Двозаміщені фталазини – антагоністи провідного шляху hedgehog








Формула / Реферат
1. Сполука формули
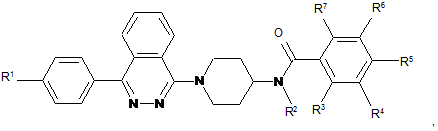
де
R1 - водень, фтор, ціаногрупа, трифторметил, метокси- або трифторметоксигрупа;
R2 - водень або метил; і
R3, R4, R5, R6 та R7 незалежно один від одного - водень, хлор, фтор, ціаногрупа, трифторметил або трифторметоксигрупа, за умови, що щонайменше два замісники з R3, R4, R5, R6 та R7 є атомами водню;
або фармацевтично прийнятна сіль такої сполуки.
2. Сполука за п. 1, яка відрізняється тим, що R1 - водень, або фармацевтично прийнятна сіль такої сполуки.
3. Сполука за п. 1, яка відрізняється тим, що R1 - фтор, або фармацевтично прийнятна сіль такої сполуки.
4. Сполука за будь-яким з пп. 1-3, яка відрізняється тим, що R2 - метил, або фармацевтично прийнятна сіль такої сполуки.
5. Сполука за будь-яким з пп. 1-3, яка відрізняється тим, що R2 - водень, або фармацевтично прийнятна сіль такої сполуки.
6. Сполука за будь-яким з пп. 1-5, яка відрізняється тим, що R3 - хлор, фтор, трифторметил або трифторметоксигрупа, або фармацевтично прийнятна сіль такої сполуки.
7. Сполука за будь-яким з пп. 1-6, яка відрізняється тим, що R5 - фтор, трифторметоксигрупа або трифторметил, або фармацевтично прийнятна сіль такої сполуки.
8. Сполука за будь-яким з пп. 1-7, яка являє собою 4-фтор-Ν-(1-(4-(4-фторфеніл)-фталазин-1-іл)-піперидин-4-іл)-N-метил-2-(трифторметил)-бензамід, або фармацевтично прийнятна сіль такої сполуки.
9. Фармацевтична композиція, яка включає сполуку за будь-яким з пп. 1-8 або фармацевтично прийнятну сіль такої сполуки у сполученні з фармацевтично прийнятним носієм, розріджувачем або наповнювачем.
10. Сполука за будь-яким з пп. 1-8 або фармацевтично прийнятна сіль такої сполуки для застосування як лікарський засіб.
11. Сполука за будь-яким з пп. 1-8 або фармацевтично прийнятна сіль такої сполуки для застосування при лікуванні раку.
12. Сполука за п. 11, де рак вибраний з групи, до складу якої входять медулобластома, базально-клітинний рак, рак стравоходу, рак шлунка, рак підшлункової залози, рак жовчних шляхів, рак простати, рак молочної залози, дрібноклітинний рак легенів, недрібноклітинний рак легенів, В-клітинна лімфома, множинна мієлома, рак яєчників, рак ободової та прямої кишки, рак печінки, рак нирки та меланома.
Текст
Реферат: Винахід стосується 1,4-двозаміщених фталазинів - антагоністів провідного шляху hedgehog, корисних при лікуванні раку. UA 102250 C2 (12) UA 102250 C2 UA 102250 C2 5 10 15 20 25 Цей винахід стосується антагоністів провідного шляху hedgehog і, більш конкретно, нових двозаміщених фталазинів та їх терапевтичного застосування. Провідний шлях Hedgehog (Hh) відіграє значну роль у формуванні ембріональної системи та підтриманні стану тканини у дорослих шляхом спрямування диференціації та проліферації клітин. Групу hedgehog-(Hh)протеїнів, до якої входять Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) та Desert Hedgehog (Dhh), складають секретовані глікопротеїни, які зазнають посттрансляційних модифікацій, в тому числі автокаталітичного розщеплення та приєднання холестерину до амінотермінального пептиду з утворенням фрагмента, який має сигнальну активність. Hh зв'язується з 12-прохідним трансмембранним протеїном Ptch (Ptch1 та Ptch2), тим самим полегшуючи опосередковане Ptch пригнічення агента Smoothened (Smo). Активація Smo запускає послідовність внутрішньоклітинних процесів, кульмінацією яких є стабілізація факторів транскрипції Gli (Gli1, Gli2 та Gli3) та експресія Gli-залежних генів, відповідальних за проліферацію клітин, виживання клітин, ангіогенез та інвазію. Передача сигналів за участю Hh останнім часом викликає значний інтерес у зв'язку з виявленням факту, що аберантна активація передачі сигналу за участю Shh призводить до виникнення різноманітних пухлин, наприклад, раку підшлункової залози, медулобластоми, базально-клітинного раку, дрібноклітинного раку легенів та раку простати. Відомі декілька антагоністів Hh, такі як ІР-609 (похідне стероїдного алкалоїда); CUR61414 (похідне амінопроліну) та JK18 (похідне 2,4-двозаміщеного тіазолу). У WO2005/033288 описані деякі 1,4двозаміщені фталазини, вказані як антагоністи hedgehog. Існує постійна потреба в ефективних інгібіторах провідного шляху hedgehog, зокрема, у таких, що мають бажані фармакодинамічні, фармакокінетичні та токсикологічні профілі. Цей винахід пропонує нові 1,4-двозаміщені фталазини, які є ефективними антагоністами згаданого провідного шляху. Цей винахід пропонує сполуку Формули I: 7 6 R R O 5 R 1 R N N N 30 35 40 45 50 55 N 2 3 4 R R R , де 1 R – водень, фтор, ціаногрупа, трифторметил, метокси- або трифторметоксигрупа; 2 R – водень або метил; і 3 4 5 6 7 R , R , R , R та R незалежно один від одного – водень, хлор, фтор, ціаногрупа, 3 4 5 трифторметил або трифторметоксигрупа, за умови, що щонайменше два замісники з R , R , R , 6 7 R та R є атомами водню; або фармацевтично прийнятну сіль такої сполуки. Для досвідченого фахівця у галузі зрозуміло, що сполуки за цим винаходом містять третинну аміногрупу та здатні реагувати з численними неорганічними та органічними кислотами, утворюючи фармацевтично прийнятні солі з кислотами. Такі фармацевтично прийнятні солі з кислотами та загальна методологія їх одержання добре відомі в галузі. Дивись, наприклад, Шталь та ін., “„Довідник з фармацевтичних солей. Властивості, добір та застосування" (P.Stahl et al., Handbook of pharmaceutical salts: properties, selection, and use, VCHA/Wiley-VCH, 2002); Берж та ін., „Фармацевтичні солі" (S.M.Berge et al., Pharmaceutical Salts, J. of Pharm. Sciences Vol. 66, No.1, January 1977). Конкретні варіанти здійснення цього винаходу охоплюють сполуки Формули I або їх фармацевтично прийнятні солі, де: 1 (a) R – водень; 1 (b) R – фтор; 2 (c) R – водень; 2 (d) R – метил; 3 (e) R – хлор, фтор, трифторметил або трифторметоксигрупа; 5 (f) R – фтор, трифторметоксигрупа або трифторметил; 1 2 (g) R – водень та R – водень; 1 2 (h) R – фтор та R – водень; 1 2 (i) R – водень та R – метил; 1 2 (j) R – фтор та R – метил; 1 UA 102250 C2 1 5 10 15 20 25 30 35 40 45 50 55 60 3 (k) R – водень та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 3 (l) R – фтор та R – хлор, фтор, трифторметил або трифторметоксигрупа; 2 3 (m) R – водень та R – хлор, фтор, трифторметил або трифторметоксигрупа; 2 3 (n) R – метил та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 5 (o) R – водень та R – фтор, трифторметоксигрупа або трифторметил; 1 5 (p) R – фтор та R – фтор, трифторметоксигрупа або трифторметил; 2 5 (q) R – водень та R – фтор, трифторметоксигрупа або трифторметил; 2 5 (r) R – метил та R – фтор, трифторметоксигрупа або трифторметил; 3 5 (s) R – хлор, фтор, трифторметил або трифторметоксигрупа та R – фтор, трифторметоксигрупа або трифторметил; 1 2 3 (t) R – водень, R – водень та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 2 3 (u) R – водень, R – метил та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 2 3 (v) R – фтор, R – водень та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 2 3 (w) R – фтор, R – метил та R – хлор, фтор, трифторметил або трифторметоксигрупа; 1 2 5 (x) R – водень, R – водень та R –фтор, трифторметил або трифторметоксигрупа; 1 2 5 (y) R – водень, R – метил та R –фтор, трифторметил або трифторметоксигрупа; 1 2 5 (z) R – фтор, R – водень та R – фтор, трифторметил або трифторметоксигрупа; 1 2 5 (aa) R – фтор, R – метил та R – фтор, трифторметил або трифторметоксигрупа; 1 2 3 (bb) R – водень, R – водень, R – хлор, фтор, трифторметил або трифторметоксигрупа та 5 R – фтор, трифторметил або трифторметоксигрупа; 1 2 3 5 (cc) R – водень, R – метил, R – хлор, фтор, трифторметил або трифторметоксигрупа та R – фтор, трифторметил або трифторметоксигрупа; 1 2 3 5 (dd) R – фтор, R – водень, R – хлор, фтор, трифторметил або трифторметоксигрупа та R – фтор, трифторметил або трифторметоксигрупа; 1 2 3 5 (ee) R – фтор, R – метил, R – хлор, фтор, трифторметил або трифторметоксигрупа та R – фтор, трифторметил або трифторметоксигрупа; 1 2 3 5 (ff) R – водень, R – водень, R –трифторметил та R – фтор; 1 2 3 5 (gg) R – фтор, R – водень, R –трифторметил та R – фтор; 1 2 3 5 (hh) R – водень, R – метил, R –трифторметил та R – фтор; 1 2 3 5 (ii) R – фтор, R – метил, R –трифторметил та R – фтор; 3 4 5 6 7 (jj) три замісники з R , R , R , R та R є атомами водню; та 3 4 5 6 7 (kk) чотири замісники з-поміж R , R , R , R та R є атомами водню. Цей винахід пропонує також фармацевтичну композицію, яка включає сполуку Формули I або її фармацевтично прийнятну сіль у сполученні з фармацевтично прийнятним наповнювачем, носієм або розріджувачем. Перевага віддається введенню сполук за цим винаходом до складу фармацевтичних композицій, які вводяться в організм різноманітними шляхами. За варіантами, яким віддається перевага, такі композиції призначені для перорального або внутрішньовенного введення. Такі фармацевтичні композиції та способи їх виготовлення добре відомі в галузі. Дивись, наприклад, Ремінгтон, „”Фармацевтична наука та практика" (Remington: The science and practice of th pharmacy, ed. by Gennaro et al., 19 Ed., Mack Publ. Co., 1995). Цей винахід пропонує також спосіб лікування медулобластоми, базально-клітинного раку, раку стравоходу, раку шлунка, раку підшлункової залози, раку жовчних шляхів, раку простати, раку молочної залози, дрібноклітинного раку легенів, недрібноклітинного раку легенів, Вклітинної лімфоми, множинної мієломи, раку яєчників, раку ободової та прямої кишки, раку печінки, раку нирки або меланоми у ссавця, який включає введення в організм ссавця, який потребує такого лікування, ефективної кількості сполуки Формули I або фармацевтично прийнятної солі такої сполуки. Мається на увазі, що реально застосовувана кількість сполуки визначається лікаремкуратором з урахуванням відповідних обставин, в тому числі патологічного стану, який підлягає лікуванню, обраного шляху введення, конкретної застосовуваної сполуки або сполук, віку, маси тіла та індивідуальної реакції пацієнта та тяжкості симптомів, які спостерігаються у пацієнта. Добові дози у звичайних випадках лежать у межах від приблизно 0,1 мг/кг до приблизно 10 мг/кг маси тіла. За деяких обставин повністю адекватними можуть бути дози, нижчі від нижчої межі вищезгаданого діапазону, тоді як в інших випадках можуть застосовуватися ще більші дози. Отже, вищезгаданий діапазон дозування не призначений для обмеження будь-яким чином обсягу винаходу. Цей винахід пропонує також сполуку Формули I або фармацевтично прийнятну сіль такої сполуки для застосування як лікарського засобу. Крім того, цей винахід пропонує застосування сполуки Формули I або фармацевтично прийнятної солі такої сполуки при виготовленні лікарського засобу для лікування раку. Зокрема, 2 UA 102250 C2 5 10 15 згадані різновиди раку вибрані з групи, яку складають медулобластома, базально-клітинний рак, рак стравоходу, рак шлунка, рак підшлункової залози, рак жовчних шляхів, рак простати, рак молочної залози, дрібноклітинний рак легенів, недрібноклітинний рак легенів, В-клітинна лімфома, множинна мієлома, рак яєчників, рак ободової та прямої кишки, рак печінки, рак нирки та меланома. Крім того, цей винахід пропонує також фармацевтичну композицію, яка включає як активний інгредієнт сполуку Формули I або її фармацевтично прийнятну сіль, для лікування медулобластоми, базально-клітинного раку, раку стравоходу, раку шлунка, раку підшлункової залози, раку жовчних шляхів, раку простати, раку молочної залози, дрібноклітинного раку легенів, недрібноклітинного раку легенів, В-клітинної лімфоми, множинної мієломи, раку яєчників, раку ободової та прямої кишки, раку печінки, раку нирки або меланоми. При вживанні в цьому описі нижчезазначені терміни мають вказані відповідні значення: „”Et2O" – діетиловий ефір; "DMF" – диметилформамід; "DMSO" – диметилсульфоксид; "TFA" – трифтороцтова кислота; "boc" або "t-boc" – трет-бутоксикарбоніл; "SCX" – сильний катіонообмінник; "PyBOP" – гексафторфосфат бензотриазол-1-ілокситрипіролідинфосфонію; "Prep" – підготовчий синтез; "Ex" – приклад; та "IC50" – концентрація засобу, яка спричиняє 50 % максимальної інгібувальної реакції, можливої для цього засобу. Схема 1 2 Стадія 1a 1 R Cl 1 R N HN N (4) BOC (2) BOC N N 2 R N N R N Стадія 2 (3a) Стадія 1b 2 HN R N 7 R 2 6 R R 1 R N O (3b) 3 R N H N N 5 R (5) 4 R Стадія 3 2 1 R N R N 7 R 6 R 5 N N 20 25 30 35 40 Формула I R O 3 R 4 R Сполуку Формули I можна одержати згідно з реакціями, показаними на Схемі 1. Проводять реакцію нуклеофільного ароматичного заміщення (SNAr) 1-хлорзаміщеного фталазину (2) з захищеним 4-аміно-boc-піперидином (3а) з одержанням заміщеного піперидином фталазину формули (4). Наприклад, можна проводити реакцію згаданого хлориду (2) з піперидином формули (3а) у диполярному апротонному розчиннику, наприклад, DMF або DMSO, у присутності органічної основи, наприклад, триетиламіну, або неорганічної основи, наприклад, карбонату калію, при нагріванні до 100-140С. Від функціональної аміногрупи, наприклад, присутньої у піперидинілфталазині формули (4), можна відщепити захисну групу, після чого проводити подальші реакції для одержання додаткових сполук за цим винаходом. Способи введення та відщеплення груп захисту азоту та кисню добре відомі в галузі. (Дивись, наприклад, Гріні та Вутс, „Групи захисту в органічному синтезі" (Greeny and Wuts, Protective groups in organic synthesis, 3rd Ed., John Wiley & Sons, New York, 1999). Наприклад, відщеплення групи boc від амінопіперидинілфталазину формули (4) можна здійснити у кислотних умовах, наприклад, у присутності хлороводню або трифтороцтової кислоти. В альтернативному варіанті НСl можна вивільнювати in situ шляхом додавання краплями ацетилхлориду до розчину спиртового розчинника, наприклад, метанолу в толуолі, при 0-20С, з подальшим доданням сполуки формули (4) та нагріванням одержаного розчину до 30-60С з одержанням сполуки формули (5). Для фахівця у цій галузі очевидно, що сполуку формули (5) можна виділити у вигляді солі, наприклад, хлористоводневої солі аміну, та 3 UA 102250 C2 5 10 15 20 застосувати на Стадії 3 або перетворити у вільний амін, застосовуючи неорганічну основу, наприклад, карбонат калію. 4-амінопіперидин можна ацилювати, застосовуючи заміщений бензоїлхлорид в інертному розчиннику, наприклад, дихлорметані або діоксані, та основу, наприклад, триетиламін або піридин, і одержати амід Формули I. В альтернативному варіанті сполуку формули (5) можна ацилювати, застосовуючи заміщену бензойну кислоту та відповідний реагент сполучення, наприклад, РуВОР, та відповідну основу, наприклад, триетиламін, в інертному розчиннику, наприклад, дихлорметані, або застосовуючи гідрохлорид 1-(3-диметиламінопропіл)-3-етилкарбодііміду та 1-гідроксибензотриазол у диполярному апротонному розчиннику, наприклад, у DMF. За альтернативним варіантом проводять реакцію нуклеофільного ароматичного заміщення (SNAr) 1-хлорзаміщеного фталазину (2) з N-піперидин-4-ілбензамідом формули (3b), як описано вище для Стадії 1, і безпосередньо одержують сполуку Формули I. Наприклад, можна проводити реакцію згаданого хлориду (2) у диполярному апротонному розчиннику, наприклад, DMF або DMSO, з N-піперидин-4-ілбензамідом формули (3b) у присутності основи, наприклад, триетиламіну, при нагріванні до 80-140С з одержанням аміду Формули I. Для фахівця у цій галузі зрозуміло, що сполуки формули (2) за Схемою 1 є наявними на ринку або можуть бути без утруднень одержані способами, аналогічними описаним у цьому документі або з застосуванням відомих у галузі методик. Наприклад, 2-фенілкарбонілбензойну кислоту, одержану шляхом реакції Гріньяра з фенілброміду та фталевого ангідриду, можна циклізувати гідразином, одержуючи 4-феніл-2Н-фталазин-1-он. Подальше оброблення оксихлоридом фосфору дає 1-хлор-4-фенілфталазин формули (2). За альтернативним варіантом, можна провести реакцію перехресного сполучення за Судзукі між 1,4дихлорфталазином та фенілборною кислотою та одержати відповідний 1-хлор-4фенілфталазин формули (2). 