Спосіб отримання таксану



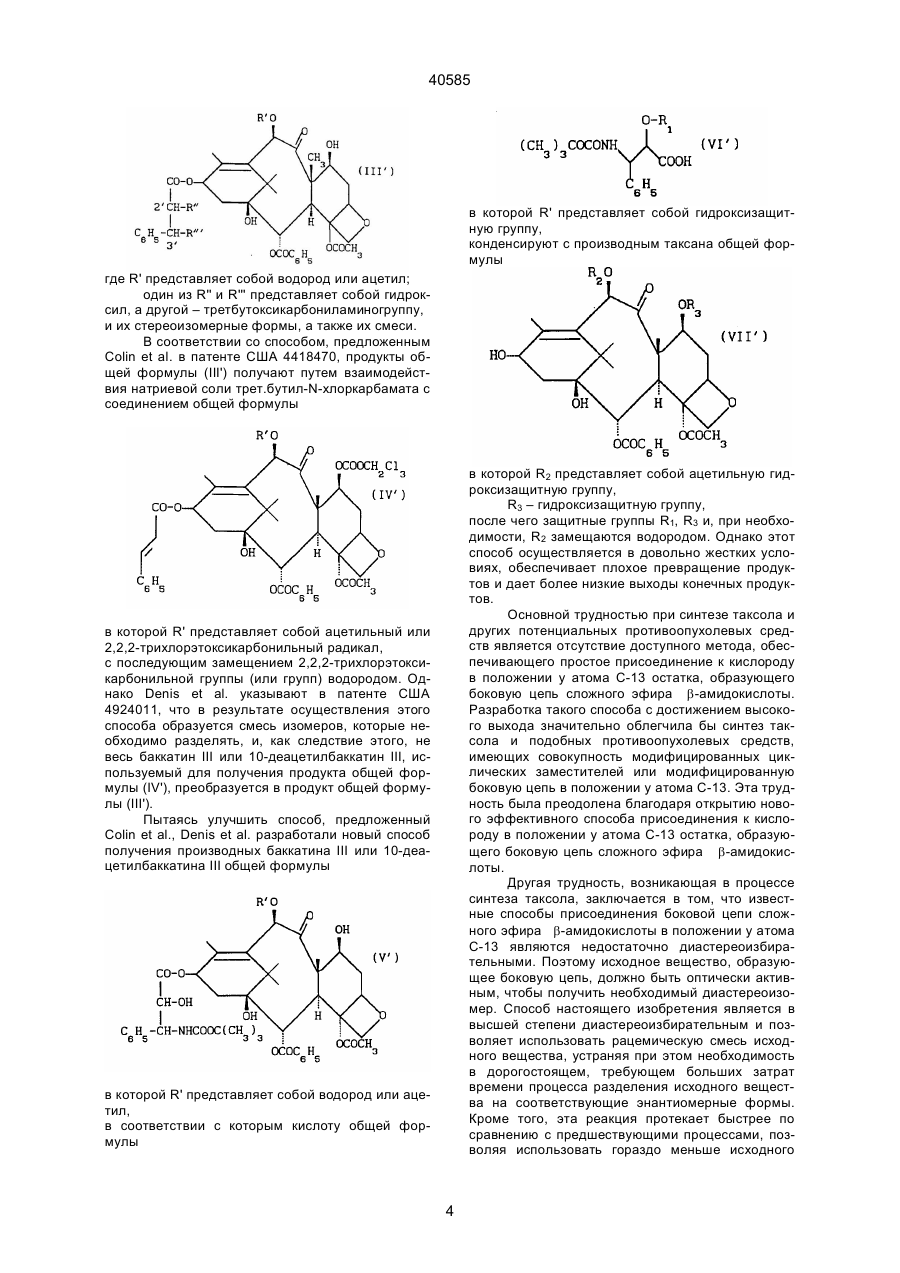



Формула / Реферат
1. Способ получения таксана формулы
где R1 означает -OR6, R3 означает водород, алкил, алкенил, алкинил, арил, гетероарил, выбранный из 5- или 6-членого гетероцикла, содержащего один или более атомов кислорода, серы или азота, или ацил;
R5 означает -COR10, -COOR10, -COS-алкил или –CONR8R10;
R6 означает водород или гидроксизащитную группу;
R8 означает водород, алкил, алкенил, алкинил, арил или гетероарил, содержащий один или два гетероатома, выбранных из серы, азота и кислорода;
R10 означает низший алкил, алкенил, алкинил, арил или гетероарил, содержащий один или два гетероатома, выбранных из серы, азота и кислорода;
R15 и R16 независимо друг от друга означают водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси, или R15 и r16 вместе образуют оксогруппу;
R17 и R18 независимо друг от друга означают водород, или вместе образуют оксогруппу;
R19 и R20 независимо друг от друга означают водород, гидрокси, защищенную гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси;
R24 означает водород, гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси;
R25 означает водород, гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси или R25 и R26 вместе взятые, образуют оксогруппу;
R26 означает водород или вместе с R25 образует оксогруппу;
R27 означает водород, гидрокси или низшую алкокси, алканоилокси, алкеноилокси, алкиноилокси или арилоилокси;
с использованием реакции этерификации с образованием сложного амидоэфира таксана, с последующим в случае необходимости удалением защитных групп, отличающийся тем, что соответствующий алкоксид металла формулы
где М означает металл, подвергают взаимодействию с β-лактамом формулы
где R1-R27 имеют указанные значения.
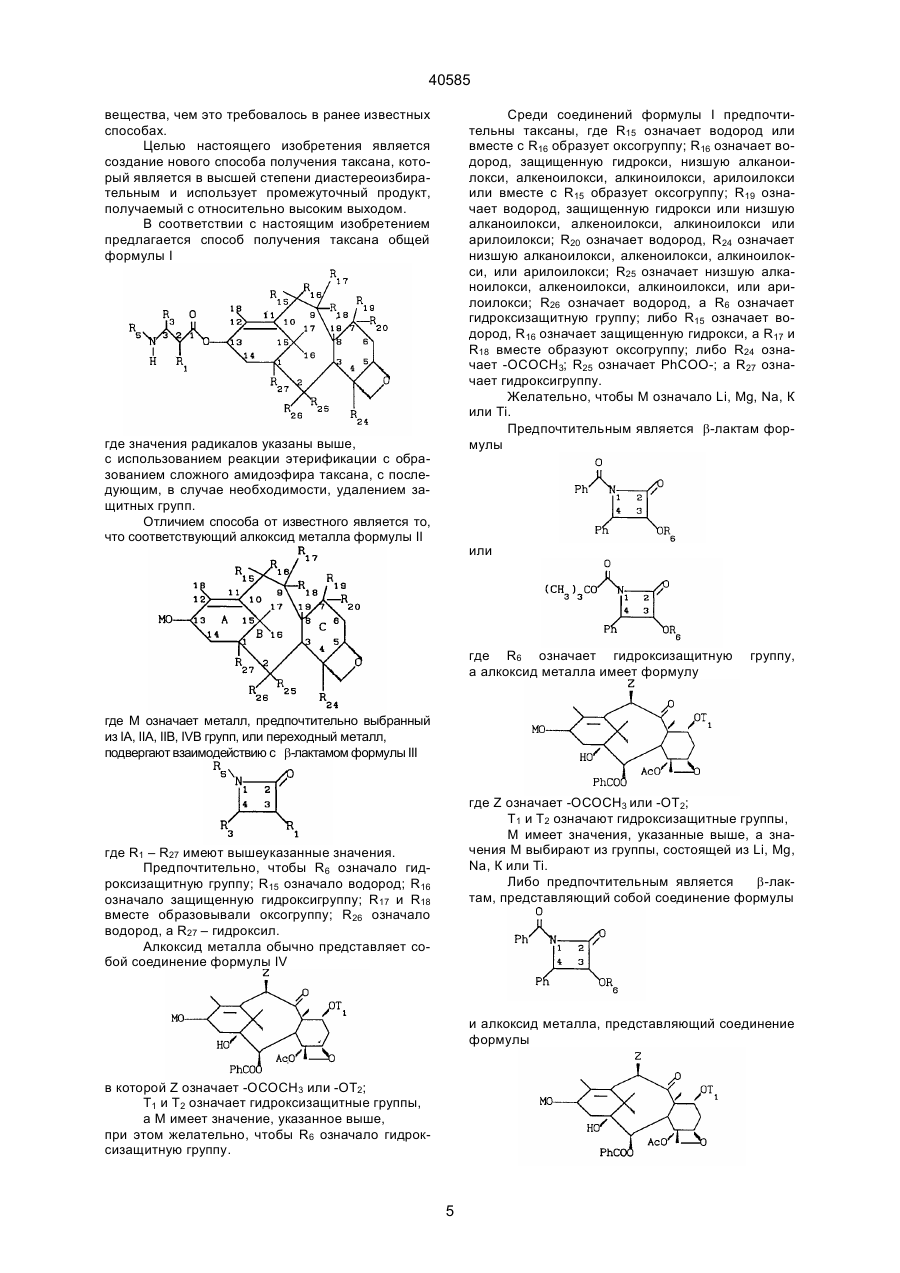
2. Способ по п.1, отличающийся тем, что R6 означает гидроксизащитную группу.
3. Способ по п.2, отличающийся тем, что R15 означает водород, R16 означает защищенную гидрокси, R17 и R18 вместе образуют оксогруппу, R26 означает водород, a R27 - гидроксил.
4. Способ по п.1, отличающийся тем, что алкоксид металла представляет собой соединение формулы
где Т1 - гидроксизащитная группа,
Z-ОТ2- или -ОСОСН3-группа, где Т2 - гидроксизащитная группа,
М имеет значение, указанное в п.1.
5. Способ по п.4, отличающийся тем, что R6 означает гидроксизащитную группу.
6. Способ по п.1, отличающийся тем, что R15 означает водород или вместе с R16 образует оксогруппу, R16 означает водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси или вместе с R15 образует оксогруппу, R19 означает водород, защищенную гидрокси или низшую алканоилокси, алкеноилокси, алкиноилоксн или арилоилокси, R20 означает водород; R24 означает низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси, R25 означает низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси, R26 означает водород.
7. Способ по п.6, отличающийся тем, что R15 означает водород, R16 означает защищенную гидрокси, а R17 и R18 вместе образуют оксогруппу.
8. Способ по п.6, отличающийся тем, что R24 означает –ОСОСН3, R25 означает PhCOO-, a R27 означает гидроксигруппу.
9. Способ по п.6, отличающийся тем, что М означает Li, Mg, Na, К или Ті.
10. Способ по п.6, отличающийся тем, что β-лактам представлен формулой
или
где R6 означает гидроксизащитную группу.
11. Способ по п.10, отличающийся тем, что алкоксид металла представлен формулой
где Т1 - гидроксизащитная группа,
Z-ОТ2- или –ОСОСН3-группа, где Т2 - гидроксизащитная группа,
М имеет значения, указанные в п.1.
12. Способ по п.11, отличающийся тем, что М выбирают из группы, состоящей из Li, Mg, Na, К и Ті.
13. Способ по п.6, отличающийся тем, что R6 означает гидроксизащитную группу.
14. Способ по п.11, отличающийся тем, что β-лактам представляет собой соединение формулы
а алкоксид металла представлен формулой
где R6 и Т1 - гидроксизащитная группа,
Z- Т2- или –ОСОСН3-группа, где Т2 - гидроксизащитная группа,
М имеет значения, определенные в п.11.
Текст
1. Способ получения таксана формулы C2 (54) СПОСІБ ОТРИМАННЯ ТАКСАНУ Зоя (11) UA где М означает металл, подвергают взаимодействию с β-лактамом формулы (19) где R1 означает -OR6, R3 означает водород, алкил, алкенил, алкинил, арил, гетероарил, выбранный из 5- или 6-членного гетероцикла, содержащего один или более атомов кислорода, серы или азота, или ацил; R5 означает -COR10, -COOR10, -COS-алкил или -CONR8R10; R6 означает водород или гидроксизащитную группу; R8 означает водород, алкил, алкенил, алкинил, арил или гетероарил, содержащий один или два гетероатома, выбранных из серы, азота и кислорода; R10 означает низший алкил, алкенил, алкинил, арил или гетероарил, содержащий один или два гетероатома, выбранных из серы, азота и кислорода; R15 и R16 независимо друг от друга означают водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси, или R15 и R16 вместе образуют оксогруппу; 40585 , , где R1-R27 имеют указанные значения. 2. Способ по п.1, отличающийся тем, что R6 означает гидроксизащитную группу. 40585 3. Способ по п.2, отличающийся тем, что R15 означает водород, R16 означает защищенную гидрокси, R17 и R18 вместе образуют оксогруппу, R26 означает водород, a R27 - гидроксил. 4. Способ по п.1, отличающийся тем, что алкоксид металла представляет собой соединение формулы где R6 означает гидроксизащитную группу. 11. Способ по п.10, отличающийся тем, что алкоксид металла представлен формулой , где Т1 - гидроксизащитная группа, Z - ОТ2- или -ОСОСН3-группа, где Т2 - гидроксизащитная группа, М имеет значение, указанное в п.1. 5. Способ по п.4, отличающийся тем, что R6 означает гидроксизащитную группу. 6. Способ по п.1, отличающийся тем, что R15 означает водород или вместе с R16 образует оксогруппу, R16 означает водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси или вместе с R15 образует оксогруппу, R19 означает водород, защищенную гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси, R20 означает водород; R24 означает низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси, R25 означает низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси, R26 означает водород. 7. Способ по п.6, отличающийся тем, что R15 означает водород, R16 означает защищенную гидрокси, а R17 и R18 вместе образуют оксогруппу. 8. Способ по п.6, отличающийся тем, что R24 означает -ОСОСН3, R25 означает PhCOO-, a R27 означает гидроксигруппу. 9. Способ по п.6, отличающийся тем, что М означает Li, Mg, Na, К или Ті. 10. Способ по п.6, отличающийся тем, что βлактам представлен формулой , где Т1 - гидроксизащитная группа, Z - ОТ2- или -ОСОСН3-группа, где Т2 - гидроксизащитная группа, М имеет значения, указанные в п.1. 12. Способ по п.11, отличающийся тем, что М выбирают из группы, состоящей из Li, Mg, Na, К и Ті. 13. Способ по п.6, отличающийся тем, что R6 означает гидроксизащитную группу. 14. Способ по п.11, отличающийся тем, что βлактам представляет собой соединение формулы а алкоксид металла представлен формулой , где R6 и Т1 - гидроксизащитная группа, Z - ОТ2- или -ОСОСН3-группа, где Т2 - гидроксизащитная группа, М имеет значения, определенные в п.11. или _______________________ Данное изобретение относится к способу получения таксана общей формулы I где R1 обозначает –OR6; R3 означает водород, алкил, алкенил, алкинил, арил, гетероарил, выбранный из 5- или 6членного гетероцикла, содержащего один или более атомов кислорода, серы или азота, или ацил; R5 означает -СОR10-, СООR10-, – СОSалкил ил -СОNН8R10; R6 означает водород или гидроксизащитную группу; R8 означает водород, алкил, алкенил, алкинил, арил или гетероарил, содержащий один или 2 40585 два гетероатома, выбранных из серы, азота и кислорода; R10 означает низший алкил, алкенил, алкинил, арил, или гетероарил, содержащий 1 или 2 гетероатома, выбранных из серы, азота и кислорода; R15 и R16 независимо друг от друга означают водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси, или R15 и R16 вместе образуют оксогруппу; R17 и R18 независимо друг от друга означают водород или вместе образуют оксогруппу; R19 и R20 независимо друг от друга означают водород, гидрокси, защищенную гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; R24 означает водород, гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; R25 означает водород, гидрокси или низшую алканоилокси, алкиноилокси или арилоилокси, или R25 и R26, вместе взятые, образуют оксогруппу; R26 означает водород или взятый вместе с R25 образует оксогруппу; R27 означает водород, гидрокси или низшую алкокси, алканоилокси, алкеноилокси, алкиноилокси или арилоилокси. Ранее установлено, что три сложных эфира N-ацилфенилизосерина и таксола, таксотера или цефаломаннина проявляют эффективное противоопухолевое действие. Предлагаемое изобретение является частичным продолжением ранее предложенных изобретений, описывающих получение таких соединений, как таксол, таксотир и других биологически активных производных с использованием алкоксидов металлов и b-лактамов. Семейство таксанов терпенового ряда, членом которого является таксол, вызвало большой интерес в области биологии и химии. Таксол является многообещающим химиотерапевтическим средством для лечения злокачественных опухолей с широким спектром антилейкозного и ингибирующего развитие опухоли действия. Таксол имеет следующую структурную формулу I': чествах, в связи с чем возникает озабоченность в том, что ограниченные запасы таксола на смогут удовлетворить растущий спрос. Поэтому в последние годы химики направили свои усилия на поиски надежного синтетического способа получения таксола, но до сих пор полученные результаты не были достаточно удовлетворительными. Одним известным синтетическим способом получения вышеуказанного вещества является синтез тетрациклического таксонового ядра из химических продуктов. Синтез таксузина, относящегося к тому же семейству, что и таксол, был описан Holton et al. в JACS 110, 6558 (1988). Несмотря на прогресс, достигнутый в этом направлении, окончательный полный синтез таксола тем не менее продолжает оставаться многостадийным, сложным и дорогостоящим процессом. Полусинтетический способ получения таксола был предложен Greene et al в JACS 110, 5917 (1988). Этот способ включает применение представителя семейства таксола – 10-деацетил-баккатина III, который имеет приведенную ниже формулу II' 10-Деацетилбаккатин III является более доступным, чем таксол, так как его можно получать из хвои Taxus baccata. В соответствии с методом, предложенным Greene et al., 10-деацетилбаккатин III превращают в таксол путем присоединения ацетильной группы в положение С-10 и присоединения боковой цепи сложного эфира b-амидокислоты в положении С-13 в результате этерификации спиртовой группы С-13 производным b-амидокарбоновой кислоты. Хотя для этого синтеза характерно сравнительно небольшое количество стадий, синтез производного b-амидокарбоновой кислоты представляет собой многостадийный процесс с низким выходом конечного продукта, а реакция присоединения требует больших затрат времени и также характеризуется низким выходом. Однако указанная реакция является ключевым моментом процесса, выполнение которой требуется в любом предлагаемом способе синтеза таксола или биологически активного производного таксола, так как Wani et al. продемонстрировали в журнале JACS 93, 2325 (1971), что присутствие боковой цепи сложного эфира b-амидокислоты в положении у атома С13 является обязательным для получения противоопухолевого действия. где Ph представляет собой фенил; Ас – ацетил. Благодаря такому многообещающему действию таксол проходит клинические испытания во Франции и Соединенных Штатах. Таксол для этих клинических испытаний в настоящее время получают из коры Taxus brevifollia (тис коротколистный). Однако в коре этих медленно растущих вечнозеленых растений таксол встречается лишь в незначительных коли Colin et al. в патенте США 4814470 недавно показали, что производные таксола приводимой ниже формулы III' обладают гораздо большей активностью по сравнению с таксолом (I'). 3 40585 в которой R' представляет собой гидроксизащитную группу, конденсируют с производным таксана общей формулы где R' представляет собой водород или ацетил; один из R'' и R''' представляет собой гидроксил, а другой – третбутоксикарбониламиногруппу, и их стереоизомерные формы, а также их смеси. В соответствии со способом, предложенным Colin et al. в патенте США 4418470, продукты общей формулы (III') получают путем взаимодействия натриевой соли трет.бутил-N-хлоркарбамата с соединением общей формулы в которой R2 представляет собой ацетильную гидроксизащитную группу, R3 – гидроксизащитную группу, после чего защитные группы R1, R3 и, при необходимости, R2 замещаются водородом. Однако этот способ осуществляется в довольно жестких условиях, обеспечивает плохое превращение продуктов и дает более низкие выходы конечных продуктов. Основной трудностью при синтезе таксола и других потенциальных противоопухолевых средств является отсутствие доступного метода, обеспечивающего простое присоединение к кислороду в положении у атома С-13 остатка, образующего боковую цепь сложного эфира b-амидокислоты. Разработка такого способа с достижением высокого выхода значительно облегчила бы синтез таксола и подобных противоопухолевых средств, имеющих совокупность модифицированных циклических заместителей или модифицированную боковую цепь в положении у атома С-13. Эта трудность была преодолена благодаря открытию нового эффективного способа присоединения к кислороду в положении у атома С-13 остатка, образующего боковую цепь сложного эфира b-амидокислоты. Другая трудность, возникающая в процессе синтеза таксола, заключается в том, что известные способы присоединения боковой цепи сложного эфира b-амидокислоты в положении у атома С-13 являются недостаточно диастереоизбирательными. Поэтому исходное вещество, образующее боковую цепь, должно быть оптически активным, чтобы получить необходимый диастереоизомер. Способ настоящего изобретения является в высшей степени диастереоизбирательным и позволяет использовать рацемическую смесь исходного вещества, устраняя при этом необходимость в дорогостоящем, требующем больших затрат времени процесса разделения исходного вещества на соответствующие энантиомерные формы. Кроме того, эта реакция протекает быстрее по сравнению с предшествующими процессами, позволяя использовать гораздо меньше исходного в которой R' представляет собой ацетильный или 2,2,2-трихлорэтоксикарбонильный радикал, с последующим замещением 2,2,2-трихлорэтоксикарбонильной группы (или групп) водородом. Однако Dеnis et al. указывают в патенте США 4924011, что в результате осуществления этого способа образуется смесь изомеров, которые необходимо разделять, и, как следствие этого, не весь баккатин III или 10-деацетилбаккатин III, используемый для получения