Спосіб одержання заміщеного імідазопіридину








Формула / Реферат
1. Спосіб одержання заміщеного імідазопіридину формули (1)
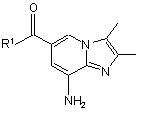 ,(1)
,(1)
де R1 є С1-С6алкоксилом або NH2,
який включає етап реагування сполуки формули (2)
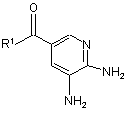 (2)
(2)
з 3-галоген-2-бутаноном в циклогексаноні.
2. Спосіб за п. 1, де 3-галоген-2-бутаноном є 3-бром-2-бутанон або 3-хлор-2-бутанон.
3. Спосіб за пп. 1 або 2, де кількість 3-галоген-2-бутанону складає 1,1-5 молярних еквівалентів.
4. Спосіб за п. 1, де температура реакції є 80-100°С.
5. Спосіб за п. 1, де циклогексанон розріджують інертним розчинником.
6. Спосіб за п. 1, де R1 є С1-С6алкоксилом.
7. Спосіб за п. 1, де R1 є NН2.
8. Спосіб за п. 1, який відрізняється тим, що сполуку (2) одержують способом, який включає етап гідрогенізації сполуки формули (4)
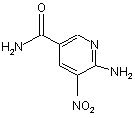 (4)
(4)
у водно-спиртовому розчині, використовуючи каталізатор.
9. Спосіб за п. 8, де каталізатором є паста Pd-Ru/C.
10. Спосіб за будь-яким з пп. 8 або 9, який відрізняється тим, що сполуку (4) одержують способом, який включає етап реагування сполуки формули (3)
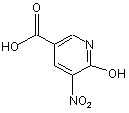 (3)
(3)
з тіонілхлоридом для одержання відповідної хлоридної сполуки, яку потім оброблюють аміаком.
Текст
Цей винахід стосується нового способу отримання заміщеного імідазопіридину, зокрема нового способу отримання 2,3-диметилімідазо[1,2-а]піридину, заміщеного в 6-позиції карбоксамідною або карбоксиалкіловою групою. В інших аспектах, винахід також стосується нових інтермедіатів, використовуваних в цьому способі. Винахід стосується нового способу, придатного для великомасштабного отримання заміщеного імідазопіридину формули (1), де R1 є С1-С6алкоксилом або NH2, і який включає етап реагування сполуки формули (2) де R1 є С1-С6алкоксилом або NH2, з 3-галоген-2-бутаноном в циклогексаноні. Подібну реакцію описано в ЕР 33094, ЕР 204285, ЕР 228006, ЕР 308917, і WO 99/55706, де заміщений амінопіридин загальної формули (X) реагує зі сполукою формули де X є, зокрема, Н, СН3 або естеровою групою, як-то СООСН3 або СООС2Н5, Υ є, зокрема, СН3, СН2СН3, і Ζ є відщеплю вальною групою, як-то галоген, мезил або тозил, для отримання сполуки загальної структури де X і Υ є як описано вище. Реакцію проводять в інертному розчиннику, як-то ацетон, спирти, бензол, Ν,Ν-диметилформамід, тетрагідрофуран, хлороформ, або діетиловий етер, бажано при підвищеній температурі, і, як варіант, у присутності неорганічної або органічної основи. Реакція характеризується довгими годинами виконання, наприклад, 16-84 години, високими температурами і відносно низькими виходами, наприклад, 22%-55%. Таким чином, така реакція не придатна для великомасштабного отримання заміщених імідазопіридинів. Нами було несподівано виявлено, що якщо спосіб згідно з винаходом здійснювати як описано тут, час реакції можна скоротити, температуру реакції знизити і вихід збільшити. Винахід стосується нового способу для великомасштабного отримання заміщеного імідазопіридину формули (1) де R1 є С1-С6алкоксилом або ΝΗ2, і який включає етап реагування сполуки формули (2) з 3-галоген-2-бутаноном в циклогексаноні. У першому втіленні винаходу сполука формули (2) де R1 є С1-С6алкоксилом, реагує з 3-галоген-2-бутаноном в циклогексаноні для