25 Схема 2 2 R Cl Cl + HN 2 Стадія 1 N Cl BOC N (7) HO (3a) BOC N N N N (6) R N 1 R B HO Стадія 5 Стадія 2 2 1 R N R N BOC N N Cl H N N (4) N N 2 R (8) 7 6 R Стадія 3 R O 5 R Стадія 6 z 3 4 R 1 R N H N N N 6 Cl R O N N 3 2 R R N N 5 R z 5 R 7 R Стадія 4 Z = Cl, OH 6 R O 2 R (5) R 7 R 3 R 4 R (9) 4 R Стадія 7 7 R HO 6 R O B 1 R HO 5 R 1 R N N N N 3 2 R R 4 R Формула I 30 Сполуку Формули I можна одержати згідно з реакціями, показаними на Схемі 2. На Стадії 1 можна провести реакцію 1,4-дихлорфталазину (6) з захищеним 4-аміно-boc-піперидином (3а) у диполярному апротонному розчиннику, наприклад, N-метилпіролідоні або DMSO, у присутності відповідної основи, наприклад, карбонату калію або триетиламіну. Суміш витримують при 70 4 UA 102250 C2 5 10 15 20 25 30 95С, і одержують сполуку формули (7). За одним зі способів, ілюстрованим Стадією 5, від сполуки формули (7) відщеплюють групу захисту, після чого ацилюють по аміногрупі на Стадії 6, застосовуючи заміщений бензоїлхлорид в інертному розчиннику, наприклад, у дихлорметані, у присутності основи, наприклад, триетиламіну, і одержують амід формули (9). За альтернативним способом сполуку формули (7) ацилюють, застосовуючи заміщену фенілкарбонову кислоту та відповідний реагент сполучення, наприклад, РуВОР, та відповідну основу, наприклад, триетиламін, в інертному розчиннику, наприклад, дихлорметані, при кімнатній температурі, або застосовуючи 1-гідроксибензотриазол та гідрохлорид 1-(3диметиламінопропіл)-3-етилкарбодііміду та диполярний апротонний розчинник, наприклад, DMF. На Стадії 7 проводять реакцію фталазинілхлориду формули (9) з фенілборною кислотою в умовах перехресного сполучення за Судзукі-Міяура. Для фахівця у цій галузі зрозуміло, що існують різноманітні умови, корисні для полегшення протікання таких реакцій перехресного сполучення. При проведенні реакції застосовують відповідний розчинник, наприклад, суміш діоксану з водою. Реакцію проводять у присутності основи, наприклад, моногідрату триосновного фосфату калію, карбонату натрію або карбонату цезію. Реакція відбувається у присутності паладієвого каталізатора, наприклад, трис(дибензиліденацетон)дипаладію(0) з трициклогексилфосфіном або (SP-4-1-)-біс[біс(1,1-диметилетил)(4-метоксифеніл)фосфінкP]дихлорпаладію (виготовленого за методикою синтезу каталізатора D, як описано в J.Org.Chem. 2007, 72, 5104-5112) в інертній атмосфері при температурі приблизно 80-160С, з одержанням сполуки Формули I. За альтернативним варіантом фталазинілхлорид формули (7) спочатку сполучають з фенілборною кислотою, як ілюстровано Стадією 2, в умовах реакції Судзукі-Міяура, як описано вище, та одержують фенілфталазин формули (4). На Стадії 3 від сполуки формули (4) відщеплюють групу захисту, після чого ацилюють по аміногрупі, як ілюстровано Стадією 4, застосовуючи заміщений бензоїлхлорид або фенілкарбонову кислоту, як описано вище, і одержують сполуку Формули I. Подані нижче описи підготовчих синтезів та Приклади запропоновані з метою додаткового більш детального ілюстрування винаходу, і вони представляють типові синтези сполук Формули I. Назви сполук за цим винаходом, як правило, утворено з використанням програми ChemDraw Ultra 10.0. Підготовчий синтез 1 Трет-бутиловий ефір {1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}метилкарбамінової кислоти N F N N N 35 40 45 50 O O Додають 1-хлор-4-(4-фторфеніл)фталазин (3,00 г, 11,6 ммоль) до розчину трет-бутилового ефіру метилпіперидин-4-ілкарбамінової кислоти (2,98 г, 13,9 ммоль) та триетиламіну (3,52 г, 34,8 ммоль) у DMF (30 мл). Витримують при 130С протягом 3 діб. Розчиняють реакційну суміш у дихлорметані, та промивають розсолом. Органічну фазу сушать сульфатом натрію, фільтрують та концентрують під зниженим тиском. Очищають одержаний залишок флешхроматографією (елюент суміш гексан: етилацетат: 2 М аміак у метанолі, 20:5:1), і одержують вказану в заголовку сполуку у вигляді твердої речовини (4,45 г, 88 %). Мас-спектроскопія з електророзпиленням (ES/MS) m/z 437,2 (M+1). Альтернативна методика Змішують трет-бутиловий ефір метилпіперидин-4-ілкарбамінової кислоти (75 г, 349 ммоль), 1-хлор-4-(4-фторфеніл)фталазин (75 г, 289 ммоль) та карбонат калію (80 г, 579 ммоль) у диметилсульфоксиді (500 мл), і витримують цю суміш при 110С протягом 3 год. Охолоджують реакційну суміш до температури навколишнього середовища, та виливають одержану суспензію у воду (1,0 л). Тверду речовину відділяють фільтруванням, та сушать у вакуумній шафі протягом 3 діб, одержуючи вказану в заголовку сполуку у вигляді білої твердої речовини (120 г, 95 %). ES/MS m/z 437,3 (M+1). Піперидинілфталазини, вказані у поданій нижче таблиці, одержують практично за методикою, описаною у Підготовчому синтезі 1, застосовуючи відповідні хлорфталазини та 5 UA 102250 C2 захищені t-boc амінопіперидини. Підг. синтез 2 3 4 5 Хімічна назва трет-бутиловий ефір метил-[1-(4-фенілфталазин-1-іл)піперидин-4419,2 (M+1) іл]карбамінової кислоти трет-бутил-1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-ілкарбамат 423,2 (M+1) трет-бутил-1-(4-фенілфталазин-1-іл)піперидин-4-ілкарбамат 405,2 (M+1) Підготовчий синтез 5 {1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}-метиламін N F N H N N 10 15 20 25 ES/MS m/z Додають трифтороцтову кислоту (100 мл) до розчину трет-бутилового ефіру {1-[4-(4фторфеніл)фталазин-1-іл]піперидин-4-іл}метилкарбамінової кислоти (11,2 г, 10,2 ммоль) у дихлорметані (100 мл). Перемішують реакційну суміш при кімнатній температурі протягом ночі, та концентрують під зниженим тиском. Одержаний залишок розчиняють у дихлорметані, і промивають 1н. NaOH та розсолом. Органічну фазу сушать сульфатом натрію, фільтрують та концентрують під зниженим тиском. Кристалізують вказану в заголовку сполуку з суміші гексану з дихлорметаном і одержують вказану в заголовку сполуку (8,46 г, 98 %). ES/MS m/z 337,2 (M+1). Альтернативна методика (виділення у формі гідрохлорида): Змішують толуол (500 мл) з метанолом (30мл) при 10С. Додають краплями ацетилхлорид (29 мл, 410 ммоль) протягом 20 хв. Під час додавання підтримують температуру нижче 15С. Додають трет-бутиловий ефір {1-[4-(4-фторфеніл)фталазин-1-іл]-піперидин-4-іл}-метилкарбамінової кислоти (71 г, 164 ммоль). Витримують суспензію при 35С протягом 2 год. Охолоджують реакційну суміш до температури навколишнього середовища, та відділяють тверду речовину фільтруванням. Сушать у вакуумній шафі при 40С протягом 12 год., і одержують вказану в заголовку сполуку у вигляді білої твердої речовини (58 г, 95 %). ES/MS m/z 337 (M+1). Незахищені амінопіперидинілфталазини, вказані у поданій нижче таблиці, одержують практично за методикою, описаною у Підготовчому синтезі 5, застосовуючи відповідні хлорфталазини та захищені t-boc амінопіперидини. Підг. синтез 6 7 8 Хімічна назва метил-[1-(4-фенілфталазин-1-іл)піперидин-4-іл]-амін 1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-амін 1-(4-фенілфталазин-1-іл)піперидин-4-амін ES/MS m/z 319,2 (M+1) 323,2 (M+1) 305,2 (M+1) 30 Підготовчий синтез 9 Трет-бутил-1-(4-хлорфталазин-1-іл)піперидин-4-іл(метил)карбамат N Cl N O N N O 35 Змішують 1,4-дихлорфталазин (5,00 г, 24,6 ммоль), трет-бутил-метил(піперидин-4іл)карбамат (5,54 г, 25,8 ммоль) та карбонат калію (4,08 г, 29,5 ммоль) в N-метилпіролідоні (50,0 мл). Витримують реакційну суміш при 80С протягом 3 діб, після чого виливають у льодяну 6 UA 102250 C2 5 10 15 воду. Фільтрують під вакуумом, і одержують жовтувату тверду речовину, яку сушать при кімнатній температурі у вакуумній шафі. Очищають хроматографією на силікагелі (елюент суміш гексану з етилацетатом, 1:1), і одержують бажаний продукт (5,79 г, 62 %). ES/MS m/z 377,2 (M+1). Альтернативна методика Змішують 1,4-дихлорфталазин (7,04 г, 35,4 ммоль), трет-бутил-метил(піперидин-4іл)карбамат (7,97 г, 37,2 ммоль), триетиламін (7,4 мл, 