О-метильні похідні азитроміцину а, що мають антибактеріальну активність, спосіб їх отримання та проміжні сполуки
Номер патенту: 27105
Опубліковано: 28.02.2000
Автори: Кобрехел Габріела, ДЬОКІС Слободан, Лазаревскі Горяна





Формула / Реферат
1. О-Метильные производные азитромицина А общей формулы (I)
обладающие антибактериальной активностью.
2. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=CH3, R4=R5=H.
3. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R4=CH3, R5=H.
4. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R5=H, R4=CH3.
5. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R4=R5=CH3.
6. Соединение по п.1, отличающееся тем, что R1=R2=R4=R5=H, R3=CH3.
7.Соединение по п.1, отличающееся тем, что R1=R2=R5=H, R3=R4=CH3.
8. Соединение по п.1, отличающееся тем, что R1=R2=R3=R5=CH , R4=CH3.
9. Соединение по п.1, отличающееся тем, что R1=R2=H, R3=R4=R5=CH3.
10. Соединение по п.1, отличающееся тем, что R1=R4=R5=H, R2=R3=CH3.
11. Соединение по п.1, отличающееся тем, что R1=R5=H, R2=R3=R4=CH3.
12. Соединение по п.1, отличающееся тем, что R1=R3=R5=H, R2=R4=CH3.
13. Соединение по п.1, отличающееся тем, что R1=H, R2=R3=R4=R5=CH3.
14. Производное азитромицина А общей формулы (II)
где (IIb) R1=R2=CO2CH2C6H5 в качестве промежуточных продуктов для получения О-метильных производных азитромицина А общей формулы (I)
15. Способ получения О-метильных производных азитромицина А общей формулы (I)
отличающийся тем, что азитромицин или его дигидрат формулы (II)
где (IIa) R1=H, R2=CH3, подвергают взаимодействию с бензилхлорформиатом в инертном растворителе, например в бензоле, в присутствии избыточного количества соответствующего основания, например бикарбоната натрия, при температуре 25 - 60°С в течение 3 - 24 часов, с последующим О-метилированием гидроксильных групп по С-6, С-11, С-4 положениям промежуточного продукта, 2', О, 3'-N-бис(бензилоксикарбонил)-N-деметилазитромицина А формулы (II), где R'=R2=CO2CH2C6H5, 1 - 18 кратным избытком метилирующего агента, например, метилйодида, диметилсульфата, в присутствии соответствующего основания, например, гидроксида натрия, в подходящем растворителе, например диметилсульфоксиде или N,N-диметилформамиде или в их смеси с инертным растворителем, например, тетрагидрофураном, ацетонитрилом, этилацетатом, при температуре от 0°С до комнатной 3 - 30 часов с образованием смеси О-метил-2', О, 3'-N-бис(бензилоксикарбонил)-N-деметилазитромицина А общей формулы (I), где
которую при необходимости:
А) разделяют на колонке силикагелем с получением хроматографически однородных соединений la-Id, в которых затем удаляют защитные группы в положениях 2 и 3 посредством гидрогенолиза в низших спиртах, например в метаноле или этаноле, в присутствии катализатора, например палладия на углероде, в атмосфере водорода при давлении 1 - 20бар, при непрерывном перемешивании реакционной среды в течение 2 - 10 часов при комнатной температуре, с образованием О-метил-N-деметилазитромицинов А общей формулы (I), где le R1=R2=R4=R5=H, R3=CH3; If R1=R2=R5=H, R3=R4=CH3; Ig R1=R2=R3=R5=H, R4=CH3; Ih R1=R2=H, R3=R4=R5=CH3, которые затем подвергают восстановительному метилированию по 3-метиламиногруппе 1 - 3 эквивалентами формальдегида и равным или двойным количеством муравьиной кислоты при температуре перегонки реакционной смеси в течение 2 - 8 часов с образованием О-метилазитромицина А в форме производных общей формулы (I), где li R1=R4=R5=H, R2=R3=CH3; Ij R1=R5=H, R2=R3=R4=CH3; Ik R1=R3=R5=H, R2=R4=CH3; II R1=H, R2=R3=R4=R5=CH3, или
В) удаляют защитную бензилоксикарбонильную группу в положениях 2 и 3 посредством гидрогенолиза, как описано в пункте А, с образованием смеси О-метилазитромицинов А в форме производных общей формулы (I), где li R1=R4=R5=H, R2=R3=CH3; Ij R1=R5=H, R2=R3=R4=CH3; Ik R1=R3=R5=H, R2=R4=CH3; II R1=H, R2=R3=R4=R5=CH3, которую разделяют на колонке с силикагелем с получением хроматографически однородных производных О-метилазитромицинов А Іі-II.
Текст
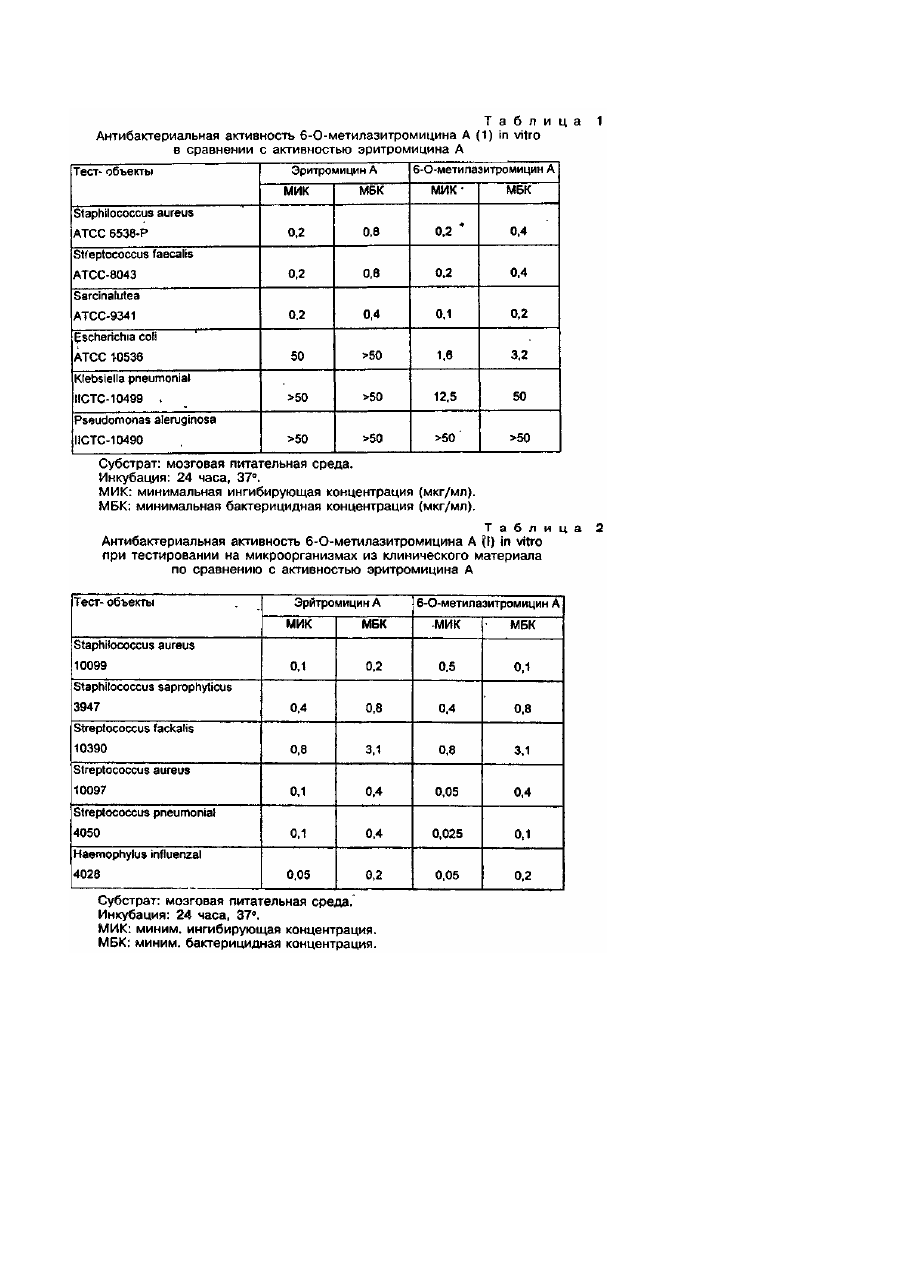
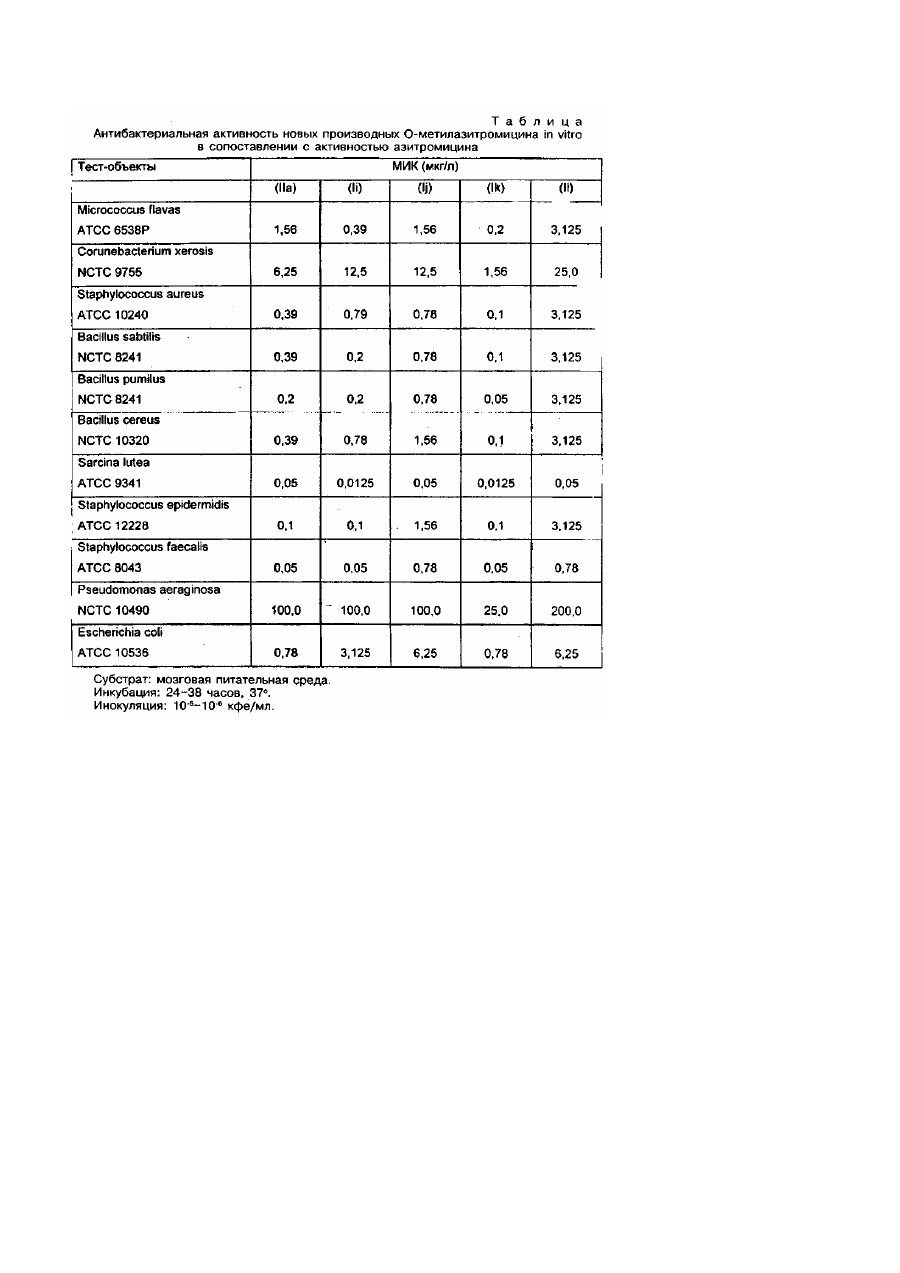
