням за допомогою РХВЕ: (колонка: Chiralpak AD 836 см20 мкм; елюент: 100% етиловий спирт 3A; швидкість потоку: 400 мл/хв, детектування УФ на довжині хвилі 250 нм). Ізомер, який елюювався другим, виділяють, одержуючи енантіомерно збагачену вказану в заголовку сполуку (вихід 38,5%): MS (m/z): 415, 417 (M+1). Підготовчий синтез 43 (+/-)-Трет-бутил-(4S,6S)-6-(гідроксиметил)-4-метил-4-(3-(піримідин-5-іл)феніл)-5,6-дигідро4H-1,3-тіазин-2-ілкарбамат OH N N S N O N H O 45 До нагрітого до 110(C розчину (+/-)-трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(гідроксиметил)-4метил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамату (150 мг, 0,36 ммоль) у 1,2-диметоксіетані 27 UA 102251 C2 5 10 (4,5 мл), етанолі (1,5 мл) та воді (2,3 мл) додають піримідин-5-борну кислоту (112 мг, 0,90 ммоль, 2,5 екв.), карбонат цезію (353 мг, 1,08 ммоль, 3 екв.) та хлорид біс(трифенілфосфін)-паладію(II) (25 мг, 0,036 ммоль, 0,1 екв.). Реакційну суміш перемішують при 110(C. Через 20 хв реакційну суміш розводять EtOAc та H2O. Шари розділяють, та водну фазу екстрагують EtOAc. Об’єднані органічні шари сушать над сульфатом натрію, фільтрують, та концентрують під зниженим тиском. Неочищений залишок очищають хроматографією на колонці із силікагелем (5% MeOH/DCM), та одержують вказану в заголовку сполуку (вихід 29%): MS (m/z): 415 (M+1). Сполуку, наведену нижче в Таблиці 19, одержують по суті як описано у підготовчому синтезі (+/-)-трет-бутил-(4S,6S)-6-(гідроксиметил)-4-метил-4-(3-(піримідин-5-іл)феніл)-5,6-дигідро-4H1,3-тіазин-2-ілкарбамату. Таблиця 19 Підг. синт. № 43a 15 Хімічна назва трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(гідроксиметил)-4-метил-5,6дигідро-4H-1,3-тіазин-2-ілкарбамат Фізичні дані MS (m/z) 415 (M+1) Підготовчий синтез 44 (+/-)-Трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(2-гідроксипроп-2-іл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-ілкарбамат OH O S Br 20 25 30 N N H O До охолодженого до 0(C розчину етил-2-аміно-4-(3-бромфеніл)-4-метил-5,6-дигідро-4H-1,3тіазин-6-карбоксилату (250 мг, 0,55 ммоль) у тетрагідрофурані (5,5 мл) додають метилмагнійхлорид (0,58 мл, 1,75 ммоль, 3,2 екв.). Через 15 хв додають додаткову кількість метилмагнійхлориду (0,38 мл, 1,2 ммоль, 2 екв.). Через 30 хв реакційну суміш гасять насиченим водним розчином NH4Cl, та розводять етилацетатом. Шари розділяють, та водний шар екстрагують етилацетатом. Об’єднані органічні шари промивають насиченим водним розчином NaCl, сушать над Na2SO4, фільтрують, та концентрують під зниженим тиском. Неочищений залишок очищають хроматографією на колонці із силікагелем (80 г), елююючи градієнтом 0100% етилацетат/гексан, та одержують вказану в заголовку сполуку (вихід 26%): MS (m/z): 443, 445 (M+1). Сполуки, наведені нижче в Таблиці 20, одержують по суті як описано у підготовчому синтезі (+/-)-трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(2-гідроксипропан-2-іл)-4-метил-5,6-дигідро-4H-1,3тіазин-2-ілкарбамату. Таблиця 20 Підг. синт. № 44a 44b 35 Хімічна назва (+/-)-трет-бутил-(4S,6S)-4-(3-бром-4-фторфеніл)-6-(2-гідроксипропан-2іл)-4-метил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат трет-бутил-(4S,6S)-4-(3-бромфеніл)-6-(2-гідроксипропан-2-іл)-4-метил5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат Фізичні дані MS (m/z) 461, 463 (M+1) 443, 445 (M+1) Підготовчий синтез 45 (+/-)-Трет-бутил-(4S,6S)-4-(4-фтор-3-(піримідин-5-іл)феніл)-6-(2-гідроксипропан-2-іл)-4метил-5,6-дигідро-4H-1,3-тіазин-2-ілкарбамат 28
ДивитисяДодаткова інформація
Назва патенту англійськоюAminidihydrothiazine derivatives as bace inhibitors for the treatment of alzheimer's disease
Автори англійськоюAudia, James, Edmund, Mergott, Dustin, James, Sheenan, Scott, Martin, Watson, Brian, Morgan
Назва патенту російськоюПроизводные аминодигидротиазина как ингибиторы васе для лечения болезни альцгеймера
Автори російськоюОдиа Джеймс Эдмунд, Мерготт Дастин Джеймс, Шихан Скотт Мартин, Уотсон Брайан Морган
МПК / Мітки
МПК: A61K 31/541, C07D 279/06, A61P 25/28
Мітки: інгібітори, хвороби, альцгеймера, похідні, лікування, васе, амінодигідротіазину
Код посилання
<a href="https://ua.patents.su/40-102251-pokhidni-aminodigidrotiazinu-yak-ingibitori-vase-dlya-likuvannya-khvorobi-alcgejjmera.html" target="_blank" rel="follow" title="База патентів України">Похідні амінодигідротіазину як інгібітори васе для лікування хвороби альцгеймера</a>
Похідні ізоіндоліну, фармацевтична композиція на їх основі, спосіб лікування хвороби альцгеймера, спосіб інгібування агрегації амілоїдних білків та спосіб виявлення амілоїдних відкладень