53,1 ммоль) та DMSO (85 мл). Витримують реакційну суміш при 85С протягом 3 діб або до повного вичерпання вихідного матеріалу. Після охолодження переносять реакційну суміш у ділильну лійку з діетиловим ефіром і промивають водою. Відділяють органічний шар, сушать MgSO 4, фільтрують і концентрують у вакуумі. Очищають одержаний залишок флеш-хроматографією (елюювання градієнтом 0-10 % метанолу в дихлорметані), і одержують вказану в заголовку сполуку (7,6 г, 57 %). ES/MS m/z 377,2 (M+1). Підготовчий синтез 10 Трет-бутил-метил-(1-(4-(4-трифторметил)феніл)фталазин-1-іл)піперидин-4-іл)карбамат F N F F N O N N O 20 25 30 35 40 45 50 У реактор для роботи в мікрохвильовій печі завантажують трет-бутил-1-(4-хлорфталазин-1іл)піперидин-4-іл(метил)карбамат (0,201 г, 0,534 ммоль), 4-(трифторметил)фенілборну кислоту (122 мг, 0,640 ммоль), моногідрат трьохосновного фосфату калію (209 мг, 0,907 ммоль), трициклогексилфосфін (19 мг, 0,064 ммоль), 1,4-діоксан (3,5 мл) та воду (1,5 мл). Продувають через реакційну суміш азот протягом 5 хв. Додають трис(дибензиліденацетон)дипаладій(0) (25 мг, 0,027 ммоль). Знову продувають через реакційну суміш азот протягом 5 хв. Витримують герметизований реактор у мікрохвильовій печі при 150С протягом 1 год. Пропускають реакційну суміш через шар силікагелю з елююванням етилацетатом. Концентрують у вакуумі, та очищають одержаний залишок хроматографією на силікагелі (елюент суміш етилацетату з гексаном, 30:70), одержуючи вказану в заголовку сполуку (0,170 г, 65 %). ES/MS m/z 486,8 (M+1). Підготовчий синтез 11 N-метил-(1-(4-(4-трифторметил)феніл)фталазин-1-іл)піперидин-4-іл)-4-амін, дигідрохлорид Додають хлороводень (4,0н. у діоксані, 20 мл, 80,0 ммоль) до трет-бутил-метил-(1-(4-(4трифторметил)феніл)фталазин-1-іл)піперидин-4-іл)карбамату (0,158 г, 0,325 ммоль). Перемішують при кімнатній температурі протягом ночі. Видаляють розчинники під зниженим тиском. Використовують неочищений матеріал (0,169 г, >100 %). ES/MS m/z 387,0 (M+1). Підготовчий синтез 12 Трет-бутил-1-(4-(4-ціанофеніл)фталазин-1-іл)-піперидин-4-іл)-(метил)карбамат У трубку для роботи під тиском завантажують трет-бутил-1-(4-хлорфталазин-1-іл)піперидин-4-іл(метил)карбамат (400 мг, 1,06 ммоль), 1,4-діоксан (12 мл), воду (4 мл), 4-ціанофенілборну кислоту (467 мг, 3,18 ммоль) та карбонат цезію (1,40 г, 4,24 ммоль). Продувають через реакційну суміш азот протягом 5 хв. Додають (SP-4-1)-біс[біс(1,1-диметилетил)(4метоксифеніл)фосфін-κP]дихлорпаладій (J.Org.Chem. 2007, 72, 5104-5112) (36,0 мг, 0,053 ммоль). Продувають через реакційну суміш азот протягом кількох хвилин, та герметично закривають реакційну посудину. Витримують реакційну суміш при 90С протягом ночі. Пропускають реакційну суміш через шар силікагелю з елююванням етилацетатом. Видаляють розчинники під зниженим тиском, та очищають одержаний залишок хроматографією на силікагелі (елюент суміш етилацетату з гексаном, 30:70), одержуючи вказану в заголовку сполуку (0,392 г, 83 %). ES/MS m/z 444,2 (M+1). Підготовчий синтез 13 4-(4-(4-метиламіно)піперидин-1іл)фталазин-1-іл)бензонітрила, дигідрохлорид Вказану в заголовку сполуку одержують практично за методикою, описаною у Підготовчому синтезі 11, застосовуючи трет-бутил-1-(4-(4-ціанофеніл)фталазин-1-іл)-піперидин-4-іл)(метил)карбамат (0,385 г, 0,868 ммоль). У подальших реакціях застосовують неочищений матеріал (0,378 г, >100 %). ES/MS m/z 344,2 (M+1). 7 UA 102250 C2 5 Підготовчий синтез 14 1-(4-хлорфталазин-1-іл)-N-метилпіперидин-4-аміна, дигідрохлорид Додають хлороводень (4,0н. у діоксані, 100 мл, 400,0 ммоль) до розчину трет-бутил-1-(4хлорфталазин-1-іл)піперидин-4-іл(метил)карбамату (7,60 г, 1,00 екв, 20,2 ммоль) у метанолі (100 мл). Перемішують при кімнатній температурі протягом однієї години. Видаляють розчинники під зниженим тиском, і одержують вказану в заголовку сполуку (7,05 г, 100 %). ES/MS m/z 277,2 (M+1). Підготовчий синтез 15 N-(1-(4-хлорфталазин-1-іл)піперидин-4-іл)-N-метил-2-(трифторметил)бензамід 10 N Cl N N N O F F F 15 20 Змішують дигідрохлорид 1-(4-хлорфталазин-1-іл)-N-метилпіперидин-4-аміну (1,01 г, 2,89 ммоль) та триетиламін (1,2 мл, 8,61 ммоль) у дихлорметані (30 мл). Продувають реакційну посудину азотом, і додають 3-трифторметилбензоїлхлорид (0,46 мл, 3,12 ммоль). Створюють над реакційною сумішшю атмосферу азоту, та перемішують при кімнатній температурі протягом ночі. Концентрують, одержуючи залишок, та очищають його хроматографією на силікагелі (елюювання градієнтом 0-10 % метанолу в дихлорметані), одержуючи вказану в заголовку сполуку (1,11 г, 86 %). ES/MS m/z 449,2 (M+1). Аміди, вказані у поданій нижче таблиці, одержують практично за методикою, описаною у Підготовчому синтезі 15, з застосуванням відповідних хлорангідридів кислот. Підг. синтез 16 17 18 25 Хімічна назва ES/MS m/z N-(1-(4-хлорфталазин-1-іл)піперидин-4-іл)-3-ціано-N-метилбензамід N-(1-(4-хлорфталазин-1-іл)піперидин-4-іл)-5-фтор-N-метил-2(трифторметил)бензамід N-(1-(4-хлорфталазин-1-іл)піперидин-4-іл)-N-метил-4(трифторметокси)бензамід 406,2 (M+1) 467,2 (M+1) 465,2 (M+1) Приклад 1 N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N-метил-4-(трифторметокси)бензаміду, гідрохлорид HCl F F N F F N N N O O 30 35 40 Змішують метил-{1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}-метиламін (100 мг, 0,300 ммоль), триетиламін (0,12 мл, 0,89 ммоль) та дихлорметан (2 мл) при кімнатній температурі. Додають до цієї суміші 4-(трифторметокси)-бензоїлхлорид (100 мг, 0,45 ммоль), та перемішують при кімнатній температурі протягом ночі. Концентрують реакційну суміш, та очищають залишок флеш-хроматографією (елюент – суміш гексан: етилацетат: 2 М аміак у метанолі, 20:5:1). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі, та видаляють розчинники у струмені азоту, одержуючи вказану в заголовку сполуку у вигляді твердої речовини (98 мг, 58 %). ES/MS m/z 525,0 (M+1). Альтернативна методика Додають гідрохлорид {1-[4-(4-фторфеніл)фталазин-1-іл]-піперидин-4-іл}-метиламіну (58 г, 155 ммоль) до 1,4-діоксану (580 мл). Додають триетиламін (86 мл, 622 ммоль), та перемішують 8 UA 102250 C2 5 10 15 протягом 20 хв. Додають краплями 4-(трифторметокси) бензоїлхлорид (24 мл, 155 ммоль) протягом 20 хв. Перемішують протягом 1 год. при кімнатній температурі. Додають воду (100 мл), екстрагують етилацетатом (200 мл), та концентрують органічну фазу під зниженим тиском. Очищають одержаний залишок флеш-хроматографією з елююванням етилацетатом, застосовуючи шар силікагелю масою 1 кг, і одержують продукт у вигляді безбарвного масла (58 г, 71 %). Змішують толуол (586 мл) з етанолом (117 мл), та охолоджують суміш до 3С. Додають ацетилхлорид (8 мл, 111 ммоль) протягом 20 хв. перемішують протягом 20 хв, після чого додають N-{1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}-N-метил-4трифторметоксибензамід (58 г, 111 ммоль) у толуолі (40 мл) однією порцією. перемішують протягом 12 год., концентрують суміш до 1/3 об'єму. Відділяють тверду речовину фільтруванням. Сушать твердий продукт у вакуумній шафі при 40С протягом ночі, і одержують вказану в заголовку сполуку у вигляді білої твердої речовини (42 г, 67 %). ES/MS m/z 525,0 (M+1). Аміди, вказані у поданій нижче таблиці 1, одержують по суті за методикою, описаною у Прикладі 1, застосовуючи відповідні хлорангідриди кислот. Таблиця 1 N F N 2 3 N N 4 O Приклад 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 6 5 Замісники бензаміду Хімічна назва N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N4-CF3 метил-4-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N3-CF3 метил-3-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N2-CF3 метил-2-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N2-OCF3 метил-2-(трифторметокси)бензаміду гідрохлорид 2,6-дифтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин2-F, 6-F 4-іл)-N-метилбензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 6-CF3 N-метил-6-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N2-CF3, 