1,3-діоксолани з протикашльовою активністю
Номер патенту: 73789
Опубліковано: 15.09.2005
Автори: Пеллєгріні Луіджи, Мелілло Габріелла, Курті Роберто, Честа Марія Кандіда, Аллєгретті Марчело




Формула / Реферат
1. Сполука формули (I)
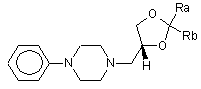 , (I)
, (I)
де кожний з Ra та Rb, які можуть бути однаковими або різними, являє собою водень, С1-С6-алкіл, феніл; або
Ra та Rb разом з атомом С, до якого вони приєднані, утворюють необов'язково заміщене 4-7-членне карбоциклічне кільце.
2. Сполука за п. 1, де Ra та Rb є однаковими.
3. Сполука за п. 1 або 2, де Ra та Rb являють собою метил або етил.
4. Сполука за п. 1, вибрана з наступних сполук:
S(-)-1,2-циклопентиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол,
S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол;
S(-)-1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол;
S(-)-1,2-(3-пентиліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол;
S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолмалеат;
S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол-L-тартрат;
S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолфумарат;
S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол-D-10-камфорсульфонат.
5. Сполука за пп. 1-4 для одержання лікарських засобів з протикашльовою активністю.
6. Спосіб одержання сполук формули (I) взаємодією фенілпіперазину з (R)-1,2-гліцерилдіоксоланом формули (II):
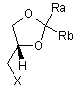 , (II)
, (II)
де X вибраний з групи, що включає Сl, Br, I і відповідний сульфоновий ефір (R-SO3-), R являє собою С1-С3-алкіл, трифторметил, феніл, п-толіл або п-метоксифеніл.
7. Спосіб за п. 6, де взаємодію проводять в присутності основи при використанні толуолу або ксилолу як розчинників.
8. Фармацевтична композиція, яка містить сполуки за пп. 1-4 в суміші з відповідними носіями.
Текст
Даний винахід відноситься до (S)-3-(4-феніл-1-піперазиніл)-1,2-пропандіолових циклічних ацеталей, які можуть використовува тися як протикашльові засоби і як проміжні продукти при одержанні леводропропізину та його солі. Винахід відноситься також до способу одержання вказаних ацеталей. Більш конкретно, даний винахід відноситься до (S)-2,2-замішених-1.3-діоксоланів формули (1): де: кожний з Ra та Rb, які можуть бути однаковими або різними, являє собою водень, С1- С6-алкіл, феніл; або Ra та Rb разом з атомом С, до якого вони приєднані, утворюють необов'язково заміщене 4-7-членне карбоциклічне кільце. Переважними сполуками формули (1) є сполуки, в яких Ra та Rb являють собою алкільні групи, що містять менше 6 атомів С. Переважно, Ra та Rb є однаковими; більш переважно, Ra та Rb являють собою метил або етил або разом з атомом С, до якого вони приєднані, утворюють кільце, що містить від 5 до 6 атомів вуглецю. Винахід відноситься також до енантіомерно чистих моноосновних солей (S)-2,2-заміщених-1,3-діоксоланів формули (1) з фармацевтично прийнятними кислотами. Особливо переважними фармацевтично прийнятними кислотами є оцтова, пропіонова, бурштинова, фумарова, малеїнова, L-яблучна, D- і L-винна, D- і L-мигдалева, L- і D-камфорсульфонова кислоти. Особливо переважними сполуками за даним винаходом є S(-)-1,2-циклопентиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-(3-пентиліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол малеат; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол