(s)-альфа-феніл-2-піридинетанамін або його сіль з неорганічною кислотою, які мають протисудомну активність, спосіб їх отримання, лікарський препарат на їх основі та спосіб лікування судом
Номер патенту: 29437
Опубліковано: 15.11.2000
Автори: Бейлестра Майкл, Мюррей Роберт Джон, Гріффіт Роналд Конрад







Текст
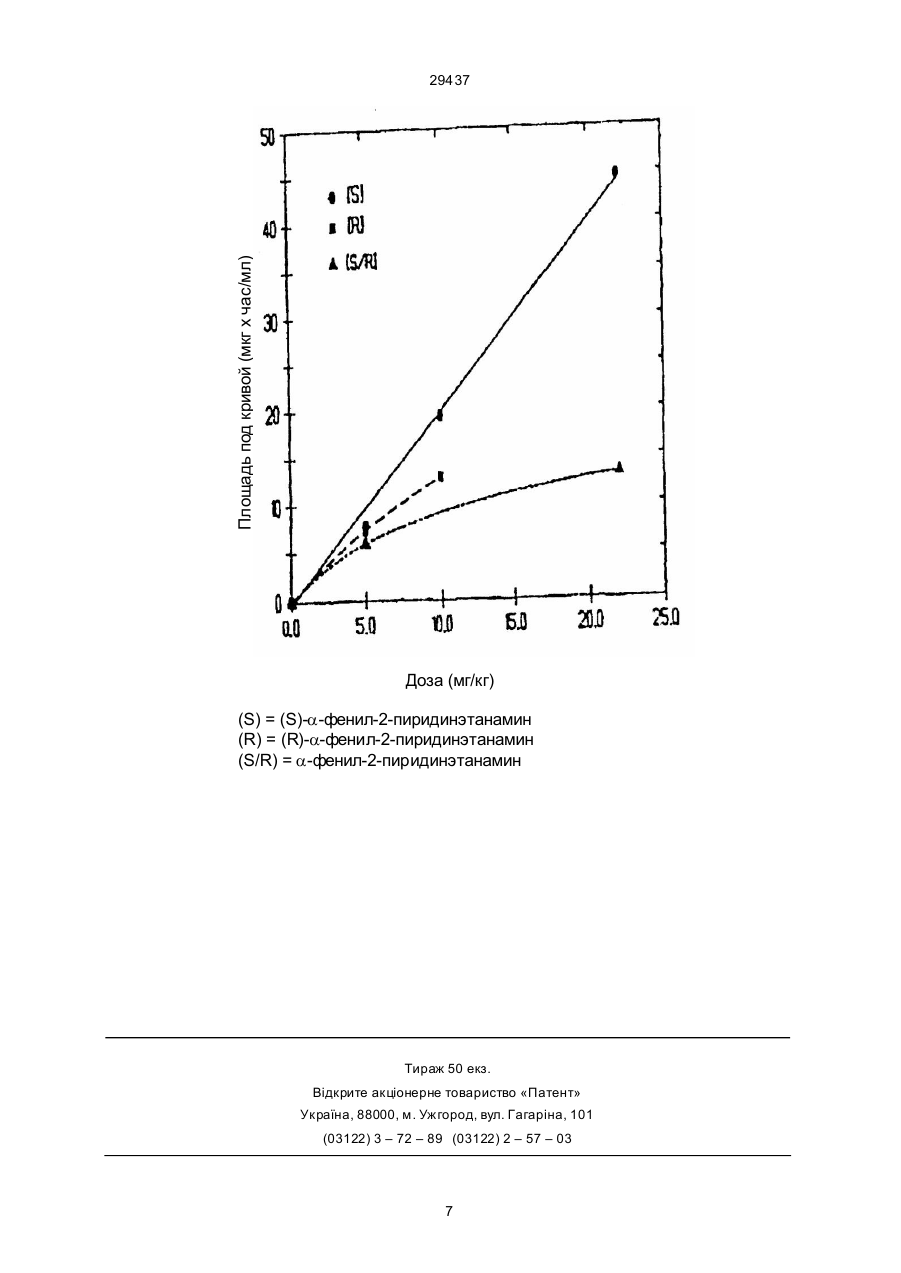
1. (S)-a-фенил-2-пиридинэтанамин, отличающийся тем, что является более чем на 90% энантиочистым, или его соль с неорганической кислотой, обладающие противосудорожной активностью. 2. (S)- a-фенил-2-пиридинэтанамин по п. 1, отличающийся тем, что является более чем на 99% энантиочистым, или его соль с неорганической кислотой. 3. (S)-a-фе нил-2-пиридинэтанамин по п. 2 или его соль с органической кислотой. 4. Лекарственный препарат, обладающий противосудорожным действием, содержащий активный ингредиент в смеси с фармацевтически приемлемыми добавками, разбавителями или носителями, отличающийся тем, что в качестве активного ингредиента он содержит эффективное количество (S)- a-фе нил-2-пиридинэтанамина более чем на 90% энантиочистого или его соли с неорганической кислотой. 5. Лекарственный препарат по п. 4, отличающийся тем, что в качестве активного ингредиента он содержит эффективное количество (S)- aфе нил-2-пиридинэтанамина более чем на 99% C2 (54) (S)-АЛЬФА-ФЕНIЛ-2-ПIРИДИНЕТАНАМIН АБО ЙОГО СIЛЬ З НЕОРГАНIЧНОЮ КИСЛОТОЮ, ЯКI МАЮТЬ ПРОТИСУДОМНУ АКТИВНIСТЬ, СПОСIБ ЇХ ОТРИМАННЯ, ЛIКАРСЬКИЙ ПРЕПАРАТ НА ЇХ ОСНОВI ТА СПОСIБ ЛIКУВАННЯ СУДОМ 29437 В настоящее время для лечения нейродегенеративных расстройств применяются лекарства, не обладающие линейными фармакокинетическими свойствами: при введении пациенту определенной дозы лекарства трудно предсказать соответствующую дозе концентрацию лекарства в его крови. Высказывалось мнение, что идеальным лекарством в этой области будет средство, для которого будет соблюдаться линейное соотноше ние дозы и концентрации в плазме крови пациента, т.е. изменение дозы будет приводить к предсказуемому изменению концентрации лекарства в крови пациента ("Pharmacokinetics of old, new and yet-to-be discovered antiepileptic drugs", R.H. Levy and B.M. Kerr, Epilepsia, vol. 30, Supp 1, S35–S41, 1989). В европейской патентной заявке 356035 описано значительное количество веществ, применимых для лечения нейродегенеративных расстройств, в том числе и a-фенил-2пиридинэтанамин (в настоящем документе мы будем называть его 1-фенил-2-(2-пиридинил)этиламин), соответствующий формуле H мера, а в наиболее предпочти тельном случае (S)-энантиомер является чистым. К производным, приемлемым для фармацевтического применения, относятся соединения – биологические предшественники (пролекарства) (S)-a-фенил-2-пиридинэтанамина, а также – они представляют особый интерес – соли от соединения с кислотами. К приемлемым биологическим предшественникам (+) -a-фенил-2-пиридинэтанамина относятся амидные производные аминогруппы от связи с аминокислотами, в частности, a-аминокислотами, например глицином. Их можно получать стандартными способами, например, амидные производные аминокислот можно готовить способами, описанными в "Ad vanced Organic Chemistry" by J. March, 2 издание, опубликованное McGraw-Hill p 1171 (Органическая химия для специалистов". Дж. Марч). Соли от соединения (S) -a-фенил-2пиридинэтанамина с кислота ми включают соли неорганических кислот (например, дигидрохлорид и дигидробромид) и соли, образованные с органическими кислотами: формат, ацетат, малат, бензоат и фумарат. a-Фенил-2-пиридинэтанамин может быть получен одним из стандартных способов (например, путем добавления аниона 2-пиколина к Nтриметилсилил-бензальдимину). В этом случае (S) -a-фе нил-2-пиридинэтанамин можно получить в результате одной или нескольких реакций aфе нил-2-пиридинэтанамина с хиральной кислотой, при которой выпадает в осадок