продукта общей формулы (IV'), преобразуется в продукт общей формулы (III'). Пытаясь улучшить способ, предложенный Colin et al., Denis et al. разработали новый способ получения производных баккатина III или 10-деацетилбаккатина III общей формулы в которой R' представляет собой водород или ацетил, в соответствии с которым кислоту общей формулы 4 40585 вещества, чем это требовалось в ранее известных способах. Целью настоящего изобретения является создание нового способа получения таксана, который является в высшей степени диастереоизбирательным и использует промежуточный продукт, получаемый с относительно высоким выходом. В соответствии с настоящим изобретением предлагается способ получения таксана общей формулы I Среди соединений формулы I предпочтительны таксаны, где R15 означает водород или вместе с R16 образует оксогруппу; R16 означает водород, защищенную гидрокси, низшую алканоилокси, алкеноилокси, алкиноилокси, арилоилокси или вместе с R15 образует оксогруппу; R19 означает водород, защищенную гидрокси или низшую алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; R20 означает водород, R24 означает низшую алканоилокси, алкеноилокси, алкиноилокси, или арилоилокси; R25 означает низшую алканоилокси, алкеноилокси, алкиноилокси, или арилоилокси; R26 означает водород, а R6 означает гидроксизащитную группу; либо R15 означает водород, R16 означает защищенную гидрокси, а R17 и R18 вместе образуют оксогруппу; либо R24 означает -ОСОСН3; R25 означает PhCOO-; а R27 означает гидроксигруппу. Желательно, чтобы М означало Li, Mg, Na, К или Ti. Предпочтительным является b-лактам формулы где значения радикалов указаны выше, с использованием реакции этерификации с образованием сложного амидоэфира таксана, с последующим, в случае необходимости, удалением защитных групп. Отличием способа от известного является то, что соответствующий алкоксид металла формулы II или где R6 означает гидроксизащитную а алкоксид металла имеет формулу группу, где М означает металл, предпочтительно выбранный из IA, IIA, IIB, IVB групп, или переходный металл, подвергают взаимодействию с b-лактамом формулы III где Z означает -ОСОСН3 или -ОТ2; Т1 и Т2 означают гидроксизащитные группы, М имеет значения, указанные выше, а значения М выбирают из группы, состоящей из Li, Mg, Na, К или Ti. Либо предпочтительным является b-лактам, представляющий собой соединение формулы где R1 – R27 имеют вышеуказанные значения. Предпочтительно, чтобы R6 означало гидроксизащитную группу; R15 означало водород; R16 означало защищенную гидроксигруппу; R17 и R18 вместе образовывали оксогруппу; R26 означало водород, а R27 – гидроксил. Алкоксид металла обычно представляет собой соединение формулы IV и алкоксид металла, представляющий соединение формулы в которой Z означает -ОСОСН3 или -ОТ2; Т1 и Т2 означает гидроксизащитные группы, а М имеет значение, указанное выше, при этом желательно, чтобы R6 означало гидроксизащитную группу. 5 40585 где R6, Z означает -ОСОСН3 или -ОТ2; Т1 и Т2 означают гидроксизащитные группы, М имеет значения, указанные выше. Краткое изложение сущности изобретения. Авторы данного изобретения установили, что при взаимодействии b-лактама с алкоксидом металла, имеющем формулу МОСЕ1Е2Е3, можно получить соответствующий сложный эфир изосерина общей формулы где Х представляет собой Cl, Br, F, CH3O- или NO2-; R2 и R4 представляют собой водород, R3 предпочтительно представляет собой арил, предпочтительно нафтил, фенил, где Х имеет указанные выше значения, Ме представляет собой метил, Ph представляет собой фенил, R1 означает -ОR6, где R6 представляет собой группы, соответственно защищающие гидроксильную группу, а R8 представляет собой водород, алкил, алкенил, алкинил, арил или гетероарил, указанный выше; R1 предпочтительнее всего представляет собой -ОR6, где R6 является триэтилсилилом ("TES"), 1-этоксиэтилом ("ЕЕ") или 2,2,2трихлорэтоксиметилом. Алкильные группы b-лактама, отдельно или с различными заместителями, указанными выше, предпочтительно представляют собой низший алкил, содержащий от 1 до 6 атомов углерода в основной цепи и всего до 15 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают метил, этил, пропил, изопропил, бутил, изобутил, третбутил, амил, гексил и др. Алкенильные группы b-лактама, отдельно или с различными заместителями, указанными выше, предпочтительно представляют собой низший алкенил, содержщий от 2 до 6 атомов углерода в основной цепи и всего до 15 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил, гексенил и др. Алкинильные группы b-лактама, отдельно или с различными заместителями, указанными выше, предпочтительно представляют собой низший алкенил, содержащий от 2 до 6 атомов углерода в основной цепи и всего до 15 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают этинил, пропинил, изопропинил, бутинил, изобутинил, пентенил, гексенил и др. Арильные группы b-лактама, описанные выше, отдельно или с различными заместителями, содержат от 6 до 15 атомов углерода и включают фенил, a-нафтил или b -нафтил и т.д. Заместители включают алкокси, защищенный гидрокси, галоген, алкил, арил, алкенил, ацил, ацилокси, нитро, амино, амидо и т.д. Предпочтительным арилом является фенил. Как указывалось выше, R1 в b-лактаме (II) может представлять собой – OR6, причем R6 представляет собой алкил, ацил, этоксиэтил где Е1, Е2 и Е3 независимо друг от друга представляют собой водород, алкил или циклическую группу при условии, что по крайней мере один из Е1, Е2 и Е3 не является водородом. Предпочтительно, чтобы два из Е1, Е2 и Е3 вместе с атомом углерода, к которому они присоединяются, образовывали моно- или полициклическую структуру, а R1, R3, R5 имеют значения, например, указанные для соединений формулы I, R2, R – означают водород. Подбирая соответствующий алкоксид металла и b-лактам, таким образом можно получить таксол, такостер и другие биологически активные производные таксана, в частности, производные таксана общей формулы I. Таким образом, при взаимодействии b-лактама (II) с алкоксидом металла, имеющим би-, триили тетрациклическое таксановое ядро, можно получить промежуточный продукт, представляющий собой сложный эфир b-амидокислоты. Этот промежуточный продукт можно затем превратить в производное таксана. Алкоксид металла обычно имеет таксановое ядро, соответствующее общей формуле в которой М представляет собой металл, R15 – R27 имеют указанные выше значения. Согласно настоящему изобретению алкоксид металла имеет тетрациклическое таксановое ядро, соответствующее алкоксиду металла формулы (II), в которой R22 и R23 вместе образуют оксетановое кольцо, а R21 означает водород. В соответствии с настоящим изобретением R5 в формуле (III) b-лактама предпочтительно представляет собой -СОR10, причем R10 является арилом, гетероарилом, п-замещенным фенилом или низшим алкоксилом, а предпочтительнее всего фенилом, метокси, этокси, третбутокси ("tBuO"; (СН3)3СО-) или 6 40585 Рацемические смеси b-лактамов можно разделить на чистые энантиомеры до стадии защиты путем перекристаллизации 2-метокси-2-(трифторметил)фенилуксусных эфиров. Однако описываемая ниже реакция, в процессе осуществления которой происходит присоединение боковой цепи сложного эфира b-амидокислоты, обладает тем преимуществом, что она является в высшей степени диастереоизбирательной, что позволяет использовать рацемическую смесь исходного вещества, образующего боковую цепь. 3-(1-этоксиэтокси)-4-фенилазетидин-2-он, получаемый по схеме А, и 3-(1-триэтилсилилокси)4-фенилазетидин-2-он, получаемый по схеме В, можно превратить в b-лактам (II) путем обработки основанием, предпочтительно н-бутиллитием, и ацилхлоридом, алкилхлорформиатом, сульфонилхлоридом, фосфинилхлоридом или фосфорилхлоридом при температуре –78оС или ниже. Способ, предлагаемый по настоящему изобретению, можно успешно использовать для этерификации моно- или полициклических алкоксидов металлов общей формулы ("ЕЕ"), триэтилсилил ("TES"), 2,2,2-трихлорэтоксиметил и другие гидроксизащитные группы, такие как ацетали и простые эфиры, то есть метоксиметил ("МОМ"), бензилоксиметил; сложные эфиры, такие как ацетаты; карбонаты, такие как метилкарбонаты; алкил и арилсилил, такой как триэтилсилил, триметилсилил, диметил-трет.бутилсилил, диметиларилсилил, диметилгетероарилсилил, триизопропилсилил и т.п. С различными защитным группами для гидроксильной группы и их синтезом можно ознакомиться в работе "Protective Groups in Organic Synthesis" T.W. Greene, John Wiley and Sons, 1981. Выбранная гидроксизащитная группа должна легко удаляться в достаточно мягких условиях, например, в 48%-ной HF, ацетонитриле, пиридине или 0,5% HCl/воде/этаноле и/или цинк /уксусной кислоте, не разрушающих сложноэфирные связи или другие заместители в промежуточном продукте таксола. Предпочтительно R6 является триэтилсилилом, 1-этоксиэтилом или 2,2,2-трихлорэтоксиметилом, а предпочтительнее всего триэтилсилилом. Поскольку b-лактам (III) включает несколько асимметричных атомов углерода, то, как известно специалистам в этой области, соединения по настоящему изобретению с асимметричными атомами углерода могут существовать в диастереоизомерных, рацемических или оптически активных формах. Все эти формы входят в объем настоящего изобретения. В частности, настоящее изобретение включает энантиомеры, диастереоизомеры, рацемические смеси и другие смеси этих соединений. b -Лактам (III) можно получать из легкодоступных соединений, как это показано на приводимых в конце текста схемах реакций А и В. Реагенты и условия для стадий: (а) триэтиламин, СН2Cl2, 25оС, 18 часов; (b) 4 эквивалента церийаммонийнитрита, СН3CN, –10оС, 10 минут; (с) КОН, ТГФ, Н2О, 0оС, 30 минут; (d) простой этилвиниловый эфир, ТГФ, толуолсульфокислоты (катализатор), 0оС, 1,5 часа; (е) н-бутиллитий, простой эфир, –78оС, 10 минут; бензоилхлорид, –78оС, 1 час; (f) диизопропиламид лития, ТГФ, от –78оС до –50оС; (g) гексаметилдисилазид лития, ТГФ, от –78оС до 0оС; (h) тетрагидрофуран (ТГФ), от –78оС до 25оС, 12 часов. Исходные вещества являются легко доступными продуктами. В схеме реакций А a-ацетоксиацетилхлорид получают из гликолевой кислоты, после чего в присутствии третичного амина его подвергают циклоконденсации с иминами, полученными из альдегидов и параметоксианилина, с образованием 1-(п-метоксифенил)-3-ацилокси- 4арилазетидин-2-онов. п-Метоксифенильную группу можно легко удалить путем окисления церийаммонийнитратом, а ацилоксигруппу можно гидролизовать в обычных условиях, аналогичных тем, которые имеют место при получении 3-гидрокси-4-арилазетидин-2-онов. Гидроксильная группа в 3-ем положении защищается 1-этоксиэтилом, но может защищаться различными известными защитными группами, такими как триэтилсилильная группа или другие триалкил(или арил)силильные группы. В схеме реакций В этил aтриэтилсилилоксиацетат легко получают из гликолевой кислоты. в которой Е1, Е2 и атом углерода, к которому они присоединяются, образуют карбоциклическую и/или гетероциклическую группу, которая может быть моно- или полициклической, а Е3 представляет собой водород или алкил, предпочтительно низший алкил. Предпочтительнее всего, если карбоциклический и/или гетероциклический остаток имеет от 6 до 20 атомов и гетероатомами являются атомы кислорода. Циклическая структура может быть углеводородной и/или замещенной гетерозаместителями, выбранными, например, из группы: сложные эфирные группы, простые эфирные группы, амины, спирты, защищенные спирты, карбонильные группы, галогены, кислород, замещенный кислород или замещенный азот. В частности, способ можно успешно использовать для получения различных производных таксана, многие из которых, как было установлено, обладают значительной биологической активностью. Таким образом, можно использовать алкоксид металла, включающий бициклическое таксановое ядро, имеет карбоциклическую структуру, соответствующую кольцам А и В алкоксида металла формулы (3) 7 40585 в которой М и R15–R27 имеют указанные выше значения. Алкоксид металла, имеющий трициклическое таксановое ядро, имеет карбоциклическую структуру, соответствующую кольцам А, В и С алкоксида металла формулы (3). Алкоксид металла, включающий тетрациклическое таксановое ядро, имеет карбоциклические кольца А, В и С алкоксида металла формулы (3) и оксетановое кольцо, определяемое R22, R23 и атомами углерода, к которым они присоединяются. Заместитель М в алкоксиде металла (III) обычно представляет собой элементы группы IA, IIA, IIВ, IVB либо переходный металл группы IIIA, IVA, VA или VIA. Таким элементом предпочтительно является элемент группы IA, IIA или переходный металл, и в наиболее предпочтительном случае им является литий, магний, натрий, калий или титан. Алкильные группы в алкоксиде металла, отдельно или с различными заместителями, указанными выше, предпочтительно представляют низший алкил, содержащий от 1 до 6 атомов углерода в основной цепи, всего до 10 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают метил, этил, пропил, изопропил, бутил, изобутил, третбутил, амил, гексил и т.п. Алкильные группы в алкоксиде металла, отдельно или с различными заместителями, указанными выше, предпочтительно представляют низший алкенил, содержащий от 2 до 6 атомов углерода в основной цепи, всего до 10 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил, гексенил и т.п. Алкильные группы в алкоксиде металла, отдельно или с различными заместителями, указанными выше, предпочтительно представляют низший алкинил, содержащий от 2 до 6 атомов углерода в основной цепи, всего до 10 атомов углерода. Они могут образовывать прямую или разветвленную цепь и включают этинил, пропинил, изобутинил, пентенил, гексенил и т.п. Типичная алканоилоксигруппа включает ацетат, пропионат, бутират, валерат, изобутират и т.п. Более предпочтительной алканоилоксигруппой является ацетат. в которой Т1 представляет собой гидроксизащитную группу, а Z представляет собой -ОТ2, где Т2 является ацилом, предпочтительно ацетилом, или другой гидроксизащитной группой. Предпочтительнее всего, если спиртом является защищенный баккатин III, в частности, 7-О-триэтилсилилбаккатин III (который можно получить в соответствии со способом, описанным Greene et al., в JACS 110, 5917 (1988), или другим методом), или 7,10-О-триэтилсилилбаккатин III. Green et al. указывают на то, что 10-деацетилбаккатин III можно превратить в 7-О-деацетилбаккатин III в соответствии со следующей схемой реакций: При оптимальных условиях 10-деацетилбаккатин III взаимодействует с 20 эквивалентами (С2Н5)3SiCl в атмосфере аргона при температуре 23оС в течение 20 часов в присутствии 50 мл пиридина/ммоль 10-деацетилбаккатин III, в результате чего после очистки образуется 7-триэтилсилил-10-деацетилбаккатин (III) (6а) с 84–86% выходом. Продукт реакции затем ацетилируют 5 эквивалентами СН3СОСl и 25 мл пиридина/ммоль соединения (6а) в атмосфере аргона при температуре 0оС в течение 48 часов с достижением 86% выхода 7-О-триэтилсилилбаккатин III (6b). Greene et al в JACS 110, 5917-5918 (1988). Альтернативно в 7-триэтилсилил-10-деацетилбаккатине III (6а) можно защитить атом кислорода в положении С-10 кислотонеустойчивой гидроксизащитной группы. Например, обработка соединения формулы (6а) н-бутиллитием в ТГФ, а затем триэтилсилилхлоридом (1,1 молярный эквивалент) при температуре 0оС позволяет получить 7,10-бис-О-триэтилсилилбаккатин III (6с) с 95% выходом. Кроме того, соединение формулы (6а) можно превратить в 7-О-триэтилсилил-10-(1этоксиэтил)баккатин III (6d) с 90% выходом путем обработки избытком простого этилвинилового Арильные части в алкоксиде металла, отдельно или с различными заместителями, имеют от 6 до 10 атомов углерода и включают фенил, aнафтил или b-нафтил и т.