L-тартрат; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол фумарат; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол D-10-камфорсульфонат. Сполуки за даним винаходом формули (1) одержують взаємодією фенілпіперазину з (R)-1,2гліцерилдіоксоланом формули (2): де X вибраний з групи, що включає СІ, Вr, І і відповідний складний сульфоновий ефір (R-SO3-), де R являє собою С1-С3-алкіл, трифторметил, феніл, п-толіл або п-метоксифеніл. Діоксолани формули (2) є відомими сполуками і/або можуть бути одержані відомими методами. Більш конкретно, складні сульфонові ефіри формули (2) (X=R-SO3-), одержують стандартними методами з використанням ангідриду або хлорангідриду алкіл- і/або арилсульфонової кислоти формули (3): R-SO3H (3) для етерифікації (R)-2,2- заміщеного-1,3-діоксолан-4-метанолу формули (4): де Ra та Rb приймають значення, визначені вище. 1,3-Діоксолан-4-метаноли формули (4) також є відомими сполуками, і їх одержання досить детально описане в літературі. Наприклад, вони можуть бути одержані ферментативним поділом рацематів формули (4) відповідно до способу, описаного в US 5190867 (02.03.1993) або, переважно, окислювальним розкладом 1,2;5,6-біс-діоксоланів D-маніту відповідно до способу, описаного Borsa et al., в ЕР 147847 (07.03.1990) і використаного для одержання (+)-1,2-ізопропіліден-sn-гліцерину (формула (4), де Ra та Rb являють собою метил) і його тозилату (формула (3), де Ra та Rb являють собою метил і R являє собою п-толіл). Зручні способи одержання 1,2;5,6-біс-діоксоланів D-маніту і відповідних D-гліцеральдегідацеталей можна знайти також в публікаціях J. Org. Chem. 56, 4056 (1991) і Synthesis 587 (1992), де детально описане одержання 2,3-О-(3-пентиліден)-D-гліцеральдегіду з 55% виходами з D-маніту. 4-Галогенметилдіоксолани формули (2), де X являє собою СІ, Вr або І, в свою чергу, можуть бути одержані, виходячи з відповідних ефірів формули (2) (Х= RSO3-, де R приймає значення, визначені вище), взаємодією з відповідним галогенідом лужного або лужноземельного металу (Na, К або Са) в інертному розчиннику, вибраному з групи, що включає ацетон, метилетилкетон, тетрагідрофуран, діоксан, диметилсульфоксид, ацетонітрил, С1-С4-спирт та їх суміші. Альтернативно, діоксолани формули (2) (Х=Сl, Вr) можуть бути одержані введенням діоксоланових груп у відповідні 3-галоген-1,2-пропандіоли, як описано в ЕР 0930311 (21.07.1999). Особливо переважним є 3 хлорпропандіол. Переважними регентами введення ацетальних груп є формальдегід, ацетальдегід та бензальдегід, ацетон, діетилкетон, бензофенон, циклогексанон та ацеталі або їх прості фенольні ефіри, такі як 2,2-диметоксипропан, 2,2-диметоксіетан і 2-метоксипропен. Альтернативно, діоксолани формули (2) (Х=С 1, Вr) можуть бути одержані введенням ацетальної групи в хіральні епіхлоргідрини або епібромгідрини або відповідні хіральні 3-галогенпропандіоли за допомогою циклоалканону відповідно до способу, описаного для одержання (±)2-хлорметил-1,4-діоксаспіро[4,5]декану в FR 1552153 або описаними в більш загальному вигляді в публікаціях Blicke F.F. et al, J.A.C.S, 74, 1735 (1972) та ibidem, 76, 1226 (1954). Вказані хіральні епіхлоргідрини або епібромгідрини і відповідні 3-галогенпропандіоли, в свою чергу, являють собою доступні проміжні продукти, які можуть бути одержані, наприклад, кінетичним поділом відповідних рацематів [відповідно до публікації Furrow et al., J. Org. Chem., 63, 6776, 1998 або, альтернативно, відповідно