Похідне тетрагідрокарбазолу або його сіль, сольват або гідрат як агоніст 5-нт1-подібного рецептора, спосіб їх одержання та фармацевтична композиція
Номер патенту: 41254
Опубліковано: 17.09.2001
Автори: Кауманн Альберто Джуліо, Кінг Френсіс Девід, Янг Родні Крістофер, Гастер Ларамі Мері








Формула / Реферат
1. Производное тетрагидрокарбазола формулы (I)
в которой R1 представляет собой нитрогруппу, -CO2R4, CN, -(CH2)nCONR5R6, -(CH2)nSO2NR5R6 или С1-6алкилсульфониламино(СН2)n, R4 представляет собой арилС1-6алкил, R5 и R6 каждый независимо представляет собой водород или С1-6алкил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо, n представляет собой 0,1 или 2, и NR2R3-это NH2, или в которой R1 представляет собой карбоксамидо-(-СОNH2), и NR2R3 представляет собой метиламино-, этиламино-, н-пропиламино-, изопропиламино-, диметиламино-, бензиламино-, пирролидин-1-ил или N-(метил)этиламиногруппу, или его соль, сольват и/или гидрат в качестве агониста или частичного агониста 5-НТ1-подобного рецептора.
2. Производное тетрагидрокарбазола по п.1, отличающееся тем, что R1 представляет собой -(CH2)nCONR5R6, n является 0, а R5 и R6 каждый независимо является атомом водорода, метилом, этилом или пропилом, или его соль, сольват и/или гидрат.
3. Производное тетрагидрокарбазола по п.2, отличающееся тем, что R5 и R6 каждый независимо представляет собой атом водорода или метил, или его соль, сольват и/или гидрат.
4.Производное тетрагидрокарбазола по п.3, отличающееся тем, что выбрано из следующих соединений:
3-амино-6-циано-1,2,3,4-тетрагидрокарбазол,
(+)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
(-)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(N-метилкарбоксамидо)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(N-метилсульфонамидометил)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазол,
3-амино-6-нитро-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(N,N-диметилкарбоксамидо)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(пиперидин-1-илкарбонил)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(пирролидин-1-илкарбонил)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(N,N-диэтилкарбоксамидо)-1,2,3,4-тетрагидрокарбазол,
3-амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазол,
3-амино-6-карбоксамидометил-1,2,3,4-тетрагидрокарбазол,
3-амино-6-(2-карбоксамидоэтил)-1,2,3,4-тетрагидрокарбазол, или его соль, сольват и/или гидрат.
5. Производное тетрагидрокарбазола по п.1, отличающееся тем, что выбрано из следующих соединений:
3-этиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-н-пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-изопропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-диметиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-бензиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-пирролидинил-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол,
3-(N-(метил)этиламино)-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол, или его соль, сольват и/или гидрат.
6. 3-метиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол или его соль, сольват и/или гидрат.
7. Соль по любому из пп.1-6, отличающаяся тем, что она является солью соляной, серной, фосфорной, янтарной, малеиновой, уксусной или фумаровой кислоты.
