Спосіб одержання багатокомпонентних фторидів
Номер патенту: 19567
Опубліковано: 25.12.1997
Автори: Комарь Віталій Корнійович, Нестеренко Юрій Олександрович, Кобзар-Зленко Валентин Андрійович, Іванов Микола Петрович



Формула / Реферат
1. Способ получения многокомпонентных фторидов, включающий смешивание фторидов исходных металлов во фторирующей атмосфере и нагрев полученной смеси, отличающийся тем, что смешивание осуществляют в водной среде при температуре 100-150°С.
2. Способ по п. 1, отличающийся тем, что смешивание осуществляют во фторопластовых емкостях.
Текст
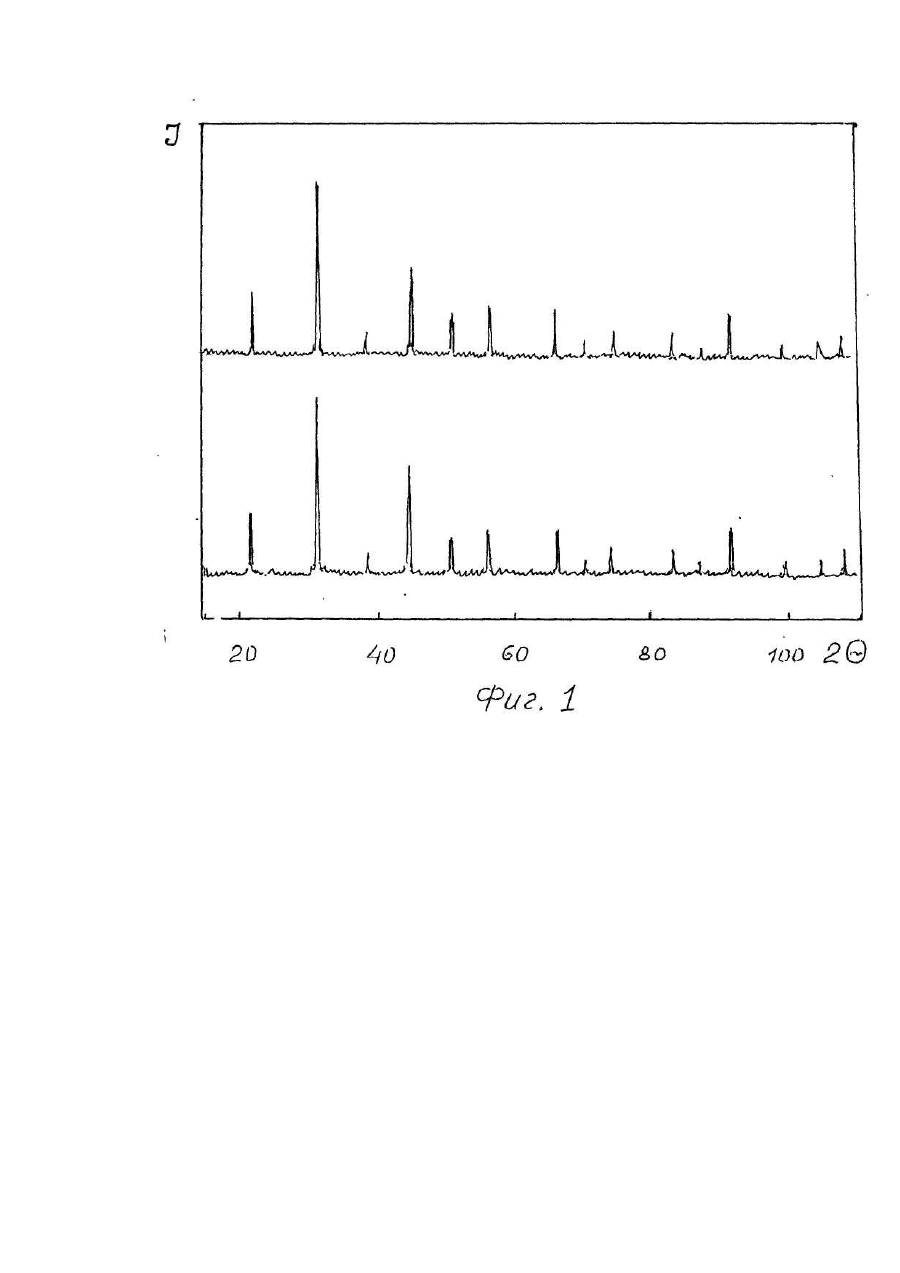
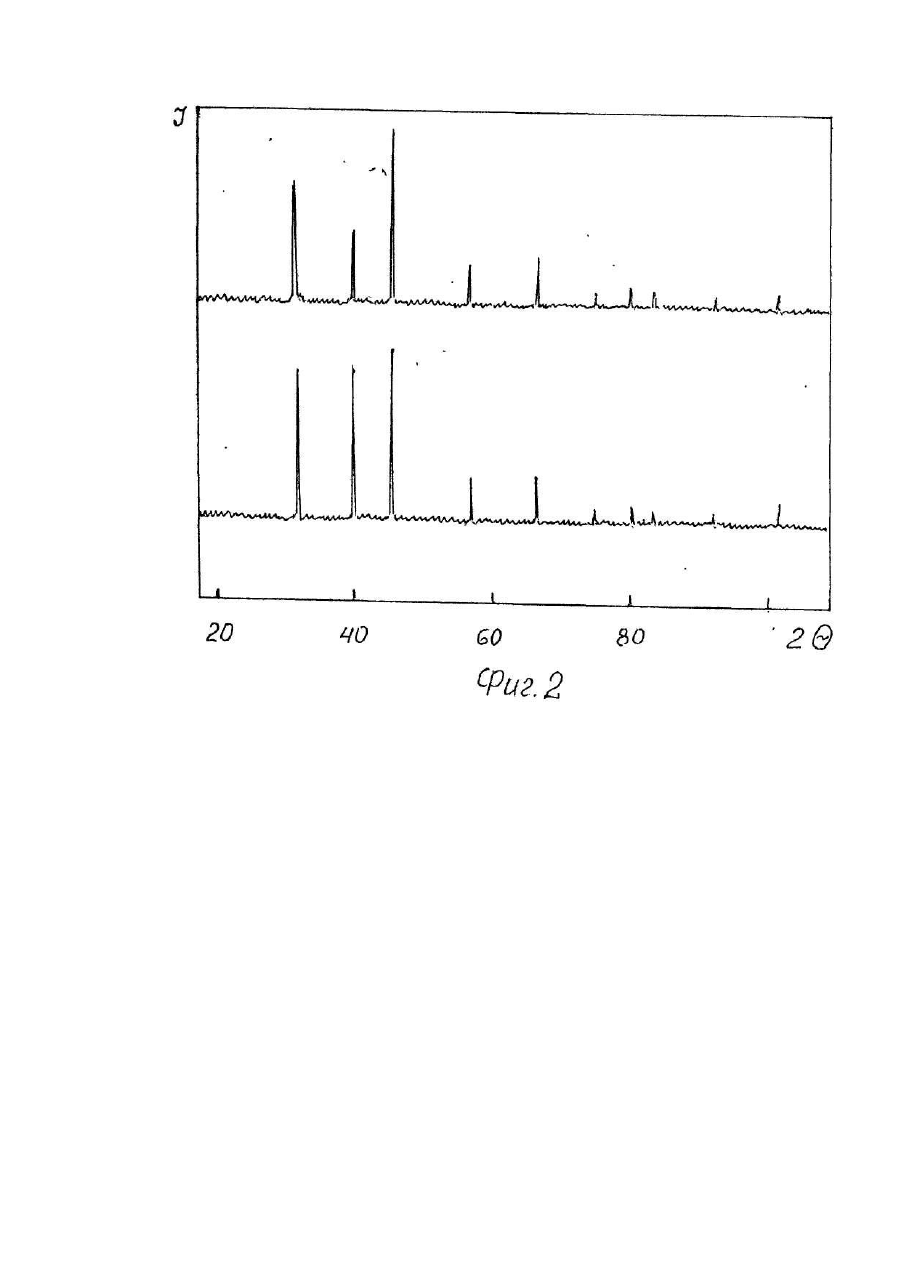
1. Способ получения многокомпонентных фторидов, включающий смешивание фторидов исходных металлов во фторирующей атмосфере и нагрев полученной смеси, отличающийся тем, что смешивание осуществляют в водной среде при температуре 100-150°С. 2. Способ по п. 1, отличающийся тем, что смешивание осуществляют во фторопластовых емкостях. Изобретение относится к области химии, в частности, получению многокомпонентных солей фтористоводородной кислоты (соединений двух и более металлов, например, КМgF3 или LiCaAIF6), служащих исходным сырьем для выращивания кристаллов, изделия из которых используются в оптическом приборостроении (лазеры, сцинтилляторы и т.п.). В отличие от простых фторидов, многокомпонентные, помимо расширения ассортимента используемых материалов класса фторидов, позволяют получить новые сочетания и значения характеристик изделий из кристаллов, обуславливающих области их практического применения в науке и технике [1-3]. Известны способы получения [4] фторидов типа MeZnF3, где Me-Na, К, Rb, Cs при нагревании фторида щелочного металла с оксидом цинка до 500-800°С в платиновом контейнере по уравнению реакции Практически вместо оксида цинка можно использовать и оксид другого металла [5]. Аналогичные соединения получают [6] сплавлением хлорида щелочного металла с фторидом двухвалентного металла где Me-Mg, Zn, Co, Ni. Общим недостатком этих способов является образование, помимо целевого продукта, оксида щелочного металла или хлорида двухвалентного металла, которые нарушают фазовый состав получаемого фторида и требуют затрат на отделение и утилизацию. Известны также способы получения сложных фторидов путем совместного сплавления простых фторидов. Сплавлением стехиометрических количеств простых фторидов KF и LaF3 или NdF3 при 900-1000°С были получены [7] фториды калий-лантана и калий-неодима по уравнению реакции Одновалентный металл является катионом, а трехвалентный образует анионный комплекс [MeF4]-. При отношении радиусов катионов Ме +/Ме3+ в интервале 0,77-1,40 существуют комплексы типа МеIМеIIIF4. Если МеI/МеIII больше 1,43, образуются