Изобретение относится к новым полусинтетическим макролидным антибиотикам азалидного ряда, в частности, к О-метиль-ным производным азитромицина А и их пригодным для фармацевтического применения солям, а также к способу получения этих соединений и промежуточным продуктам для этой цели, к их использованию в производстве лекарственных средств, особенно антибактериальных препаратов. Эритромицин А представляет собой макролидный антибиотик, химическая структура которого характеризуется наличием 14-членного агликонового кольца с кето-группой в положений С-9 (Патент США №2653899, 9/153). До настоящего времени этот макролидный антибиотик является одним из основных лекарственных препаратов,лля лечения инфекционных заболеваний у человека. Однако, в кислой среде этот антибиотик быстро превращается в ангидроэритромицин, представляющий собой неактивный С-6/С-12 метаболит со спирокетальной структурой (Kurath Р et. a!., Experientu, 1971, 26, 362). Известно, что спироциклизация эритромици-на А эффективно ингибируется посредством химической трансформации C-9(S) и С-9(Р)-кетонов при получении С-9 окси-мов (Djokic S. et. al., Tetrahedron Lett, 1967, 1945) или С-9(Р)-аминов (Egan K., J. Org, Chem, 1974, 39, 2492) или посредством элиминирования С-9 кетона при расширении агликонового кольца (Патент США № 4328334, 5/1982). В результате перегруппировки Бекманна в оксиме эритроми-цина А с последующим восстановлением образующегося иминового эфира (Djokic S. et. al., Chem. Soc., Perkin. Trans 1, 1986, 1881) происходило образование 11-аза-10-дезоксо-10дигидроэритромицина А (9-дезоксо-9а-аза-9а-гомоэритромицина А), который стал первым 15-членным макролидным антибиотиком азалидного ряда. В результате метилирования вновь введенной вторичной аминогруппы в аглико-новое кольцо формальдегидом в присутствии муравьиной кислоты с помощью модифицированного метода Eschweiles-Clask (Kobrehel G., & Djokic S., Бельгийский патент №892357, 7/1982) или после предварительной защиты аминогрупп путем их превращения в соответствующие N-оксиды с .последующим алкилированием и восстановлением этих N-оксидов (Bright, патент США №4474768, 10/1984) был получен N-метил-11-аза-10-дезоксо-10-дигидроэритромицин А (9дезоксо-9а-аза-9а-метил-9а-гомоэритромицин А, Номенклатура по органической химии IUPAC, 1979, 68 - 70, 459, 500 503), который проходит клинические испытания под незапатентованным названием азитромицин. В сравнении с исходным антибиотиком азитромицин, помимо повышенной стабильности в киспой среде, характеризуется более выраженной активностью по отношению к грамотрицательным микроорганизмам in vitro и более высокой концентрацией в тканях. Оценивается даже возможность его применения в разовой суточной дозе (Ratschema J. et. at., Antimicsob. Agents Chemothec, 1987, 31, 1939). Известно также, что С-6/С-12 спирокристаллизация эритромицина А эффективно подавляется посредством Ометилирования гидроксильной группы в положении С-6 агликонового кольца (Watanabe Y. et. аl., патент США №4331803, 5/1982). Реакция эритоомицина А с бензилхлоро-формиатом и последующее метилирование образующегося при этом 2'-0,3'-N-бис(бензилоксикарбонильного)производного при удалении защитной группы в положениях 2'- и 3'-, а также N-метилирование 3'-метил-аминогруппы в условиях восстановления, приводили к образованию больших количеств 11-O-метилированного и 6,11-ди-О-метилированного эритромицина А, помимо 6-Oметилированного эритромицина A (Morimoto S. et. al., J. Antibiot., 1984, 37, 187). Более высокая степень избирательности достигается посредством предварительного оксими-рования С-9 кетонов и О-метилирования соответствующих замещенных и незамещенных бензилоксиминовых производных (Morimoto S. et. al., патент США №4680368, 7/1987). 6-О-метилированный эритромицин А проходит клинические испытания под непатентованным названием кларитромицин. В сопоставлении с эритромицином А кларитромицин характеризуется более высокой активностью по отношению к грамположительным микроорганизмам in vitro (Kirist Н. A. et. al., Antimicrobial Agents and Chemotrfer., 1989, 1419). Проведенный заявителем поиск не выявил изобретений, касающихся О-метильных производных азитромицина А. Таким образом предметом настоящего изобретения являются новые О-метиль-ные производные азитромицина формулы (I), а также их пригодные для фармацевтического применения соли. Кроме того предметом настоящего изобретения является способ получения О-метильных производных азитромицина А формулы (I) и их пригодных для фармацевтического применения солей, согласно которому азитромицин или его дигидрат (Djokic S. et. al., J. Chem. Res. (S), 1988, 1239 - 12621 (M) 1988, 1239 - 12621) формулы (II): в которой На R'=H, R2=CH3, реагирует с бензилхлороформиатом в присутствии избыточного количества подходящего основания, например бикарбоната натрия, в инертном растворителе, например в бензоле, при температуре от 25 до 60° в течение 3 - 24 часов, в зависимости от температуры реакции, с последующим