Спосіб синтезу клофарабіну
Номер патенту: 115206
Опубліковано: 25.09.2017
Автори: Сипченко Володимир, Матвієнко Віктор, Забудкін Олександр, Матвієнко Ярослав






Формула / Реферат
1. Спосіб одержання клофарабіну, який включає:
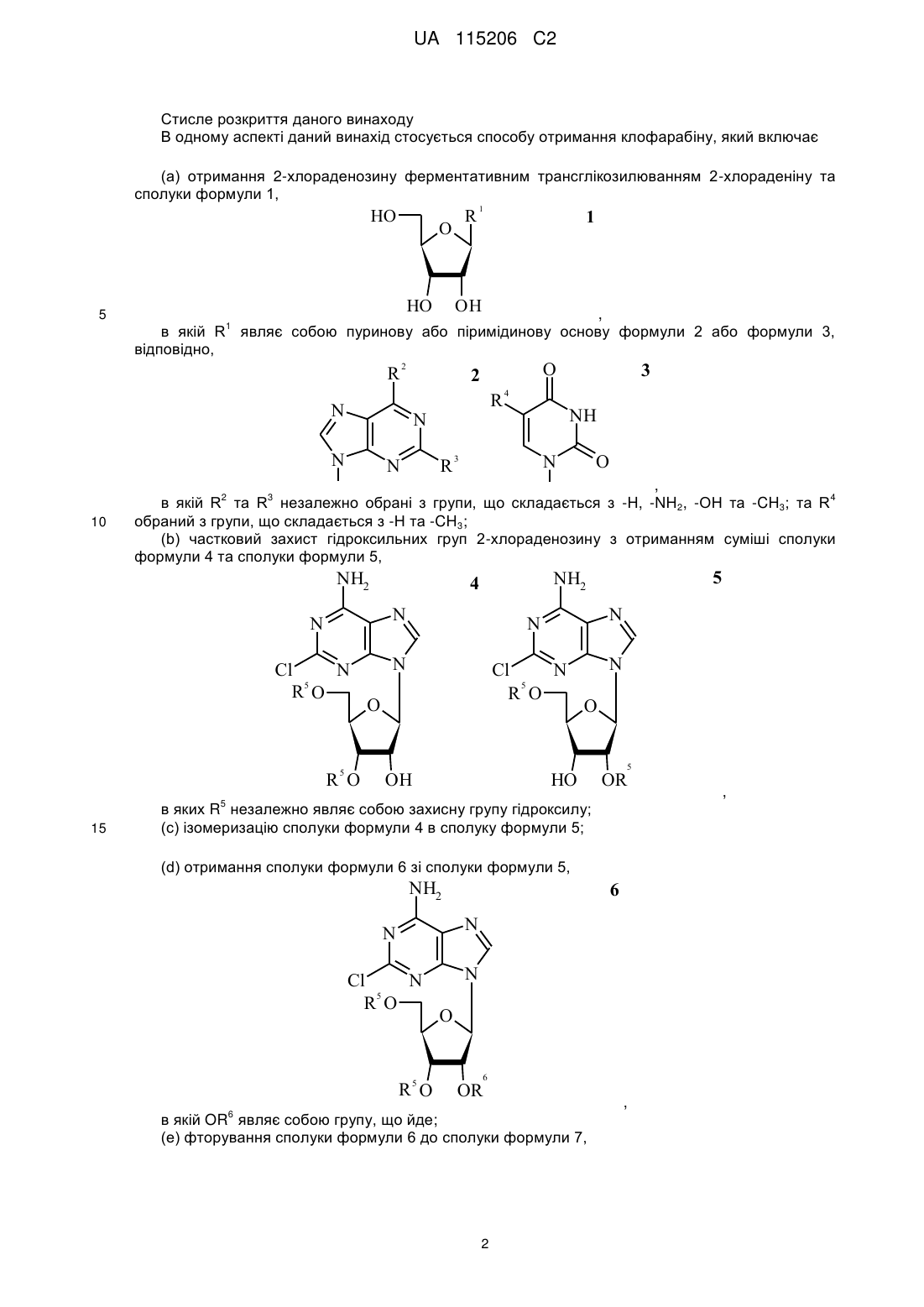
(а) одержання 2-хлораденозину ферментативним трансглікозилюванням 2-хлораденину та сполуки формули 1,
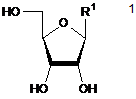 ,
,
в якій R1 являє собою пуринову або піримідинову основу формули 2 або формули 3, відповідно,
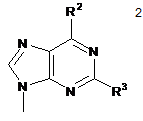 ,
,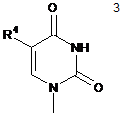 ,
,
в яких R2 та R3 незалежно вибрані з групи, яка складається з -Н, -NH2, -ОН та -СН3; та R4 вибраний з групи, яка складається з -Н та -СН3;
(b) частковий захист гідроксильних груп 2-хлораденозину з одержанням суміші сполуки формули 4 та сполуки формули 5,
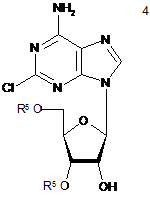 ,
,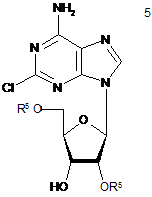 ,
,
в яких R5 незалежно являє собою захисну групу гідроксилу;
(c) ізомеризацію сполуки формули 4 в сполуку формули 5;
(d) одержання сполуки формули 6 зі сполуки формули 5,
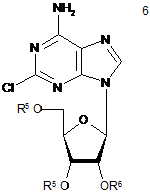 ,
,
в якій OR6 являє собою відхідну групу;
(e) фторування сполуки формули 6 до сполуки формули 7,
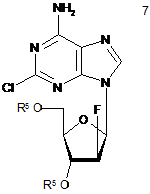 , та
, та
(f) зняття захисту зі сполуки формули 7 з одержанням клофарабіну.