до публікації Т. Takeichi et al., Tetrahedron, 36, 3391 (1980)], або ферментативним поділом (див. публікації Kasai N. et al., JP 02257895 (1990); CA.: 114,41064q, 1991). Фенілпіперазини алкілуються 1,2-гліцерилдіоксоланом формули (2) в стандартних умовах реакції перетворення вторинного аміну в третинний при використанні на кожний моль алкілуючого агента формули (2), щонайменше, одного моля або невеликого молярного надлишку фенілпіперазину в присутності, щонайменше, одного моля протиоснови. Протиоснова вибрана з групи, що включає тонко подрібнені неорганічні основи, такі як карбонати або гідрокарбонати лужних або лужноземельних металів (Na, К, Mg, Са), оксиди Са або Mg, третинні аміни, такі як триетиламін, диметил-або діетиланілін, ароматичні аміни, такі як піридин, піколін або етилметилпіридин і, якщо це бажано, безпосередньо фенілпіперазин, який потім може повторно циклізуватися з одержанням циклу. Реакцію алкілування можна провести при нагріванні, необов'язково в присутності інертних розчинників, таких як толуол і/або ксилол, які при кипінні розчинника будуть переважно зменшува ти тривалість реакції. Після завершення реакції алкілування будь-які нерозчинні речовини відфільтровують або видаляють центрифугуванням, потім органічні фази повторно промивають водою для простого видалення домішок і побічних продуктів і розчинник відганяють і одержують з високими виходами залишок, який складається з, по суті, чистого 1,3-діоксолану за даним винаходом формули (1), який виділяють прямою кристалізацією або одержанням солі бажаної фармацевтично прийнятної кислоти. Сполуки (1) та їх солі надзвичайно легко кристалізуються із звичайних розчинників: отже, спосіб за даним винаходом знижує до мінімуму будь-який ризик забруднення продукту слідовими кількостями гліцидолів і/або епігалогенгідринів як можливих домішок. Моноосновні солі сполук формули (1) одержують звичайними стандартними способами, такими як одержання солей з еквімолекулярними кількостями бажаної кислоти у відповідному розчиннику з подальшою кристалізацією одержаної солі. Крім того, сполуки за даним винаходом формули (1) і їх моноосновні солі є, по суті, позбавленими смаку і не мають залишковий гіркий присмак леводропропізину. Сполуки (1) є ефективними проліками леводропропізину, у водних розчинах яких 1,3-діоксолани формули (1) або їх солі перетворюються в леводропропізин в результаті гідролізу в теплому середовищі при молярному надлишку розбавленої мінеральної кислоти, такому як соляна кислота, або розчинних у воді карбонових кислот, таких як оцтова, малонова або лимонна кислоти. Сполуки формули (1) і водні розчини їх моноосновних солей є стабільними при фізіологічних значеннях pH. Сполуки за даним винаходом і їх солі мають протикашльову активність, що доведено результатами порівняльних біологічних випробувань при внутрішньовенному способі введення і в порівнянні з самим леводропропізином. Випробування проводилися на самцях морської свинки Dunkin-Hartley (4-6 тварин в кожній групі), їх обробляли аерозолем 0,0045% (г./об.) водного розчину капсацину (capsacin) [Lavezzo Α., Pulm. Pharmacol., 5, 143-147, 1992; Galileo L. et al., Br. J. Pharmacol., 112, 795-800, 1994] протягом 5 хвилин після внутрішньовенного введення 0,5мл розчину (-)-1,2-циклогексилідин-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу фумарату (DF 1689A, 10мг/кг), або, для порівняння, 0,5мл розчину