диастереомерная соль, и одной или нескольких рекристаллизаций. Таким образом, настоящее изобретение предлагает также способ получения соединения, предложенного настоящим изобретением, который предусматривает селективную преципитацию диастереомерной соли, образованной в результате реакции между a-фе нил-2-пиридинэтанамином и хиральной кислотой. В связи с этим стоит упомянуть хи ральные кислоты: D-или Lвинную, и особенно S(+) и R(-) миндальную. Преципитацию можно провести в орга ническом растворите ле, который серьезно не повлияет на ход реакции (например, в этилацетате), при температуре, близкой к комнатной. Соединения, предложенные настоящим изобретением, имеют лекарственное назначение, в частности, пригодны для использования в качестве антиконвульсантов и нейропротектантов при лечении нейродегенеративных расстройств. В числе эти х расстройств инсульт, це ребральная ишемия, церебральный паралич, последствия гипогликемии, эпилепсия, вызванное СПИДом слабоумие, болезнь Альцгеймера, хорея Хантингтона, оливопонтоцеребеллярная атрофия, перинатальная асфиксия, болезнь Паркинсона, гипоксия, повреждения нейронов, связанные со злоупотреблением различными веществами (например, наркоти ками или кокаином), рети нопатия, ши зофрения, ишемические состояния после оста новки сердца или хирур гических операций, интоксикации, повреждения спинного мозга, боковой амиотрофический склероз. NH2 N Это вещество не обладает линейной фармакокинетикой, а его (S)-энантиомер, как ни странно, проявляет линейные фармакокинетические свойства. Итак, предметом настоящего изобретения является (S)-a-фенил-2-пиридинэтанамин, соответствующий формуле H NH2 N практически без примеси (R)-энантиомера, и его производные, приемлемые к фармацевти ческому применению (далее мы будем называть их "соединения/вещества, предложенные настоящим изобретением"). Выражение "практически без примеси (R)энантиомера" означает, что в порции (S)-энантиомера содержится не более 10% по весу (R)энантиомера, т.е. чистота (S)-энантиомера превышает 90%; в предпочтительном случае в смеси содержится не более 1% по весу (R)-энантио 2 29437 Одно из предположений о природе нейродегенеративных расстройств гласит, что дегенерацию нервной ткани вызывает или ускоряет деятельность аминокислот возбуждения, присутствующи х обычно в центральной нервной системе (ЦНС). Медиатором быстрого возбуждения в мозгу млекопитающих счи тается глута мат – эндогенная аминокислота. Глутамат известен также как сильный нейротоксин, способный уничтожать нейроны ЦНС при условиях, связанных с инсуль том и остановкой сердца. Выяснилось, что чувствительность центральных нейронов к гипоксии и ишемии можно снизить при помощи антагонисти ческого действия по отношению к постсинаптическим глутаматным рецепторам. Глутамат является агонистом широкого спектра и действуе т на четыре нейронных рецептора возбуждения. Эти рецепторы названы по аналогии с аминокислотами, возбуждающими их: каинатные (КА), N-метил-D-аспараги натные (NMDA), кискалатные (QUIS) и 2-амин-4-фосфо нобутиратные (АРВ). Глутамат считается смешанным агонистом, способным связываться с рецепто рами всех четырех ти пов и возбуждать их. Та ким образом, средства, блокирующие действие глута мата на эти рецепторы или проявляющие антагонизм по отноше нию к нему, способны предотвратить токсическое поражение нервной системы, связанное с гипоксией или ишемией. В частности, средства, связывающиеся с площадками рецепторов NMDA и блокирующие действие глутамата на эти рецепто ры, полезны для лечения и предуп реждения нейродегенеративных заболеваний. Фармакологическую активность соединений, предложенных настоящим изобретением, можно оценить по результатам опытов, описанных ниже. а) Блокирующее действие по отноше нию к NMDA оценивается по способности снимать у мышей судороги, вызванные внутривенным введением 