Похідні 2(1н)-хінолінону як антагоністи серотоніну, спосіб їх одержання (варіанти), медичний препарат та фармацевтична композиція на їх основі
Номер патенту: 44332
Опубліковано: 15.02.2002
Автори: Алетрю Мішель, Деллак Женев'єв, Маккорт Гері, Хорнарт Крістіан


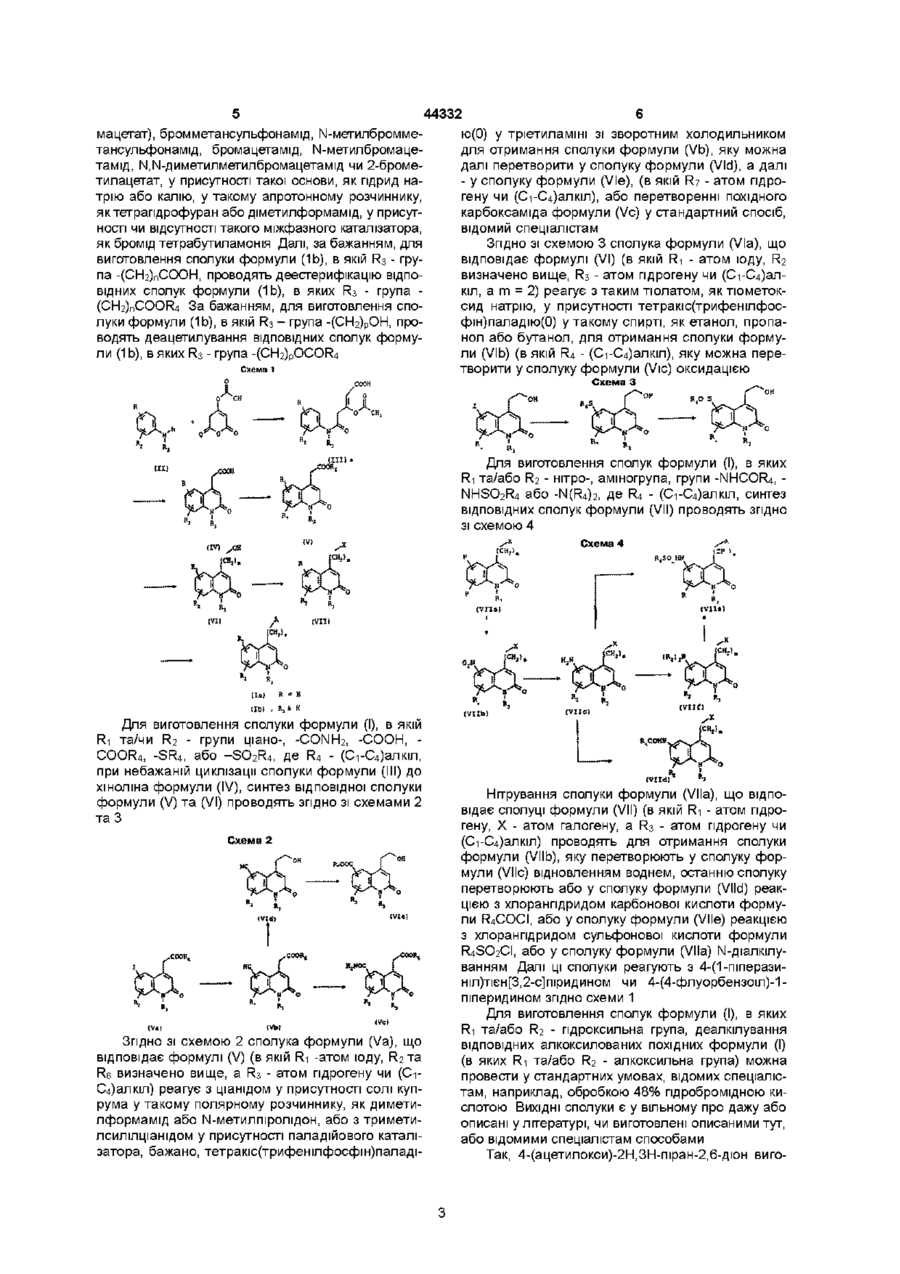





Формула / Реферат
Похідні 2(1Н)-хінолінону загальної формули (1)
в якій
А - 4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл або 4-(4-флуорбензоїл)-1-піперидил, r1 та R2 кожний, незалежно один від одного - атом гідрогену, галогену, аміно-, гідпрокси-, нітро- чи ціаногрупа, або (С1-С6)алкіл, (С1-С6)алкоксил, трифлуорметил, трифлуорметоксил або групи -СООН, -COOR4, -CONH2, -CONHR4, -CONR4R5, -SR4, SО2R4, -NHCOR4, -NHSO2R4 або -N(R4)2, де R4 та R5 кожний -(С1-С4)алкіл, R3 - атом гідрогену або (С1-С4)алкіл, або групи -(СН2)рОН, -(CH2)pNH2, (СН2)nСООН, (СН2)nСООR4, (СН2)nСОNН2, -(СН2)nСОNHОН, -(CH2)pSH, -(СН2)nSO3H, (CH2)nSO2NH2, -(CH2)nSO2NHR4, -(CH2)nSO2NR4R5, -(CH2)pNHSO2R4, -(CH2)pNHCOR4, (CH2)pOCOR4, де R4 та R5 кожний -(С1-С4)алкіл, n - 1,2,3 чи 4, р - 2,3 чи 4, a m - 2,3 чи 4,
а також їх солі приєднання фармацевтично придатних кислот чи основ.
2. Сполуки за п. 1, які відрізняються тим, що m = 2.
3. Сполуки за п. 1 або 2, які відрізняються тим, що R1 у положенні 6 або 7 хіноліну є атомом гідрогену, флуору чи хлору, або аміно-, гідрокси-, нітро- чи ціаногрупою, (С1-С6)алкілом, метоксилом, трифлуорметоксилом, ацетиламіно-, метилсульфоніламіно- або диметиламіногрупою, a R2 - атом гідрогену.
4. Сполуки за будь-яким з пп. 1, 2 або 3, які відрізняються тим, що R3 - атом гідрогену або (С1-С4)алкіл, або групи -(СН2)рОН, (СН2)nСООН, (CH2)nCOOR4, -(CH2)nCONH2, -(CH2)nCONHR4, (CH2)pOCOR4, де R4 та R5 кожний -(С1-С4)алкіл, n -1,2,3 чи 4, р - 2,3 чи 4.
5. Сполуки за будь-яким з пп. 1, 2, 3 чи 4, які відрізняються тим, що n = 1, а р = 2.
6. Спосіб одержання сполук за п. 1, який полягає у тому, що 4-(1-піперазиніл)тієн[3,2-с]піридин реагує зі сполукою формули (VII):
в якій R1, R2, R3 та m позначено у п. 1, а Х - група, що відщеплюється.
7. Спосіб одержання сполук формули (Іb):
в якій A, R1, R2, R3 та m позначено у п. 1, a R3 - не атом гідрогену, який полягає у тому, що сполука формули (Іа):
реагує з електрофільним агентом.
8. Медичний препарат, у склад якого входить сполука за будь-яким одним з пп. 1, 2, 3, 4 чи 5.
9. Фармацевтична композиція, яка містить сполуку за будь-яким одним з пп. 1, 2, 3, 4 чи 5 у комбінації з будь-яким фармацевтичнo придатним наповнювачем.
