Спосіб одержання 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону

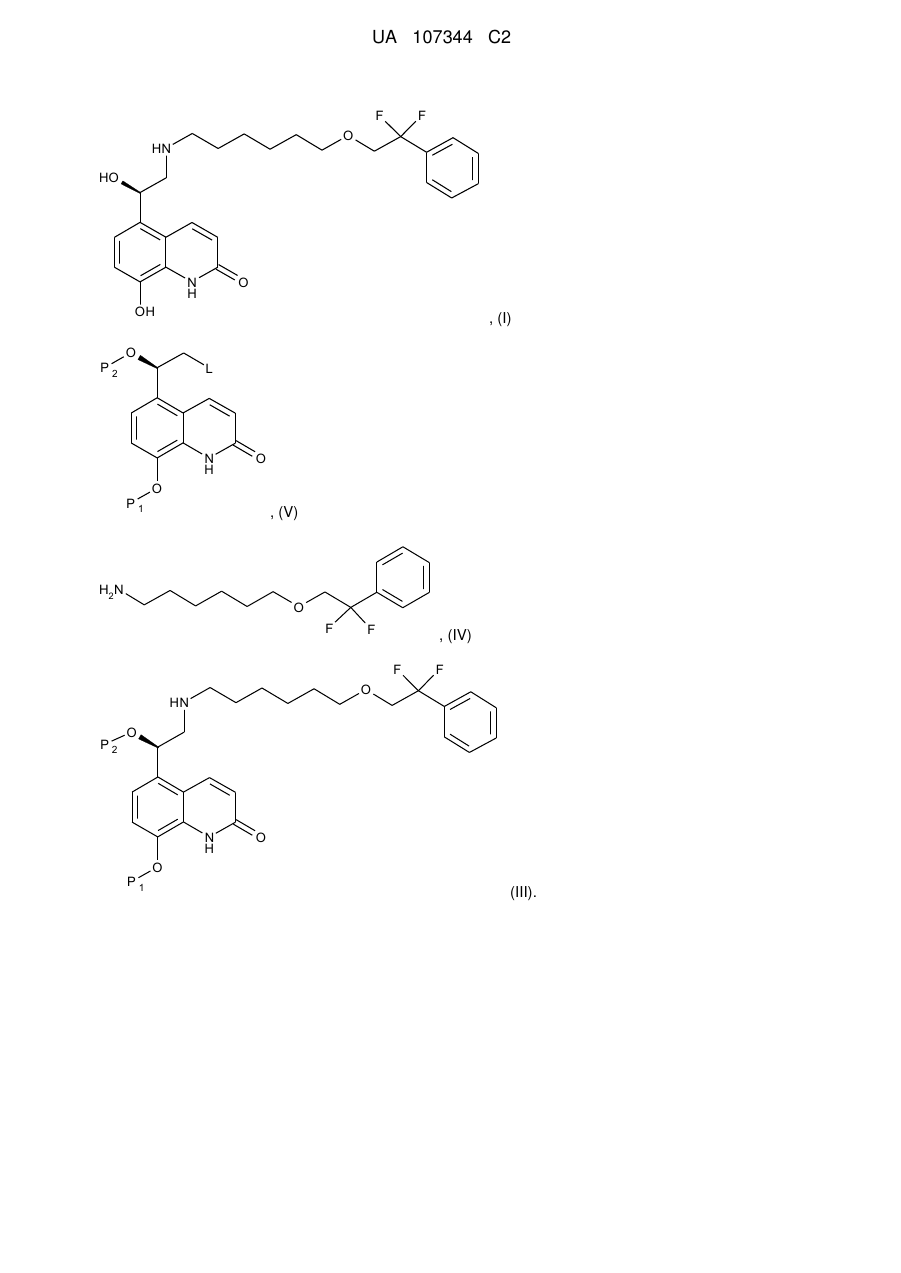











Формула / Реферат
1. Спосіб одержання сполуки 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону формули (І) або її фармацевтично прийнятної солі:
 , (I)
, (I)
який полягає в тому, що:
a) вводять в реакцію у ксилольному розчиннику сполуку формули (V):
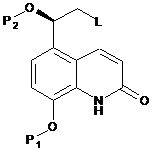 , (V)
, (V)
де P1 і Р2 являють собою захисні групи для гідроксигруп, і L являє собою відхідну групу,
з 6-(2,2-дифтор-2-фенілетокси)гексан-1-аміном формули (IV):
 (IV)
(IV)
з одержанням сполуки формули (III):
 (III);
(III);
b) здійснюють стадію зняття захисної групи Р1 і стадію зняття захисної групи Р2 для видалення захисних груп P1 і Р2 і одержання сполуки формули (І).
2. Спосіб за п. 1, у якому:
(і) стадію зняття захисної групи Р2 здійснюють при температурі у діапазоні близько 30-60 C протягом до 8 годин, і/або
(іі) стадію зняття захисної групи Р1 здійснюють у присутності розчинника, що являє собою оцтову кислоту або суміш оцтової кислоти зі спиртом або зі складним ефіром.
3. Спосіб за п. 1 або п. 2, у якому на стадії (b):
здійснюють зазначену стадію зняття захисної групи Р2 у сполуці формули (III) з одержанням сполуки формули (II):
 , (II)
, (II)
у якій Р1є таким, як визначено вище; і
здійснюють зазначену стадію зняття захисної групи P1 у сполуці формули (II) з одержанням сполуки формули (І):
 (I).
(I).
4. Спосіб за будь-яким із попередніх пунктів, у якому (а) Р1 являє собою бензильну групу, і стадію зняття захисної групи Р1 здійснюють гідруванням, і/або (b) Р2 являє собою тpет-бутилдиметилсилільну групу, і стадію зняття захисної групи Р2 здійснюють реакцією з тригідратом фториду тетра-н-бутиламонію або з хлоридом водню.