мети-лированием гидроксильных групп в положениях С-6, С-11 и С-4" нового, еще не описанного промежуточного продукта, 2'-О, 3'-Nбис(бензилоксикарбонил)-N-деметилазитромицина А формулы (II), в котором lib R1=RZ=CO2CH2C6H5, 1-18 кратным избыточным количеством соответствующего метилирующего агента, например, метил-йодида, диметилсульфата, метилметансульфоната или метил-n-толуолсульфоната, в Присутствии соответствующего основания, например, гидрида натрия, в водной гидроокиси калия или гидроокиси натрия, в соответствующем растворителе, например в диметилсульфоксиде или N,N-диметилформамиде, или в их смесях с инертным растворителем, например, с тетрагидрофураном, ацетонитрилом, этила-цетатом, 1,2-диметоксиэтаном, при температуре от 0°С до комнатной з течение 3 - 30 часов, с образованием смеси О-метил-2', О, 3'-N-бис(бензилоксикарбонил)-N-деметил-азитромицина А формулы (І), в котором Которую в случае необходимости подвергают: А) разделению на колонках из сили-кагеля (силикагель 60, 70 - 230меш, компании Мерк) в системе растворителей CH2CI2/CH3OH/NH4OH (90:9:0,5) с образованием хроматографически гомогенных соединений (la) с Rf0,660, (Ib) с Rf 0,811, (Ic) с R, 0,843 и (Id) с R, 0,881, в которых затем элиминируют защитные бензилок-сикарбонильные группы в положениях 2'-и 31- посредством гидрирования в растворе низкоатомного спирта, например метанола или этанола, в присутствии катализатора, например палладия черного или палладия на углероде, в атмосфере водорода при давлении 1-20бар при непрерывном перемешивании реакционной смеси, на протяжении 2 - 10 часов, при комнатной температуре, с образованием (после фильтрации для удаления катализатора и выделения продукта посредством обычных методов экстракции в градиенте рН (рН 5,0 и рН 9,0) из воды соответствующим гидрофобным растворителем, например хлороформом, етилацетатом и т.п.) О-метил-N-диметил-азитромицина А в форме производных формулы (І), в которых и которые затем подвергают восстановительному N-метилированию по 3'-метила-миногруппе 1 - 3-кратными эквивалентными количествами формальдегида (37%) в присутствии равного или удвоенного количества муравьиной кислоты (98 - 100%) или иного источника водорода в инертном растворителе из числа галогенизирован-ных углеводородов, таких как хлороформ, или низкоатомных спиртов, таких как метанол или этанол, низкоатомных кетонов, таких как ацетон, в условиях кипячения с обратным холодильником, в течение 2 - 8 часов, с образованием, после выделения продукта с помощью обычных методов экстракции в градиенте рН (рН 5,0 и рН 9,0), О-метилазитромицина А в форме производных формулы (I), в которых Б) элиминированию защитной бензилоксикарбонильной группы в 2'- и 3'- положениях посредством гидрирования, как описано в пункте А, с образованием смеси 6-О-метил-(Іе), 6,11-ди-О-метил-(1f), 11-О-метил-(Іg) и 6,11,4"-три-Ометил-О-деметил-(1h)азитромицинов А, которую подвергают восстановительному N-метилированию формальдегидом (37%) в присутствии муравьиной кислоты (98 - 100%) или некоторых других источников водорода, как описано в пункте А, с образованием смеси 6-О-метил-(le), 6,11-ди-О-метил-(lj), 11-О-метил-(1k) и 6,11,4"-три-О-метил-(ll)-азитромицинов А, которую разделяют на колонке из силикагеля в системе растворителей CH2CI2/CH3OH/NH4OH (90:9:0,5), с образованием хроматографически гомогенных (тонкослойная хроматография в той же системе растворителей) Ометильных производных азитромицина А (li) с Rf 0,346, (Ij) с Rf 0,393, (Ik) с Rf 0,428 и (II) с Rf 0,456. Пригодные для фармацевтических целей соли соединений формулы (I), получают в процессе взаимодействия Ометильных производных азитромицина А (I) по крайней мере с "эквимолярными концентрациями соответствующих органических и неорганических кислот, например из числа хлористого водорода, йодистого водорода, серной кислоты, фосфорной кислоты, уксусной кислоты, пропионовой кислоты, трифторуксусной кислоты, малеиновой кислоты, лимонной кислоты, янтарной, кислоты, этилянтарной кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, n-толуолсульфоновой кислоты, лаурилсульфоновой кислоты и т.