3-азиридино-антрациклінові похідні, спосіб отримання та фармацевтична композиція
Номер патенту: 43839
Опубліковано: 15.01.2002
Автори: Суарато Антоніо, ГРАНДІ Марія, Барджотті Альберто, Карузо Мікеле, Ріпамонті Маріна







Формула / Реферат
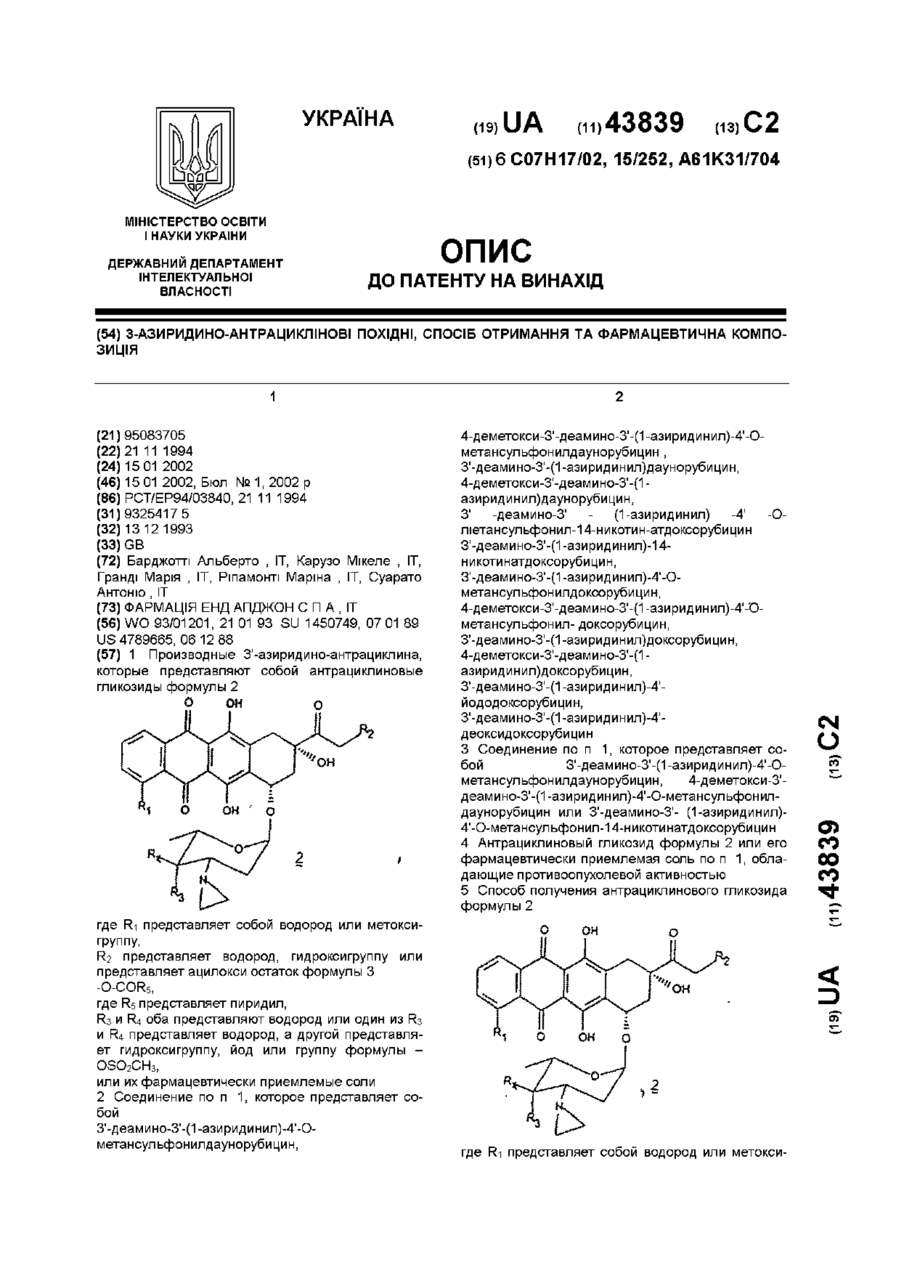
1. Производные 3'-азиридино-антрациклина, которые представляют собой антрациклиновые гликозиды формулы 2:
где R1 представляет собой водород или метоксигруппу;
R2 представляет водород, гидроксигруппу или представляет ацилокси остаток формулы 3:
-O-COR5,
где R5 представляет пиридил;
R3 и R4 оба представляют водород или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу, йод или группу формулы –ОSО2СН3;
или их фармацевтически приемлемые соли.
2. Соединение по п. 1, которое представляет собой:
3'-деамино-3'-(1-азиридинил)-4'-O-метансульфонилдаунорубицин;
4-деметокси-З'-деамино-3'-(1-азиридинил)-4'-O-метансульфонилдаунорубицин ;
3'-деамино-3'-(1-азиридинил)даунорубицин;
4-деметокси-3'-деамино-3'-(1-азиридинил)даунорубицин;
3' -деамино-3' - (1-азиридинил) -4' -O-ліетансульфонил-14-никотин-атдоксорубицин.
З'-деамино-3'-(1-азиридинил)-14-никотинатдоксорубицин;
З'-деамино-3'-(1-азиридинил)-4'-O-метансульфонилдоксорубицин;
4-деметокси-3'-деамино-3'-(1-азиридинил)-4'-Ό-метансульфонил- доксорубицин;
3'-деамино-3'-(1-азиридинил)доксорубицин;
4-деметокси-3'-деамино-3'-(1-азиридинил)доксорубицин;
3'-деамино-3'-(1-азиридинил)-4'-йододоксорубицин;
3'-деамино-3'-(1-азиридинил)-4'-деоксидоксорубицин.
3. Соединение по п. 1, которое представляет собой З'-деамино-3'-(1-азиридинил)-4'-O-метансульфонилдаунорубицин, 4-деметокси-3'-деамино-3'-(1-азиридинил)-4'-O-метансульфонил-даунорубицин или 3'-деамино-3'- (1-азиридинил)-4'-O-метансульфонил-14-никотинатдоксорубицин.
4. Антрациклиновый гликозид формулы 2 или его фармацевтически приемлемая соль по п. 1, обладающие противоопухолевой активностью.