5. Споcіб за будь-яким із попередніх пунктів, де L являє собою бром.
6. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р2 здійснюють за допомогою тригідрату фториду тетра-н-бутиламонію у тетрагідрофурані.
7. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р2 здійснюють при температурі 40-50 C протягом періоду часу, що не перевищує 6 годин.
8. Спосіб за п. 7, у якому період часу не перевищує 4 годин.
9. Спосіб за будь-яким із попередніх пунктів, в якому одержують гемінападисилатну або мезилатну сіль сполуки формули (І).
10. Спосіб за п. 9 для одержання гемінападисилатної солі сполуки формули (І), у якому після стадії (b) додають нафтален-1,5-дисульфонову кислоту без виділення 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1Н)-ону формули (І).
11. Спосіб за п. 3, у якому проміжну сполуку формули (II), отриману зі стадії зняття захисної групи Р2, очищають кристалізацією з тетрагідратом 1,5-нафталендисульфонової кислоти в етанолі.
12. Спосіб за будь-яким із попередніх пунктів, у якому Р1 являє собою бензил, і стадію зняття захисної групи Р1 здійснюють гідруванням у присутності каталізатора паладію на вугіллі у кількості менше 10% (мас.) щодо кількості сполуки формули (II).
13. Спосіб за п. 12, у якому використовують кількість каталізатора, що становить менше 5%.
14. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р1 здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або зі складним ефіром, переважно розчинником є оцтова кислота або суміш метанол/оцтова кислота (1:1).
15. Спосіб за п. 14, у якому як розчинник використовують суміш метанол/оцтова кислота (1:1).
16. Спосіб за будь-яким із попередніх пунктів, у якому
(і) стадію зняття захисної групи Р2 здійснюють при температурі у діапазоні 30-60°C протягом періоду до 8 годин, і
(іі) стадію зняття захисної групи Р1 здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або зі складним ефіром.
Текст
Реферат: Спосіб одержання сполуки 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону формули (І) або її фармацевтично прийнятної солі, що включає взаємодію сполуки формули (V) зі сполукою формули (IV) у ксилольному розчиннику, з одержанням сполуки формули (III), і наступне здійснення стадії зняття захисної групи Р1 і стадії зняття захисної групи Р2. UA 107344 C2 (12) UA 107344 C2 F F O HN HO O N H OH P2 , (I) O L N H P1 O O , (V) H2N O F F , (IV) F HN P2 F O O N H P1 O O (III). UA 107344 C2 5 10 15 Даний винахід відноситься до поліпшеного способу одержання 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону та її фармацевтично прийнятних солей. 5-(2-{[6-(2,2-Дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-он (сполука (I)), а також спосіб її одержання, описаний у міжнародній заявці на патент WO2006122788A1. Нападисилатна сіль 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону, а також спосіб її одержання, описаний у міжнародній заявці на патент WO2008095720A1. Винахідники зненацька виявили, що можливо (a) підвищити вихід сполуки (I) та її солі, (b) мінімізувати кількість домішок у кінцевому продукті й/або (c) знизити час реакції, шляхом модифікації синтетичних методик, описаних у WO2006122788A1 і WO2008095720A1. Ці мети можуть бути досягнуті шляхом підбору певних розчинників і/або зміни або навіть виключення деяких стадій очищення, у такий спосіб знижуючи час реакції, збільшуючи загальний вихід кінцевого продукту. Крім того, спосіб згідно з даним винаходом є більше придатним для великомасштабного одержання. У WO2006122788A1 описаний тристадійний спосіб одержання 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону: Стадія a) 20 Стадія b) 25 Стадія c) У WO2008095720A1 описаний спосіб одержання нападисилатної солі сполуки формули (Ia): Стадія d) 30 35 * e.e.: енантіомерний надлишок Як наслідок, для одержання нападисилатної солі сполуки формули (Ia) з проміжних сполук (V) і (IV), відповідно до методів, описаних у попередньому рівні техніки, повинні бути виконані чотири стадії реакції, на якій кожна проміжна сполука, при її одержанні, була виділена й, очищена перед її використанням як вихідний матеріал на наступній стадії. Стадії очищення у методиках попереднього рівня техніки здійснювали, використовуючи звичайні методи очищення, відомі з рівня техніки, такі як, наприклад екстракція розчинником або 1 UA 107344 C2 5 10 хроматографічні методики. Розрахований загальний вихід одержання нападисилатної сполуки (Ia) становив близько 9,5 %, у той час як рівень домішок, визначених аналізом ВЕРХ, становив близько 5-6 %. Зненацька було виявлено, що спосіб одержання 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону та її солей може бути значно поліпшений шляхом модифікації умов реакції, особливо модифікацією або навіть виключенням процесів очищення на деяких стадіях, у такий спосіб спрощуючи численні стадії реакції, у той же час підвищуючи загальний вихід реакцій. Крім того, було виявлено, що залежно від вибору розчинників, необхідний продукт може бути отриманий з більше високим виходом і у більше чистій формі у порівнянні з відомим раніше способом. Відповідно, даний винахід відноситься до способу одержання сполуки 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону формули (I) або її фармацевтично прийнятної солі: F F O HN HO (I) N OH H 15 O який включає: а) реакцію