Спосіб синтезу праміпексолу і його фармацевтично прийнятних солей
Номер патенту: 93945
Опубліковано: 25.03.2011
Автори: Гобец Станіслав, Зівец Матей, Коленц Іванка, Зупет Рок, Анзіц Борут






Формула / Реферат
1. Спосіб одержання праміпексолу, в якому здійснюють:
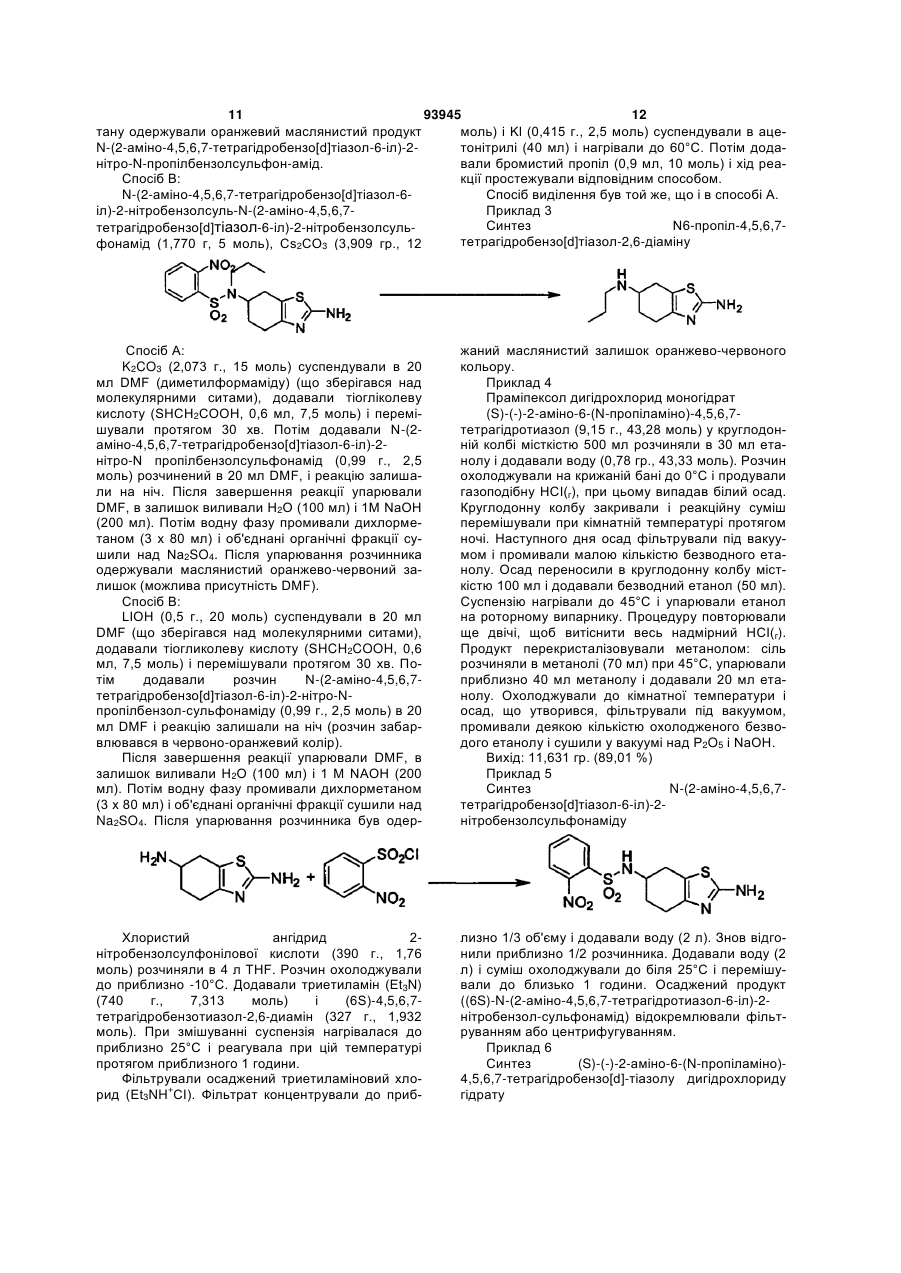
(і) реакцію 4,5,6,7-тетрагідробензо[d]тіазол-2,6-діаміну формули І
 (I)
(I)
з нітро- або галогензаміщеним або незаміщеним арилсулфонілхлоридом формули II
![]() , (II)
, (II)
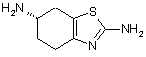
в результаті якої одержують проміжну сульфонамідну сполуку формули III
 , (III)
, (III)
(іі) реакцію проміжної сполуки формули III із сполукою формули IV
![]() , (IV)
, (IV)
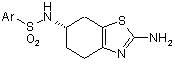
де Х можна вибрати з Сl, Вr, І або інших груп, здатних до нуклеофільного заміщення, такого як OTs, OMs, в результаті якої одержують проміжну сполуку формули V
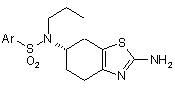 , (V)
, (V)
(iii) перетворення проміжної сполуки формули V в праміпексол за допомогою використання селективного реагенту для відщеплення сульфонільної захисної групи;
(iv) необов'язкове перетворення праміпексолу у фармацевтично прийнятну сіль.