5. Способ получения антрациклинового гликозида формулы 2:
где R1 представляет собой водород или метоксигруппу;
R2 представляет водород, гидроксигруппу или представляет ацилокси остаток формулы 3:
-O-COR5 ,
где R5 представляет пиридил;
R3 и R4 оба представляют водород или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу, йод или группу формулы –ОSО2СН3;
или его фармацевтически приемлемой соли, отличающийся тем, что включает обработку антрациклинового гликозида- общей формулы 6:
где R1, R2, R3, R4 имеют значения, указанные выше, и R9 - гидрокси или атом галогена, растворенного в безводном органическом растворителе, который представляет собой сухой метиленхлорид, метанол или их смесь в объемном соотношении 1:1-1:3, при перемешивании при температуре 0-30°С от 15 до 2 часов силикагелем, и при желании, превращение полученного таким образом антрациклинового гликозида формулы 2 в его фармацевтически приемлемую соль.
6. Способ по п.5, отличающийся тем, что размеры частиц силикагеля находятся в интервале от 230 до 400 мешей.
7. Способ получения антрациклинового гликозида формулы 2:
где R1 представляет собой водород или метоксигруппу;
R2 представляет водород, гидроксигруппу или представляет ацилокси остаток формулы 3:
-O-COR5,
где R5 представляет пиридил;
R3 и R4 оба представляют водорода или один из R3 и R4
представляет водород, а другой представляет гидроксигруппу,
иод или группу формулы –ОSО2СН3;
или его фармацевтически приемлемой соли, отличающийся тем, что включает взаимодействие производного формулы 5:
где R1, R3 и R4 имеют значения, указанные выше, с производным соли формулы 3:
X+ -OCOR5, 3’
где R5 имеет значения, указанные выше.
8. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая фармацевтически приемлемый разбавитель или носитель, отличающаяся тем, что в качестве активного агента она содержит антрациклиновый гликозид формулы 2:
где R1 представляет собой водород или метоксигруппу;
R2 представляет водород, гидроксигруппу или представляет ацилркси остаток формулы 3:
-O-COR5 ,
где R5 представляет пиридил;
R3 и R4 оба представляют водорода или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу йод или группу формулы –ОSО2СН3;
или его фaрмацевтически приемлемую соль.
Текст
1 Производные З'-азиридино-антрациклина, которые представляют собой антрациклиновые гликозиды формулы 2 о он о где Ri представляет собой водород или метоксигруппу, F 2 представляет водород, гидроксигруппу или ? представляет ацилокси остаток формулы 3 -O-CORs, где R5 представляет пиридил, F 3 и R4 оба представляют водород или один из R3 ? и R4 представляет водород, а другой представляет гидроксигруппу, йод или группу формулы OSO2CH3, или их фармацевтически приемлемые соли 2 Соединение по п 1, которое представляет собой 3'-деамино-3'-(1-азиридинил)-4'-Ометансульфонилдаунорубицин, 4-деметокси-3'-деамино-3'-(1-азиридинил)-4'-Ометансульфонилдаунорубицин , 3'-деамино-3'-(1-азиридинил)даунорубицин, 4-деметокси-3'-деамино-3'-(1 азиридинил)даунорубицин, 3' -деамино-3' - (1 -азиридинил) -4' -Оліетансульфонил-14-никотин-атдоксорубицин 3'-деамино-3'-(1 -азиридинил)-14никотинатдоксорубицин, 3'-деамино-3'-(1-азиридинил)-4'-Ометансульфонилдоксорубицин, 4-деметокси-3'-деамино-3'-(1-азиридинил)-4'-'Ометансульфонил- доксорубицин, 3'-деамино-3'-(1-азиридинил)доксорубицин, 4-деметокси-3'-деамино-3'-(1 азиридинил)доксорубицин, 3'-деамино-3'-(1-азиридинил)-4'йододоксорубицин, 3'-деамино-3'-(1-азиридинил)-4'деоксидоксорубицин 3 Соединение по п 1, которое представляет собой 3'-деамино-3'-(1-азиридинил)-4'-Ометансульфонилдаунорубицин, 4-деметокси-З'деамино-3'-(1-азиридинил)-4'-О-метансульфонилдаунорубицин или З'-деамино-З'- (1-азиридинил)4'-О-метансульфонил-14-никотинатдоксорубицин 4 Антрациклиновый гликозид формулы 2 или его фармацевтически приемлемая соль по п 1, обладающие противоопухолевой активностью 5 Способ получения антрациклинового гликозида формулы 2 где Ri представляет собой водород или метокси О го 00 го 43839 группу, F 2 представляет водород, гидроксигруппу или ? представляет ацилокси остаток формулы 3 -O-CORs, где R5 представляет пиридил, F 3 и R4 оба представляют водород или один из R3 ? и R4 представляет водород, а другой представляет гидроксигруппу, йод или группу формулы OSO2CH3, или его фармацевтически приемлемой соли, отличающийся тем, что включает обработку антрациклинового гликозида- общей формулы 6 где R-i, R2, R3, R4 имеют значения, указанные выше, и Rg - гидрокси или атом галогена, растворенного в безводном органическом растворителе, который представляет собой сухой метиленхлорид, метанол или их смесь в объемном соотношении 1 1-1 3, при перемешивании при температуре 0-30°С от 15 до 2 часов силикагелем, и при желании, превращение полученного таким образом антрациклинового гликозида формулы 2 в его фармацевтически приемлемую соль 6 Способ по п 5, отличающийся тем, что размеры частиц силикагеля находятся в интервале от 230 до 400 мешей 7 Способ получения антрациклинового гликозида формулы 2 где Ri представляет собой водород или метоксигруппу, R2 представляет водород, гидроксигруппу или представляет ацилокси остаток формулы 3 -O-CORs, 4 где R5 представляет пиридил, F 3 и R4 оба представляют водорода или один из ? R3 и R4 представляет водород, а другой представляет гидроксигруппу, иод или группу формулы -OSO2CH3, или его фармацевтически приемлемой соли, отличающийся тем, что включает взаимодействие производного формулы 5 О ОН О где R-і, R3 и R4 имеют значения, указанные выше, с производным соли формулы 3 Х+ OCOR5, 3' где Rs имеет