8. Способ получения производного тетрагидрокарбазола формулы (I) или его соли, сольвата и/или гидрата
в которой R1 представляет собой нитрогруппу, -CO2R4, CN, -(CH2)nCONR5R6, -(CH2)nSO2NR5R6 или C1-6алкилсульфониламино(СН2)n, R4 представляет собой арилС1-6алкил, R5 и R6-каждый независимо представляет собой водород или С1-6алкил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо, n представляет собой 0, 1 или 2, и NR2R3 - это NH2, или в которой R1 представляет собой карбоксамидо-(CONH2), a NR2R3 представляет собой метиламино-, этиламино-, н-пропиламино-, изопропиламино-, диметиламино-, бензиламино-, пирролидин-1-ил или N-(метил)этиламиногруппу, отличающийся тем, что проводят реакцию соединения формулы (II)
где R1 описан выше, или его кислой соли с соединением формулы (III)
где R2 и R3 описаны выше, или его N-защищенного производного, или реакцию соединения формулы (IV)
где R1 описан выше, a Z представляет собой уходящую группу с соединением формулы HNR2R3, как описано выше, или реакцию соединения формулы (V)
где n=0, 1 или 2 и HNR2R3 описана выше, с ацилирующим или сульфонилирующим агентом, или превращение одного соединения формулы (I) в другое соединение формулы (I), например для получения соединения формулы (I), где R1 представляет собой -(СН2)nСОNH2 (например -СОNH2) или CO2R4, осуществляют гидролиз соединения формулы (I), где R1 представляет собой (СН2)nСN, или его N-защищенную производную, или для получения соединения формулы (I), где R1 представляет собой -CONR5R6, например -CONH2, осуществляют аминирование соединения формулы (I), где R1-это -CO2Н, или его N-защищенного производного; или для получения соединения формулы (I), где один из R2 и R3 представляет собой атом водорода, а другой представляет собой C1-6алкил, осуществляют алкилирование соединения формулы (I), где R2 и R3 являются атомами водорода, после чего при необходимости подвергают депротекции любой из защищенных атомов азота и при желании образуют соль.
9. Фармацевтическая композиция, включающая биологически активное вещество и физиологически приемлемый носитель, отличающаяся тем, что в качестве биологически активного вещества она содержит фармацевтически эффективное количество производного тетрагидрокарбазола формулы (I), как описано выше, по любому из пп.1-7, или физиологически приемлемую соль, сольват и/или гидрат этого соединения.
10. Фармацевтическая композиция, включающая биологически активное вещество и физиологически приемлемый носитель, отличающаяся тем, что в качестве биологически активного вещества она содержит фармацевтически эффективное количество соединения (I) по п.6 или его физиологически приемлемую соль, сольват и/или гидрат этого соединения.
Текст
1. Производное тетрагидрокарбазола формулы (I) C2 (54) ПОХІДНЕ ТЕТРАГІДРОКАРБАЗОЛУ АБО ЙОГО СІЛЬ, СОЛЬВАТ АБО ГІДРАТ ЯК АГОНІСТ 5-НТ1ПОДІБНОГО РЕЦЕПТОРА, СПОСІБ ЇХ ОДЕРЖАННЯ ТА ФАРМАЦЕВТИЧНА КОМПОЗИЦІЯ 41254 где R1 описан выше, a Z представляет собой уходящую группу с соединением формулы HNR2R3, как описано выше, или реакцию соединения формулы (V) 7. Соль по любому из пп. 1-6, отличающаяся тем, что она является солью соляной, серной, фосфорной, янтарной, малеиновой, уксусной или фумаровой кислоты. 8. Способ получения производного тетрагидрокарбазола формулы (I) или его соли, сольвата и/или гидрата , (V) , где n=0, 1 или 2 и HNR2R3 описана выше, с ацилирующим или сульфонилирующим агентом, или превращение одного соединения формулы (I) в другое соединение формулы (I), например для получения соединения формулы (I), где R1 представляет собой -(СН2)nСОNH2 (например -СОNH2) или CO2R4, осуществляют гидролиз соединения формулы (I), где R1 представляет собой (СН2)nСN, или его N-защищенную производную, или для получения соединения формулы (I), где R1 представляет собой -CONR5R6, например -CONH2, осуществляют аминирование соединения формулы (I), где R1 - это -CO2Н, или его N-защищенного производного; или для получения соединения формулы (I), где один из R2 и R3 