Текст
ПОХІДНІ 2(1Н)-хшолінону загальної формули (1) гену, флуору чи хлору, або аміно-, гідрокси-, нггрочи ціаногрупою, (Сі-Сє)алкілом, метоксилом, трифлуорметоксилом, ацетиламіно-, метилсульфоніламіно- або диметиламіногрупою, a R2 - атом гідрогену 4 Сполуки за будь-яким з пп 1,2 або 3, які відрізняються тим, що R3 - атом гідрогену або (С-і-С4)алкіл, або групи -(СН2)РОН, (СН2)ПСООН, (CH2)nCOOR4, -(CH2)nCONH2, -(CH2)nCONHR4, (CH2)POCOR4, де R4 та Rs кожний -(Сі-С4)алкіл, п 1,2,3 чи 4, р-2,3 чи 4 5 Сполуки за будь-яким з пп 1, 2, 3 чи 4, які відрізняються тим, щоп = 1,ар = 2 6 Спосіб одержання сполук за п 1, який полягає у тому, що 4-(1-піперазиніл)тієн[3,2-с]піридин реагує зі сполукою формули (VII) X / А (VII) О R в якій Ri, R2, R3Ta m позначено у п 1, а X - група, що відщеплюється 7 Спосіб одержання сполук формули (ІЬ) в якій А - 4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл або 4-(4флуорбензоіл)-1-піперидил, Ri та R2 кожний, незалежно один від одного - атом гідрогену, галогену, аміно-, пдпрокси-, нггро- чи ціаногрупа, або ( d Сє)алкіл, (Сі-Сб)алкоксил, трифлуорметил, трифлуорметоксил або групи -СООН, -COOR4, -CONH2, CONHR4, -CONR4R5, -SR4, SO2R4, -NHCOR4, NHSO2R4 або -N(R4)2, де R4 та R5 кожний -(Сі-С4)алкіл, R3 - атом гідрогену або (Сі-С4)алкіл, або групи (СН2)РОН, -(CH2)PNH2, (CH2)nCOOH, (CH2)nCOOR4, (CH2)nCONH2, -(CH2)nCONHOH, -(CH2)PSH, (CH2)nSO3H, (CH2)nSO2NH2, -(CH2)nSO2NHR4, (CH2)nSO2NR4R5, -(CH2)PNHSO2R4, -(CH2)PNHCOR4, (CH2)POCOR4, де R4 та Rs кожний -(Сі-С4)алкіл, п 1,2,3 чи 4, p - 2,3 чи 4, a m - 2,3 чи 4, а також їх солі приєднання фармацевтично придатних кислот чи основ 2 Сполуки за п 1, які відрізняються тим, що m = 2 3 Сполуки за п 1 або 2, які відрізняються тим, що Ri у положенні 6 або 7 хіноліну є атомом гідро А (ІЬ) в якій А, R-і, R2, R3Ta m позначено у п 1, a R3 - не атом гідрогену, який полягає у тому, що сполука формули (Іа) А (іа) реагує з електрофільним агентом 8 Медичний препарат, у склад якого входить сполука за будь-яким одним з пп 1, 2, 3, 4 чи 5 со го З 44332 4 9 Фармацевтична композиція, яка містить сполуку будь-яким фармацевтично придатним наповнюваза будь-яким одним з пп 1, 2, 3, 4 чи 5 у комбінації з чем Винахід відноситься до похідних 2(1Н)-хінолона, їх виготовлення та застосування у терапії Сполуки згідно з винаходом відповідають формулі (І) А в якій А - 4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл або 4-(4-флуорбензоіл)-1-піперидил, RiTa R2 кожний, незалежно один від одного - атом пдрогена, галогена, аміно-, гідроксильна, нггро- або ціаногрупа, (С-і-Сб)алкіл, (Сі-Сб)алкоксил, трифлуорметил, трифлуорметоксил або групи -СООН, -COOR4, CONH2, -CONHR4, -CONR4R5, -SR4, SO2R4, NHCOR4, -NHSO2R4 або -N(R4)2, де R4Ta R5 кожний (Сі-С4)алкіл, R3 - атом пдрогена або (Сі-С4)алкіл, або групи -(CH2)DOH, -(CH2)DNH2, (CH2)nCOOH, (CH2)nCOOR4, -(CH2)nCONH2, -(CH2)nCONHOH, (CH2)PSH, -(CH2)nSO3H, (CH2)nSO2NH2, (CH2)nSO2NHR4, -(CH2)nSO2NR4R5, -(CH2)PNHSO2R4, -(CH2)DNHCOR4, (CH2)pOCOR4, де R4Ta R5 кожний (Сі-С4)алкіл, n -1, 2, 3 чи 4, p - 2, 3 чи 4, a m - 2, 3 чи 4, а також їх солі приєднання фармацевтичне придатних кислот чи основ Згідно винаходу сполуки формули (І) можна синтезувати як показано на схемі 1 4-(ацетилокси)-2Н, ЗН-піран-2,6-дюн реагує зі сполукою формули (II) (в якій Ri та R2 визначено вище, a R2 - атом пдрогена або (Сі-С4)алкіл) при кімнатній температурі у такому полярному розчиннику, як оцтова кислота Після висушування отриманої сполуки формули (III) и циклізують у присутності такої бажано обезводненої неорганічної чи органічної кислоти, як концентрована сульфатна, фосфатна чи трифлуорметилсульфатна кислота, при температурі у межах 10 - 150°С, отримуючи заміщену чи незаміщену 2-оксо-1,2-дипдро-4-хшоліноцтову кислоту формули (IV), яку естерифікують спиртом формули ReOH (де R6 - (Сі-С4)алкіл) з використанням, бажано, тюнілхлориду Отриманий естер формули (V) далі відновлюють гідридом у апротонному розчиннику, наприклад, алюмопдридом лгтію у діоксані, чи боропдридом натрію у надлишку тетрапдрофурану зі зворотним холодильником, або боропдридом лгтію у теграгідрофурані при кімнатній температурі, одержуючи спирт формули (VI) (в якій m = 2), сполуки формули (VI), в яких m = 3 чи 4, отримують зі сполук, в яких m = 2, відомими спеціалістам гомологічними способами Сполуки формули (VI) (в яких m = 2, 3 чи 4) далі активують до сполук формули (VII) (в яких X - така група, що відщеплюється, як атом хлора чи брому), наприклад, реакцією зтюнілхлоридом у хлороформі зі зворотним холодильником або дибромтрифенілфосфораном при кімнатній температурі у дихлорметані, або до сполук формули (VII) (в яких X - така група, що відщеплюється, як метансульфонілоксильна, тр ифлуор метан сул ьфонілоксильна, птолуолсульфонілоксильна), наприклад, реакцією з сульфоновим ангідридом чи хлорангідридом сульфонової кислоти у присутності такої основи, як піридин або тріетиламш Наприкінці, сполука формули (VII) реагує з 4-(1-піперазиніл)тієн[3,2-с]піридином або з 4-(4-флуорбензоіл)-1-піперидином у присутності чи відсутності апротонного розчинника, у присутності неорганічної основи при температурі у межах 20 - 150°С, краще в ацетонггрилі або диметилформаміді у контакті з гідрокарбонатом натрію, отримуючи сполуку формули (І) Для виготовлення сполуки формули (ІЬ) (в якій R3 відрізняється від атома гідрогену) можна алкілувати відповідну сполуку формули (Іа) (в якій R3 атом гідрогену), використовуючи електрофільний агент типу F?3Br чи R3I, наприклад, третбутил (бро мацетат), бромметансульфонамід, N-метилбромметансульфонамід, бромацетамід, N-метилбромацетамід, N.N-диметилметилбромацетамід чи 2-брометилацєтат, у присутності такої основи, як гідрид натрію або калію, у такому апротонному розчиннику, яктетрапдрофуран або діметилформамід, у присутності чи відсутності такого міжфазного каталізатора, як бромід тетрабутиламонія Далі, за бажанням, для виготовлення сполуки формули (1Ь), в якій R3 - група -(СН2)ПСООН, проводять деестерифікацію ВІДПОВІДНИХ сполук формули (1Ь), в яких R3 - група (CH2)nCOOR4 За бажанням, для виготовлення сполуки формули (1Ь), в якій R3 - група -(СН2)РОН, проводять деацетилування ВІДПОВІДНИХ сполук формули (1Ь), в яких R3 - група -(CH2)DOCOR4 Схема 1 44332 ю(0) у тріетиламші зі зворотним холодильником для отримання сполуки формули (Vb), яку можна далі перетворити у сполуку формули (Vld), а далі - у сполуку формули (Vie), (в якій R7 - атом гідрогену чи (Сі-С4)алкіл), або перетворенні похідного карбоксаміда формули (Vc) у стандартний спосіб, відомий спеціалістам Згідно ЗІ схемою 3 сполука формули (Via), що відповідає формулі (VI) (в якій Ri - атом юду, R2 визначено вище, R3 - атом гідрогену чи (Сі-С4)алкіл, а т - 2 ) реагує з таким тюлатом, як тюметоксид натрію, у присутності тетракіс(трифенілфосфш)паладію(О) у такому спирті, як етанол, пропанол або бутанол, для отримання сполуки формули (Vlb) (в якій R4 - (Сі-С4)алкіл), яку можна перетворити у сполуку формули (Vic) оксидацією Схема З Для виготовлення сполук формули (І), в яких Ri та/або R2 - нітро-, аміногрупа, групи -NHCOR4, NHSO2R4 або -N(R4)2, де R4 - (Сі-С4)алкіл, синтез ВІДПОВІДНИХ сполук формули (VII) проводять згідно зі схемою 4 Для виготовлення сполуки формули (І), в якій Ri та/чи R2 - групи ціано-, -CONH2, -COOH, COOR4, -SR4, або -SO 2 R 4 , де R4 - (С г С 4 )алкіл, при небажаній циклізації сполуки формули (III) до хіноліна формули (IV), синтез відповідної сполуки формули (V) та (VI) проводять згідно зі схемами 2 та З Схема 2 coo?. Згідно ЗІ схемою 2 сполука формули (Va), що відповідає формулі (V) (в якій Ri -атом юду, R2 та R6 визначено вище, a R3 - атом гідрогену чи ( d С4)алкіл) реагує з ціанідом у присутності солі купрума у такому полярному розчиннику, як диметилформамід або N-метилпіролідон, або з триметилсилілціанідом у присутності паладійового каталізатора, бажано, тетракіс(трифенілфосфш)паладі Нітрування сполуки формули (Vila), що відповідає сполуці формули (VII) (в якій Ri - атом гідрогену, X - атом галогену, a R3 - атом гідрогену чи (Сі-С4)алкіл) проводять для отримання сполуки формули (Vlib), яку перетворюють у сполуку формули (Vile) відновленням воднем, останню сполуку перетворюють або у сполуку формули (Vlld) реакцією з хлорангідридом карбонової кислоти формули R4COCI, або у сполуку формули (Vile) реакцією з хлорангідридом сульфонової кислоти формули R4SO2CI, або у сполуку формули (Vila) N-діалкілуванням Далі ці сполуки реагують з 4-(1-піперазиніл)тієн[3,2-с]піридином чи 4-(4-флуорбензоіл)-1піперидином згідно схеми 1 Для виготовлення сполук формули (І), в яких Ri та/або R2 - гідроксильна група, деалкілування ВІДПОВІДНИХ алкоксилованих похідних формули (І) (в яких Ri та/або R2 - алкоксильна група) можна провести у стандартних умовах, відомих спеціалістам, наприклад, обробкою 48% пдробромідною кислотою ВИХІДНІ сполуки є у вільному про дажу або описані улггературі, чи виготовлені описаними тут, або відомими спеціалістам способами Так, 4-(ацетилокси)-2Н,ЗН-піран-2,6-дюн виго 44332 8 товляють з 3-оксоглутаровоі кислоти згідно з зниженим тиском і залишок вносили до 400мл дихЕ G Frandsen & N Jacobsen, J Chem Soc Perkm лорметану Суміш промивали насиченим розчиI, pp 933-6(1978) ном гідрокарбонату натрію, а далі водою, органічну фазу сушили сульфатом натрію Після фільтруСпосіб циклізації, пристосований для цього, вання та концентрування було отримано 12,6г суописано у європейських патентних заявках ЕР міші двох естерів (71%) Обидва естери відокрем0364327 та ЕР 0577325 лювали флешхроматографією на дюксиді СИЛІЦІЯ, Введення нггрилу у сполуки формули (V) проелюючи сумішшю метанол/дихлорметан 3 97 водять згідно зі способом, описаним N Chantam & T/Hanafusa, J Org Chem 514714-4716(1986) Отримали 4,0г метил(6-метокси-1-метил-2-оксо-1,2-дипдро-4-хінолінацетату), т плавл 129 Ароматичне нуклеофільне заміщення юдова130°С, та 7,8г метил(6-метоксі-2-оксо-1,2-дипдроних арилів тюлатими базується на способі, описа4-хінолінацетату), т плавл 223 - 224°С ному Т Migital et al , Bull Chem Soc Japan, 53 1385(1980) 1 4 4-(2-пдроксіетил)-6-метокси-2(1Н)-хшолон 4-(1-піперазиніл)тієн[3,2-с]піридин синтезують 1,4г (37ммоль) боропдриду натрію додали при згідно J S New et al , J Med Chem 32 № 6 1147 кімнатній температурі до суспензії 3,1г 56(1989) (12,5ммоль) метил(6-метоксі-2-оксо-1,2-дипдрохінолінацетату) у ЮОмл сухого тетрапдрофурану та Нижченадані приклади ілюструють винахід без 1мл метанолу, нагрівали зі зворотним холодильнийого обмеження Мікроаналіз, ІК, ЯМРта масспекком 16 годин Після охолодження до 5°С по краптри підтверджують структуру отриманих сполук лям додали 1мл метанолу, а через ЗО хвилин 0,5г Хімічну будову та фізичні властивості ряду боропдриду натрію і знов нагрівали реакційну сусполук згідно з винаходом проілюстровано у нижміш подальші 8 годин Після охолодження та оброченаданих таблицях бки 5мл метанолу розчинник випарювали та залиСпіввідношення (х у) відповідає співвідношок вносили у 200мл дихлорметану та ЮОмл 1Н шенню (кислота/основа) соляної кислоти Органічну фазу відокремлювали, Приклад 1 (Сполука 27) промивали водою та сушили сульфатом натрію Пдрохлорид (2 1) 6-метокси-4-[2-[4-(тієн[3,2Після фільтрування та концентрування під вакуус]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хінолону мом отримали 1,95г очікуваного спирту 1 1 3-(ацетилокси)-5-[(4-метоксифеніл)метиламшо]-5-оксо-2-пентенова кислота 27г (158ммоль) Вихід 72% 4-(ацетилокси)-2Н,ЗН-піран-2,6-дюну при енергій1 5 4-(2-хлоретил)-6-метокси-2(Н)-хінолон ному перемішуванні при кімнатній температурі до3,4мл (46,6ммоль) тюнілхлориду при перемідавали до розчину 20г (146ммоль) І\І-метил-4-мешуванні додавали при кімнатній температурі до сутоксіаніліну у ЮОмл оцтової кислоти Через 5 госпензії 3,11г (14,2ммоль) 4-(2-пдроксіетил)-6-методин перемішування при кімнатній температурі докси-2(Н)-хінолону у 50мл хлороформу та Змл дидавали 700мл льодяної води і перемішували суметилформаміду Суспензію нагрівали зі зворотміш ЗО хвилин Отриману бежеву тверду речовину ним холодильником 14 годин (повне розчинення) ВІДДІЛЯЛИ від розчину, промивали водою, подрібПісля охолодження до кімнатної температури до нювали у діетилетері, сушили над пентоксидом реакційної суміші по краплям додали 50мл води і фосфору 24 год при 40°С продовжували перемішування ЗО хвилин Органічну фазу зібрали, відокремили відстоюванням після Отримали 28,1г твердого продукта Т плавл утворення осаду, промили водою, висушили суль85 - 88°С Вихід 76% фатом магнію та профільтрували Фільтрат конце1 2 6-метоксі-2-оксо-1,2-дипдро-4-хіноліноцтонтрували у вакуумі Отримали 3,2г блідожовтого ва кислота твердого продукту 41 г (1 ЗЗммоль) 3-(ацетил окси)-5-[(4-мето кс ифеніл)метиламіно]-5-оксо-2-пентеновоі кислоти Т плавл 231 -232°С невеликими порціями додавали при кімнатній темВихід 94% пературі до 70мл сульфатної кислоти (96 - 97%) і 16 Пдрохлорид 6-метокси-4-[2-[4-(тієн[3,2суміш при перемішуванні нагрівали до 80°С протяс]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хінолону гом 1,5 години Після охолодження реакційну су(2 1) міш виливали у 100г льоду та ЮОмл води, перемі1,2г (5ммоль) 4-(2-хлоретил)-6-метокси-2(Н)-хішували 15 хвилин, тверду речовину ВІДДІЛЯЛИ від нолону додали до суспензії 1,2г (5,5ммоль) 4-(1-пірозчину, рясно промивали водою і сушили 48 гоперазиніл)тіен[3,2-с]піридину та 0,44г (5,25ммоль) дин при 50°С гідрокарбонату натрію у 15мл ацетонггрилу і суміш нагрівали зі зворотним холодильником 10 годин Зібрали 14,9г суміші 6-метокси-1-метил-2-окПісля випарювання розчинника під вакуумом залисо-1,2-дипдро-4-хіноліноцтовоі кислоти та 6-метокшок внесли до ЮОмл дихлоретану і послідовно сі-2-оксо-1,2-дипдро-4-хіноліноцтовоі кислоти Випромивали насиченим розчином гідрокарбонату хід 45% натрію, а далі водою Після висушування сульфа1 3 Метил(6-метоксі-2-оксо-1,2-дипдро-4-хінотом натрію, фільтрування та конденсування фільтлінацетат) рату сирий продукт очищали флеш-хроматографі1 бмл (219ммоль) тюнілхлориду по краплям єю на дюксиді СИЛІЦІЮ, елюючи сумішшю метапри перемішуванні додавали при кімнатній темпенол/дихлорметан (5 95), що містила сліди водноратурі до суспензії 16,8г (68ммоль) суміші 6-метокго аміаку У вигляді основи було отримано 0,50г си-1-метил-2-оксо-1,2-дипдро-4-хіноліноцтовоі киспродукту Вихід 24% лоти та 6-метоксі-2-оксо-1,2-дипдро-4-хінолшоцтовоі