значения, указанные выше 8 Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая фармацевтически приемлемый разбавитель или носитель, отличающаяся тем, что в качестве активного агента она содержит антрациклиновый гликозид формулы 2 О где Ri представляет собой водород или метоксигруппу, R2 представляет водород, гидроксигруппу или представляет ацилркси остаток формулы 3 -O-CORs, где Rs представляет пиридил, R3 и R4 оба представляют водорода или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу йод или группу формулы OSO2CH3, или его фармацевтически приемлемую соль 43839 Настоящее изобретение относится к новым антрациклиновым гликозидам, которые обладают противоопухолевой активностью, к способам их получения и к содержащим их фармацевтическим композициям В настоящем изобретении предложены антрациклиновые гликозиды, родственные даунорубицину и доксорубицину, в которых 3-аминогруппа сахарного остатка замкнута в азиридиновое кольцо, и, необязательно, гидроксигруппа у С-4' сахара может быть защищена в форме сультоната В настоящем изобретении предложены также их растворимые в воде производные и фармацевтически приемлемые соли присоединения кислот В настоящем изобретении предложено соединение, которое является антрациклиновым гликозидом формулы 1 или 2 о где Ri представляет водород или метоксигруппу, F 2 представляет водород, гидроксигруппу ? или представляет ацилоксиостаток формулы 3 -O-CORs где R5 представляет линейный или разветвленный C-i-Cs алкил, моно- или би-циклический арил, предпочтительно фенил, или гетеро моноили би-циклическое кольцо, предпочтительно, пиридил, причем каждая из групп может быть необязательно замещена (а) аминогруппой -NR6R7, где R6 и R7 независимо представляют водород или С1-С4 алкил, или (Ь) карбоксигруппой, R3 и R4 оба представляют водород, или один из R3 и R4 представляет водород, а другой представляет гидроксигруппу или группу формулы -OS2R8, где Rs может быть линейной или разветвленной ал кильной группой, содержащей от 1 до 6 атомов углерода, например, от 1 до 4 атомов углерода, Rs может представлять, в частности, метил, этил, нпропил или изопропил В другом варианте Rs может представлять арильную группу, такую, как фенил, незамещенный или замещенный 1 - 3 заместителями, каждый из которых может быть независимо линейной или разветвленной алкильной или алкоксигруппой, содержащей от 1 до 6 атомов углерода, например, 1 - 3 атома углерода, атомом галоида или нитрогруппой Примеры атомов галоида включают фтор, хлор, бром и йод, предпочтительно, фтор или хлор, более предпочтительно, хлор В настоящем изобретении арильная группа представляет моноциклический или бициклический ароматический углеводород, содержащий от 6 до 10 атомов углерода, например, фенил или нафтил Гетероциклическое кольцо представляет 5- или 6-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее, по крайней мере, один гетероатом, выбранный из О, S и N, которое необязательно конденсировано со второй 5- или 6-членной, насыщенной и ненасыщенной гетероциклической группой Примеры насыщенных и ненасыщенных гетероциклических колец включают пиразол ильное, имидазолильное, пиридильное, пиразильное, пиримидильное, пиридазинильное, морфолино, тилморфолино, фурильное и тиенильное кольца Предпочтительно, чтобы R2 представлял гидрокси или О-никотинил, R3 представлял гидрокси или -OSCbRs, где Rs представлял бы СгС4алкил, a R4 представлял бы водород Примеры соединений настоящего изобретения включают (1а)3'-диамино-3'-[1-азиридинил]-4-Ометансульфонил-даунорубицин (Ri = ОСН, R4 = Н, R3 = OSO2CH3) (1Ь)4-деметокси-3'-деамино-3'[1-азиридинил]4'-О-метансульфонил—даунорубицин (Ri = R4 = Н, R3 = OSO2CH3) (1с)3'-деамино-3'[1-азиридинил]-даунорубицин (R = ОСНз, R4 = Н, R3=OH) (1 ф4-деметокси-3'-деамино-3'-[1 -азиридинил]даунорубицин (Ri = R4 = Н, R3 = ОН) (2а)3'-деамино-3'-[1-азиридинил]-4'-Ометансульфонил-14~никотинат-даунорубицин (Ri = ОСНз, R2 = О-никотинил, R4 = Н, R3 =OSO2 CH3) (2Ь)3'-деамино-3'-[1 -азиридинил]-14никотинат-доксорубицин (Ri = ОСНз, R2 - Оникотиноил, R4 = Н, R3 = ОН) (2с)3'-деамино-3'-[1-азиридинил]-4'-Ометансульфонил-доксорубицин (Ri = ОСНз, R2 ОН, R4 = Н, R3 = OSO2CH3 (2сІ)4-деметокси-3' -деамино-3'-[1 -азиридинил]4'-О-метансульфонил-доксорубицин (Ri = R4 = Н, R2 = ОН, R3 = OSO2CH3) (2е)3'-деамино-3'-[1-азиридинил]доксорубицин (Ri = ОСНз, R4 = Н, R2 = R3 = ОН) (21^4-деметокси-3'-деамино-3'-[1-азиридинил]доксорубицин (Ri = R4 = Н, R2 = R3 - ОН) (2д)3'-деамино-3'-[1-азиридинил]-4' 43839 иододоксорубицин = ОСНз, R2 - ОН, R4 = Н, R3 (2п)3'-деамино-3'-[1-азиридинил]-4'деоксидоксорубицин (Ri = ОСН3, R2 - ОН, R3 = R4 = Н) и такие их фармацевтически приемлемые соли, как соли хлористоводородной кислоты (соляной кислоты) Далее, в настоящем изобретении предложен способ получения азиридиноантрациклинового гликозида формулы 1 или 2, как указано ранее, или его фармацевтически приемлемой соли, который включает (а) превращение антрациклина общей формулы 4 ОН R< О ОН О 8 ния (которое можно получить из соединения формулы 1 по способу, описанному в патенте США № 3803124) до получения азиридиноантрациклинового производного формулы 2, где R2 представляет гидроксильную группу, и, при желании, (с) осуществление взаимодействия соединения формулы 5, как определено ранее, с солью производного формулы 3' X+-OCOR5 где Rs имеет указанные ранее значения, при условии, что Rs не представляет остатка, содержащего первичную аминогруппу, а Х+ представляет ион, предпочтительно ион натрия или калия, и, при желании, превращение полученного таким образом