д. Заместители включают алкилокси, гидрокси, галоген, алкил, арил, алкенил, ацил, ацилокси, нитро, амино, амидо и т.д. Наиболее предпочтительным арилом является фенил. Алкоксиды металлов (III) получают путем взаимодействия спирта, имеющего у таксанового ядра в положении С-13 гидроксильную группу, с металлоорганическим соединением в приемлемом растворителе. Спиртом предпочтительно является производное баккатина III или 10-деацетилбаккатина III формулы 8 40585 эфира и каталитическим количеством метансульфоновой кислоты. Эти методы иллюстрируются на приводимой ниже схеме реакций: группа в положении у атома С-7 защищена триэтилсилильной или 1-этоксиэтильной группой. 8 b-d b, R = -COCH3 c, R = -Si(C2H5)3 d, R = –EE Промежуточное соединение (8b) легко превращается в таксол, если R1 представляет собой -ОR6, R2 и R3 – водород, R4 – фенил, R5 – бензоил, а R6 представляет собой гидроксизащитную группу, такую как триэтилсилил. Промежуточное соединение (8с) легко превращается в таксотер, если R1 представляет собой -ОR6, R2 и R3 – водород, R4 – фенил, R5 – тетрабутоксикарбонил, а R6 представляет собой гидроксизащитную группу, такую как триэтилсилил. Промежуточное соединение (8d) легко превращается в 10-деацетилтаксол, если R1 представляет собой -ОR6, R2 и R3 – водород, R4 – фенил, R5 – бензоил, а R6 представляет собой гидроксизащитную группу, такую как триэтилсилил. Промежуточные соединения (8b, 8c и 8d) можно превратить в указанные соединения путем гидролиза триэтилсилильной и 1-этоксиэтильной групп в мягких условиях, не вызывающих разрушение сложноэфирной связи или заместителей производного таксана, как показано на схеме 7-О-триэтилсилилбаккатин III (6b), 7-10-Отриэтилсилилбаккатин III (6с) или 7-О-триэтилсилил-10-(1-этоксиэтил)баккатин III (6d) взаимодействуют с металлоорганическим соединением, таким как н-бутиллитий, в таком растворителе, как тетрагдирофуран (ТГФ), с образованием алкоксида металла 13-О-литий-7-О- триэтилсилилбаккатина III (7b), 13-О-литий-7,10-О- триэтилсилилбакката III (7с) или 13-О-литий-7,10-О-триэтилсилил- 10(1-этоксиэтил)баккатина III (7d), как это показано на следующей схеме реакций: Другие производные таксана можно легко получить путем выбора соответствующих заместителей R1 – R5 b-лактама (III) или R15 – R27 алкоксида металла (III). Получение других аналогичных соединений иллюстрируется в приводимых ниже примерах. Превращение спирта в алкоксид металла и конечный синтез таксола могут проводиться в одном реакторе. b-Лактам предпочтительно вводят в реактор после образования в нем алкоксида металла. Для превращения спирта в соответствующий алкоксид металла предпочтительно используется такое металлоорганическое соединение, Как показано на следующей схеме реакций, приемлемый алкоксид металла по настоящему изобретению, например, производное 13-О- литий-7-О-триэтилсилилбаккатина III (6b, 7с или 7d), взаимодействует с b-лактамом по настоящему изобретению с образованием промежуточного продукта (8b, 8с или 8d), в котором гидроксильная 9 40585 как н-бутиллитий, но также можно использовать другие источники металлического заместителя, например, диизопропиламид лития, другие амиды лития или магния, бромид этилмагний, бромид металмагния, другие литийорганические соединения, другие магнийорганические соединения, натрийорганические соединения, титанорганические соединения, цирконийорганические соединения, цинкорганические соединения, кадмийорганические соединения или калийорганические соединения, или соответствующие амиды. Металлорганические соединения легко доступны, либо их можно получить известными методами, включая восстановление органических галогенидов металлов. Предпочтение отдается низшим алкилгалогенидам. Например, бутилбромид может взаимодействовать с металлическим литием в простом диэтиловом эфире с образованием раствора н-бутиллития, причем эта реакция протекает следующим образом: СН3СН2СН2СН2Br+2Li например, на 60–70% по сравнению с ранее известными способами. Растворимость соединений формулы (I) в воде можно улучшить в том случае, если R1 представляет собой -ОR6, R19 представляет собой -ОТ1, а R6 и/или Т1 являются функциональной группой, увеличивающей растворимость в частности, -COGCOR1, при этом G представляет собой этилен, пропилен, CHCH, 1,2-циклогексан или 1,2-фенилен, R1 – ОН основания, NR2R3, ОR3, SR3, OCH2CONR4R5, OH, R2 – водород, метил, R3-(CH2)nNR6R7; (CH2)nNÅ R6R7R8X1Θ , n = от 1 до 3, R4 – водород, низший алкил, включающий от 1 до 4 атомов углерода, R5 – водород, низший алкил, включающий от 1 до 4 атомов углерода, бензил, гидроксиэтил, СН2СО2Н, диметиламиноэтил, R6R7 – низший алкил, включающий 1 или 2 атома углерода, бензил или R6, и R7 вместе с атомом азота в группе NR6R7 образует следующие кольца СН3СН2СН2СН2Li+LiBr Альтернативно, алкоксид лития может вступать в реакцию обмена с галогенидами металлов, в результате чего образуются алкоксиды алюминия, бора, церия, кальция, циркония или цинка. Хотя предпочтительным растворителем для реакционной смеси является тетрагидрофуран, также можно использовать другие эфирные растворители, такие как диметоксиэтан, или ароматические растворители. Определенные растворители, включая некоторые галогенированные растворители и некоторые углеводороды с прямой цепью, в которых реагенты плохо растворяются, являются неприемлемыми. Другие растворители являются неприемлемыми по другим причинам. Например, сложные эфиры нельзя использовать с определенными металлорганическими соединениями, такими как н-бутиллитий, вследствие их несовместимости. Хотя рассматриваемая здесь схема реакций служит для синтеза определенных производных таксола, она может применяться при использовании другого b-лактама или тетрациклического алкоксида металла. Поэтому в соответствии со способом по настоящему изобретению для получения промежуточного продукта таксола, помимо 13-Олитий-7-О-триэтилсилилбаккатина III, можно использовать и другие алкоксиды металлов. b-Лактам и тетрациклический алкоксид металла для получения других синтетических таксолов, производных таксола, 10-деацетилтаксолов могут быть естественного или искусственного происхождения, причем их энантиомеры и диастереоизомеры также входят в объем настоящего изобретения. Преимуществом настоящего способа является его высокая диастереизобретательность. Поэтому можно использовать рацемические смеси исходных веществ боковой цепи. При этом может быть достигнута значительная экономия благодаря тому, что не нужно разделять рацемические смеси b-лактамов на чистые энантиомеры. Дополнительная экономия может иметь место вследствие уменьшения используемого количества исходного вещества, образующего боковую цепь, R8 – низший алкил, включающий 1 или 2 атома углерода, бензил Х1Θ – галогенид, основания: NH3, (HOC2H4)3N, N(CH3)3, CH3N(C2H4OH)2, NH2(CH2)6-NH2, N-метилгликамин, NaOH, KOH. Получение соединений, в которых R6 или Т1 представляют собой -COGCO-R1, рассматривается в патенте США 4942184, выданном Handwitz. Ниже приведены примеры, иллюстрирующие изобретение, но не органичивающие его. Используемые сокращения: Am – трет.-амилокси Bn – бензоил Br – бром Cl – хлор Et – этил F – фтор Н – водород iBu – изобутил iPr – изопропил Ме – метил N – азот Np – нафтил nBu – н-бутил nPr – н-пропил О – кислород о – орто Р – фосфор р – пара Ph – фенил S – сера TES – триэтилсилил TMS – триметилсилил tBu – трет.-бутил Tol – толуол 10 40585 Пример 1. Получение 2'-этоксиэтил-7-триэтилсилилтаксола, а затем таксола из рацемической смеси bлактама. К раствору из 7-триэтилсилилбаккатина III (20 мг, 0,028 ммоля) в 1 мл тетрагидрофурана при температуре –78оС по каплям добавляли 0,17 мл 0,164М раствора n-BuLi в гексане. После выстаивания в течение 30 минут при –78оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-(1этоксиэтокси)-4-фенилазетидин-2-она (47,5 мг 0,14 моля) в 1 мл тетрагидрофурана. Полученный раствор медленно нагревали в течение 1,5 часов до 0оС, а затем перемешивали при 0оС в течение 1 часа и добавляли 1 мл 10%-ного раствора уксусной кислоты в тетрагидрофуране. Эту смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали посредством высокоскоростной хроматографии с образованием 23 мг (80%) (2'R,3'S)-2'этоксиэтил-7-триэтилсилилтаксола и 3,5 мг (13%) 2',3'-эпи(2'R,3'S)-2'-этоксиэтил-7-триэтилсилилтаксола. 5 мг пробы (2'R,3'S)-2'-этоксиэтил-7-триэтилсилилтаксола растворяли в 2 мл этанола и добавляли 0,5 мол 0,5% водного раствора HCl. Эту смесь перемешивали при 0оС в течение 30 часов и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали посредством высокоскоростной хроматографии с образованием 4,5 мг (примерно 90%) таксола, что во всех отношениях соответствует достоверной пробе. 5 мг 2',3'-эпи(2'R,3'S)-2'-этоксиэтил-7-триэтилсилилтаксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5% водного раствора HCl. Эту смесь перемешивали при 0оС в течение 30 часов и разбавляли 50 мл этилацетата. Полученный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали посредством высокоскоростной хроматографии с образованием 4,5 мг (примерно 90%) 2',3'-эпитаксола. Пример 2. Получение 2',7-(бис)триэтилсилилтаксола, а затем таксола из рацемической смеси b-лактама. К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,087 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилокси-4-фенилазетидин-2-она (274 мг 0,715 моля) в 1 мл тетрагидрофурана. Полученный раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа. Затем добавляли 1 мл 10%-ного раствора уксусной кислоты в тетрагидрофуране. Эту смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очи щали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 131 мг (85%) (2'R,3'S)-2,7(бис)триэтилсилилтаксола и 15 мг (10%) 2',3'эпи(2'S,3'R)-2',7-(бис)триэтилсилилтаксола. К раствору из 121,3 мг (0,112 ммоля) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола в 6 мл ацетонитрила и 0,3 мл пиридина при температуре 0оС добавляли 0,9 мл 48% водного раствора HF. Эту смесь перемешивали при 0оС в течение 8 часов, а затем при 25оС в течение 6 часов. Полученную смесь распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора образовывалось 113 мг вещества, которое очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 94 мг (98%) таксола, который во всех отношениях был идентичен достоверной пробе. К раствору из 5 мг (2'R,3'S)-3',7-(бис)триэтилсилилтаксола в 0,5 мл ацетонитрила и 0,03 мл пиридина при температуре 0оС добавляли 0,09 мл 48% водного раствора HF. Эту смесь перемешивали при 0оС в течение 8 часов, а затем при 25оС в течение 6 часов. Полученную смесь распределяли между насыщенным водным раствором бикарбоната натрия и этилацетата. В результате выпаривания этилацетатного раствора было получено 5 мг вещества, которое очищали посредством высокоскоростной хроматографии и перекристаллизовывали с образованием 4,6 мг (примерно 95%) 2',3'-эпитаксола. Пример 3. Получение 2',7-(бис)триэтилсилилтаксола, а затем таксола из оптически активного b-лактама. К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,087 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли (+)-цис-1-бензоил-3триэтилсилокси-4-фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл тетрагидрофурана. Этот раствор нагревали до 0оС и выдерживали при этой температуре в течение 2 часов. Добавляли 1 мл 10%-ного раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 145 мг (94%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. К раствору из 121,3 мг (0,112 ммоля) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола в 6 мл ацетонитрила и 0,3 мл пиридина при температуре 0оС добавляли 0,9 мл 48% водного раствора HF. Эту смесь перемешивали при 0оС в течение 8 часов, а затем при 25оС в течение 6 часов. Полученную смесь распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 113 мг вещества, которое очищали посредством высокоскоростной хроматографии и перекристаллизовывали с образова 11 40585 нием 94 мг (98%) таксола, который во всех отношениях был идентичен достоверной пробе. Пример 4. Получение таксотера К раствору из 7,10-триэтилсилилбаккатина III (200 мг, 0,248 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-(трет.