п. в инертном растворителе. Соли выделяют посредством фильтрации, если они нерастворимы в используемом инертном растворителе, или посредством осаждения (наиболее часто - с использованием лиофилизации) отличающимся от растворителя соединением. О-метилированные производные азитромицина А формулы (li)-(ll) и их пригодные для фармацевтического применения соли обладают высокой антибактериальной активностью. Предварительное определение противобактериальной активности 6-О-метилазитромицина A (li) проводили in vitro на нескольких видах грамположительных и грамотрицательных микроорганизмов, а также на выделенных в клинических условиях возбудителях, в сопоставлении с аналогичной активностью эритромицина А. Определения проводили методом "разведения в пробирках". При этом использовали "мозговой питательный бульон", стандартные штаммы микроорганизмов и штаммы, свежеизолированные из клинического материала. Результаты пересчитывали на минимальную ингибирующую концентрацию или на бактерицидную концентрацию (соответственно МИК и МБК, в мкг/мл). Эти данные представлены в табл.1 и 2 и свидетельствуют о том, что 6-О-метилазитромицин А характеризуется несколько более высокой активностью по отношению к изученным штаммам микроорганизмов по сравнению с эритромицином А. В табл.3 представлены результаты тестирования 6-О-метил-(li), 6,11-ди-О-метил-(lj), 11-О-метил-(Іk) и 6,11,4"-Ометил (II) азитромицина А в сопоставлении с азитромицином. Определение минимальных ингибирующих концентраций (МИК, мкг/мл) на серии стандартных бактериальных штаммов показало, что 6-О-метил-азитромицин А (Іі) в два раза более активен по отношению к Bacillus subtilis NCTC 8241 и Sarcina lutea ATCC 9341 и в 4 раза более активен по отношению к Micrococcus flavus ATCC 6538 Р нежели азитромицин. Активность 11-О-метил-азитромицина A (Ik) также была значительно выше чем у азитромицина, поскольку большинство исследованных штаммов микроорганизмов были в 2 - 4 раза более чувствительны к действию этого производного по сравнению с исходным материалом. Также предметом настоящего изобретения являются лекарственные формы, обеспечивающие эффективную и в то же время физиологическую дозировку новых соединений согласно данному изобретению. Препараты (li)-(ll) и их пригодные для фармацевтического применения соли могут использоваться в качестве терапевтических средств при лечении инфекционных заболеваний человека и животных, вызванных грамположительными бактериями, микоплазмами и патогенными микроорганизмами, обладающими чувствительностью к этим препаратам (li)-(ll). Эти препараты и их пригодные для фармацевтического использования соли можно вводить перорально или парентерально, в частности в форме подножных или внутримышечных инъекций, в виде таблеток, капсул, порошков и т.п., которые получают общепринятыми в фармацевтической практике методами. Пример 1. 2'-О,3'-N-бис(бензилоксикарбонил)-N-деметил-азитромицин А (llb). Способ А. 48г NaHCO3 вводят в раствор дигидрата азитромицина (30г, 0,038моль) в 140мл сухого бензола и нагревают полученную смесь, перемешивая при 55 - 60°С, после чего по каплям добавляют в нее 75мл бензилхлороформиата (89,63г, 0,53моль) на протяжении часа. Реакционную смесь непрерывно перемешивают при указанной температуре в течение 3 часов, после чего оставляют на ночь при комнатной температуре. Бензольную суспензию трижды экстрагируют 150мл 0,25Н HCI, полученный бензольный раствор упаривают над CaCl2, фильтруют и снова упаривают при пониженном давлении в густое масло. Полученный остаток по каплям добавляют в 500мл охлажденного петролейного эфира при непрерывном перемешивании, суспензию продолжают перемешивать на холоде в течение 4 часов, осадок отфильтровывают, промывают петролейным эфиром и высушивают, получая 27,5г (71,6%) искомого продукта, который после перекристаллизации из смеси эфира с петролейным эфиром имеет точку плавления 148 154°, ЕИ-МС м/с 1003 (М+); ТСХ, СН2СІ2/СН3ОН/NН4ОН (90:9:0,5) Rf 0,704; ИК (СНСІ3): 3510, 3350, 2960, 1704, 1690, 1605, 1450, 1380, 1330, 1290, 1255, 1160, 1115, 1050, 995 см-1; Н ЯМР (CDCl3): 2,301 (3Н, 9a-NCH3), 2,844, 2,802 (3Н, 3'-NCH3), 3,397 (3Н, 3'-OCH3; 13 С ЯМР (CDCl3): 177,260 (С-1), 100,115 (С-1'), 95,149 (С-1"), 75,028 (С-6), 74,607, (С-12), 69,415 (С-9), 64,617 (С10), 36,964 (9a-NCH3). Способ Б. 22г NaHCO3 добавляют в раствор бензилхлороформиата (30мл 0,21моль) в 50мл сухого бензола при непрерывном перемешивании. Затем, постепенно, на протяжении 3 часов в полученную смесь добавляют 15г азитромицина (0,019моль). После добавления 3/4 общего количества азитромицина в систему вводят еще 15мл (0,106моль) бензилхлороформиата. Реакционную смесь оставляют на 24 часа при комнатной температуре в условиях непрерывного перемешивания, фильтруют, фильтрат трижды экстрагируют 15мл 0,25 НNCI, высушивают над MgSO4 и упаривают при пониженном давлении. Добавление петролейного эфира приводит к осаждению неочищенного 