конгруэнтно плавящиеся комплексы типа Ме3IМеIIIF6 [8]. Аналогичным образом при сплавлении YF3 с KF в течение 600 часов при 600°С получали калий-иттрий фторид [9], а также литий-редкоземельные фториды [10], калий-магний фторид [11,12] и калий-марганец фторид [12]. Путем сплавления стехиометрических количеств простых фторидов получали шихту для выращивания монокристаллов и более сложных составав, например, LiMeII[MeIIIF6]1 где MeII-Mg, Ca, Mn, Co, Ni, Zn; МеIII-АІ, Ga, V, Cr, Fe. Сплавление осуществляли в атмосфере аргона в платиновом контейнере при 700-800°С [3,13]. Таким образом, из приведенных аналогов следует, что при сплавлении простых фторидов в расплаве происходит образование новых фторидов типа MeI[MeIIF3], MeI[MeIIIF4] или MelMell[MeIIIF6]. Существенные недостатки известных аналогов заложены в самом технологическом процессе, который возможно осуществить практически только в расплаве исходных компонентов, когда обеспечиваются условия контактирования взаимодействующих соединений на молекулярном уровне. Использование высоких температур сопряжено с разложением фторидов под действием влаги (пирогидролиз) [14] с образованием фтористого водорода и оксида металла, что нарушает фазовый состав продуктов синтеза и снижает качество выращиваемых кристаллов. Большая длительность (сотни часов) высокотемпературного воздействия на фториды также способствует протеканию пирогидролиза. Подавление пирогидролиза достигается использованием дополнительной фторирующей атмосферы, создаваемой обычно фтористым водородом. Поэтому инженерно-техническое обеспечение способа связано с большими энергетическими затратами при достижении высоких температур синтеза, использованием платиновых тиглей, стойких в условиях высокой химической активности расплавленных фторидов, необходимостью применения фторирующей или защитной атмосферы, исключающей нарушение фазового состава продуктов синтеза, защитой персонала и окружающей среды. Все это обусловливает большие материальные затраты на технологическое осуществление процессов синтеза. В качестве прототипа выбран способ получения фторидов типа ABF3, где А-К, Rb, Cs; B-Mg, Ca, Zn, а также BaZnF4 (2) - соединений для выращивания кристаллов. Способ заключается в смешивании и сплавлении в платиновом тигле стехиометрических количеств соответствующих простых фторидов в атмосфере сухого фтористого водорода. В технологическом отношении фтористый водород удобно получать в качестве побочного продукта синтеза при использовании бифторидов щелочных металлов [9, 12] что не требует поступления HF из внешнего источника. К существенным недостаткам прототипа относится использование исходных индивидуальных фторидов, что требует энергетических затрат извне, расходуемых на структурные преобразования молекул простых фторидов и образование комплексных анионов в молекуле синтезируемого соединения (теплота плавления, энергия образования и т.