бутоксикарбонил)-3-триэтилсилилокси-4-фенилазетидин- 2-она (467 мг, 1,24 ммоля) в 2 мл тетрагидрофурана. Этот раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа до добавления 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Эту смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 280 мг сырого 2',7,10-трис-триэтилсилилтаксотера. К раствору 280 мг неочищенного продукта, полученного на предыдущей стадии, в 12 мл ацетонитрила и 0,6 мл пиридина при 0оС добавляли 1,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов, и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 215 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 190 мг (95%) таксотера, который перекристаллизовывали из смеси метанола и воды. Все аналитические и спектральные данные были идентичны данным, приведенным для таксотера в патенте США 4814470. Пример 5 при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 320 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дезфенил-3'-(2-нафтил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 320 мг (0,283 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 225 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 166 мг (34%) 3'дезфенил-3'-(2-нафтил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 164–165оС; [a]Na25 o –52,6 (c 0,005, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,14 (д., J = 7,3 Гц, 2Н, ортобензоат), 7,96 (м., 1Н, ароматический), 7,90 (м., 1Н, ароматический), 7,85 (м., 3Н, ароматический), 7,76 (м., 2Н, ароматический), 7,60 (м., 3Н, ароматический), 7,52 (м., 4Н, ароматический), 7,41 (м., 2Н, ароматический), 7,01 (д., J = 8,8 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,26 (двойной дублет, J = 9,2 9,2 Гц, 1Н, Н13), 5,97 (двойной дублет, J = 8,8, 2,5 Гц, 1Н, H3'), 5,68 (д., J = 7,1 Гц, 1Н, Н2b), 4,93 (м., 1Н, Н5), 4,2 (м., 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,30 (д., J = 8,5 Гц, 1Н, Н20 a ), 4,20 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,81 (д., J = 7,1 Гц, 1Н, Н3), 3,60 (д., J = 5 Гц, 1Н, 2'ОН), 2,48 (м., 1Н, Н6 a), 2,45 (широкий, 1Н, 70Н), 2,39 (с., 3Н, 4Ас), 2,30 (м., 2Н, Н14), 2,24 (с., 3Н, 10Ас), 1,83 (м., 1Н, Н6 b), 1,82 (широкий синглет, 3Н, Ме18), 1,68 (с., 1Н, 10Н), 1,68 (с., 3Н, Ме19), 1,24 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 6 Получение Nр2 представляет собой в которой Npt представляет собой Получение 3'-дезфенил-3'-(2-нафтил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(2-нафтил)азетидин2-она (620 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Этот раствор нагревали до 0оС и выдерживали Получение 3'-дезфенил-3'-(1-нафтил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой 12 40585 выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(4- метоксифенил)азетидин-2-она (590 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 320 мг смеси, содержащей (2'R,3'S)2',7-(бис)триэтилсилил-3'-дезфенил-3'- (4-метоксифенил)таксола и небольшое количество (2'S,3'R)изомера. К раствору из 320 мг (0,288 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 255 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 172 мг (68%) 3'дезфенил-3'-(4-метоксифенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 174–176оС; [a]Na25 –48,86o (c 0,05, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 7,1 Гц, 2Н, ортобензоат), 7,72 (м., 2Н, ароматический), 7,59 (м., 1Н, ароматический), 7,53–7,36 (м., 8Н, ароматический), 6,96 (д., J =8,8 Гц, 1Н, NН), 6,90 (м., 2Н, ароматический), 6,26 (с., 1Н, Н10), 6,21 (двойной дублет, J = 9,3, 9,36 Гц, 1Н, H13), 5,70 (двойной дублет, J = 8,8, 2,76 Гц, 1Н, Н3'), 5,66 (д., J = 6,8 Гц, 1Н, Н2 b ), 4,93 (двойной дублет, J = 9,9, 2,2 Гц, 1Н, Н5), 4,74 (двойной дублет, J = 5,5, 2,7 Гц, 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,29 (д., J = 8,8 Гц, 1Н, Н20 a ), 4,18 (д., J = 8,8 Гц, 1Н, Н20 b ), 3,78 (д., J = 6,8 Гц, 1Н, Н3), 3,78 (д., J = 6,8 Гц, 1Н, Н3), 3,78 (с., 3Н, ArOMe), 3,67 (д., J = 5,5 Гц, 1Н, 2'ОН), 2,61 (м., 1Н, Н6 a ), 2,50 (д., J = 4,4 Гц, 1Н, 7ОН), 2,37 (с., 3Н, 4Ас), 2,31 (м., 2Н, Н14), 2,22 (с., 3Н, 10Ас), 1,84 (м., 1Н, Н6 b ), 1,79 (широкий синглет, 3Н, Ме18), 1,79 (с., 1Н, 10Н), 1,67 (с., 3Н, Ме19), 1,22 (с., 3Н, Ме17), 1,13 (с., 3Н, Ме16). Пример 8 смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(1-нафтил)азетидин2-она (620 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Этот раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа до добавления 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 325 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил3'-десфенил-3'-(1-нафтил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 325 мг (0,287 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 260 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 166 мг (64%) 3'-(1нафтил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 164–165оС; [a]Na25 –52,6o (c 0,005, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,11 (д., J = 7,1 Гц, 2Н, ортобензоат), 8,11 (м., 3Н, ароматический), 7,91 (м., 3Н, ароматический), 7,70 (м., 2Н, ароматический), 7,63–7,46 (м., 7Н, ароматический), 6,75 (д., J = 8,8 Гц, 1Н, NH), 6,52 (двойной дублет, J = 8,8, 1,6 Гц, 1Н, Н3'), 6,27 (с., 1Н, Н10), 6,27 (двойной дублет, J = 9,1, 9,1 Гц, 1Н, H13), 5,68 (д., J = 7,1 Гц, 1Н, Н2 b ), 4,85 (двойной дублет, J = 7,6, 2,2 Гц, 1Н, Н5), 4,97 (двойной дублет, J = 1,6 Гц, 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,24 (д., J = 8,5 Гц, 1Н, Н20 a), 4,17 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,80 (д., J = 7,1 Гц, 1Н, Н3), 3,65 (широкий, 1Н, 2'ОН), 2,55 (м., 1Н, Н6 a ), 2,48 (широкий, 2Н, 70Н), 2,41 (с., 3Н, 4Ас), 2,38 (м., 2Н, Н14), 1,96 (с., 3Н, 10Ас), 1,86 (м., 1Н, Н6 b ), 1,80 (широкий синглет, 3Н, Ме18), 1,76 (с., 1Н, 10Н), 1,69 (с., 3Н, Ме19), 1,28 (с., 3Н, Ме17), 1,16 (с., 3Н, Ме16). Пример 7 Получение 3'-дезфенил-3'-(4-метоксифенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После Получение 3'-дезфенил-3'-(4-хлорфенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана 13 40585 при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(4-хлорфенил)азетидин-2-она (595 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 320 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-десфенил-3'-(4хлорфенил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 320 мг (0,287 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 255 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 158 мг (62%) 3'десфенил-3'-(4-хлорфенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 173–175оС; [a]Na25 o –50,8 (c 0,01, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,13 (д., J = 7,1 Гц, 2Н, ортобензоат), 7,72 (м., 2Н, ортобензамид), 7,65–7,35 (м., 10Н, ароматический), 6,97 (д., J = 8,8 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,25 (двойной дублет, J = 8,3, 8,3 Гц, 1Н, H13), 5,78 (двойной дублет, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,67 (д., J = 7,1 Гц, 1Н, Н2 b), 4,95 (двойной дублет, J = 8,8, 2,2 Гц, 1Н, Н5), 4,77 (широкий синглет, 1Н, Н2'), 4,40 (м., 1Н, Н7), 4,31 (д., J = 8,2 Гц, 1Н, Н20 a), 4,19 (д., J = 8,2 Гц, 1Н, Н20 b ), 3,80 (д., J = 7,1 Гц, 1Н, Н3), 3,61 (широкий синглет, 1Н, 2'ОН), 2,54 (м., 1Н, Н6 a ), 2,38 (с., 3Н, 4Ас), 2,32 (м., 2Н, Н14), 2,24 (с., 3Н, 10Ас), 1,85 (м., 1Н, Н6 b ), 1,80 (широкий синглет, 3Н, Ме18), 1,68 (с., 3Н, Ме19), 1,23 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 9 смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(4- бромфенил)-азетидин-2-она (660 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 330 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дезфенил-3'-(4-бромфенил)таксола и небольшое количество (2'S,3'R)изомера. К раствору из 330 мг (0,284 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 265 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 186 мг (64%) 3'дезфенил-3'-(4-бромфенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 170–172оС; [a]Na25 –50,94o (c 0,01, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 7,2 Гц, 2Н, ортобензоат), 7,71 (м., 2Н, ароматический), 7,61 (м., 1Н, ароматический), 7,50–7,47 (м., 6Н, ароматический), 7,38 (м., 3Н, ароматический), 7,04 (д., J = 8,8 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,23 (двойной дублет, J = 8,2, 8,2 Гц, 1Н, H13), 5,75 (двойной дублет, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,66 (д., J = 7,1 Гц, 1Н, Н2 b ), 4,94 (двойной дублет, J = 9,3, 1,7 Гц, 1Н, Н5), 4,75 (двойной дублет, J = 2,2 Гц, 1Н, Н2'), 4,38 (м., 1Н, Н7), 4,29 (д., J = 8,2 Гц, 1Н, Н20 a ), 4,18 (д., J = 8,2 Гц, 1Н, Н20 b ), 3,79 (д., J = 7,1 Гц, 1Н, Н3), 3,7 (широкий, 1Н, 2'ОН), 2,53 (м., 1Н, Н6 a ), 2,38 (широкий, 1Н, 7ОН), 2,37 (с., 3Н, 4Ас), 2,30 (м., 2Н, Н14), 2,23 (с., 3Н, 10Ас), 1,87 (м., 1Н, Н6 b ), 1,80 (широкий синглет, 3Н, Ме18), 1,80 (с., 1Н, 10Н), 1,67 (с., 3Н, Ме19), 1,22 (с., 3Н, Ме17), 1,13 (с., 3Н, Ме16). Пример 10 Получение 3'-дезфенил-3'-(4-бромфенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой Получение 3'-дезфенил-3'-(3,4-метилендиоксифенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана 14 40585 при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(3,4-метилендиоксифенил)азетидин-2-она (610 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 320 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил-3'-(3,4-метилендиоксифенил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 320 мг (0,284 ммоля) смеси, полученной выше, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 113 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 165 мг (64%) 3'десфенил-3'-(3,4-метилендиоксифенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 178–180оС; [a]Na25 –46,6o (c 0,005, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 7,2 Гц, 2Н, ортобензоат), 7,72 (м., 2Н, ароматический), 7,15 (м., 1Н, ароматический), 7,50 (м., 2Н, ароматический), 7,38 (м., 2Н, ароматический), 7,0 (м., 1Н, ароматический), 6,94 (м., 2Н, ароматический), 6,88 (д., J = 9,1 Гц, 1Н, NH), 6,83 (м., 1Н, ароматический), 6,28 (с., 1Н, Н10), 6,23 (двойной дублет, J = 9,1, 9,1 Гц, 1Н, Н13), 5,97 (с., 2Н, метилен), 5,69 (двойной дублет, J = 9,1, 2,5 Гц, 1Н, Н3'),5,68 (д., J = 6,9 Гц, 1Н, Н2 b ), 4,95 (двойной дублет, J = 9,6, 2,2 Гц, 1Н, Н5), 4,72 (двойной дублет, J = 2,5 Гц, 1Н, Н2'), 4,41 (м., 1Н, Н7), 4,31 (д., J = 8,4 Гц, 1Н, Н20 a ), 4,20 (д., J = 8,4 Гц, 1Н, Н20 b ), 3,81 (д., J = 6,9 Гц, 1Н, Н3), 3,60 (широкий, 1Н, 2'ОН), 2,56 (м., 1Н, Н6 a ), 2,43 (д., J = 4,1 Гц, 1Н, 7ОН), 2,39 (с., 3Н, 4Ас), 2,31 (м., 2Н, Н14), 2,24 (с., 3Н, 10Ас), 1,88 (м., 1Н, Н6 b ), 1,82 (широкий синглет, 3Н, Ме18), 1,69 (с., 1Н, 1ОН), 1,68 (с., 3Н, Ме19), 1,24 (с., 3Н, Ме17), 1,15 (с., 3Н, Ме16). Пример 11 Получение 3'-дезфенил-3'-(3,4-диметоксифенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(3,4-диметоксифенил)азетидин-2-она (630 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 330 мг смеси, содержащей (2'R,3'S)2',7-(бис)триэтилсилил-3'-дезфенил-3'-(3,4-диметоксифенил)таксола и небольшое количество (2'S,3'R)изомера. К раствору из 330 мг (0,286 ммоля) смеси, полученной на предыдущей стадии, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 260 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 175 мг (67%) 3'-десфенил-3'-(3,4-диметоксифенил)таксона, который перекристаллизовывали из смеси метанола/воды. Температура плавления 165–167оС; [a]Na25 o –42,0 (c 0,005, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 8,3 Гц, 2Н, ортобензоат), 7,73 (д., J =8,2 Гц, ортобензамид), 7,65–7,35 (м., 6Н, ароматический), 7,1–7,0 (м., 2Н, ароматический), 6,94 (д., J = 8,8 Гц, 1Н, NH), 6,88 (д., J = 8,3 Гц, 2Н, ароматический), 6,27 (с., 1Н, Н10), 6,21 (двойной дублет, J = 9,3, 9,3 Гц, 1Н, Н13), 5,69 (м., 2Н, Н3, Н2 b ), 4,94 (двойной дублет, J = 9,9, 2,2 Гц, 1Н, Н5), 4,77 (д., J = 2,8 Гц, 1Н, Н2'), 4,39 (двойной дублет, J = 11,0, 6,6 Гц, 1Н, Н7), 4,30 (д., J = 8,5 Гц, 1Н, Н20 a ), 4,19 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,88 (с., 3Н, ArOMe), 3,87 (с., 3Н, ArOMe), 3,80 (д., J = 7,1 Гц, 1Н, Н3), 3,59 (д., J = 4,4 Гц, 1Н, 2'ОН), 2,54 (м., 1Н, Н6 a ), 2,38 (с., 3Н, 4Ас), 2,36 (м., 2Н, Н14 a Н14 b), 2,23 (с., 3Н, 10Ас), 1,86 (м., 1Н, Н6 b), 1,80 (широкий синглет, 3Н, Ме18), 1,68 (с., 3Н, Ме19), 1,23 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 12 15 40585 Получение N-дебензоил-N-этоксикарбонилтаксола К раствору из 7-триэтилсилилбаккатина III (150 мг, 0,221 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,136 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор цис-1этоксикарбонил-3-триэтилсилилокси-4-фенилазетидин-2-она (386 мг, 1,11 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 252 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензил-N-этоксикарбонилтаксол и небольшое количество (2'S,3'R)изомера. К раствору из 252 мг (0,112 ммоля) смеси, полученной на предыдущей стадии, в 12 мл ацетонитрила и 0,6 мл пиридина при 0оС добавляли 1,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 216 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 155 мг (85%) N-дебензоил-N-этоксикарбонилтаксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 161,5–162,5оС; 25 [a]Na –62,2o (c 0,51, CHCl 3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 7,7 Гц, 2Н, ортобензоат), 7,65–7,3 (м., 8Н, ароматический), 6,28 (м., 1Н, Н10), 6,27 (м., 1Н, Н13), 5,67 (д., J = 7,1, 1Н, Н2 b ), 5,53 (д., J = 9,3 Гц, 1Н, Н3'), 5,29 (д., J = 9,3 Гц, 1Н, NH), 4,94 (двойной дублет, J = 9,3, 2,2 Гц, 1Н, Н5), 4,64 (двойной дублет, J = 5,0, 2,8 Гц, 1Н, Н2'), 4,41 (м., 1Н, Н7), 4,29 (д., J = 8,5 Гц, 1Н, Н20 a ), 4,17 (д., J = 8,5 Гц, 1Н, Н20 b ), 4,01 (кв., J = 7,1 Гц, 2Н, СООСН2СН3), 3,79 (д., J = 7,1 Гц, 1Н, Н3), 3,45 (д., J = 5 Гц, 1Н, 2'ОН), 2,54 (м., 1Н, Н6 a ), 2,47 (д., J = 3,9 Гц, 1Н, 70Н), 2,36 (м., 3Н, 4Ас), 2,24 (с., 3Н, 4Ас), 2,22 (м., 2Н, Н14 a , Н14 b ), 1,87 (м., 1Н, Н6 b), 1,83 (широкий синглет, 3Н, Ме18), 1,77 (с., 1Н, 1ОН), 1,68 (с., 3Н, Ме19), 1,27 (с., 3Н, Ме17), 1,15 (с., 3Н, Ме16), 1,14 (т., J = 7,1 Гц, 2Н, СООСН2СО3). Пример 13 Получение К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(4-нитрофенил)-азетидин-2-она (610 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 320 мг смеси, содержащей (2'R,3'S)-2,'7(бис)триэтилсилил-3'-дезфенил-3'-(4-нитрофенил)таксола и небольшое количество (2'S,3'R)изомера. К раствору из 320 мг (0,284 ммоля) смеси, полученной на предыдущей стадии, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 255 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 147 мг (57%) 3'-дезфенил-3'-(4-нитрофенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 188–190оС; [a]Na25 o –63,7 (c 0,01, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,26 (д., J = 8,8 Гц, 2Н, ортобензоат), 8,20 (м., 2Н, ароматический), 7,73 (м., 4Н, ароматический), 7,60 (м., 1Н, ароматический), 7,52 (м., 1Н, ароматический), 7,41 (м., 1Н, ароматический), 7,15 (м., J = 8,8 Гц, 1Н, NH), 6,26 (с., 1Н, Н10), 6,26 (двойной дублет, J = 9,3, 9,3, Гц, 1Н, H13), 5,93 (двойной дублет, J = 8,8, 2,8 Гц, 1Н, Н3'), 5,66 (д., J = 6,6 Гц, 1Н, Н2 b ), 4,94 (двойной дублет, J = 9,3, 1,7 Гц, 1Н, Н5), 4,85 (двойной дублет, J = 3,9, 2,8 Гц, 1Н, Н2'), 4,38 (м., 1Н, Н7), 4,30 (д., J = 8,8 Гц, 1Н, Н20 a), 4,19 (д., J = 8,8 Гц, 1Н, Н20 b ), 3,86 (д., J = 3,9 Гц, 1Н, 2'ОН), 3,79 (д., J = 6,6 Гц, 1Н, Н3), 2,55 (м., 1Н, Н6 a), 2,46 (д.,J = 3,8 Гц, 1Н, 70Н), 2,41 (с., 3Н, 4Ас), 2,38 (м., 2Н, Н14), 2,23 (с., 3Н, 10Ас), 1,82 (м., 1Н, Н6 b), 1,80 (широкий синглет, 3Н, Ме18), 1,74 (с., 1Н, 10Н), 1,68 (с., 3Н, Ме19), 1,21 (с., 3Н, Ме17), 1,13 (с., 3Н, Ме16). Пример 14 Получение 3'-дезфенил-3'-(2-фурил)таксола К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл тетрагидрофурана 3'-дезфенил-3'-(4-нитрофенил)так сола 16 40585 при температуре –45оС по каплям добавляли 0,087 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(2-фурил)азетидин-2-она (266 мг, 0,715 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 143 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-десфенил-3'-(2фурил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 143 мг (0,112 ммоля) смеси, полученной на предыдущей стадии, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 115 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 98 мг (81%) 3'-дезфенил-3'-(2-фурил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 174–176оС; [a]Na25 –47,8o (c 0,045, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,14 (д., J = 7,0 Гц, 2Н, ортобензоат), 7,74 (м., 2Н, ароматический), 7,51 (м., 7Н, ароматический), 6,86 (д., J = 9,2 Гц, 1Н, NH), 6,40 (д., J =1,2 Гц, 2Н, фурил), 6,29 (с., 1Н, Н10), 6,24 (двойной дублет, J =9,2, 9,2 Гц, 