отримання сполуки формули (1) де R1 є С1-С6алкоксилом. У другому втіленні винаходу сполука формули (2) де R1 є NH2, реагує з 3-галоген-2-бутаноном в циклогексаноні для отримання сполуки формули (1) де R1 є NH2. Спосіб згідно з винаходом здійснюють розчиненням або суспендуванням сполуки формули (2) де R1 є С1-С6 алкоксилом або NH2, в циклогексаноні та додаванням 3-галоген-2-бутанону, нагріванням реакції кілька годин, а потім виділенням сполуки формули (1) де R є С1-С6алкоксилом або NH2, з високими виходами. Кількість циклогексанону не є вирішальним у виконанні цього винаходу, і тому в практичних умовах її можна встановлювати відповідно до потреб та використовуваного обладнання. Також можна змішувати циклогексанон з інертними розчинниками, як-то етери. До приданих інертних розчинників входить, але не обмежується цим, тетрагідрофуран (ТГФ). Кількість інертного розчиннику може складати приблизно 50%, за об'ємом, не спричиняючи зменшення у виході. Кількість 3-галоген-2-бутанону не є вирішальним у виконанні цього винаходу. Саме для практичних та економічних цілей краще додавати 1,1-5 молярних еквівалентів, краще 1,1-2еквівалентів. До придатних 3галоген-2-бутанонів належать, але не обмежуються цим, 3-бром-2-бутанон і 3-хлор-2-бутанон, останній з яких є бажаним. Температури та час реакції можна змінювати для задоволення актуальних потреб. Бажано, щоб температура реакції знаходилася у межах 80-100°С. Така температура реакції дає повну реакцію у межах кількох годин, наприклад, 1-4 години. Перетворення є звичайно вище 95%, а вихід виділення є звичайно вище 70%. Вихідний матеріал, використовуваний згідно з винаходом, може бути отриманий як описано в WO 99/55706 або, альтернативно, як описано нижче на Схемі 1. Етап і Сполуку (3) на Схемі 1 оброблюють тіонілхлоридом, або будь-яким еквівалентним реагентом, при підвищеній температурі у відповідному розчиннику декілька годин для отримання відповідної хлоридної сполуки. Реакцію проводять, використовуючи приблизно 1-5екв. тіонілхлориду, бажано 1-2,5екв., в толуолі при приблизно 100°С 2-8 годин. Після чого, відповідний хлорид оброблюють 2-25екв. аміаку, бажано 312екв., у тому ж самому розчиннику як описано вище при приблизно температурі навколишнього середовища для отримання сполуки (4). Етап її Сполуку (4) на Схемі 1 гідрогенізують у водно-спиртовому розчині, використовуючи каталізатор для отримання сполуки (5). До відповідних каталізаторів належать, але не обмежуються цим, паладій, рутеній або їх суміші. Паста Pd-Ru/C є бажаним каталізатором. До відповідних спиртів належать, але не обмежуються цим, метанол, етанол і пропанол, з яких метанол є бажаним. Заміщений імідазопіридин формули (1), де R1 є С1-С6алкоксилом або NH2, отриманий згідно з винаходом, може отже бути використаний для отримання певних похідних заміщеного імідазопіридину, що зокрема ефективні як інгібітори шлунковокишкової Н+, К+-АТФази, і таким чином як інгібітори секреції шлункової кислоти. Сполуки Формули (1) можуть реагувати зі сполукою Формули (6) де R3 є Н, С1-С6алкілом, гідроксильованим С1-С6алкілом або галогеном; R4 є Н, С1-С6алкілом, гідроксильованим С1-С6алкілом або галогеном; R5 є Н, або галогеном; і Υ є відщеплю вальною групою, як-то галогенід, тозил або мезил, для отримання сполуки Формули (7) де R1, R3, R4, і R5 є як визначено вище. Зручно