150 мг/кг NMDA в соответствии с процедурой Czuczwar'a et al. (Neurotransmitters, Seizures and Epilesy III, edited by G. Nistico et al. Raven Press, New York, 1986, pages 235–246 – "Нейромедиаторы, судороги и эпилепсия"). Мышам интраперитонеально вводят испытываемое вещество, а через 30 минут дают N MD A. Действие NMD A проявляется в утрате животными рефлекса направления тонических/клонических судорогах. За проявлением этих признаков наблюдают в течение 60 минут после введения NMDA, затем подсчитывают смертность. d) Антигипоксическое действие удобно проверять на мышах. Группы мышей исследуют через различные промежутки времени после интраперитонеального введения возрастающи х доз испытываемого вещества. За писывают вре мя жизни животных в гипоксической среде (96% азота и 4% кислорода) с регулируе мой температурой. Проводят статическое сравнение между экспериментальной и контрольной (вводят только носитель) группами. Записывают реакцию на дозу и минимальную эффективную до зу испытываемого вещества (A.A. Artu and J.D. Michenfelder, Anaesthesia and Analgesia, 1981, 60, 867). Можно вводить испытываемые вещества и другими способами. e) Антиэпилепти ческое действие оценивается по способности испытываемого ве щества предупреждать тоническое разгибание задних конечностей, вызванное у мышей и крыс максимальным электрошоком, при пероральном, интраперитонеальном, внутривенном или подкожном введении. Процедура разработана отделением эпилепсии NINCDS (R.J. Porter et al. Cleve. Clin. Quarterly 1984, 51, 293). Проводят сравнение с действием известных препаратов диланти на и фе нобарбитала. f) Инсульт с закупоркой четырех сосудов вызывает у крыс глобальную ише мию и представляет собой основной способ оценки эффективности испытываемых соединений по предупреждению поражения уязвимых участков мозга, обычно пирамидальных СА1 – нейронов или гипокампа. Эти участки отвечают за кратковременную память, как у ла бораторных животных, так и у че ловека. В первый день крысам прижигают под анастезией вертебральные арте рии, а каротиды изолируют. На второй день каротиды зажимают на различные промежутки времени. Для уничтожения нейронов СА1 достаточно 10 минут. Зажимы снимают, кровоток восстанавливается, и испытываемое вещество вводят через различные промежутки времени после восста новления кровотока. В период ишемии и выздоровления температуру те ла поддерживают на уровне 37оС. Нейроны СА1 обычно погибают в течение 48–72 часов, поэтому крысы получают лечение на протяжении хотя бы трех дней (интраперитонеально, внутривенно или перорально), а на 7-й день у них извлекают мозг для гисто логического анализа. Определение состояния нейронов СА1 производится двумя способами: считают жизнеспособные нейроны и определяют степень тяжести патологии (W.A. Pulsinelli and A. Buchan, "The NMD A receptor/ion channel: lts importance to in vivo ischemia injury to selectively vulnerable neurons", Pharmacology of Cerebral lschemia, еdited by J. Krieglstein and H. Oberpichler, published by Wissenschaftliche Verlagsgesellschaft, Stuttgart, 1990, p. 169). g) Фокальный инсульт. При этом используются крысы со спонтанной гипертензией, так как у них слабое коллатеральное мозговое кровообраще ние. У них вызывают 2-часовую фокальную ишемию мозга путем пережимания средней мозговой артерии и каротиды, расположенной на той же стороне (под анестезией). Лекарство вводят (обычно интраперитонеально) до b) Антагонистическое действие по отношению к NMDA-рецепто рам можно оценивать in vitro путем измерения активности испытываемого вещества при ингибировании связывания антагониста рецепторов, 10, 11-дигидро-5-метил-5Ндибензо[a,b]- циклогептен-5,10-имина (МК801), с рецепто рами. Постановка опыта описана у Foster and Wong, Br.J.Pharmacol 91, 403–409 (1987). с) Аффинность с NMDA-и гли циновыми рецепторами можно оценить в опытах по связыванию [3H]L-глутамата и [3H]-глицина, поставленных по способу Monaghan and Cotman, PNAS, 83, 7532 (1986) и Watson et al., Neurosci. Res. Comm, 2, 169, (1988). 