Індазоли, які мають анальгетичну активність
Номер патенту: 80605
Опубліковано: 10.10.2007
Автори: Гугліелмотті Анджело, Алісі Марія Алессандра, Каццолла Нікола, Поленцані Лоренцо, Фурлотті Гвідо







Формула / Реферат
1. Сполука формули
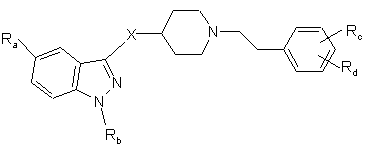 (I),
(I),
у якій
X є C(O)NHCH2, NHC(O) або NHC(O)CH2,
Ra є H, NH2C(O), CH3C(O)NH, CH3SO2, CH3SO2NH, лінійним або розгалуженим С1-С3алкілом, лінійним або розгалуженим С1-С3алкокси або галогеном,
Rb є Н, лінійним або розгалуженим C1-С6алкілом; арил-(С1-С3)алкілом, необов'язково заміщеним 1 або 2 атомами галогену, С1-С3алкільною групою або С1-С3алкоксигрупою,
і в якій
а) якщо X є C(O)NHCH2,
Rc є гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" є Н, або лінійним або розгалуженим С1-С6алкілом,
Rd є Н, гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення,
за умови, що коли Ra та Rd обидва є Н, і Rb є ізопропілом, то Rc не є гідрокси;
б) коли X є NHC(O) або NHC(O)CH2,
Rc та Rd, які можуть бути однаковими або різними, є Н, гідрокси, С1-С3алкокси, галогеном, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення,
та її кислі адитивні солі з фармацевтично прийнятними органічними та неорганічними кислотами.
2. Сполука за п. 1, яка відрізняється тим, що Ra є Н або С1-С3алкілом.
3. Сполука за пп. 1 або 2, яка відрізняється тим, що Rb є Н або С1-С3алкілом.
4. Сполука за будь-яким з пп. з 1 по 3, яка відрізняється тим, що Rc є Н, NO2, NH2, OH або С1-С3алкокси.
5. Сполука за будь-яким з пп. з 1 по 4, яка відрізняється тим, що Rd є Н.
6. Кисла адиційна сіль сполуки за будь-яким з пп. з 1 по 5, яка відрізняється тим, що кислоту вибирають із групи, яка включає щавлеву, малеїнову, метансульфонову, паратолуолсульфонову, бурштинову, лимонну, винну, молочну, хлористоводневу, фосфорну та сірчану кислоту.
7. Фармацевтична композиція, яка містить ефективну кількість сполуки формули (І):
(I),
у якій
X є C(O)NHCH2, NHC(O) або NHC(O)CH2,
Ra є H, NH2C(O), CH3C(O)NH, CH3SO2, CH3SO2NH, лінійним або розгалуженим С1-С3алкілом, лінійним або розгалуженим С1-С3алкокси або галогеном,
Rb є Н, лінійним або розгалуженим С1-С6алкілом; арил-(С1-С3)алкілом, необов'язково заміщеним 1 або 2 атомами галогену, С1-С3алкільною групою або С1-С3алкоксигрупою,
і в якій
а) якщо X є C(O)NHCH2,
Rc є гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" є Н, або лінійним або розгалуженим C1-С6алкілом,
Rd є Н, гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення,
за умови, що коли Ra та Rd обидва є Н, і Rb є ізопропілом, то Rc не є гідрокси,
б) якщо X є NHC(O) або NHC(O)CH2,
Rc та Rd, які можуть бути однаковими або різними, є Н, гідрокси, С1-С3алкокси, галогеном, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення,
або її фармацевтично прийнятну адитивну сіль з органічною або неорганічною кислотою та принаймні один фармацевтично прийнятний інертний інгредієнт.
8. Фармацевтична композиція за п. 7, яка відрізняється тим, що містить сполуку за будь-яким з пп. з 2 по 6.
Текст
1. Сполука формули C2 2 (19) 1 3 80605 4 або 2 атомами галогену, С1-С3алкільною групою або С1-С3алкоксигрупою, і в якій а) якщо X є C(O)NHCH 2, Rc є гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" є Н, або лінійним або розгалуженим C1-С6алкілом, Rd є Н, гідрокси, аміно, ді-(С1-С3)алкіламіно, три(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення, за умови, що коли Ra та R d обидва є Н, і Rb є ізопропілом, то Rc не є гідрокси, б) якщо X є NHC(O) або NHC(O)CH 2, Rc та Rd, які можуть бути однаковими або різними, є Н, гідрокси, С1-С3алкокси, галогеном, аміно, ді(С1-С3)алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення, або її фармацевтично прийнятну адитивну сіль з органічною або неорганічною кислотою та принаймні один фармацевтично прийнятний інертний інгредієнт. 8. Фармацевтична композиція за п. 7, яка відрізняє ться тим, що містить сполуку за будьяким з пп. з 2 по 6. Даний винахід стосується індазолу, який має анальгетичну активність, способу його одержання та фармацевтичної композиції, яка його містить. Хронічний біль є дуже поширеною проблемою. У середньому приблизно 20% дорослого населення страждає від нього, і його зазвичай пов'язують із клінічними станами, які характеризуються хронічними та/або дегенеративними пошкодженнями. Типовими прикладами патологій, які характеризуються хронічним болем, є ревматоїдний артрит, остеоартрит, фіброміальгія, невропатії і т. ін. [Ashburn MA, Staats PS. Management of chronic pain. Lancet 1999; 353: 1865-69]. Хронічний біль часто забирає багато сил і буває причиною втрати працездатності та низької якості життя. Таким чином, він також має негативні економічні та соціальні наслідки. Анальгетичні медикаменти, які нині застосовують для лікування від хронічного болю, належать здебільшого до двох класів: нестероїдних протизапальних ліків (NSAID), які поєднують анальгетичну активність та протизапальну активність, і опіоїдних анальгетиків. Ці класи складають основу триступінчастої "анальгетичної шкали", запропонованої Всесвітньою організацією охорони здоров'я для медикаментозного лікування від болю [Textbook of Pain. 4th edition. PD Wall and R Melzack Eds. Churchill Livingstone, 1999]. Загальновідомо, що хронічний біль важко піддається лікуванню з застосуванням наявних на даний час засобів. Отже, розробка нових анальгетиків завжди була однією з головних цілей фармацевтичної промисловості. Незважаючи на широкі дослідження, спрямовані на пошук прийнятної анальгетичної сполуки, все ще існує значна кількість пацієнтів, больовий стан яких і досі неможливо належним чином вилікувати [Scholz J, Woolf CJ. Can we conquer pain? Nat Neusci. 