у ксилольному розчиннику сполуки формули (V): P2 O P1 L N O O H (V) де P1 і P2 являють собою захисні групи для гідроксигрупи, і L являє собою відхідну групу, з 6(2,2-дифтор-2-фенілетокси)гексан-1-аміном формули (IV): H2N 20 O F F (IV) з одержанням сполуки формули (III): P2 O P1 O HN F F N O O H (III) 25 30 35 b) здійснення стадії зняття захисної групи P1 і стадії зняття захисної групи P2, для видалення захисних груп P1 і P2 і одержання сполуки формули (I). Звичайно, (i) стадію зняття захисної групи P2 здійснюють при температурі у діапазоні 30-60ºC протягом періоду до 8 годин, і/або (ii) стадію зняття захисної групи P1 здійснюють у присутності розчинника, яким є оцтова кислота, або суміш оцтової кислоти зі спиртом або складним ефіром. Стадію (a) проводять у ксилольному розчиннику. Навпаки, відповідну стадію реакції, описану у WO2006/122788, проводять у ДМСО. Несподіваним відкриттям даного винаходу є використання конкретного ксилольного розчинника, здатного істотно поліпшити чистоту сполуки формули (III). У кращому варіанті здійснення описана вище стадія (b) включає: - здійснення описаної вище стадії зняття захисної групи P2 у сполуці формули (III) з одержанням сполуки формули (II), 2 UA 107344 C2 HO P1 F F O HN N O O H (II) де P1 є таким, як визначено вище; і - здійснення описаної вище стадії зняття захисної групи P1 у сполуці формули (II) з одержанням сполуки формули (I): F F O HN HO 5 N OH H O (I) . Таким чином, у цьому варіанті здійснення, даний винахід відноситься до способу одержання сполуки 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)ону формули (I) або її фармацевтично прийнятної солі: F F O HN HO (I) N OH H 10 O який включає: а) реакцію у ксилольному розчиннику сполуки формули (V): P2 O P1 L N O O H (V) де P1 і P2 являють собою захисні групи для гідроксигрупи, і L являє собою відхідну групу, з 6(2,2-дифтор-2-фенілетокси)гексан-1-аміном формули (IV): H2N 15 O F F (IV) з одержанням сполуки формули (III): P2 O P1 O HN F F N O O H (III) b) зняття захисної групи зі сполуки формули (III) з одержанням сполуки формули (II): HO P1 20 O HN F F N O O H (II) де P1 є таким, як визначено вище; і с) зняття захисної групи зі сполуки формули (II) з одержанням сполуки формули (I). 3 UA 107344 C2 F F O HN HO N OH H 5 O (I) Звичайно, у даному варіанті здійснення: (i) стадію b) здійснюють при температурі у діапазоні 30-60ºC протягом періоду до 8 годин, і/або (ii) стадію c) здійснюють у присутності розчинника, яким є оцтова кислота, або суміш оцтової кислоти зі спиртом або складним ефіром. В іншому варіанті здійснення, спосіб згідно з даним винаходом включає: a) реакцію у ксилольному розчиннику сполуки формули (V): P2 O P1 L N O O H (V) 10 де P1 і P2 являють собою захисні групи для гідроксигрупи, і L являє собою відхідну групу, з 6(2,2-дифтор-2-фенілетокси)гексан-1-аміном формули (IV): H2N O F F (IV) з одержанням сполуки формули (III): P2 O P1 O HN F F N O O H (III) 15 b) зняття захисної групи зі сполуки формули (III) з одержанням сполуки формули (II'): O P2 O HN F F N O OH H (II') де P2 є таким, як визначено вище; і c) зняття захисної групи зі сполуки формули (II') з одержанням сполуки формули (I). F F O HN HO N OH H 20 25 O (I) Звичайно, у даному варіанті здійснення: (i) стадію b) здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або складним ефіром, і/або (ii) стадію c) здійснюють при температурі у діапазоні 30-60ºC протягом періоду до 8 годин. P1 і P2 являють собою захисні групи для гідроксигрупи. P1 і P2 можуть бути однаковими або різними. Переважно, вони є різними. Фахівець у даній галузі техніки легко підбере придатні захисні групи для гідроксигруп для положень P1 і P2. Наприклад, придатні захисні групи описані у книзі T. W. Greene і G. M. Wuts, Protecting Groups in Organic Synthesis, 3-е видання, Wiley, New York, 1999, і наведених у ній посиланнях. Приклади придатних захисних груп для гідроксигруп включають алкільні групи, такі як метил, 4 UA 107344 C2 5 10 15 20 25 30 35 40 45 50 55 60 етил і трет-бутил; ацильні групи, наприклад алканоїльні групи, такі як ацетил; арилметильні групи, такі як бензил (Bn), п-метоксибензил (PMB), 9-флуоренілметил (Fm) і дифенілметил (бензгідрил, DPM); силільні групи, такі як триметилсиліл (TMS) і трет-бутилдиметилсиліл (TBS); та їм подібні. Звичайно P1 являє собою бензильну групу. У даному варіанті здійснення, стадію зняття захисної групи P1 звичайно здійснюють гідруванням, переважно у присутності каталізатора, такого як гідроксид паладію (II) (Pd(OH)2) або паладій (0) (Pd(0)). Переважно, каталізатором є паладій (0) на вугіллі. Звичайно, у даному варіанті здійснення, реакцію гідрування стадії зняття захисної групи P 1 здійснюють у присутності каталізатора у кількості менше 10 %, переважно менше 5 %, найбільше переважно близько 4 % за масою від кількості використовуваного реагенту. Використання каталізатора у цих кількостях звичайно дозволяє знизити рівень домішок, що утворюються. Зокрема, можна знизити утворення дефторованих домішок, тобто 5-(2-{[6-(2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону. Також можна знизити утворення дигідрохінолінових домішок. Звичайно, P2 являє собою трет-бутилдиметилсилільну групу. У цьому варіанті здійснення стадію зняття захисної групи P2, як правило, здійснюють