2. Спосіб за п. 1, в якому трансглікозилювання на стадії (а) виконують з використанням пурин-нуклеозидфосфорилази або комбінації пурин-нуклеозидфосфорилази та уридинфосфорилази.
3. Спосіб за п. 1 або п. 2, в якому фторування на стадії (e) виконують з використанням фторуючого засобу.
4. Спосіб за п. 3, в якому фторуючий засіб вибирають з групи, яка складається з фтористоводневої кислоти та суміші фтористоводневої кислоти та органічної основи Льюїса.
5. Спосіб згідно з п. 4, в якому органічна основа Льюїса являє собою амін.
6. Спосіб за будь-яким з пп. 1-5, в якому R1 являє собою піримідинову основу, яка являє собою уридин.
7. Спосіб за будь-яким з пп. 1-6, в якому R5 являє собою захисну групу гідроксилу, яка являє собою бензоїл.
8. Спосіб за будь-яким з пп. 1-7, в якому OR6 являє собою відхідну групу, яка являє собою трифторметансульфонат.
Текст
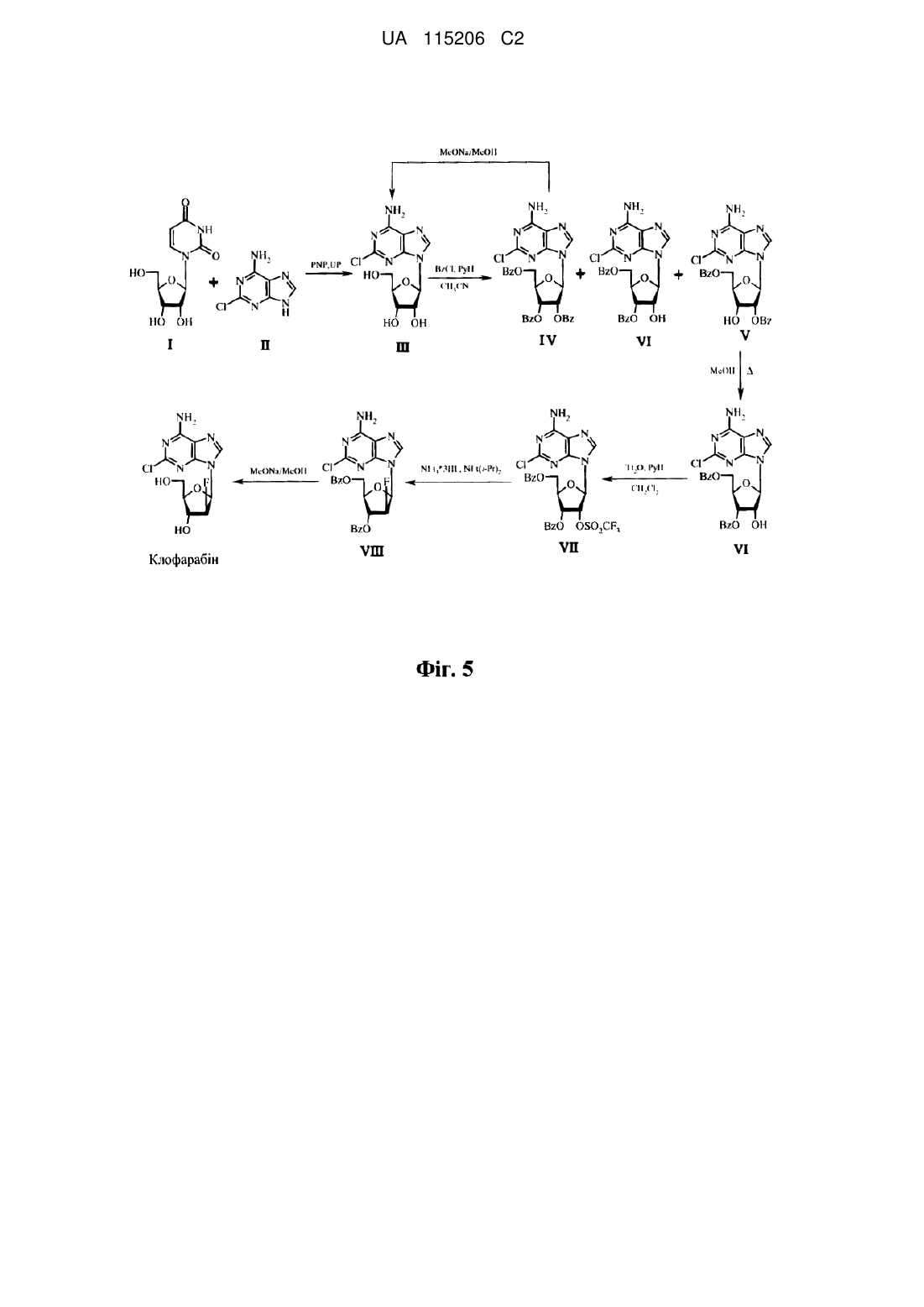
Реферат: Даний винахід стосується високоефективного способу одержання протиракового нуклеозиду клофарабіну, при цьому спосіб передбачає одержання 2-хлораденозину ферментативним трансглікозилюванням 2-хлораденину та нуклеозидів, бензоїлювання, ізомеризацію, утворення сульфонату складного ефіру, фторування та зняття захисту. UA 115206 C2 (12) UA 115206 C2 UA 115206 C2 5 10 15 20 25 30 35 40 45 50 55 60 Область техніки, до якої належить даний винахід Даний винахід стосується способу отримання клофарабіну з високим виходом та без утворення небажаних α-N9 стереоізомерів. Попередній рівень техніки даного винаходу Клофарабін (систематична назва IUPAC-5-(6-аміно-2-хлорпурин-9-іл)-4-фтор-2(гідроксилметил)оксолан-3-ол) являє собою антиметаболіт пуринового нуклеотиду, який використовується для лікування різних типів лейкозів, зокрема гострого лімфобластного лейкозу. На даний момент в рівні техніки відомі декілька способів отримання клофарабіну. Шляхи синтезу, як правило, мають в основі реакцію сполучення пуринових похідних та арабінофуранозильного фрагменту. В залежності від способу введення атому фтору такі схеми синтезу можуть бути розділені на дві групи: (i) способи, в яких атом фтору включається в рибофуранозний фрагмент та вводиться в молекулу на стадії сполучення; (ii) способи, в яких атом фтору вводиться після стадії сполучення шляхом хімічних перетворень попередньо отриманого нуклеозиду. В патенті США № 5034518 розкривається оригінальна схема синтезу для отримання клофарабіну, яка розроблена Montgomery зі співавторами. Шлях синтезу показаний на фіг. 1. Якщо стисло, 1-бром-2-фторцукор 1 поєднують з 2,6-дихлорпурином 2 з наступними стадіями амінування та зняття захисту. Ця стадія сполучення призводить до утворення суміші α- и βаномерів продукту 3. Після хроматографічного розділення можна виділити бажаний β-аномер з виходом всього лише 32 %. Головні недоліки цього способу полягають в низькому загальному виході бажаного продукту (13 % виходячи з 1), низькій стабільності вихідного броміду 1 та складній очистці продукту 3 реакції сполучення через наявність α-ізомеру. В патенті США № 6949640 розкривається модифікація вищевказаного шляху синтезу, яка передбачає (i) застосування солей, утворених шляхом обробки 2-хлор-6-заміщенного пурину сильними основами (такими як NaH) замість NH-форм пуринів, та (ii) застосування 2-дезоксі-2фтор-3,5-ди-O-бензоїл-α-D-арабінофуранозилу броміду 4 замість О-ацетильної похідної 1 (цей шлях синтезу показаний на фіг. 2). Ці модифікації дозволили збільшити загальний вихід клофарабіну до приблизно 40 % виходячи зі сполуки 4. Однак, схема синтезу ускладнюється низькою стабільністю броміду 4, який важко отримати, та утворенням суміші аномерів на стадії сполучення, що потребує складної колонкової хроматографії для відокремлення бажаного продукту. Додаткова модифікація способу Montgomery та співавторів розкривається в патенті США № 6680382 та додатково ілюструється на фіг. 3. Ця модифікація передбачає застосування калієвої солі 2-хлораденіну 5, яку отримують шляхом обробки 2-хлораденіну за допомогою t-BuOK, замість калієвої солі 2,6-дихлорпурину, застосування трьохкомпонентної суміші розчинників (наприклад, трет-амілового спирту, CH2Cl2, CH3CN) та додавання CaH2 для повного видалення води з розчинників. Застосування розчинників з низькою полярністю на стадії сполучення дозволило досягти стереоселективного протікання реакції (з відношенням аномеру β до α, що доходить до 15:1). Крім того, наявність аміногрупи в вихідному матеріалі 2-хлораденіні дозволяло уникнути стадії амінування продукту реакції сполучення. В результаті загальний вихід очищеного клофарабіну з фторованого цукру 4 складає приблизно 32 %. Однак, даний спосіб як і до того має два недоліки, які полягають у складному отриманні фторованого цукру 4 (який отримують у чотири стадії з 