д.), что реализуется в расплаве исходных фторидов при контактировании взаимодействующих компонентов на молекулярном уровне. В основу изобретения поставлена задача обеспечить уменьшение энергетических затрат и экологической вредности процесса синтеза путем снижения температуры синтеза, что исключает необходимость использования изделий из драгметаллов, протекание процессов пирогидролиза и применение фторирующей атмосферы. Решение поставленной задачи обеспечивается тем, что в способе получения многокомпонентных фторидов, включающем смешивание фторидов исходных металлов во фторирующей атмосфере и нагрев полученной смеси, согласно изобретению, смешивание осуществляют в водной среде при температуре 100150°С. Поставленная задача обеспечивается также тем, что смешивание осуществляют во фторопластовых емкостях. Использование в процессе синтеза солей аммония типа NH4 [MeF3], NH4[MeF4] или (ΝΗ4)3[AlF6], где Me двух- или трехвалентный металл в качестве промежуточных продуктов синтеза в водной среде исключает энергетические затраты на структурные перестройки атомов из отдельных индивидуальных молекул в расплаве при образовании анионных комплексов, т.е. нагрев до получения расплава исходных компонентов со всеми вытекающими отсюда последствиями, которые рассматривались ранее при оценке аналогов и прототипа. Замещение катиона аммония на катион щелочного или щелочноземельного металла достигается также в водной среде и не требует повышения температуры до плавления исходных компонентов. При этом существенно снижаются требования по инженерно-техническому обеспечению заявляемого способа потому, что интенсивность проявления химической активности фторидов при реализации данного способа ниже, чем при температурах их плавления и соответствует экологически более чистому производству. Доступность материалов, необходимых для инженерно-технического обеспечения заявляемого способа, по сравнению с известными, делает его экономически выгодным. Осуществление процесса в области рабочих температур, допускающих использование изделий из фторопласта, исключает необходимость применения тиглей из платины и способствует решению поставленной задачи с наименьшими затратами и реализации синтеза в экологически чистом режиме. Отделение получаемых продуктов синтеза от побочных продуктов и реакционной среды осуществляют на стадии выпаривания и сушки при температуре 100-150°С. В качестве побочных продуктов выделяются газообразные NH3, СО2и Н2О, образующиеся при разложении (NН4)2СО3 (температура разложения равна 58°С). Для удаления воды требуется температура выше 