проводити цю реакцію в інертному розчиннику, як-то ацетон, ацетонітрил, диметоксиетан, метанол, етанол або диметилформамід з або без основи. Основою є, наприклад, гідроксид лужного металу, як-то гідроксид натрію і гідроксид калію, карбонат лужного металу, як-то карбонат калію і карбонат натрію; або органічний амін, як-то триетиламін. Сполуки Формули (7), де R1 є С1-С6алкоксилом, можуть згодом далі реагувати з аміносполукою загальної Формули (8) де R6 і R7 є однаковим або різними і вибрані з групи, що складається з Н, С1-С6алкілу, гідроксильованого С1-С6алкілу, С1-С6алкокси-заміщеного С1-С6алкілу, гідроксильованого С1-С6алкоксизаміщеного С1-С6алкілу, арилу, для отримання відповідної амідосполуки. R6 і R7 можуть разом з атомом нітрогену, до якого вони приєднані, утворювати насичене або ненасичене кільце, що, як варіант, містить один або більше додаткових гетероатомів з утворенням таким чином, наприклад, морфоліну, піперазину, піролідину, або піперидину. Реакцію можна проводити нагріванням реагентів в чистій аміносполуці або розчиненням в інертному розчиннику в стандартних умовах. З іншого боку, сполуки Формули (7) де R3, R4, і R5 є як визначено вище, a R1 є NН2, можуть бути гідролізовані в стандартних умовах з відповідними сполуками карбонової кислоти Формули (9) де R3, R4, і R5 є як визначено вище. Сполуки Формули (9) можуть згодом реагувати з аміносполуками Формули (8) де R6 і R7 є як визначено вище, у присутності зв'язуючого реагента для отримання відповідної амідосполуки. Реакцію можна проводити в інертному розчиннику в стандартних умовах. Приклади Приклад 1.1 Отримання Бромбутанону У реакторі, бромід натрію (84кг) суспендують в диметилформаміді (125л). 3-Хлор-2-бутанон (85кг) додають при 15-30°С. Перемішування продовжують протягом 4 годин, а тоді фільтрують: Осад на фільтрі промивають циклогексаноном (38л). Отриманий таким чином бромбутанон готовий для використання на етапі циклізації. Приклад 1.2 Синтез метил 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалату До суспензії метилового естеру 5,6-діаміно-нікотинової кислоти (1екв., 5,1г) в циклогексаноні (50мл) протягом 10хв. додавали бромбутанон (1,2екв., 3,9мл). Суміш нагрівали до 100°С (внутрішня температура) та перемішували протягом 2,5 годин при цій температурі. Суміш охолоджували до кімнатної температури, а блідий твердий продукт відфільтровували і промивали ТВМЕ (3´10мл). Сушили під зниженим тиском при 45°С. Вихід: 6,53г (75%). Приклад 1.3 Синтез етил 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалату До суспензії етилового естеру 5,6-діаміно-нікотинової кислоти (1екв., 5,0г) в циклогексаноні (50мл) протягом 15хв. додавали бромбутанон (1,4екв., 5,95г). Суміш темно-коричневого кольору нагрівали до 100°С (внутрішня температура) та перемішували 1,5 години при цій температурі. Суміш охолоджували до кімнатної температури, а твердий продукт світло-коричневого кольору відфільтровували і промивали ТВМЕ (20мл). Сушили під зниженим тиском при 45°С. Вихід: 5,06г (65%). Приклад 1.4 Синтез ізопропіл 8-аміно-2,3-диметилімідазо[1,2-а]піридин -6-карбоксилату До суспензії ізопропілового естеру 5,6-діаміно-нікотинової кислоти (1екв., 5,1г) в циклогексаноні (50мл) протягом 10хв. додавали бромбутанон (1,2екв., 3,4мл). Суміш темно-коричневого кольору нагрівали до 100°С (внутрішня температура) та перемішували 