1-O-ацетил-2,3,5-три-O-бензоїл-β-D-рибофуранози) та в його низькій стабільності. Спосіб, розкритий в публікації заявки на видачу патенту США № 2012/0010397, стосується вищевказаної другої групи способів отримання клофарабіну. Він показаний на фіг. 4. На першій стадії отримують 2',3',5'-три-O-бензоїл-2-хлораденозин 8 шляхом сполучення цукру 6 та TMSзахищеного 2-хлораденіну 7. Подальше дебензоїлювання за допомогою гідразин-гідрату призводить до утворення суміші 3',5'- и 2',5'’-ди-O-бензоїльних похідних. Після ізомеризації 2',5'ди-O-бензоїльної похідної в 3',5'-похідну 9 отримують трифлат 10 шляхом обробки сполуки 9 трифторметансульфоновим ангідридом. Трифлат 10 після його фторування та зняття захисту дає кінцевий продукт клофарабін. Однак, недоліки цього способу полягають в великій кількості ізомерів (які важко розділяти), що утворюються на стадії сполучення між 6 та 7 (β-N9-, α-N7-, βN7-ізомери), у застосуванні високотоксичного гідразин-гідрату, у виході очищеного клофарабіну, що складає всього лише 15 %. Отже, залишається необхідність у покращених способах синтезу клофарабіну, які долають обмеження відомих схем синтезу. Зокрема, необхідна схема синтезу, яка забезпечує високий вихід продукту та не передбачає утворення небажаних стереоізомерів. Відповідно, метою даного винаходу є забезпечення такого способу отримання клофарабіну. 1 UA 115206 C2 Стисле розкриття даного винаходу В одному аспекті даний винахід стосується способу отримання клофарабіну, який включає (a) отримання 2-хлораденозину ферментативним трансглікозилюванням 2-хлораденіну та сполуки формули 1, HO 5 HO 1 1 OH , в якій R являє собою пуринову або піримідинову основу формули 2 або формули 3, відповідно, 1 R 2 O 2 N R N N 10 R O N R 3 4 NH N 3 O , 2 3 4 в якій R та R незалежно обрані з групи, що складається з -H, -NH2, -OH та -CH3; та R обраний з групи, що складається з -H та -CH3; (b) частковий захист гідроксильних груп 2-хлораденозину з отриманням суміші сполуки формули 4 та сполуки формули 5, NH2 N N Cl N 5 RO 5 R O 5 NH2 4 N Cl N 5 R O N O OH N N O HO 5 OR 5 15 в яких R незалежно являє собою захисну групу гідроксилу; (c) ізомеризацію сполуки формули 4 в сполуку формули 5; (d) отримання сполуки формули 6 зі сполуки формули 5, NH2 6 N N Cl N 5 R O 5 R O N O 6 OR 6 в якій OR являє собою групу, що йде; (e) фторування сполуки формули 6 до сполуки формули 7, 2 , , UA 115206 C2 7 NH2 N N Cl N 5 RO N O F 5 RO 5 10 15 20 25 30 35 40 45 , та (f) зняття захисту зі сполуки формули 7 з отриманням клофарабіну. Згідно з переважними варіантами виконання трансглікозилювання на стадії (a) виконують з використанням пурин-нуклеозидфосфорилази або комбінації пурин-нуклеозидфосфорилази та уридинфосфорилази. Згідно з наступними переважними варіантами здійснення фторування на стадії (e) виконують з використанням фторуючого засобу. Переважно фторуючий засіб обраний з групи, що складається з фтористоводневої кислоти та суміші фтористоводневої кислоти та органічної основи Льюїса. Особливо переважною органічною основою Льюїса (що використовується в суміші з фтористоводневою кислотою) є амін. 1 Згідно з наступними переважними варіантами здійснення заступник R являє собою 5 піримідинову основу, що є уридином; та/або замісник R являє собою захисну групу гідроксилу, 6 що є бензоїлом; та/або замісник OR являє собою групу, що йде, що є трифторметансульфонатом. Стислий опис графічних матеріалів На фіг. 1 показана схема синтезу для отримання клофарабіну, яка розкрита в патенті США № 5034518. На фіг. 2 показана схема синтезу для отримання клофарабіну, яка розкрита в патенті США № 6949640. На фіг. 3 показана схема синтезу для отримання клофарабіну, яка розкрита в патенті США № 6680382. На фіг. 4 показана схема синтезу для отримання клофарабіну, яка розкрита в публікації заявки на видачу патенту США № 2012/0010397. На фіг. 5 показана схема синтезу для отримання клофарабіну згідно з даним винаходом. Докладне розкриття даного винаходу Даний винахід має в основі несподіване відкриття того, що, якщо починається з 2хлораденозину, виконання каскаду реакцій, що включає стадії ферментативного трансглікозилювання, бензоїлювання, ізомеризації, утворення сульфонату складного ефіру (тобто сульфонилювання), фторування та зняття захисту, призводить до утворення клофарабіну з високим виходом без супутнього утворення небажаних стереоізомерів, таким чином долаючи головні недоліки відомих способів та забезпечуючи більш ефективну і менш трудомістку схему синтезу. Далі даний винахід буде описаний відносно конкретних варіантів здійснення та відносно конкретних графічних матеріалів, але слід брати до уваги, що даний винахід обмежений не ними, а лише формулою винаходу, що додається. Графічні матеріали, що описуються, є виключно схематичними та ілюстративними та вважаються