леводропропізину (pH 4,5, 10мг/кг), або рівного об'єму сольового розчину (контроль). Кашель підраховувався протягом 4 хвилин обробки аерозолем капсицину, і таким чином були одержані докази значного інгібування (47,3%) відповіді на стимулювання кашлю в порівнянні з 43,6% інгібуванням, обчисленим для контрольної групи. Фактично, одержані наступні кількісні показники кашлю: 9,17±1,01 для контрольних тварин, 5,17±0,54 для тварин, оброблених леводропропізином, 4,84±0,54 для тварин, оброблених сполукою за винаходом DF 1689A. Протикашльова дія додатково характеризується тривалим ефектом: фактично, показники інгібування кашлю, стимульованого аерозольним введенням розчину капсацину, які дорівнюють 34,6 і 27,3%, були обчислені через 15 і 30 хвилин після внутрішньовенного введення. Сполуки формули (1) та їх солі можуть відповідним чином бути введені у вигляді аерозолю; дійсно, самці морської свинки Dunkin-Hartley, що зазнали обробки аерозолем 1% (мас/об.) водних розчинів сполуки DF 1689A і леводропропізину протягом 10 хвилин, показали значне зменшення кількості приступів кашлю, індукованих цими лікарськими засобами, в порівнянні з контролем (аерозоль сольового розчину), при приблизно 35% інгібіторної дії для обох лікарських засобів. Для застосування як протикашльових засобів сполуки (1) будуть вводитися в фармацевтичні композиції стандартними методами в поєднанні зі стандартними наповнювачами для введення пероральним, парентеральним способами або за допомогою аерозолю, наприклад, у вигляді капсул, таблеток, що не розкладаються в шлунку, сиропів, препаратів з виділенням, що контролюється, діючої речовини. Величина добової дози буде залежати від різних факторів, таких як частота і сила кашлю і загальний стан пацієнта (вік, стать і маса тіла). Добова доза для дорослого суб'єкта масою 60кг буде коливатися в інтервалі від 10мг до 1500мг сполук формули (1) на добу, при цьому необов'язково може бути розбита на декілька введень. Сполуки за даним винаходом можуть також вводитися дітям, навіть протягом тривалого періоду, в прийнятних дозах завдяки їх низькій токсичності. І нарешті, сполуки за даним винаходом можуть використовуватися як проміжні продукти при одержанні леводропропізину та його солі. Наведені далі приклади додатково ілюструють даний винахід. Приклад 1 До розчину 13,8г вологого 1,2;5,6-ди-О-(пентиліден)-D-маніту (одержаного відповідно до способу, описаного в публікації Synthesis, 587, 1992) в тетрагідрофурані (ТГФ), охолодженому до 20-25°С, протягом 10 хвилин додають суспензію періодату калію (7,95г) та бікарбонату калію (0,32г) у воді (50мл). Суміш енергійно перемішують протягом трьох годин, охолоджують до 5°С і фільтрують. Осаджений йодат калію промивають етилацетатом і дві об'єднані фази залишають нагріватися до кімнатної температури. Водну фазу обробляють NaCl і повторно екстрагують етилацетатом. До об'єднаних органічних фаз при енергійному перемішуванні додають розчин 3,2г NaBH4 і 1,88г броміду тетрабутиламонію в 120мл води. Суміш взаємодіє при кімнатній температурі протягом 3 годин, фази розділяють, водну фазу екстрагують етилацетатом (2x30мл), фільтрують, сушать над сульфатом натрію і розчинник випаровують у вакуумі. Одержаний маслянистий залишок відганяють при зниженому тиску з одержанням 7,12г 2,3-О-(3-пентиліден)-D-гліцерину, [a]D=±17,2 (EtOH). До розчину 6,45г одержаного спирту в етилацетаті (18мл) додають 6мл триетиламіну, потім