100°С, что ограничивает нижний предел температуры. Водная среда способствует гомогенизации при смешивании компонентов, равномерному распределению температуры и образованию комплексных анионов [BIIF3]- (15), [BIIIF4]- или [ВIIIF6]3- (8). "Сухие" способы проведения процессов требуют тщательного размола компонентов, продолжительных стадий перемешивания, при нагревании сопровождаются локальными перегревами и нарушениями стехиометрических соотношений за счет испарения более летучего компонента. Поэтому предлагаемый способ осуществляется в водной среде. Так как соединения синтезируют с целевым назначением для выращивания кристаллов и термообработка получаемых порошков не имеет существенного значения, а сырье пригодно для выращивания уже после обработки до 150°С, то более высокие температуры сушки энергетически не целесообразны. К тому же более высокий нагрев фторопластовых изделий способствует их деформации и разрушению. Этим обусловлен верхний предел температуры. В качестве емкости для синтеза выбран фторопласт. Его химическая стойкость по отношению к агрессивным фторсодержащим средам, а также достаточно высокая термостабильность позволяют использовать данный полимерный материал в условиях предлагаемого способа. Для более быстрого удаления побочных продуктов синтеза процесс выпаривания и сушки солей целесообразно осуществлять под вакуумом. Использование вакуума, создаваемого форвакуумным или водоструйным насосом, позволяет сократить продолжительность синтеза до 4-6 часов и способствует утилизации газообразных побочных продуктов. Заявляемый способ включает следующие операции: 1. Смешивание исходных компонентов в водной среде. 2. Нагревание смеси. 3. Выпаривание и сушка продукта. Примеры осуществления способа. Пример 1. Синтез KZnF3 осуществляли по суммарному уравнению реакции из которого следует, что для получения 0,5 г - моля KZnF3 (80,7 г) необходимо 51,7 г ZnF2; 18.5 г NH4F и 34,6 г К2СО3. Навески ZnF2 и NH4F помещали во фторопластовый конус и перемешивали с 80 мл дистиллированной воды до получения однородной массы. Так как NH4F гигроскопичен, его брали на 10-30% больше теоретически необходимого количества. Сверхстехиометрическое количество NH4F в дальнейшем сублимируется при синтезе или выращивании кристаллов, создавая фторирующую среду, подавляя процессы пирогидролиза. Как известно, в системе ZnF2, NH4F, Н2О образуется комплексный анион [ZnF3j-, что характерно и для Cd, и для Mg [15]. После добавления в смесь навески К2СО3 ион аммония замешается катионами калия, которые с комплексным анионом имеют более прочные связи. Фторопластовый конус закрывали крышкой и нагревали при 100°С в течение 6 часов на песчаной бане под вакуумом, создаваемым форвакуумним или водоструйным насосом. В