кислоти у 250мл метанолу і продовжували пеДипдрохлорид виготовляли у суміші метаремішування 16 годин Розчинник випарювали під нол/хлороводнева кислота/етер Т плавл 254°С (з 44332 10 розкладанням) З 4 6-хлор-4-(2-пдроксіетил)-1 -метил-2(1 Н)-хінолон Приклад 2 (Сполука 28) Гідрохлорид (1 1) 4-[2-[4-(4-флуорбензоіл)-13,0г (79ммоль) боропдриду натрію додали до піпериділ]етил]-6-метокси-2(1Н)-хшолону суспензії 5,9г (23,4ммоль) метил(6-хлор-1-метил1,2-дипдро-4-хінолінацетату) у ЮОмл сухого тетСуміш 1,1г (4,6ммоль) 4-(2-хлоретил)-6-метокрапдрофурану та Юмл метанолу і нагрівали зі си-2(Н)-хінолону, 1,0г (5,5ммоль) 4-(4-флуорбензозворотним холодильником 9 годин Після охолоіл)піперидину та 0,38г (4,6ммоль) гідрокарбонату дження розчинник випарювали під вакуумом та занатрію у 20мл ацетонгтрилу нагрівали зі зворотним лишок вносили у 400мл дихлорметану та ЮОмл холодильником 8,5 годин Після випарювання реаЗН соляної кислоти Органічну фазу промивали вокційної суміші досуха сирий продукт очищали дою та сушили сульфатом натрію, фільтрували та флеш-хроматографією на дюксиді СИЛІЦІЮ, елююфільтрат конденсували Сирий продукт очищали чи сумішшю метанол/дихлорметан (5 95), що місфлеш-хроматографією на дюксиді СИЛІЦІЮ, елюютила сліди водного аміаку чи сумішшю метанол/дихлорметан (5 95) У вигляді основи було отримано 0,53г очікуваного продукту Отримали 5,9г очікуваного спирту Вихід 30% Вихід 92% Гідрохлорид виготовляли у суміші метаТ плавл 169- 170°С нол/хлороводнева кислота З 5 6-хлор-4-(2-хлоретил)-1-метил-2(1Н)-хінолон Т плавл 237°С (з розкладанням) Приклад 3 (Сполука 4) 5,5мл (75ммоль) тюнілхлориду при перемішуванні додавали по краплям до суспензії 5,9г Гідрохлорид (1 1) 6-хлор-4-[2-[4-(4-флуорбен(24,8ммоль) 6-хлор-4-(2-пдроксіетил)-1-метилзоіл)-1-піпериділ]етил]-1-метил-2(1Н)-хінолону 2(1Н)-хінолону у 120мл хлороформу з двома крап3 1 3-(ацетилокси)-5-[(4-хлорфеніл)метиламілями піридину та двома краплями диметилформано]-5-оксо-2-пентенова кислота міду Реакційне середовище обережно нагрівали зі 19,8г (116ммоль) 4-(ацетилокси)-2Н,ЗН-піранзворотним холодильником 2,5 години, а далі обро2,6-дюну при перемішуванні до давали малими бляли, як описано у прикладі 1 5 порціями до розчину 15г (Юбммоль) 4-хлор-М-метилбензамшу у 40мл чистої оцтової кислоти РеакОтримали 5,4г очікуваного продукту ційну суміш 3 години перемішували при 35°С, охоТ плавл 120- 122°С лоджували до кімнатної температури і розбавляли Вихід 86% Юмл льодяної води Тверду речовину відокремлю3 6 Гідрохлорид (1 1) 6-хлор-4-[2-[4-(4-флуовали відстоюванням, рясно промивали водою та рбензоіл)-1-піпериділ]етил]-1-метил-2(1Н)-хінолосушили при 40°С 48 годин ну 25,5г очікуваної сполуки отримали у вигляді Суміш 0,90г (3,5ммоль) 6-хлор-4-(2-хлоретил)аморфної твердої речовини, яку без подальшої об1-метил-2(1Н)-хінолону, 0,71г (4,0ммоль 4-(4-флуробки використовували у наступній операції орбензоіл)піперидину та 0,60г (7,0ммоль) гідрокарбонату натрію у 15мл ацетонгтрилу нагрівали зі Вихід 77% зворотним холодильником 11 годин Реакційну суЗ 2 6-хлор-1-метил-2-оксо-1,2-дипдро-4-хіноліміш випарювали досуха і сирий продукт очищали ноцтова кислота флеш-хроматографією на дюксиді СИЛІЦІЮ, елюю25,5г (81,8ммоль) 3-(ацетилокси)-5-[(4-хлорфечи сумішшю метанол/дихлорметан (4 96), що місніл)метиламшо]-5-оксо-2-пентеновоі кислоти неветила сліди водного аміаку ликими порціями при енергійному перемішуванні уводили до 40мл концентрованої сульфатної кисУ вигляді основи було отримано 0,86г очікувалоти при кімнатній температурі Реакційну суміш ного продукту далі нагрівали до 85°С 60 хвилин, охолоджували і Вихід 62% виливали у суміш 500г льоду та 500мл води ТверГідрохлорид виготовляли у суміші метаду речовину відокремлювали відстоюванням, пронол/хлороводнева кислота/етер мивали водою та сушили при 40°С 24 години Т плавл 244°С (з розкладанням) Отримані 9,47г очікуваної сполуки без подальПриклад 4 (Сполука 5) шої обробки використовували у наступній операції Гідрохлорид (2 1) 6-флуор-1-метил-4-[2-[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хіВихід 46% нолону З 3 Метил(6-хлор-1-метил-1,2-дипдро-4-хінолінацетат) 41 3-(ацетилокси)-5-[(4-флуорфеніл)метиламшо]-5-оксо-2-пентенова кислота 11мл (147ммоль) тюнілхлориду приблизно ЗО хвилин по краплям при перемішуванні додавали 9,93г (58,4ммоль) 4-(ацетилокси)-2Н,ЗН-пірандо суспензії 12,5г (49ммоль) 6-хлор-1-метил-2-ок2,6-дюну при перемішуванні не великими порціями со-1,2-дипдро-4-хіноліноцтовоі кислоти у 150мл додавали до розчину 6,64г (53,1ммоль) ІЧ-метил-4метанолу Суміш перемішували 17 годин при кімфлуораніліну у ЮОмл чистої оцтової кислоти Реанатній температурі і розчинник видаляли під вакуукційну суміш перемішували 2 години при 35°С, мом Залишок розчиняли у 400мл дихлорметану і охолоджували до кімнатної температури і розбавдалі промивали насиченим розчином гідрокарболяли 500мл льодяної води Отриману тверду речонату натрію, а далі водою Органічну фазу сушили вину ВІДДІЛЯЛИ від розчину, рясно промивали восульфатом натрію і фільтрат та конденсували Будою і сушили у сушильній шафі (40°С) 48 годин ло отримано 11,16г очікуваної сполуки Отримали 28,1г очікуваного продукта у вигляді аморфної твердої речовини, що плавиться нижче Т плавл 99-101 °С 50°С Вихід 85% 11 44332 12 Вихід 76%, під вакуумом Отримали 2,36г очікуваного хлори4 2 6-хлор-1-метил-2-оксо-1,2-дипдро-4-хіноліДУ ноцтова кислота Т плавл 141 -142°С 31,8г (107ммоль) 3-(ацетилокси)-5-[(4-флуорВихід 98% феніл)метиламіно]-5-оксо-2-пентеновоі кислоти 4 6 Гідрохлорид (2 1) 6-флуор-1-метил-4-[2~ невеликими порціями при інтенсивному перемішу[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]ванні додали при кімнатній температурі до 130мл 2(1Н)-хінолону концентрованої сульфатної кислоти, реакційну су1,4г (5,8ммоль) 4-(2-хлоретил)-6-флуор-1-меміш нагрівали до 90°С протягом 1,5 години Після тил-2(1 Н)-хінолону додали до суміші 1,3г охолодження цей розчин виливали у суміш 500г (5,9ммоль) 4-(1-піперазиніл)тієн[3,2-с]піридину та льоду та 500мл води Одержану сіру тверду речо0,50г (5,95ммоль) гідрокарбонату натрію у 20мл вину ВІДДІЛЯЛИ від розчину, промивали водою, поацетонітрилу і нагрівали реакційну суміш до 55 дрібнювали у діетиловому етері і сушили 24 годи60°С 18 годин Розчинник випарювали і залишок ни при 40°С внесли до ЮОмл дихлоретану, промивали насиченим розчином гідрокарбонату натрію, а далі водоОтримали 11,37г продукту ю Органічну фазу висушували сульфатом натрію, Т плавл 230°С фільтрували та конденсували фільтрат Сирий Вихід 45% продукт очищали флеш-хроматографією на дюк43 метил(6-флуор-1-метил-2-оксо-1,2-дипдсиді СИЛІЦІЮ, елюючи сумішшю метанол/дихлормеро-4-хінолінацетат) тан (5 95), що містила сліди водного аміаку 1 бмл (219ммоль) тюнілхлориду по краплям при перемішуванні протягом приблизно ЗО хвилин У вигляді основи отримано 0,70г очікуваного додавали до суспензії 11,37г (49,38ммоль) суміші продукту 6-флуор-1-метил-2-оксо-1,2-дипдро-4-хінолшоцтоВихід 27% воі кислоти у 120мл метанолу Суміш перемішуваОснову розчиняли у Юмл метанолу і обробляли протягом ночі (13 годин) при кімнатній темперали надлишком 2Н розчину пдрогенхлориду у етері турі і розчинник випарювали під зниженим