соединения формулы 2, в его фармацевтически приемлемую соль, или (d) осуществление взаимодействия соединения формулы 5, как определено ранее, с солью производного формулы 3', где Rs представляет первичную аминогруппу, защищенную чувствительной к кислоте защитной группой, с последующим удалением защитной группы, и, при желании, превращение полученного таким образом соединения формулы 2 в его фармацевтически приемлемую соль В настоящем изобретении предложен другой способ получения азиридиноантрациклинового гликозида формулы 2, как определено ранее, или его фармацевтически приемлемой соли, который включает (а) обработку атрациклина общей формулы 6 ОН N где R-і, R2 и R3 имеют указанные ранее значения, a Rg представляет сульфонатную группу или атом галоида, предпочтительно, атом хлора, в антрациклин формулы 1, причем соединение формулы 4, предпочтительно, растворяют в безводном органическом растворителе в присутствии безводной соли щелочного металла и мягкого основания, и, при желании, (Ь) гидролиз производного формулы 5 О 1 0 ОН О где R-і, R2, R4 имеют указанные ранее значе где R-і, R2, R3, R4 и Rg имеют указанные ранее значения (такие соединения были раскрыты также в WO 93/01201), силикагелем, и, при желании, превращение полученного таким образом соединения формулы 2, в его фармацевтически приемлемую соль Следует отметить, что антрациклины формулы 4 или 6 также могут образовывать азиридиновое кольцо, если их обработать силикагелем Для такой обработки можно использовать мягкие условия, что позволяет получать соединения формулы 2, исходя из чувствительных к основаниям сложноэфирных производных, таких как те, которые имеют формулу 6 В соответствии со способом настоящего изобретения предпочтительные условия реакции получения азиридиноантрациклинов формулы 1 43839 включают растворение соединения формулы 4, как было определено ранее, в безводном органическом растворителе, таком как безводный метиленхлорид, в присутствии безводной щелочной соли металла, например, безводного карбоната или бикарбоната натрия или калия, при перемешивании, при температуре от 0 до 30°С, предпочтительно при комнатной температуре, и в течение от 15 минут до 2 часов, предпочтительно, около 30 минут В другом способе соединения формулы 4 растворяют в смеси таких органических растворителей, как сухой метиленхлорид и метанол, в соотношении от 1 1 до 1 3 по объему, затем этот раствор обрабатывают силикагелем, предпочтительно 230 - 400 мешей, при перемешивании при температуре от 0 до 30°С, предпочтительно, при комнатной температуре, в течение промежутка времени от 15 минут до 2 часов, предпочтительно, около 30 минут В аналогичном процессе условия реакции для превращения соединений формулы 6, как определено ранее, в азиридиноатрациклины формулы 2, предпочтительно, включают растворение соединений формулы 6 в безводном органическом растворителе, например, в сухом метиленхлориде и метаноле, и обработку полученного раствора силикагелем, предпочтительно, 230-400 мешей, при перемешивании при температуре от 0 до 30°С предпочтительно, при комнатной температуре в течение промежутка времени от 15 минут до 2 часов, предпочтительно, в течение около 30 минут Такой полярный растворитель, как метанол, в способе обработки силикагелем используют для удаления антрациклина из силикагеля В другом способе получения азиридиноантрациклннового гликозида формулы 2 или его фармацевтически приемлемой соли, где F 2 представ? ляет группу формулы 3, в которой R5 не представляет остатка, содержащего первичную аминогруппу, предпочтительные условия реакции включают осуществление взаимодействия соединения формулы 5 с производным соли кислоты формулы 3', как было определено ранее, в безводном полярном растворителе, предпочтительно ацетоне или диметилформамиде, при температуре от 20 до 60°С, предпочтительно, при комнатной температуре, в течение от 4 до 15 часов, предпочтительно, от 5 до 12 часов Условия реакции для получения азиридиноантрациклинового гликозида общей формулы 2, где F 2 представляет группу формулы 3, в которой Rs ? представляет первичную аминогруппу, включают осуществление взаимодействия соединений формулы 5, как было определено ранее, с производным соли кислоты формулы 3', в которой аминогруппа защищена группой, чувствительной к кислоте, например, аминогруппа защищена Шиффовым основанием, в полярном растворителе, таком как ацетон или диметилформамид, при температуре от 20 до 60°С, предпочтительно, при комнатной температуре, в течение от 4 до 15 часов, предпочтительно от 5 до 12 часов, затем полученное N-защищенное-сложноэфирное производное деблокируют, растворяя его, например, в 10 метиленхлориде, и добавляя дистиллированную воду и водную соляную кислоту, предпочтительно, примерно такой же объем воды, что и метиленхлорида, а соляную кислоту в количестве, соответствующем примерно трем эквивалентам 0,1 н НСІ Полученную смесь интенсивно перемешивают при температуре от О до 20°С, предпочтительно, около 15°С, в течение промежутка времени от 30 минут до 2 часов, предпочтительно, от 45 до 90 минут, выделяют, и водную фазу сушат вымораживанием до получения растворимой соли соляной кислоты С-15 сложноэфирного производного формулы 2 Предпочтительно, защищать первичную аминогруппу группой метилендифенила В следующем аспекте в настоящем изобретении предложены Фармацевтические композиции, содержащие антрациклиновый гликозид формулы 1 или 2, или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым разбавителем или носителем Можно использовать обычные носители и разбавители