3 29437 или через различные промежутки времени после пережимания артерий, либо после восстановления кровото ка в течение 2 часов. Через 24 часа после опыта мозг удаляют, замораживают и разрезают на части. Эффективность лекарства по сокраще нию объема инфаркта коры головного мозга подсчитывают при помощи специально оборудованного компьютера (A.M. Buchan, D. Xue and A. Slivka, Stroke, 1992, 23, 273). Токсичность веществ, предложенная настоящим изобретением, определяется следующими способами: а) Опыты по дозировке на основе описанных у N.W. Spurling and P.F. Carey, "A protocol for dose selection in repeat dose toxicity studies", poster presentation 974 at the Society of Toxicology annual meeting, Seattle, USA, 23–27 February 1992 (стенд 974 на ежегодном заседании Общества токсикологов в Сиэтле, США, 23–27, февраля 1992). Крысам вводят ежедневно внутривенно возрастающие дозы испытываемых веществ, пока не нахо дят максимальную повторяемую дозу, при превыше нии которой конвульсии и другие отклонения от нормы неприемлемы. b) Опыты с пово ротной площадкой (L.L. Cougenour, J.R. McLean, and R.B. Parker, Pharmacol Biochem Behav, 1977, 6, 351). Мышам вводят испытываемое вещество и через 30 минут са жают их на небольшую сетчатую платформу, которую поворачивают на угол 180о. Мы шам, которые за 30 секунд не выберутся, засчитывают поражение. При достаточном числе доз и животных ЕД50 (до за, дающая 50% поражений) определяется легко. с) Тест на 28 поведенческих признаков по S. Iwin [Psychopharmacology 1968, 13, 222]. Мышам (по 3 в гр уппе) вводят возрастающие от груп пы к группе дозы испыты ваемого вещества в пределах 25–400 мг/кг и наблюдают 28 симптомов сразу же, че рез 30 минут, че рез 3 и 24 часа после вве дения. d) Тест на фенциклидиновое поведение. "Фенциклидиновое" поведение – это побочные эффекты действия конкурентных и не конкурентных анта гонистов NMD A-рецепторов. Чтобы выяснить, оказывает ли испытываемое вещество такое побочное действие, крысам вводят его перорально (в дозах кратных ED50 (50% антиэпилептического действия) из опыта с электрошоком). Затем животных помещают в индивидуальные камеры из прозрачного пластика и наблюдают за их поведением в течение 4 часов в ожидании проявления одного из 5 характерных признаков: повышенной активности, атаксии, кружения, качания головой и ретропульсии. В экспериментальную гр уппу взяли 5 крыс, поведение которых сравнивали с поведением контрольных животных, получавши х фенциклидин. Общий счет встречаемости составил 25, т.е. 5 крыс по 5 признаков. Фенциклидин дает такой счет при дозе, в 10 раз большей ED50 (W. Kock, J.H. Woods. P. Ornstein, 1987, Phychopharmacology, 91, 279). e) Опыт с узкой доской для измерения степени повреждения нервной системы у крыс [G.E. Garske et al., Epilepsy Research, 1991, 9, 161]. Крыс сажают на узкую доску (1,25 см шириной, на высоте 40 см над столом), которая начинается в хорошо освещенной входной камере, прохо дит через постепенно темнеющий коридор и кончается в темной выходной камере. Длина доски составляет 63 см. Крыса считается ненормальной, если не может пройти по доске. Здесь учитываются две ха рактерные особенности поведения крыс: они боятся высоты и любят темноту. Линейную фармакокинетику можно обнаружить у крыс путем оценки площа ди под кривыми зависимости концентрации лекарства в плазме крови от времени, полученными при однократном введении испытываемого вещества внутривенно возрастающи ми дозами (Smith et. al., Xenobiotica, 20, 1187 – 1199, 1990). Кровь берут катетером из яремной вены в разное время в течение 24 часов. Плазму отделяют центрифугирова нием и определяют концентрацию испытываемого ве щества