необмежуючими. Термін "який включає" ("який містить"), що використовується в даному описі та формулі винаходу, не виключає інші елементи або стадії. Для цілей даного винаходу термін "який складається з" вважається переважним варіантом терміну "який містить". Якщо далі в даному документі група визначається як така, що містить щонайменше деяку кількість варіантів здійснення, то це також слід розуміти як розкриття групи, яка переважно складається лише з цих варіантів здійснення. Використання іменника в однині передбачає множину цього іменника, якщо спеціально не вказано інше. У випадку, коли вказані числові значення в контексті даного винаходу, спеціалісту в даній області буде зрозуміло, що технічний ефект ознаки, що розглядається, забезпечують в діапазоні точності, який, як правило, охоплює відхилення числового значення в межах ± 10 % та переважно ± 5 %. 3 UA 115206 C2 5 10 Крім того, терміни перший, другий, третій, (a), (b), (c) та подібні в даному описі та в формулі винаходу використовують для розрізнення подібних елементів та необов'язково для опису послідовного або хронологічного порядку. Слід розуміти, що терміни, які використовуються таким чином, є взаємозамінними за відповідних умов, та що варіанти здійснення даного винаходу, які описуються в даному документі, можуть працювати в послідовностях, відмінних від тих, що описуються або ілюструються в даному документі. Наступні визначення термінів будуть наведені далі в контексті використання цих термінів. Наступні терміни або визначення представлені виключно з метою пояснення даного винаходу. Ці визначення не повинні бути розтлумачені в об'ємі, який менше, ніж відомий рядовому спеціалісту в даній області. В одному аспекті даний винахід стосується способу отримання клофарабіну, який включає (a) отримання 2-хлораденозину ферментативним трансглікозилюванням 2-хлораденіну та сполуки формули 1, HO HO 15 1 1 OH , в якій R являє собою пуринову або піримідинову основу формули 2 або формули 3, відповідно, 1 R N 2 O 2 R N N 20 R O N R 3 4 NH N 3 O , 2 3 4 в яких R та R незалежно обрані з групи, що складається з -H, -NH2, -OH та -CH3; та R обраний з групи, що складається з -H та -CH3; (b) частковий захист гідроксильних груп 2-хлораденозину з отриманням суміші сполуки формули 4 та сполуки формули 5, NH2 N N Cl N 5 RO 5 R O N O 5 NH2 4 N N Cl N 5 R O N OH HO 5 O в яких R незалежно являє собою захисну групу гідроксилу; (c) ізомеризацію сполуки формули 4 в сполуку формули 5; 25 (d) отримання сполуки формули 6 зі сполуки формули 5, 4 5 OR , UA 115206 C2 NH2 6 N N Cl N 5 R O N O 6 5 R O OR , 6 в якій OR являє собою групу, що йде; (e) фторування сполуки формули 6 до сполуки формули 7, 7 NH2 N N Cl N 5 RO N O F 5 RO 5 10 15 20 25 30 35 , та (f) зняття захисту зі сполуки формули 7 з отриманням клофарабіну. Спосіб згідно з даним винаходом схематично наведений на фіг. 5. Далі нумерація сполук відповідає номенклатурі, яка використана на фіг. 5. 2-Хлораденозин (ΙΙΙ) отримують шляхом реакції ферментативного трансглікозилювання між 2-хлораденіном (ΙΙ) та будь-яким нуклеозидом, який містить рибозу, переважно уридином (Ι) (оскільки він легко розчиняється та легко доступний), з використанням ферментів, таких як переважно пурин-нуклеозидфосфорилаза (PNP) та уридинфосфорилаза (UP). Ця реакція є зворотною. Для зсуву рівноваги в сторону продукту (ΙΙΙ) використовують надлишок уридину від 1,5 до 5 еквівалентів, переважно 3,3. Реакція може бути проведена при температурі від 40 до 70 °C, переважно при температурі від 58 до 61 °C. Через стереоспецифічність ферментативного трансглікозилювання єдиним продуктом глікозилювання є β-аномер (ΙΙΙ). 2-Хлораденозин може бути очищений від вихідних реагентів (Ι), (ΙΙ) та урацилу (побічного продукту), наприклад, з використанням препаративної хроматографії низького тиску. Як правило, обирають умови, які забезпечують утримування 2-хлораденозину сорбентом, тоді як більша частина домішок не утримується в колонці. Це дозволяє спростити процес очистки шляхом включення лише стадій "внесення" та "промивання". Потім отриманий 2-хлораденозин переважно бензоїлюють бензоїлхлоридом в присутності піридину. В цій реакції бензоїлхлорид може бути використаний в кількості, яка змінюється від 1,8 до 2,5 еквівалента, переважно 2,3 еквівалента, виходячи з триольної сполуки формули (ΙΙΙ). Реакція може бути проведена при температурі від -10 до 20 °C, переважно при температурі від 0 до 5 °C. В якості побічного продукту може утворюватись до 35 % 2',3',5'-три-O-бензоїл-2хлораденозину (ΙV). Однак, присутність цього побічного продукту не впливає на наступну стадію ізомеризації, а також його легко можна відокремити при кристалізації 3',5'-ди-O-бензоїльної похідної. Таким чином, майже вся кількість вихідної сполуки 2-хлораденозину в підсумку