2. Спосіб за п. 1, який відрізняється тим, що реакцію між проміжними продуктами І і II проводять у присутності основи.
3. Спосіб за п. 1, який відрізняється тим, що як нітрозаміщений арилсульфонілхлорид формули II застосовують моно-, ди- або тринітрозаміщений бензолсульфонілхлорид.
4. Спосіб за п. 1, який відрізняється тим, що як нітрозаміщений арилсульфонілхлорид формули II застосовують 2-нітробензолсульфонілхлорид.
5. Спосіб за п. 1, який відрізняється тим, що реакцію між проміжною сполукою III і сполукою формули IV проводять у присутності основи.
6. Спосіб за п. 1, який відрізняється тим, що як селективний реагент застосовують тіол у присутності основи або алкоксиду.
7. Спосіб за п. 6, який відрізняється тим, що як тіол застосовують тіофенол або тіогліколеву кислоту у присутності основи.
8. Спосіб за п. 1, який відрізняється тим, що спосіб є безперервним процесом.
9. Сполука формули III
, (III)
де Аr є нітро- або галогензаміщений арил.
10. Сполука формули V
, (V)
де Аr є нітро- або галогензаміщений арил.
Текст
1. Спосіб одержання праміпексолу, в якому здійснюють: (і) реакцію 4,5,6,7-тетрагідробензо[d]тіазол-2,6діаміну формули І H2N S C2 2 , (V) де Аr є нітро- або галогензаміщений арил. (19) 1 3 Даний винахід належить до області органічної хімії і відноситься до нового способу синтезу праміпексолу і його фармацевтично прийнятних солей. Праміпексол, переважно його дигідрохлорид моногідрат є сполукою, що застосовується при лікуванні хвороби Паркінсона. Існувала необхідність в новому економічному способі, який би забезпечив високий вихід і в той же час був би промислово придатним. Праміпексол, що має хімічну назву S(-)-2аміно-4,5,6,7-тетрагідро-6пропіламінобензотиазол, належить до сімейства біоізостерів гідрокситетраліну і ерголіну, які розроблялися як антагоністи допамінових рецепторів. Праміпексол застосовують для лікування ознак і симптомів хвороби Паркінсона і синдрому неспокійних ніг. Праміпексол є об'єктом Патенту EP 186087, що описує деякі можливі способи його синтезу: а) реакцією циклогексанону формули 93945 4 де щонайменше один з R1', R2', R3', R4' є захисною аміногрупою; г) відновленням ацильної або фенілацильної групи із застосуванням гідриду металу в розчиннику, унаслідок чого утворюється відповідна алкільна або фенілалкильна група; д) реакцією сполуки формули де щонайменше один із замінників є атомом водню, із сполукою формули R5-Z, де R5 є алкілом і Z є групою, здатною до нуклеофільного заміщення. Також є ряд патентів, що описують розділення рацематів праміпексолу, наприклад, EP 1318987, де описується розділення діастереомірних солей кристалізацією, WO 2006/003471, де описується розділення хиральною хроматографією, WO 2006/012276, де описується розділення рідинною хроматографією, WO 2006/120268, де описується розділення виннокислих похідних. Способи одержання праміпексолу відновним амінуванням проміжної діамін сполуки формули де X є галогеном, з тіосечовиною формули пи; де R1 і R2 можуть бути H, алкіл, аліл і інші груб) реакцією циклогексанону формули з формамідин дисульфідом; в) відщепленням захисної групи від сполуки формули описуються в WO 2005/092871, WO 2006/003677, WO 2006/070349, WO 2006/097014. Спосіб за даним винаходом є новим винахідницьким підходом до вирішення аспектів синтезу, що пропонує високу швидкість перетворення проміжних сполук, і спосіб, де небажані побічні продукти не утворюються. Перевага способу згідно даного винаходу полягає в тому, що на всіх стадіях зберігається висока оптична чистота і рацемізація на будь-якій стадії не відбувається. Об'єктом даного винаходу є спосіб одержання праміпексолу, що включає процес синтезу, представлений на схемі 1 5 Спосіб згідно даного винаходу включає: (i) реакцію 4,5,6,7-тетрагідробензо[d]тіазол2,6-діаміну формули І з нітро- або галогензаміщеним або незаміщеним арилсульфонілхлоридом формули II, в результаті якої одержують проміжну сульфонамідну сполуку формули