5-CF3 метил-2,5-біс(трифторметил)бензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 4-CF3 N-метил-4-(трифторметил)бензаміду гідрохлорид 2,4-дихлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин2-Cl, 4-Cl 4-іл)-N-метилбензаміду гідрохлорид 2,4,6-трифтор-N-(1-(4-(4-фторфеніл)фталазин-12-F, 4-F, 6-F іл)піперидин-4-іл)-N-метилбензаміду гідрохлорид 5-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 5-F N-метил-2-(трифторметил)бензаміду гідрохлорид 3-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)3-F, 4-CF3 N-метил-4-(трифторметил)бензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 3-CF3, N-метил-3-(трифторметил)бензаміду гідрохлорид 2-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-Cl, 5-CF3 N-метил-5-(трифторметил)бензаміду гідрохлорид 3-хлор-2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1іл)піперидин-4-іл)-N-метил-6-(трифторметил)бензаміду 2-F, 3-Cl, 6-CF3 гідрохлорид 9 ES/MS m/z 509,2 (M+1) 509,2 (M+1) 509,2 (M+1) 525,2 (M+1) 477,2 (M+1) 527,2 (M+1) 576,8 (M+1) 527,0 (M+1) 509,0 (M+1) 495,0 (M+1) 527,0 (M+1) 527,0 (M+1) 527,0 (M+1) 543,0 (M+1) 561,0 (M+1) UA 102250 C2 Продовження таблиці 1 N F N 2 3 N N 4 O Приклад 17 18 19 20 21 22 23 24 25 26 27 28 29 30 5 10 15 20 6 5 Замісники бензаміду Хімічна назва N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N3-OCF3 метил-3-(трифторметокси)бензаміду гідрохлорид 2-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-Cl N-метилбензаміду гідрохлорид 3-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)3-Cl N-метилбензаміду гідрохлорид 4-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)4-Cl N-метилбензаміду гідрохлорид 2,4-дифтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин2-F, 4-F 4-іл)-N-метилбензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 5-CF3 N-метил-5-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-N2-CF3, 4-CF3 метил-2,4-біс(трифторметил)бензаміду гідрохлорид 4-ціано-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)4-CN N-метилбензаміду гідрохлорид 2,6-дихлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин2-Cl, 6-Cl 4-іл)-N-метилбензаміду гідрохлорид 3-ціано-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)3-CN N-метилбензаміду гідрохлорид 4-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 4-Cl N-метил-2-(трифторметил)бензаміду гідрохлорид 2,4-дихлор-5-фтор-N-(1-(4-(4-фторфеніл)фталазин-12-Cl, 4-Cl, 5-F іл)піперидин-4-іл)-N-метилбензаміду гідрохлорид 5-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 5-Cl N-метил-2-(трифторметил)бензаміду гідрохлорид 4-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 4-F N-метил-2-(трифторметил)бензаміду гідрохлорид ES/MS m/z 525,2 (M+1) 475,0 (M+1) 475,0 (M+1) 475,0 (M+1) 477,0 (M+1) 527,0 (M+1) 577,0 (M+1) 466,2 (M+1) 509,0 (M+1) 466,2 (M+1) 542,6 (M+1) 526,6 (M+1) 543,2 (M+1) 527,0 (M+1) Альтернативна методика до Прикладу 30 Додають гідрохлорид {1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}метиламіну (80 г, 240 ммоль) до води (500 мл) для утворення суспензії. Додають карбонат калію до досягнення рН 10. Додають дихлорметан (400 мл). Інтенсивно перемішують до повного розчинення твердої фази. Відділяють органічний шар, і концентрують до одержання прозорого масла (74 г, 220 ммоль), одержуючи {1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}метиламін. Змішують {1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}метиламін (12 г, 35 ммоль), піридин (20 мл, 247 ммоль) та 1,4-діоксан (120 мл). Перемішують реакційну суміш протягом 20 хв. Додають дихлорметан (25 мл). Перемішують суспензію протягом 20 хв. Додають краплями 4-фтор-2-(трифторметил)бензоїлхлорид (6,5 мл, 43 ммоль) протягом 20 хв. Перемішують протягом 2 год. Виливають суміш у воду (100 мл), екстрагують дихлорметаном (200 мл), і концентрують суміш під зниженим тиском. Очищають залишок флеш-хроматографією (елюювання сумішшю етилацетату з гексаном, 1:1), і одержують продукт у вигляді білої твердої речовини (10,3 г, 55 %). Додають 4-фтор-N-{1-[4-(4-фторфеніл)фталазин-1-іл]піперидин-4-іл}-Nметил-2-трифторметилбензамід (10 г, 19,85 ммоль) до толуолу (125 мл) для утворення суспензії. Додають метанол (30 мл), і одержують однорідний розчин. Додають однією порцією хлороводень (5,21 мл, 4,0н. у діоксані, 20 ммоль). Залишають при перемішуванні на 1 год., і концентрують до 1/3 об'єму. Відділяють тверду речовину, і сушать її у вакуумній шафі протягом 10 UA 102250 C2 5 12 год. при 35С, одержуючи вказану в заголовку сполуку у вигляді білої твердої речовини (9,5 г, 85 %). ES/MS m/z 527,0 (M+1). Приклад 31 4-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2-(трифторметил)бензаміду гідрохлорид HCl N N N N F O F F F 10 15 Змішують метил-[1-(4-фенілфталазин-1-іл)піперидин-4-іл]амін (800 мг 2,51 ммоль), триетиламін (1,05 мл, 7,54 ммоль) та дихлорметан (20 мл) при кімнатній температурі. Додають до цієї суміші 4-фтор-2-(трифторметил)бензоїлхлорид (683 мг, 3,01 ммоль) і перемішують при кімнатній температурі протягом ночі. Концентрують реакційну суміш і очищають одержаний залишок флеш-хроматографією (елюент суміш гексан: етилацетат: 2 М аміак у метанолі, 20:5:1). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі. Відділяють одержану тверду речовину фільтруванням, і одержують вказану в заголовку сполуку (1,13 г, 88 %). ES/MS m/z 509,2 (M+1). Аміди, вказані у поданій нижче таблиці 2, одержують по суті за методикою, описаною у Прикладі 31, застосовуючи відповідні хлорангідриди кислот. Хлористоводневі солі виділяють шляхом фільтрування або випарювання розчинника. 20 Таблиця 2 N N 2 3 N N 4 O Приклад 32 33 34 35 36 37 38 39 40 41 6 5 Хімічна назва N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2(трифторметокси)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-4(трифторметокси)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2(трифторметил)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-3(трифторметил)бензаміду гідрохлорид 2-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)6-(трифторметил)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-4(трифторметил)бензаміду гідрохлорид 2-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)4-(трифторметил)бензаміду гідрохлорид 2,4-дихлор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2,4біс(трифторметил)бензаміду гідрохлорид 4-ціано-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид 11 Замісники бензаміду 2-OCF3 4-OCF3 2-CF3 3-CF3 2-F, 6-CF3 4-CF3 2-F, 4-CF3 2-Cl, 4-Cl 2-CF3, 4-CF3 4-CN ES/MS m/z 507,2 (M+1) 507,2 (M+1) 491,2 (M+1) 491,2 (M+1) 509,2 (M+1) 491,2 (M+1) 509,2 (M+1) 491,0 (M+1) 559,2 (M+1) 448,2 (M+1) UA 102250 C2 Продовження таблиці 2 N N 2 3 N N 4 O Приклад 42 43 44 45 46 47 48 6 5 Замісники бензаміду Хімічна назва 3-хлор-2-фтор-N-метил-N-(1-(4-фенілфталазин-12-F, 3-Cl, 6-CF3 іл)піперидин-4-іл)-6-(трифторметил)бензаміду гідрохлорид 5-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)2-CF3, 5-F 2-(трифторметил)бензаміду гідрохлорид N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2,52-CF3, 5-CF3 біс(трифторметил)бензаміду гідрохлорид 2-хлор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-52-Cl, 5-CF3 (трифторметил)бензаміду гідрохлорид 2-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)2-F, 5-CF3 5-(трифторметил)бензаміду гідрохлорид 3-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)3-F, 4-CF3 4-(трифторметил)бензаміду гідрохлорид 2-фтор-N-метил-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)2-F, 3-CF3 3-(трифторметил)бензаміду гідрохлорид ES/MS m/z 543,0 (M+1) 509,2 (M+1) 559,2 (M+1) 525,2 (M+1) 509,2 (M+1) 509,2 (M+1) 509,2 (M+1) Приклад 49 N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-4-(трифторметил)бензаміду, гідрохлорид 5 HCl O F N H F F F N N N 10 15 Змішують 1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-амін (110 мг, 0,34 ммоль), триетиламін (0,14 мл, 1,02 ммоль) та дихлорметан (2 мл) при кімнатній температурі. Додають до цієї суміші 4-(трифторметокси)бензоїлхлорид (85 мг, 0,41 ммоль), та перемішують при кімнатній температурі протягом ночі. Концентрують реакційну суміш, та очищають залишок флеш-хроматографією (елюент суміш гексан: етилацетат: 2 М аміак у метанолі, 20:5:1). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі, та видаляють розчинники у струмені азоту, одержуючи вказану в заголовку сполуку у вигляді твердої речовини (57 мг, 32 %). ES/MS m/z 495,2 (M+1). Аміди, вказані у поданій нижче таблиці 3, одержують по суті за методикою, описаною у Прикладі 49, застосовуючи відповідні хлорангідриди кислот. 12 UA 102250 C2 Таблиця 3 N F H N 2 3 N N 4 O Приклад 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 6 5 Замісники бензаміду Хімічна назва 4-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-44-F іл)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-33-CF3 (трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-44-OCF3 (трифторметокси)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-22-CF3 (трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-22-OCF3 (трифторметокси)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-33-OCF3 (трифторметокси)бензаміду гідрохлорид 4-ціано-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-44-CN іл)бензаміду гідрохлорид 2,6-дифтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин2-F, 6-F 4-іл)бензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 6-CF3 6-(трифторметил)бензаміду гідрохлорид 2,4,6-трифтор-N-(1-(4-(4-фторфеніл)фталазин-12-F, 4-F, 6-F іл)піперидин-4-іл)бензаміду гідрохлорид 2-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-F, 5-CF3 5-(трифторметил)бензаміду гідрохлорид 4-фтор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 4-F 2-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)-2,42-CF3, 4-CF3 біс(трифторметил)бензаміду гідрохлорид 2-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-42-Cl іл)бензаміду гідрохлорид 3-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-43-Cl іл)бензаміду гідрохлорид 4-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-44-Cl іл)бензаміду гідрохлорид 2,4-дихлор-5-фтор-N-(1-(4-(4-фторфеніл)фталазин-12-Cl, 4-Cl, 5-F іл)піперидин-4-іл)бензаміду гідрохлорид 4-хлор-N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)2-CF3, 4-Cl 2-(трифторметил)бензаміду гідрохлорид ES/MS m/z 445,2 (M+1) 495,2 (M+1) 511,2 (M+1) 495,2 (M+1) 511,0 (M+1) 511,0 (M+1) 452,0 (M+1) 463,0 (M+1) 513,0 (M+1) 481,0 (M+1) 513,0 (M+1) 513,0 (M+1) 563,0 (M+1) 461,0 (M+1) 461,0 (M+1) 461,0 (M+1) 512,6 (M1) 528,6 (M+1) Приклад 68 N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-4-(трифторметил)бензаміду, гідрохлорид 5 HCl O F N H F F N N N 13 UA 102250 C2 5 10 Змішують 1-(4-фенілфталазин-1-іл)-піперидин-4-амін (110 мг, 0,34 ммоль), триетиламін (0,14 мл, 1,02 ммоль) та дихлорметан (2 мл) при кімнатній температурі. Додають до цієї суміші 4-(трифторметокси)-бензоїлхлорид (85 мг, 0,41 ммоль), та перемішують при кімнатній температурі протягом ночі. Концентрують реакційну суміш, та очищають залишок флешхроматографією (елюент суміш гексан: етилацетат: 2 М аміак у метанолі, 20:5:1). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі, та видаляють розчинники у струмені азоту, одержуючи вказану в заголовку сполуку у вигляді твердої речовини (116 мг, 67 %). ES/MS m/z 477,2 (M+1). Аміди, вказані у поданій нижче таблиці 4, одержують по суті за методикою, описаною у Прикладі 68, застосовуючи відповідні хлорангідриди кислот. Таблиця 4 N H N 2 3 N N 4 O Приклад 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 6 5 Хімічна назва 4-фтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-3(трифторметил)бензаміду гідрохлорид N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2(трифторметил)бензаміду гідрохлорид N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2(трифторметокси)бензаміду гідрохлорид N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-3(трифторметокси)бензаміду гідрохлорид 4-ціано-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид 3-ціано-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид 2,4-дифтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид 2,6-дифтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид 2-фтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-6(трифторметил)бензаміду гідрохлорид 2,4,6-трифтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид 2-фтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-5(трифторметил)бензаміду гідрохлорид 4-фтор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2(трифторметил)бензаміду гідрохлорид N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)-2,4біс(трифторметил)бензаміду гідрохлорид 2-хлор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид 3-хлор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид 4-хлор-N-(1-(4-фенілфталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид Замісники бензаміду 4-F 3-CF3 2-CF3 2-OCF3 3-OCF3 4-CN 3-CN 2-F, 4-F 2-F, 6-F 2-F, 6-CF3 2-F, 4-F, 6-F 2-F, 5-CF3 2-CF3, 4-F 2-CF3, 4-CF3 2-Cl 3-Cl 4-Cl Приклад 86 N-(1-(4-(4-фторфеніл)фталазин-1-іл)піперидин-4-іл)бензаміду, гідрохлорид 14 ES/MS m/z 427,2 (M+1) 477,2 (M+1) 477,2 (M+1) 493,0 (M+1) 493,0 (M+1) 434,0 (M+1) 434,0 (M+1) 445,0 (M+1) 445,0 (M+1) 495,0 (M+1) 463,0 (M+1) 495,0 (M+1) 495,0 (M+1) 545,0 (M+1) 443,0 (M+1) 443,0 (M+1) 443,0 (M+1) UA 102250 C2 HCl F N N H N N 5 10 15 O Змішують 1-хлор-4-(4-фторфеніл)фталазин (150 мг, 0,58 ммоль), N-(піперидин-4-іл)бензамід (178 мг, 0,87 ммоль), триетиламін (0,404 мл, 2,9 ммоль) та диметилформамід (1 мл) при кімнатній температурі. Нагрівають до 100С та перемішують протягом ночі. Виливають реакційну суміш у колонку об'ємом 60 мл з 10 г сильної катіонообмінної смоли Fenomenex Strata SCX (55 мкм, 70 Å) (смола містить функціональні групи бензолсульфонової кислоти). Елююють цільовий продукт 2н. метанольним розчином аміаку (40 мл), і концентрують. Очищають одержаний залишок флеш-хроматографією (градієнт від 20 % до 30 % суміші [10 % 2н. метанольного розчину аміаку в етилацетаті] з гексаном). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі, та видаляють розчинники у струмені азоту, одержуючи вказану в заголовку сполуку у вигляді твердої речовини (137 мг, 51 %). ES/MS m/z 427,2 (M+1). Приклад 87 N-(1-(4-(4-ціанофеніл)фталазин-1іл)піперидин-4-іл)-4-фтор-N-метил-2(трифторметил)бензаміду, гідрохлорид N N N F N N O HCl F F F 20 25 30 Завантажують у реакційну посудину місткістю 4 мл дигідрохлорид 4-(4-(4(метиламіно)піперидин-1-іл)фталазин-1-іл)бензонітрилу (44,7 мг, 0,107 ммоль), дихлорметан (1 мл) та триетиламін (0,0598 мл, 0,429 ммоль). Продувають посудину азотом, та додають 4фтор-2-(трифторметил)бензоїлхлорид (0,033 г, 0,14 ммоль). Закривають посудину, і залишають реакційну суміш для перемішування протягом ночі при кімнатній температурі. Випарюють до одержання залишку, та очищають хроматографією на силікагелі (елюювання сумішшю етилацетату з гексаном 40:60, потім етилацетатом). До розчину виділеного продукту в суміші дихлорметану з метанолом додають 1н. розчин HCl у діетиловому ефірі, та видаляють розчинники у струмені азоту. Сушать залишок у вакуумній шафі при 50С і одержують вказану в заголовку сполуку (36,0 мг, 59 %). ES/MS m/z 533,8 (M+1). Аміди, вказані у поданій нижче таблиці 5, одержують по суті за методикою, описаною у Прикладі 87, застосовуючи відповідні вихідні матеріали, одержані за Підготовчим синтезом 11 або Підготовчим синтезом 13, та 2- (трифторметокси)бензоїлхлорид. 