при перемішуванні із зовнішнім охолоджуванням розчин 7,76г п-толуолсуль фохлориду в AcOEt (18мл). Суміш перемішують протягом 12 годин при кімнатній температурі, потім розбавляють водою (10мл), фази розділяють, промивають водою (3x10мл), суша ть над сульфатом натрію, фільтрують і випаровують досуха при зниженому тиску з одержанням 11,98г 2,3-О-(3-пентиліден)-D-гліцерину тозилату, [a]D= -4 (ДМФА). До розчину тозилату в н-бутанолі (70мл) послідовно додають при енергійному перемішуванні 4,5г тонко подрібненого карбонату натрію і 6мл фенілпіперазину. Суміш кип'ятять із зворотним холодильником при перемішуванні, потім залишають для взаємодії при температурі кипіння розчинника ще на 20 годин. Потім бутанол випаровують при зниженому тиску, залишок обробляють водою і повторно екстрагують етилацетатом. Об'єднані органічні фази сушать і фільтрують, потім розчинник випаровують у вакуумі. Одержаний залишок кристалізують з водного метанолу з одержанням 8,95г S(-)-1,2-(3-пентиліден)-3-(4-фенілпіперазин-1іл)пропан-1,2-діолу, іншою назвою якого є 4-фенілпіперазин, 1-(2,2-діетил-1,3-діоксолан-4-ілметил). Приклад 2 Відповідно до методики прикладу 1 при використанні 2,3-О-(2-пропіліден)-D-гліцерину тозилату одержують S(-)-1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол, іншою назвою якого є 4-фенілпіперазин, 1(2,2-диметил-1,3-діоксолан-4-ілметил). Приклад 3 До розчину 3,1г 2,3-О-(3-циклогексиліден)-D-гліцерину [a]D=+15,6 (EtOH) в дихлорметані (10мл) в атмосфері інертного газу додають 2,8мл триетиламіну і потім після охолоджування до приблизно 15°С по краплях розчин 1,44мл метансульфохлориду в 2мл дихлорметану, не допускаючи підвищення температури вище 30°С. Одержану суспензію перемішують протягом 1 години при кімнатній температурі, потім розбавляють крижаною водою. Органічну фазу відділяють, промивають водою, знебарвлюють активним вугіллям і випаровують у вакуумі з одержанням залишку 4,3г 2,3-O-(3-циклогексиліден)-D-гліцерину мезилату, [a]D =-3,3 (СНСl3). 1 Н ЯМР: δ 4.37(м, 1Н); δ 4.24 (м, 2Н); δ 4.1 (дд, 1Н, J1=8.7 Гц, J2=6.4 Гц); δ 3.8 (дд, 1H, J1=8.7 Гц); δ 3.1(c, 3Н); δ 1.7,1.3 (м, 10Η) До розчину мезилату в толуолі (16мл) додають до 5,45мл фенілпіперазину, суміш кип'ятять із зворотним холодильником до завершення реакції (приблизно 8 годин). Реакційну суміш охолоджують до приблизно 50°С, додають 10мл води і енергійно перемішують протягом, щонайменше, 10 хвилин. Фази розділяють і органічну фазу повторно промивають водою. Розчинник випаровують з одержанням густого масла, яке розчиняють в теплому ізопропанолі (15мл). Потім розчин повільно охолоджують для відділення твердого кристалічного S(-)1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу. Т. пл. 63-64°С, [a]D=-7,8 (1% МеОН). 1 Н ЯМР: δ 7.26(т, 2Н, J=7.4 Гц); δ 6.95(д, 2Н, J=8.9 Гц); δ 6.85(т, 1Η, J=7.28 Гц); δ 4.3 (м, 1Η, J=6.1 Гц); δ 4.1 (дд, 1Η, J1=8.0 Гц, J2=6.1 Гц); δ 3.65 (дд, 1Η, J1=8.0, J2=7.1 Гц); δ 3.2(т, 4Η, J=5.05 Гц); δ 2.9, 2.55 (м, 6Н); δ 1.7, 1.3 (м, 10Н) Приклад 4 Відповідно до методики, описаної у прикладах 1 і 2, взаємодією 1,3-діоксолану, вибраного з групи, що включає 2S-1,4-діоксаспіро[4.4]нонан-2-метанол п-толуолсульфонат; 2S-1,4-діоксаспіро[4.4]нонан-2-хлорметил; 2S-1,4-діоксаспіро[4.5]декан-2-метанол трифторметансульфонат; 2S-1,4-діоксаспіро[4.5]декан-2-хлорметил; 2S-1,4-діоксолан-4-хлорметил-2,2-диметмл; 2S-1,4-діоксолан-4-йодметил-2,2-диметил, з