реакцією з тригідратом тетра-нбутилфториду амонію (TBAF), переважно у розчиннику, такому як тетрагідрофуран (ТГФ), або з хлоридом водню у розчиннику, вибраному з простих ефірів, складних ефірів і спиртів. Переважно, у цьому варіанті здійснення даного винаходу, стадію зняття захисної групи P2 здійснюють з хлоридом водню у розчиннику, вибраному з діетилового ефіру, третбутилметилового ефіру (TBME), етанолу та ізопропілацетату. Альтернативно, у цьому варіанті здійснення даного винаходу, стадію зняття захисної групи P2 переважно здійснюють за допомогою TBAF у тетрагідрофурані (ТГФ) або 2метилтетрагідрофурані, переважно у ТГФ. Альтернативно, у цьому варіанті здійснення даного винаходу, стадію зняття захисної групи P2 переважно здійснюють за допомогою нафтален-1,5-дисульфонової кислоти у тетрагідрофурані (ТГФ). L являє собою відхідну групу.Фахівець-хімік легко здатний підібрати придатні відхідні групи для L-положення. Приклади придатних відхідних груп включають атоми галогену, мезилатні групи (-O-S(O)2-CH3) і трифлатні (-OS(O)2-CF3) групи. Переважно, L являє собою атом галогену. Більше переважно, L являє собою атом брому. Звичайно, розчинник, який використовують на стадії (a), по суті не містить ДМСО. Більше переважно, він по суті не містить ДМСО і діоксан. Використання ксилольного розчинника, докладно описане вище на стадії (a), здатне у цілому поліпшити чистоту й/або вихід, у порівнянні з аналогічними процесами, в яких стадію (a) проводять у розчинниках, таких як ДМСО. В іншому варіанті здійснення даного винаходу, стадію зняття захисної групи P2 здійснюють при температурі у межах від 40-50ºC протягом періоду часу, що не перевищує 6 годин, переважно не довше 4 годин, більше переважно не довше 2 годин, найбільше переважно до однієї години. Скорочення часу реакції для стадії зняття захисної групи P2 дозволяє зненацька знизити утворення небажаних побічних продуктів. В іншому варіанті здійснення даного винаходу, гідрування на стадії зняття захисної групи P 2 необов'язково здійснюють у присутності фториду тетрабутиламонію у кількості близько 0,3-0,9 г TBAF на грам реагенту. Звичайно, реагентом є сполука формули (II). В іншому варіанті здійснення даного винаходу сполуку, отриману на стадії зняття захисної групи P2, очищають кристалізацією. Звичайно, кристалізацію здійснюють за допомогою 1,5нафталендисульфонової кислоти у спирті, переважно етанолі. Очищення сполуки, отриманої на стадії зняття захисної групи P2, перекристалізацією, на відміну від хроматографії, дозволяє підвищити чистоту й/або вихід. Переважно, у цьому варіанті здійснення даного винаходу, стадію зняття захисної групи P2 здійснюють перед стадією зняття захисної групи P1, і сполука, отримана на стадії зняття захисної групи P2, тому являє собою сполуку формули (II). У кращому варіанті здійснення даного винаходу, стадію зняття захисної групи P1 здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або зі складним ефіром. Переважно, у цьому варіанті здійснення, розчинник являє собою оцтову кислоту окремо або суміш оцтової кислоти/метанолу (1:1), більше переважно суміш оцтової кислоти/метанолу (1:1). Звичайно, зазначений розчинник містить менше 5 % (об/об), переважно менше 3 %, більше переважно менше 1 % будь-яких рідин, відмінних від оцтової кислоти, спирту й складного ефіру, переважно будь-яких рідин, відмінних від оцтової кислоти й метанолу. 5 UA 107344 C2 5 10 15 20 25 30 35 40 45 50 55 60 У кращому варіанті здійснення винаходу, одержують фармацевтично прийнятну сіль сполуки формули (I). Переважно зазначена сіль являє собою нападисилатну сіль або мезилатну сіль. Нападисилатні солі звичайно є тими, які описані у WO2008/095720. Переважно нападисилатна сіль являє собою гемінападисилатну сіль або мононападисилатну сіль. Мононападисилатна сіль звичайно містить від близько 0,8 і 1,2 молярних еквівалентів нафтален-1,5-дисульфонової кислоти на молярний еквівалент вільної основи, звичайно близько 1,0 молярного еквівалента нафтален-1,5-дисульфонової кислоти на молярний еквівалент вільної основи. Гемінападисилатна сіль звичайно містить від близько 0,35 до 0,65 молярних еквівалентів нафтален-1,5-дисульфонової кислоти на молярний еквівалент вільної основи, звичайно близько 0,5 молярних еквівалента нафтален-1,5-дисульфонової кислоти на молярний еквівалент вільної основи. Даний винахід також відноситься до 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил] аміно}-1гідроксіетил)-8-гідроксихінолін-2(1H)-ону або її фармацевтично прийнятної солі, отриманої способом за даним винаходом. Переважно, даний винахід відноситься до нападисилатної солі або мезилатної солі 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил] аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону, отриманої способом за даним винаходом. Більше переважно, сіль являє собою нападисилатну сіль. Описані вище молярні співвідношення можуть визначатися стандартними методиками, 1 наприклад H ЯМР, елементним аналізом і методами ВЕРХ. Коли одержують нападисилатну сіль сполуки формули (I), звичайно після стадії (b) додають нафтален-1,5-дисульфонову кислоту без виділення 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону формули (I). Одержуючи кінцевий продукт одностадійної реакції цим способом, без виділення вільної основи, можна підвищити чистоту й/або вихід. Крім того, така одностадійна реакція також краща, оскільки здатна підвищити ефективність способу. У кращому варіанті здійснення даного винаходу, стадію (b), і при необхідності наступну стадію солеутворення, кожну, проводять без очищення проміжної сполуки, отриманої на попередній стадії реакції. Сполуки формули (V) можуть бути отримані відомими способами або аналогічно відомим способам. Наприклад, сполука, в якій P1 являє собою бензил і P2 являє собою TBS, може бути отримана методиками синтезу, описаними у документах US2004059116 (приклад 9C), WO2004/011416 (приклад 2) і WO2004/016578 (приклад 1ii). 