2002; 5: 1062-76]. Несподівано було виявлено, що такі властивості має нова група індазолів. Таким чином, у першому аспекті даний винахід стосується індазолу загальної формули: де X є C(O)NHCH2, NHC(O) або NHC(O)CH2; Ra є Η, NH2C(O), CH3C(O)NH, CH3SO2 , CH3SO2NH, лінійним або розгалуженим С 1-С3 алкілом, лінійним або розгалуженим С 1-С3 алкокси або галогеном; Rb є Н, лінійним або розгалуженим С 1-С6 алкілом; арил-(С1-С3)алкілом, необов'язково заміщеним 1 або 2 атомами галогену, С1С3алкільною групою або С1-С3алкокси-групою; і а) якщо X є C(O)NHCH 2 Rс є гідрокси, аміно, ді-(С1-С3)алкіл-аміно, три-( С1-С3)алкіл-амоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R та R" є Н, або лінійним або розгалуженим С1-С6алкілом, Rd є Н, гідрокси, аміно, ді-(С1-С3)алкіламіно, три-(С1-С3)алкіл-амоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення, однак, за умови, що коли Ra та Rd обидва є Н, і Rb є ізопропілом, то Rс не є гідрокси; б) коли X є NHC(O) або NHC(O)CH 2 Rс та Rd, які можуть бути однаковими або різними, є Н, гідрокси, С1-С3алкокси, галогеном, аміно, дЦСгС^алкіламіно, три-(С1-С3)алкіламоніометилом, нітро, трифторометилом, нітрилом, CH3C(O)NH, CH3SO2NH, CH3SO2, R'R"NSO2, де R' та R" мають вищезазначені значення, 5 80605 та їх кислими адиційними солями з фармацевтично прийнятними органічними та неорганічними кислотами. Типовими прикладами фармацевтично прийнятних кислот є: щавлева, малеїнова, метансульфонова, паратолуолсульфонова, бурштинова, лимонна, винна, молочна, хлористоводнева, фосфорна, сірчана. Оптимальними значеннями для R3 є Η та С1С3алкіл. Оптимальними значеннями для Rb є Η та С1С3алкіл. Оптимальними значеннями для Rс є Η, ΝΟ2, ΝΉ2, ОН та С 1-С3алкокси. Оптимальним значенням для Rd є Н. Анальгетичну активність сполук формули (І) визначали за допомогою двох експериментальних моделей на щура х: механічної гіпералгезії, викликаної CFA, та механічної гіпералгезії при діабетичній невропатії, викликаної стрептозоцином. Як відомо спеціалістам у даній галузі, вищезгадані експериментальні моделі можна з вважати такими, що дозволяють прогнозувати активність у людському організмі. Викликана CFA гіпералгезія є синдромом, який характеризується активацією кругообігів з метою контролювання запальної реакції, і є пов'язаним з виникненням станів, які перешкоджають сприйняттю болю. Ін'єкція CFA фактично є здатною периферично викликати вивільнення специфічних речовин (посередників запальної реакції та альгогенних агентів), які відповідають за локальне пошкодження, і центрально, на рівні спинного мозку, визначати біохімічні зміни, які підтримують посилення сприйняття болю. Загальновідомо, що ця модель являє собою дійсний засіб дослідження ліків для застосування у лікуванні запального болю в людини і, зокрема, для боротьби з такими станами, як гіпералгезія та алодинія. Типовими прикладами людських патологій, які характеризуються цим типом болю, пов'язаного з дегенеративними запальними процесами, є ревматоїдний артрит та остеоартрит. У свою чергу, діабетична невропатія, викликана стрептозоцином у щура, представляє інсулін-залежний синдром, який характеризується суп утнім зниженням швидкості провідності рухових та сенсорних нервів та виникненням багатьох відхилень у сприйнятті болю. Загальновідомо, що ця експериментальна модель є корисним засобом для дослідження ліків для застосування у лікуванні невропатичного болю в людини. Зокрема, ця модель є дійсним прикладом організму, що страждає від невропатичних болів, які характеризуються такими явищами, гіпералгезії та алодинії внаслідок первинних пошкоджень або дисфункцій нервової системи. Типовими прикладами людських патологій, які характеризуються дисфункціями цього типу та наявністю невропатичного болю, є діабет, рак, імунодефіцитні хвороби, травми, ішемії, розсіяний склероз, невралгії сідничного нерва, невралгія трійчастого нерва та постгерпетичні синдроми. 6 У другому аспекті даний винахід стосується способу одержання сполуки формули (І), у якій X C(O)NHCH2, та її кислих адиційних солей з фармацевтичне прийнятними органічними або неорганічними кислотами, який характеризується тим, що включає такі етапи: а) реакція аміну формули (II) де Rc та Rd мають однакові значення, як було зазначено вище, або, якщо Rc або Rd є аміно- або спиртовою групою, Rc та Rd можуть бути аміноабо спиртовою групою, захищеною захисною групою традиційного типу, з похідною індазолкарбонової кислоти формули (IIIа) де Ra та Rb мають вищезазначені значення, і Υ є атомом СІ або Вг, або OR або OC(O)R групою, де R є лінійним або розгалуженим алкілом, який має від 1 до 6 атомів вуглецю, або з похідною індазол-карбонової кислоти формули (IIIb) де Ra має вищезгадане значення, б) розщеплення будь-якої можливої захисної групи ви щезгаданої аміно- або спиртової групи і в) необов'язкове утворення кислої адиційної солі індазоламіду формули (І) з фармацевтичне прийнятною органічною або неорганічною кислотою. У третьому аспекті даний винахід стосується способу одержання сполуки формули (І), у якій X = NH(CO) або NH(CO)CH2 та її кислих адиційних солей з фармацевтично прийнятними органічними 7 80605 або неорганічними кислотами, який характеризується тим, що включає такі етапи: а') реакція аміну формули (IV) де Ra та Rb мають вищезазначені значення, конденсують з похідною карбонової кислоти формули (V) де Rc та Ra мають однакові значення, як було зазначено вище, або, якщо Rс або Rd є аміно- або спиртовою групою, Rc та Rd можуть бути аміно- або спиртовою групою, захищеною захисною групою традиційного типу, і Ζ є групою