фенілпіперазином одержують такі сполуки: S(-)-1,2-циклопентиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол; S(-)-1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол. Приклад 5 0,232г Фумарової кислоти при перемішуванні додають до розчину S-(-)-1,2-циклогексиліден-3-(4фенілпіперазин-1-іл)пропан-1,2-діолу (0,632г) в абсолютному етанолі (8мл). Суміш перемішують до повного розчинення речовин, потім розчинник випаровують у вакуумі і твердий залишок кристалізують з ацетону з одержанням твердого кристалічного S(-)-1,2-циклогексиліден-2-(4-фенілпіперазин-1-іл)пропан-1,2-діолу фумарату (DF1689A, 0,52г), т.пл. 162-164°С, [a]D=-15,7(c=1%;CH3OH). 1 H ЯМР: δ 7.5 (дд, 2Н, J1=8.7 J2=7.3); δ 7.2 (м, 3Н); δ 6.75(с, 2Н); δ 4.8 (м, 1H); δ 4.35 (дд, 1H, J1=8.9 Гц, J2=6.7Гц); δ 3.86 (дд, 1Н, J1=8.9 Гц, J 2=5,8 Гц); δ 3.6-3.4 (м, 10Н); δ 1.8-1.48 (м, 10Н) Приклад 6 Відповідно до способу одержання солі S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу і L-винної, малеїнової, D(+)-10-камфорсульфонової, L-мигдалевої кислот, одержують такі сполуки: S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол L-тартрат, т.пл. 130-132°С, [a]D=-9,7° (c=1%; МеОН); S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол малеат, т.пл. 160-162°С, [a]D=-23,6 (с=1%; СН3ОН); S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол, D(+)-10-камфорсульфонат, т.пл. 110115°С, [a]D -3,9 (с=1%; СН3ОН); S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діол, L-манделат, [a]D =+26,8 (с=1%; СН3ОН). Приклад 7 Суспензію 3,5г S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу в 70мл водної оцтової кислоти (10% мас/об.) кип'я тять із зворотним холодильником протягом 2 годин, потім в неї барботують пар для відгонки циклогексанону, який відділяють. Водну фазу нейтралізують до pH 7 додаванням 10% розчину NaOH, потім охолоджують до 5-10°С з одержанням 2,05г (-)3-(4-фенілпіперазин-1-іл)пропандіолу, т.пл. 102-103°С, [a]D=-23,5° (2,8% СН2Сl2). Приклад 8 Альтернативно, 0,35 моль-еквівалентів одного з 1,3-діоксолан-похідних, описаних в прикладах 1-4, а саме S(-)-1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу; S(-)-1,2-(циклопентиліден)-3-(4фенілпіперазин-1-іл)пропан-1,2-діолу; S(-)-1,2-циклогексиліден-3-(4-фенілпіперазин-іл)пропан-1,2-діолу; S(-) 1,2-(2-пропіліден)-3-(4-фенілпіперазин-1-іл)пропан-1,2-діолу, при перемішуванні додають частинами до 36% розчину соляної кислоти (36мл) в 45мл води; суспензію нагрівають до 80°С з одержанням прозорого розчину, який витримують при цій температурі протягом 30 хвилин, потім охолоджують до 20-25°С. Водну фазу екстрагують дихлорметаном (3x15мл), потім додають н-бутанол (0,5л). Двофазну суміш кип'ятять для відгонки азеотропної суміші вода:н-бутанол, виділяючи приблизно 300мл дистиляту, потім охолоджують для прискорення кристалізації (-)-3-(4-фенілпіперазин-1-іл)пропандіолу гідрохлориду (85г). Розчин гідрохлориду в 125мл води знебарвлюють активованим вугіллям (2,12г), нагріваючи до 50°С і витримуючи при цій температурі протягом 15 хвилин, фільтрують і потім нейтралізують додаванням водного розчину гідроксиду амонію (30% мас/мас). Розчин нагрівають до 50°С і ініціюють кристалізацію додаванням кристалів (-)-3-(4-фенілпіперазин-1-іл)-1,2-пропандіолу. Суспензію залишають для мимовільного охолоджування, потім витримують протягом 2 годин при +2 - +4°С і фільтрують з одержанням 70-72г (-)3-(4фенілпіперазин-1-іл)-1,2-пропандіолу.