2О,3'-N-бис(бензилоксикарбонил)-N-деметил-азитромицина А, который отфильтровывают и сразу же суспёндируют в 50мл охлажденного эфира при непрерывном перемешивании. Суспензию продолжают перемешивать при комнатной температуре на протяжении часа, осадок отфильтровывают и высушивают. Выход гомогенного (по данным тонкослойной хроматографии конечного продукта составляет 8,67г (43,09%). Продукт имеет те же физико-химические свойства, что и при получении по способу А. Пример 2. О-Метилирование 2'-О,3'-N-бис(бензилоксикарбонил)-N-деметил-азитромицина A, (la, Ib, Ic и Id). Способ А. 6мл (0,106моль) метилированного йода добавляют в раствор продукта, полученного, как описано в примере (6г, 0,006моль), в 64мл диметилсульфоксида с тетрагидрофураном (1:1.). После этого в полученную смесь добавляют еще 6,6мл (0,106моль) метилированного йода, а затем, постепенно 2,4f (приблизительно 0,06моль) NaH (55 - 60%) в масле, при комнатной температуре. Реакционную смесь оставляют при непрерывном перемешивании на 5 часов, затем оставляют на ночь и выливают в насыщенный раствор NaCI (100мл) и дважды экстрагируют 100мл этилацетата. Объединенные органические экстракты трижды промывают 100мл насыщенного раствора NaCI, высушивают над К2СО3 и упаривают, получая 6,35г неочищенного искомого продукта. Последний подвергают гидрированию, как описано в примере 9, или, в случае необходимости, очищают посредством хроматографии на колонке из силикагеля (силикагель 60, 70 - 230меш, компании Мерк) с использованием системы растворителей CH2CI2/CH3OH/NH4OH (90:9:0,5). Посредством концентрации и упаривания фракций Rf 0,881 (тонкослойная хроматография в идентичной системе растворителей) из 1,5г неочищенного продукта получают 0,12г хроматографически чистого 2'-О,3'-Nбис(бензилоксикарбонил)-N-деметил- 6,11,4"-три-О-метилазитромицина A (Id): 'Н ЯМР (CDCI3): 2,246 (3Н, 9a-NCH3), 2,831, 2,798 (3Н, 3'-МСН3), 3,367 (3Н, 3''-ОСН3), 3,305 (3Н, 6-ОМе), 3,465 (3Н, 4"-ОСН3), и 3,485 (3Н, 11-ОСН3) м.ч.; 13 С ЯМР (CDCI3): 176,975 (С-1), 69,920 (С-9), 35,967 (9а-NСН3), 79,1 (С-6), 52,8 (6-ОСН3), 89,0 (С-11), 62,0 (11ОСН3), 87,357 (С-4"), 61,131 (4"-ОСН3), 49,176 и 49,526 (3"-ОСН3) и 36,457 (3'-11СН3) м.ч. После объединения и упаривания фракций с Rf 0,843, получают 0,32г хроматографически чистого 2'-О,3'-Nбис(бензилоксикарбонил)-N-деметил-11-О-метилазитромицина A, (Ic): ЕИ-МС мс/с 1016 (M+); 1 Н ЯМР (CDCl3): 2,239 (3Н, 9a-NCH3 ), 2,805, 2,847 (3Н, 3'-NCH3), 3,374 (3Н, 3"-ОСН3) и 3,573 (3Н, 11-ОСН3) м.ч. После упаривания фракций c Rf 0,811 получают 0,316г 2'-О,3'-N-бис(бензилоксикарбонил)-N-деметил-6,11-Ометилазитромицина A, (Ib): ИК (CHCI3): 3570, 3490, 1740, 1690, 1455, 1380, 1330, 1295, 1260, 1200, 1160, 1120, 1095, 1055, 1005, 990, 980 см-1; 1 Н ЯМР (CDCI3): 2,292 (ЗН, 9а-NСН3), 2,838, 2,795 (3Н, 3'-NCH3), 3,380 (6Н, 6-ОСН3 и 3"-ОСН3) и 3,488 (3Н, 11ОСН3) м.ч.; 13 С ЯМР (CDCI3): 177,939 (С-1), 69,471 (С-9), 35,271 (9а-NСН3), 88,994 (С-11), 52,892 (6-ОСН3), 61,09 (11-ОСН3), 36,851 (3'-NCH3) и 49,549, 49,154 (3"-ОСН3) м.ч. После комцентрирования и упаривания до сухого остатка фракций с Rf 0,661 получают 0,384г 2'-О,3'-Nбис(бензилоксикарбонил)-N-деметил-6-О-метилазитромицина A, (la): ЕИ-МС м/с 1016 (М+); ИК (CHCI3): 3570, 3500, 2960, 1740, 1690, 1450, 1380, 1325, 1290, 1255, 1200, 1160, 1120, 1050, 995 см-1; 'Н ЯМР (CDCl3): 2,288 (ЗН, 9a-NCH ), 2,805, 2,847 (3Н, 3'-М-ОН3), 3,380 (6Н, 6-ОСН3 и 3"-ОСН3) м.ч. Способ Б. В раствор продукта, полученного, как описано в примере 1 (6г), в 60мл ди-метилсульфоксида с тетрагидрофураном (1:1) постепенно добавляют метилированный йод (3мл) и 2,1 г NaOH (55 - 60%) при непрерывном перемешивании в течение 2 часов при температуре 0 - 5°. Затем реакционную смесь перемешивают еще в течение часа при 0 - 5°, полученную суспензию выливают в насыщенный раствор NaCI и экстрагируют этилацетатом. Органические экстракты промывают насыщенным раствором NaCI, высушивают над К2СО3 и упаривают до сухого остатка при пониженном давлении. Конечный продукт (2 г) очищают посредством хроматографии на силикагеле в системе растворителей CH2CI2/CH3OH/NHpH (90:9:0,5) с выходом 0,89г 6-С-метилпроизводного-(Іа), 0,11г 6,11-ди-Ометилпроизводного (Ib) и 0,48г 11-О-метилпроизводного (lс). Способ B. После добавления 6мл метилированного йода в раствор продукта, полученного, как описано в примере 1, в N,Nдиметилформамиде (6г в 60мл) в реакционную смесь постепенно вводят 2,4г (55 - 60%) NaH, при непрерывном перемешивании на протяжении 2 часов при комнатной-температуре. Затем перемешивание