1,5 години при цій температурі. Суспензію охолоджували до кімнатної температури та блідо-жовтий твердий продукт відфільтровували і промивали ТВМЕ (3´10мл). Сушили під зниженим тиском при 45°С. Вихід: 6,0г (74%). Приклад 1.5 Синтез 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксаміду 5,6-Діаміно-нікотинамід (50г, 0,313моль (аналіз: 95,4%), 1,0екв.) суспендували в циклогексаноні (250мл). Суспензію нагрівали до 100°С. Фільтрат (бромбутанон в циклогексаноні) додавали при 100°С протягом 3 годин 10хв. Нагрівання продовжували 3 години, а тоді джерело нагрівання видаляли. Реакційній суміші давали можливість охолонути до 20°С і перемішували при цій температурі ще 2 години. Твердий продукт відфільтровували, обережно промивали ТВМЕ (2´330мл) і висушували для виходу 70,3г названої сполуки. Вихід: 70%. Приклад 1.6 Синтез 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксаміду NaBr (27,0г; 0,259моль; 1,33екв.) суспендували в циклогексаноні (220мл) і додавали однією порцією 3хлор-2-бутанон (25,7мл; 0,242моль; 1,24екв.). Суміш нагрівали до 80°С і перемішували 3 години. Суміш охолоджували до 50°С, білий твердий продукт відфільтровували і промивали циклогексаноном (60мл). 5,6Діаміно-нікотинамід (30г; 0,1946моль; 1,0екв.) додавали до фільтрату і суміш нагрівали до 100°С 4 години, після чого ВЕРХ визначали 98% перетворення. Реакційну суміш охолоджували до 20°С, перемішування продовжували 2 години при 20°С. Твердий продукт відфільтровували, промивали ТВМЕ (220мл) і висушували для виходу 46,6г названої сполуки . Вихід: 73%. Приклад 1.7 Синтез 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксаміду 5,6-Діаміно-нікотинамід (30,0г; 0,183моль; 1,0екв.) суспендували в циклогексаноні (280мл). 3-Бром-2бутанон (24мл; 0,22моль; 1,2екв.) додавали і суміш нагрівали 4 години до 100°С. Реакційну суміш охолоджували до 20°С і перемішували ще 2 години. Твердий продукт відфільтровували, промивали ТВМЕ (200мл) і висушували для виходу 48,4г названої сполуки. Вихід: 78%. Приклад 1.8 Синтез метил 2,3-диметил-8-(2,6-диметилбензиламіно)-імідазо [1,2-а]піридин-6-карбоксалату Метил 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалат (0,8г, 3,6ммоль), 2,6диметилбензилхлорид (0,57г, 3,7ммоль), карбонат натрію (1,0г, 9,4ммоль) і каталітичну кількість йодиду калію додавали до ацетонітрилу (10мл) і нагрівали під зворотним холодильником 20 годин. Після фільтрації, солі промивали метиленхлоридом і розчинники випарювали під зниженим тиском. Залишок очищували хроматографією на колонці з силікагелем, використовуючи метиленхлорид: етилацетат (75:25) як елюент. Залишок жовтого кольору оброблювали гексаном для отримання 0,23г (19%) названого продукту. Приклад 1.9 Синтез етил 2,3-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбоксалату Етил 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалат (0,7г, 3,0ммоль), 2-етил-6метилбензилхлорид (0,5г, 3,0ммоль), карбонат натрію (0,64г, 6,0ммоль) та каталітичну кількість йодиду калію додавали до ацетону (50мл) і нагрівали під зворотним холодильником 20 годин. Після фільтрації, ацетон випарювали під зниженим тиском для отримання масла. Масляний продукт очищували хроматографією на колонці з силікагелем, використовуючи діетиловий етер:нафтовий етер (1:1) як елюент для отримання 0,12г (9%) названого продукту. 