1Н, Н13), 5,89 (двойной дублет, J =9,2, 2,4 Гц, 1Н, Н3'), 5,69 (д., J =7,0 Гц, 1Н, Н2 b ), 4,96 (двойной дублет, J =9,5, 1,8 Гц, 1Н, Н5), 4,83 (двойной дублет, J =2,4 Гц, 1Н, Н2'), 4,42 (двойной дублет, J =10,7, 6,7 Гц, 1Н, Н7), 4,31 (д., J =8,6 Гц, 1Н, Н20 a ), 4,20 (д., J =8,6 Гц, 2Н, Н20 b ), 3,83 (д., J =7,0 Гц, 1Н, Н3), 2,56 (м., 1Н, Н6 a), 2,35 (м., 2Н, Н14), 2,24 (с., 3Н, 4Ас), 1,89 (м., 1Н, Н6 b ), 1,87 (широкий синглет, 3Н, Ме18), 1,87 (с., 1Н, 1ОН), 1,69 (с., 3Н, Ме19), 1,25 (с., 3Н, Ме17), 1,15 (с., 3Н, Ме16). Пример 15 Получение 0,174 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(4- фторфенил)-азетидин-2-она (570 мг, 1,43 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 315 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дезфенил-3'-(4-фторфенил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 315 мг смеси, полученной на предыдущей стадии, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 250 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 160 мг (64%) 3'-дезфенил-3'-(4-фторфенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 171–173оС; [a]Na25 o –49,0 (c 0,005, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,13 (д., J = 7,5 Гц, 2Н, ортобензоат), 7,25 (м., 2Н, ароматический), 7,61 (м., 1Н, ароматический), 7,50 (м., 4Н, ароматический), 7,43 (м., 2Н, ароматический), 7,10 (м., 2Н, ароматический), 6,96 (д., J = 8,7 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,25 (двойной дублет, J = 8,7, 8,7 Гц, 1Н, Н13), 5,79 (двойной дублет, J = 8,7, 2,4 Гц, 1Н, Н3'),5,67 (д., J = 7,1 Гц, 1Н, Н2 b), 4,45 (двойной дублет, J = 7,9, 1Н, Н5), 4,76 (двойной дублет, J = 4,8 Гц, 2,4 Гц, 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,31 (д., J = 8,9 Гц, 1Н, Н20 a), 4,20 (д., J = 8,9 Гц, 1Н, Н20 b ), 3,80 (д., J = 7,1 Гц, 1Н, Н3), 3,57 (д., J = 4,8 Гц, 1Н, 2'ОН), 2,58 (м., 1Н, 6Н a), 2,43 (в., J = 4,3 Гц, 1Н, 7ОН), 2,38 (с., 3Н, 4Ас), 2,30 (м., 2Н, Н14), 2,24 (с., 3Н, 10Ас), 1,85 (м., 1Н, Н6 b), 1,80 (широкий синглет, 3Н, Ме18), 1,69 (с., 1Н, 10Н), 1,55 (с., 3Н, Ме19), 1,23 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 16 Получение 3'-дезфенил-3'-(2-тиенил)таксола К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,087 мл 1,63М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре 3'-дезфенил-3'-(4-фторфенил)так сола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 17 40585 –45оС к этой смеси по каплям добавляли раствор цис-1-(4-бензоил)-3-триэтилсилилокси-4-(2-тиенил)азетидин-2-она (277 мг, 0,715 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 169 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-десфенил-3'-(2тиенил)таксол и небольшое количество (2'S,3'R)изомера. К раствору из 169 мг (0,112 ммоля) смеси, полученной на предыдущей стадии, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 140 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 93 мг (76%) 3'-дезфенил-3'-(2-тиенил)таксола, который перекристаллизовывали из смеси метанола/воды. Температура плавления 173–175оС; [a]Na25 o –42,1 (c 0,515, CHCl3). 1 Н-ЯМР (CDCl3, 300 МГц) d 8,14 (д., J = 7,1 Гц, 2Н, ортобензоат), 7,72 (д., J = 8,7 Гц, 2Н, ортобензамид), 7,65–7,35 (м., 6Н, ароматический), 7,31 (двойной дублет, J = 5,5, 1,1 Гц, 1Н тиенил), 7,19 (двойной дублет, J = 3,9, 1,1 Гц, 1Н, тиенил), 7,03 (двойной дублет, J = 5,5 3,9 Гц, 1Н, тиенил), 6,96 (д., J = 8,8 Гц, 1Н, NH), 6,28 (с., 1Н, Н10), 6,24 (двойной дублет, J = 8,8, 7,7 Гц, 1Н, Н13), 6,05 (двойной дублет, J = 8,8, 1,7 Гц, 1Н, Н3'), 5,68 (д., J = 7,1 Гц, 1Н, Н2), 4,95 (двойной дублет, J = 9,3, 1,6 Гц, 1Н, Н5), 4,78 (д., J = 2,2 Гц, 1Н, Н2'), 4,40 (двойной дублет, J = 11,0, 6,6 Гц, 1Н, Н7), 4,31 (д., J = 8,5 Гц, 1Н, Н20 a), 4,20 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,81 (д., J = 7,1 Гц, 1Н, Н3), 3,72 (широкий синглет, 1Н, 2'ОН), 2,54 (м., 1Н, Н6 a ), 2,41 (с., 3Н, 4Ас), 2,37 (м., 2Н, Н14 a, Н14 b ), 2,23 (с., 3Н, 10Ас), 1,88 (м., 1Н, Н6 b ), 1,82 (широкий синглет, 3Н, Ме18), 1,68 (с., 3Н, Ме19), 1,23 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 148 мг (96%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. Пример 18 Получение 2',7-гидроксизащищенного таксола с использованием алкоксида К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,286 мл 0,5 М раствора гексаметилдисилазида калия в толуоле. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-бензоил-3-триэтилсилилокси-4-фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 3 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя образовался остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 139 мг (90%) (2'R,3'S)-2',7(бис)триэтилсилилтаксола. Пример 19 Получение 2',7-гидроксизащищенного таксола с использованием алкоксида лития из гексаметилдисилазида лития К раствору из 7-триэтилсилилбаккатина (III) (100 мг, 0,143 ммоля) 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,143 мл 1,0 М раствора гексаметилдисилазида лития в тетрагидрофуране. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-бензоил-3-триэтилсилилокси4-фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 2 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя образовался остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, что позволило получить 151 мг (98%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. Пример 20 Получение таксола с использованием алкоксида лития (из гексаметилдисилазида лития) К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,143 мл 1,0 М раствора гексаметилдисилазида лития в толуоле. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-бензоил-3-(2-метокси-2-пропилокси)-4фенилазетидин-2-она (58 мг, 0,172 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и Пример 17 Получение 2'7-гидроксизащищенного таксола с использованием алкоксида магния К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,048 мл 3,0 М раствора бромметилмагния в простом эфире. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-бензоил-3-триэтилсилилокси-4-фенила зетидин-2-она (82 мг, 0,215 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 4 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную 18 40585 гидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 2 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток 320 мг сырого (2'R,3'S)-2',7-(бис)триэтилсилил-N-дебензоил-N-(4- хлорбензоил)таксола. К раствору из 320 мг (0,286 ммоля) указанного сырого продукта в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 252 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 213 мг (84%) N-дебензоил-N-(4-хлорбензоил)таксола, который перекристаллизовывали из смеси метанола и воды. Температура плавления 179–181оС. [a]Na25 o –49,8 (c 0,01, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,12 (д., J = 7,1 Гц, 2Н, ортобензоат), 7,64 (м., 2Н, ароматический), 7,60 (м., 1Н, ароматический), 7,49 (м., 9Н, ароматический), 7,03 (д., J = 8,8 Гц, 1Н, NH), 6,26 (с., 1Н, Н10), 6,21 (двойной дублет, J = 8,2, 28,2 Гц, 1Н, Н13), 5,76 (двойной дублет, J = 8,8, 8,2 Гц, 1Н, Н3'), 5,66 (д., J = 7,1 Гц, 1Н, Н2 b), 4,92 (двойной дублет, J = 9,9, 1,1 Гц, 1Н, Н5), 4,77 (двойной дублет, J = 5,5, 2,2 Гц, 1Н, Н2'), 4,38 (м., 1Н, Н7), 4,29 (д., J = 8,8 Гц, 1Н, Н20 a), 4,18 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,78 (д., J = 6,6 Гц, 1Н, Н3), 3,35 (д., 5,5 Гц, 1Н, 2'ОН), 2,55 (м., 1Н, Н6 a), 2,49 (д., J = 4,2 Гц, 1Н, 7ОН), 2,36 (с., 3Н, 4Ас), 2,28 (м., 2Н, Н14), 2,22 (с., 3Н, 10Ас), 1,85 (м., 1Н, Н6 b), 1,77 (широкий синглет, 3Н, Ме18), 1,76 (с., 1Н, 1ОН), 1,67 (с., 3Н, Ме19), 1,22 (с., 3Н, Ме17), 1,13 (с., 3Н, Ме16). Пример 23 выдерживали при этой температуре в течение 2 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя образовался остаток, который очищали путем перекристаллизации, что позволило получить 147 мг (99%)(2'R,3'S)-2'-(2-метокси-2-пропилокси)-7-триэтилсилилтаксола. К раствору из 116 мг (0,112 ммоля) (2'R,3'S)2'-(2-метокси-2-пропилокси)-7-триэтилсилилтаксола в 6 мг ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 8 часов, а затем при 25оС в течение 10 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. В результате выпаривания этилацетатного раствора было получено 113 мг вещества, которое очищали посредством перекристаллизации с образованием 95 мг (99%) таксола, который во всех отношениях был идентичен достоверной пробе. Пример 21 Получение 2',7-гидроксизащищенного таксола с использованием алкоксида натрия К раствору из 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) 1 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,143 мл 1 М раствора гексаметилдисилазида натрия в тетрагидрофуране. После выстаивания в течение 1 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-бензоил-3-триэтилсилилокси-4фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 3 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. Выпаривание органического слоя позволило получить остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией, в результате чего было получено 108 мг (70%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. Пример 22 Получение N-дебензоил-N-(4-трет.бутилбензоил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63 М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при –45оС к этой смеси по каплям добавляли раствор (+)-цис-1-(4трет.бутилбензоил)-3-триэтилсилилокси-4- фенилазетидин-2-она (226 мг, 0,515 ммоля) в 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 2 часов перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным Получение N-дебензоил-N-(4-хлорбензоил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,174 мл 1,63 М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор (+)-цис1-(4-хлорбензоил)-3-триэтилсилилокси-4-фенилазетидин-2-она (215 мг, 0,515 ммоля) 2 мл тетра 19 40585 раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, который очищали фильтрованием через силикагель с образованием 330 мг сырого (2'R,3'S)-2',7(бис)триэтилсилил-3'-дебензоил-N-(4- трет.бутилбензоил)таксола. К раствору из 330 мг (0,289 ммоля) указанного сырого продукта в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. Выпаривание этилацетатного раствора позволило получить 260 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 240 мг (92%) N-дебензоил-N-(4-трет.бутилбензоил)таксола, который перекристаллизовывали из смеси метанола и воды. Температура плавления 171–173оС. [a]Na25 –49,1o (c 0,05, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,13 (д., J = 7,1 Гц, 2Н, ортобензоат), 7,76–7,25 (м., 12Н, ароматический), 6,98 (д., J = 8,8 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,21 (двойной дублет, J = 8,8, 8,8 Гц, 1Н, Н13), 5,77 (двойной дублет, J = 8,8, 2,7 Гц, 1Н, Н3'), 5,67 (д., J = 6,6 Гц, 1Н, Н2 b ), 4,94 (двойной дублет, J = 9,3, 1,2 Гц, 1Н, Н5), 4,78 (двойной дублет, J = 4,4, 2,7 Гц, 1Н, Н2'), 4,38 (м., 1Н, Н7), 4,29 (д., J = 8,2 Гц, 1Н, Н20 a ), 4,20 (д., J = 8,2 Гц, 1Н, Н20 b ), 3,79 (д., J = 6,6 Гц, 1Н, Н3), 3,65 (д., J = 4,4 Гц, 1Н, 2'ОН), 2,57 (м., 1Н, Н6 a ), 2,48 (д., J = 4,1 Гц, 1Н, 7ОН), 2,37 (с., 3Н, 4Ас), 2,31 (м., 2Н, Н14), 2,22 (с., 3Н, 10Ас), 1,85 (м., 1Н, Н6 b ), 1,79 (широкий синглет, 3Н, Ме18), 1,68 (с., 1Н, 1ОН), 1,68 (с., 3Н, Ме19), 1,29 (с., 9Н, Ar-tBu), 1,23 (с., 3Н, Ме17), 1,13 (с., 3Н, Ме16). Пример 24 который очищали фильтрованием через силикагель с образованием 362 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-N-дебензоил-N-(4метоксибензоил)-3'-дезфенил-3'-(4-фторфенил)таксола и небольшое количество (2'S,3'R) изомера. К раствору из 362 мг смеси, полученной в результате выполнения предыдущей реакции, в 12 мл ацетонитрила и 0,6 мл пиридина при 0оС добавляли 1,8 мл 48% водного раствора HF. Полученную смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. Выпаривание этилацетатного раствора позволило получить 269 мг вещества, которое очищали посредством высокоскоростной хроматографии с образованием 183 мг (71%) N-дебензоил-N-(4-метоксибензоил)-3'-дезфенил-3'-(4- фторфенил)таксола, который перекристаллизовывали из смеси метанола и воды. Температура плавления 172,5–174,5оС; 25 [a]Na –47,0o (c 0,0044, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,13 (д., J = 7,2 Гц, 2Н, ортобензоат), 7,7–7,4 (м., 9Н, ароматический), 7,10 (двойной дублет, J = 8,8, 8,8 Гц, 2Н, ароматический), 6,97 (д., J = 8,8 Гц, 1Н, NH), 6,27 (с., 1Н, Н10), 6,23 (двойной дублет, J = 8,8, 8,8 Гц, 1Н, Н13), 5,76 (двойной дублет, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,67 (д., J = 7,1 Гц, 1Н, Н2 b ), 4,94 (двойной дублет, J = 9,9, 2,2 Гц, 1Н, Н5), 4,75 (двойной дублет, J = 4,4, 2,2 Гц, 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,31 (д., J = 8,5 Гц, 1Н, Н20 a), 4,19 (д., J = 8,5 Гц, 1Н, Н20 b ), 3,79 (д., J = 7,1 Гц, 1Н, Н3), 3,59 (д., = 4,4 Гц, 1Н, 2'ОН), 2,54 (м., 1Н, Н6 a ), 2,47 (д., J = 4,4 Гц, 1Н, 7ОН), 2,36 (с., 3Н, 4Ас), 2,30 (м., 2Н, Н14 a, Н14 b), 2,24 (с., 3Н, 10Ас), 1,88 (м., 1Н, Н6 a), 1,78 (широкий синглет, 3Н, Ме18), 1,74 (с., 1Н, 1ОН), 1,68 (с., 3Н, Ме19), 1,23 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 25 Получение (2'R,3'S)-2',7-(бис)триэтилсилилтаксола с использованием алкоксида цинка К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при температуре –45оС по каплям добавляли 0,286 мл 0,5 М раствора гексаметилдисилазида калия в толуоле. Через 15 минут добавляли 32 мг (0,143 ммоля) комплекса хлорида цинка с диметоксиэтаном. Еще через 1 час после выдержки при –45оС нагревали до 0оС и к ней по каплям добавляли раствор (+)-цис1-бензоил-3-триэтилсилилокси-4-фенилазетидин2-она (82 мг, 0,215 ммоля) в 1 мл ТГФ. Раствор перемешивали при 0оС в течение 3 часов, а затем добавляли 1 мл 10%-ного раствора АсОН в ТГФ. Смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетат/гексан 60/40. После упаривания органического слоя получали остаток, который очищали посредством высокоскоростной хроматографии с последующей перекристаллизацией с получением 139 мг (90%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. Пример 26 Получение (2'R,3'S)-2',7-(бис)триэтилсилилтаксола с использованием алкоксида кадмия К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в ТГФ при температуре Получение N-дебензоил-N-(4-метолксибензоил)-3'-(4- фторфенил)таксола К раствору из 7-триэтилсилилбаккатина III (200 мг, 0,285 ммоля) в 2 мл тетрагидрофурана при температуре –45оС по каплям добавляли 0,175 мл 1,63 М раствора n-BuLi в гексане. После выстаивания в течение 0,5 часа при температуре –45оС к этой смеси по каплям добавляли раствор цис-1-(4-метоксибензоил)-3-триэтилсилилокси-4(4-фторфенил)азетидин- 2-она (614 мг, 1,43 ммоля) 2 мл тетрагидрофурана. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа перед добавлением 1 мл 10% раствора уксусной кислоты в тетрагидрофуране. Полученную смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетата и гексана с соотношением 60/40. В результате выпаривания органического слоя был получен остаток, 20 40585 –45оС по каплям добавляли 0,286 мл 0,5 М раствора гексаметилдисилазида калия в толуоле. Через 15 минут добавляли 26 мг (0,143 ммоля) безводного хлорида кадмия. Еще через 1 час после выдержки при –45оС нагревали до 0оС и к ней по каплям добавляли раствор (+)-цис-1-бензоил-3триэтилсилилокси-4-фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл ТГФ. Раствор перемешивали при 0оС в течение 3 часов, а затем добавляли 1 мл 10%-ного раствора АсОН в ТГФ. Смесь распределяли между насыщенным водным раствором NaНСО3 и смесью этилацетат/гексан 60/40. После упаривания органического слоя получали остаток, который очищали высокоскоростной хроматографией с последующей перекристаллизацией с получением 131 мг (85%) (2'R,3'S)-2',7-(бис)триэтилсилилтаксола. Пример 27 Получение (2'R,3'S)-2',7-(бис)триэтилсилилтаксола с использованием алкоксида натрия. К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в ТГФ при температуре –45оС по каплям добавляли к суспензии 3,5 мг гидрида натрия в 0,5 мл ТГФ. Раствор нагревался до 0оС и к нему по каплям добавляли раствор (+)цис-1-бензоил-3-триэтилсилилокси-4-фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл ТГФ. Затем раствор нагревался до 0оС и выдерживался при этой температуре в течение 10 часов, после чего добавляли 1 мл 10%-ного раствора АсОН в ТГФ. Смесь распределяли между насыщенным водным раствором NaНСО3 и этилацетат/гексан 60/40. После упаривания органического слоя получали остаток, который очищали высокоскоростной хроматографией с последующей перекристаллизацией с получением 98 мг (64%) (2'R,3'S)-2',7(бис)триэтилсилилтаксола. Пример 28 Получение (2'R,3'S)-2'7-(бис)триэтилсилилтаксола с использованием алкоксида титана. К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в ТГФ при температуре –45оС по каплям добавляли 0,087 мл 1,63 М раствора n-BuLi в гексане. Через 1 час после выдержки при –45оС раствор охлаждали до –78оС и к нему добавляли 0,140 мл 1,0 М раствора тетрахлорида титана в хлористом метилене. Затем раствор нагревали до –45оС и к нему по каплями добавляли (+)-цис-1-бензоил-3-триэтилсилилокси-4- фенилазетидин-2-она (82 мг, 0,215 ммоля) в 1 мл ТГФ. Затем раствор нагревался до 0оС и выдерживался при этой температуре в течение 10 часов, после чего добавляли 1 мл 10%-ного раствора АсОН в ТГФ. Смесь распределяли между насыщенным водным раствором NaНСО3 и этилацетат/гексан 60/40. После упаривания органического слоя получали остаток, который очищали высокоскоростной хроматографией с последующей перекристаллизацией с получением 129 мг (84%) (2'R,3'S)-2',7(бис)триэтилсилилтаксола. Пример 29 Получение N-дебензоил-N-(4-метилбензоил)-3'-дезфенил-3'-(4- нитрофенил)таксола К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при температуре –45оС по каплям добавляли 0,104 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(4-метилбензиол)-3триэтилсилилокси-4-(4-нитрофенил)азетидин-2она (377 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и смесью этилацет/гексан 60/40.После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 195 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(4-метилбензоил)-3'-дезфенил-3'-(4-нитрофенил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 195 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. После выпаривания этилацетатного раствора получали 156 мг вещества, которое очищали посредством высокоскоростной хроматографией, получая 93,6 мг (60%) N-дебензоил-N-(4-метилбензоил)-3'-дезфенил-3'-(4-нитрофенил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 184–185оС; [a]Na25 o –55,0 (c 0,0048, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,26 (д., J = 8,79 Гц, 2Н, Ar-NO2), 8,12 (д., J = 7,1 Гц, 2Н, орто-бензоат), 7,7–7,2 (м., 9Н, ароматический), 7,07 (д., J = 9,3 Гц, 1Н, NH), 6,25 (с., 1Н, Н10), 6,26 (дд., J = 8,8, 8,8 Гц, 1Н, Н13), 5,93 (дд., J = 9,3, 2,2 Гц, 1Н, Н3'), 5,66 (д., J = 6,6 Гц, 1Н, Н2 b ), 4,95 (дд., J = 9,3, 1,6 Гц, 1Н, Н5), 4,82 (дд., J = 3,3, 2,2 Гц, 1Н, Н2'), 4,39 (м., 1Н, Н7), 4,30 (д., J = 8,2 Гц, 1Н, Н20 a), 4,19 (д., J = 8,2 Гц, 1Н, Н20 b ), 3,84 (д., J = 3,85 Гц, 1Н, 2'ОН), 3,8 (д., J = 6,7 Гц, 1Н, Н3), 2,55 (м., 1Н, Н6 a ), 2,45 (м., 1Н, 7ОН), 2,4 (с., 3Н, 4Ас), 2,38 (с., 3Н, ArMe), 2,29 (м., 2Н, Н14 a), 2,23 (с., 3Н, 10Ас), 1,88 (м., 1Н, Н6 b ), 1,80 (широкий синглет, 3Н, Ме18), 1,71 (с., 1Н, 1ОН), 1,68 (с., 3Н, Ме19), 1,21 (с., 3Н, Ме17), 1,14 (с., 3Н, Ме16). Пример 30 Получение N-дебензоил-N-(фуроил)-3'дезфенил-3'-(4- нитрофенил)таксола К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(фуроил)-3-триэтилсилилокси-4- (4нитрофенил)азетидин-2-она (596 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацет-гексаном 60/40.После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 320 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(фуроил)-3'дезфенил-3'-(4-нитрофенил)таксол и небольшое количество (2'S,3'R) изомера. 21 40585 Температура плавления 178–179оС; [a]Na25 –56,0 (c 0,0064, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,25 (d., J = 8,7 Гц, 2Н, Ar-NO2), 8,12 (d., J = 7,2 Гц, 2Н, орто-бензоат), 7,72–7,49 (m, 7Н, ароматический), 7,05 (br, 1Н, NH), 6,88 (m, 2Н, ароматический), 6,27 (s, 1Н, Н10), 6,25 (dd, J = 9,1, 9,1 Гц, 1Н, Н13), 5,91 (д, J = 8,7 Гц, 1Н, H3'), 5,66 (d, J = 6,8 Гц, 1Н, Н2 b ), 4,95 (d, J = 9,6 Гц, 1Н, Н5), 4,81 (d, J = 2,7, Гц, 1Н, Н2'), 4,38 (m, 1H, H7), 4,3 (d, J = 8,2 Гц, 1Н, Н20 a), 4,19 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,8 (br s, 1Н, 2'ОН), 3,82 (s, 3H, OMe), 3,80 (d, J = 6,8 Гц, 1Н, Н3), 2,54 (m, 1H, H6 a ), 2,48 (m, 1H, 7OH), 2,40 (s, 3H, 4Ac), 2,32 (m, 2H, H14), 2,23 (s., 3Н, 10Ас), 1,89 (m, Н6 b ), 1,81 (br s, 3Н, Ме18), 1,77 (s., 1Н, 10Н), 1,69 (s., 3Н, Ме19), 1,22 (s., 3Н, Ме17), 1,14 (s., 3Н, Me16). Пример 32 Получение N-дебензоил-N-(4-трет-бутилбензоил)-3'дезфенил-3'-(4- нитрофенил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(4-трет-бутилбензоил)-3- триэтилсилилокси-4-(4-нитрофенил)азетидин-2-она (413 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 202 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтил-силил-Nдебензоил-N-(4- трет-бутилбензоил)-3'-дезфенил3'-(4-нитрофенил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 202 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После выпаривания этилацетатного раствора получали 163 мг вещества, которое очищали быстрой хроматографией, получая 102,0 мг (63%) N-дебензоил-N-(4-трет-бутилбензоил)-3'- дезфенил- 3'-(4-нитрофенил)таксола, который перекристаллизовывали из водного метанола. К раствору 320 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. После выпаривания этилацетатного раствора получали 254 мг вещества, которое очищали быстрой хроматографией, получая 187 мг (74%) N-дебензоил-N-(фуроил)-3'-дезфенил- 3'-(4- нитрофенил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 184–185оС; [a]Na25 o –60,0 (c 0,006, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,26 (d., J = 8,79 Гц, 2Н, Ar-NO2), 8,12 (d., J = 7,2 Гц, 2Н, орто-бензоат), 7,68 (d, J = 8,8 Гц, 2Н, орто-бензамид), 7,7– 7,47 (m, 6Н, ароматический), 7,3 (d, J = 9,3 Гц, 1Н, NH), 7,02 (d, J = 3,3 Гц, 1Н, фурил), 6,48 (dd, J = 3,3 Гц, 1,65 Гц, 1Н, фурил), 6,27 (s, 1Н, Hz, Н10), 6,26 (dd, J = 8,5 Гц, 1Н, Н13), 5,87 (dd, J = 8,8, 1,65 Гц, 1Н, H3'), 5,65 (d, J = 6,6 Гц, 1Н, Н2 b ), 4,93 (d, J = 8,2 Гц, 1Н, Н5), 4,79 (dd, J = 2,7, 1,4 Гц, 1Н, Н2'), 4,38 (m, 1H, H7), 4,29 (d, J = 8,4 Гц, 1Н, Н20 a), 4,18 (d, J = 8,4 Гц, 1Н, Н20 b), 3,97 (d, J = 3,3 Гц, 1Н, 2'ОН), 3,79 (d, J = 6,6 Гц, 1Н, Н3), 2,5 (m, 1H, H6 a), 2,4 (m, 1H, 7OH), 2,38 (s, 3H, 4Ac), 2,27 (m, 2H, H14), 2,22 (s., 3Н, 10Ас), 1,88 (m., 1Н, Н6 b ), 1,81 (br s, 3Н, Ме18), 1,78 (s., 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,21 (s., 3Н, Ме17), 1,13 (s., 3Н, Ме16). Пример 31 Получение N-дебензоил-N-(4-метоксибензоил)-3'дезфенил-3'-(4- нитрофенил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(4-метоксибензоил)-3- триэтилсилилокси-4-(4-нитрофенил)азетидин-2-она (390 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 198 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтил-силил-Nдебензоил-N-(4-метоксибензоил)-3'-дезфенил-3'(4-нитрофенил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 197 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 159 мг вещества, которое очищали быстрой хроматографией, получая 106,0 мг (66%) N-дебензоил-N-(4-метоксибензоил)-3'- дезфенил- 3'-(4-нитрофенил)таксола, который перекристаллизовывали из водного метанола. o Температура плавления 189–190оС; [a]Na25 –62,0o (c 0,0021, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,3 (d., J = 8,8 Гц, 2Н, Ar-NO2), 8,11 (d., J = 7,2 Гц, 2Н, орто-бензоат), 7,7–7,41 (m, 9Н, ароматический), 7,11 (d, J = 8,8 Гц, 1Н, NH), 6,27 (s, 1Н, Н10), 6,25 (dd, J = 8,3, 8,3 Гц, 1Н, Н13), 5,92 (dd, J = 8,8, 1,6 Гц, 1Н, H3'), 5,66 (d, J = 7,1 Гц, 1H, Н2 b ), 4,94 (d, J = 9,3 Гц, 1Н, Н5), 4,82 (dd, J = 3,0, 2,2, Гц, 1Н, Н2'), 4,39 (m, 1H, H7), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a ), 4,19 (d, J = 8,2 Гц, 1Н, Н20 b), 3,91 (d, J = 3,85 Гц, 1Н, 2'ОН), 3,79 (d, J = 7,1 Гц, 1Н, Н3), 2,55 (m, 1H, H6 a ), 2,47 (m, 1H, 70H), 2,39 (s, 3H, 4Ac), 2,29 (m, 2H, 22 40585 H14), 2,22 (s., 3Н, 10Ас), 1,89 (m, 1Н, Н6 b ), 1,81 (br s, 3Н, Ме18), 1,79 (s., 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,30 (s, 9Н, t-Бу), 1,21 (s., 3Н, Ме17), 1,14 (s., 3Н, Me16). Пример 33 Получение 3'-дезфенил-3'-(изопропил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-бензоил-3- триэтилсилилокси-4-(4изопропил)азетидин-2-она (296 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 179 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил3'- (изопропил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 179 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 140 мг вещества, которое очищали быстрой хроматографией, получая 112,0 мг (80%) 3'-дезфенил3'-(изопропил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 162–163оС; [a]Na25 o –33,0 (c 0,0068, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,16 (d., J = 7,2 Гц, 2Н, орто-бензоат), 7,65 (d, J = 8,4 Гц, 2Н, ортобензамид), 7,62–7,32 (m, 6Н, ароматический), 6,33 (d, J = 9,3 Гц, 1Н, NH), 6,26 (s, 1Н, Н10), 6,19 (dd, J = 8,2, 8,2 Гц, 1Н, Н13), 5,67 (d, J = 7,1 Гц, 1H, Н2 b ), 4,96 (d, J = 9,8 Гц, 1Н, Н5), 4,59 (d, J = 2,7, Гц, 1Н, Н2'), 4,40 (m, 1H, H7), 4,31 (d, J = 9,2 Гц, 1Н, Н20 a ), 4,22 (m, 2Н, Н3', Н20 b ), 3,79 (d, J = 6,6 Гц, 1Н, Н3), 3,64 (d, J = 4,4 Гц, 1Н, 2'ОН), 2,54 (m, 1Н, Н6 a ), 2,49 (s, 3Н, 4Ас), 2,47 (m, 1H, 70H), 2,40 (m, 1H, СН-Ме), 2,29 (m, 2Н, Н14), 2,21 (s., 3Н, 10Ас), 1,88 (m, 1Н, Н6 b), 1,84 (br s, 3Н, Ме18), 1,78 (s., 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,20 (s., 3Н, Ме17), 1,14–1,04 (m, 9Н, Me16 + Меизопропил). Пример 34 Получение 3'-дезфенил-3'-(5-метил-2-тиенил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-бензоил-3- триэтилсилилокси-4-(5метил-2-тиетил)-азетидин-2-она (343 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между на сыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 188 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил3'- (5-метил-2-тиенил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 181 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 150 мг вещества, которое очищали быстрой хроматографией, получая 120,0 мг (80%) 3'-дезфенил- 3'-(5-метил-2-тиенил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 162–163оС; [a]Na25 o –32,0 (c 0,0054, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,12 (d., J = 7,1 Гц, 2Н, орто-бензоат), 7,7 (d, J = 8,3 Гц, 2Н, ортобензамид), 7,61–7,34 (m, 6Н, ароматический), 7,2 (d, J = 5,5 Гц, 1Н, тиенил), 6,86 (d, J = 5,5 Гц, 1Н, тиенил), 6,79 (d, J = 8,8 Гц, 1Н, NH), 6,27 (s, 1Н, Н10), 6,19 (dd, J = 9,3, 9,3 Гц, 1Н, Н13), 6,07 (dd, J = 8,2, 2,7 Гц, 1Н, Н3'), 5,67 (d, J = 7,1 Гц, 1Н, 2 b ), 4,93 (d, J = 9,9 Гц, 1Н, Н5), 4,67 (dd, J = 6,7, 2,7 Гц, 1Н, Н2'), 4,34 (m, 1H, Н7), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a), 4,20 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,81 (d, J = 7,1 Гц, 1Н, Н3), 3,40 (d, J = 6,6 Гц, 1Н, 2'ОН), 2,55 (m, 1Н, Н6 a ), 2,46 (d, J = 4,4 Гц, 1Н, 70Н), 2,40 (m, 2Н, Н14), 2,35 (s, 3Н, 4Ас), 2,32 (s, 3Н, Ме тиэтил), 2,23 (s, 3Н, 10Ас), 1,83 (m, 1Н, Н6 b ), 1,82 (br s, 3Н, Ме18), 1,80 (s, 1Н, 10Н), 1,68 (s, 3Н, Ме19), 1,24 (s, 3Н, Ме17), 1,14 (s, 3Н, Ме16). Пример 35 Получение N-дебензоил-N-(пивалоил)таксола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(пивалоил)-3- триэтилсилилокси-4фенилазетидин-2-она (517 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 304 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(пивалоил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 304 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 238 мг вещества, которое очищали быстрой хроматографией, получая 23 40585 Пример 37 Получение N-дебензоил-N-(1-нафтоил)так 216 мг (90%) N-дебензоил-N-(пивалоил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 173–175оС; [a]Na25 o –58,6 (c 0,0103, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,12 (d., J = 7,7 Гц, 2Н, орто-бензоат), 7,63–7,30 (m, 8Н, ароматический), 6,50 (d, J = 9,3 Гц, 1Н, NH), 6,27 (s, 1Н, Н10), 6,17 (dd, J = 8,3, 8,3 Гц, 1Н, Н13), 5,67 (d, J = 7,2 Гц, 1Н, Н3'), 5,67 (d, J = 8,2 Гц, 1Н, Н2 b ), 4,93 (d, J = 8,3 Гц, 1Н, Н5), 4,68 (br s, 1Н, Н2'), 4,39 (m, 1H, Н7), 4,29 (d, J = 8,2 Гц, 1Н, Н20 a), 4,17 (d, J = 8,2 Гц, 1Н, Н20b ), 3,77 (d, J = 6,6 Гц, 1Н, Н3), 3,47 (d, 1Н, 2'ОН), 2,51 (m, 1Н, Н6 a ), 2,47 (br s, 1Н, 70Н), 2,35 (s, 3Н, 4Ас), 2,30 (m, 2H, H14), 2,24 (s, 3Н, 10Ас), 1,85 (m, 1Н, Н6 b ), 1,79 (br s, 3Н, Ме18), 1,67 (s, 3Н, Ме19), 1,25 (s, 3Н, Ме17), 1,16 (s, Мепивалоил), 1,14 (s, 3Н, Ме16). Пример 36 Получение N-дебензоил-N-(2-фуроил)таксола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(2-фуроил)-3-триэтилсилилокси-4-фенилазетидин-2-она (531 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 306 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(2-фуроил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 306 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 240 мг вещества, которое очищали быстрой хроматографией, получая 192 мг (80%) Nдебензоил-N-(2-фуроил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 172–174оС; [a]Na25 o –49,6 (c 0,0103, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,12 (d., J = 7,7 Гц, 2Н, орто-бензоат), 7,64–7,33 (m, 9Н, ароматический), 7,19 (d, J = 8,8 Гц, 1Н, NH), 7,01 (d, J = 3,3 Гц, 1Н, фурил), 6,45 (dd, 1Н, фурил), 6,26 (s, 1Н, Н10), 6,21 (dd, J = 8,8, 8,8 Гц, 1Н, Н13), 5,72 (dd, J = 8,8, 1,7 Гц, 1Н, Н3'), 5,66 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,92 (d, J = 8,2 Гц, 1Н, Н5), 4,75 (br s, 1Н, Н2'), 4,40 (m, 1H, Н7), 4,28 (d, J = 8,2 Гц, 