15 UA 102250 C2 Таблиця 5 1 N R N OCF3 N N O Приклад 88 89 5 Хімічна назва N-(1-(4-(4-ціанофеніл)фталазин-1-іл)піперидин-4-іл)-Nметил-4-(трифторметокси)бензаміду гідрохлорид N-метил-4-(трифторметокси)-N-(1-(4-(4(трифторметил)феніл) фталазин-1-іл)піперидин-4іл)бензаміду гідрохлорид Структура 1 R =CN 1 R =CF3 ES/MS m/z 532,2 (M+1) 575,2 (M+1) Приклад 90 N-метил-2-(трифторметил)-N-(1-(4-(4-(трифторметил)феніл)фталазин-1-іл)піперидин-4-іл)бензаміду, гідрохлорид HCl F N F F N N N O F F F 10 15 20 У реактор для роботи в мікрохвильовій печі завантажують N-(1-(4-хлорфталазин-1-іл)піперидин-4-іл)-N-метил-2-(трифторметил)бензамід (0,101 г, 0,23 ммоль), 4(трифторметил)фенілборну кислоту (0,171 г, 0,9 ммоль), карбонат цезію (0,295 г, 0,91 ммоль), 1,4-діоксан (3 мл) та воду (1 мл). Двічі продувають реактор азотом. Додають (SP-4-1)-біс[біс(1,1диметилетил)(4-метоксифеніл)фосфін-κP]дихлорпаладій (J.Org.Chem. 2007, 72, 5104-5112) (0,002 г, 0,003 ммоль), та витримують реакційну суміш при 90С протягом 16 год. Після охолодження розділяють два шари та видаляють воду. Випарюють органічний розчинник у струмені азоту. Залишок органічного шару очищають хроматографією на силікагелі (елюювання градієнтом 0-10 % метанолу в дихлорметані). До розчину виділеного продукту в метанолі додають 4н. розчин HCl у діоксані, та видаляють розчинники у вакуумі, одержуючи вказану в заголовку сполуку (0,100 г, 75 %). ES/MS m/z 559,2 (M+1). Сполуки, вказані у поданій нижче таблиці 6, одержують по суті за методикою, описаною у Прикладі 90, застосовуючи відповідні вихідні матеріали, одержані за Підготовчими синтезами 15-18, та відповідні борні кислоти. 16 UA 102250 C2 Таблиця 6 7 R 1 N R 6 R N 5 R N N O 3 R Приклад 91 92 93 94 95 96 97 98 99 100 101 102 5 10 15 4 R R групи x (R =H якщо не зазначено інше) Хімічна назва 3-ціано-N-метил-N-(1-(4-(41 4 (трифторметил)феніл)фталазин-1-іл)піперидин-4R =CF3; R =CN іл)бензаміду гідрохлорид N-(1-(4-(4-ціанофеніл)фталазин-1-іл)піперидин-4-іл)-N1 3 R =CN; R =CF3 метил-2-(трифторметил)бензаміду гідрохлорид N-(1-(4-(4-ціанофеніл)фталазин-1-іл)піперидин-4-іл)-51 3 6 фтор-N-метил-2-(трифторметил)бензаміду R =CN; R =CF3; R =F гідрохлорид 3-ціано-N-(1-(4-(4-ціанофеніл)фталазин-11 4 R =CN; R =CN іл)піперидин-4-іл)-N-метилбензаміду гідрохлорид 5-фтор-N-метил-N-(1-(4-(41 3 R =OCF3; R =CF3; (трифторметокси)феніл)фталазин-1-іл)піперидин-46 R =F іл)-2-(трифторметил)бензаміду гідрохлорид N-метил-N-(1-(4-(4-(трифторметокси)феніл)фталазин1 3 1-іл)піперидин-4-іл)-2-(трифторметил)бензаміду R =OCF3; R =CF3 гідрохлорид 3-ціано-N-(1-(4-(4-метоксифеніл)фталазин-11 4 R =OCH3; R =CN іл)піперидин-4-іл)-N-метилбензаміду гідрохлорид 3-ціано-N-метил-N-(1-(4-(41 4 (трифторметокси)феніл)фталазин-1-іл)піперидин-4R =OCF3; R =CN іл)бензаміду гідрохлорид N-метил-4-(трифторметокси)-N1 5 (1-(4-(4-(трифторметокси)феніл) R =OCF3; R =OCF3 фталазин-1-іл)піперидин-4-іл)бензаміду гідрохлорид N-(1-(4-(4-метоксифеніл)фталазин-1-іл)піперидин-41 5 R =OCH3; R =OCF3 іл)-N-метил-4-(трифторметокси)бензаміду гідрохлорид 5-фтор-N-(1-(4-(4-метоксифеніл)фталазин-11 3 R =OCH3; R =CF3; іл)піперидин-4-іл)-N-метил-2-(трифторметил)бензамід 6 R =F гідрохлорид N-(1-(4-(4-метоксифеніл)фталазин-1-іл)піперидин-41 3 R =OCH3; R =CF3 іл)-N-метил-2-(трифторметил)бензамід гідрохлорид ES/MS m/z 516,2 (M+1) 516,2 (M+1) 534,2 (M+1) 473,2 (M+1) 593,2 (M+1) 575,2 (M+1) 478,2 (M+1) 532,2 (M+1) 591,2 (M+1) 537,2 (M+1) 539,2 (M+1) 521,2 (M+1) Провідний шлях Sonic Hedgehog (Shh) відіграє вирішальну роль на під час ембріогенезу, але пригнічується у більшості тканин після раннього постнатального розвитку.Навпаки, у понад 30 % людських медулобластом виявлено високі рівні експресії Gli1 (пов'язаного з гліомою онкогенного гомолога 1); цей факт свідчить про те, що аномальне активування провідного шляху Shh відіграє важливу роль у підгрупі дитячих мозкових пухлин. Shh, секретований мозковими клітинами Пуркіньє, сприяє проліферації клітин-попередників гранул; це свідчить, що нерегульована активація провідного шляху hedgehog (Hh) може сприяти розвитку медулобластоми. Це припущення підтверджується розвитком медулобластом у мишей з геном -/+ плямистості (Patched gene, Ptch ). Лікування цих мишей антагоністом hedgehog пригнічувало ріст пухлини. Крім того, є документальні відомості, що лікування антагоністом hedgehog забезпечувало інгібування експресії Gli1 у цих мозкових пухлинах. Нерегульована активація провідного шляху hedgehog зазначена також при численних інших видах раку. Наприклад, виявлено, що hedgehog діє як фактор виживання при таких видах раку: 17 UA 102250 C2 5 10 15 20 25 30 35 40 45 50 55 60 базально-клітинний рак; ракові захворювання верхньої частини шлунково-кишкового тракту (стравоходу, шлунка, підшлункової залози та жовчних шляхів); рак простати; рак молочної залози; дрібноклітинний рак легенів; недрібноклітинний рак легенів; В-клітинна лімфома; множинна мієлома; рак шлунка; рак яєчника; рак ободової та прямої кишки; рак печінки; меланома; рак нирки; та медулобластома. З'ясовано, що елементи провідного шляху hedgehog є потенціальними мішенями для фармацевтичних засобів лікування раку. Знайдена у медулобластомних пухлинах (АТСС, НТВ186) клітинна лінія Daoy реагує на ліганди Hh. При обробленні цих клітин введеними екзогенно Shh агентами активується провідний шлях Hh, що призводить до посиленої експресії Gli1. Циклопамін – алкалоїд, виділений з чемериці каліфорнійської Veratrum californicum, є слабким антагоністом hedgehog, і було показано, що він пригнічує експресію Gli1 як реакцію на стимуляцію Shh. Дослідження останнього часу свідчать, що циклопамін інгібує ріст культивованих клітин та ксенотрансплантатів медулобластоми. Застосовуючи цю модель клітин Shh, можна ідентифікувати ефективні інгібітори провідних шляхів hedgehog. Оскільки сполуки за цим винаходом є антагоністами hedgehog, вони є придатними для лікування вищезазначених типів пухлин. Визначення ІС50 біологічної активності Описані нижче методика випробувань та їх результати, які демонструють корисність те ефективність сполук та способів їх використання за цим винаходом, подані з ілюстративною метою та не призначені для будь-якого обмеження обсягу винаходу. Функціональні проби підтверджують, що сполуки за цим винаходом виявляють здатність до інгібування провідного шляху Shh. Усі ліганди, розчинники та реагенти, використовувані в описаних нижче випробуваннях, є наявними на ринку або можуть бути без утруднень виготовлені фахівцем у цій галузі. Біологічну активність визначали, застосовуючи функціональну пробу з використанням нейронних ракових клітин Daoy та вимірювання рівнів рибонуклеїнової кислоти Gli1 з використанням системи випробування на bDNA (розгалужену деоксирибонуклеїнову кислоту) (Panomics, Inc., Fremont, CA). Gli1 був раніше відкритий у лінії клітин гліобластоми, і він кодує протеїн zinc finger, який активується провідним шляхом Shh. Максимальний відгук одержують шляхом індукування транскрипції Gli1 у клітинах Daoy за допомогою кондиціонованого середовища (клітин НЕК-293, які стабільно експресують рекомбінантний Shh) протягом 24 год. з подальшим визначенням кількості стимульованого транскрипту Gli1. Мінімальним відгуком є кількість транскрипту Gli1, інгібованого досліджуваною сполукою у клітинах Daoy, які були стимульовані кондиціонованим середовищем (клітинами нирки людського ембріона (НЕК-293), які стабільно експресують рекомбінантний Shh) протягом 24 год. Функціональна проба для визначення інгібування Gli1 у клітинах Daoy У системі випробування на bDNA застосовується технологія з використанням розгалуженої ДНК для забезпечення ампліфікації рибонуклеїнової кислоти-мішені (транскрипту). У цій технології використовуються три типи синтетичних гібридних Gli1-специфічних зондів сDNA, які визначають специфічність транскрипту-мішені (розширювачів захоплення (СЕs), розширювачів мічення (LЕs) та блокаторів (BLs), які у формі комплекса гібридизуються з транскриптамимішенями з метою підсилення сигналу гібридизації. Додання хемілюміногенного субстрату на стадії ампліфікації дозволяє для детектування використовувати люмінесценцію. Фізіологічно релевантна пухлинна лінія клітин Daoy, одержана з Американської колекції типових культур (АТСС), являє собою лінію клітин людської нейронної пухлини, які реагують на Shh, і була виділена у 1985 р. з десмопластичної мозочкової медулобластомної пухлини. Ендогенні рівні транскриптів