призводить до утворення 3',5'-ди-O-бензоїльної похідної. Далі виконують ізомеризацію 2',5'-ди-O-бензоїльного ізомеру (V) в 3',5'-ди-O-бензоїльний ізомер (VΙ). Перегрупування переважної бензоїльної групи з положення 2' в 3' в нуклеозидах відоме в рівні техніки (дивіться, зокрема, Maruyama, T. et al. (1999) Chem. Pharm. Bull. 47, 966970). Переважно реакцію ізомеризації виконують шляхом кип'ятіння в метанолі протягом 40-45 годин при енергійному перемішуванні. Отримані в результаті кристали після гарячої фільтрації реакційної суміші характеризуються чистотою >98 % (за HPLC), в той час як майже всі домішки 5 UA 115206 C2 5 10 15 20 25 30 35 40 45 50 55 60 залишаються в залишковій рідині, з якої після повторної обробки може бути виділений 2хлораденозин. Наступне сульфонілювання сполуки формули VΙ виконують шляхом обробки за допомогою Tf2O з використанням піридину в якості основи. В цій реакції трифторметансульфоновий ангідрид може бути використаний в кількості, яка змінюється від 1,3 до 2,0 еквівалентів, переважно 1,6 еквівалента, виходячи зі сполуки формули (VΙ). Розчинником може бути органічний розчинник, такий як ацетонітрил, тетрагідрофуран, дихлорметан, хлороформ, переважно дихлорметан. Реакція може бути проведена при температурі від -20 до 30 °C, переважно при температурі від -10 до 0 °C. На наступній стадії можна отримати фторпохідну (VΙΙΙ) шляхом обробки трифлата (VΙΙ) фторуючими засобами, такими як NEt3*3HF, TBAF або TBAF*(t-BuOH)4, переважно NEt3*3HF, наприклад, з використанням диізопропілетиламіну в якості основи. Розчинником може бути, наприклад, етилацетат, тетрахлорметан, толуол, ацетонітрил, переважно толуол. Реакцію можна проводити при температурі від 0 до 100 °C, переважно при температурі від 35 до 40 °C. Отриманий захищений клофарабін можна кристалізувати з суміші етилацетату та метанолу. За вищезгаданих умов вихід продукту реакції після кристалізації складає приблизно 60 %. В кінці із захищеного клофарабіну знімають захист за м'яких умов. Такі м'які умови (30 хвилин при 30 °C) дозволяють виконувати реакцію без будь-яких побічних реакцій. Утворений метилбензоат відокремлюють шляхом екстракції метиленхлоридом, а клофарабін, який міститься в водній фазі, очищають з використанням препаративної хроматографії низького тиску з наступною кристалізацією. Отриманий клофарабін характеризується високою чистотою без таких кількостей домішок, які можна виявити. Загальний вихід клофарабіну складає приблизно 30-40 %. Даний винахід далі описується за допомогою графічних матеріалів та наступних прикладів, які приведені виключно з метою ілюстрації конкретних варіантів здійснення даного винаходу і не повинні вважатись обмежуючими будь-яким чином для об'єкта винаходу, що заявляється. Приклади Приклад 1a. Отримання 2-хлораденозину (ΙΙΙ) з 2-хлораденіну та уридину 400 г уридину та 150 г KH2PO4 розчиняли при перемішуванні в суміші води (52 л) та DMSO (1,8 л) при 58-61 °C. В отриманий розчин додавали першу порцію (0,75 л) розчину, отриманого з 2-хлораденіну (85 г), води (7 л) та KOH (120 г). Регулювали pH отриманої в результаті суміші до 7,1-7,2 за допомогою водного розчину KOH. Додавали розчини уридинфосфорилази та пуриннуклеозидфосфорилази при перемішуванні при 58-61 °C. Другу частину розчину 2-хлораденіну, що залишилась, поступово додавали в реакційну суміш при перемішуванні протягом 3годинного періоду при 58-61 °C з підтримуванням pH в діапазоні 7,1-7,2 за допомогою водного розчину HCl. Потім реакційну суміш перемішували протягом 1 години при 58-61 °C та додавали NaOH для доведення pH до 11. Отриманий в результаті розчин, який містив 2-хлораденозин, очищали за допомогою препаративної хроматографії з наступним виділенням продукту, що включав кристалізацію в воді, з отриманням 120 г названої сполуки. Як правило, чистота 2-хлораденозину складала > 99 %, а вихід виходячи з 2-хлораденіну складав > 85 %. Приклад 1b. Отримання 2-хлораденозину (ΙΙΙ) з 2-хлораденіну та гуанозину 229 г гуанозину та 75 г KH2PO4 розчиняли при перемішуванні в суміші води (52 л) та DMSO (1,8 л) при 58-61 °C. В отриманий розчин додавали першу порцію (0,75 л) розчину, отриманого з 2-хлораденіну (42 г), води (7 л) та KOH (60 г). Регулювали pH отриманої в результаті суміші до 7,1-7,2 за допомогою водного розчину KOH. В реакційну суміш додавали розчин пуриннуклеозидфосфорилази при перемішуванні при 58-61 °C. Другу частину розчину 2-хлораденіну, що залишилась, поступово додавали в реакційну суміш при перемішуванні протягом 3годинного періоду при 58-61 °C з підтримуванням pH в діапазоні 7,1-7,2 за допомогою водного розчину HCl. Потім реакційну суміш перемішували протягом 1 години при 58-61 °C та додавали NaOH для доведення pH до 11. Отриманий в результаті розчин, який містив 2-хлораденозин, очищали за