6-(2,2-Дифтор-2-фенілетокси)гексан-1-амін (IV) одержують за методиками синтезу, описаними у WO2006122788A1 (проміжна сполука 9). Реагенти й розчинники, які використовують у даному винаході, є комерційно доступними, наприклад від Aldrich Chemical Company, Inc. або Fluka Chemie GmbH. Кращими умовами способу стадії (a) є наступні: До розчину 10,30-11,30 г (40-44 ммоля) 6-(2,2-дифтор-2-фенілетокси)гексан-1-аміну (IV) у 15-25 мл ксилольного розчинника додавали 19,9 г (40 ммолей) (R)-8-(бензилокси)-5-(2-бром-1(трет-бутилдиметилсилілоксі)етил)хінолін-2(1H)-ону (V) і 9-12 г бікарбонату натрію або 15-20 г карбонату калію. Реакційну суміш нагрівали при кипінні зі зворотним холодильником протягом 46 год. Після охолодження до кімнатної температури осаджені неорганічні солі відфільтровували й промивали 80-120 мл ксилолу. Розчинник видаляли, одержуючи маслянистий залишок, що використовували на наступній стадії без додаткового очищення. Кращими умовами стадії зняття захисної групи P2 є наступні: Маслянистий залишок, отриманий на попередній стадії, розчиняли у 300-350 мл ТГФ. Потім до реакційної суміші додавали 20-25 г TBAF. Реакційну суміш перемішували протягом 1-2 годин при 40-50ºC. Після охолодження до кімнатної температури розчинник видаляли у вакуумі. Всього 250-300 мл суміші вода/органічний розчинник (1:1) додавали до залишку. Органічний шар відокремлювали, і водний шар екстрагували двічі органічним розчинником (2 × 20-30 мл). Органічні шари поєднували й концентрували у вакуумі для видалення розчинника. Кращими органічними розчинниками, які використовували для екстракції, є толуол, дихлорметан, ізопропілацетат або метилізобутилкетон (MIK), більше переважно толуол, ізопропілацетат або дихлорметан, найбільше переважно ізопропілацетат або дихлорметан. В альтернативному способі отриманий залишок, коли видаляють розчинник реакції (ТГФ), може використовуватися безпосередньо на наступній стадії кристалізаційного очищення без водної екстракції. Залишок очищали кристалізацією з 8-9 г тетрагідрату 1,5-нафталендисульфонової кислоти у 300-400 мл етанолу. Отриманий продукт відфільтровували й промивали 50-70 мл етанолу. Отриманий сирий залишок обробляли 250-260 мл суміші метанол/ дихлорметан (1:2), 6 UA 107344 C2 5 10 15 20 25 30 35 40 45 50 55 60 метанол/ізопропілацетат (1:2) або метанол/толуол (1:2). До цієї суспензії додавали розчин 3,5-4 г NaOH у 170-190 мл води. Реакційну суміш перемішували при 20-30ºC протягом 40-50 хвилин. Органічну фазу відокремлювали, і розчинник видаляли у вакуумі. Кращими умовами способу стадії зняття захисної групи P1 є наступні: Проміжну сполуку (II) розчиняли у загальному об'ємі 160-170 мл суміші оцтова кислота/спирт (1:1), переважно оцтова кислота/метанол. До розчину додавали 1-1,5 г 10 % Pd/C, 50 % води. Потім до розчину необов'язково додавали близько 5-15 г TBAF. Після декількох насичень азотом реакційну суміш гідрували при температурі 20-30ºC, при тиску менше 4 бар, переважно при тиску 1-2 бар, протягом 6-8 год. Каталізатор потім відфільтровували й промивали 190-200 мл метанолу. До фільтрату додавали близько 200-250 мл оцтової кислоти, і до цього фільтрату додавали розчин 6-6,5 г тетрагідрату 1,5-нафталендисульфонової кислоти у 50-70 мл суміші метанол/оцтова кислота (1:1). Суміш нагрівали при кипінні зі зворотним холодильником протягом 30 хвилин. Після охолодження до кімнатної температури продукт відфільтровували й промивали 25-30 мл метанолу. Отриманий продукт необов'язково може бути очищений суспендуванням у метанолі у гарячих умовах, таких як при температурі кипіння метанолу. Кінцевий продукт (Ia) сушили у вакуумі при 50ºC. Спосіб синтезу, описаний у даному винаході, далі ілюструється наступними прикладами. Приклади наведені тільки для ілюстрації, і не повинні розглядатися як обмежуючі. 1 Структури отриманих сполук підтверджували даними H-ЯМР і MS. ЯМР записували за допомогою ЯМР-спектрометра Varian Gemini-200 при частоті 200 або 300 МГц. Тетраметилсилан використовували як стандарт, і зразки розчиняли у дейтерованому диметилсульфоксиді (ДМСО-d6) або дейтерованому хлороформі (CDCl3). Їхню чистоту визначали за даними ВЕРХ, за допомогою встаткування Alliance 2795 Waters, оснащеного детектором з діодною матрицею (DAD) і ZMD або ZQ мас-детектором (іонізація електроспреєм). Метод ВЕРХ за допомогою колонки Symmetry C18 (3,5 мкм, 21 × 100 мм) і рухливої фази, що складається з двох фаз: Фаза A: Забуференний (мурашина кислота/аміак) водний розчин при pH: 3. Фаза B: 50:50 суміш ацетонітрил/метанол з форміатом амонію. Градієнт становив від 0 % до 95 % фази B протягом 10 хвилин. Експерименти препаративної ВЕРХ-MS здійснювали за