тиском Отриманий осад відокремлювали, перекристалізоЗалишок розчиняли у 400мл дихлорметану і далі вували з метанолу і сушили під вакуумом Отримапромивали насиченим розчином гідрокарбонату ли 0,38г дипдрохлориду натрію, а потім водою Після сушки сульфатом наТ плавл 280°С (з розкладанням) трію, фільтрування та концентрування фільтрату Приклад 5 (Сполука 10) отримали 9,6г очікуваного продукту Гідрохлорид (2 1) 7-флуор-2-оксо-4-[2-[4-(тіТ плавл 134- 135°С єн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-1,2-дипдро-1-хіноліноцтовоі кислоти Вихід 78% 4 4 6-флуор-4-(2-пдроксіетил)-1 -метил-2(1 Н)2,9мл 0,5М розчину трет-бути л (б ром ацетату) у хінолон тетрапдрофурані по краплям додали до суміші 0,50г (1,23ммоль) 7-флуор-4-[2-[4-(тієн[3,2-с]піри3,78г (ЮОммоль) боропдриду натрію додали дин-4-іл)-1-піперазиніл]етил]-2(1Н)-хінолону (вигодо суспензії 8,0г (32ммоль) метил(6-флуор-1-метитовленого з 3-флуораніліну, як описано у прикладі лі-2-оксо-1,2-дипдрохінолінацетату) у ЮОмл сухо4), 0,10г 1,79ммоль) свіжорозмолотого гідроксиду го тетрапдрофурану і суміш нагрівали зі звороткалію та 0,12г 0,37ммоль) броміду тетрабутиламоним холодильником 20 годин Після охолодження нію у 20мл тетрапдрофурану при 0 - 5°С Через ЗО до 5°С по краплям додали 2мл метанолу та Зг бохвилин витримки при 0 - 5°С температурі дали підропдриду натрію і нагрівали реакційну суміш зі нятися до кімнатної і продовжували перемішуванзворотним холодильником 12 годин Розчинник виня 6 годин Розчинник випарювали під вакуумом і парювали під вакуумом та залишок вносили у залишок вносили до ЮОмл дихлорметану, органіч400мл дихлорметану та 150мл 2Н соляної кислону фазу промивали водою, сушили сульфатом нати, органічну фазу промивали водою, сушили сутрію та конденсували Сирий продукт очищали льфатом натрію, фільтрували та фільтрат конценфлеш-хроматографією на дюксиді СИЛІЦІЮ, елюютрували чи сумішшю метанол/дихлорметан (5 95), що місОтримали 1,95г очікуваного спирту тила сліди водного аміаку Вихід 66% Т плавл 153- 154°С 4 5 4-(2-хлоретил)-6-флуор-1 -метил-2(1 Н)-хінолон Змл (41 ммоль) тюнілхлориду додавали по краплям при перемішуванні до суспензії 2,2г (9,95ммоль) 6-флуор-4-(2-пдроксіетил)-1-метил2(1Н)-хінолону у ЮОмл хлороформу з двома краплями піридину та двома краплями диметилформаміду Реакційну суміш обережно нагрівали зі зворотним холодильником 4,5 години Після охолодження до кімнатної температури до реакційної суміші по краплям додали 50мл води і продовжували перемішування ЗО хвилин Органічну фазу зібрали, відокремили відстоюванням після утворення осаду, промили водою, висушили сульфатом магнію та профільтрували Фільтрат концентрували Отримали 0,48гтрет-бутил(г\І-ацетату) у вигляді густого безбарвного масла Вихід 75% 50мл ЗН розчину пдрогенхлориду у етилацетаті додали до цього масла і суміш при кімнатній температурі перемішували 4 години, випарювали досуха і отриману білу тверду речовину подрібнювали у етері та сушили під вакуумом Отримали 0,47г очікуваної кислоти у вигляді дипдрохлориду Вихід 87% Т плавл 218 - 220°С (з розкладанням) Приклад 6 (Сполука 12) Гідрохлорид (2 1) 7-флуор-2-оксо-4-[2-[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-1,2-дипдро-1-хінолінацетаміда 3,9мл 0,5М розчину бромацетаміду у тетрапд 13 44332 14 рофурані по краплям додали до суміші 0,53г Юмл тетра-пдрофурану, а потім по краплям дода(1,3ммоль) 7-флуор-4-[2-[4-(тієн[3,2-с]піридин-4-ли 0,16мл (1,6ммоль) етилхлорформіату Після пеіл)-1-піперазиніл]етил]-2(1Н)-хшолону, 0,10г ремішування при -10°С протягом 45 хвилин реакційну суміш фільтрували і тверду речовину проми1,79ммоль) розмолотого гідроксиду калію та 0,13г вали 3 х 8мл тетрапдрофурану Далі до фільтрату 0,4ммоль) броміда тетрабутиламонія у 25мл тетпри 5 - 10°С додали 0,25г (6,61 ммоль) боропдриду рапдрофурану при 0 - 5°С Через ЗО хвилин темпенатрію, а потім 0,94мл метанолу Після пе ремішуратурі дали піднятися до кімнатної і продовжували вання протягом 2 годин при 5 - 10°С до суміші доперемішування 20 годин Реакційну суміш випарюдали 13мл 1Н соляної кислоти Суміш екстрагувавали під вакуумом досуха і залишок вносили до ли дихлорметаном, а потім етилацетатом ОрганіЮОмл дихлорметану, органічну фазу промивали чні фази сушили сульфатом натрію і концентруваводою, сушили сульфатом магнію та концентрували у вакуумі Отримали 0,315г продукту ли Сирий продукт подрібнювали у суміші етер/дихлорметан (1 3), тверду речовину відбирали та Вихід 92% очищали флеш-хроматографією на дюксиді СИЛІТ плавл 231 - 233°С ЦІЮ, елюючи сумішшю метанол/дихлорметан (10 7 3 4-(2-брометил)-6-ціан-1-метил-2(1Н)-хіно90), що містила сліди водного аміаку лон Отримали 0,303г білої твердої речовини, яку 0,24г (1,05ммоль) 6-ціан-4-(2-пдроксіетил)-1 перетворили у дипдрохлорид у суміші 2М соляна метил-2(1Н)-хінолону додали невеликими порціякислота/етер/метанол ми до 0,48г (1,14ммоль) дибромтрифенілфосфорану у 14мл дихлорметану при кімнатній темпераОтримали 0,32г дипдрохлориду турі Через 75 хвилин перемішування при кімнатній Вихід 75% температурі реакційну суміш виливали у 200мл диТ плавл 280°С (з розкладанням) хлорметану і промивали її водою Органічну фазу Приклад 7 (Сполука 20) сушили сульфатом натрію, фільтрували та конденГідрохлорид (2 1) 1-метил-2-оксо-4-[2-[4-(тісували під вакуумом Білий залишок розтирали у єн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-1,2-дипддіетилетері Отриману тверду речовину вносили у ро-6-хінолінкарбонггрил мінімум дихлорметану і суміш швидко фільтрували 7 1 Метил(6-ціан-1 -метил-2-оксо-1,2-дипдрокрізь шар дюксиду СИЛІЦІЮ, елюючи етером, фільт4-хінолінацетат) 1,1 мл триметилсилілціаніду рат випарювали (0,84ммоль) разом з 0,15г (0,13ммоль) тетракістрифенілфосфінпаладію додали до розчину 0,50г Отримали 0,20г продукту, який використовува(1,4ммоль) метил(6-юд-1-метил-2-оксо-1,2-дипдли без подальшої очистки Вихід 65% ро-4-хінолінацетату) (виготовленого з ІЧ-метил-4-7 4 Гідрохлорид (2 1) 1-метил-2-оксо-4-[2-[4~ юданіліну, як описано у прикладі 1) у бмл безвод(тієн[3,2-с]піридин-4-іл)-1 -піперазиніл]етил]-2(1 Н)ного тріетиламіну Реакційну суміш нагрівали зі хінолону зворотним холодильником у атмосфері азоту 4 гоСуміш 0,19г (0,65ммоль) 4-(2-брометил)-6-цідини Після охолодження до кімнатної температуан-1-метил-2(1Н)-хшолону, 0,15г (0,65ммоль) 4-(1ри суміш виливали у 60мл толуолу та 60мл води піперазиніл)тієн[3,2-с]піридину та 0,09г Органічну фазу промивали водою, сушили суль(0,11 ммоль) гідрокарбонату натрію у Юмл ацетоніфатом натрію та концентрували під вакуумом Затрилу нагрівали 36 годин при 55°С Реакційну сулишок очищали флеш-хроматографією на дюксиді міш випарювали досуха, залишок вносили до СИЛІЦІЮ, елюючи сумішшю мета нол/дихлорметан ЮОмл хлороформу і промивали органічну фазу (5 95) водою, сушили сульфатом натрію, концентрували і сирий продукт очищали флеш-хроматографією на Отримали 0,313г очікуваного нітрилу Вихід дюксиді СИЛІЦІЮ, елюючи сумішшю метанол/дихло87% рметан (10 90), що містила сліди водного аміаку Т плавл 202 - 203°С 7 2 6-ціан-4-(2-пдроксіетил)-1 -метил-2(1 Н)-хіОтримали 0,211г основи у вигляді безбарвного нолон масла 7 2 1 6-ціан-1-метил-2-оксо-1,2-дипдро-4-хіноВихід 48% ліноцтова