1 Н-ЯМР (500МГц, CDCI 3): d 1,25 (t, 3H), 1,5 (t, 3Н), 2,35 (s, 3Н), 2,42 (s, 3Н), 2,44 (s, 3Н), 2,75 (q, 2H), 4,45-4,5 (m, 4H), 4,9 (bs, 1H), 6,8 (s, 1H), 7,05-7,2 (m, 3Н), 8,1 (s, 1H). Приклад 1.10 Синтез 2,3-диметил-8-(2-етил-6-метилбензиламіно)-N-пропіл-імідазо[1,2-а]піридин-6-карбоксаміду Етил 2,3-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбоксалат (0,12г, 0,33 моль), пропіламін (1,0г, 17ммоль) і каталітичну кількість ціаніду натрію нагрівали під зворотним холодильником в метанолі (20мл) 24 години. Додаткову кількість пропіламіну (1,0г, 17ммоль) додавали і реакційну суміш нагрівали під зворотним холодильником 24 години. Розчинник випарювали під зниженим тиском і залишок очищували хроматографією на колонці з силікагелем, використовуючи діетиловий етер як елюент. Кристалізація з діетилового етеру дала 0,053г (42%) названої сполуки. 1 Н-ЯМР (300МГц, CDCI 3): d 1,0 (t, 3Н), 1,2 (t, 3Н), 1,65-1,75 (m, 2H), 2,3 (s, 3Н), 2,35 (s, 3Н), 2,38 (s, 3Н), 2,7 (q, 2Н), 3,4-3,5 (m, 2H), 4,35 (d, 2H), 4,9 (bs, 1H), 6,2 (bs, 1H), 6,35 (s, 1H), 7,0-7,2 (m, 4H), 7,85 (s, 1H). Приклад 1.11 Синтез 2,3-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбоксаміду 8-Аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксамід (3,3г, 16,2ммоль), 2-етил-6-метилбензилхлорид (2,73г, 16,2ммоль), карбонат калію (8,0г, 58ммоль) і йодид калію (1,1г, 6,6ммоль) додавали до ацетону (150мл) і нагрівали під зворотним холодильником 20 годин. Додаткову кількість 2-етил-6метилбензилхлориду (1,0г, 5,9ммоль) додавали і реакційну суміш нагрівали під зворотним холодильником 7 годин. Метиленхлорид (60мл) і метанол (30мл) додавали. Реакційну суміш фільтрували і розчинники випарювали під зниженим тиском. Залишок очищували хроматографією на колонці з силікагелем, використовуючи метиленхлорид: метанол (100:7) як елюент. Кристалізація з етилацетату дала 2,8г (50%) названої сполуки. 1Н-ЯМР (300МГц, CDCI 3): d 1,2 (t, 3Н), 2,34 (s, 3Н), 2,36 (S, 3Н), 2,38 (S, 3Н), 2,7 (q, 2H), 4,4 (d, 2H), 4,9 (bs, 1H), 6,0 (bs, 2H), 6,45 (s, 1H), 7,0-7,2 (m, 3Н), 7,9, (s, 1H). Приклад 1.12 Синтез 2,3-диметил-8-(2-етил-6-метил6ензиламіно)-імідазо[1,2-а]піридин-6-карбонової кислоти 2,3-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбоксаміду мезилат (11,0г, 0,025моль) та гідроксид натрію (7,0г, 0,17моль) розчиняли в етанолі (95%) (120мл) і нагрівали під зворотним холодильником 20 годин. Розчинник випарювали під зниженим тиском і до залишку додавали воду (150мл). Рівень рН прилаштовували до 5, додаванням концентрату. НСІ, оцтову кислоті і осад твердого продукту відділяли фільтрацією, промивали водою і ацетоном, і висушували для отримання 7,6г (88%) названої сполуки. 1Н-ЯМР (500МГц, DMSO-d6): d 1,15 (t, 3Н), 2,26 (s, 3Н), 2,34 (s, 3Н), 2,39 (S, 3Н), 2,69 (q, 2H), 4,38 (d, 2H), 5,2 (bs, 1H), 6,73 (s, 1H), 7,07-7,2 (m, 3Н), 8,12 (s, 1H). Приклад 1.13 Синтез 2,3-диметил-8-(2-етил-6-метилбензиламіно)-6-(морфолінокарбоніл)-імідазо[1,2-а]піридину 2,3-Диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбонову кислоту (0,15г, 0,44ммоль) і тетрафлуорборат о-Бензотриазол-і-іл-Ν,Ν,Ν',Ν'-Тетраметилуронію (TBTU) (0,14г, 0,44ммоль) додавали до метиленхлориду (10мл). Морфолін (0,12г, 1,4ммоль) додавали і реакційну суміш перемішували при температурі навколишнього середовища 1,5 години. Реакційну суміш додавали до колонки з силікагелем і очищення хроматографією, використовуючи етилацетат: метиленхлорид (1:1) як елюент, дало 0,12г (66%) бажаного продукту. 