допомогою обернено-фазової колонкової хроматографії низького тиску з наступним виділенням продукту, що включав кристалізацію в воді, з отриманням 53 г названої сполуки. Як правило, чистота 2хлораденозину складала > 99 %, а вихід виходячи з 2-хлораденіну складав > 70 %. Приклад 2. Бензоїлювання 2-хлораденозину Розчин 2-хлораденозину (750 г) в піридині (7,5 л) охолоджували до -5-0 °C. Потім в реакційну суміш повільно додавали розчин бензоїлхлориду (720 г) в ацетонітрилі (1440 мл) при перемішуванні та охолодженні. При цьому внутрішня температура реакційної суміші не повинна була перевищувати 5 °C. Суміш утримували протягом 30 хвилин за тих саме умов. Після цього 6 UA 115206 C2 5 10 15 20 25 30 35 40 45 50 55 60 розчинники випаровували під пониженим тиском та при температурі 60 °C. Залишок розчиняли в CH2Cl2 та послідовно промивали 1 M водним розчином H 2SO4, насиченим водним розчином NaHCO3 та водою. Органічну фазу випаровували під пониженим тиском з отриманням суміші 2хлор-9-(2',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденіну та 2-хлор-9-(3',5'-ди-O-бензоїл-β-Dрибофуранозил)-аденіну (обидва в сумі складали приблизно 65 % за HPLC), а також 2',3',5'-триO-бензоїл-2-хлораденозину (приблизно 30 % за HPLC). Приклад 3. Ізомеризація 2-хлор-9-(2',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденіну (V) в 2хлор-9-(3',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденін (VΙ) Суміш 2-хлор-9-(2',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденіну та 2',3',5'-три-O-бензоїл-2хлораденозину, отриману в прикладі 2, додавали в 25 л MeOH. Отриману в результаті суміш нагрівали зі зворотним холодильником протягом 40-45 годин при енергійному перемішуванні. Після цього суміш піддавали гарячому фільтруванню в вакуумі, а відфільтрований залишок промивали за допомогою MeOH. Фільтрат, який містив 2',3',5'-три-O-бензоїл-2-хлораденозин та 2-хлор-9-(2',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденін, направляли на повторну оборку для отримання 2-хлораденозину шляхом гідролізу. Тверду речовину сушили в вакуумі при 50 °C з отриманням 880 г 2-хлор-9-(3',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденіну з чистотою за HPLC, що складає > 98 %. Приклад 4. Отримання 2-хлор-9-(3',5'-ди-O-бензоїл-2'-O-трифторметилсульфоніл-β-Dрибофуранозил)-аденіну (VΙΙ) 2-Хлор-9-(3',5'-ди-O-бензоїл-β-D-рибофуранозил)-аденін (880 г) суспендували в суміші піридину (0,8 л) та CH2Cl2 (8 л). В атмосфері N2 розчин трифторметансульфонового ангідриду (455 мл) в CH2Cl2 (1500 мл) повільно додавали в реакційну суміш при перемішуванні та охолоджували при -10-0 °C. Реакційну суміш утримували протягом 30 хвилин за тих самих умов. Потім додавали насичений водний розчин NaHCO3 при перемішуванні для доведення pH отриманої в результаті суміші до 7. Органічну фазу відокремлювали, промивали водою та випаровували під пониженим тиском з отриманням 1000 г (вихід 95 %) названого продукту, який характеризується чистотою за HPLC, що складає > 95 %. Приклад 5. Отримання 2-хлор-9-(3',5'-ди-O-бензоїл-2'-дезоксі-2'-фтор-β-Dарабінофуранозил)-аденіну (VΙΙΙ) 2-Хлор-9-(3',5'-ди-O-бензоїл-2'-O-трифторметилсульфоніл-β-D-рибофуранозил)-аденін, отриманий в прикладі 4, суспендували в 5 л толуолу. Поступово додавали N, Nдиізопропілетиламін (350 мл) та триетиламінтригідрофторид (1,2 л) при перемішуванні. Реакційну суміш перемішували протягом 48 годин при 35-40 °C та випаровували під пониженим тиском. Залишок обробляли етилацетатом та відфільтровували. До фільтрату додавали насичений водний розчин NaHCO3 при перемішуванні для доведення pH отриманої в результаті суміші до 7. Органічну фазу відокремлювали, промивали водою та випаровували під пониженим тиском. Залишок розчиняли в метанолі (1,5 л) при перемішуванні та при 60 °C. Отриманий розчин витримували при 2-6 °C протягом 2-3 годин до повного осадження. Утворений осад відфільтровували, промивали метанолом та сушили з отриманням 540 г (вихід 60 %) названого продукту, який характеризувався чистотою по HPLC, що складала > 95 %. Приклад 6. Отримання 2-хлор-9-(2'-дезоксі-2'-фтор-β-D-арабінофуранозил)-аденіну (тобто клофарабіну) 2-Хлор-9-(3',5'-ди-O-бензоїл-2'-дезоксі-2'-фтор-β-D-арабінофуранозил)-аденін, отриманий в прикладі 5, суспендували в 1,5 л метанолу. Додавали 30 мл 10 % метанольного розчину MeONa та реакційну суміш перемішували протягом 30 хвилин при 30 °C. Після цього в реакційну суміш додавали воду (1,5 л) та CH2Cl2 (1,5 л). Після перемішування протягом 10 хвилин водну фазу відокремлювали та випаровували під пониженим тиском до об'єму приблизно 1,5 л. Отриманий в результаті розчин, який містив клофарабін, очищували за допомогою препаративної колонкової хроматографії низького тиску з наступним виділенням продукту, що включав кристалізацію в суміші