при помощи жидкостной хроматографии высокого давления с ультрафиолетовым облучением. Строят график по значениям концентрации во времени для каждой дозы и вычисляют площадь под графи ком. Если средство обладает линейной фармакокинети кой, площадь под графи ком будет прямо пропорциональна дозе. Если линейная фармакокинети ка проявляется у крыс, значит, она будет проявляться у человека (Leander et al., Epilepsia, 33, 696–704, 1992, at p 703). Помимо этого, настоящим изобрете нием предложен способ лечения нейродегенеративного расстройства, предусматривающий введение пациенту те рапевтически эффективного количества соединения, предложенного настоящим изобретением. Особый интерес представляет способ, при котором введенная доза прямо пропорциональна концентрации лекарственного вещества в плазме крови. В упо мянутых вы ше случаях до зы будут меняться в зависимости от состава соединения, способа введения и схемы лечения. Однако, как правило, результат оказывается удовлетворительным, если вво дить соединения, предложенные настоящим изобретением, животным в дневной дозировке от 0,1 мг до 20 мг на килограмм животного веса, предпочтительно, дробными дозами от 1 до 4 раз в день или в виде препаратов длительного действия. Людям следует вводить от 5 до 1400 мг в день, предпочтительно, от 10 до 100 мг, в формах и дозировках, пригодных для перорального вве дения и содержащи х от 2 мг до 1400 мг активного соединения в смеси с жидким или твердым фармацевтическим носителем или разбавите лем. Соединения, предложенные настоящим изобретением, можно использовать в чистом виде и в виде соответствующих ле карственных форм, пригодных для энтерального и парентерального применения. В соответствии с настоящим изобретением, следует применять лекарственный состав, содержащий предпочти тельно не более 80% по весу, а более предпочтительно – не более 50% по весу, соединения, предложенного настоящим изобретением, в смеси с приемлемыми к фармацевтическому применению добавкой, разбавителем или носителем. Примерами разбави телей и носите лей могут служить 4 29437 К раствору рацемической смеси a -фенил2-пиридинэтанамина (свободное основание продукта эта па (а), по луче ние путем нейтрализации водного раство ра продук та этап(а) 25%ным раство ром NaOH в во де и экстраги рования хло рофор мом) (10,96 г; 0 ,0553 моль) в этилацета те (400 мл) до бавили раствор S(+)-миндальной кислоты (8,41 г; 0 ,0553 моль) в эти лацета те (300 мл). По лучен ный преципитат рекристаллизова ли из го ряче го эти лацета та (500 мл) еще три ра за. Соль подще ло чили 25% раство ром NaOH в во де, экстрагирова ли хлорофор мом 3х100 мл, вы суши ли над MgSO 4 , профильтрова ли и сконцентрирова ли в вакууме. Остаток раство рили в эти ла цетате (300 мл) и подкислили насыщенным раствором HCl/EtOAc. По лученное белое твер дое вещество о тфильтрова ли и высуши ли в ва кууме. Получили (-)-a -фе нил-2-пиридинэта намина ди гидрохлорид (5 ,5 г), тем пература плавления 220– 222оС, [a ]D = -87,3 о (c = 1,0 CH 3 OH). Филь трат о т пер во началь ной преципита ции ней трализова ли 25% ра створом Na OH в во де, экстраги рова ли в 2 х250 мл CHCl 3, вы суши ли над MgSO 4, про филь трова ли и сконцентриро вали в ва кууме. Оста ток раство рили в эти ла цета те (500 мл) и до бави ли к это му раство р у раствор R(-)-мин дальной кисло ты (6,5 г, 0 ,043 моль) в эти ла цета те (500 мл). Преципитат о тфиль трова ли и рекристаллизова ли еще три ра за. Соль подще ло чили 25% раство ром NaOH в во де, экстраги рова ли хло рофор мом 3х100 мл, вы суши ли над MgSO 4 , профи льтро вали и скон центрирова ли в ва кууме. Оста ток раство рили в эти лаце та те (300 мл) и подкисли ли на сыщен ным раство ром HCl/EtOAc. По лученное бе лое твер дое ве ще ство отфиль трова ли и высуши ли в ва кууме. Получи ли соеди нение, ука занное в заголовке (3,84 г), тем