C(O)Y або CH2C(O)Y, у якій Υ є атомом СІ або Вг, або OR або OC(O)R групою, де R є лінійним або розгалуженим алкілом, який має від 1 до 6 атомів вуглецю, б') розщеплення будь-якої можливої захисної групи ви щезгаданої аміно- або спиртової групи і с') необов'язкове утворення кислої адиційної солі індазоламіду формули (І) з фармацевтично прийнятною органічною або неорганічною кислотою. Звичайному спеціалістові у даній галузі легко стане зрозуміло, що деякі сполуки формули (І) також можуть бути одержані з іншої сполуки формули (І) традиційними способами. Наприклад, якщо Rc та/або Rd є NO2 групою, остання може бути відновлена для забезпечення відповідної сполуки формули (І), у якій Rc та/або Rd є NH2. Амін формули (II) одержують згідно з традиційними способами. Наприклад, шляхом алкілування ізоніпекотаміду прийнятним галідом з наступним відновленням аміду до первинного аміну [WO 9807728] або шляхом захисту амінометилпіперидину бензальдегідом [Synthetic Communications 22(16), 2357-2360, 1992], алкілування прийнятним галідом та депротекції. Проміжна сполука формули (II), у якій Rc та Rd мають вищезазначені значення, є новою. Ця проміжна сполука, таким чином, є іншим аспектом даного винаходу. Сполуки формули (IIIа) та (IIIb) також одержують згідно з традиційними способами. Наприклад, сполуки формули (IIIа), у яких Υ є хлором, одержують із відповідної кислоти тіонілхлоридом [J. Med. Chem., 1976, Vol. 19 (6), 8 стор. 778-783], тоді як сполуки формули (IIIа), у яких Υ є OR або OC(O)R, одержують за допомогою відомих реакцій етерифікації або утворення змішаних ангідридів [R.C. Larok, Comprehensive Organic Transformations, VCH, стор. 965-966]. У свою чергу, сполуки формули (IIIb) можуть бути одержані згідно з [J.O.C. 1958, Vol. 23 стор. 621]. У свою чергу, сполуки формули (IV) можуть бути одержані згідно з традиційними способами, описаними у літературі, наприклад, у [п ублікаціях J. of Heterocyclic Chemistry 1979 (16) 783-784, або J.A.C.S., 1943 (65) 1804-1806]. Сполуки формули (V) також можуть бути одержані згідно з традиційними способами. Наприклад, сполуки формули (V), у якій Υ є хлором, можуть бути одержані шляхом омилення відповідних естерів з наступною обробкою тіонілхлоридом. В оптимальному варіанті етапи (а) та (а') здійснюють шляхом реакції - сполуки формули (II) зі сполукою формули (IIIа), у якій Υ є хлором, або - сполуки формули (II) зі сполукою формули (IIIb), або - сполуки формули (IV) зі сполукою формули (V), у якій Υ є хлором, у присутності прийнятного розріджувача і при температурі від 0 до 140°С протягом часу від 0,5 до 20 годин. В оптимальному варіанті температура реакції становить від 15 до 40°С. В оптимальному варіанті час реакції становить від 1 до 18 годин. В оптимальному варіанті розріджувач є апротонним, полярним або неполярним. У ще кращому варіанті він є апротонним неполярним. Прикладами прийнятних апротонних неполярних розріджувачів є ароматичні вуглеводні, наприклад, толуол. Прикладами прийнятних апротонних полярних розріджувачів є диметилформамід та диметилсульфоксид. У варіантах втілення, у яких сполуку формули (II) піддають реакції зі сполукою формули (IIIа), у якій Υ є хлором, або у яких сполуку формули (IV) піддають реакції зі сполукою формули (V), у якій Υ є хлором, вищезгадані етапи (а) та, відповідно, (а') в оптимальному варіанті здійснюють у присутності органічного або неорганічного акцептора кислот. Прикладами прийнятних органічних акцепторів кислот є піридин, триетиламін та інші подібні сполуки. Прикладами прийнятних неорганічних акцепторів кислот є лужні карбонати та бікарбонати. На етапах (б) та (б') розщеплення захисної групи аміно- або спиртової групи в оптимальному варіанті здійснюють способами, відомими у хімії захисних гр уп. У свою чергу, перед етапами (в) та (в') в оптимальному варіанті здійснюють етап виділення індазоламіду формули (І). У ще одному аспекті даний винахід стосується фармацевтичної композиції, яка містить ефективну кількість сполуки формули (І) або її адиційної солі з фармацевтичне прийнятною кислотою та принаймні один фармацевтично прийнятний інертний інгредієнт. 9 80605 Типовим прикладом патологічного стану, який може бути поліпшений шляхом лікування з застосуванням фармацевтичної композиції згідно з даним винаходом, є хронічний біль. Як правило, цей хронічний біль є зумовленим хронічними пошкодженнями або дегенеративними процесами, такими як ревматоїдний артрит, остеоартрит, фіброміальгія, онкологічний біль, невропатичний біль і т. ін. В оптимальному варіанті фармацевтичні композиції згідно з даним винаходом одержують у прийнятній дозованій формі. Прикладами прийнятних дозованих форм є таблетки, капсули, вкриті таблетки, гранули, розчини та сиропи для перорального введення; креми, мазі та лікарські пластирі для місцевого застосування; супозиторії для ректального застосування та стерильні розчини для ін'єкційного, аерозольного або очного введення. В оптимальному варіанті ці дозовані форми рецептують таким чином, щоб забезпечувати контрольоване вивільнення з часом сполуки формули (І) або її солі з фармацевтичне прийнятною кислотою. Дійсно, залежно від типу лікування, потрібний час вивільнення може бути дуже коротким, нормальним або тривалим. Дозовані форми також можуть містити інші традиційні інгредієнти, такі як: консерванти, стабілізатори, поверхнево-активні речовини, буфери, солі для регулювання осмотичного тиску, емульгатори, підсолоджувачі, барвники, ароматизатори і т. ін. Крім того, якщо це вимагається конкретними способами терапії, фармацевтична композиція згідно з даним винаходом може містити