итоге получали 80,2 г белого мелкокристаллического порошка KZnF3. Изложенная последовательность операций отражает направленность химического взаимодействия исходных компонентов, которая способствует достижению цели, а именно, формированию в водной среде промежуточного соединения с анионом [ZnF3]-, в котором катион аммония при добавлении карбоната калия замещается катионом калия с образованием основного продукта синтеза KZnF3 и побочного продукта (МН4)гСОз. разлагающегося при выпаривании и сушке на легко удаляемые на утилизацию газообразные NН3, СО2 и Н2О. В этом отношении приведенное стехиометрическое уравнение (5) является суммарным, так как отражает лишь исходные компоненты и конечные продукты синтеза. Пример 2. Синтез КСdF3 осуществляли по суммарному уравнению реакции, аналогичному (5), в котором фторид цинка заменяли CdF2. Для получения 0,5 г-моля KCdF3 (104,4 г) брали навески CdF2 - 75,2 г; NH2F - 20,3 г (110%); К2СО3 - 34,5 г и 80 мл дистиллированной воды. Последовательность операций такая же, как и в примере №1. Стадию выпаривания и сушки продукта проводили при 150°С в течение 4 часов. Получали 104,2 г белого мелкокристаллического порошка со структурой перовскита. Пример 3. Синтез KMgF3 осуществляли аналогично описанным процессам в предыдущих примерах. Для получения 0,5 г-моля KMgF3 (60,2 г) брали MgF2 - 31,2 г; NH4F - 24,0 г (130%); К2СО3 - 34,5 г и 60 мл дистиллированной воды. Выпаривание и сушку продукта проводили при 140°С в течение 5 часов. Получали 58,9 г белого мелкокристаллического порошка со структурой KMgF3 (перовскита). Пример 4. Для получения 0,5 г-моля (129.5 г) KNdF4 брали 100 г NdF3, 22,4 г (120%) NH4F, 34,5 г К2СО3 и 150 мл дистиллированной воды, которые смешивали, как и в предыдущих примерах. После выпаривания и сушки продукта при 130°С в течение 6 часов получали 129 г светло-сиреневого мелкокристаллического порошка KNdF4 со структурой перовскита. Пример 5. Для получения 0,1 г-моля (25,4 г) KLaF4 брали навески 19,6 г LаF3; 4,5 г (120%) NH4F; 6,9 г К2СО3 и 30 мл дистиллированной воды. После выпаривания и сушки при 130°С в течение 5 часов получали 25,4 г белого мелкокристаллического порошка KLaF4, имеющего структуру перовскита. Пример 6. Синтез LiCa[AIF6] осуществляли по уравнению реакции: Для получения 0,1 г/моля LiCa[AIF 6] (18,8 г) брали 19,5 r(NH4)3[AIF6]; 3,7 г Li2СО3; 10,0 г СаСО3. Навеску гексафторалюмината аммония растворяли в 100 мл дистиллированной воды, куда добавляли навески карбонатов кальция и лития. После выпаривания и сушки при 120°С в течение 6 часов получали 18,7 г белого мелкокристаллического порошка LiCa[AIF 6]. В приведенных примерах процессы выпаривания и сушки осуществляли при минимальной температуре 100°С, соответствующей температуре