реакционной смеси продолжают в течение еще 2 часов при той же температуре, после чего оставляют ее на ночь. Продукт реакции выделяют, как описано в способе А, получая смесь (4,54г) 6,11-ди-О-метилированного (Ib) и 6,11,4"-три-Ометилированного (Id) производных. Эту смесь подвергают гидрированию в метаноле (6мл) в присутствии NaOAc/HOAc буфера (рН 5) и палладия на угле (2г, 5%) в качестве катализатора, как описано в примере 3. После выделения продукта и упаривания растворителя при рН 9,0 получали смесь (2,33г) 6,11-ди-Ометилазитрофицина A (Ih) с R, 0,220 и 6,11,4"-три-О-метил-N-деметилазитромицина A (In) с Rf 0,263, которую разделяли на колонке из силикагеля в системе растворителей CH2CI2/CH3OH/NH4OH (90:9:1), получая хроматографически чистые продукты (II) и (In). Пример 3. 6-О-Метил-N-деметилазитромицин А (lе). 2,0г (0,002моль) 2'-О,3'-N-бис(бензилоксикарбонил)-N-деметил-6-О-метилазитромицина А (lа) разводили в 30мл этанола. В полученный раствор добавляли 10мл воды, содержащей 0,185мл уксусной кислоты и 0,3 г уксуснокислого натрия (рН 5), а также 0,7г палладия на угле (10%). Реакционную смесь перемешивали при пониженном давлении (10бар) на протяжении 10 часов, катализатор удаляли фильтрованием и упаривали смесь до сухого остатка. Последний растворяли в CHCI3 (30мл), добавляли воду (30мл), доводили рН до 5,0 добавляя 1 Н НСl, разделяли отдельные слои и водный слой экстрагировали дважды CHCI3 (оба раза по 15мл). В реакционную смесь вводили CHCI3 (30мл), рН доводили до 9,0, доливая при помешивании 2 Н·NaOH, разделяли отдельные слои и снова экстрагировали водный слой CHCI3 (дважды по 15мл). Объединенные органические экстракты (рН 9,0) сушили над К2СО3, фильтровали и упаривали, получая 1,03г (70%) искомого продукта. ЕИ-МС м/е 748. ТСХ, Rf 0,182. ИК (CHCL): 3670, 3500, 2960, 2920, 1725, 1460, 1375, 1345, 1320, 1280, 1260, 1165, 1120, 1085, 1045, 1010, 995, 900см-1. 1 Н ЯМР (CDCI3): 2,278 (3Н, 9a-NCH3),' 2,406, (3Н, 3'-NCH3), 3,312 (3Н,3"-ОСН3), 3,384 (3Н, 6-ОСН3) м.ч. Пример 4. 6,11-Ди-О-метил-N-деметилазитромицин A (If). Следуя процедуре, описанной в примере 3, посредством гидрирования с использованием палладия на угле (10%) в этаноле, в присутствии буферного раствора (уксуснокислый натрий/уксусная кислота, рН 5,0) из 0,165 г (0,16моль) 2'-О, 3'-N-бис(бензилоксикарбонил)-N-деметил-6,11-О-метилазитромицина А (lb) получали 0,093г (76,2%) хроматографически однородного 6,11-ди-О-метил-N-деметилазитромицина А с точкой плавления 95 - 98°. ЕИ-МС м/е 762. ТСХ, Rf 0,331. 1 Н NHP (CDCl3): 2,265 (3Н, 9а-СН3), 2,422 (3Н, 3'-NCH3), 3,312 (3Н, 3"-ОСН3), 3,374 (3Н, 6-ОСН3) и 3,521 (3Н, 11OCH3) м.ч. 13 С ЯМР (CDCl3): 177,7 (С-1), 65,9 (С-9), 36,8 (9a-NCH3), 79,3 (С-6), 88,9 (С-1), 52,7 (6-OCHJ, 33,1 (3'-NCHJ, и 49,7 (3’’-ОСН3) м.ч. Пример 5. 11-О-Метил-N-деметилазитромицин A (Ig). В соответствии с процедурой, описанной в примере 3, из 0,250г (0,246ммоль) 2'-О,3'-N-бис(бензилоксикарбонил)N-деметил-11-О-метилазитромицина А (Iс) посредством гидрирования в присутствии палладия на угле (10%) в метаноле и буферного раствора (уксуснокислый натрий/уксусная кислота, рН 5,0) получали 0,168г (69,5%) 11-Ометил-N-деметилазитромицина A (Ig). ТСХ, Rf 0,244. ИК (CDCL): 3500, 2970, 2940, 1736, 1460, 1380, 1165 см-1. 'Н ЯМР (CDCI3): 2,44 (3Н, 9a-NCH3), 2,458 (3Н, 3'-NCH3), 3,336 (3Н, 3"-ОСН3), и 3,590 (3Н, 11-ОСН3) м.ч. 13 С ЯМР (CDCI3): 177,6 (С-1), 70,7 (С-9), 35,8 (9а-МСН3), 74,4 (С-6), 85,0 (С-1), 33,1 (3'-NCH3) и 50,7 (3"-ОСН3) м.ч. Пример 6. 6-О-Метил-азитромицин А (l). Способ А. В раствор 0,78г (0,00104моль) 6-О-метил-N-деметилазитромицина А (Іе) в CDCI3 (50мл) добавляли 0,085мл (0,00113моль) формальдегида (37%) и 0,078мл (0,00203моль) муравьиной кислоты (98 - 100%). Реакционную смесь кипятили с обратным холодильником при перемешивании на протяжении 8 часов, затем охлаждали до комнатной температуры и переливали в 50мл воды. После доведения рН реакционной смеси до 5,0 добавлением 1 H НСІ разделяли образующиеся слои и водный слой дважды экстрагировали СНС13 (каждый раз по 20мл). К водной фракции доливали CHCI3 (20мл), рН доводили до 9,0 при непрерывном перемешивании 2 Н NaON, образующиеся слои разделяли и водную фракцию снова дважды экстрагировали CHCI3 (по 20мл). Объединенные экстракты CHCI3 (рН 9,0) подсушивали над К2СО3 и упаривали с выходом 0,495г (62,74%) конечного продукта, который в случае необходимости дополнительно очищали на колонке из силикагеля в системе растворителей CH2CI2/CH3OH/NH4OH (90:9:0,5), получая хроматографически гомогенный материал (1) с точкой плавления 103 - 109°. ЕИ-МС м/е 762. ТСХ R, 0,346. ИК (КВг): 3500, 2980, 2940, 1740, 1462, 1385, 1330, 1280, 1260, 1170, 1112, 1059, 1018 и 1055 см-1. 