кислота Дипдрохлорид виготовили у суміші 2М соляна кислота/етер/метанол 10,4мл 0,5Н розчину гідроксиду л т ю по краплям додали до 1,21г (4,7ммоль) метил(6-ціан-1~ Отримали 0,182г дипдрохлориду метил-2-оксо-1,2-дипдро-4-хінолінацетату) у Юмл Т плавл 200°С (з розкладанням) метанолу при 0 - 5°С Температурі дали піднятися Приклад 8 (Сполука 17) до кімнатної і реакційну суміш перемішували 2 гоГідрохлорид (2 1) 6-пдрокси-1-метил-4-[2-[4~ дини, потім вилили у 250мл льодяної води і підкис(тієн[3,2-с]піридин-4-іл)-1 -піперазиніл]етил]-2(1 Н)лили до рН 2 - З 4Н соляною кислотою Утворений хінолону білий осад відбирали, промивали водою і сушили 0,47г (1,08ммоль) 6-метокси-1-метил-4-[2-[4~ у вакуумі при 40°С (тієн[3,2-с]піридин-4-іл)-1 -піперазиніл]етил]-2(1 Н)хінолону (отриманого з метил(6-метокси-1-метилОтримали 0,85г очікуваного продукту 2-оксо-1,2-дипдро-4-хінолінацетату), як у прикладі Вихід 75% 1) додали до 25мл 48% пдрогенбромідної кислоти Т плавл 238°С і нагрівали суміш зі зворотним холодильником З 7 2 2 6-ціан-4-(2-пдроксіетил)-1 -метил-2(1 Н)години Після охолодження сірий осад відфільтрохінолон вували, промивали холодною водою і сушили під 0,22мл тріетиламіну (1,58ммоль) додали при вакуумом при 40°С У вигляді диброміду одержали 10°С до суспензії 0,365г (1,51ммоль) 6-ціан-1-ме0,444г продукту тил-2-оксо-1,2-дипдро-4-хіноліноцтовоі кислоти у 15 44332 16 Вихід 71 % дукт очищали флеш-хроматографіею на дюксиді СИЛІЦІЮ, елюючи спочатку сумішшю метанол/ети0,14г (0,24ммоль) цього продуїсгу внесли до лацетат (6,5 93,5), що містила сліди тріетиламшу, 20мл 3,7Н розчину пдрогенхлориду у безводному а потім сумішшю метанол/дихлорметан (6,5 93,5), метанолі і суміш перемішували при кімнатній темщо містила сліди водного аміаку пературі 3 години Осад відокремили, промили діетилетером та сушили у сушильній шафі 0,14г продукту отримали у вигляді основи Отримали 0,112г очікуваного продукту Вихід Вихід 26% 95% Три гідрохлорид далі виготовили у стандартних умовах Т плавл 227°С (з розкладанням) Приклад 9 (Сполука 18) Т плавл 233°С (з розкладанням) Пдрохлорид (2 1) 6-нітро-4-[2-[4-(тієн[3,2-с]піПриклад 11 (Сполука 33) ридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хшолону Гідрохлорид (2 1) 6-ацетиламшо-4-[2-[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хі9 1 4-(2-хлоретил)-6-нітро-2(1 Н)-хінолон нолону 20,0г (96,4ммоль) 4-(2-хлоретил)-2(1Н)-хшолону невеликими порціями додавали до охолодженої 111 Гідрохлорид (1 1) 6-ацетиламшо-4-(2~ до 5°С суміші 120мл 65% нггратної та 80мл концехлоретил)-2(1 Н)-хінолону 0,75мл (5,39ммоль) трінтрованої сульфатної кислоти, і суміш нагрівали етиламшу, а потім 0,35мл (4,9ммоль) ацетилхлопри 45°С 2 години Реакційну суміш виливали у риду при кімнатній температурі додали до суспен600мл льодяної води, блідожовтий осад відокремзії 1,0г (4,49ммоль) 6-амшо-4-(2-хлоретил)-2(1Н)лювали, промивали водою і сушили у вакуумі хінолону у 50мл хлороформу Суміш перемішували 16 годин, а потім розбавили 200мл хлорофорОтримали 22,5г очікуваного продукту му Суспензію промивали 1Н соляною кислотою і Вихід 92% відокремлювали осад Т плавл 239 - 237°С 9 2 Пдрохлорид (2 1) 6-нітро-4-[2-[4-(тієн[3,2Отримали 0,72г очікуваного продукту Вихід с]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хінолону 60% Суміш 1г (3,96ммоль) 4-(2-хлоретил)-6-нітро112 Гідрохлорид (2 1) 6-ацетиламіно-4-[2-[42(1Н)-хінолону, 0,87г (4ммоль) 4-(1-піперазиніл)ті(тієн[3,2-с]піридин-4-іл)-1 -піперазиніл]етил]-2(1 Н)єн[3,2-с]піридинута 0,5г (5,95ммоль) гідрокарбонахінолону ту натрію у Юмл диметилформаміду нагрівали 20 Суміш 0,35г (1,32ммоль) пдрохлориду 6-ацегодин при 50°С Залишок далі відфільтровували і тиламшо-4-(2-хлоретил)-2(1Н)-хінолону, 0,38г промивали водою, 200мл води додавали до фільт(1,75ммоль) 4-(1-піперазиніл)тієн[3,2-с]піридину та рату і утворений осад відокремлювали і сушили у 0,17г (2ммоль) гідрокарбонату натрію у Юмл дивакуумі метилформаміду нагрівали 24 години при 60°С Після охолодження до кімнатної температури реак22,5г очікуваного продукту отримали у вигляді ційну суміш розбавляли ЮОмл води і залишали на основи ніч при 5°С Утворений продукт відокремлювали і Вихід 74% сушили у вакуумі Сирий продукт очищали флешГідрохлорид виготовили у суміші етер/метахроматографією на дюксиді СИЛІЦІЮ, елюючи спонол/соляна кислота чатку сумішшю метанол/етилацетат (5 95), а поТ плавл 242°С (з розкладанням) тім сумішшю метанол/дихлорметан (10 90), що Приклад 10 ( Сполука 16) містила сліди водного аміаку Гідрохлорид (3 1) 6-аміно-4-[2-[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хшолону 0,20г продукту отримали у вигляді основи 10 1 Гідрохлорид (1 1) 6-аміно-4-(2-хлореВихід 34% тил)-2(1 Н)-хінолону Дипдрохлорид далі виготовили у стандартних умовах 0,70г паладію на активованому вугіллі (5% Pd) додали при кімнатній температурі до суспензії 3,5г Т плавл 225°С (з розкладанням) (13,8ммоль) 4-(2-хлоретил)-6-нітро-2(1Н)-хінолону Пояснення до таблиці у ЗООмл метанолу, суміш перемішували при тиску - у колонці "Сіль" НСІ означає гідрохлорид, водню О.ОбМПа 3 години Відфільтровували катаспіввідношення (х у) відповідає співвідношенню лізатор і фільтрат конденсували (кислота основа), відсутність позначки означає речовину у вигляді основи 2,97г продукту отримали у вигляді основи Гідрохлорид виготовили у суміші етер/метау колонці "Т плавл " d означає плавління з нол/соляна кислота розкладанням Т плавл > 242°С 10 2 Гідрохлорид (3 1) 6-амшо-4-[2-[4-(тієн[3,2-с]піридин-4-іл)-1-піперазиніл]етил]-2(1Н)-хінолону Суміш 0,35г (1,35ммоль) пдрохлориду 6-аміно(•Є) 4-(2-хлоретил)-2(1Н)-хшолону, 0,33г (1,5ммоль) 4(1-піперазиніл)тієн[3,2-с]піридину та 0,17г > (5,95ммоль) гідро карбонату натрію у Юмл димеа І) тилформаміду нагрівали 24 години при 60°С Після охолодження до кімнатної температури реакційну суміш розбавляли 50мл води і сирий продукт (! SI екстрагували хлороформом Органічну фазу суши.. ли сульфатом натрію і концентрували Сирий про 44332 18 Сполуки згідно з винаходом піддавали фармацевтичному дослідженню, що показало їх властивості антагоністів серотоніну та їх ЦІННІСТЬ як терапевтичне активних Так, сполуки згідно з винаходом тестували на здатність інгібувати вазопресорну дію серотоніну Використовували самців пацюків (Sprague-Dawley, Charles River France) масою 250 - 300г, їх анестезували пентабарбіталом натрію (бОмг/кг, внутрішньочеревним шляхом) та підтримували штучним диханням (респіратор Harvard™ - рівень респірації 70мл/хв , об'єм повітря - 1мл на 100г маси тіла) Тварин забивали металевим прутом, уведеним в орбіту правого ока вздовж спинного хребта Правий та лівий блукаючі нерви секцюнували (біваготомія), а праву сонну артерію перев'язували, ліву сонну артерію катетерізували з метою вимірювання артеріального кров'яного тиску, використовуючи камеру для вимірювання тиску (типу Statham™ P23Db) Стегнову вену катетерізували з метою введення різних сполук Підвищення кров'яного тиску, індуковане внутрішньовенним введенням се 20 19 44332 ротоншу у дозі 30мг/кг, вимірювали Сполуки згідно зок пацюків і його кору відокремлювали та гомогез винаходом або розбавитель для ЛІКІВ ВВОДИЛИ за нізували при 0°С у 20 об'ємах суміші, що