кипения воды и началу ее интенсивного удаления. Дальнейшее снижение температуры приводит к неоправданному увеличению длительности процесса. С другой стороны, повышение температуры сокращает продолжительность процессов выпаривания и сушки. Однако выше 150°С наблюдается деформация фторопластовых емкостей, что ограничивает верхний температурный предел процесса. Дифрактометрический анализ синтезированных по предлагаемому способу продуктов показал, что все они обладают структурой перовскита, характерной для данного класса соединения (11), На фиг. 1 и 2 представлены примеры дифрактограмм синтезированных (1) и эталонных (2) образцов KZnF3 и KMgF3 соответственно, которые свидетельствуют об идентичности сопоставляемых между собой соединений, что является доказательством получения именно данных продуктов. Дифрактограммы остальных синтезированных соединений имеют аналогичный вид. Таким образом, по сравнению с прототипом, заявляемый способ обеспечивает снижение энергозатрат. Так, необходимая для проведения синтеза температура в прототипе составляет 800-1100°С, в то время как в заявляемом способе всего 100-150°С, т.е, энергозатраты - в 8 раз меньше. Снижение температуры, в свою очередь, позволяет исключить из процесса потребление драгметаллов (платины) и заменить их более дешевым фторопластом. Кроме того, заявляемый способ является экологически чистым, так как при температуре его осуществления отсутствуют процессы пирогидролиза фторидов, служащие источником фтористого водорода.
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for obtaining multicomponent fluorides
Автори англійськоюKobzar-Zlenko Valentyn Andriiovych, Komar Vitalii Korniiovych, Ivanov Mykola Petrovych
Назва патенту російськоюСпособ получения многокомпонентных фторидов
Автори російськоюКобзарь-Зленко Валентин Андреевич, Комар Виталий Корнеевич, Иванов Николай Петрович
МПК / Мітки
Мітки: спосіб, багатокомпонентних, фторидів, одержання
Код посилання
<a href="https://ua.patents.su/5-19567-sposib-oderzhannya-bagatokomponentnikh-ftoridiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання багатокомпонентних фторидів</a>
Спосіб одержання монофториду хлору

Номер патенту: 4267
Опубліковано: 27.12.1994
Автори: Буряк Микола Іванович, Волков Сергій Васильович, Бандур Віктор Аркадійович
МПК: C01B 7/00
Мітки: одержання, хлору, спосіб, монофториду
Формула / Реферат:
Способ получения монофторида хлора, включающий пропускание газообразного хлора через расплав фторидов лития и калия, отличающийся тем, что, с целью повышения степени использования хлора, процесс осуществляют путем пропускания хлора через эвтектический расплав фторидов лития и калия в присутствии фторида щелочноземельного металла, взятого в количестве 1-10 мас. %.