Композиции можно выполнять и вводить обычными способами Подходящие способы введения включают парэнеральное введение- для парентерального введения жидкую композицию можно приготовить, используя активное соединение и стерильный разбавитель или носитель, в котором активное соединение либо может раствориться, либо в нем получают его суспензию Парентеральную композицию можно приготовить в форме стерильного твердого вещества, которое необходимо снова растворить перед употреблением в таком подходящем носителе, как физиологический раствор, стерильная вода или другие стерильные носители Соединения настоящего изобретения пригодны для терапевтического лечения организмов человека и животных Они полезны как противоопухолевые агенты, особенно для лечения лейкемии или аденокарциномы толстой кишки Терапевтически эффективное количество соединения вводят пациенту, у которого имеется опухоль, для улучшения состояния пациента Можно вводить такое количество соединения, которого достаточно для ингибирования роста опухоли Вводимые дозы можно оценить, зная интервалы доз для доксорубицина и даунорубицина, модифицированные относительно активности, демонстрируемой соединениями настоящего изобретения в тестах против опухолей ин витро и ин виво Обычно, подходящие дозы попадают в интервал от 1 до 200мг/м2 площади поверхности тела, предпочтительно, от 1 до 100мг/м2, в зависимости от характера и степени серьезности заболевания, которое нужно лечить, и от общего состояния пациента Нижеследующие примеры иллюстрируют изобретение Пример 1 Получение 3'-деамино-3'-[1-азиридинил]-4'-0метансульфонилдаунорубицина (Ri = ОСНз, R4 Н, R3 = OSO2CH3) 3'-М-(2-хлорэтил)-4'-Ометансульфонилдаунорубицин (4а, Ri = ОСНз, R4 = Н, R3 = OSO2CH3, R9 = СІ) (0,33г, 0,05ммоля), 12 11 43839 полученный по способу WO/93/012001, растворяводного бромистого водорода (15мл) Полученную ют в смеси 10мл безводного метиленхлорида и смесь выдерживают при 30°С в течение 20 часов, 20мл метанола, и перемешивают с силикагелем затем экстрагируют Н-бутанолом (50мл) Органи(Мегск, 200 - 400 мешей, 2г) при комнатной темпеческий растворитель удаляют при пониженном ратуре в течение 30 минут Полученный раствор давлении, а остаток, растворенный в сухом ацефильтруют, концентрируют досуха и сырой матетоне (200мл), обрабатывают никотинатом калия риал обрабатывают с помощью флешхромато(2г) при кипении с обратным холодильником в теграфии на колонке с силикагелем, используя в чение часа Растворитель удаляют при пониженкачестве злюента смесь метнленхлорида и метаном давлении, а сырой материал обрабатывают нола (95 5 по объему), до получения указанного в хроматографически на колонке с силикагелем, заглавии соединения 1а (выход 0,22г) используя в качестве элюирующей системы смесь метиленхлорида и метанола (95 5 по объему) до ТСХ на пластинах Kieselgel F254 (Мегск) с исполучения указанного в заглавии соединения 2а пользованием в качестве элюента системы мети(0,35г), Т плавления 148 - 149°С (с разложением) ленхлорид метанол (93 2 по объему) дает Rf = + 0,65 Масс-спектр с полевой десорбцией м/z [М ] ТСХ на пластинах Kieselgel F254 (Мегск), с ис631 пользанием в качестве элюирующей системы метиленхлорида и метанола (10 1 по объему) дает 2Н-ЯМР (400МгГц, CDCI) 5 Rf = 0,37 Масс-спектр с полевой десорбцией M/Z 1,16, 1,25 (м, 2Н, водороды азиридина), 1,36 (М+) 752 (д, J=6,4I~4, ЗН, СНз-51), 1,52 (м, 1Н, Н-31), 1,73 (м, Пример 4 2Н, водороды азиридина), 1,80 (м,1Н, Н-2'экв), 2,09 (м, 1Н, Н-2'акс), 2,12 (м, 1Н, Н-8акс), 2,31 (м, Получение 3'-деамино-3'-[1-азиридинил]-4'-О1Н, Н-8экв), 2,39 (с, ЗН, СОСН3), 2,98 (д, J = метансульфонилдоксорубицина 19,2Гц, 1Н, Н-10акс), 3,21 (дд, J = 1,7, 19,2Гц, 1Н, (2с Ri = ОСНз, R2 = ОН, R4 = Н, R3 = Н-Юэкв), 3,22 (с, ЗН, CH3SO2), 4,09 (кв, и-6,4Гц, OSO2CH3) 1Н, Н-51), 4,10 (с, ЗН, ОСНз), 4,44 (с, 1Н, ОН-9), 3'-1\1-(2-хлорэтил)-4'4,75 (с, 1Н, Н-41), 5,28 (м, 1Н, Н-7), 5,55 (д, J = метансульфонилдоксорубицин (6а Ri = ОСНз, R2 3,4Гц, 1Н, Н-Г), 7,41 (д, J = 8,1 Гц, 1Н, Н-3), 7,80 = ОН, R9 = СІ, R3 = OSO2CH3, R4 = H), полученный (дд, J = 7,7, 8,1 Гц, 1Н-Н-2), 3,05 (д, J = 7,7Гц, 1Н, по способу патента Великобритании 9114549, Н-1), 13,30 (с, 1Н, ОН-11), 14,00 (с, 1Н, ОН-6) превращают в указанное в заглавии соединение 2с по способу примера 1 ТСХ на пластинах Пример 2 Kieselgel F254 (Мегск) с использованием в качеПолучение 4-деметокси-3'-деамино-3'-[1стве элюирующей системы метиленхлориде и азирндинил]-4'-О-метансульфонилдаунорубицина ацетона (3 2 по объему) дает Rf = 0,35 Масс(1 b R1 = R4 = Н, R3 = OSO2CH3) спектр с полевой десорбцией м/z (М+) 647 4-деметокси-І\І-(2-гидроксизтил)даунорубицин Пример 5 (4b R1 = R4 = H, R3 = OSO2CH3, Rg = OH, 0,3r, Получение 3'-деамино-3'-[1-азиридинил]-4'0,5ммоля) растворяют в смеси метиленхлорида иододоксорубицина (10мл) и метанола (5мл), и встряхивают при комнатной температуре с Зг силикагеля в течение 30 (2g Ri = ОСНз, R2 = ОН, R4 = Н, R3 = 1) минут Затем органический раствор отфильтровы3'-1\1-(2-хлорэтил)-4'-иододоксорубицин (6b Ri вают, а растворитель удаляют при пониженном = ОСНз, R2 = ОН, R9 = СІ, R3 = I, R4 = H, получендавлении-остаток обрабатывают с помощью ный по способу патента Великобритании 9114549, флеш-хроматографии на колонке с силикагелем, превращают в указанное в заглавии соединение используя в качестве системы элюента смесь ме2д по способу примера 1 тиленхлорида и метанола (95 5 по объему), до ТСХ на пластинах Kieselgel F254 (Мегск) с исполучения указанного в заглавии соединения 1в пользованием в качестве элюирующей системы (0,1 