представляет собой атом водорода, а другой представляет собой C1-6алкил, осуществляют алкилирование соединения формулы (I), где R2 и R3 являются атомами водорода, после чего при необходимости подвергают депротекции любой из защищенных атомов азота и при желании образуют соль. 9. Фармацевтическая композиция, включающая биологически активное вещество и физиологически приемлемый носитель, отличающаяся тем, что в качестве биологически активного вещества она содержит фармацевтически эффективное количество производного тетрагидрокарбазола формулы (I), как описано выше, по любому из пп. 1-7, или физиологически приемлемую соль, сольват и/или гидрат этого соединения. 10. Фармацевтическая композиция, включающая биологически активное вещество и физиологически приемлемый носитель, отличающаяся тем, что в качестве биологически активного вещества она содержит фармацевтически эффективное количество соединения (I) по п. 6 или его физиологически приемлемую соль, сольват и/или гидрат этого соединения. (I) в которой R1 представляет собой нитрогруппу, -CO2R4, CN, -(CH2)nCONR5R6, -(CH2)nSO2NR5R6 или C1-6алкилсульфониламино(СН2)n, R4 представляет собой арилС1-6алкил, R5 и R6 - каждый независимо представляет собой водород или С1-6алкил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо, n представляет собой 0, 1 или 2, и NR2R3 - это NH2, или в которой R1 представляет собой карбоксамидо-(CONH2), a NR2R3 представляет собой метиламино-, этиламино-, н-пропиламино-, изопропиламино-, диметиламино-, бензиламино-, пирролидин-1-ил или N-(метил)этиламиногруппу, отличающийся тем, что проводят реакцию соединения формулы (II) , (II) где R1 описан выше, или его кислой соли с соединением формулы (III) , (III) где R2 и R3 описаны выше, или его N-защищенного производного, или реакцию соединения формулы (IV) , (IV) назначение эрготамина, дигидроэрготамина или метисергида, которые также используются как профилактические средства. Эти лекарства подавляют 5-НТ1-подобные рецепторы, но обладают и другим действием; лечение ими связано с рядом неблагоприятных побочных эффектов. Кроме того, некоторые пациенты испытывают "головные боли при снятии лекарств", которые появляются после прекращения лечения продуктом спорыньи, таким, как эрготамин, заставляя их повторять лечение, и Настоящее изобретение относится к некоторым производным тетрагидрокарбазола для применения в лечении расстройств, характеризуемых избыточным расширением просвета сосудов, в частности, лечения мигрени. Мигрень - несмертельное заболевание, которым страдает один из десяти человек; основной симптом - головная боль; другие симптомы включают рвоту, светобоязнь. В настоящее время наиболее широко используемым лечением является 2 41254 азота, к которому они присоединены, образуют кольцо; n представляет 0, 1 или 2 и R2 и R3, каждый независимо, представляют водород, С1-6алкил или бензил, или, вместе с атомом азота, к которому они присоединены, образуют кольцо пирролидино, пиперидино или гексагидроазепино; и их физиологически приемлемых солей в производстве лекарства для лечения состояния, где показан 5-НТ1-подобный антагонист, в частности, мигрени, а также ее профилактики. Изобретение также предусматривает способ лечения состояния, в котором показан 5-НТ1-подобный антагонист, в частности, мигрени, который включает назначение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I) или его физиологически приемлемой соли. R1 представляет водород, галоген, циано, гидрокси, С1-6алкокси, арилС1-6алкокси, -CO2R4, -(СН2)nCONR5R6 или –(СН2)nSO2NR5R6, и R2 и R3, каждый независимо, представляют водород или С1-6алкил. Следует сказать, что соединения формулы (I) могут содержать один или несколько асимметричных центров, и такие соединения существуют как оптические изомеры (энантиомеры). Итак, изобретение включает все такие энантиомеры и смеси, включая их рацемические смеси. В соединениях формулы (I) атомом галогена может быть фтор, хлор, бром или йод. Алкильная группа или половина