1Н-ЯМР (300МГц, CDCI 3): d 1,2 (t, 3Н), 2,32 (s, 3Н), 2,35 (s, 3Н), 2,37 (s, 3Н), 2,7 (q, 2H), 3,7 (s, 8H), 4,35 (d, 2H), 4,95 (bs, 1H), 6,15 (s, 1H), 7,0-7,2 (m, 3Н), 7,4 (s, 1H). Приклад 1.14 Синтез (2-етил-6 метилбензиламіно)-N(2-(2-гiдроксиетокси)етил)-2,3-диметилімідазо[1,2-а]піридин-6карбоксаміду 2,3-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин-6-карбонову кислоту (0,3г, 0,88ммоль) та тетрафлуорборат о-Бензотриазол-і-іл-Ν,Ν,Ν',Ν'-Тетраметилуронію (TBTU) (0,29г, 0,90ммоль) додавали до метиленхлориду (10мл). 2-(2-аміноетокси)етанол (0,2г, 1,9ммоль) додавали і реакційну суміш перемішували при температурі навколишнього середовища 2 години. Розчинник випарювали під зниженим тиском і залишок очищували хроматографією на колонці з силікагелем, використовуючи метиленхлорид: метанол (9:1) як елюент. Кристалізація з діетилового етеру дала 0,24г (80%) бажаного продукту. 1Н-ЯМР (500МГц, CDCI 3): d 1,25 (t, 3Н), 2,25 (s, 3H), 2,3 (s, 3Н), 2,35 (S, 3Н), 2,75 (q, 2H), 3,4-3,45 (m, 2H), 3,55-3,7 (m, 6H)3 4,35 (d, 2H), 5,05 (t, 1H), 6,45 (s, 1H), 7,0-7,2 (m, 4H), 7,5 (s, 1H). Приклад 1.15 Синтез ізопропіл 8-[(2,6-диметил6ензил)аміно]-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалату Ізопропіл 8-аміно-2,3-диметилімідазо[1,2-а]піридин-6-карбоксалат (9,85кг, 1,0екв., 29,71моль) суспендували в ізопропанолі (59л); Nal (0,6екв., 2,68кг, 17,88моль) і К2СО3 (2,5екв., 10,29кг, 74,48моль) додавали і суміш нагрівали до приблизно 70°С. 2,6-Диметилбензилхлорид (1,1екв., 5,22кг, 32,77моль) розчиняли в ізопропанолі (~60л) і цей розчин додавали до реакційної суміші. По завершенні додавання, температуру підтримували 60°С ще 1,5 години. Додатковий К2СО3 додавали (9,15кг) і отриману суспензію перемішували ще 2 години при 60°С. Додатковий 2,6-диметилбензилхлорид (2,76кг) в ізопропанолі (22л) додавали повільно при температурі 60°С; після додавання реакційну суміш перемішували ще 4 години при цій температурі. Суспензію розводили з водою (124л), охолоджували, перемішували і фільтрували. Осад на фільтрі промивали водою, а тоді охолодженим ізопропанолом, висушували під зниженим тиском при 40°С для отримання 11,37кг вологого матеріалу, вихід: 90%. Приклад 1.16 Синтез 8-[(2,6-диметилбензил)аміно]-N-(2-гідроксиетил)-2,3-диметилімідазо[1,2-а]піридин-6карбоксаміду У реактор завантажували ізопропіл 8-[(2,6-диметилбензил)аміно]-2,3диметилімідазо[1,2-а]піридин-6карбоксалат (11,30кг, 1екв., 27,02моль) і ТГФ (45л), етаноламін (18,97кг, 11екв., 309,2моль) додавали при температурі приблизно 20°С. Суспензію нагрівали до приблизно 100°С. Розчинник дистилювали, а тоді ТГФ (35л) додавали і продовжували дистиляцію. Процедуру додавання ТГФ і його дистиляції повторювали до завершення перетворення. До суспензії додавали етанол (140л) і суспензію нагрівали під зворотним холодильником. Для отримання прозорого розчину, довали ще етанол (13л). Нагрітий розчин фільтрували, а тоді охолоджували. Білий твердий продукт відфільтровували, промивали етанолом і висушували для виходу продукту як білого порошку. (8271г). 