ДивитисяДодаткова інформація
Назва патенту англійською1,3-dioxolans with antitussive activity
Назва патенту російською1,3-диоксоланы с противокашлевой активностью
МПК / Мітки
МПК: A61K 31/496, C07D 317/28, A61P 11/14
Мітки: 1,3-діоксолани, активністю, протикашльовою
Код посилання
<a href="https://ua.patents.su/4-73789-13-dioksolani-z-protikashlovoyu-aktivnistyu.html" target="_blank" rel="follow" title="База патентів України">1,3-діоксолани з протикашльовою активністю</a>
Фенілфенантридини з інгібувальною щодо фде-4 активністю

Номер патенту: 69436
Опубліковано: 15.09.2004
Автори: Бойме Рольф, Хацельманн Армін, Грюндлер Герхард, Амшлер Германн (помер), Флокерзі Дітер, Клей Ханс-Петер, Босс Хільдегард, Бундшух Даніела
МПК: A61P 19/02, A61P 17/04, A61K 31/5377, A61P 13/04, A61P 25/28, A61P 15/10, C07D 221/12, A61P 17/14, A61P 11/02, A61P 25/24, A61P 37/04, C07D 401/12, A61K 31/496, A61P 17/16, A61P 37/06, A61P 9/04, A61P 17/08, C07D 403/10, A61P 1/00, A61P 37/08, A61P 11/08, A61P 11/06, A61P 17/06, C07D 401/10, A61P 11/00, A61P 31/00, A61P 11/16, A61P 13/12, A61K 31/473, A61P 27/14, A61P 17/10, A61P 1/04, A61P 29/00, A61P 25/04
Мітки: інгібувальною, фде-4, фенілфенантридини, активністю
Формула / Реферат:
1. Фенілфенантридини формули І, (І)у якійR1 означає гідроксил, С1-С4алкокси, С3-С7циклоалкокси, С3-С7циклоалкілметокси або повністю чи переважно фторзаміщений С1-С4алкокси,R2 означає гідроксил, С1-С4алкокси, С3-С7циклоалкокси, С3-С7циклоалкілметокси або повністю чи переважно фторзаміщений С1-С4алкокси або в якійR1 і R2 спільно означають...
Індазольні сполуки, фармацевтична композиція на їх основі, спосіб модулювання активності рецептора протеїнкіназ, спосіб лікування захворювань, опосередкованих активністю протеїнкінази

Номер патенту: 66933
Опубліковано: 15.06.2004
Автори: Томас Крістіна, Браганза Джон Ф., Х'юа Є., Рейч Зігфрід Хайнц, Борчардт Аллен Дж., Каніа Роберт Стівен, Варней Мікаель Девід, Джонсон Теодор Отто Джр., Коллінз Міхаел Раймонд, Валлейс Мікаель Бреннан, Бендер Стівен Лі, Кріппс Стефан Джеймс, Темпчик-Расселл Анна Марі, Джонсон Міхаель Девід, Л'юю Хєп Те, Палмер Цінтіа Луіза, Тенг Мін
МПК: A61K 31/4178, A61P 43/00, A61P 27/02, C07D 405/14, C07D 403/04, C07D 417/12, C07D 471/04, C07D 409/12, A61K 31/454, C07D 209/18, C07D 403/12, C07D 417/14, A61K 31/496, A61K 31/416, C07D 401/12, C07D 413/12, C07D 405/06, A61K 31/4709, C07D 409/06, A61P 3/10, C07D 403/14, C07D 401/06, A61P 35/00, A61P 17/06, A61P 19/02, A61K 31/427, A61P 29/00, A61K 31/4196, C07D 405/04, A61K 31/4245, A61K 31/4439, A61K 31/444, C07D 401/14, A61P 27/06, C07D 231/56, A61P 9/10, C07D 491/056, A61K 31/4184, A61K 31/437, C07D 401/04, A61K 31/4188, A61K 31/5377, C07D 409/14, C07D 413/14