Номер патенту: 64842
Опубліковано: 15.03.2004
Автори: Уокер Лері Кресвелл, Секкаб Аннетт Тереза, Лай Йінгджіє, Оджеллі-Шафран Корінн Елізабет
МПК: C07D 401/10, C07D 209/44, C07D 403/10, C07D 403/12, A61K 31/4035, A61P 43/00, C07D 401/12
Мітки: альцгеймера, спосіб, похідні, білків, лікування, ізоіндоліну, фармацевтична, інгібування, агрегації, відкладень, виявлення, амілоїдних, композиція, хвороби, основі
Формула / Реферат:
1. Похідні ізоіндоліну формули Іабо їх фармацевтично прийнятні солі,де Χ - феніл або заміщений феніл,Υ - феніл, заміщений феніл, піридил або заміщений піридил,де заміщений феніл та заміщений піридил можуть мати 1-4 замісники, кожний з яких незалежно вибраний з -ОС1-С12алкілу, галогену, -С1-С6алкілу, фенілу, -C(O)NHR",...
Похідні терфенілу для лікування хвороби альцгеймера

Номер патенту: 95766
Опубліковано: 25.08.2011
Автори: Буссар Сірілл, Сунозе Міхіро, Мейджор Джеремі, Вільсон Френсіс, Леформал Аделін, Рід Елісон, Жанг Ян, Херрісон Річард Джон, Ернадес-Перні Ремедіос, Бурккхардт Свеня, Смелт Катрін, Рідер Валері, Тейлор Джесс, Хо Чіх Юнг, Кенсфілд Ендрю
МПК: C07C 57/00, C07C 59/00, A61P 25/28, A61K 31/192, C07C 51/09, C07D 257/00
Мітки: лікування, терфенілу, похідні, хвороби, альцгеймера
Формула / Реферат:
1. Сполука, що має загальну формулу (І), (I) в якій Х означає зв'язок або групу -CR5R6, в якій R5 і R6, незалежно один від іншого, вибрані з групи, яку складають Н; алкіл, вибраний з групи СН3, С2H5, і-С3Н7, н-С3Н7, i-C4H9, н-C4H9, втор-С4Н9, трет-С4Н9; алкеніл, вибраний з групи С2Н3, i-С3Н5, н-С3Н5, н-C4Н7, i-C4Н7,...
Похідні піримідину та їх застосування у терапії та у виробництві лікарського засобу для запобігання та/або лікування хвороби альцгеймера