допомогою встаткування Gilson, оснащеного подвійним насосом (поршневий насос Gilson 321); вакуумним дегазатором (Gilson 864); інжектором-збірником фракцій (рідинний маніпулятор Gilson 215); двома ін'єкційними модулями, аналітичним і препаративним (Gilson 819); клапаном (Gilson Valvemate 7000); 1/1000 розгалужувачем (Acurate by LC Packings); приставним насосом (Gilson 307); детектором з діодною матрицею (Gilson 170) і MS-детектором (Thermoquest Finnigan aQa, квадрупольний мас-спектрометр із ES і APCI видами іонізації). Устаткування ВЕРХ-MS контролювали за допомогою IBM PC. Експериментальний розділ Порівняльний приклад I (відповідно до WO2006122788 і WO2008095720): Проміжна сполука III. 8-(Бензилокси)-5-((1R)-1-{[трет-бутил(диметил)силіл]окси}-2-{[6-(2,2дифтор-2-фенілетокси)гексил]-аміно}етил)хінолін-2(1H)-он До розчину 8-(бензилокси)-5-((1R)-2-бром-1-{[трет-бутил(диметил)силіл]окси}етил)хінолін2(1H)-ону (V) (4,80 г, 9,83 ммоля) і 6-(2,2-дифтор-2-фенілетокси)гексил]аміну (IV) (3,04 г, 11,8 ммоля) у диметилсульфоксиді (13,5 мл) додавали бікарбонат натрію (2,49 г, 29,4 ммоля) та йодид натрію (2,22 г, 14,8 ммоля). Суміш нагрівали при 140ºC протягом 2 годин. Після охолодження реакційну суміш розбавляли водою (40 мл) і екстрагували діетиловим ефіром (2 × 20 мл). Об'єднані органічні екстракти промивали водою (2 × 10 мл) і насиченим розчином хлориду натрію (20 мл), сушили (Na2SO4), і розчинник видаляли при зниженому тиску. Зазначену у заголовку сполуку одержували (6,40 г, 98 %) у вигляді масла. Проміжна сполука (II). 8-(Бензилокси)-5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}1-гідроксіетил)хінолін-2(1H)-он До розчину проміжної сполуки (III) (6,4 г, 9,63 ммоля) у тетрагідрофурані (60 мл) додавали TBAF (5,02 г, 19,26 ммоля). Суміш перемішували при кімнатній температурі протягом ночі. Розчинник видаляли при зниженому тиску. Очищення колонковою хроматографією за допомогою метиленхлориду/метанолу (від 95:5 до 85:15) в якості елюенту приводило до одержання 8-(бензилокси)-5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1гідроксіетил)хінолін-2(1H)-ону (II) (1,1 г, 20 %) у вигляді масла. 5-((1R)-2-{[6-(2,2-Дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін2(1H)-он (I) До проміжної сполуки (II) (1,10 г, 2,0 ммоля) у метанолі (50 мл) додавали 20 % паладію на вугіллі (300 мг). Суміш гідрували при 2 бар протягом 3 годин. Каталізатор відфільтровували 7 UA 107344 C2 5 10 15 20 25 30 35 40 45 50 55 через целіт, і розчинник концентрували. Отримане масло очищали колонковою хроматографією на силікагелі, елюючи сумішшю метиленхлорид/метанол (95:5) з одержанням зазначеної у заголовку сполуки (0,50 г, 54 %) у вигляді масла. Нападисилат 5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону (Ia) 5-((1R)-2-{[6-(2,2-Дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін2(1H)-он (6,63 г; 14,4 ммоля) розчиняли у 134 мл метанолу з одержанням 1,075 M розчину, що нагрівали до температури близько 50ºC. Потім до нагрітого розчину додавали 7,74 ммоля тетрагідрату нафтален-1,5-дисульфонової кислоти. Суміш потім перемішували протягом 30 хвилин при температурі кипіння, і потім охолоджували до 20/25ºC, і перемішували при цій температурі протягом 1 години. Отриманий осад виділяли фільтрацією, промивали метанолом і сушили у вакуумі при температурі 50ºC (15,67 г, 90 %). Приклад II (відповідно до даного винаходу) Проміжна сполука III. 8-(Бензилокси)-5-((1R)-1-{[трет-бутил(диметил)силіл]окси}-2-{[6-(2,2дифтор-2-фенілетокси)гексил]-аміно}етил)хінолін-2(1H)-он До розчину [6-(2,2-дифтор-2-фенілетокси)гексил]аміну (IV) (11,0 г, 42,8 ммоля) у ксилолі (20 мл) додавали 8-(бензилокси)-5-((1R)-2-бром-1-{[трет-бутил(диметил)силіл]оксі}етил)хінолін2(1H)-он (V) (19,9 г, 40,7 ммоля) і бікарбонат натрію (10,4 г, 123 ммоля). Суміш нагрівали при кипінні зі зворотним холодильником протягом 6 годин. Після охолодження до кімнатної температури до реакційної суміші додавали додатково ксилол (176 мл), і осаджені неорганічні солі відфільтровували й промивали ксилолом (100 мл). Отриманий фільтрат концентрували у вакуумі для видалення розчинника, одержуючи маслянистий залишок (проміжна сполука (III)), який використовували на наступній стадії без очищення. Проміжна сполука (II). 8-(Бензилокси)-5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}1-гідроксіетил)хінолін-2(1H)-он Проміжну сполуку (III) розчиняли у тетрагідрофурані (330 мл). Потім до цього розчину додавали TBAF (23,3 г, 73 ммоля). Суміш перемішували при 45ºC протягом 1 години. Після охолодження до кімнатної температури розчинник видаляли у вакуумі, і отриманий залишок необов'язково екстрагували 266 мл суміші вода/дихлорметан (1:1). Органічні шари витягали, і потім упарювали у вакуумі. Потім додавали 352 мл 96 % етанолу, і суміш нагрівали до 50-60ºC. При цій температурі додавали розчин 8,5 г тетрагідрату 1,5-нафталендисульфонової кислоти у 35 мл 96 % етанолу протягом 1 години. Систему додавання промивали 29 мл 96 % етанолу, який додавали до реакційної суміші. Реакційну суміш перемішували при кипінні зі зворотним холодильником протягом 30 хвилин, і потім охолоджували до кімнатної температури. Продукт відфільтровували й промивали 60 мл етанолу. Сирий продукт на фільтрі обробляли 252 мл суміші метанол/дихлорметан (1:2). Потім додавали