Зг) ТСХ на пластинах Kieselgel F254 (Мегск) с метиленхлорида и ацетона (9 1 по объему), дает использованием в качестве элюируюшей системы R f =0,45 метиленхлорида и метанола (20 1 по объему) Масс-спектр с полевой десорбцией м/z (М+) дает Rf = 0,42 Масс-спектр с полевой десорбцией 679 м/z (М+) 601 Пример 6 Получение 3'-деамино-3'-[1-азиридинил]-4'Пример 3 деокендаксорубицина Получение 3'-деамино-3'-[1-азиридинил]-4'-Ометансульфонил-14-никотинатдоксорубицина (2h R1 = ОСНЗ, R2 = ОН, R3 = R4 = Н) 3'-1\1-(2-хлорэтил)-4'-деоксидоксорубицин (6с (2а Ri = ОСНз, R2 = О-никотиноил, R4 = H, R3 Ri = ОСНз, R2 = ОН, R9 = СІ, R3 = R4 = H) получен= OSO2CH3) ный по способу патента Великобритании 9114549, 3'-деамино-3'-[1-азиридинил]-4'-Опревращают в указанное в заглавии соединение метансульфонилдаунорубицин (1а, 0,63г, 2п по способу примера 1 ТСХ на пластинах 1ммоль), полученный по способу примера 1, расKieselgel F245 (Мегск), с использованием в качетворяют в смеси 6мл безводного метанола и 13мл стве элюирующей системы метиленхлорида и диоксана, добавляют 0,5мл зтилортоформата, и ацетона (20 1 по объему), дает Rf = 0,33 Массзатем полученную смесь обрабатывают раствоспектр с полевой десорбцией м/z (М+) 553 ром брома (1г) в 5мл безводного метнленхлорида при 10°С в течение 1,5 часов Затем реакционную Биологическая активность смесь осаждают смесью этилового эфира (100мл) 3'-деамино-3'-[1-азиридинил 3-4'и петролейного эфира (50мл) Осадок собирают и О~метансульфонилдаунарубицин (1а), 4снова растворяют в смеси ацетона (15мл) и 0,25н деметокси-3'-деамнно-3'-[1-азиридинил]-4'-О 14 13 43839 метансульфонилдаунорубицин (1в), контрольных х 100 3'-деамино-3'-[1-азиридинилъ-4'-ОЖивотные метансульфонил-14-никотинатдоксорубицин (2а) и Для оценки противоопухолевой активности 3'-деамино-3'-[1-азиридинил]-4'-Оиспользовались инбредные DBA/2 и C57BI/6, гибметансульфонилдоксорубицин (2с), были тестиридные C57BL/6 х DBA/2 (B6D2F1) и BALB/c x рованы ин витро на двух человеческих клеточных DBA/2 (CD2F1) взрослые самки мыши Животным линиях, LoVo (аденокарцинома ободочной кишки было по 2 - 3 месяца, их вес составлял 20 - 22г, человека) и LoVo/DX (аденокарцинома, устойчисодержались в обычных лабораторных условиях вая к доксорубицину), в сравнении с доксорубициВ экспериментах с ксенотрансплантатом опухоли ном человека использовали швейцарских мышей Nu/Nu в возрасте 4-6 недель, весом 20 - 25г, котоЦитотоксическая активность приводится в вирых содержали в клетках с подстилкой из фильтде ИК50, т е концентрация, ингибирующая 50% ровальной бумаги, корм и подстилку стерилизоваобразования колоний, рассчитываемая на основали, а воду подкисляли Колонию мышей нии кривых концентрация реакция Показателем подвергали обычному тестированию на отсутстустойчивости, R J является отношение ИК50 для вие антител к ряду патогенов, включая гепатит устойчивых клеток к ИК50 для чувствительных клемышей, вирус Сендай и Mycoplasma pulmoms ток Соединения 1а, 1в, 2а и 2с демонстрируют Всех животных поставляли Charles River (Calco, высокую активность против обоих клеточных лиComo, Italy) и Harlan Nossan (Correzzana, Milano, ний, и низкий показатель устойчивости (таблица Italy) 1) Лейкозы Соединения 1а, 1в, 2а и 2с оценивали также Сублинию P388/DX Johnson поддерживали ин виво против Р388 мышиной лейкемии, устойчипосредством еженедельных и/п пассажей на мывой к доксорубицину (105 клеток/мышь транспланшах BD2F1 в дозе 106 клеток на мышь В эксперитируют внутривенно мышам BD2F1) в сравнении с менте мышам той же линии инъецировали по 105 доксорубнцином клеток в/в Соединения 1а, 1в, 2а и 2с демонстрируют также поразительно более высокую активность, Лейкоз мышей L1210 (первоначально получем доксорубицин (таблица 11) ченный из NCI) и сублинии, резистентные к L-PAM (L1210/L-PAM, первоначально полученный из Таблица 1 NCI), к cDDP (L1210/CDDP, первоначально полуЦитотоксическая активность in vitro (1С м ) соченный из NCI), и к DX (L1210/DX) поддерживали единений Іа, їв, 2а, 2с на LoVo и LoVo/DX клетках посредством еженедельных и/п пассажей на мыпо сравнению с активностью доксорубицина шах DBA/2N в дозе 106 клеток на мышь Животных с резистентными опухолями лечили И К50 (нг/мл) Соединение следующим образом LoVo LoVo/DX R\{Z) мышам L1210/L-PAM вводили и/п 7,5мг/кг L1а 13 22 1,7 РАМ через 48 часов после трансплантации опухо1в 27 26 0,9 ли 2а 14 40 2,9 мышам L1210/CDDP вводили и/п 5мг/кг цис2с 2,7 24 9,2 платина через 96 часов после трансплантации доксорубицин 82,5 4975 60,3 опухоли мышам L1210/DX вводили и/п бмг/кг DX через Анализ колоний 4 часа обрабатки 48 часов после трансплантации опухоли (1) ИС50 — концентрация, ингибирующая 50% В эксперименте мышам CD2F1 в/в инъецирообразования колоний 5 вали по 10 клеток (2) R J — показатель устойчивости = (ИС50 Солидные опухоли человека LoVo/DX)/(HK50 LoVo) Карциному молочной железы МХ1 (первонаТаблица 2 чально полученную из Национального института Противоопухолевая активность соединений рака [NCI], NHI, Bethesda, MD), карциномы яичника 1а, 1 в, 2а и 2с против лейкемии P388/DX по сравА2780 (любезно предоставленные д-ром Ozols, нению с доксорубицином Национальный институт рака, NHI, Bethesda, MD) и Н207 (первоначально полученную из каталога 1 T/CUJ од " АТСС), и мелкоклеточную карциному легких № Соединение % (мг/кг) 592 трансплантировали п/к бестимусным мышам, 1а 2,2 190 используя 15 - 20мг опухолевой взвеси Для экс1в 3,8 240 периментов опухоли вырезали и их фрагменты 2а 2,5 200 имплантировали п/к в область левого бока 2с 1,8 195 Введение лекарственных