містить 5 хвилин (для вивчення при внутрішньовенному на л 50ммоль буферу Трис-НСІ з рН 7,4, введенні) або за 75 хвилин (для вивчення при ора120ммоль NaCI та 5ммоль КСІ Гомогенну суміш льному введенні) до введення серотоніну Сполуцентрифугували при 40000д 10 хвилин, а потім пеки згідно з винаходом застосовували у дозах в мелету ДВІЧІ видаляли, промивали суспендуванням у жах 0,001 - 10мг/кг Процент інгібування відносно такому ж буфері, знов гомогенізували і центрифусеротоніну використовували для оцінки потенційгували Наприкінці, фінальну пелету розбавляли ної активності сполук згідно з винаходом як антаготим же буфером у пропорції 500мг вологої тканини ністів серотоніну на Юмл буферу Тканину далі піддавали попередньому інкубуванню протягом 10 хвилин при 37°С Сполуки згідно з винаходом також тестували в у присутності 10имоль/л парпліну, а після цього шрамках моделі звужування судин суматриптаном кубували протягом 10 хвилин при 37°С у присутнона ізольованих підшкірних венах собак (антагоніссті [зН]спіроперидолу (активність 19Кі/имоль) з тична активність на рецепторах типу 5-НТ-і, згідно концентрацією О.ЗнМ та досліджуваної сполуки з з Humpherey et al , Br J Pharmacol 1988 94 1123) концентрацією у межах 0,0001 -100иМ Підшкірні вени гончаків або собак AngloPoitevm видаляли під анестезією внутрішньовенною ІН'ЄКЦІЄЮ пента барбітал у Судину розрізали на спіралі шириною 0,4см, а потім ділили на сегменти довжиною 0,5см Кожний фрагмент, уставлений між двома затискачами, розміщали у ізольованій камері для органу, яка вміщувала 20мл фізіологічного розчину Кребса такого складу (у мМ) NaCI 118, RCI4.7, MgCI 2 1,2, CaCI2 2,6, NaHCO3 25, глюкоза 11,1, аскорбінова кислота 0,11 Орган, витриманий при 37°С у струмі карбогену (95%02/5%С02) при рН 7,4, було поєднано з ізометричним вимірювальним приладом типу 351 Hugo Sachs з базовим навантаженням 2г і зв'язано з поліграфом Gould 2400S, здатним визначати зміну напруження Отримані дані обробляли за допомогою мікрокомп'ютера Через 90 хвилин спокою, що різноманггили частими промиваннями, під час яких змінювали базове навантаження, орган стимулювали 3 иМ норадреналіном з метою контролю Його життєздатності Потім будували графік залежності скорочення під дією суматриптану від його концентрації у межах ЮнМ - ЮиМ При досягненні максимального скорочення (плато при дії двох послідовних концентрацій суматриптану), препарат рясно промивали, чергуючи періоди відпочинку для того, щоб дати органу змогу повернутися до початкового напруження Сполуку при ДОСЛІДІ далі додавали у камеру для органу за 15 хвилин до початку побудови другого графіку залежності скорочення під дією суматриптану Скорочення, отримане у присутності сполуки виражали як процент від максимального скорочення, яке було визначено за першим графіком Графіки аналізували нелінійною регресією, щоб визначити Енакс (максимальна ВІДПОВІДЬ) та ЕК50 (концентрація, що визиває 50% максимальної ВІДПОВІДІ) Антагоністичну здатність сполуки оцінювали розрахунком константи дисоціації Кв згідно з рівнянням Kg = [молярна концентрація сполуки] / (CR-1), де CR - відношення величини ЕКбо суматриптану у присутності та відсутності сполуки Результат виражали як рА2 = -logKg Величина рА2 сполук згідно з винаходом більша за 6 Відбирали 1-мл аліквоти, відфільтровували їх під вакуумом, промивали ДВІЧІ 5МЛ ХОЛОДНОГО бу феру, сушили і вимірювали радіоактивність Для визначення активності сполук будували графік залежності процента інгібування специфічного зв'язування [зН]спіроперидолу від концентрації витісняючої речовини ІК50 - концентрацію, при якій інгібується 50% специфічного зв'язування, визначали графічно Специфічне зв'язування визначали як зв'язування витіснення 100uM 5-HT ІК50 сполук згідно з винаходом менше за 1 иМ Результати цих випробувань показують, що сполуки згідно з винаходом виявляють антагоністичні властивості по відношенню до серотоніна На основі цього їх можна використовувати при лікуванні та попередженні таких різних форм патолопй з участю серотоніну, як артеріальні, венозні, легеневі, воротньовенозні, ниркові, очні або церебральні ішемії або ішемія нижніх КІНЦІВОК, серцева недостатність, інфаркт міокарду, стенокардія, коронарний чи периферійний вазоспазм, тромбоз (сполуки самі по собі або як синерпсти при тромболізі) артеріїт, синдром Шарко, рестеноз після анпопластики та різноманггні патологічні стани, споріднені з атеросклерозом, розлади мікрокровообігу та легенева дисфункція, їх також можна використовувати поодинці або у сполученні з іншими речовинами при операціях імплантації судин Сполуки згідно з винаходом можна використовувати у сполученні з такими іншими речовинами, що мають серцевосудинну та серцеволегеневу активність, як ан-титромботичні, тромболітичні, рблокуючі, антагоністи кальцію, антагоністи тромбоксанута інгібітори тромбоксан-синтетази Для цієї мети ці сполуки можуть бути у всіх формах, придатних для орального чи парентерального застосування таблетках, драже, капсули, включаючи тверді желатинові, та очні рецептури для місцевого застосування, у сполученні з придатними розріджувачами Дози, присутні у цих формах, такі, щоб забезпечити прийом від 0,1 мг до 1г один чи кілька разів на добу їх також можна застосовувати в усіх формах, придатних для трансдермального застосування Сполуки згідно з винаходом також тестували на інгібування зв'язку [зН]спіроперидолу з серотонінерпчними рецепторами кори головного мозку пацюків типу 5-НТ2 Для цього тесту видаляли мо ДП "Український інститут промислової власності "(Укрпатент) Україна, 04119, Киів-119, вул сім'ї Хохлових, 15 (044) 456-20-90 10
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61P 7/02, A61K 31/435, C07D 495/04, A61K 31/4353, A61K 31/00, A61K 31/4365, A61P 7/00, A61P 9/00, A61K 31/47, C07D 401/06, A61K 31/4709, A61P 43/00
Мітки: одержання, препарат, композиція, фармацевтична, антагоністи, медичний, спосіб, серотоніну, похідні, основі, 2(1н)-хінолінону, варіанти
Код посилання
<a href="https://ua.patents.su/10-44332-pokhidni-21n-khinolinonu-yak-antagonisti-serotoninu-sposib-kh-oderzhannya-varianti-medichnijj-preparat-ta-farmacevtichna-kompoziciya-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні 2(1н)-хінолінону як антагоністи серотоніну, спосіб їх одержання (варіанти), медичний препарат та фармацевтична композиція на їх основі</a>
Похідні метилпіперазиназепіну, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі

Номер патенту: 40569
Опубліковано: 15.08.2001
Автори: Льєжуа Жан-Франсуа Фернан, Деларж Жак Елі
МПК: C07D 498/04, A61P 25/02, C07D 513/04, C07D 517/00, C07D 471/04, A61K 31/496, A61K 31/4353
Мітки: похідні, варіанти, метилпіперазиназепіну, спосіб, композиція, основі, одержання, фармацевтична
Формула / Реферат:
1. Производные метилпиперазиназепина формулы (I):в которой X означает атом кислорода, серы или селена, или группу ΝΗ или NR3, где R3 является группой -СОН, -СОСF3 или алкилом с 1-4 атомами углерода, разветвленным или неразветвленным, R1 - атом водорода, атом галогена или алкил с 1-4 атомами углерода, разветвленный или неразветвленный, R2- атом водорода, атом галогена или алкил с 1-4 атомами углерода, разветвленный или...