розчин 3,6 г NaOH у 116 мл води, і реакційну суміш перемішували при 20-25ºC протягом 45 хвилин. Водний шар відокремлювали й екстрагували дихлорметаном (3 × 42 мл). Органічні фази витягали й перемішували разом із розчином 4,2 г NaCl у 168 мл води. Органічну фазу відокремлювали, і розчинник видаляли у вакуумі, одержуючи 8-(бензилокси)-5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1гідроксіетил)хінолін-2(1H)-он (II) (16,8 г, 75 %) у вигляді масла. Нападисилат 5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону (Ia) До розчину проміжної сполуки (II) (16,8 г, 30,5 ммоля) у суміші метанолу (69 мл) і оцтової кислоти (77 мл) додавали суспензію 10 % паладію на вугіллі, 50 % води, (1,33 г) у суміші метанолу (15 мл) і оцтової кислоти (7 мл). Суміш гідрували при 1-2 бар протягом 8 годин. Каталізатор відфільтровували через целіт і промивали метанолом (193 мл). До цього фільтрату додавали оцтову кислоту (220 мл). Потім до фільтрату повільно додавали розчин тетрагідрату 1,5-нафталендисульфонової кислоти (6,33 г) у суміші метанолу (54 мл) і оцтової кислоти (27 мл). Реакційну суміш нагрівали при кипінні зі зворотним холодильником протягом 30 хвилин, і потім охолоджували до кімнатної температури. Осад відфільтровували й промивали метанолом (27 мл). Сирий продукт на фільтрі розчиняли у метанолі (800 мл) і нагрівали при кипінні зі зворотним холодильником протягом 30 хв. Продукт відфільтровували й промивали додатково метанолом (34 мл). Отриману тверду речовину сушили у вакуумі при 50ºC, одержуючи нападисилат 5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону (14,9 г, 81 %). Загальний вихід нападисилату 5-((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил] аміно}-1гідроксіетил)-8-гідроксихінолін-2(1H)-ону (Ia) становив близько 60,7 % (75 % x 81 %), та його чистота становила ВЕРХ домішок = 1,5 %, e.e. > 98 %. 60 8 UA 107344 C2 Таблиця 1 Порівняльні результати Стадія реакції Стадія (a) Стадія (b)* Стадія (c)** Стадія (d) Загальний вихід Домішки у кінцевому продукті (Ia) Порівняльний приклад Отриманий продукт містив 39 % домішок Вихід: 20 % Вихід: 54 % Вихід: 90 % 9,5 % Приклад за винаходом Отриманий продукт містив 7 % домішок Вихід: 75 % 5,8 % 1,5 % Вихід: 81 % 60,75 % * стадія зняття захисної групи P2 ** стадія зняття захисної групи P1 5 Як видно з Таблиці 1, застосування ксилольного розчинника на стадії (a) істотно знижує кількість домішок у проміжній сполуці формули (III). Крім того, загальний вихід нападисилату 5((1R)-2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону істотно підвищується при зниженні домішок до низького рівня у порівнянні з порівняльним прикладом. Це досягається модифікацією деяких методик очищення, у такий спосіб спрощуючи реакційні стадії й знижуючи кількість різних реагентів. ФОРМУЛА ВИНАХОДУ 10 1. Спосіб одержання сполуки 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8гідроксихінолін-2(1H)-ону формули (І) або її фармацевтично прийнятної солі: F F O HN HO O N H OH 15 , (I) який полягає в тому, що: a) вводять в реакцію у ксилольному розчиннику сполуку формули (V): P2 O L N H P1 O O , (V) де P1 і Р2 являють собою захисні групи для гідроксигруп, і L являє собою відхідну групу, з 6-(2,2-дифтор-2-фенілетокси)гексан-1-аміном формули (IV): H2N O F 20 F (IV) з одержанням сполуки формули (III): 9 UA 107344 C2 F HN P2 O N H P1 5 10 F O O O (III); b) здійснюють стадію зняття захисної групи Р1 і стадію зняття захисної групи Р2 для видалення захисних груп P1 і Р2 і одержання сполуки формули (І). 2. Спосіб за п. 1, у якому: (і) стадію зняття захисної групи Р2 здійснюють при температурі у діапазоні близько 30-60 °C протягом до 8 годин, і/або (іі) стадію зняття захисної групи Р1 здійснюють у присутності розчинника, що являє собою оцтову кислоту або суміш оцтової кислоти зі спиртом або зі складним ефіром. 3. Спосіб за п. 1 або п. 2, у якому на стадії (b): здійснюють зазначену стадію зняття захисної групи Р2 у сполуці формули (III) з одержанням сполуки формули (II): F F O HN HO N H P1 15 O O , (II) у якій Р1 є таким, як визначено вище; і здійснюють зазначену стадію зняття захисної групи P1 у сполуці формули (II) з одержанням сполуки формули (І): F F O HN HO N H 20 25 30 O OH (I). 4. Спосіб за будь-яким із попередніх пунктів, у якому (а) Р 1 являє собою бензильну групу, і стадію зняття захисної групи Р1 здійснюють гідруванням, і/або (b) Р2 являє собою тpетбутилдиметилсилільну групу, і стадію зняття захисної групи Р2 здійснюють реакцією з тригідратом фториду тетра-н-бутиламонію або з хлоридом водню. 5. Споcіб за будь-яким із попередніх пунктів, де L являє собою бром. 6. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р 2 здійснюють за допомогою тригідрату фториду тетра-н-бутиламонію у тетрагідрофурані. 7. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р2 здійснюють при температурі 40-50 °C протягом періоду часу, що не перевищує 6 годин. 8. Спосіб за п. 7, у якому період часу не перевищує 4 годин. 9. Спосіб за будь-яким із попередніх пунктів, в якому одержують гемінападисилатну або мезилатну сіль сполуки формули (І). 10. Спосіб за п. 9 для одержання гемінападисилатної солі сполуки формули (І), у якому після стадії (b) додають нафтален-1,5-дисульфонову кислоту без виділення 5-(2-{[6-(2,2-дифтор-2фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1Н)-ону формули (І). 