інші фармакологічне активні інгредієнти, супутнє застосування яких є терапевтично корисним. Кількість сполуки формули (І) або її фармацевтичне прийнятної кислої солі у фармацевтичній композиції згідно з даним винаходом може бути різною в широких межах, залежно від відомих чинників, таких як, наприклад тип хвороби, яку піддають лікуванню, тяжкість хвороби, маса тіла пацієнта, дозована форма, вибраний шлях введення, кількість введень на день та ефективність вибраної сполуки формули (І). Однак оптимальна кількість легко і звично може бути визначена спеціалістом у даній галузі. Як правило, кількість сполуки формули (І) або її солі з фармацевтичне прийнятною кислотою у фармацевтичній композиції згідно з даним винаходом має бути такою, щоб забезпечувати рівень введення від 0,001 до 100мг/кг/день. У ще кращому варіанті - від 0,1 до 10мг/кг/день. Дозовані форми фармацевтичної композиції згідно з даним винаходом одержують згідно зі способами, відомими хімікам-фармацевтам, і ці способи включають змішування, гранулювання, пресування, розчинення, стерилізацію і т. ін. Представлені нижче приклади пояснюють винахід, жодним чином його не обмежуючи. У представлених нижче прикладах замісники на ароматичному кільці (Re та Rd) вказано жирними цифрами. Приклад 1 10 (N(( 1 -(2-(4-нітрофеніл)етил)-4піперидил)метил-1Н-індазол-3-карбоксамід гідро хлорид (AF3R298) (І, Ra = R b = Rd = Η; Rc = 4-ΝΟ2; Χ = C(O)NHCH2) a) N-гeкcaгiдpo-4-пipидинiдмeтил-Nфeнiлмeтилiдeн-aмiн Бензальдегід (4,6г; 0,044моль) по краплях додавали до розчину 4-амінометилпіперидину (5,0г; 0,044моль) у толуолі (20 мл). Одержаний таким чином розчин перемішували протягом 3год. при кімнатній температурі. Потім розчинник видаляли шляхом випарювання під зниженим тиском і залишок двічі захоплювали толуолом для одержання потрібного продукту, який використовували як такий без подальшого очищення. б) 1-(2-(4-нітрофеніл)етил)-4-піперидилметанамін Продукт з Прикладу 1а) (8,8г; 0,044моль) розчиняли в абсолютному етанолі (50 мл) і додавали до суспензії, яка містила 2-(4нітрофеніл)етилбромід (10,0г; 0,044моль) та безводний карбонат калію (12,1г; 0,088моль) в абсолютному етанолі (100мл). Одержану таким чином суспензію кип'ятили з дефлегмацією протягом 16 годин. Реакційну суміш після цього залишали для охолодження до кімнатної температури і фільтрували. Фільтрат випарювали під зниженим тиском. Одержаний таким чином залишок після цього суспендували у 3N НСІ (50мл) і перемішували протягом 3год. при кімнатній температурі. Розчин після цього переносили до ділильної лійки і кислотну водну фаз у промивали етилацетатом (4х50мл), водну фаз у після цього робили лужною шляхом додавання 6Ν NaOH і екстрагували дихлорометаном. Органічну фазу висушували над NaSO4 і , розчинник видаляли шляхом випарювання під зниженим тиском для одержання потрібного продукту (9г). 1 H-ЯМР (δ, CDCI3 + D2O): 1,43-1,50 (m, 3Н); 1,76 (d J=12 Гц, 2Н); 2,03 (t, J=12 Гц, 2Н); 2,67-2,52 (m, 4Н); 2,82-3,06 (m, 4Н); 7,39 (d, J=9 Гц, 2Н); 8,12 (d, J=9 Гц, 2Н); 7,95 (квінтет, J=1 Гц, 1H). в) N((1 -(2-(4-нітрофеніл)етил )-4піперидил)метил)-1Н-індазол-3-карбоксамід гідрохлорид Розчин продукту з Прикладу 1б) (8,4г; 0,032моль) у толуолі (85мл) додавали, застосовуючи крапельну лійку, до суспензії, яка містила 7Н,14Н-індазол(2',3':4,5)піразин(1,2b)індазол-7,14,діон (4,6г; 0,016моль), одержаної, як описано у [J.O.C., 1958, Vol. 23, стор. 621], у толуолі (60 мл). Реакційну суміш перемішували при кімнатній температурі протягом 38 годин, а потім фільтрували. Тверду речовину відокремлювали і додавали до перемішуваного насиченого розчину NаНСО3 (200мл) протягом 2год. Реакційну суміш фільтрували і, таким чином, одержаний твердий продукт перетворювали на відповідний гідрохлорид шляхом розчинення в абсолютному етанолі, додавання хлористого водню в етанолі та рекристалізації з етанолу для одержання потрібного продукту (4,2г). т. пл.:251-252.5°С 11 80605 Елементний аналіз для C22H25N5O 3. HCl Виявлено % Розраховано % С 59,60 59,52 Η 5,96 5,90 Ν 15,77 15,78 1 H-ЯМР (δ, D MSO): 1,52-2,11 (m, 5H); 2,85-302 (m, 2H); 3,17-3,64 (m, 8H); 7,19-7,28 (m, 1H); 7,367,46 (m, 1H); 7,53-7,56 (m, 3H); 8,13-8,26 (m, 3H); 8,55 (t, J=6 Гц, 1H); 10,82 (s широк., 1H); 13,70 (s, 1H). Приклад 2 N((1 -(2-(4-амінофеніл)етил)-4піперидил)метил)-1Н-індазол-3-карбоксамід дигідрохлорид (AF3R02) (І, Ra = R b = Ra = Η, Re = 4-NH2, X = C(O)NHCH2) Розчин продукту з Прикладу 1в) у формі основи (3г; 0,007моль) у 95° етанолі (200мл) гідрогенізували на 10% Pd-C (0,3г) при 40 psi протягом 3 годин. Суміш після цього фільтрували і фільтрат концентрували під зниженим тиском. Одержаний таким чином продукт кристалізували з етилацетату і перетворювали на відповідний гідрохлорид шляхом розчинення у суміші етилацетату: етанолу = 9:1 та додавання хлористого водню в етанолі для одержання потрібного продукту (1,2г). т. пл.: 271-273°С (декомп.) Елементний аналіз для C22H27N5O. 2HCI ½ Н2О Виявлено % Розраховано % С 57,31 57,52 Η 6,68 6,58 Ν 15,05 15,24 12 стехіометричної кількості щавлевої кислоти та рекристалізації з етилацетату: етанолу = 9:1, двічі, для одержання потрібної солі (3,5г). т. пл.: 98°С (декомп.) Елементний аналіз для C25H31N5O 3. C2H204 1/2 Н2О Виявлено % Розраховано % С 59,27 59,11 Η 6,15 6,25 Ν 12,72 12,77 1 H-ЯМР (δ, D MSO + D2O): 1,55 (d, J=7 Гц, 6H); 1,44-1,66 (m, 2H); 1,83-2,02 (m, 3Н); 2,98 (t, J=12 Гц, 2H); 3,10-3,40 (m, 6H); 3,55 (d, J=12 Гц, 2H); 5,07 (гептет, J=7 Гц, 1H); 7,28 (t, J=8 Гц, 1Н); 7,46 (t, J=7 Гц, 1H); 7,59 (d, J=9 Гц; 2Н); 7,79 (d, J=8 Гц; 1Н); 8,11-8,26 (m, 3H); 8,42 (t, J=6 Гц, 1Н). Приклад 4 