Gli1 у клітинах Daoy є низькими, але можуть стимулюватися шляхом застосування кондиціонованого середовища, взятого з клітин, які стабільно надлишково експресують людський Shh (клітин НЕК-293, стабільно трансфектованих hShh). Клітини Daoy вирощують до злиття у склянках для тканинних культур типу Т225 у середовищі для вирощування Daoy, яке містить мінімальне базове середовище (МЕМ) плюс 10 % сироватки крові плоду корови (FBS) з 0,1 нМ замінних амінокислот та 1 мМ пірувату натрію. Клітини видаляють із склянок Т225 з застосуванням трипсину та етилендіамінтетраоцтової кислоти (EDTA), центрифугують, знов суспендують у середовищі та підраховують. Потім клітини Daoy висівають у кількості 50000 клітин на лунку у ростовому середовищі у 96лункові прозорі планшети Costar для культивування тканин та піддають інкубуванню протягом ночі при 37С в атмосфері 5 % діоксиду вуглецю (СО2). Промивають клітини один раз сольовим розчином з фосфатним буфером (PBS), після чого додають 100 мкл кондиціонованого середовища Shh (Shh-СМ) для стимулювання рівнів експресії Gli1. Розводять Shh-СМ для 18 UA 102250 C2 5 10 15 20 25 30 досягнення максимальної стимуляції, застосовуючи стандартне ростове середовище – 0,1 % FBS/DMEM (модифіковане за Дюльбекко середовище Eagle). Потім клітини Daoy, оброблені Shh-СМ, обробляють різними концентраціями інгібіторів hedgehog у діапазоні концентрацій від приблизно 1 мкМ до 0,1 нМ. Суміші з досліджуваними сполуками інкубують протягом 24 год. при 37С в атмосфері 5 % СО2. Визначення кількості транскрипту Gli1 виконують, застосовуючи пробу на Gli1 типу Quantigene 2.0 згідно з описом виробника (Panomics, Inc.) Виготовляють розведений буфер лізисної суміші (DLM), який містить протеїназу-К. Після інкубування протягом 24 год. з досліджуваною сполукою клітини промивають один раз PBS та додають до клітин 180 мкл DLM. Планшет з клітинами, який містить лізисний буфер, закривають та витримують при 55С протягом 30-45 хв. Одержані лізати клітин потім розтирають 5 разів. Робочий набір зондів, який містить зонди Gli1, виготовляють шляхом розведення зондів у DLM згідно з інструкціями виробника, після чого додають у планшети для проби на bDNA 20 мкл робочого набору зондів разом з 80 мкл лізатів Daoy. Планшети закривають та інкубують при 55С протягом ночі. Потім обробляють планшети для bDNA згідно з інструкціями виробника. Сигнал кількісно вимірюють шляхом сканування планшетів на приладі Perkin Elmer Envision reader, який детектує люмінесценцію. Сигнал люмінесценції є прямо пропорційним кількості транскрипту-мішені, присутнього у пробі. Дані про сигнал люмінесценції, одержані при функціональній пробі, застосовують для обчислення ІС50 при випробуванні in vitro. Ці дані обчислюють на основі максимальних контрольних значень (для клітин Daoy, оброблених Shh-СМ) та мінімального контрольного значення (для клітин Daoy, оброблених Shh-СМ та інгібувальною концентрацією контрольної сполуки – 1 мкМ N-(3-(1Н-бензо[d]імідазол-2-іл)-4-хлорфеніл)3,5-диметоксибензаміду). Чотирьохпараметрова логістична крива застосовується для отримання значень ІС50 з використанням програм ActivityBase, версія 5.3, рівняння 205 (за Guidance for Assay Development and HTS, vs 5, Copyright 2005, Eli Lilly and Co., та Nhe National Institutes of Health Chemical Genomics Center). Чотирьохпараметрове рівняння має таку форму: Fit = (A + ((B - A) / (1 + ((C / x)D))) де А – найнижче значення, В – найвище значення, С = ІС50 та D – коефіцієнт Гілла. При випробуванні за вищезазначеною методикою сполуки, взяті за приклади, виявили ІС50
ДивитисяДодаткова інформація
Назва патенту англійськоюDisubstituted phthalazine hedgehog pathway antagonists
Автори англійськоюHipskind, Philip, Arthur, Takakuwa, Takako
Назва патенту російськоюДвухзамещенные фталазины - антагонисты проводящего пути hedgehog
Автори російськоюХипскинд Филип Артур, Такакува Такако
МПК / Мітки
МПК: A61K 31/502, A61P 35/00, C07D 401/04
Мітки: двозаміщені, провідного, фталазини, антагоністи, hedgehog, шляху
Код посилання
<a href="https://ua.patents.su/22-102250-dvozamishheni-ftalazini-antagonisti-providnogo-shlyakhu-hedgehog.html" target="_blank" rel="follow" title="База патентів України">Двозаміщені фталазини – антагоністи провідного шляху hedgehog</a>
Двозаміщені фталазини – антагоністи провідного шляху hedgehog

Номер патенту: 102115
Опубліковано: 10.06.2013
Автори: Уілсон Такако, Хіпскінд Філіп Артур, Бастіан Джолі Ен, Селл Даніель Джон
МПК: A61K 31/502, A61P 35/00, C07D 403/04
Мітки: антагоністи, шляху, hedgehog, фталазини, провідного, двозаміщені
Формула / Реферат:
1. Сполука вказаної нижче формули:,деR1 - водень, фтор, ціаногрупа, трифторметил або метоксигрупа;R2 - водень, фтор або трифторметил;R3 - водень або хлор, за умови, що принаймні один замісник з R2 та R3 - водень;R4 - хлор, фтор, ціаногрупа, трифторметил, дифторметил, метокси- або трифторметоксигрупа;
Похідні бензилу та піридинілу як модулятори сигнального шляху hedgehog

Номер патенту: 100684
Опубліковано: 25.01.2013
Автори: Келлехер Джозеф, ІІІ, Перез Лоренс Блес, Джаін Ріші Кумар, Лей Джон, Лламас Луїс, Пейкерт Стефан, Міллер-Мослін Керен, Даі Міао, Каркі Раджеш, Маківан Майкл А., Юсуфф Наіїм, Хі Фенг
МПК: C07D 417/14, A61K 31/4545, C07D 491/048, C07D 403/12, A61K 31/502, C07D 401/06, C07F 9/58, C07D 237/28, C07D 405/14, C07D 401/12, C07D 401/04, C07D 487/04, C07D 401/14, C07D 237/30
Мітки: піридинілу, сигнального, похідні, hedgehog, шляху, модулятори, бензилу
Формула / Реферат:
1. Сполука формули І (I)або її фармацевтично прийнятні солі, у якій R1 являє собою необов'язково заміщений феніл; R2 являє собою гетероцикл, у якому щонайменше один гетероатом являє собою N і який є необов'язково заміщеним; L являє собою нижчий алкіл, (СН2)1-2-А, А-(СН2)1-2...
Спосіб контрацепції (варіанти) та набір, що містить антиестроген і прогестин

Номер патенту: 68365
Опубліковано: 16.08.2004
Автори: Міллер Кристофер Пол, Гест Майкл Джей
МПК: A61P 15/18, A61K 31/138, A61K 31/57, A61K 31/565
Мітки: набір, прогестин, контрацепції, антиестроген, містить, варіанти, спосіб
Формула / Реферат:
1.Спосіб забезпечення контрацепції, який включає введення жінці дітородного віку ефективної у відношенні контрацепції кількості комбінації антиестрогену формул І або II, що мають структуру або ,де:R1 являє собою Н, ОН, алкоксикарбоніл або аралкоксикарбоніл, що містять 2-12 атомів вуглецю, алкокси або аралкокси, що містять 1-12 атомів вуглецю, циклоалкілокси, що містить 3-12 атомів вуглецю, галоген або моно- або...
Похідні індол-2-ону, двозаміщені в положенні 3, їх одержання і їх застосування в терапії

Номер патенту: 101162
Опубліковано: 11.03.2013
Автори: БАРОНІ Марко, Пулео Летиція
МПК: A61K 31/496, C07D 209/40, C07D 209/08, A61P 3/04, A61K 31/454, A61P 3/10, C07D 401/12, A61P 3/00, C07D 211/26
Мітки: індол-2-ону, терапії, застосування, двозаміщені, похідні, положенні, одержання
Формула / Реферат:
1. Сполука формули (І):, (I) в якій:--- означає простий або подвійний зв'язок,X означає -N<, -CH< або ;Y означає >N- або >СН-, за умови, що щонайменше один з X, Y означає N;Аr означає (С6-10)арильну або (С2-10)гетероарильну...
Антагоністи ванілоїдного рецептора підтипу 1 (vr1) і їх застосування

Номер патенту: 94940
Опубліковано: 25.06.2011
Автори: Кеніг Джон Р., Гомтсян Артур Р., Браун Брайан С., Лі Чіх-Хунг
МПК: C07D 231/56, C07D 493/00, A61K 31/352, A61K 31/416
Мітки: ванілоїдного, підтипу, застосування, антагоністи, vr1, рецептора
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, проліки, сіль проліків або їх комбінація, (I) деX1 є -(CR1aR1b)m-, -(CR1aR1b)nG1- або -(CR1aR1b)p-G1-C(R1aR1b)-; m дорівнює 1, 2, 3 або 4; n дорівнює 1, 2 або 3; р дорівнює 1 або 2; G1 являє собою О,...
Попередній патент: Спосіб виділення оцтової кислоти
Наступний патент: Похідні амінодигідротіазину як інгібітори васе для лікування хвороби альцгеймера
Випадковий патент: Інспекційний риштак шахтного скребкового конвеєра