пература плавления 220– 222оС , [a ]D = +87,1о (c =1 ,1 CH 3OH ). Чистоту энантиомера можно определить путем воздействия на миндальную кислоту или дигидрохлорид энантиочистым (более 99,5%) метилбензил изоцианатом с последующей жидкостной хроматографией высокого давления с нормальной последовательностью фаз и этанолом/гексаном [6:94] в качестве растворителя. Чистота энантиомеров, полученных так, как описано выше, составила более 99,5%. Рентгеновская кристаллография показала, что (+)-энантиомер имеет абсолютную-(S) стереохи мию. для таблеток и драже: лактоза, крахмал, тальк, стеариновая кислота; для капсул: вин ная кислота или лактоза; в растворах для инъекций: вода, спирты, глицерин, растительные масла; для суппозиториев: натуральные или сгущенные масла и воски. При лечении болезни Паркинсона предпочтительно в качестве добавки к веществам, предложенным настоящим изобретением, использовать L-бора. Кроме того, настоящим изобретением предусмотрено использование вещества, предложенного настоящим изобретением, в качестве активного начала при изготовлении медикамента для лечения нейродегенеративного расстройства. Достоинства веществ, предложенных настоящим изобретением, заключаются в том, что они менее токсичны, более эффективны, дольше действуют, имеют более широкий спектр действия, являются более сильными, дают меньше побочных эффектов, легче усваиваются и имеют другие фармакологические свойства, выгодно отличающие их от уже известных и используемых в соответствующей области ве ществ. Изобретение иллюстрируется следующи ми примерами. Пример 1. Приготовление (S)-a-фенил-2пиридинэтанамина дигидрохлорида: а) a-фенил-2-пиридинэтанамин дигидрохлорид. К охлажденному (0оС) раствору бензальдегида (34,24 г, 0,323 моль) в 600 мл тетрагидрофурана добавили лития бис(триметилсилил)-амид (323 мл 1,0 М раствора в тетрагидрофуране, 0,323 моль) по каплям, в течение 30 минут. Смесь перемеши вали при 0оС в течение трех часов. В отдельную колбу с круглым дном, содержащую охлажденный (минут 78оС) раствор 2-пиколина (30,0 г; 0,323 моль) в тетрагидрофуране (600 мл), добавили n-бутиллития (129,2 мл 2,5 М раствора в гексане) понемногу в те чение двадцати минут. Первой смеси позво лили нагреться до 0оС и оставили так еще на 40 минут. Вторую смесь (содержащую соединившийся с литием анион 2пиколина) каплями добавляли в первую в те чение 20 минут. За тем, еще через 30 минут, ледяную баню убрали и смеси позволили нагреться до температуры окружающей среды. Еще че рез час смесь вылили в разделительную во ронку со льдом (1л) и 12 NHCl (200 мл). Водный слой промыли 3х200 мл диэтилового эфи ра (Et2O) и подще лочили 25% NaOH в воде. Водный слой экстрагировали 2х200 мл хлороформа, экстракт высушили над MgSO4, профильтровали и сконцентрировали в вакууме. Остаток растворили в этилацетате (EtOAc) и подкислили насыщенным раствором HCl/EtOAc. Раствор разбавили Et2O, а полученное белое твердое вещество отфильтровали и высуши ли в вакууме. Получили соединение, названное в подзаголовке (43%, 37,08 г), температура плавления 206–208оС. b) a-фенил-2-пиридинэтанамина дигидрохлорид. Пример 2. Соединение из примера 1 обнаруживало активность (ED50) в до зе 3,7 мг/кг при предупреждении тонического разгибания задних конечностей у крыс, вызванного максимальным электрошоком (см. выше), при пероральном введении. Для энантиомера доза RD50 составляла 20,2 мг/кг. Ме тоды проведения экспериментов Фармакокинетика a-фенил-2-пиридинэтанамина (соединения, упоминаемого в ЕР– А– 0356035 под названием 1-фенил-2-(2-пиридинил)этиламин), (S)- a-фенил-2-пиридинэтанами 5 29437 на (соединения по настояще му изобретению и его энантиомера (R) a-фенил-2-пиридинэтанамин) были изучены ( в виде дигидрохлоридов) с помощью способов, описанных на стр. 6 строка 30 по стр. 7 строка 8 настоящей заявки. Результаты Соотношение между концентрацией