води та ацетону, з отриманням 260 г названої сполуки. Як правило, чистота клофарабіну складала > 99,9 %, а загальний вихід (після 6 стадій) складав 32 %. Даний винахід, що ілюстративно описується в даному документі, може бути відповідним чином здійснений за відсутності будь-якого елементу або елементів, обмеження або обмежень, спеціально не розкритих в даному документі. Так, наприклад, терміни "який містить", "який включає", "який має" тощо слід розуміти розширено та без обмеження. Крім того, терміни та вирази, які використовуються в даному документі, були використані в якості термінів опису, а не обмеження, і також терміни та вирази не призначені для використання з метою виключення будь-яких еквівалентів показаних та описаних ознак або їх частин, але слід відзначити, що можливі різні модифікації, що входять до об'єму даного винаходу, що заявляється. Таким чином, слід розуміти, що, хоча даний винахід був конкретно розкритий за допомогою варіантів 7 UA 115206 C2 5 10 здійснення та необов'язкових ознак, спеціалістами в даній області можуть бути використані модифікації та варіації даного винаходу, які передбачені в ньому, і що такі модифікації та варіації підпадають під об'єм даного винаходу. Даний винахід був описаний в даному документі в широкому сенсі та загалом. Кожні з більш вузьких об'єктів та субгенеричних груп, що потрапляють в загальне розкриття, також складають частину даного винаходу. Це стосується загального опису даного винаходу з умовою або негативним обмеженням, яке виключає будь-який об'єкт, що заявляється, з родового поняття, незалежно від того, вказаний чи ні виключений матеріал конкретно в даному документі. Інші варіанти виконання підпадають під об'єм наступної формули винаходу. Крім того, оскільки ознаки або аспекти даного винаходу описані в термінах груп Маркуша, спеціалістам в даній області повинно бути зрозуміло, що даний винахід тим самим також описується за допомогою будь-якого окремого представника або підгрупи представників групи Маркуша. ФОРМУЛА ВИНАХОДУ 15 1. Спосіб одержання клофарабіну, який включає: (а) одержання 2-хлораденозину ферментативним трансглікозилюванням 2-хлораденину та сполуки формули 1, HO O HO 20 R 1 OH , в якій R являє собою пуринову або піримідинову основу формули 2 або формули 3, відповідно, 1 R 2 4 NH N N N R 3 O 2 R N 25 1 N 3 O , , 2 3 4 в яких R та R незалежно вибрані з групи, яка складається з -Н, -NH2, -ОН та -СН3; та R вибраний з групи, яка складається з -Н та -СН3; (b) частковий захист гідроксильних груп 2-хлораденозину з одержанням суміші сполуки формули 4 та сполуки формули 5, NH2 N N Cl NH2 4 N N R5 O R5 O N N Cl N N R5 O O 5 O HO OR , , в яких R незалежно являє собою захисну групу гідроксилу; (c) ізомеризацію сполуки формули 4 в сполуку формули 5; (d) одержання сполуки формули 6 зі сполуки формули 5, OH 5 5 8 UA 115206 C2 NH2 6 N N Cl N N R5 O O R5 O OR6 , в якій OR являє собою відхідну групу; (e) фторування сполуки формули 6 до сполуки формули 7, 6 NH2 7 N N Cl N N R5 O O F R5 O 5 10 15 , та (f) зняття захисту зі сполуки формули 7 з одержанням клофарабіну. 2. Спосіб за п. 1, в якому трансглікозилювання на стадії (а) виконують з використанням пуриннуклеозидфосфорилази або комбінації пурин-нуклеозидфосфорилази та уридинфосфорилази. 3. Спосіб за п. 1 або п. 2, в якому фторування на стадії (e) виконують з використанням фторуючого засобу. 4. Спосіб за п. 3, в якому фторуючий засіб вибирають з групи, яка складається з фтористоводневої кислоти та суміші фтористоводневої кислоти та органічної основи Льюїса. 5. Спосіб згідно з п. 4, в якому органічна основа Льюїса являє собою амін. 1 6. Спосіб за будь-яким з пп. 1-5, в якому R являє собою піримідинову основу, яка являє собою уридин. 5 7. Спосіб за будь-яким з пп. 1-6, в якому R являє собою захисну групу гідроксилу, яка являє собою бензоїл. 6 8. Спосіб за будь-яким з пп. 1-7, в якому OR являє собою відхідну групу, яка являє собою трифторметансульфонат. 9 UA 115206 C2 10 UA 115206 C2 Комп’ютерна верстка В. Мацело Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 11
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the synthesis of clofarabine
Автори англійськоюZabudkin, Aleksandr, Matvienko, Victor, Matviienko, Iaroslav, Sypchenko, Volodymyr
Автори російськоюЗабудкин Александр, Матвиенко Виктор, Матвиенко Ярослав, Сыпченко Владимир
МПК / Мітки
МПК: C12P 19/40, C12P 17/18
Мітки: спосіб, клофарабіну, синтезу
Код посилання
<a href="https://ua.patents.su/13-115206-sposib-sintezu-klofarabinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб синтезу клофарабіну</a>
Спосіб твердофазового синтезу a,b-ненасиченої алкеноатної смоли

Номер патенту: 71917
Опубліковано: 17.01.2005
Автори: Мортон Джордж К., Лабадін'єр Рішар Ф., Сальвіно Джозеф М., Мейсон Хелен Дж.