N((1-(2-(4-амінофеніл)етил)-4піперидил)метил)-1-(1-метилетил)-1Н-індазол-3карбоксамід дигідрохлорид (AF3R294) (І, Ra = Rd = Η, Rb = i-C3H7, Re = 4-NH2, X = C(O)NHCH2) Розчин продукту з Прикладу 3, у формі основи (2,7г; 0,006моль) у 95° етанолі (30мл) гідрогенізували на 10% Pd-C (0,27г) при 40 psi протягом 5 годин. Суміш після цього фільтрували і фільтрат концентрували під зниженим тиском. Одержаний таким чином продукт перетворювали на відповідний гідрохлорид шляхом розчинення в етилацетаті, додавання хлористого водню в етанолі та рекристалізації з суміші етилацетату: етанолу = 8:2, для одержання потрібного продукту (1,4г). т. пл.: 278°С (декомп.) Елементний аналіз для C25H33N5O. 2HCl Н2О 1 H-ЯМР (δ, D MSO + D2O): 1,45-1,66 (m, 2H); 1,80-2,00 (m, 3H); 2,86-3,14 (m, 4H); 3,19-3,35 (m, 4H); 3,46-3,80 (m, 2H + HDO); 7,22-7,35 (m, 3H); 7,35-7,49 (m, 3H); 7,64 (d, J=9 Гц; 1Н); 8,17 (d, J=9 Гц, 1Н). Приклад 3 N((1-(2-(4-нітрофеніл)етил)-4піперидил)метил)-1-(1-метилетил)-1Н-індазол-3карбоксамід оксалат (AF3R306) (І, Ra = Rd = Η, Rb = i-С3Н7, Re = 4-NO2, X = C(O)NHCH2) Хлорид 1-(1-метилетил)-1Н-індазол-3карбонової кислоти (2,45г; 0,011моль), одержаний, як описано [у ЕР-В1-0 975 623], порціями додавали до розчину продукту 1б) (3,0г; 0,011моль) та триетиламіну (4,6мл; 0,033моль) у толуолі (50мл). Суміш перемішували при кімнатній температурі протягом 18 год. Розчинник видаляли шляхом випарювання під зниженим тиском. Залишок захоплювали у IN NaOH та дихлорометан. Суміш переносили до ділильної лійки. Органічну фаз у відокремлювали і висушували над Na2SO4. Розчинник видаляли шляхом випарювання під зниженим тиском і одержаний таким чином залишок очищали шляхом флеш-хроматографії, елююючи з етилацетатом для одержання потрібного продукту (5,5г), який після цього перетворювали на відповідний оксалат шляхом розчинення в етилацетаті, додавання Виявлено % Розраховано % 1 С 59,05 58,82 Η 7,42 7,31 Ν 13,63 13,72 H-ЯМР (δ, DMSO): 1,55 (d, J=7 Гц, 6H); 1,452,13 (m, 5H); 2,80-3,64 (m, 10H); 5,08 (гептет, J=7 Гц, 1Н); 7,20-7,49 (m, 6H); 7,79 (d, J=9 Гц; 1Н); 8,18 (d, J=9 Гц, 1Н) 8,39 (t, J=6 Гц, 1H); 9,15-11,18 (m, 4H) Приклад 5 N-(1 -метил-1 Н-індазол-3-іл)-1 -(2фенілетил)піперидин-4-карбоксамід гідрохлорид (AF3R334) (І, Ra = Re = Ra = Η, Rb = СН3, X = NHC(O)) а) гідрохлорид 1-(2-фенілетил-4-піперидинкарбонової кислоти Суспензію 1-(2-фенілетил)-4карбетоксипіперидин (12,2г, 0,047моль), одержану, як описано у [J. Med. Chem. 1996 (39), 749-756], у IN NaOH (100мл) нагрівали з дефлегмацією протягом 4год. Після охолодження до кімнатної температури розчин робили кислотним за допомогою 6N НСІ, доводячи до рН 2, концентрували шляхом випарювання під зниженим тиском і одержану таким чином тверду речовину фільтрували і висушували в сушильній камері у вакуумі для одержання потрібного продукту (12,1г). 13 80605 1 H-ЯМР (δ, D MSO + D2O): 1,79-2,19 (m, 4H); 2,43-3,74 (m, 9H); 7,18-7,41 (m, 5H); б) 1-(2-фенілетил)-4-піперидинкарбонілхлорид гідрохлорид Суспензію продукту з Прикладу 5а) (2,0г; 0,007моль) та тіонілхлориду (0,81мл; 0,011моль) у толуолі (20мл) нагрівали з дефлегмацією протягом 3год. Розчинник після цього видаляли шляхом випарювання під зниженим тиском і залишок захоплювали у толуол (2 x20мл) для одержання потрібного продукту (2,2 г), який використовували як такий без подальшого очищення. в) N-(1 -метил-1Н-індазол-3-іл)-1-(2фенілетил)піперидин-4-карбоксамід гідрохлорид Продукт з Прикладу 5б) (1,68г; 0,006моль) додавали до розчину 1-метил-1Н-3-індазоламіну (0,86г; 0,006моль), одержаного, як описано у [журналі Journal of Heterocyclic Chemistry 1979 (16), 783-784], та триетиламіну (2,4мл; 0,018моль) у толуолі (20мл). Реакційну суміш перемішували при кімнатній температурі протягом 18год., а потім розчинник видаляли шляхом випарювання під зниженим тиском. Одержаний таким чином залишок захоплювали у IN NaOH та дихлорометан і переносили до ділильної лійки. Органічну фазу відокремлювали, висушували над Na2SO4 і розчинник видаляли шляхом випарювання під зниженим тиском. Одержаний таким чином продукт перетворювали на відповідний гідрохлорид шляхом розчинення в етанолі, додавання хлористого водню в етанолі та рекристалізації з етанолу для одержання потрібної солі (1,6г). т. пл.: 235-237Т Елементний аналіз для C22H26N4O. HCl 1/4 Н2О Виявлено % Розраховано % С 65,71 65,50 Η 6,80 6,87 Ν 13,73 13,89 1 H-ЯМР (δ, D MSO + D2O): 1,91-2,27 (m, 4H) 2,70-3,42 (m, 7H); 3,63-3,75 (m, 2H); 3,96 (s, 3Н); 7,10 (t, J=8 Гц, 1H); 7,22-7,46 (m; 6H); 7,56 (d, J=8 Гц, 1H) 7,74 (d, J-8 Гц, 1Н); 10,51 (s, 1H) Приклад 6 N-(1-метил-1Н-індазол-3-іл)-1-(2-(4метоксифеніл)етил)піперидин-4карбоксамід гідрохлорид (AF3R328) (І, Ra = Ra = Η, Rb = СН3, Re = 4-OCH 3, Χ = NHC(O)) а) гідрохлорид 1-(2-(4-метоксифеніл)етил)-4піперидин-карбонової кислоти. Названий продукт одержували (15,8г) шляхом обробки способом, подібним до описаного у Прикладі 5а), але беручи за вихідну сполуку 1-(2(4-метоксифеніл)етил)-4-карбетоксипіперидин (16,5г; 0,057моль), одержаний, як описано [у US 6 017 931], замість 1-(2-фенілетил)-4карбетоксипіперидину. 1 H ЯМР (δ, DMSO): 1,80-2,17 (m, 4Н); 2,41-3,74 (m, 7Н); 3,73- (s, 3Н); 6,89 (d, J=9 Гц, 2H); 7,19 (d, J=9 Гц, 2H) 11,00 (s широк., 1H); 12,53 (s широк., 1Н) б) 1-(2-(4-метоксифеніл)етил)-4піперидинкарбонілхлорид гідро хлорид. 