в плазме и временем выражены в виде кривых по каждой дозе, и площади участков под каждой кривой вычислены. На чертеже приведена диаграмма площа ди под кривой "концентрация в плазме/время" против до зы каждого тестированного соединения. Структурные формулы соединений приведены ниже N N (R) a-Фенил-2-пиридинэтанамин. (S) -a-Фенил-2-пиридинэтанамин проявляет линейную фармакокинетику, поскольку, как показано на чертеже, площадь под кривой "концентрация в плазме/время" для данных доз указанного соединения прямо пропорциональна назначаемой дозе. a-Фенил-2-пиридинэтанамина и (R) -a-фенил-2-пиридинэтанамин не дают линейной фармакокинетики, поскольку площадь под кривой "концентрация в плазме/время" для данных доз указанных соединений не является прямо пропорциональной назначаемой дозе. Поэтому предполагается, что (S) -a-фенил2-пиридинэтанамин будет проявлять линейную фармакокинети ку при использовании для лечения человека, в то время как a-фенил-2пиридинэтанамин и (R) -a-фенил-2-пиридинэтанамин не будут проявлять линейную фармакокинети ку при лечении человека. Линейная фармакокинетика (S) -a-фенил2-пиридинэтанамина является неожиданным и весьма благоприятным свойством соединения, поскольку позволяет прогнозировать концентрацию соединения в головном мозге человека на основе концентраций в плазме, что облегчает дозировку, и поэтому данное соединение более привлекательно в качестве лекарственного препарата, чем a-фе нил-2-пиридинэтанамин или (R) -a-фенил-2-пиридинэтанамин. NH2 a -Фенил-2-пиридинэтанамин. N NH2 NH2 (S) a -Фенил-2-пиридинэтанамин. 6 Площадь под кривой (мкг х час/мл) 29437 Доза (мг/кг) (S) = (S)-a-фенил-2-пиридинэтанамин (R) = (R)-a-фенил-2-пиридинэтанамин (S/R) = a-фенил-2-пиридинэтанамин Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03 7 29437 8
ДивитисяДодаткова інформація
Назва патенту англійською(s)-a-phenyl -2-pyridinetamine or salt thereof with inorganic acid having anticonvulsant activity, a process for preparation thereof, medicament on their basis and a method for treatment of convulsions
Автори англійськоюGriffit Ronald Conrad, Murrey Robert John, Beilestra Michael
Назва патенту російською(s)-a-фенил-2-пиридинетанамин или его соль с неорганической кислотой, которые обладают противосудорожной активностью, способ их получения, лекарственный препарат на их основе и способ лечения судорог
Автори російськоюГриффит Рональд Конрад, Мюррей Роберт Джон, Бейлестра Майкл
МПК / Мітки
МПК: A61K 31/44, C07D 213/38, A61P 25/08
Мітки: судом, отримання, активність, основі, лікарський, кислотою, протисудомну, неорганічною, лікування, спосіб, s)-альфа-феніл-2-піридинетанамін, сіль, мають, препарат
Код посилання
<a href="https://ua.patents.su/8-29437-s-alfa-fenil-2-piridinetanamin-abo-jjogo-sil-z-neorganichnoyu-kislotoyu-yaki-mayut-protisudomnu-aktivnist-sposib-kh-otrimannya-likarskijj-preparat-na-kh-osnovi-ta-sposib-likuvannya.html" target="_blank" rel="follow" title="База патентів України">(s)-альфа-феніл-2-піридинетанамін або його сіль з неорганічною кислотою, які мають протисудомну активність, спосіб їх отримання, лікарський препарат на їх основі та спосіб лікування судом</a>
Похідні піримідину, що мають активність антагоністів ангіотензину іі, спосіб їх отримання та фармацевтична композиція

Номер патенту: 27748
Опубліковано: 16.10.2000
Автори: Еллінгбой Джон Ватсон, Нікайдо Маделен, Бажлі Жак Фрамрос
МПК: C07D 487/04, C07D 471/04
Мітки: іі, фармацевтична, композиція, антагоністів, піримідину, активність, спосіб, похідні, мають, ангіотензину, отримання
Текст:
...Результаты анализа из расчеты на формулу 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 минут после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение, в соответствии с примером 1, при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от А II в среднем на 41 % в...