III; (ii) реакцію проміжної сполуки формули III із сполукою формули IV, де X можна вибрати з Cl, Br, І або інших груп, здатних до нуклеофільного заміщення, таких як OTs, OMs, в результаті якого одержують проміжну сполуку формули V; (iii) перетворення проміжної сполуки формули V з селективним реагентом з метою відщеплення сульфонільної захисної групи від праміпексолу формули VI; (iv) необов'язкове перетворення праміпексолу у фармацевтично прийнятну сіль. Спосіб може здійснюватися безперервно або періодично. Реакція сполуки І із сполукою Il проводиться в органічному розчиннику у присутності неорганічної або органічної основи. Органічну основу можна вибрати з групи третинних амінів, таких як триметиламін, триетиламін, діізопропілетиламін, три-нпропіламін, три-н-бутиламін, N,N,N',N'-тетраметил1,3-пропандиамін, N-метилморфолін, N,N'диметилпіперазін, 1,4-диазабіцикло[2.2.2]октан, 1,5-диазабіцикло[4.3.0]-5-нонен; 1,8диазабіцикло[5.4.0]-7-ундецен; гетероциклічних ароматичних амінів, які вибрані з піридинів, пірролідинів, таких як N-метилпірролідин, диметилпірролідин, 4-диметиламінопіридин, коллідин, лютидин; переважно триетиламін. Неорганічну основу можна вибрати з гідроксидів лужних і лужноземельних металів, таких як NAOH, КОН, LIOH, Mg(OH)2, карбонатів, гідрокарбонатів, четвертинних гідроксидів амонію, таких як тетрабутиламмоній гідроксид, тетраетиламмоній гідроксид, або алкоксидів. Органічний розчинник можна вибрати з кетону, таких як ацетон, метилетилкетон; простих ефірів, таких як THF (тетрагідрофуран), діетиловий ефір; нітрилу, таких як ацетонітрил; амідів, таких як диметилацетамід, диметилформамід; пірролідонів, таких як н-метилпірролідон; алканів, що галоїдуються, таких як метиленхлорид, хлороформ. Переважно реакцію проводять таким чином, що нітрозаміщений арилсульфонілхлорид розчиняють в розчиннику, охолодженому до температу 93945 Схема 1 6 ри від -20°C до 10°С, переважно нижче 0°С, потім додають основу і сполуку формули І і поступово підвищують температуру реакції до кімнатної температури. Як сполуку формули Il застосовують арилсульфонілхлорид, переважно нітро- або галогензаміщений арилсульфонілхлорид. Як нітрозаміщений арилсульфонілхлорид можна застосовувати моно-, ди- або тринітрозаміщені бензолсульфонілхлориди, такі як 2нітробензолсульфонілхлорид, 4нітробензолсульфонілхлорид, 2,4-, 2,3-, 2,5-, 2,6динітробензолсульфонілхлорид, 2,4,6тринітросульфонілхлорид, переважно 2нітробензолсульфонілхлорид. Як галогензаміщені арилсульфонілхлориди можна застосовувати, 4Br-, 4-СІ- і 4-F-бензолсульфонілхлориди. Виділення проміжної сполуки формули III проводять таким чином, що після знесолювання розчин упарюють насухо і залишок заливають водою, і після перемішування випадає жовтий осад, який необов'язково можна промити водою. Альтернативно, розчин можна упарити до приблизно 1/3 об'єму, після чого можна додати воду. Реакцію проміжної сполуки III з проміжною сполукою IV проводять в органічному розчиннику у присутності основи, необов'язково у присутності каталізатора, такого як Kl, в результаті якої одержують проміжну сполуку V. Як основи можна застосовувати органічну або неорганічну основу. Як органічні основи можна застосовувати первинні, вторинні і третинні аміни, такі як триетиламін, диізопропіламін, метиламін, піридин, піролідин, піперазин, піримідин. Неорганічну основу можна вибрати з гідроксидів лужних і лужноземельних металів, таких як NAOH, KOH, LIOH, Mg(OH)2, карбонатів, таких як К2СО3, Cs2CO3 гідрокарбонатів, четвертинних гідроксидів амонію, таких як гідроксид тетрабутиламмонію, гідроксид тетраетиламмонію, або алкоксидів, переважно карбонат калію, карбонат цезію. Як розчинник можна застосовувати кетон, такий як ацетон, метилетилкетон, діетилкетон; прості ефіри, такі як THF, діетиловий ефір, діоксан; нітрил, такі як ацетонітрил, пропіонітрил, спирти, такі як пропанол; аміди, такі як диметилацетамід, диметилформамід; пірролідони, такі як