14 Названий продукт одержували (14,2г), беручи за вихідну сполуку продукт з Прикладу 6а) (13,8г; 0,048моль), і шляхом обробки способом, подібним до описаного у Прикладі 5б). Одержаний таким чином продукт використовували як такий без подальшого очищення. в) N-(1-метил-1Н-індазол-3-іл)-1-(4метоксифеніл)етил)піперидин-4-карбоксамід гідрохлорид Названий продукт одержували (9,2г), беручи за вихідну сполуку продукт з Прикладу 6б) (14,2г; 0,045моль) та 1-метил-1Н-3-індазоламін (6,6г; 0,045моль) і шляхом обробки способом, подібним до описаного у Прикладі 5в). Суміш етилацетату: етанолу (9:1) використовували як розчинник для кристалізації. т. пл.: 137-139°С(декомп.) Елементний аналіз для C23H28N4O 2. НСl Н2O Виявлено % Розраховано % С 61,80 61,80 Η 7,14 6,99 Ν 12,45 12,53 1 H-ЯМР (δ, D MSO): 1,95-2,25 (m, 4H) 2,69-3,48 (m, 7H); 3,57-3,70 (m, 2H); 3,74 (s, ЗН); 3,96 (s, 3Н); 6,92 (d, J=9 Hz, 2H); 7,08 (t, J=9 Гц, 1Н); 7,20 (d, J=9 Гц, 2Н); 7,38 (t, J=8 Гц, 1H); 7,56 (d, J=9 Гц, 1H); 7,76 (d, J=8 Гц, 1H); 10,36-11,07 (m, 2H) Приклад 7 N-(1-метил-1Н-індазол-3-іл)-1-2-4гідроксифеніл)етил)піперидин-4-карбоксамід гідрохлорид (AF3R33O) (І, Ra - Rd = Η, Rb = СН3, Re = 4-ОН, Χ = NHC(O)) Розчин продукту з Прикладу 6в) (6,7г; 0,017моль) у дихлорометані (300мл) по краплях додавали до розчину ВВr3 (8,5г; 0,034моль) у дихлорометані (50мл). Реакційну суміш перемішували при кімнатній температурі протягом 6 год. Потім обережно додавали воду і суміш робили лужною за допомогою IN NaOH, доводячи до рН = 9, і переносили до ділильної лійки. Органічну фазу відокремлювали, висушували над Na2SO4 і розчинник видаляли шляхом випарювання під зниженим тиском. Одержаний таким чином залишок (4,4г) очищали шляхом флеш-хроматографії, застосовуючи суміш СНСl3:МеОН = 9:1 як елюент. Таким чином, одержували 3 г продукту і перетворювали на відповідний гідрохлорид шляхом розчинення в етанолі, додавання хлористого водню в етанолі, випарювання розчинника та кристалізації з суміші етилацетату:етанолу (9:1) для одержання потрібного продукту (2,8г). т. пл.: 249-252°С Елементний аналіз для C22H26N4O2 . HCl 2/3 Н2О Виявлено % Розраховано % 1 С 62,14 61,91 Η 6,64 6,69 Ν 13,45 13,13 H-ЯМР (δ, D MSO): 1,96-2,25 (m, 4H) 2,67-3,47 (m, 7H); 3,63 (d, J=12 Гц, 2H); 3,95 (s, 3Н); 6,69-6,80 (m, 2H); 7,00-7,13 (m, 3Н); 7,38 (t, J=9 Гц, 1H); 7,56 15 80605 (d, J=9 Гц, 1Н); 7,76 (d, J=9 Гц, 1H); 9,37 (s широк., 1H); 10,35-10,90 (m, 2H) Приклад 8 N((1-(2-(4-гідроксифеніл)етил-4піперидін)метил-5-метил-1-(1-метилетил)-1Ніндазол-3-карбоксамід гідрохлорид (AF3R296) (І, Ra = СН3, Rb = і-С3Н7, Re = 4-ОН, Rd = Η, Χ C(O)NHCH2) а) 1-(2-(4-гідроксифеніл)етил)-4піперидилметан-амін Названий продукт одержували (9,3г) шляхом обробки способом, подібним до описаного у Прикладі 1б), але беручи за вихідну сполуку продукт з Прикладу 1а) (7,5г, 0,037моль) та 2-(4гідроксифеніл)етилбромід (7,5г; 0,037моль), одержаний, як описано в [Acta Chemica Scandinava (1947-1973) 1967, 21 (1), 52-62], замість 2-(4нітрофеніл)етил броміду. 1 H-ЯМР (δ, CDCI3 + D2O): 1,15-1,41 (m, 3Н); 1,74 (d, J=9 Гц, 2Н); 1,90-2,07 (m, 2H); 2,45-2,61 (m, 4Н); 2,65-2,75 (m, 2Н); 3,01 (d, J=12 Гц, 2Н); 6,75 (d, J=9 Гц, 2Н); 7,00 (d, J=9 Гц, 2Н). б) ізопропіловий естер 1-(1-метилетил)-5метил-1H-індазол-3-карбонової кислоти 60% суспензію йодиду натрію в мінеральній олії (17,1г; 0,43моль) додавали до суспензії 5-метил-1Hіндазол-3-карбонової кислоти (30г; 0,17моль), приготовленої, як описано у [J. Heterocyclic Chem. 1964, Vol. 1 (5) 239-241], у диметилформаміді (450мл), і реакційну суміш нагрівали до 70°С. Через 30 хвилин додавали ізопропілбромід (48мл, 0,51моль). Реакційну суміш перемішували протягом 6 годин при 70°С. Після охолодження додавали воду. Реакційну суміш переносили до ділильної лійки й екстрагували діетиловим етером. Органічну фазу промивали водним сатуратом з бікарбонатом натрію і, нарешті, розчинник видаляли шляхом випарювання під зниженим тиском. Таким способом одержували 20г олії, яку очищали шляхом флеш-хроматографії, елююючи з сумішшю гексану: етилацетату = 7:3 для одержання 12г потрібного продукту. 1 H-ЯМР (δ, CDCI3): 1,47 (d, J=6 Гц, 6Н); 1,64 (d, J=7 Гц, 6Н); 2,50 (d, J=l Гц, 3Н); 4,92 (гептет, J=7 Гц, 1Н); 5,39 (гептет, J=6 Гц, 1H); 7,23 (dd, J=9;1 Гц, 1Н); 7,40 (d, J=9 Гц, 1H); 7,95 (квінтет, J=1 Гц, 1H). в) 1-(1 –метилетил)-5-метил-1Н-індазол-3карбонова кислота Суспензію продукту, одержаного згідно з Прикладом 8б) (8г; 0,03моль), у 1 М NaOH (42мл) нагрівали з дефлегмацією протягом 3 годин. Потім її виливали у воду, підкислювали 2М НСІ і екстрагували дихлорометаном. Після випарювання розчинника під зниженим тиском одержували 7г потрібного продукту. 1 H-ЯМР (δ, CDCI3): 1,61 (d J=7 Гц, ОН); 2,44 (s, ЗН); 4,88 (гептет, J=7 Гц, 1Н); 7,19 (d, J=9 Гц, 1Н); 7,34 (d, J=9 Гц, 1H); 7,97 (s, 1H); 9,32 (s широк., 1Н). г) хлорид 1-(1-метилетил)-5-метил-1Н-індазол3-карбонової кислоти 16 Тіонілхлорид (6,78г; 0,057моль) додавали до суспензії продукту, приготовленого згідно з Прикладом 8в) (4,01г; 0,019моль) у толуолі (70мл), і реакційну суміш нагрівали з дефлегмацією протягом 2 годин. Розчинник видаляли шляхом випарювання під зниженим тиском і двічі захоплювали толуолом (50 мл x 2) для одержання потрібного продукту (4,3 г), який використовували як такий без подальшого очищення. д) Ν((1-(2-(4-гідроксифеніл)етил)-4піперидил)метил)-5-метил-1-(1-метилетил)-1Ніндазол-3-карбоксамід гідрохлорид Обробка способом, подібним до описаного у Прикладі 3, але з використанням продукту з Прикладу 8а) (4,0г, 0,017моль) та хлориду 1-(1метилетил)-5-метил-1Н-індазол-3-карбонової кислоти (4,0г, 0,017моль), приготовленого, як описано у попередньому Прикладі 8г), одержували 4,5г потрібного продукту і його перетворювали на відповідний гідрохлорид шляхом розчинення в абсолютному етанолі, додавання хлористого водню в етанолі та рекристалізації з етанолу для одержання потрібної солі (3,2г). т. пл.: 257,5-259,5°С Елементний аналіз для С 26Н34N4О 2 НСl Виявлено % Розраховано % С 66,20 66,30 Η 7,75 7,49 Ν 11,87 11,89 1 H-ЯМР (δ, DMSO): 1,53 (d, J=7 Гц, 6H); 1,441,76 (m, 3Н); 1,87 (d, J=12 Гц, 2Н); 2,42 (s, ЗН); 2,79-3,45 (m, 8H); 3,54 (d, J=12 Гц, 2H); 5,03 (гептет, J=7 Гц, 1Н); 6,73 (d, J=9 Гц, 2H); 7,05 (d, J=9 Гц, 2H); 7,26 (d,d J=9,2 Гц, 1H); 7,67 (d, J=9 Гц; 1H); 7,96 (s, 1H); 8,30 (t, J=6 Гц, 1H); 9,35 (s, 1H); 10,35 (s широк., 1Н). Випробування 1. Ме ханічна гіпералгезія, викликана шляхом CFA у щура Самців CD-щурів масою 150-200г використовували після надходження. Застосовуючи анальгезіометр, відбирали щурів, які мали поріг реакції на механічний ноцицептивний подразник від 150 до 180г. Через застосування поступового підвищення тиску на дорсальну зону лівої задньої лапи щура інструмент дозволяє записувати ноцифенсивну реакцію, виражену у грамах, згідно з рухом, коли тварина відсмикує лапу [Randall LO and Semite JJ. A method for the measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 1957; 111: 409-419]. Гіпералгезію викликали шляхом однобічної ін'єкції 150мкл повного ад'юванта Фройнда (Complete Freund's Adjuvant - CFA) у підошовну поверхню лівої задньої лапи тварини [Andrew D, Greenspan JD. Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. J Neurophysiol 1999; 82(5): 2649-2656; Hargreaves K, Dubner R, Brown R, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988; 32: 77-88]. 17 80605 Досліджувані сполуки випробували (доза 10"5 моль/кг) шляхом здійснення тесту через 23 години після ін'єкції CFA. Через 1 год. після введення больовий поріг, виміряний у контрольних тварин, порівнювали з виміряним у тварин, яким вводили досліджуваний продукт. Контрольним тваринам вводили такий самий індиферентний наповнювач (воду), як і той, який застосовували для досліджуваних продуктів. Результати показано у Таблиці 1. Таблиця 1 Вплив на CFA Лікування Кількість щурів Носій AF3R294 AF3R296 AF3R298 AF3R302 AF3R306 AF3R328 AF3R330 AF3R334 12 12 12 12 12 12 12 12 12 Больовий поріг (г) через 1 год. після введення 121±4,1 194±22,4 151±10,7 174±9,8 160±10,2 186±11,0 150±8,7 161±10,5 170±11,0 Больовий поріг нормальних тварин однакової маси/віку =155±2,1г. 2. Механічна гіпералгезія у щурів з діабетом, викликаним стрептозотоцином Самців CD-щурів масою 240-300г використовували після надходження. Діабетичний синдром викликали однією внутрішньочеревинною (і.р.) ін'єкцією 80 мг/кг стрептозотоцину, розчиненого у стерильному фізіологічному розчині [Courteix С, Eschalier A, Lavarenne J. Streptozotocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain, 1993; 53: 81-88; Bannon AW, Decker MW, Kim Dj, Campbell JE, Americ SP. ABT-594, a novel cholinergic channel modulator, is efficacious in nerve ligation and diabetic neuropathy models of neuropathic pain. Brain Res. 1998; 801: 158-63]. Через принаймні три тижні після ін'єкції стрептозотоцину відбирали щурів, які мали рівень глікемії >300мг/дл і мали поріг реакції на механічний ноцицептивний подразник £120г. Рівні глікемії вимірювали за допомогою рефлектометра з застосуванням реактивних смужок, просочених глюкозо-оксидазою. Больовий поріг вимірювали, застосовуючи анальгезіометр. Через застосування поступового підвищення тиску на дорсальну зону лівої задньої лапи щура інструмент дозволяє записувати ноцифенсивну реакцію, виражену у грамах, згідно з рухом, коли тварина відсмикує лапу. Через 2год. після введення больовий поріг, виміряний у контрольних тварин, порівнювали з виміряним у тварин, яким вводили досліджуваний продукт (доза 10-5моль/кг). Контрольним тваринам вводили такий самий індиферентний наповнювач (воду), як і той, який 18 застосовували для досліджуваних Результати показано у Таблиці 2. продуктів. Таблиця 2 Вплив на діабетичну невропатію Лікування Кількість щурів Носій AF3R294 AF3R296 AF3R298 AF3R302 AF3R306 AF3R328 AF3R330 AF3R334 8 8 8 8 8 8 8 8 8 Больовий поріг (г) через 2год. після введення 112±4,0 198±18,6 154±8,7 170±10,2 164±10,2 184±13,8 158±6,2 171±9,6 184±10,5 Больовий поріг нормальних тварин однакової маси/віку = 240±8,7г.
ДивитисяДодаткова інформація
Назва патенту англійськоюThe indazoles having an analgesic activity
Автори англійськоюAlisi, Maria, Alessandra, Cazzolla, Nicola, Furlotti, Guido, Guglielmotti Angelo, Polenzani, Lorenzo
Назва патенту російськоюИндазолы, которые обладают анальгетической активностью
Автори російськоюАлиси Мария Алессандра, Каццолла Никола, Фурлотти Гвидо, Гуглиэлмотти Анджело, Поленцани Лоренцо
МПК / Мітки
МПК: A61P 29/00, C07D 231/56
Мітки: анальгетичну, мають, активність, індазоли
Код посилання
<a href="https://ua.patents.su/9-80605-indazoli-yaki-mayut-analgetichnu-aktivnist.html" target="_blank" rel="follow" title="База патентів України">Індазоли, які мають анальгетичну активність</a>
Заміщені гідразиди 6-метилбензотіазоліл-2-оксамінової кислоти, які мають анальгетичну, жарознижуючу та діуретичну активність

Номер патенту: 57899
Опубліковано: 15.07.2003
Автори: Зубкова Ірина Вікторівна, Дроговоз Світлана Мефодіївна, Березнякова Алла Іллівна, Черних Валентин Петрович, Зупанець Ігор Альбертович, Банний Іван Прокопович
МПК: A61P 29/00, C07D 277/82, A61K 31/428, A61P 7/10
Мітки: жарознижуючу, 6-метилбензотіазоліл-2-оксамінової, кислоти, діуретичну, гідразиди, заміщені, активність, анальгетичну, мають
Формула / Реферат:
Замещенные гидразиды 6-метилбензотиазолил-2-оксаминовой кислоты общей формулыгде R1 - водород;R2 - ацетил или R1 и R2 вместе - о-нитробензилиден или п-нитробензилиден,обладающие анальгетической, жаропонижающей и диуретической активностью.