препаратов доксорубицин 16,9 106 Все лекарственные растворы приготавливали непосредственно перед употреблением Растворы Соединения суспендируют в Твин 80 (10%) и вводили в/в в объеме 10мг/кг веса тела Схемы вводят внутривенно через день после транспланлечения представлены в каждой таблице тации опухоли Оценка противоопухолевой активности и ток(1) ОД = оптимальная доза сичности (2) Среднее время выживания обработанных В экспериментах с моделями лейкозов активмышей/среднее время выживания ность лекарственных препаратов определяли пу 43839 16 15 тем сравнения медианы срока выживания (МСВ) в метру, D = большему диаметру) Ингибирование экспериментальной группе с тем же показателем в роста опухоли (ИРО%) рассчитывали согласно контрольной группе, и результаты, выраженные уравнению как УПЖ%, были следующими средний вес опухоли в экспериментальной группе ИРО% = хЮО (2) средний вес опухоли в контрольной группе МСВ в экспериментальной группе СВ в экспеВ экспериментах с солидными опухолями чериментальной группе ловека излеченными считались мыши, не имевМСВ в экспериментальной группе УПЖ% = 100-100 шие опухоли спустя 90 дней после ее имплантаМСВ в контрольной группе ции Количество выживших в течение длительного Токсичность оценивалась на основании маквремени (ВДВ) относится к мышам, которые выроскопических находок при вскрытии и потери вежили в течение периода времени более 60 дней са со дня имплантации опухоли Таблица 3 Противоопухолевая активность 4В экспериментах с солидными опухолями акдеметокси-3-дезамино-3'[1-азиридинил]-4'-Отивность оценивалась как ингибирование роста метансульфонил даунорубицина (1Ь) и 3опухоли через неделю после последнего введения т т дезамино-З [1-азиридинил}-4 -О-метансульфонил лекарственного препарата Рост опухоли оценидоксо-рубииина (2с) в отношении чувствительных вали с помощью кронциркуля фиксировали два и резистентних мышиных лейкозов диаметра и рассчитывали вес опухоли по следующей формуле d2x D/2 (где d = меньшему диа1Ь Опухоль од' 1 P388/DX L1210 L1210/L-PAM L1210/CDDP L1210/DX 2с УПЖ% (мг/кг/день) 3,8 3,8 3,8 3,8 3,8 А2780 Н207 МХ1 N592 Схема лечения в/в q4dx3 в/в q4dx3 в/ в q7dx3 в/в q7dx3 (мг/кг/день) 1,8 1,4 1,4 1,8 1,8 1044 101 71 48 66 1 — Опухолевые клетки инокулировали в/в на день 0 2 — Оптимальная доза Соединения суспендировали в Твине 80 в концентрации 10% и инъецировали в/в через один день после трансплантации опухоли 3 — Увеличение продолжительности жизни Опухоль1 од' 3 УПЖ%3 95 43 57 34 22 4 — 1/40 выживших в течение длительного времени Таблица 4 Противоопухолевая активность в отношении ксенотрансплантатов опухолей человека по сравнению с 4-деметокси-3-дезамино-3'[1азиридинил]-4'-О-метансульфонил доксорубицином (1Ь) Доза/ общая доза (мг/кг) ИРО2% Токсичность3 Излеченные мыши4 ЛЗРО", дни 2,5/7,5 99 0/7 0/7 20 2,5/7,5 99 0/8 2/8 45 2,66/8 99 1/8 1/8 60 2,25/9 100 0/6 1/6 47 1 —Опухолевую взвесь инокулировали п/к 2 — Ингибирование роста опухоли спустя одну неделю после последнего введения лекарственного препарата 3 — Количество мышей, умерших вследствие токсичности,/общее количество мышей 4 — Мыши, не имевшие опухоли на 90 день, через 90 дней после имплантации опухоли 5 — Задержка роста опухоли у экспериментальных мышей — задержка роста опухоли у кон трольных мышеи ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ КОМПОЗИЦИЯ Для приготовления соединений для инъекций применяли следующую процедуру Соединение 1Ь (2мг) растворяли в 1мл смеси кремофора и этанола 65 5 (по объему) при встряхивании, затем добавляли физиологический раствор (4 мл) и раствор встряхивали в течение 2 минут ДП «Український інститут промислової власності» (Укрпатент) вул Сім'ї Хохлових, 15, м Київ, 04119, Україна (044) 456 - 20 - 90
ДивитисяДодаткова інформація
Автори англійськоюBardzhotti Alberto, GRANDI MARIA, SUARATO ANTONINO
Автори російськоюБарджотти Альберто, ГРАНДИ Мария, Суарато Антонио
МПК / Мітки
МПК: C07H 17/02, A61K 31/704, A61P 35/00, A61K 31/7028, C07H 15/252, A61K 31/7034, A61K 31/70
Мітки: похідні, спосіб, фармацевтична, отримання, 3-азиридино-антрациклінові, композиція
Код посилання
<a href="https://ua.patents.su/8-43839-3-aziridino-antraciklinovi-pokhidni-sposib-otrimannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">3-азиридино-антрациклінові похідні, спосіб отримання та фармацевтична композиція</a>
Похідні 2-аміно-1,2,3,4-тетрагідронафталіну, які призначені для лікування серцево-судинних захворювань, спосіб їх отримання і фармацевтична композиція, що їх містить

Номер патенту: 26689
Опубліковано: 12.11.1999
Автори: САНТАНГЕЛО Франшеско, МАРКІНІ Франшеско, БЕРТОЛІНІ Джорджо, МОНТАНАРІ Стефанія, КАЗАГРАНДЕ Чесаре, Семераро Клаудіо
МПК: A61P 43/00, C07C 217/14, A61K 31/675, A61K 31/66, C07C 271/20, C07C 237/22, A61P 9/12, C07C 219/00, A61K 31/165, A61P 9/10, A61P 15/00, A61P 9/00, A61K 31/135, C07C 271/44, C07D 213/79, C07F 9/09, C07C 217/20, C07F 9/08, A61P 13/02, A61K 31/215
Мітки: фармацевтична, захворювань, серцево-судинних, лікування, отримання, спосіб, похідні, призначені, містить, 2-аміно-1,2,3,4-тетрагідронафталіну, композиція
Формула / Реферат:
1. Производные 2-амино-1,2,3,4-тетрагидронафталина формулы (I)в которой R1 и R2, которые одинаковы или различны, представляют атом водорода или группу OY';Y и Y', которые одинаковы или различны, представляют атом водорода или ацил, происходящий из возможно замещенной алифатической, ароматической или гетероароматической карбоновой кислоты, из возможно замещенной карбаминовой или угольной кислоты или из фосфорной кислоты...