1 Н ЯМР (CDCL3): 2,300 (3Н, 9a-NCH3), 2,316 (6Н, 3'-N(СН3)2), 3,333 (ЗН, 3'-ОСН3), и 3,384 (ЗН, 6-ОСН3) м.ч. 13 С ЯМР (CDCL3): 177,540 (С-1), 68,850 (С-9), 36,8 (9a-NCH3), 79,2 (С-6), 52,822 (6-ОСН3), 61,627 (С-10), 40,350 (З'N(СН3)2) и 49,457 (3"-ОСН3) м.ч. Биологическая активность: 1мг содержит 754мкг азитромицина: Способ Б. В раствор 0,5г (0,668ммоль) 6-О-метил-N-деметилазитромицина А в ацетоне (30мл) добавляли 0,128мл (1,71моля) формальдегида (37%) и 0,118мл (3,06ммоль) муравьиной кислоты (98 - 100%). Полученную смесь кипятили с обратным холодильником на протяжении 2 часов. Затем ее охлаждали до комнатной температуры и упаривали ацетон, получая густой сироп. В последний добавляли 20мл воды и посредством экстракции в градиенте рН метилхлоридом, как описано в способе А, выделяли конечный продукт с выходом 0,46г (90,3%). Пример 7. 6,11-Ди-О-метилазитромицин А (I). В соответствии с процедурой, описанной в примере 6, из 0,49г 6,11-ди-О-метил-N-деметилазитромицина А (II) посредством восстановительного N-метилирования формальдегидом (37%, 0,083мл) в присутствии муравьиной кислоты (98-100%) получали 0,46г (92,3%) искомого продукта. ЕИ-МС м/е 776 (М+). ТСХ, R, 0,391. 1 Н ЯМР (CDCI3): 2,295 (3Н, 9a-NCH3), 2,316 (3Н, 3'-N(CH3)2), 3,321 (3Н, 3"-ОСН3), 3,38 (3Н, 6-ОСН3) и 3,524 (3Н, 11ОСН3) м.ч. Пример 8. 11-О-метилазитромицин A (Ik). В соответствии с процедурой, описанной в примере 6, из 0,32г (0,43ммоль) 11-О-метил-М-деметилазитромицина (Ig) посредством восстановительного метилирования формальдегидом (37%) в присутствии муравьиной кислоты (98 100%) получали 0,238г (72,44%) искомого производного 11-О-метилазитромицина A (Ik). Пример 9. 6-О-метилазитромицин А (li), 6,11-ди-О-метилазитромицин A (Ij), 11-О-метилазитромицин A (Ik) и 6,11,4"-три-О-метилазитромицин А (II). 1. В раствор неочищенного продукта, полученного, как описано в примере 2, в этаноле (2,16 г в 30мл) добавляли 10мл воды, содержащей 0,185мл уксусной кислоты и 0,3 г уксуснокислого натрия, а также 0,7 палладия на угле (10%), после чего реакционную смесь подвергали гидролизу, как описано в примере 3. При рН 9,0 получали 0,98 г смеси 6,Ометил-(Іе), 6,11-ди-О-метил-(lf), 11-О-метил-(lg) и 6,11,4"-три-О-метил-11 -деметилазитромицина А (I). 2. 0,98 г смеси, полученной в соответствии с пунктом 1, разводили в 50мл CHCl3, добавляли 0,106мл формальдегида (37%) и 0,096мл муравьиной кислоты (98 - 100%) и подвергали N-метилированию, как описано в примере 6. При рН 9,0 получали 0,537 г смеси, которую хроматографировали на колонке из силикагеля (силикагель 60 компании Мерк, 70-230 меш) в системе растворителей СН2CI2/CH3OH/NH4OH (90:9:1,5), получая 0,238г хроматографически гомогенного (Ii) с Rf 0,346, 0,065 г (Ij) с Rf 0,391, 0,105 г (Ik) с Rf, 0,428 и 0,094 г (II) с Rf 0,456. Пример 10. 6,11-4"-Три-О-метил-N-деметилазитромицин A (In). В соответствии с процедурой, описанной в примере 3, из 3,35г (3,21ммоль) 2'-О,3'-11-бис-(бензилоксикарбонил)-Nдеметил-6,11,4"-три-О-метилазитромицина A (Id) посредством гидрирования с палладием на угле (10%, 1 г) в этаноле (60мл) в присутствии буферного раствора (уксуснокислый натрий/уксусная кислота, рН 5,0) получали 1,41г (56,7%) искомого продукта, который, в случае необходимости хроматографировали на колонке из силикагеля в системе растворителей CH2CI2/CH3OH/NH4OH (90:9:0,5) с выходом хроматографически гомогенного вещества (Ih). ЕИ-МС м/е 775. ТСХ Rf 0,263. 1 Н ЯМР (CDCI3): 2,262 (ЗН, 9a-NCH,), 2,393, (ЗН, 3'-NCH3), 3,308 (6Н, 3"-ОСН3 и 6-ОСН3) и 3,475 (4"-ОСН3) и 3,521 (11-ОСН3 ) м.ч. 13 С ЯМР (CDCI3): 175,0 (С-1), 64,8 (С-9), 79,8 (С-6), 50,6 (6-ОСН3), 86,1 (С-11), 59,1 (11-ОСН3), 87,7 (С-4") и 60,9 (4"ОСН3) м.ч. Пример 11. 6,11-4"-Три-О-метилазитромицина А (II). В соответствии с процедурой, описанной в примере 6, из 1,2г (1,55ммоль) 6,11,4"-три-О-метил-Nдеметилазитромицина A (Ih) с использованием 0,131мл формальдегида (37%, 1,71ммоль) и 6,121мл (3,15ммоль) муравьиной кислоты (98 - 100%) получали 0,75г (64,4%) искомого продукта. ЕИ-МС м/е 789. ТСХ, R, 0,456. 1 Н ЯМР (CDCI3): 2,216 (ЗН, 9а-NСН3), 2:311 (6Н, З'-N(CH3)2 3,321 (ЗН, 3"-ОСН3); 3,302 (6-ОСН3) 3,482 (4"-ОСН3), и 3,521 (11-ОСН3)м.ч. 13 С ЯМР (CDCI3): 177,859 (С-1), 68,6 (С-9), 36,8 (9а-NСН3), 80,7 (С-6), 51,0 (6-ОСН3), 89,0 (С-11), 62,0 (11-OCH3), 87,3 (С-4'') и 61,3 (4"-OCH3) м.ч.