Мітки: протеїнкіназ, рецептора, фармацевтична, опосередкованих, індазольні, модулювання, композиція, лікування, протеїнкінази, активності, активністю, сполуки, спосіб, основі, захворювань
Формула / Реферат:
1. Сполука формули І:, ІдеR1 є заміщеним або незаміщеним арилом або гетероарилом, або групою формули СН=СН-R3 або СН=N-R3, де R3 є заміщеним або незаміщеним алкілом, алкенілом, циклоалкілом, гетероциклоалкілом, арилом або гетероарилом, таR2 є заміщеним або незаміщеним арилом , гетероарилом або Υ-X, де Υ є О, S, С=СН2, С=О, S=О, SО2, алкіліден, NH або N-(С1-C8алкіл), та Χ є заміщений або незаміщений...
0,0-диетил-s-b-тіоціанетилдитіофосфат, володіючий акарицидною активністю

Номер патенту: 39257
Опубліковано: 15.06.2001
Автори: Мельник Ярослав Гнатович, Сікура Наталія Михайлівна, Мельник Галина Федорівна
МПК: A01N 59/26, A01N 57/10, A01P 7/02
Мітки: активністю, володіючий, 0,0-диетил-s-b-тіоціанетилдитіофосфат, акарицидною
Формула / Реферат:
O, O-Діетил-S-b-тіоціанетилдитіофосфат формули, (C2 H5O)2 P(S)SCH2CH2-SCNщо має акарицидну активність.
0,s-етилен (0,0-диетилтіофосфорил)дипропілфосфат, володіючий інсектицидною активністю

Номер патенту: 39258
Опубліковано: 15.06.2001
Автори: Гриб Олег Кузьмич, Мельник Ярослав Гнатович, Мельник Галина Федорівна
МПК: A01N 57/10, A01N 59/26, A01P 7/04
Мітки: володіючий, 0,0-диетилтіофосфорил)дипропілфосфат, активністю, інсектицидною, 0,s-етилен
Формула / Реферат:
O, S -Етилен(O, O-діетилдитіофосфорил)дипропілфосфат формули, (C2 H5O)2 P(S)CH2 CH2O (O)P(OC3 H7)2що має інсектицидну активність.
S, o-етилен (о,о-диетилтіофосфорил)-диетилтіофосфат, володіючий інсектицидною активністю

Номер патенту: 32278
Опубліковано: 15.12.2000
Автори: Гриб Олег Кузьмич, Мельник Ярослав Гнатович, Мельник Галина Федорівна
МПК: A01N 59/26, A01N 57/10, A01P 7/04
Мітки: o-етилен, о,о-диетилтіофосфорил)-диетилтіофосфат, активністю, володіючий, інсектицидною
Текст:
...сполуки доведена хроматографічним методом , склад елєментнда аналізом, а будова-спектральними методами. ПЙіКГДЦ І. До 4.28 г. / 0.02 г.моль / 0,0-диетил-3 - й -оксиетилтіо^осйату і И.О.'З г. /0.02 г.моль / триетилшіну у 10 діл е^іру при інтенсивному перемішуванні і кімнатній температурі краплинами додають 3.76 г. / 0,0^ г.моль / 0,0-диетилхлортіогТюсфату в 5 мл ефіру. Реакпілпу суміш нагрівають при кипінні протягом 2 год, і після...
Попередній патент: Спосіб одержання заміщеного імідазопіридину
Випадковий патент: Спосіб очистки 4-/н-алкокси-с3-с5/-4'-циано-1,1'біфенілів