Спосіб одержання вольфраматів або молібдатів цинку чи кадмію

Номер патенту: 5673
Опубліковано: 28.12.1994
Автори: Іванов Микола Петрович, Нагорна Людмила Лаврентіївна, Кобзар-Зленко Валентин Андрійович
МПК: C01G 39/00, C01G 41/00
Мітки: спосіб, цинку, молібдатів, кадмію, вольфраматів, одержання
Формула / Реферат:
Способ получения вольфраматов или молибдатов цинка или кадмия, включающий смешение триоксида вольфрама или молибдена с оксидом цинка или кадмия и нагрев полученной смеси, отличающийся тем, что перед смешением триоксид вольфрама или молибдена растворяют в водном растворе гидроксида аммония, оксид или гидроксид цинка или кадмия - в водном растворе нитрата аммония и, нагрев полученного после смешения раствора ведут при температуре кипения в...
Спосіб приготування шихти для одержання порошків кубічного нітриду бору

Номер патенту: 4580
Опубліковано: 28.12.1994
Автори: Якименко Валеріан Дмитрович, Боримський Олександр Іванович, Лисанов Владіслав Сєргєєвіч, Фєльдгун Лєон Ізраілєвич, Давідєнко Валєрій Міхайловіч, Новіков Микола Васильович
МПК: C01B 21/064
Мітки: порошків, кубічного, шихти, бору, нітриду, спосіб, одержання, приготування
Формула / Реферат:
(57) Способ приготовления шихты для получения порошков кубического нитрида бора, включающий обработку порошка магния водным раствором соли, высушивание порошка и смешивание его с порошком графитоподобного нитрида бора, отличающийся тем, что обработку магния проводят водным раствором соли из ряда галогенидов или сульфатов меди, цинка, алюминия, олова, хрома, марганца, железа.
Спосіб одержання германату вісмуту

Номер патенту: 3781
Опубліковано: 27.12.1994
Автори: Нагорна Людмила Лаврентіївна, Кобзар-Зленко Валентин Андрійович, Іванов Микола Петрович
МПК: C01G 17/00, C01G 29/00
Мітки: спосіб, вісмуту, одержання, германату
Формула / Реферат:
Способ получения германата висмута, включающий смешивание соединений германия и висмута и термообработку полученной смеси в тигле, отличающийся тем, что в качестве соединения германия используют германат щелочного металла, в качестве соединения висмута - нитрат-хлорид или сульфат висмута, а термообработку ведут при 300-900°С в течение 0,5-1,5 ч.
Спосіб одержання виробів із гомогенних алюмінідів титану

Номер патенту: 12530
Опубліковано: 28.02.1997
Автори: Івасишин Орест Михайлович, Демідік Олександр Миколайович, Іванова Інна Іванівна, Саввакін Дмитро Георгійович
МПК: C22C 1/04
Мітки: одержання, гомогенних, алюмінідів, виробів, спосіб, титану
Формула / Реферат:
Способ получения изделий из гомогенных алюминидов титана, предусматривающий смешивание исходных порошков, прессование полученной шихты в заготовку и нагрев заготовки, отличающийся тем, что в качестве исходных порошков используют гидрид титана и алюминий, смешивание производят, одновременно с размалыванием в среде аргона в течение 3-5 часов, прессуют заготовку при давлении не менее 4-10 Па, нагревают ее в вакууме 1,33 10'3 Па со скоростью...
Попередній патент: Потокова лінія для подання компонентів електросталеплавильного виробництва
Наступний патент: Сонячний водонагрівник
Випадковий патент: Склад електрода для електродугової хіміко-термічної обробки низькотемпературною плазмою