Nметилпірролідон, ароматичні вуглеводні, такі як толуол, ксилол; сульфоксиди, такі як диметилсу 7 93945 8 льфоксид. no.3, р.494-498, J Het Chem, 2000, vol.37, no.2, Як реагент формули IV застосовують бромисp.405-409. тий пропіл, йодистий пропіл, хлористий пропіл, Хід реакції, зазвичай, контролюють застосупропілтозилат, пропілметансульфонат, пропілсуванням аналітичних способів, таких як тонкошарольфат, переважно бромистий пропіл. ва хроматографія HPLC (високоефективна рідинна Виділення проміжної сполуки формули V прохроматографія), газова хроматографія і тому подіводять таким чином, що реакційну суміш знесобне, і реакцію завершує при підтвердженні відсутлюють, після розчин упарюють насухо і залишок ність початкової сполуки. розчиняють в дихлорметані. Даний розчин промиВиділення проводять таким чином, що до сувають водним лужним розчином, насиченим водміші вільної основи праміпексолу і розчинника доним розчином NaCI і сушать над Na2SO4. Після дають у вказаному порядку HCI в газоподібній фоупарювання одержують маслянистий продукт. рмі і розчин газоподібного HCI в розчиннику. Одержану проміжну сполуку V перетворять в Праміпексол кристалізується з розчину у вигляді праміпексол з використанням реагенту для селекдигідрохлориду гідрату. Розчинниками, придатнитивного видалення арилсульфонильної групи. Як ми для виділення, є спирти, переважно метанол, реагент для селективного видалення нітробензолетанол, ізопропанол, 2-бутанол, складні ефіри, сульфонильної групи застосовують тіол у присутпереважно метилацетат, етилацетат, кетон, переності основи або алкоксиду. В якості тіолу можна важно ацетон, трет-бутилметилкетон. Виділення застосовувати тіофенол, тіогліколеву кислоту, мепраміпексолу у формі дигідрохлориду можна проркаптоетанол, переважно тіогліколеву кислоту. Як водити переосадженням за допомогою антирозоснову можна застосовувати органічну або неорчинників. Продукт можна виділити ліофілізацією ганічну основу. Як органічні основи можуть застоабо швидким упарюванням водних розчинів прамісовуватися первинні, вторинні і третинні аміни, такі пексолу дигідрохлориду. як триметиламін, триетиламін, диізопропілетилаПраміпексол у вигляді дигідрохлориду гідрату мін, три-н-пропиламін, три-н-бутиламін, N,N,N’,N’перекристалізовують із спиртів або суміші спиртів, тетраметил-1,3-пропандиамін, N-метилморфолін, переважно з метанолу, з додаванням етанолу. N,N'-диметипіперазін, 1,4Праміпексол основа, одержана способом, що диазабіцикло[2.2.2]октан, 1,5-диазабіцикло[4.3.0]є об'єктом даного винаходу, додатково можна пе5-нонен; 1,8-диазабіцикло[5.4.0]-7-ундецен; гетеретворювати в дигідрохлорид способами, описароциклічні ароматичні аміни, вибрані з піридинів, ними в EP 186087, WO 2006/070349, ЕР1318987, пірролідинів, таких як N-метилпірролідин, диметиWO 2006/012276. лпірролідин, 4-диметиламінопіридин, коллідин, Сполука, одержана способом, що є складовим лютидин; переважно триетиламін. Як неорганічні об'єкт даного винаходу, можна застосовувати як основи можна застосовувати гідроксиди, переважтерапевтично активний інгредієнт, який разом з но гідроксид літію, карбонати, гідрокарбонати, алтерапевтично прийнятним носієм можна застосукоксиди лужних або лужноземельних металів, певати як лікарський засіб. Воно може застосовувареважно карбонат калію, карбонат цезію. Як тися для лікування симптомів, пов'язаних з хвороалкоксиду можна застосовувати алкоксиди, такі як бою Паркінсона, а також для лікування синдрому метоксиди, етоксиди, пропоксиди, переважно менеспокійних ніг. токсиди лужних або лужноземельних металів. Сполука, одержана згідно даного винаходу, Реакцію можна проводити в органічному розможна включати до складу фармацевтичної комчиннику, такому як кетон, такому як ацетон, метил позиції, яка на додаток до активного інгредієнта етил кетон, діетилкетон; прості ефіри, такі як THF, також містить фармацевтично прийнятні ад'ювандіетиловий ефір, метилетиловий