2. Отримання вихідних матеріалів Приклад 2.1 Синтез 6-аміно-5-нітро-нікотинаміду 100г 6-гідрокси-5-нітро-нікотинової кислоти (0,54моль; ВЕРХ > 98% величини площі) суспендували в толуолі (750мл). ДМФ (1мл, 0,013моль, 0,024екв.) додавали і суміш нагрівали до 110°С (внутрішня температура). Тіонілхлорид (99мл, 2,5екв.) додавали протягом 120хв. Нагрівання продовжували 4 години при 110°С. Реакційну суміш концентрували до половини об'єму (400мл розчиннику дистилювали), і додавали толуол (400мл). Цю процедуру повторювали знову (410мл толуолу дистилювали і свіжий толуол (410мл) додавали знову. Розчин тоді охолоджували до 20°С і повільно додавали до водного аміаку (25%, 440мл, 12екв.) протягом 40хв. Осадження почалося відразу. Під час додавання температура трималася нижче 15°С. По завершенні додавання реакційній суміші давали можливість нагріватися до кімнатної температури і перемішування продовжували 16 годин. Твердий продукт відфільтровували, промивали водою (500мл), етанолом (250мл), ТВМЕ (250мл) і висушували (50-10мбар, температура бані 40°С, 16 годин) для виходу 91,3г названої сполуки (0,501моль, 87%). Приклад 2.2 Синтез 5,6-діаміно-нікотинаміду 44,5г 6-аміно-5-нітро-нікотинаміду (0,24моль; ВЕРХ: 93% величини площі) суспендували у суміші метанол/вода 1:1 (500мл), 5,0г каталізатору [паста Pd(4%)-Ru(l%)/C (62% Н2O тип: 485; Johnson Matthey); тип: 485; Johnson Matthey] додавали. Гідрогенізацію проводили при 5бар і 30°С протягом 5 годин. По завершенні каталізатор відфільтровували і промивали сумішшю метанол/вода 1/1 (50мл). 480мл розчиннику дистилювали. Отриману суспензію охолоджували до 20°С і відфільтровували. Твердий продукт промивали метанолом (20мл) та ТВМЕ (30мл). Після сушіння (200-10мбар; температура бані 40°С, 16 годин) 27,3г названої сполуки (0,18моль, 73%) отримували. Приклад 2.3 Синтез 5,6-діаміно-нікотинаміду 42,3г 6-аміно-5-нітро-нікотинаміду (0,23моль, ВЕРХ: 93% величини площі) суспендували в суміші метанол/вода 1:1 (500мл). 5,2г каталізатору [Pd(5%)/C (57,8% Н2О); тип: 39, Johnson Matthey] додавали. Гідрогенізацію проводили при 5бар і 30°С протягом 4 годин. По завершенні каталізатор відфільтровували і промивали сумішшю метанол/вода 1/1 (100мл). 550мл розчиннику дистилювали. Отриману суспензію охолоджували до 20°С і відфільтровували. Твердий продукт промивали метанолом (20мл) і ТВМЕ (30мл). Після сушіння (200-10мбар; температура бані 40°С, 16 годин) 28,5г названої сполуки (0,18моль, 78%) отримували.
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for the preparation of substituted imidazopyridine
Назва патенту російськоюСпособ полученния замещенного имидазопиридина
МПК / Мітки
МПК: C07D 471/04
Мітки: одержання, імідазопіридину, заміщеного, спосіб
Код посилання
<a href="https://ua.patents.su/8-73788-sposib-oderzhannya-zamishhenogo-imidazopiridinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання заміщеного імідазопіридину</a>
Похідні імідазопіридину, які інгібують секрецію шлункової кислоти

Номер патенту: 66846
Опубліковано: 15.06.2004
Автори: Дальстрем Мікаель, Старке Інґемар, Норберґ Петер, Амін Косрат
МПК: A61K 31/496, A61K 31/5355, A61K 31/437, A61P 1/04, C07D 471/04, A61P 1/00
Мітки: імідазопіридину, шлункової, інгібують, похідні, кислоти, секрецію
Формула / Реферат:
1. Сполука формули І (І)чи її фармацевтично прийнятна сіль, деR1 -(a) Н,(b) СН3 чи(c) СН2ОН,R2 -(a) СН3 чи(b) СН2СН3,R3 -(a) Н,(b) С1-6алкіл,(c) гідроксилований С1-6алкіл чи(d) галоген,R4 -(a) Н,(b) С1-6алкіл,(c) гідроксилований С1-6алкіл...