ДивитисяДодаткова інформація
Назва патенту англійськоюO-methyl azithromicyne a derivatives, having antibacterial activity, the method for obtaining thereof and intermediate compounds
Назва патенту російськоюО-метильные производные азитромицина а, обладающие антибактериальной активностью, способ их получения и промежуточные соединения
МПК / Мітки
МПК: C07H 17/08, C07H 17/00, A61K 31/70, A61P 31/04, A61K 31/7042
Мітки: мають, активність, сполуки, азитроміцину, спосіб, антибактеріальну, похідні, проміжні, отримання, о-метильні
Код посилання
<a href="https://ua.patents.su/7-27105-o-metilni-pokhidni-azitromicinu-a-shho-mayut-antibakterialnu-aktivnist-sposib-kh-otrimannya-ta-promizhni-spoluki.html" target="_blank" rel="follow" title="База патентів України">О-метильні похідні азитроміцину а, що мають антибактеріальну активність, спосіб їх отримання та проміжні сполуки</a>
Сірковмісні похідні імідазолу, що мають гіпотензивну активність, спосіб їх отримання, проміжні сполуки і фармацевтична композиція

Номер патенту: 26130
Опубліковано: 07.06.1999
Автори: Кай Жан-Клод, Жукей Сімон, Вевер Жан-Поль, Амон Жіль, Фортен Мішель, Корб'є Ален
МПК: C07D 403/12, A61K 31/415, A61P 9/12, A61P 9/10, A61P 9/00, C07D 401/12, A61K 31/4178, C07D 233/84, A61P 15/00, C07D 233/90, A61P 13/02, A61P 43/00, A61K 31/4164, A61P 21/00, C07D 403/10, C07D 233/94
Мітки: сірковмісні, гіпотензивну, проміжні, імідазолу, отримання, сполуки, активність, фармацевтична, похідні, композиція, мають, спосіб
Формула / Реферат:
1. Серосодержащие производные имидазола общей формулы (I)где R1 - алкил;R2 - водород, гидроксиметил, карбоксигруппа, группа формулы -S(=O)2-A, где A - низший алкил или фенил,- группа формулы -S(=O)2(CH2)k-B, где k = 1 и 2, B - низшая алкокси- или гидроксигруппа,- группа -S-D, где D - незамещенный фенил или замещенный галогеном, низшей алкоксигруппой низший алкил, возможно замещенный галогеном, и...
Похідні азолу, що мають фунгіцидну або регулюючу ріст рослин активність, і проміжні сполуки для їх отримання

Номер патенту: 27101
Опубліковано: 28.02.2000
Автори: СІМІЗУ Сусуму, ЄНАРІ Хіроюкі, ІКЕДА Сусуму, САЙСОДЗІ Тосіхіде, ІТО Ацусі, Кумазава Сатору, САТО Набуо
МПК: A01N 43/50, C07C 25/00, C07C 49/697, C07D 303/00, C07C 49/657, C07C 13/00, C07C 67/00, C07C 17/00, C07C 1/00, C07C 69/716, C07C 45/67, C07D 233/60, C07D 521/00, C07C 17/26, C07C 45/00, A01N 43/653, C07D 249/08
Мітки: мають, ріст, активність, отримання, сполуки, проміжні, рослин, фунгіцидну, регулюючу, похідні, азолу
Формула / Реферат:
1. Производные азола общей формулы (I)где R1 и R2 - водород или С1-С5-алкил, при условии, что оба одновременно не могут быть водородом;X - галоген, С1-С5-алкил или фенил;n=0 - 2;А - азот или группа -СН-, обладающие фунгицидной или регулирующей рост растений активностью.2. Производные азола общей формулы (I) по п.1 отличающиеся тем, что R1 и R2 - водород или С1-С3-апкил, при условии, что оба...
Триазольні похідні, що мають протигрибкову активність, фармацевтична композиція та проміжні сполуки

Номер патенту: 26751
Опубліковано: 12.11.1999
Автори: Кеннет Річардсон, СТЕФЕН Джемс Рей
МПК: A61P 31/10, C07D 213/61, C07D 403/06, C07D 405/06, A61K 31/4427, C07D 239/30, A61P 31/04, A61K 31/443, C07D 521/00, C07D 401/06, A61K 31/4418, A61K 31/44, A61K 31/505
Мітки: похідні, фармацевтична, сполуки, протигрибкову, проміжні, активність, мають, композиція, триазольні
Формула / Реферат:
1. Триазольные производные общей формулы:или их фармацевтически приемлемые соли, где R представляет фенил, замещенный 1 - 3 заместителями, каждый независимо выбранный из галоида, -CF3 или -OCF3, R1 - C1-C4-алкил, R2 - H, X - CH или N, Y - F или Cl, обладающие противогрибковой активностью.2. Соединение по п.1, в котором R является фенилом, замещенным 1 или 2 галоидными заместителями.3. Соединение по п.1, в котором R...
Піримідинові похідні, що мають гербіцидну активність, спосіб їх отримання, гербіцидна композиція і спосіб знищення бур’янів

Номер патенту: 26890
Опубліковано: 29.12.1999
Автори: Ясухумі Тойокава, Нобухіде Вада, Содзі Кусано
МПК: C07D 239/34, C07D 403/12, A01P 13/00, A01N 43/54, C07D 239/52
Мітки: отримання, знищення, піримідинові, активність, гербіцидну, мають, спосіб, композиція, похідні, гербіцидна, бур'янів
Формула / Реферат:
1, Пиримидиновые производные формулы:В которой R представляет собой атом водорода, группу - CH2CH2S(O)nR', где R' является метильной или этильной группой, а n является целым числом от 0 до 2 илигде R1 является метильной, этильной или пропильной группой, А представляет собой атом хлора или метоксильную группу, D и Е, одинаковые или различные, представляют собой атом водорода, хлора, метильную группу, метокси- или...
Хелатні комплекси азитроміцину, які мають антацидну дію

Номер патенту: 26749
Опубліковано: 12.11.1999
Автори: Слободан Дьйоліч, Нєвєнка Лопотар, Златко Вайтрер, Хрвойє Крнієвіч, Лідія Колачні Бабіч
МПК: A61K 33/26, A61K 31/7048, A61K 31/7042, A61K 31/70, C07H 17/08, C07H 23/00, A61P 1/04, A61K 31/60, A61K 31/7135, A61K 33/24, A61K 33/06
Мітки: хелатні, азитроміцину, дію, антацидну, комплекси, мають
Формула / Реферат:
1. Хелатные комплексы азитромицина, обладающие антацидным действием, полученные взаимодействием азитромицина с гелем гидроокиси алюминия-гидрокарбоната магния, сукральфосфата или висмутсубсалицилата при массовом соотношении 1 : 1 - 4 в спиртовой среде при комнатной температуре в токе азота.2. Хелатный комплекс азитромицина по п.1, отличающийся тем, что весовое соотношение азитромицина и Al(OH)3 - MgCO3 геля составляет 1 : 1 и указанный...
Попередній патент: Похідні азолу, що мають фунгіцидну або регулюючу ріст рослин активність, і проміжні сполуки для їх отримання
Наступний патент: Спосіб витягування шляхом випарювання твердих залишків із текучого середовища і установка для його здійснення
Випадковий патент: Гербіцидна композиція, спосіб боротьби з небажаною рослинністю та спосіб захисту посівів від фітотоксичної дії 3-фенілурацилів