Номер патенту: 92181
Опубліковано: 11.10.2010
Автори: Арзель Ерван, Стааф Карін, Роттічі Дідьє, БЕРГ Стефан, Турек Домініка, Геллберґ Свен, Берроуз Джеремі, Педерсен Торбен, Рейн Тобіас, Уерта Фернандо, Андерссон Ларс
МПК: A61P 25/16, A61P 25/28, A61P 3/10, A61K 31/506, C07D 405/14, C07D 403/04, C07D 401/14
Мітки: виробництві, застосування, хвороби, альцгеймера, терапії, похідні, засобу, лікування, запобігання, лікарського, піримідину
Формула / Реферат:
1. Сполука формули І: де R1 є вибраним з водню, ціано, С1-3галоалкілу, ORa, SO2NRbRc, C0-2алкілC(O)NRbRc, С1-4алкілNRbRc, CH2ORh, SO2Ri, C(O)ORa, CH(OH)Rj та C(O)Rj; R2 та R4 є незалежно вибраними з водню, гало, ціано, NO2, С1-4алкілу, С1-3галоалкілу, ORa, C(O)NRbRc, SO2Rі та C(O)ORa;...
1-фенілалкіл-1,2,3,6-тетрагідропіридини, спосіб їх одержання та фармацевтична композиція для лікування хвороби альцгеймера

Номер патенту: 52707
Опубліковано: 15.01.2003
Автори: КАРДАМОН Розанна, ФУРНЬЄ Жаклін, БАРОНІ Марко, ГУЗЗІ Умберто
МПК: A61K 31/4418, A61K 31/444, C07D 211/70, A61P 25/28, C07D 401/04
Мітки: композиція, спосіб, фармацевтична, альцгеймера, лікування, одержання, 1-фенілалкіл-1,2,3,6-тетрагідропіридини, хвороби
Формула / Реферат:
1. Сполука формули (І): (I),в якій:Y означає -СН- або -N-,R1 означає водень, галоген, трифторметил, (С1-С4)-алкіл або (С1-С4)-алкоксил,R2 означає метил або етил,кожний із радикалів R3 та R4 означає водень або (С1-С3)-алкіл,Х означає:(а) (С1-С6)-алкіл, (С1-С6)-алкоксил, карбокси-(С3-С7)-алкіл,...
Заміщені ізоіндоли як інгібітори васе та їх застосування

Номер патенту: 96598
Опубліковано: 25.11.2011
Автори: Мюррей Крістофер, Пател Сагіл, Арнольд Джеймс, Конґрів Маілз, Сілвестер Марк, Чессарі Ґіанні, Йоверґ Ліселотте, Колмодін Карін, Роттіссі Дід'є, Ракос Лассло, Берґ Стефан, Голенс Йорґ, Керс Анніка, Едвардс Філ
МПК: C07D 401/10, A61K 31/4433, A61P 25/28, A61K 31/506, A61K 31/4439, C07D 403/10, A61K 31/4035, C07D 209/44, C07D 407/14
Мітки: застосування, ізоіндоли, інгібітори, заміщені, васе
Формула / Реферат:
1. Сполука формули І , I де R1 вибрано з групи: гідроген, нітро, ціано, -Q-С1-6алкіл, -Q-С2-6алкеніл, -Q-C2-6алкініл, -Q-С3-6циклоалкіл, -Q-С5-7циклоалкеніл, -Q-C1-6алкілС3-6циклоалкіл, -Q-арил, -Q-гетероарил, -Q-C1-6алкіларил, -Q-С1-6алкілгетероарил, -Q-гетероцикліл, та -Q-С1-6алкілгетероцикліл, де вказані...
Попередній патент: Двозаміщені фталазини – антагоністи провідного шляху hedgehog
Наступний патент: Вдосконалений спосіб гасіння небажаних побічних реакцій при патогенній інактивації еритроцитів
Випадковий патент: Композиція інгредієнтів для виготовлення горілки "прима м'яка"