ефір, діоксан, ти, які, як правило, відомі рядовим фахівцям у фадиметоксиетан; нітрил, такі як ацетонітрил, пропірмації, описані, наприклад, в EP 1318987, WO онітрил; аміди, такі як диметилацетамід, димети2006/015944, WO 2006/015942, WO 03/053402, лформамід; пірролідони, такі як NIPCOM000145762D. У цих композиціях середній метилпірролідон, ароматичні вуглеводні, такі як розмір частинок активного інгредієнта може бути толуол, ксилол, сульфоксиди, такі як диметилсувід 5 до 500 мікрометрів, переважно від 10 до 300 льфоксид, галогеналкани. мікрометрів, переважніше від 50 до 170 мікрометРеакція протікає при температурі реакційної рів. Розподіл частинок по розмірах можна визнасуміші від кімнатної до температури флегми, печати способом розсіяння при лазерному збудженреважно при температурі 40-70°С. ні, застосовуючи, наприклад, апарат Malvern Переважний варіант втілення реакції згідно Mastersizer 2000 Apparatus з синтетичним ізопаданого способу полягає в тому, що реагент для рафіном lsopar L як середовище розбавлення. видалення захисної групи заздалегідь активують Фармацевтичну композицію можна одержати таким чином, що його (реагент) перемішують распособами, відомими рядовому фахівцеві у фарзом з основою в продовж від 10 хвилин до 1 годимації, такими як пряме ущільнення, сухе гранулюни, переважно протягом 30 хвилин, до додавання вання або вологе гранулювання. проміжної сполуки формули V. Хід реакцій і чистоту продуктів можна контроЯк початкову сполуку І можна застосовувати лювати наступними двома способами: продукт, що одержується за будь-яким способом, а) хроматографічна чистота відомим з, наприклад, EP 186087, WO Високоефективна рідинна хроматографія, 2004/041797, EP 1731514. Початкову сполуку можHPLC, спосіб процентних відношень на також одержати промисловим способом, розкКолонка: ритим в CN 1772744, J Med Chem, 1987, vol.30, Gemini С-18 110А 150 мм х 4,6 мм, розмір час 9 93945 10 тинок 5 мкм (альтернативно можна застосовувати - буферний розчин А: 0,01 M КН2РO4 (рН = 10) Alltima С-18, 250 мм х 4.6 мм, розмір частинок 5 - буферний розчин В: CH3CN : CH3OH : H2O = мкм) 600 : 300: 100 (рН=10) Рухома фаза: - градієнт: Час (хвилини) 0,0 18 24 40 41 Опорний час %А 80 50 30 30 80 %В 20 50 70 70 20 7 хвилини Потік: приблизно 1,0 мл/хв Детектор: УФ, довжина хвилі: 263 Інжекція: 20 мкл Температура: 25°С (кімнатна температура) б) енантіомірна чистота Високоефективна рідинна хроматографія, HPLC Метод вимірювання: % площі Колонка: Chiralpack IA 250 мм х 4,6 мм, розмір частинок 5 мкм Рухома фаза: гексан/етанол/ДЕА (діетиламін) відносно 75/25/0,05 Температура колонки: 25°C Потік 0,8 мл/хв Детектор: УФ, 263 нм Інжекція: 20 мкл Альтернативно можна застосовувати наступні хроматографічні умови: Колонка: Chiralpack AD-H 250 мм х 4,6 мм, розмір частинок 5 мкм Рухома фаза: етанол/гексан/ДЕА відносно 90/10/0,1 Температура колонки: 25°C Потік: 0,5 мл/хв Детектор: УФ, 263 нм Інжекція: 5 мкл Даний винахід ілюструється наступними прикладами, що не обмежують даний винахід. Приклади Приклад 1 Синтез N-(2-аміно-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2нітробензолсульфонаміду Хлорангідрид орто-нітробензолсульфонілової кислоти (8,865 гр., 40 моль) розчиняли в 100 мл THF і при перемішуванні охолоджували до -10°C (лід + сіль). Потім додавали спочатку 3 еквіваленти триетиламіну (Et3N) (120 моль, 16,8 мл) і потім також 1,1 еквівалент 4,5,6,7тетрагідробензо[d]тіазол-2,6-діаміну (7,605 гр., 45 моль). Суспензію, що утворилася, при перемішуванні поступово нагрівали до кімнатної температури і витримували до закінчення реакції. В ході реакції разом з розчинним продуктом утворювався нерозчинний в THF Et3NH+CI1 який після закінчення реакції був відфільтрований під вакуумом, і реакційна суміш упарювалася насухо на роторному випарнику. У залишок виливали H2O (300 мм), внаслідок чого на дні круглодонної колби утворилася оранжева в'язка рідина. Після розтирання скляною паличкою сформувався