МПК: C07D 307/33, C07C 317/44, C07C 269/00, C07C 45/00, C07C 249/00, C07C 233/87, C07C 57/00, C07C 231/00, C07C 259/00, C08F 12/00, C07C 239/00, C07C 47/20, C07C 49/213, C07C 211/03, C07D 311/68, C07C 51/347, C07C 317/46, C07C 315/00, C07C 45/65, C07C 251/54, C07C 233/33, C07C 45/61, C07C 47/52, C07C 271/18, C07C 49/807, C07C 209/40, C07C 209/66, C07C 209/30, C07D 313/00, C07C 45/41
Мітки: алкеноатної, твердофазового, синтезу, a,b-ненасиченої, смолі, спосіб
Формула / Реферат:
1. Спосіб одержання -ненасиченої алкеноатної смоли формули,де являє собою твердий носій;L відсутній або являє собою зв'язувальну групу;Rm являє собою Н або аліфатичну групу іRn являє собою аліфатичну або ароматичну групу, що включає(а) обробку суміші першого розчинника і полімерної фосфонацетоксисмоли формули,де R20 і R21 незалежно являють собою алкіл;надлишком...
Спосіб синтезу хіральних n-арилпіперазинів

Номер патенту: 79273
Опубліковано: 11.06.2007
Автори: Фейгельсон Грегг Брайан, Зелдіс Джозеф, Джирковскі Іво
МПК: C07D 319/00, C07D 405/12
Мітки: n-арилпіперазинів, синтезу, хіральних, спосіб
Формула / Реферат:
1. Спосіб отримання сполуки формули VII, VIIдеR являє собою С1-С3 алкіл,Y являє собою фрагмент, вибраний з групи, що включає С1-С6 алкокси, С1-С6 алкіл, С3-С7 циклоалкіл або С3-С7 циклоалкокси,Аr являє собою 2,3-дигідробензодіоксин-5-іл або феніл, який необов'язково заміщений і який містить до трьох замісників, незалежно вибраних з...
Спосіб синтезу ефірів тіофенкарбонової кислоти та його застосування

Номер патенту: 79157
Опубліковано: 25.05.2007
Автори: Лекув Жан-П'єр, Вейс-Людо Люсіль, Ланглуа Паскаль
МПК: C07B 61/00, C07D 333/38
Мітки: ефірів, синтезу, спосіб, кислоти, тіофенкарбонової, застосування
Формула / Реферат:
1. Спосіб промислового синтезу сполук формули (І):, (І)де R і R', які є однаковими або різними, кожний являє собою лінійну або розгалужену (С1-С6)алкільну групу,який відрізняється тим, що сполуку формули (IIІ):, (III)де R є таким, як визначено вище, піддають...
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Рок Майкл Харольд, Петерсен Ханс, Еллегор Петер
МПК: C07C 255/56, C07C 253/30, C07D 307/87
Мітки: варіанти, проміжні, s-циталопраму, рацемічних, спосіб, кетони, сполук, одержання, циталопраму
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, реактор гетерогенного екзотермічного синтезу та спосіб здійснення гетерогенних екзотермічних реакцій синтезу з високою продуктивністю

Номер патенту: 73466
Опубліковано: 15.08.2005
Автори: ПАГАНІ Джорджіо, Філліппі Ерманно
МПК: B01J 8/00, B01J 35/00, B01J 8/04, C01C 1/04
Мітки: екзотермічних, модернізації, продуктивністю, гетерогенних, реакцій, синтезу, екзотермічного, реактор, гетерогенного, високою, здійснення, реактора, спосіб
Формула / Реферат:
1. Спосіб модернізації in situ реактора гетерогенного екзотермічного синтезу, що включає зовнішній кожух, в якому розміщені один на одному в просторовому взаємозв'язку каталітичні шари, за яким попередньо встановлюють принаймні перший каталітичний шар у верхній частині згаданого кожуха та принаймні другий шар каталізатора в нижній частині цього кожуха, потім перший та другий шари завантажують першим каталізатором із завчасно...
Попередній патент: Застосування 7-бром-5-(о-хлорфеніл)-3-пропокси-1,2-дигідро-3н-1,4-бенздіазепін-2-ону для гальмування нейропатичного болю та судом різної етіології
Наступний патент: Дістанційно- і самокерований агрегат бойової автономної модульної платформи високої прохідності для прихованого транспортування військових вантажів
Випадковий патент: Спосіб виготовлення ксенотрансплантатів