1Н, Н20 a), 4,18 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,78 (d, J = 6,6 Гц, 1Н, Н3), 3,69 (d, J= 3,8, 1Н, 2'ОН), 2,53 (m, 1Н, Н6 a), 2,47 (br s, 1Н, 70Н), 2,36 (s, 3Н, 4Ас), 2,32 (m, 2H, H14), 2,22 (s, 3Н, 10Ас), 1,84 (m, 1Н, Н6 b ), 1,79 (br s, 3Н, Ме18), 1,67 (s, 3Н, Ме19), 1,22 (s, 3Н, Ме17), 1,13 (s, 3Н, Ме16). сола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора n-BuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(1-нафтоил)-3- триэтилсилилокси-4фенилазетидин-2-она (617 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 323 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебен зоил-N-(1-нафтоил)таксол и небольшое количество (2'S,3'R) изомера. К раствору 323 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 259 мг вещества, которое очищали быстрой хроматографией, получая 192 мг (74%) N-дебензоил-N-(1-нафтоил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 179–181оС; [a]Na25 o –49,2 (c 0,0108, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,22 (w, J = 7,7 Гц, 1Н, 2Н-нафоиловое кольцо), 8,10 (d, J = 7,7 Гц, 2Н, орто-бензоат), 7,90–7,40 (m, 14Н, ароматический), 6,80 (d, J = 9,3 Гц, 1Н, NH), 6,33 (dd, 1Н, J = 7,7, 7,7 Гц, 1Н, Н13), 6,28 (s, 1Н, Н10), 5,88 (dd, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,69 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,94 (d, J = 7,7 Гц, 1Н, Н5), 4,81 (dd, J = 5,8, 2,8 Гц, 1Н, Н2'), 4,40 (m, 1H, Н7), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a), 4,20 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,80 (d, J = 6,6 Гц, 1Н, Н3), 3,48 (d, J= 5,5, 1Н, 2'ОН), 2,54 (m, 1Н, Н6 a), 2,46 (d, J = 4,4 Гц, 1Н, 7-ОН), 2,40 (s, 3Н, 4Ас), 2,24 (s, 3H, 10Ac), 2,18 (m, 2Н, H14), 1,90 (m, 1Н, Н6 b ), 1,83 (br s, 3Н, Ме18), 1,79 (s, 1H, 10H), 1,67 (s, 3Н, Ме19), 1,27 (s, 3Н, Ме17), 1,14 (s, 3Н, Ме16). Пример 38 Получение N-дебензоил-N-(2-нафтоил)таксола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(2-нафтоил)-3- триэтилсилилокси-4-фенилазетидин-2-она (617 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, ко 24 40585 дезфенил- 3'-(2-фурил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 152–153оС; [a]Na25 o –61,0 (c 0,006, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,11 (d., J = 7,1 Гц, 2Н, орто-бензоат), 7,65–7,47 (m, 5Н, ароматический), 7,42 (s, 1Н, фурил), 6,36 (m, 2Н, фурил), 6,29 (s, 1Н, Н10), 6,26 (dd, J = 8,2, 8,2 Гц, 1Н, Н13), 5,67 (d, 7,1 Гц, Н2 b ), 5,37 (br s, 2H, NH, H3'), 4,94 (d, J = 8,2 Гц, 1Н, Н5), 4,72 (d, J = 3,3, Гц, 1Н, Н2'), 4,41 (m, 1H, H7), 4,30 (d, J = 8,8 Гц, 1Н, Н20 a ), 4,18 (d, J = 8,8 Гц, 1Н, Н20 b ), 4,03 (dd, J = 14,2, 7,1 Гц, 2Н, О-СН2-), 3,81 (d, J = 7,1 1H, H3), 3,32 (br s, 1H, 2'OH), 2,54 (m, 1H, H6 a ), 2,46 (d, 1H, 70H), 2,38 (s, 3H, 4Ac), 2,31 (m, 2H, H14), 2,24 (s, 3H, 10Ac), 1,89 (m, 1H, H6 b), 1,87 (br s, 3H, Me18), 1,76 (s, 1H, 10H), 1,67 (s, 3H, Me19), 1,26 (s, 3H, Me17), 1,15 (m, 6H, Me16 + Этил). Пример 40 Получение 3'-дезфенил-3'-(4-диметиламинофенил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-бензоил-3- триэтилсилилокси-4-(4диметиламинофенил)азетидин-2-она (363 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 172 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'дезфенил-3'- (4-диметиламинофенил)таксол и небольшое количество (2'S,3'R)- изомера. К раствору 192 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 153 мг вещества, которое очищали быстрой хроматографией, получая 131 мг (86%) 3'-дезфенил-3'-(4- диметиламинофенил)таксола, который перекристаллизовывали из водного метанола. торый очищали фильтрацией через силикагель, получая 323 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(2-нафтоил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 323 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 259 мг вещества, которое очищали быстрой хроматографией, получая 195 мг (75%) N-дебензоил-N-(1-нафтоил)таксола, который перекристаллизовывали из водного метанола. Температура плавления 181–183оС; [a]Na25 o –49,2 (c 0,0108, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,22 (br s, 1H, Н1-нафоиловое кольцо), 8,16 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,86–7,36 (m, 14Н, ароматический), 7,12 (d, J = 9,3 Гц, 1Н, NH), 6,26 (s, 1Н, Н10), 6,22 (dd, J= 7,7, 7,7 Гц, 1Н, Н13), 5,86 (dd, J = 9,3, 2,2 Гц, 1Н, Н3'), 5,68 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,95 (dd, J = 9,3, 1,6 Гц, 1Н, Н5), 4,84 (dd, J = 5,5, 2,8 Гц, 1Н, Н2'), 4,41 (m, 1H, Н7), 4,31 (d, J = 8,8 Гц, 1Н, Н20 a ), 4,20 (d, J = 7,7 Гц, 1Н, Н20 b ), 3,80 (d, J = 7,1 Гц, 1Н, Н3), 3,57 (d, J = 5,0, 1Н, 2'ОН), 2,53 (m, 1Н, Н6 a), 2,46 (d, J = 4,4 Гц, 1Н, 7-ОН), 2,41 (s, 3Н, 4Ас), 2,33 (m, 2H, H14), 2,26 (s, 3H, 10Ac), 1,86 (m, 1Н, Н6 b ), 1,80 (br s, 3Н, Ме18), 1,76 (s, 1H, 10H), 1,68 (s, 3Н, Ме19), 1,23 (s, 3Н, Ме17), 1,14 (s, 3Н, Ме16). Пример 39 Получение N-дебензоил-N-(этоксикарбонил)-3'-дезфенил-3'-(2- фурил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(этоксикарбонил)-3-триэтилсилилокси-4-(2-фурил)азетидин-2-она (290 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 179 мг смеси, содержащей(2'R,3'S)-2',7-(бис)триэтил-силил-N-дебензоил-N–(этоксикарбонил)3’-дезфенил-3’-(2-фурил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 179 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 139 мг вещества, которое очищали быстрой хроматографией, получая 125 мг (90%) N-дебензоил-N-(этоксикарбонил)-3' Температура плавления 171–172оС; [a]Na25 –49,0o (c 0,005, CHCl3). 1 H-ЯМР (CDCl3, 300 МГц) d 8,14 (d., J = 7,1 Гц, 2Н, орто-бензоат), 7,70–7,30 (m, 12Н, ароматический), 6,77 (br, 1Н, NH), 6,28 (m, 1Н, Н10), 6,20 (dd, J = 8,1, 8,1 Гц, 1Н, Н13), 5,61 (m, 2H, H3' + H2 b), 4,95 (d, J = 9,8 Гц, 1Н, Н5), 4,74 (m, 1Н, Н2'), 4,41 (m, 1H, H7), 4,31 (d, J = 8,8 Гц, 1Н, Н20 a ), 4,19(d, J=8,8 Гц, 1Н, Н20 b), 3,80 (d, J = 7,1 Гц, 1Н, Н3), 3,48 (br s, 1Н, 2'OН), 2,95 (s, 6H, диметиламино), 2,54 (m, 1Н, Н6 a), 2,45 (d, 1H, 70H), 2,39 (s, 3H, 4Ac), 2,33 (m, 2Н, Н14), 2,23 (s., 3Н, 10Ас), 1,90 (m, 1Н, Н6 b ), 1,82 (br s, 3Н, Ме18), 1,79 (s., 25 40585 торый очищали фильтрацией через силикагель, получая 331 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дебензоил-N-(2-тиеноил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 311 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 246 мг вещества, которое очищали быстрой хроматографией, получая 195 мг (80%) N-дебензоил-N-(2-тиеноил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 218–219оС; [a]Na25 –47,6o (c 0,0103, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,12 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,64–7,31 (m, 10Н, ароматический), 7,05 (dd, 1Н, тиениловое кольцо), 6,86 (d, J = 9,3 Гц, 1Н, NH), 6,23 (s, 1H, H10), 6,21 (dd, J = 8,8, 8,8 Гц, 1Н, Н13), 5,75 (dd, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,66 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,93 (dd, J = 9,3, 1,65 Гц, 1Н, Н5), 4,78 (dd, J = 5,0, 2,7 Гц, 1Н, Н2'), 4,41 (m, 1H, H7), 4,28 (d, J = 8,2 Гц, 1Н, Н20 a), 4,18 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,78 (d, J = 6,6 Гц, 1Н, Н3), 3,63 (d, J = 4,9 Гц, 1Н, 2'ОН), 2,53 (m, 1H, Н6 a), 2,48 (d, J = 4,4 Гц, 1H, 70H), 2,36 (s, 3Н, Ас), 2,30 (m, 2Н, Н14), 2,23 (s., 3Н, 10Ас), 1,85 (m, 1Н, Н6 b ), 1,78 (br s, 3Н, Ме18), 1,68 (s., 3Н, Ме19), 1,23 (s., 3Н, Ме17), 1,13 (s, 3Н, Me16). Пример 43 Получение N-дебензоил-N-(4-фенилбензоил)таксола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(4-фенилбензоил)-3-триэтилсилилокси-4фенилазетидин-2-она (654 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 331 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N-(4- фенилбензоил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 311 мг (0,286 ммоля) смеси, полученной из предыдущей реакции, в 18 мл ацетонитрила и 0,93 мл пиридина при 0оС добавляли 2,8 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 266 мг вещества, которое очищали быстрой хроматографией, получая 213 мг (80%) N-дебензоил-N(4-фенилбензоил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 183–184оС; [a]Na25 –48,4o (c 0,0109, CHCl3). 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,24 (s., 3Н, Ме17), 1,14 (s, 3Н, Me16). Пример 41 Получение 3'-дезфенил-3'-(циклогексил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-бензоил-3-триэтилсилилокси-4-(циклогексил)азетидин-2-она (331 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 186 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил3'- (циклогексил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 186 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 147 мг вещества, которое очищали быстрой хроматографией, получая 107 мг (73%) 3'дезфенил-3'-(циклогексил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 163–164оС; [a]Na25 –32,0o (c 0,0049 CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,16 (d, J = 7,2 Гц, 2Н, орто-бензоат), 7,65 (d, J = 7,4 Гц, 2Н, ортобензамид), 7,64–7,33 (m, 6Н, ароматический), 6,29 (s, J = 9,9 Гц, 1Н, NH), 6,26 (s, 1Н, Н10), 6,20 (dd, J = 9,35, 9,35 Гц, 1Н, Н13), 5,68 (d, J = 7,7 Гц, 1Н, Н2 b), 4,97 (d, J = 7,7 Гц, 1Н, Н5), 4,60 (dd, J = 4,9, 1,4 Гц, 1Н, Н2'), 4,40 (m, 1H, H7), 4,27 (m, 1H, H3'), 4,26 (d, J = 8,2 Гц, 1Н, Н20 a), 4,21 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,78 (d, J = 7,7 Гц, 1Н, Н3), 3,66 (d, J = 4,9 Гц, 1Н, 2'ОН), 2,58 (m, 1H, Н6 a ), 2,50 (s, 3H, 4Ac), 2,45 (d, J = 3,9 Гц, 1H, 70H), 2,31 (m, 2Н, Н14), 2,22 (s., 3Н, 10Ас), 1,86 (m, 1Н, Н6 b), 1,83 (br s, 3Н, Ме18), 1,75 (s., 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,27–1,20 (m, 11Н, циклогексил), 1,20 (s., 3Н, Ме17), 1,11 (s, 3Н, Me16). Пример 42 Получение N-дебензоил-N-(2-тиеноил)таксола. К раствору 7-триэтилсилилбаккатина III (200 мг, 0,286 ммоля) в 2 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(2-тиеноил)-3- триэтилсилилокси-4-фенилазетидин-2-она (554 мг, 1,43 ммоля) в 2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, ко 26 40585 1 H ЯМР (CDCl3, 300 МГц) d 8,14 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,81 (d, J = 8,2 Гц, 2Н, 4-фенил бензамид орто), 7,64–7,33 (m, 15Н, ароматический), 7,02 (dd, J = 8,8 Гц, 1Н, NH), 6,27 (s, 1H, H10), 6,24 (dd, J = 8,3, 8,3 Гц, 1Н, Н13), 5,81 (dd, J = 8,8, 2,7 Гц, 1Н, Н3'), 5,69 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,93 (dd, J = 8,3, 1,1 Гц, 1Н, Н5), 4,80 (dd, J = 5,0, 2,2 Гц, 1Н, Н2'), 4,41 (m, 1H, H7), 4,30 (d, J = 8,0 Гц, 1Н, Н20 a ), 4,19 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,80 (d, J = 7,1 Гц, 1Н, Н3), 3,57 (d, J = 5 Гц, 1Н, 2'ОН), 2,53 (m, 1H, Н6 a), 2,46 (d, J = 4,4 Гц, 1H, 70H), 2,39 (s, 3Н, Ас), 2,31 (m, 2Н, Н14), 2,23 (s., 3Н, 10Ас), 1,87 (m, 1Н, Н6 b), 1,80 (br s, 3Н, Ме18), 1,76 (s, 1Н, 10Н), 1,68 (s., 3Н, Ме19), 1,24 (s., 3Н, Ме17), 1,14 (s, 3Н, Me16). Пример 44 Получение N-дебензоил-N-(трет-бутоксикарбонил-3'-дезфенил-3'- (2-фурил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(трет-бутоксикарбонил)-3- триэтилсилилокси-4-(2-фурил)азетидин-2-она (314 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 182 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дебензоил-N-(трет-бутоксикарбонил)-3'-дезфенил-3'(2-фурил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 182 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, а затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 143 мг вещества, которое очищали быстрой хроматографией, получая 133 мг (93%) N-дебензоил-N-(трет-бутоксикарбонил)-3'- дезфенил-3'-(2-фурил)таксола. Т.пл. 155–156оС; [a]Na25 –73,0o (c 0,0046, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,11 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,63–7,46 (m, 3Н, ароматический), 7,41 (в, J = 1,1 Гц, 1Н, фурил), 6,38–6,31 (m, 2Н, фурил), 6,30 (s, 1H, H10), 6,23 (dd, J = 9,4, 9,4 Гц, 1Н, Н13), 5,67 (d, J = 7,1 Гц, 1Н, Н2 b), 5,3 (d, J = 9,3 Гц, 1Н, NH), 5,22 (d, J = 9,9 Гц, 1Н, Н3'), 4,95 (d, J = 9,3 Гц, 1Н, Н5), 4,71 (br s, 1Н, Н2'), 4,42 (m, 1H, H7), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a ), 4,16 (d, J = 8,2 Гц, 1Н, Н20 b), 3,82 (d, J = 7,1 Гц, 1Н, Н3), 3,29 (d, J = 5,5 Гц, 1Н, 2'ОН), 2,56 (m, 1H, Н6 a), 2,47 (d, J = 3,6 Гц, 1H, H3), 2,39 (s, 3Н, 4Ас), 2,34 (m, 2Н, Н14), 2,24 (s., 3Н, 10Ас), 1,90 (m, 1Н, Н6 b), 1,88 (br s, 3Н, Ме18), 1,70 (s, 1Н, 10Н), 1,67 (s., 3Н, Ме19), 1,34 (s, 9Н, t-бутил), 1,25 (s., 3Н, Ме17), 1,14 (s, 3Н, Me16). Пример 45 Получение 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N- (циклогексилкарбонил)таксола К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,094 мл 1,68 М раствора nBuLi в гексане. После выдержки в течение 1 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(циклогексилкарбонил)-3- триэтилсилилокси-4-(2-тиенил)азетидин-2-она (275 мг, 0,672 ммоля) в 1 мл ТГФ. Раствор постепенно нагревали до 0оС в течение 6 часов, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 149 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил-3'- (тиенил)-N-дебензоил-N-(циклогексилоксикарбонил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 149 мг смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 110 мг вещества, которое очищали быстрой хроматографией, получая 95 мг (81%) 3'-дезфенил-3'(2-тиенил)-N-дебензоил-N-(циклогексилоксикарбонил)таксола, который перекристаллизовывали из смеси эфира-гексан. Т.пл. 160–163оС; [a]Na25 –44,9o (c 0,265, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,14 (d, J = 7,2 Гц, 2Н, орто-бензоат), 7,62 (m,1Н, пара-бензоат), 7,51 (t, J = 7,2 Гц, 2Н, мета-бензоат), 7,29 (dd, J = 4,8, 1,2 Гц, 1Н, тиенил), 7,10 (d, J = 3,3 Гц, 1Н, тиенил), 7,01 (dd, J = 4,8, 3,3 Гц, 1Н, тиенил), 6,29 (s, 1H, H10), 6,28 (t, J = 9,0 Гц, 1Н, Н13), 5,67 (d, J = 7,2 Гц, 1Н, Н2 b), 5,56 (d, J = 9,3 Гц, 1Н, NH), 5,41 (dd, J = 9,3, 0,9 Гц, 1Н, Н3'), 4,94 (dd, J = 9,3, 1,8 Гц, 1Н, Н5), 4,66 (d, J = 0,9 Гц, 1Н, Н2'), 4,49 (m, 1Н, циклогексил), 4,41 (dd, J = 11,1, 6,6 Гц, 1H, H7), 4,30 (d, J = 8,1 Гц, 1Н, Н20 a), 4,17 (d, J = 8,1 Гц, 1Н, Н20 b ), 3,81 (d, J = 7,2 Гц, 1Н, Н3), 3,42 (m, 1Н, 2'ОН), 2,54 (m, 2H, Н6 a, 70Н), 2,40 (s, 3Н, Ас), 2,31 (m, 2Н, Н14), 2,24 (s., 3Н, 10Ас), 1,86 (s, 3Н, Ме18), 1,68 (br s., 3Н, Ме19), 1,26 (s., 3Н, Ме17), 1,15 (m, 3Н, Me16). Пример 46 27 40585 Получение N-дебензоил-N-(дифенилкарбамил)таксола К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,087 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(дифенилкарбамил)-3- триэтилсилилокси-4фенилазетидин-2-она (676 мг, 1,43 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 168 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-N-дебензоил-N- (дифенилкарбамил)таксол и небольшое количество (2'S,3'R)изомера. К раствору 168 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 135 мг вещества, которое очищали быстрой хроматографией, получая 121 мг (89%) N-дебензоил-N- (дифенилкарбамиол)таксола, который перекристаллизовывали из водного метанола. Т.