жовтий осад (N(2-аміно-4,5,6,7-тетрагідробензо[d]тіазол-6-іл)-2нітробензолсульфонамід). Осад фільтрували і промивали 100 мл холодного етилового ефіру і сушили. Вихід реакції склав 95%. Приклад 2 Синтез N-(2-аміно-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2-нітро-nпропілбензолсульфонаміду Спосіб А: N-(2-аміно-4,5,6,7-тетрагідробензо[d]тіазол-6іл)-2-нітробензол-N-(2-аміно-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2-нітробензолсульфонамід (1,770 гр., 5 моль) K2CO3 (2,856 гр., 20 моль) суспендували в ацетонітрилі (40 мл) і при перемішуванні нагрівали до 60°С. Потім додавали бромистий пропіл (1,65 мл, 18 моль) і реакцію за лишали на ніч (хід реакції простежували відповідним способом). Після завершення реакції присутній осад фільтрували під вакуумом. Реакційну суміш упарювали насухо, і залишок розчиняли в дихлорметані (150 мл). Органічну фазу промивали 1 M NAOH (3 х 50 мл), насиченим розчином NaCI (2 х 50 мл) і сушили над Na2SO4. Після упарювання дихлорме 11 93945 12 тану одержували оранжевий маслянистий продукт моль) і Kl (0,415 г., 2,5 моль) суспендували в ацеN-(2-аміно-4,5,6,7-тeтpaгiдpoбeнзo[d]тiaзoл-6-iл)-2тонітрилі (40 мл) і нагрівали до 60°С. Потім доданiтpo-N-пpoпiлбeнзoлcyльфoн-aмiд. вали бромистий пропіл (0,9 мл, 10 моль) і хід реаСпосіб В: кції простежували відповідним способом. N-(2-aмiнo-4,5,6,7-тeтpaгiдpoбeнзo[d]тiaзoл-6Спосіб виділення був той же, що і в способі А. iл)-2-нiтpoбeнзoлcyль-N-(2-аміно-4,5,6,7Приклад 3 Синтез N6-пропіл-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2-нітробензолсультетрагідробензо[d]тіазол-2,6-діаміну фонамід (1,770 г, 5 моль), Cs2СО3 (3,909 гр., 12 Спосіб А: K2CO3 (2,073 г., 15 моль) суспендували в 20 мл DMF (диметилформаміду) (що зберігався над молекулярними ситами), додавали тіогліколеву кислоту (SHCH2COOH, 0,6 мл, 7,5 моль) і перемішували протягом 30 хв. Потім додавали N-(2аміно-4,5,6,7-тетрагідробензо[d]тіазол-6-іл)-2нітро-N пропілбензолсульфонамід (0,99 г., 2,5 моль) розчинений в 20 мл DMF, і реакцію залишали на ніч. Після завершення реакції упарювали DMF, в залишок виливали Н2O (100 мл) і 1М NaOH (200 мл). Потім водну фазу промивали дихлорметаном (3 х 80 мл) і об'єднані органічні фракції сушили над Na2SO4. Після упарювання розчинника одержували маслянистий оранжево-червоний залишок (можлива присутність DMF). Спосіб В: LIOH (0,5 г., 20 моль) суспендували в 20 мл DMF (що зберігався над молекулярними ситами), додавали тіогликолеву кислоту (SHCH2COOH, 0,6 мл, 7,5 моль) і перемішували протягом 30 хв. Потім додавали розчин N-(2-аміно-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2-нітро-Nпропілбензол-сульфонаміду (0,99 г., 2,5 моль) в 20 мл DMF і реакцію залишали на ніч (розчин забарвлювався в червоно-оранжевий колір). Після завершення реакції упарювали DMF, в залишок виливали H2O (100 мл) і 1 M NAOH (200 мл). Потім водну фазу промивали дихлорметаном (3 х 80 мл) і об'єднані органічні фракції сушили над Na2SO4. Після упарювання розчинника був одер жаний маслянистий залишок оранжево-червоного кольору. Приклад 4 Праміпексол дигідрохлорид моногідрат (S)-(-)-2-аміно-6-(N-пропіламіно)-4,5,6,7тетрагідротиазол (9,15 г., 43,28 моль) у круглодонній колбі місткістю 500 мл розчиняли в 30 мл етанолу і додавали воду (0,78 гр., 43,33 моль). Розчин охолоджували на крижаній бані до 0°С і продували газоподібну НСІ(г), при цьому випадав білий осад. Круглодонну колбу закривали і реакційну суміш перемішували при кімнатній температурі протягом ночі. Наступного дня осад фільтрували під вакуумом і промивали малою кількістю безводного етанолу. Осад переносили в круглодонну колбу місткістю 100 мл і додавали безводний етанол (50 мл). Суспензію нагрівали до 45°C і упарювали етанол на роторному випарнику. Процедуру повторювали ще двічі, щоб витіснити весь надмірний НСІ(г). Продукт перекристалізовували метанолом: сіль розчиняли в метанолі (70 мл) при 45°С, упарювали приблизно 40 мл