может иметь прямую или разветвленную цепь. Подходящие ариловые группы включают, например, ненасыщенные моноциклические или бициклические кольца и частично насыщенные бициклические кольца с до 12 атомами углерода, такие как фенил, нафтил и тетрагидронафтил. Если R5 и R6, вместе с атомом азота, образуют кольцо, это, предпочтительно, 57-членное насыщенное гетероциклическое кольцо, которое может содержать другой гетероатом, выбранный из кислорода, серы или азота. Подходящие кольца включают пирролидино, пиперидино, пиперазино и морфолино. В вышеуказанных соединениях R1 предпочтительно представляет галоген (например, бром), СF3, С1-6алкокси (например, метокси), (СН2)nCN, -(СН2)nСОNR5R6, -(CH2)nSO2NR5R6 или С1-6алканоиламино. Наиболее предпочтительно R1 представляет группу -(СН2)nСОNR5R6, в которой n представляет 0, и R5 и R6, каждый независимо, представляют водород, метил, этил или пропил. Желательно, чтобы R5 и R6, независимо, представляли водород или метил. Если R1 представляет –CO2R4, R4, предпочтительно, представляет С1-6алкил. R2 и R3, каждый, предпочтительно представляют водород, метил или этил. Наиболее предпочтительно NR2R3- -NН2. Для использования в соответствии с настоящим изобретением соединение формулы (I) является, предпочтительно, частичным антагонистом. Подходящие физиологически приемлемые соли очевидны для специалистов данной области и включают, например, соли кислотного присоединения, такие как те, которые образованы с неорганическими кислотами, например, хлористоводо приводит к привыканию. Совсем недавно для потенциального использования в лечении мигрени были предложены различные производные триптамина. Ввиду вышесказанного, существует необходимость получить эффективные и безопасные медикаменты для лечения мигрени. В патентах США №№ 4257952, 4172834, 4062864 и 3059309 раскрыт большой класс тетрагидрокарбазолов с формулой: Q1 N=B Q2 Q3 N Q4 R , в которой N=В является, помимо прочего, -NHR' или -NR'R", где R' и R" - низший алкил, арилнизший алкил или вместе образуют гетероциклическое кольцо; R, помимо прочего, - водород; Q1, помимо прочего, - водород, галоген, низший алкокси, циано, -СО2R1 или –СОNR2R3 (где R1 может быть водородом, низшим алкилом, или –CH2Ar и R2 и R3 - водород, низший алкил или вместе образуют гетероциклическое кольцо); Q2, помимо прочего, - водород, арил-(низший алкокси), гидрокси, тригалометил, нитро или алканоиламино, и Q3 и Q4 могут каждый быть, помимо прочего, водородом. Эти соединения известны как обладающие обезболивающим, психотропным и антигистаминным действием. Неожиданно было выявлено, что некоторые тетрагидрокарбазолы противодействуют и частично противодействуют 5-окси-триптамин-подобным рецепторам и могут найти применение в лечении состояний, в которых показан 5-НТ1-подобный антагонист или частичный антагонист, в особенности, состояний, связанных с головной болью, таких, как мигрень, общая головная боль и боль, связанная с изменениями в сосудах. В этом описании термин "5-НТ1-подобный антагонист" используется для обозначения частичных антагонистов на этом рецепторе. Поэтому настоящее изобретение предусматривает использование соединений общей формулы (I): R 2 1 NR R 3 N H , (I) в которой: R1 представляет водород, галоген, трифторометил, нитро, гидрокси, C1-6алкил, С1-6алкокси, арил С1-6алкокси, -CO2R4, -(CH2)nCN, -(СН2)nCONR5R6, -(CH2)nSO2NR5R6, С1-6алканоиламино(СН2/n или С1-6алкилсульфониламино(СН2)n; R4 представляет водород, С1-6алкил или арил С1-6алкил; R5 и R6, каждый независимо, представляют водород или С1-6алкил, или R5 и R6, вместе с атомом 3 41254 3-амино-6-карбоксамидометил-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-метиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-этиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-н-пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-i-пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-диметиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-бензиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-пирролидинил-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат и 3-/N-/метил/этиламино/-6-карбоксамидо-1,2,3,4тетрагидрокарбазол оксалат, 3-амино-6-/2-карбоксамидоэтил/-1,2,3,4-тетрагидрокарбазол оксалат. В другом аспекте настоящее изобретение предусматривает новое соединение формулы (I), т. е. соединение (IA) или какое-либо из упомянутых соединений (в виде свободного основания или физиологически приемлемой соли) для использования в качестве терапевтического агента, в частности, как 5-НТ1 антагониста или частичного антагониста, например, в лечении мигрени. Изобретение также предусматривает способ получения новых соединений формулы (I). Соединения формулы (I) можно получить методами, известными в области техники для получения тетрагидрокарбазолов, например: А. Реакцией соединения формулы (II): родной, серной или фосфорной кислотами и органическими кислотами, например, янтарной, малеиновой, уксусной или фумаровой кислотами. Другие неприемлемые физиологически соли, например, оксалаты, могут использоваться, например, в изоляции соединений формулы (I), и включены в объем этого изобретения. В объем изобретения также включены сольваты и гидраты соединений формулы (I). R 1 NH2 N H , (IA) в которой R1, как определено выше, с оговоркой, что R1 - не водород, гидрокси, метокси или бензилокси и их соли. Настоящее соединение далее предусматривает следующие специфические соединения, которые также считаются новыми: 3-амино-6-циано-1,2,3,4-тетрагидрокарбазол гидрохлорид, (+)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид, (-)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-амино-6-бромо-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-амино-6-метил-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-/N-метилкарбоксамидо/-1,2,3,4-тетрагидрокарбазол полуоксалат, 3-амино-6-цианометил-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-/N-метилсульфонамидометил/-1,2,3,4тетрагидрокарбазол оксалат, 3-амино-6-хлоро-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-трифторметил-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-н-бутилокси-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-нитро-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-/N,N-диметилкарбоксамидо/-1,2,3,4тетрагидрокарбазол полуоксалат, 3-амино-6-нитро-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-/N,N-диметилкарбоксамидо/-1,2,3,4тетрагидрокарбазол полуоксалат, 3-амино-6-/пиперидин-1-илкарбонил/-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-амино-6-/пирролидин-1-илкарбонил/-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-амино-6-/N,N-диэтилкарбоксамидо/-1,2,3,4-тетрагидрокарбазол гидрохлорид, 3-амино-6-/ацетамидо/-1,2,3,4-тетрагидрокарбазол оксалат, 3-амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат, R 1 NHNH2 , (II) (в которой R1 - как определен выше) или его соли кислотного присоединения с соединением формулы (III): 2 NR R 3 O , (III) (в которой R2 и R3 - как определены выше) или его N-защищенным производным, или В. Реакцией соединения формулы (IV): R 1 Z N H , (IV) (в которой R1 имеет то же значение, как определено для формулы (I) и Z - уходящая группа), с соединением формулы HNR2R3; 4 41254 С.Реакцией соединения формулы (V): или метансульфонилхлорид), алкилэстеры, активированные эстеры и симметричные и смешанные ангидриды. Реакцию можно проводить в органическом растворителе, таком как галоалкан (например, дихлорметане), амиде (например, N,N-диметилформамиде; эфире (например, тетрагидрофуране) или третичном амине, таком как пиридин. В общем, также используется основание, например, триэтиламин, диметиламинопиридин или карбонат или бикарбонат щелочного металла. Реакция проводится при температуре в пределах от -10 до 100°С. Соединения формулы (V) можно получить способами, аналогичными процессам (А) и (В), описанным выше. Еще соединение (V) можно получить восстановлением соединения формулы (I), в котором R1 - нитро, например, каталитическим