Спосіб одержання похідних заміщеного 2-нітрогуанідину

Номер патенту: 69387
Опубліковано: 15.09.2004
Автор: Майєнфіш Петер
МПК: C07D 261/08, C07D 277/28, C07D 405/14, C07D 213/89, C07D 277/32, C07D 251/08, C07D 413/14, C07D 277/20, C07D 213/40, C07D 261/10, C07D 417/14, C07D 307/14, C07D 213/61, C07D 401/14
Мітки: одержання, похідних, заміщеного, спосіб, 2-нітрогуанідину
Формула / Реферат:
1. Спосіб одержання заміщеного 2-нітрогуанідину формули, (I)та, у відповідних випадках, його E/Z-ізомерів, сумішей E/Z-ізомерів та/або таутомерів, відповідно, у вільній формі або у формі солей, де R1 є воднем або С1-С4-алкілом,R2 є воднем, С1-С6-алкілом, С3-С6-циклоалкілом або радикалом -СН2В, Неt є незаміщеним або - залежно від здатності...
Спосіб та проміжні сполуки для одержання похідних заміщеного хроманолу

Номер патенту: 58520
Опубліковано: 15.08.2003
Автори: Касталді Майкл Джей., Раггері Селлі Г., Даггер Роберт В., Хокінс Джоел М., Келлі Сара Е., Реггон Джеффрі В., Піскопіо Ентоні Д., Карон Стефан
МПК: B01J 23/44, C07C 33/00, C07D 311/22, C07F 5/00, B01J 31/16
Мітки: сполуки, спосіб, проміжні, одержання, хроманолу, похідних, заміщеного
Формула / Реферат:
1. Спосіб одержання похідних заміщеного хроманолу формули (Х) (X)або енантіомера цієї сполуки, причому у сполуці формули (Х) R3 - заміщена частина бензойної кислоти приєднана до атома вуглецю в положенні 6 або 7 хроманового кільця;R1 означає -(CH2)qCHR5R6, де q означає число від 0 до 4;кожен R2 і R3 незалежно вибирають із групи, що складається з водню, фтору, хлору, С1-С6-алкілу, С1-С6-алкокси, фенілсульфінілу,...
Спосіб одержання заміщеного орто-нітробензонітрилу

Номер патенту: 71932
Опубліковано: 17.01.2005
Автори: Віой Агнесса, Раттон Патрік, Стефан Домінік, Касадо Майкл
МПК: C07C 255/49, C07C 205/00, C07B 61/00, C07C 253/14
Мітки: орто-нітробензонітрилу, заміщеного, спосіб, одержання
Формула / Реферат:
1. Спосіб одержання сполуки формули І:, (І)де R1 - С1-4-галогеналкіл,R2 - водень або С1-4-алкокси,який включає взаємодію відповідного орто-нітрогалогенбензолу формули II:, (ІІ)де R1 та R2 мають вищевказані значення, а Х - атом фтору або брому,у випадку,...
Бензамід 3- або 4-заміщеного 4-(амінометил)піперидину, спосіб його одержання (варіанти), фармацевтична композиція на його основі, спосіб її одержання, проміжна сполука та спосіб її одержання

Номер патенту: 67745
Опубліковано: 15.07.2004
Автори: Босманс Жан-Поль Рене Марі Андре, Суркін Мішель, де Клеін Мішель Анна Жозеф
МПК: C07D 211/42, A61K 31/443, A61K 31/4427, A61K 31/453, A61K 31/497, A61K 31/4525, A61P 1/10, A61K 31/4545, A61K 31/502, C07D 405/12, A61K 31/501, C07D 405/06, C07D 405/14, A61P 1/00, C07D 211/44, A61K 31/445, A61K 31/454, A61P 43/00, C07D 401/06
Мітки: проміжна, 4-(амінометил)піперидину, одержання, композиція, фармацевтична, основі, спосіб, 4-(заміщеного, бензамід, сполука, варіанти
Формула / Реферат:
1. Сполука бензаміду 3- або 4-заміщеного 4-(амінометил)піперидину формули (I), (I)її стереоізомерні форми, N-оксиди або фармацевтично прийнятні солі з кислотами або лугами, деR1 і R2, взяті разом, утворюють бівалентний радикал формули-O-CH2-O- (a-1),-O-CH2-CH2- (a-2),-O-CH2-CH2-O- (a-3),-O-CH2-CH2-CH2-...
Попередній патент: Перетворювач енергії хвиль
Наступний патент: 1,3-діоксолани з протикашльовою активністю
Випадковий патент: Спосіб і пристрій для одержання надглибокого вакууму в населеному відсіку космічного об'єкта