2-метилтіо-7-(4-хлорфеніл)-4,5,6,7-тетрагідро-1,2,4-триазоло[1,5-а]піримідин-5-он, що проявляє протизапальну та анальгетичну активність

Номер патенту: 68302
Опубліковано: 15.07.2004
Автори: ЛІПСОН ВІКТОРІЯ ВІКТОРІВНА, Рожовецька Ірина Андріївна, Бородіна Вікторія Василівна, Божко Тетяна Степанівна, Ладогубець Олена Василівна
МПК: A61K 31/01, A61K 31/33, C07D 487/04
Мітки: проявляє, активність, 2-метилтіо-7-(4-хлорфеніл)-4,5,6,7-тетрагідро-1,2,4-триазоло[1,5-а]піримідин-5-он, протизапальну, анальгетичну
Формула / Реферат:
2-Метилтіо-7-(4-хлорфеніл)-4,5,6,7-тетрагідро-1,2,4-триазоло[1,5-а]піримідин-5-он формули,що проявляє протизапальну та анальгетичну активність.
Хлор- та нітрозаміщені n-фенілантранілової кислоти, що проявляють протизапальну, анальгетичну активність

Номер патенту: 65875
Опубліковано: 15.04.2004
Автори: Ісаєв Сергій Григорович, Брунь Лідія Володимирівна, Павлій Олег Олександрович, Зупанець Ігор Альбертович
МПК: C07C 229/58, A61K 31/196, A61P 29/00
Мітки: n-фенілантранілової, активність, анальгетичну, проявляють, протизапальну, кислоти, нітрозаміщені, хлор
Формула / Реферат:
Хлор- та нітрозаміщені N-фенілантранілової кислоти, загальної формули:де:R1, R3, R4-H, R2-Cl, R5-COOHабо R1, R3-NO2; R2, R5-H; R4-Cl,або R1, R3-NO2; R2, R4-H; R5-Cl.
4-хлор-n-(2′-карбоксифініл) антранілова кислота, яка виявляє протизапальну, анальгетичну активність
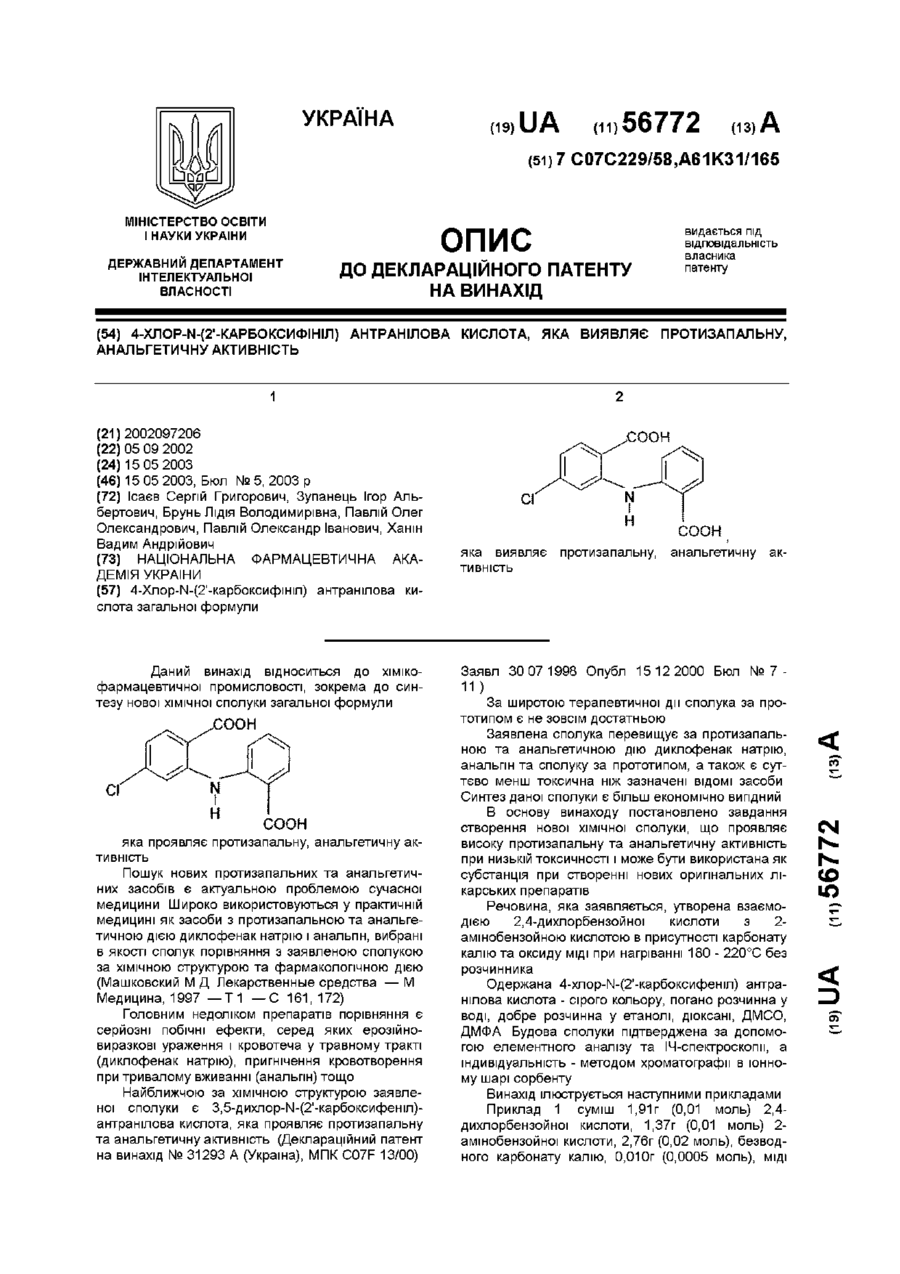
Номер патенту: 56772
Опубліковано: 15.05.2003
Автори: Павлій Олександр Іванович, Зупанець Ігор Альбертович, Павлій Олег Олександрович, Ісаєв Сергій Григорович, Ханін Вадим Андрійович, Брунь Лідія Володимирівна
МПК: A61P 29/00, A61K 31/196, C07C 229/58
Мітки: протизапальну, кислота, анальгетичну, виявляє, активність, антранілова, яка, 4-хлор-n-(2'-карбоксифініл
Формула / Реферат:
4-Хлор-N-(2'-карбоксифініл) антранілова кислота загальної формули:,яка виявляє протизапальну, анальгетичну активність.
D-(+)-глюкозиламонієва сіль 4-хлор-n-(4′-карбоксифеніл) антранілової кислоти, яка виявляє протизапальну, анальгетичну активність

Номер патенту: 55973
Опубліковано: 15.04.2003
Автори: Яковлева Лариса Василівна, Ісаєв Сергій Григорович, Павлій Олег Олександрович, Зупанець Ігор Альбертович, Брунь Лідія Володимирівна
МПК: A61K 31/7004, A61K 31/196, C07C 229/58, A61P 29/00
Мітки: 4-хлор-n-(4'-карбоксифеніл, антранілової, d-(+)-глюкозиламонієва, анальгетичну, кислоти, сіль, яка, протизапальну, активність, виявляє
Формула / Реферат:
D-(+)-глюкозиламонієва сіль 4-хлор-N-(4’-карбоксифеніл) антранілової кислоти загальної формули,яка виявляє протизапальну, анальгетичну активність.
Попередній патент: Фармацевтична композиція і лікарський засіб седативної та кардіологічної дії
Наступний патент: Спосіб виготовлення холоднотягненого дроту
Випадковий патент: Сушарка для сипких матеріалів