метанолу і додавали 20 мл етанолу. Охолоджували до кімнатної температури і осад, що утворився, фільтрували під вакуумом, промивали деякою кількістю охолодженого безводого етанолу і сушили у вакуумі над P2O5 і NaOH. Вихід: 11,631 гр. (89,01 %) Приклад 5 Синтез N-(2-аміно-4,5,6,7тетрагідробензо[d]тіазол-6-іл)-2нітробензолсульфонаміду Хлористий ангідрид 2нітробензолсулфонілової кислоти (390 г., 1,76 моль) розчиняли в 4 л THF. Розчин охолоджували до приблизно -10°С. Додавали триетиламін (Et3N) (740 г., 7,313 моль) і (6S)-4,5,6,7тетрагідробензотиазол-2,6-диамін (327 г., 1,932 моль). При змішуванні суспензія нагрівалася до приблизно 25°С і реагувала при цій температурі протягом приблизного 1 години. Фільтрували осаджений триетиламіновий хлорид (Et3NH+CI). Фільтрат концентрували до приб лизно 1/3 об'єму і додавали воду (2 л). Знов відгонили приблизно 1/2 розчинника. Додавали воду (2 л) і суміш охолоджували до біля 25°C і перемішували до близько 1 години. Осаджений продукт ((6S)-N-(2-аміно-4,5,6,7-тетрагідротиазол-6-іл)-2нітробензол-сульфонамід) відокремлювали фільтруванням або центрифугуванням. Приклад 6 Синтез (S)-(-)-2-аміно-6-(N-пропіламіно)4,5,6,7-тетрагідробензо[d]-тіазолу дигідрохлориду гідрату 13 93945 У 4,1 л ацетонітрилу суспендували карбонат калію (1890 гр., 13,675 моль), (6S)-N-(2-аміно4,5,6,7-тетрагідробензотиазол-6-іл)-2-нітробензолсульфонамід (590 гр., 1,665 моль) і бромистий пропіл (1,09 л, 12 моль). Суміш нагрівали при перемішуванні до приблизно 60°C і перемішували при цій температурі в продовж близько 12 годин. Суміш охолоджували до біля 25°С. Бромід калію витягували фільтруванням. Розчин концентрували до приблизно 1/4 об'єму (не перевищуючи 60°C) і охолоджували до кімнатної температури. Додавали хлористий метилен (2 л) і водний 1 M NaOH розчин (2,43 л) і суміш перемішували приблизно 30 хвилин. Фази розділяли і водну фазу знов промивали дихлорметаном (1,46 л). Органічні фази збирали і концентрували до близько 1/10 об'єму. Додавали 0,83 л етанолу і розчин концентрували до 1/10 об'єму. Додавали 3,35 л етанолу і етанольний розчин (6S)-1N-(2-аміно-4,5,6,7тетрагідробензотиазол-6-іл)-2-нітро-Нпропілбензолсульфонаміду зберігали для подальшої реакції. Етанол (2,35 л) і LIOH (288 г., 12 моль) завантажували в реактор і суспензію охолоджували до 0-5°С. В продовж близько 30 хвилин додавали тіогліколеву кислоту (SHCH2COOH) (720 г., 7.816 моль) (температура не повинна перевищувати 25°С). Суспензію нагрівали до біля 25°С і перемішували в продовж близько 45 хвилин. Додавали етанольний розчин (6S)-1М-(2-аміно-4,5,6,7 Комп’ютерна верстка А. Крулевський 14 тетрагідробензотіазол-6-іл)-2-нітро-Нпропілбензолсульфонаміду. Повітря в реакторі заміщали азотом. Суміш нагрівали до приблизно 50°C і змішували при цій температурі в продовж близько 4 годин. Суміш охолоджували до приблизно 25°C і фільтрували. Фільтрат концентрували при 40°C до приблизно 1/4 об'єму і охолоджували до температури навколишнього середовища. Додавали дихлорметан (4,23 л) і водний 1 M розчин NaOH (2,53 л) і суміш перемішували в продовж близько 30 хвилин. Фази розділяли і водну фазу знов промивали 4,23 л дихлорметану. Органічні фази, що містять праміпексол, збирали і концентрували до приблизно 1/4 об'єму. Додавали 5 л етанолу. До етанольного розчину праміпексолу додавали воду (27,6 мл, 1,53 моль) і розчин охолоджували до приблизно -10°С. У розчин вводили газоподібні НСІ(г) (200 гр). Під час додавання газоподібного НСІ(г) температура розчину, і пізніше суспензії, не повинна перевищувати 25°C. Після додавання суспензію нагрівали до приблизно 40°C І концентрували до 2/3 об'єму. Додавали 2,65 л етанолу і суспензію концентрували до 1/2 об'єму. Знов додавали 3,5 л етанолу і концентрували суспензію до 1/2 об'єму. Розчин охолоджували до приблизно -15°С і продукт виділяли фільтруванням. Продукт сушили на повітрі при 25°С і остаточно при 40°C. Підписне Тираж 23 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for synthesis of pramipexole and pharmaceutically acceptable salts thereof