Похідні азолів, їх фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб їх отримання, фармацевтична композиція
Номер патенту: 27343
Опубліковано: 15.09.2000
Автори: Стріт Леслі Дж., Матасса Віктор Г., Бейкер Реймонд
МПК: C07D 409/14, C07D 417/06, C07D 403/06, C07D 401/14, C07D 521/00, C07D 413/06, A61P 25/04, A61K 31/415, A61K 31/42, A61K 31/41, A61K 31/4433, C07D 413/14, C07D 409/06, C07D 403/04, A61K 31/425, C07D 409/04, A61K 31/4427, C07D 403/14, A61K 31/445, C07D 417/14
Мітки: похідні, азолів, отримання, фармацевтична, спосіб, рецепторів, проліки, прийнятні, 5-нt1-подібних, агоністами, фармацевтично, солі, композиція
Текст:
...2-15-(1-бензилтетразол-5-илметил)-1Н-индол-3ил]этиламин М,М-диметил-2-{5-(2-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(1-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(тетрээол-2-ил метил )• 1 Н-индол3-ил]этиламин N,N-димeтил-2-[5-(тeтpaзoл-1-илмeтил)-1 Н-индол3-ил]этиламин N,N-flHMeTHn-2-tS-{1 -метил-1,2,4-триазол-5-и л метил) 1Н-индол-3-ил]этиламин...
Похідні 1,5 – бензодіазепіну, що є антагоністами гастрину та холецистокініну, спосіб їх одержання, фармацевтична композиція

Номер патенту: 29416
Опубліковано: 15.11.2000
Автори: Феріані Алдо, Тріст Девід Гордон, Тарзія Джорджіо, Фінч Гаррі
МПК: A61P 43/00, A61P 1/04, A61P 25/18, A61P 25/24, C07D 243/12, A61P 25/16, A61P 25/00, A61K 31/55, A61P 25/22, A61P 1/00
Мітки: одержання, похідні, композиція, гастрину, бензодіазепіну, фармацевтична, спосіб, антагоністами, холецистокініну
Текст:
...R2 - замещенный или незамещенный фенил, где заместителями могут быть галоген, Сі-Сд- апкуіл, нитрогруппа, цианогруппа, трифторметоксигруппа, СгСм-алкилтиогруппа или группа (CH3)nR\ в которой R4 - гидрокси гр уппа, СгС ^а лкоксигр уппа , CO2R5 или NR6R7, R3 - фенил, возможно замещенный одним или двумя атомами галогена, R5- водоро д или Сі Оалкил, R6 и R представляют независимо Сі-Саалкил, RB - водород или галоген, m = 0, 1 или 2, п...
Похідні піримідину, що мають активність антагоністів ангіотензину іі, спосіб їх отримання та фармацевтична композиція

Номер патенту: 27748
Опубліковано: 16.10.2000
Автори: Бажлі Жак Фрамрос, Еллінгбой Джон Ватсон, Нікайдо Маделен
МПК: C07D 471/04, C07D 487/04
Мітки: отримання, піримідину, іі, фармацевтична, ангіотензину, спосіб, похідні, мають, композиція, антагоністів, активність
Текст:
...Результаты анализа из расчеты на формулу 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 минут после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение, в соответствии с примером 1, при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от А II в среднем на 41 % в...
Похідні 11-b-бензальдоксиместра-4,9-дієну, спосіб їх отримання та фармацевтична композиція

Номер патенту: 41309
Опубліковано: 17.09.2001
Автори: Оеттел Міхаель, ЕЛЬГЕР Вальтер, ШУБЕРТ Герд, Кауфманн Гюнтер, Шобек Лотар, Курішко Анатолі
МПК: A61P 35/00, A61P 15/00, A61P 13/02, C07J 75/00, A61K 31/56, C07J 41/00
Мітки: 11-b-бензальдоксиместра-4,9-дієну, похідні, композиція, спосіб, фармацевтична, отримання
Формула / Реферат:
1. Производные 11-b-бензальдоксимэстра-4,9-диена формулы (I): гдеR1 обозначает атом водорода или алкильный остаток с 1-6 С-атомами;R2 обозначает атом водорода; алкильную, арильную, аралкильную или алкиларильную группу с 1-10 С-атомами; ацильный остаток с 1-10 С-атомами или остаток -CONHR4 или -COOR4, причем R обозначает атом водорода; алкильный, арильный, аралкильный или алкиларильный остаток с 1-10...
Попередній патент: Клейова композиція для кріплення гуми до металу
Наступний патент: Пінопласт, що біологічно розкладається, та спосіб його одержання
Випадковий патент: Спосіб електроосмотичного зневоднення вугілля перед коксуванням