10 UA 107344 C2 5 10 15 11. Спосіб за п. 3, у якому проміжну сполуку формули (II), отриману зі стадії зняття захисної групи Р2, очищають кристалізацією з тетрагідратом 1,5-нафталендисульфонової кислоти в етанолі. 12. Спосіб за будь-яким із попередніх пунктів, у якому Р1 являє собою бензил, і стадію зняття захисної групи Р1 здійснюють гідруванням у присутності каталізатора паладію на вугіллі у кількості менше 10 % (мас.) щодо кількості сполуки формули (II). 13. Спосіб за п. 12, у якому використовують кількість каталізатора, що становить менше 5 %. 14. Спосіб за будь-яким із попередніх пунктів, у якому стадію зняття захисної групи Р1 здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або зі складним ефіром, переважно розчинником є оцтова кислота або суміш метанол/оцтова кислота (1:1). 15. Спосіб за п. 14, у якому як розчинник використовують суміш метанол/оцтова кислота (1:1). 16. Спосіб за будь-яким із попередніх пунктів, у якому (і) стадію зняття захисної групи Р2 здійснюють при температурі у діапазоні 30-60 °C протягом періоду до 8 годин, і (іі) стадію зняття захисної групи Р1 здійснюють у присутності розчинника, яким є оцтова кислота або суміш оцтової кислоти зі спиртом або зі складним ефіром. Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 11
ДивитисяДодаткова інформація
Автори російськоюMarchueta Hereu, Iolanda, Moyes Valls, Enrique
МПК / Мітки
МПК: A61P 11/00, A61K 31/4704, C07D 215/26
Мітки: одержання, 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону, спосіб
Код посилання
<a href="https://ua.patents.su/13-107344-sposib-oderzhannya-5-2-6-22-diftor-2-feniletoksigeksilamino-1-gidroksietil-8-gidroksikhinolin-21n-onu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону</a>
Мезилат 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону як агоніст b2-адренергічного рецептора

Номер патенту: 105913
Опубліковано: 10.07.2014
Автори: Каррера Каррера Франсеск, Марчуета Ереу Іоланда, Пуіг Дуран Карлос, Моес Вальс Енріке
МПК: C07D 215/26, A61P 11/00, A61K 31/4704
Мітки: b2-адренергічного, агоніст, 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону, рецептора, мезилат
Формула / Реферат:
1. Мезилат 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону та його фармацевтично прийнятні сольвати. 2. Сіль за п. 1, вибрана з групи, що включає:мезилат (R,S)-5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону,мезилат...
Застосування 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-он для нормалізації легеневої функції пацієнта

Номер патенту: 103370
Опубліковано: 10.10.2013
Автори: Массана Монтехо Ерік, Руф Торстен
МПК: A61P 11/08, A61K 31/4704, A61P 11/06
Мітки: 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-он, функції, застосування, легеневої, нормалізації, пацієнта
Формула / Реферат:
1. Застосування сполуки, яка являє собою 5-(2-{[6-(2,2-дифтор-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1Н)-он, у формі рацемату, стереоізомеру або суміші стереоізомерів, або її фармацевтично прийнятної солі або сольвату, для виготовлення лікарського засобу для застосування шляхом інгаляції для нормалізації легеневої функції пацієнта - людини, що страждає від астми, хронічного обструктивного захворювання легенів (COPD),...
Нападизилат 5-(2-{[6-(2,2-дифторо-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону як агоніст b2 адренергічного рецептора

Номер патенту: 97829
Опубліковано: 26.03.2012
Автори: Пуіг Дуран Карлос, Мойєс Валлс Енріке
МПК: C07D 215/26
Мітки: 5-(2-{[6-(2,2-дифторо-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1н)-ону, нападизилат, рецептора, агоніст, адренергічного
Формула / Реферат:
1. Кристалічна сіль мононападизилат або гемінападизилат 5-(2-{[6-(2,2-дифторо-2-фенілетокси)гексил]аміно}-1-гідроксіетил)-8-гідроксихінолін-2(1H)-ону або її фармацевтично прийнятні сольвати. 2. Сіль за п. 1, що описується формулою (I):,(I)у якій n дорівнює 1 або 2,і її фармацевтично прийнятні...
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Рок Майкл Харольд, Петерсен Ханс, Еллегор Петер
МПК: C07D 307/87, C07C 255/56, C07C 253/30
Мітки: сполук, s-циталопраму, кетони, циталопраму, одержання, проміжні, варіанти, спосіб, рацемічних
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Спосіб одержання 4-{4-[({[4-хлор-3-(трифторметил)феніл]аміно}карбоніл)аміно]фенокси}-n-метилпіридин-2-карбоксаміду

Номер патенту: 90691
Опубліковано: 25.05.2010
Автори: Герінг Райнхольд, Ленц Яна, Берве Матіас, Мюллер-Гліманн Маттіас, Штіль Юрген, Кун Олівер, Морс Клаус, Льогерс Міхаель, Маттхойс Майк, Хайльманн Вернер
МПК: C07D 213/81
Мітки: спосіб, 4-{4-[({[4-хлор-3-(трифторметил)феніл]аміно}карбоніл)аміно]фенокси}-n-метилпіридин-2-карбоксаміду, одержання
Формула / Реферат:
1. Спосіб одержання сполуки формули (І), (І)при якому на першій стадії піддають реакції сполуку формули (V) (V)з 4-хлор-3-трифторметилфенілізоціанатом у нехлорованому органічному розчиннику, інертному по відношенню до ізоціанатів, спочатку завантажують сполуку формули (V) при...
Попередній патент: Спосіб і пристрій для отримання вуглецевмісного палива
Наступний патент: Композиція, що містить частинки авермектину, покриті фотозахисним агентом
Випадковий патент: Процес створення художньої паркетної підлоги