Автори англійськоюZivec, Matej, Gobec, Stanislav, Anzic, Borut, Zupet, Rok, Kolenc, Ivanka
Назва патенту російськоюСпособ синтеза прамипексола и его фармацевтически приемлемых солей
Автори російськоюЗивец Матей, Гобец Станислав, Анзиц Борут, Зупет Рок, Коленц Иванка
МПК / Мітки
МПК: C07D 277/82
Мітки: праміпексолу, спосіб, солей, фармацевтично, прийнятних, синтезу
Код посилання
<a href="https://ua.patents.su/7-93945-sposib-sintezu-pramipeksolu-i-jjogo-farmacevtichno-prijjnyatnikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб синтезу праміпексолу і його фармацевтично прийнятних солей</a>
Спосіб промислового синтезу периндоприлу і його фармацевтично прийнятних солей

Номер патенту: 75070
Опубліковано: 15.03.2006
Автори: Ланглуа Паскаль, Турбе Юге
МПК: C07D 209/42, C07K 5/06
Мітки: промислового, фармацевтично, солей, спосіб, периндоприлу, синтезу, прийнятних
Формула / Реферат:
1. Спосіб промислового синтезу периндоприлу формули (I) (І)і його фармацевтично прийнятних солей, який відрізняється тим, що бензиловий ефір формули (IV): ,(IV)де Вn представляє бензильну групу,реагує зі сполукою формули (V):
Спосіб синтезу периндоприлу і його фармацевтично прийнятних солей

Номер патенту: 83269
Опубліковано: 25.06.2008
Автори: Ланглуа Паскаль, Дюбюффе Тьєррі
МПК: C07C 229/36, C07C 309/00, C07K 5/06
Мітки: периндоприлу, фармацевтично, спосіб, синтезу, прийнятних, солей
Формула / Реферат:
1. Спосіб синтезу периндоприлу формули (І):і його фармацевтично прийнятних солей, який відрізняється тим, що сполуку формули (II), конфігурації (S):в якій R являє собою атом водню або захисну групу кислотної функції, піддають реакції зі сполукою...
Спосіб синтезу периндоприлу і його фармацевтично прийнятних солей

Номер патенту: 80905
Опубліковано: 12.11.2007
Автори: Дюбюффе Тьєррі, Ланглуа Паскаль, Фюжьє Клод
МПК: C07D 209/42, C07K 5/06
Мітки: спосіб, периндоприлу, синтезу, фармацевтично, солей, прийнятних
Формула / Реферат:
1. Спосіб синтезу сполуки формули (І): (I)та її фармацевтично прийнятних солей, який відрізняється тим, що сполуку формули (II): (II),в якій R являє собою атом водню або бензил, або лінійну, або розгалужену (С1-С6)алкільну групу,піддають реакції зі...
Спосіб синтезу периндоприлу і його фармацевтично прийнятних солей

Номер патенту: 84898
Опубліковано: 10.12.2008
Автори: Лекув Жан-П'єр, Дюбюффе Тьєррі
МПК: C07K 5/06
Мітки: прийнятних, спосіб, синтезу, солей, фармацевтично, периндоприлу
Формула / Реферат:
1. Спосіб промислового синтезу периндоприлу формули (І)і його фармацевтично прийнятних солей, який відрізняється тим, що бензиловий ефір формули (IIa) або (IIb):або адитивну сіль ефіру формули (IIа) або (IIb) з мінеральною кислотою або органічною кислотою піддають реакції зі...
Спосіб синтезу (1s)-4,5-диметокси-1-(метиламінометил)бензоциклобутану і його адитивних солей і застосування у синтезі івабрадину і його адитивних солей з фармацевтично прийнятною кислотою

Номер патенту: 79482
Опубліковано: 25.06.2007
Автори: Лекув Жан-П'єр, Гонсалез Бланко Ісаак, Лерестіф Жан-Мішель, Брігот Даніель
МПК: C07C 217/56, C07C 217/58, A61P 9/06, C07B 61/00, C07C 213/00, A61K 31/55, C07D 223/00, A61P 9/10, C07B 57/00
Мітки: спосіб, застосування, івабрадину, 1s)-4,5-диметокси-1-(метиламінометил)бензоциклобутану, синтезу, фармацевтично, прийнятною, адитивних, солей, кислотою, синтезі
Формула / Реферат:
1. Спосіб розщеплення аміну формули (IV): (IV)шляхом реакції з оптично активною двокислотною сполукою, з наступним фільтруванням, або фільтруванням відсмоктуванням, одержаної таким чином суспензії, з тим, щоб одержати сполуку формули (VII): (VII),яку піддають реакції з...
Попередній патент: Комбінований агрегат для догляду за рослинами
Наступний патент: Фазована антенна решітка
Випадковий патент: Витратний порошковий електрод для електрошлакової технології