пл. 159–162оС; [a]Na25 –89,0o (c 0,0103, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,05 (d, J = 7,7 Гц, 2Н, орто-бензоат), 7,57–7,14 (m, 18H, ароматический), 6,29 (s, 1H, H10), 6,23 (m, 1Н, Н13), 5,67 (d, J = 6,6 Гц, 1Н, Н2 b), 5,51 (dd, J = 9,3, 2,2 Гц, 1Н, Н3'), 5,36 (d, J = 9,3 Гц, 1Н, NH), 4,94 (d, J = 8,2 Гц, 1Н, Н5), 4,63 (dd, J = 5,5, 2,2 Гц, 1Н, Н2'), 4,42 (m, 1H, H7), 4,25 (d, J = 8,8 Гц, 1Н, Н20 a ), 4,18 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,77 (d, J = 7,1 Гц, 1Н, Н3), 3,38 (d, 1Н, 2'ОН), 2,53 (m, 2H, Н6 a), 2,47 (d, J = 3,9 Гц, 1Н, 70Н), 2,39 (s, 3Н, 4Ас), 2,24 (s., 3Н, 10Ас), 1,87 (m, 1Н, Н6 b), 1,84 (br s, 3Н, Ме18), 1,70 (s, 1Н, 10Н), 1,67 (s, 3Н, Ме19), 1,28 (s, 3Н, Ме17), 1,15 (m, 3Н, Me16). Пример 47 нил)азетидин-2-она (281 мг, 0,715 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 156 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дезфенил-3'-(2-тиенил)-Nдебензоил-N-(2-тиеноил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 156 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 124 мг вещества, которое очищали быстрой хроматографией, получая 103 мг (83%) 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N(2-тиеноил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 206–208оС; [a]Na25 –38,6o (c 0,0095, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,14 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,64–7,01 (m, 9H, ароматический), 6,81 (d, J = 9,3 Гц, 1Н, NH), 6,28 (s, 1H, H10), 6,23 (dd, J = 8,2, 8,2 Гц, 1Н, Н13), 6,00 (dd, J = 9,3, 1,1 Гц, 1Н, Н3'), 5,67 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,94 (dd, J = 10,4, 2,2 Гц, 1Н, Н5), 4,76 (br s, 1Н, Н2'), 4,41 (m, 1H, H7), 4,29 (d, J = 8,8 Гц, 1Н, Н20 a ), 4,20 (d, J = 8,3 Гц, 1Н, Н20b ), 3,80 (d, J = 6,6 Гц, 1Н, Н3), 3,69 (br s, 1Н, 2'ОН), 2,54 (m, 1H, Н6 a), 2,39 (s, 3Н, 4Ас), 2,35 (br s, 1H, 7OH), 2,30 (m, 2H, H14), 2,23 (s., 3Н, 10Ас), 1,91 (br s, 1H, 10H), 1,87 (m, 1Н, Н6 b ), 1,82 (br s., 3Н, Ме18), 1,68 (s, 3Н, Ме19), 1,22 (s, 3Н, Ме17), 1,11 (m, 3Н, Me16). Пример 48 Получение 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N-(2- фуроил)таксола К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,087 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 1 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(2-фуроил)-3триэтилсилилокси-4-(2-тиенил)азетидин-2-она (270 мг, 0,715 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, Получение 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N-(2- тиеноил)таксола К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,087 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 1 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(2-тиеноил)-3триэтилсилилокси-4-(2-тие 28 40585 нитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 120 мг вещества, которое очищали быстрой хроматографией, получая 105 мг (87%) 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N(аллилоксикарбонил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 137–139оС; [a]Na25 –58,81o (c 0,006, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,13 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,60–7,00 (m, 6H, ароматический), 6,28 (s, 1H, H10), 6,25 (m, 1Н, Н13), 5,80 (m, 1H, аллиловый), 5,67 (d, J = 7,1 Гц, 1Н, Н2 b), 5,56 (br s, 2H, NH & H3'), 5,21 (m, 2H, аллиловый), 4,94 (dd, J = 9,3, 1,7 Гц, 1Н, Н5), 4,67 (br s, 1Н, Н2'), 4,47–4,38 (m, 3H, H7 & аллиловый), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a), 4,18 (d, J = 8,2 Гц, 1Н, Н20 b), 3,80 (d, J = 7,1 Гц,1Н, Н3), 3,46 (br s, 1H, 2'ОН), 2,56 (m, 1H, Н6a ), 2,38 (s, 3H, 4Ac), 2,31 (m, 2H, H14), 2,24 (s., 3Н, 10Ас), 1,89 (m, 1Н, Н6 b ), 1,84 (br s., 3Н, Ме18), 1,74 (br s, 1H, 7OH), 1,68 (s, 3Н, Ме19), 1,60 (br s, 1H, 1OH), 1,26 (s, 3Н, Ме17), 1,14 (m, 3Н, Me16). Пример 50 получая 156 мг смеси, содержащей (2'R,3'S)-2',7(бис)триэтилсилил-3'-дезфенил-3'-(2-тиенил)-Nдебензоил-N-(2-фуроил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 156 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 122 мг вещества, которое очищали быстрой хроматографией, получая 110 мг (90%) 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N(2-фуроил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 217–218оС; [a]Na25 –39,6o (c 0,0062, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,14 (d, J = 6,6 Гц, 2Н, орто-бензоат), 7,65–7,18 (m, 7H, ароматический), 7,12 (d, J = 9,3 Гц, 1Н, NH), 7,04 (dd, 1Н, тиенил), 6,46 (dd, 1Н, фуроил), 6,28 (s, 1H, H10), 6,22 (dd, J = 9,3, 9,3 Гц, 1Н, Н13), 5,99 (dd, J = 8,8, 2,2 Гц, 1Н, Н3'), 5,68 (d, J = 7,1 Гц, 1Н, Н2 b ), 4,94 (dd, J = 9,9, 2,2 Гц, 1Н, Н5), 4,76 (d, 1Н, Н2'), 4,42 (m, 1H, H7), 4,33 (d, J = 8,2 Гц, 1Н, Н20 a ), 4,20 (d, J = 8,8 Гц, 1Н, Н20 b ), 3,81 (d, J = 7,1H, Н3), 3,60 (d, J = 4,9 Гц, 1Н, 2'ОН), 2,53 (m, 1H, Н6 a), 2,42 (br s, 1H, 7OH), 2,40 (s, 3H, 4Ac), 2,31 (m, 2H, H14), 2,23 (s., 3Н, 10Ас), 1,87 (m, 1Н, Н6 b), 1,83 (br s., 3Н, Ме18), 1,68 (s, 3Н, Ме19), 1,24 (s, 3Н, Ме17), 1,14 (m, 3Н, Me16). Пример 49 Получение N-дебензоил-N-(диметилкарбамил)таксола. К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1(диметилкарбамил)-3- триэтилсилилокси-4-фенилазетидин-2-она (249 мг, 0,715 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 150 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-Nдебензоил-N- (диметилкарбамил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 150 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 117 мг вещества, которое очищали быстрой хроматографией, получая 105 мг (90%) N-дебензоил-N-(диметилкарбамил)таксола, Получение 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N- (аллилоксикарбонил)таксола К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,087 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 1 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(аллилоксикарбонил)-3- триэтилсилилокси4-(2-тиенил)азетидин-2-она (263 мг, 0,715 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО 3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 153 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N-(аллилоксикарбонил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 153 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацето 29 40585 ческий), 6,28 (s, 1H, H10), 6,23 (m, 1Н, Н13), 5,70 (m, 2H, H3' & H2 b ), 5,19 (d, J = 8,8 Гц, 1Н, NH), 4,95 (d, J = 9,8, 7,7 Гц, 1Н, Н5), 4,68 (br s, 1Н, Н2'), 4,41 (m, 1H, H7), 4,30 (d, J = 8,2 Гц, 1Н, Н20 a ), 4,19 (d, J = 8,2 Гц, 1Н, Н20 b ), 3,81 (d, J = 7,1 Гц, 1Н, Н3), 3,20 (m, 4H, диэтилкарбамил), 2,53 (m, 1H, Н6 a), 2,40 (s, 3H, 4Ac), 2,31 (m, 2H, H14), 2,23 (s., 3Н, 10Ас), 1,88 (m, 1Н, Н6b ), 1,82 (br s., 3Н, Ме18), 1,67 (s, 3Н, Ме19), 1,25 (s, 3Н, Ме17), 1,14 (m, 3Н, Me16), 1,07 (t, 6Н, диэтилкарбамил). Пример 52 который перекристаллизовывали из водного метанола. Т.пл. 179–181оС; [a]Na25 –54,36o (c 0,00195, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,11 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,64–7,20 (m, 8H, ароматический), 6,27 (s, 1H, H10), 6,19 (m, 1Н, Н13), 5,67 (d, J = 7,1 Гц, 1Н, Н2 b), 5,40 (dd, J = 7,7, 3,3 Гц, 1H, H3'), 5,20 (d, J = 7,7 Гц, 1Н, NH), 4,94 (dd, J = 9,8, 2,2 Гц, 1Н, Н5), 4,66 (d, 1Н, Н2'), 4,40 (m, 1H, H7), 4,29 (d, J = 8,2 Гц, 1Н, Н20 a ), 4,17 (d, J = 8,2 Гц, 1Н, Н20b ), 3,77 (d, J = 7,1 Гц, 1Н, Н3), 2,89 (s, 6H, диметилкарбамид), 2,53 (m, 1H, Н6 a), 2,46 (br s, 1H, 70H), 2,36 (s, 3H, 4Ac), 2,29 (m, 2H, H14), 2,23 (s., 3Н, 10Ас), 1,87 (m, 1Н, Н6 b ), 1,79 (br s., 3Н, Ме18), 1,67 (s, 3Н, Ме19), 1,25 (s, 3Н, Ме17), 1,14 (m, 3Н, Me16). Пример 51 Получение N-дебензоил-N-(тиоэтоксикарбонил)таксола. К раствору 7-триэтилсилилбаккатина III (120 мг, 0,171 ммоля) в 1,2 мл ТГФ при –45оС добавляли по каплям 0,104 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 0,5 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(тиоэтоксикарбонил)-3-триэтилсилилокси-4-фенилазетидин-2-она (323 мг, 0,885 ммоля) в 1,2 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 179 мг смеси, содержащей (2'R,3'S)-2',7-(бис)триэтилсилил-N-дебензоилN- (тиоэтоксикарбонил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 179 мг (0,171 ммоля) смеси, полученной из предыдущей реакции, в 11 мл ацетонитрила и 0,55 мл пиридина при 0оС добавляли 1,7 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 148 мг вещества, которое очищали быстрой хроматографией, получая 127 мг (86%) N-дебензоил-N(тиоэтоксикарбонил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 168–170оС; [a]Na25 –35,7o (c 0,0051, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,12 (d, J = 8,7 Гц, 2Н, орто-бензоат), 7,64–7,33 (m, 8H, ароматический), 6,28 (s, 1H, H10), 6,24 (m, 2Н, Н13, NH), 5,66 (d, J = 7,1 Гц, 1Н, Н2 b ), 5,58 (br, 1H, H3'), 4,93 (d, J = 7,1 Гц, 1Н, Н5), 4,66 (d, J = 2,7 Гц, 1Н, Н2'), 4,40 (m, 1H, H7), 4,29 (d, J = 8,8 Гц, 1Н, Н20 a), 4,22 (d, J = 8,8 Гц, 1Н, Н20 b ), 3,79 (d, J = 7,1 Гц, 1Н, Н3), 3,42 (d, J = 4,5 Гц, 1Н, 2'OH), 2,74 (m, 2H, S-CH2-), 2,55 (m, 1H, Н6 a), 2,49 (m, 1H, 7OH), 2,36 (s, 3H, 4Ac), 2,26 (m, 2H, H14), 2,24 (s, 3Н, 10Ас), 1,89 (m, 1Н, Н6 b), 1,75 (s, 1H, 10H), 1,67 (s, 3Н, Получение 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N- (диэтилкарбамил)таксола. К раствору 7-триэтилсилилбаккатина III (100 мг, 0,143 ммоля) в 1 мл ТГФ при –45оС добавляли по каплям 0,174 мл 1,63 М раствора nBuLi в гексане. После выдержки в течение 1 часа при –45оС к полученной смеси по каплям добавляли раствор цис-1-(диэтилкарбамил)-3-триэтилсилилокси-4(2-тиенил)азетидин-2-она (273 мг, 0,715 ммоля) в 1 мл ТГФ. Раствор нагревали до 0оС и выдерживали при этой температуре в течение 1 часа, после чего к нему добавляли 1 мл 10% раствора АсОН в ТГФ. Смесь распределяли между насыщенной водной NaНСО3 и этилацетат-гексаном 60/40. После упаривания органического слоя получали остаток, который очищали фильтрацией через силикагель, получая 155 мг смеси, содержащей (2'R,3'S)2',7-(бис)триэтилсилил-3'-дезфенил-3'-(2-тиенил)N-дебензоил-N-(диэтилкарбамил)таксол и небольшое количество (2'S,3'R)-изомера. К раствору 155 мг (0,143 ммоля) смеси, полученной из предыдущей реакции, в 6 мл ацетонитрила и 0,3 мл пиридина при 0оС добавляли 0,9 мл 48% водного раствора HF. Смесь перемешивали при 0оС в течение 3 часов, затем при 25оС в течение 13 часов и распределяли между насыщенным водным раствором бикарбонатом натрия и этилацетатом. После упаривания этилацетатного раствора получали 122 мг вещества, которое очищали быстрой хроматографией, получая 114 мг (93%) 3'-дезфенил-3'-(2-тиенил)-N-дебензоил-N(диэтилкарбамил)таксола, который перекристаллизовывали из водного метанола. Т.пл. 149–151оС; [a]Na25 –66,6o (c 0,00975, CHCl3). 1 H ЯМР (CDCl3, 300 МГц) d 8,12 (d, J = 7,1 Гц, 2Н, орто-бензоат), 7,64–6,99 (m, 6H, аромати 30
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for obtaining taxane
Автори англійськоюHOLTON Robert A.
Назва патенту російськоюСпособ получения таксана
Автори російськоюХолтон Роберт А.
МПК / Мітки
МПК: C07D 205/00, C07D 407/12, C07D 409/12, C07C 231/00, C07C 229/34, C07D 317/60, C07C 233/87, C07D 305/00, C07C 227/00, C07F 7/00, C07C 229/26, C07C 229/22, C07F 7/18
Мітки: спосіб, отримання, таксану
Код посилання
<a href="https://ua.patents.su/85-40585-sposib-otrimannya-taksanu.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання таксану</a>
Спосіб отримання розчину целаніду

Номер патенту: 14464
Опубліковано: 25.04.1997
Автори: Стрельцова Тетяна Романівна, Конєв Федір Андрійович, Литовкіна Олена Георгіївна
МПК: A61K 9/08
Мітки: спосіб, целаніду, отримання, розчину
Формула / Реферат:
Способ получения раствора целанида путем обработки кристаллического целанида растворителем, отличающийся тем, что, с целью увеличения сроков хранения, целанид растворяют в пропиленгликоле-1,2, содержащем в 100 мл 0,25-0,35 г хлорида натрия,
Похідні піримідину, що мають активність антагоністів ангіотензину іі, спосіб їх отримання та фармацевтична композиція

Номер патенту: 27748
Опубліковано: 16.10.2000
Автори: Нікайдо Маделен, Бажлі Жак Фрамрос, Еллінгбой Джон Ватсон
МПК: C07D 471/04, C07D 487/04
Мітки: фармацевтична, композиція, похідні, ангіотензину, іі, мають, антагоністів, спосіб, отримання, активність, піримідину
Текст:
...Результаты анализа из расчеты на формулу 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 минут после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение, в соответствии с примером 1, при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от А II в среднем на 41 % в...
Спосіб отримання солей омепразолу

Номер патенту: 3639
Опубліковано: 27.12.1994
Автор: Арне Елоф Брендстрьом
МПК: C07D 401/12, A61K 31/4427, A61P 1/04, A61K 31/44
Мітки: солей, спосіб, отримання, омепразолу
Формула / Реферат:
Способ получения солей омепразола общей формулигде n =1,2 или 4; КАТn+ - Li+, Nа+, К+, Мg2+, Са2+, Ті4+, N+(К)4 илигде R - низший алкил, отличающийся тем, что проводят взаимодействие омепразола формулыс основанием, способным выделять катион КАТn+ , с выделением целевого продукта.
Похідні дипептидів та спосіб їх отримання
Номер патенту: 27254
Опубліковано: 15.08.2000
Автори: Масаакі Матсуо, Дайдзіро Хагівара, Хіросі Міяке
МПК: A61P 27/02, C07K 5/097, C07K 5/078, A61K 38/00, C07K 1/113, A61P 29/00, A61P 17/00, C07K 5/06, A61P 11/00, C07K 5/02
Мітки: спосіб, похідні, дипептидів, отримання
Текст:
...ІРЕ - изопропиловый эфир; Me - метил; 1 Nal - 3-(1-нафтил)аланин; 2 Nal - 3-(2-нафтил)аланин; NMM - N-метилморфолин, 4N-HCI/DOX - 4Ы-хлористый водород в 1,4-диоксане; Ph - фенил; Pri - изопропил; Pro (40H) - 4-гидроксипролин; ^ • ' Pro (40 Me) - 4-метоксипролин; TEA -триэтиламин; IFA - трифторуксусная кислота; THF - тетрагидрофуран; WSC - 1-этил-3-(3-диметиламинопропил)-карбодиимид. В таблице даны исходные соединения и целевые...
Спосіб отримання пантокрину для ін’єкцій

Номер патенту: 14451
Опубліковано: 25.04.1997
Автори: Шитов Геннадій Гаврилович, Вакушин Борис Іванович, Тимофєєв Віктор Васильович, Донштрубова Ганна Георгіївна, Борщевська Марина Іллівна, Попов Юрій Павлович
МПК: A61K 35/36
Мітки: пантокрину, ін'єкцій, отримання, спосіб
Формула / Реферат:
Способ получения пантокрина для инъекций путем инкубации концентрата пантокрина при температуре 1-5°С в течение 7-10 дней, фильтрации, разведения дистиллированной водой, подкисления раствора до рН 3,5-5,0 введением стабилизатора, обработки активированным углем в течение 30 мин с последующей стерилизующей фильтрацией и ампулированием, отличающийся тем, что, с целью упрощения способа, после разведения концентрата вводят ацидин в концентрации...
Попередній патент: Спосіб одержання танталового порошку, танталовий порошок та електрод на його основі
Наступний патент: Клиновий патрон
Випадковий патент: Горілка "тещин продукт"