Похідне тетрагідробензимідазолу або його фармацевтично прийнятна сіль, що проявляють активність антагоніста 5-нт3-рецептора, та фармацевтична композиція на його основі

Номер патенту: 27290
Опубліковано: 15.09.2000
Автори: Кейдзі Міята, Ісао Янагісава, Акіра Матсухіса, Токуо Коіде, Дзунйа Охморі, Такесі Сузукі, Мітсуакі Охта
МПК: C07D 403/08, C07D 403/12, A61K 31/40, A61K 31/415, C01B 7/00, C07D 235/06
Мітки: фармацевтична, проявляють, тетрагідробензимідазолу, прийнятна, фармацевтично, сіль, антагоніста, композиція, похідне, активність, основі, 5-нт3-рецептора
Текст:
...%: C 71,77; H 6,13; N 15,13. Масс-спектр (Е1): m/z; 358 (M+). Примеp 17. Гидрохлорид N-[(4,5,6,7- тетрагидробензимидазол-5-ил)карбонил]-фенотиазина Физико-химические свойства: Т.пл. 268 – 270оС. Элементный анализ для C20H17N3О × HCl × 0,5 х х х H2O: Рассчитано, %: C 61,14; H 4,87; N 10,69; Cl 9,02. Найдено, %: C 61,15; H 4,64; N 10,60; Cl 8,59. Масс-спектр (Е1): m/z; 347 (M+, сво бодное соединение). Пример 18....
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: основі, композиція, одержання, солі, активність, таутомерні, форми, фармацевтична, протизапальну, спосіб, похідні, 3-циклоалкілпропен-2-аміду, мають
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...
Лікарський препарат імунокоригуючої дії на основі клітинної суспензії, спосіб лікування цукрового діабету з використанням цього препарату

Номер патенту: 27048
Опубліковано: 28.02.2000
Автори: Смикодуб Олександр Іванович, Новицька Алла Володимирівна, Єфімов Андрій Семенович
МПК: A61K 35/407, A61K 35/28, A61K 35/54, A61K 35/14, A61K 35/48
Мітки: використанням, лікарський, суспензії, препарат, спосіб, основі, клітинної, діабету, імунокоригуючої, препарату, цього, цукрового, дії, лікування
Формула / Реферат:
1. Лекарственный препарат иммунокоррегирующего действия на основе клеточной суспензии, приготовленной из нативных или криоконсервированных гемопоэтических клеток эмбриональной печени и/или селезенки, отличающийся тем, что количество ядросодержащих клеток в такой суспензии составляет от 5 до 90×106/мл, количество колониеобразующих единиц грануломоноцитарного ряда от 20 до 80×103/мл, количество колониеобразующих единиц бластов - от...
Лікарський препарат імунозамінної дії на основі клітинної суспензії та спосіб лікування синдрому набутого імунодефіциту (віл-інфекції) з використанням цього препарату

Номер патенту: 27055
Опубліковано: 28.02.2000
Автори: Смикодуб Олександр Іванович, Пилипчак Олена Макарівна, Марков Ігор Семенович
МПК: A61K 35/14, A61K 35/407, A61K 35/54, A61K 35/48, A61K 35/28
Мітки: лікарський, основі, спосіб, препарату, синдрому, цього, набутого, препарат, використанням, лікування, клітинної, імунозамінної, імунодефіциту, віл-інфекції, суспензії, дії
Формула / Реферат:
1. Лекарственный препарат иммунозамещающего действия на основе клеточной суспензии, приготовленной из нативных или криоконсервированных гемопоэтических клеток эмбриональной печени и/или селезенки, отличающийся тем, что количество ядросодержащих клеток в такой суспензии составляет от 5 до 200×106/мл, количество колониеобразующих единиц грануломоноцитарного ряда от 20 до 200×103/мл, количество колониеобразующих единиц бластов - от...
Попередній патент: Спосіб визначення водорозчинних та рухомих форм мікроелементів в грунті
Наступний патент: Спосіб збереження або збільшення щільності кістки або інгібування втрати кісткової маси
Випадковий патент: Запірно-пломбуючий пристрій