Похідні 2-ціано-3-гідроксипропенаміду, спосіб їх одержання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 40573
Опубліковано: 15.08.2001
Автор: Кюо Елізабет Анн
МПК: A61K 31/44, A61P 35/00, A61P 29/00, A61K 31/4406, C07D 213/75, A61K 31/4402, C07D 213/84, A61K 31/4409, A61K 31/4418
Мітки: варіанти, композиція, 2-ціано-3-гідроксипропенаміду, основі, фармацевтична, спосіб, одержання, похідні
Формула / Реферат:
1. Производные 2-циано-З-гидроксипропенамида, отвечающие общей формуле (I):в которой:А, В и Ε каждый представляет собой группу =СН- или атом азота, причем следует иметь в виду, что как минимум один из А, В или Ε является атомом азота,R представляет собой циклоалкильный радикал, включающий от 3 до 6 атомов углерода, алкиловый радикал, включающий от 2 до 6 атомов углерода, алкинильный радикал, включающий от...
Похідні n-алкіленпіперидинів як антагоністи рецептора нейрокініну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 26894
Опубліковано: 29.12.1999
Автори: Мартінез Серж, Емонд-Альт Ксавьє, Пруаєтто Венсанзо, ван Броєкк Дідьє
МПК: C07D 211/54, C07D 401/12, C07D 211/58, A61K 31/4427, C07D 211/46, C07D 409/12, A61K 31/4465, A61K 31/4433, A61K 31/4468, A61K 31/44, A61K 31/4545, A61P 43/00, C07D 401/06, A61K 31/445, A61K 31/454, C07D 403/12
Мітки: похідні, рецептора, антагоністи, композиція, нейрокініну, проміжні, сполуки, спосіб, одержання, n-алкіленпіперидинів, фармацевтична
Текст:
...где Alk является (С,-Сэ)алкильной группой, R представляет собой атом водорода 40 или С^-С^алкил, Z представляет собой фенил, незамещенный или моно- или дизамещенный галогеном или (С^-Сзіалкоксигруппой, наф 26894 7 тил, замещенный галогеном, фенилметильной группой, замещенной на фениле (С,С4}алкоксигруппой, * обозначает хиральный центр, в виде рацемата или оптически чистых изомеров, или их соли с минеральными или органическими кислотами,...
Похідні оксазолідин-2-ону, спосіб їх одержання і фармацевтична композиція на їх основі.

Номер патенту: 43848
Опубліковано: 15.01.2002
Автори: Раддатц Петер, Вурцігер Ханнс, Бернотат-Данієловскі Сабіне, Юращік Хорст, Мельцер Гвідо, Ганте Йоахім
МПК: A61K 31/4166, C07D 233/32, A61K 31/495, C07D 413/06, A61P 9/10, A61P 9/08, A61P 19/10, A61P 7/02, A61P 29/00, C07D 413/14, A61K 31/445, A61K 31/496, C07D 413/10, C07D 277/14, A61P 15/00, A61P 13/02, C07D 263/20, A61K 31/415
Мітки: похідні, спосіб, оксазолідин-2-ону, основі, фармацевтична, композиція, одержання
Формула / Реферат:
1. Производные оксазолидин-2-она формулы I:где Х обозначает О,Y обозначаетилиR1 обозначаетилиR2 и R3 каждый, независимо друг от друга, обозначает Н, А или бензил, А обозначает алкил с 1-6 С-атомами,D обозначает амидиногруппу, аминометил, аминогидроксиминометил, 5-метил-1,2,4-оксадиазолидин-3-ил или гуанидинометил,r и s, независимо друг от друга, обозначают...
Похідні еритроміцину, спосіб їх одержання та фармацевтична композиція на їх основі
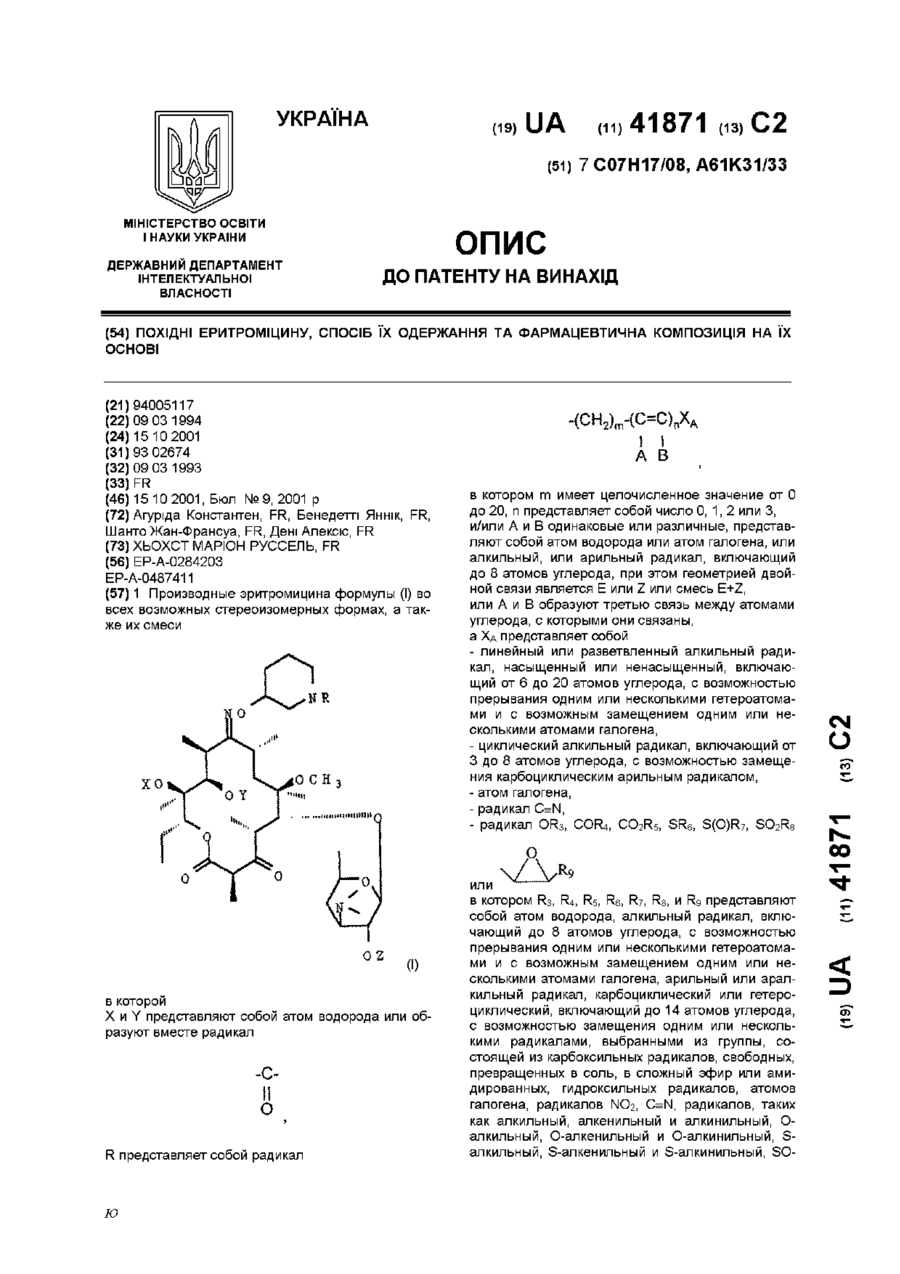
Номер патенту: 41871
Опубліковано: 15.10.2001
Автори: Бенедетті Яннік, Дені Алексіс, Агуріда Константен, Шанто Жан-Франсуа
МПК: A61K 31/443, A61K 31/4523, C07D 405/14, C07H 17/08, A61K 31/445, A61P 31/04
Мітки: одержання, фармацевтична, спосіб, основі, похідні, еритроміцину, композиція
Формула / Реферат:
1. Производные эритромицина формулы (I) во всех возможных стереоизомерных формах, а также их смесив которой:Х и Y представляют собой атом водорода или образуют вместе радикал:R представляет собой радикал:в котором m имеет целочисленное значение от 0 до 20, n представляет собой число 0, 1, 2 или 3,и/или А и В одинаковые или различные, представляют собой атом водорода или атом галогена,...
Попередній патент: Похідні 2-(4-заміщеного)бензиламіно-2-метилпропанаміду, спосіб їх одержання та фармацевтична композиція
Наступний патент: Спосіб пасивно-активного корегування хребта яриги м.п.
Випадковий патент: Лікарська форма протипухлинного препарату та спосіб її одержання