гидрированием. В химии хорошо известно, что гидролиз нитрила первоначально дает амид, который затем можно гидролизовать в кислоту. Поэтому точный продукт способа (Di) будет зависеть от условий реакции, выбранных для гидролиза. Чтобы получить соединение, в котором R1 представляет Н2NСО-, гидролиз предпочтительно проводится с использованием перекиси водорода в присутствии гидроксида щелочного металла, например, гидроксида натрия, в растворителе, таком как спирт, метаноле. Другие подходящие средства гидролиза включают уксусную кислоты и BF3; или муравьиную кислоту и бромистоводородную или хлористоводородную кислоты. Чтобы получить соединение, в котором R1 представляет -СООН кислоту или основание, можно использовать каталитический гидролиз. Способ (Dii) можно осуществить реакцией соединения формулы (I), в котором R1 - -CO2H, с амидом HNR5R6, в присутствии связующего агента, например, дициклогексилкарбодиимида или N,N-карбонилдиимидазола. В другом варианте исходный материал карбоновой кислоты можно вначале подвергнуть реакции для получения активированного производного карбоксильной группы, например, кислотного хлорида, кислотного ангидрида или активированного эстера, которое затем непосредственно реагирует с амином HNR5R6. Карбоновую кислоту можно также активировать на месте обработкой гексаметилфосфоротриамидом. Алкилирование в соответствии со способом (Diii) можно осуществить реакцией амина формулы (I) с ацилирующим агентом, например, ангидридом, таким, как уксусный или пропионовый ангидрид, чтобы получить промежуточное соединение, в котором один из R2 или R3 является –С(О)С1-6алкилом, с последующим восстановлением данного промежуточного соединения для получения нужного продукта. Специалистам также известны другие реагенты и условия. Расщепление по способу (Div) можно провести восстановлением с использованием хорошо известных методов. Во многих из указанных реакций необходимо защитить группу -NR2R3, когда одна или обе из групп представляют водород. Подходящие N-защитные группы хорошо известны в области техники и включают, например, ацильные группы, такие 2 H2N(CH2) n NR R 3 N H , (V) с ацилирующим или сульфонилирующим агентом; D. Преобразованием одного соединения формулы (I) в другое соединение формулы (I), например: (i) чтобы получить соединение формулы (I), в котором R1 представляет –(CH2)nCONH2 или СО2R4, проводят гидролиз соединения формулы (I), в котором R1 представляет –(СН2)nСN, или его N-защищенного производного; (ii) чтобы получить соединение формулы (I), в котором R1 представляет -СОNR5R6, аминируют соединение формулы (I), в котором R1 представляет –СО2Н, или его N-защищенного производного или (iii) чтобы получить соединение формулы (I), в котором один из R2 и R3 - водород, а другой – С1-6алкил, алкилируют соединение (I), в котором R2 и R3, оба, - водороды; (iv) чтобы получить соединение формулы (I), в котором R1 представляет гидрокси, расщепляют соединение, в котором R1 представляет алкокси или аралкокси; при необходимости, с последующим освобождением от защиты любых защитных атомов азота и образованием соли при желании. Способ (А), который является формой синтеза индола Фишера, может проводиться с использованием методов, хорошо известных в химии. Так, реакцию можно провести в растворителе, например, в спирте, таком как этанол или бутанол; или уксусной кислоте, и при температуре в пределах 0-150°С. Гидразины формулы (II), которые обычно используются в качестве соли гидрохлорида, - соединения известные, и их можно получить известными способами. Циклогексанон формулы (III) можно получить окислением соответствующего циклического спирта с использованием окисляющего агента, такого, как пиридин хлорхромат, пиридин дихромат, дипиридин Сr(VI)оксид, гипохлорит натрия, гипохлорит кальция или диоксид марганца. Уходящей группой Z в соединениях формулы (IV) может быть, например, атом галогена или сульфонилокси группа, например, n-толуолсульфонилокси или метансульфонилокси. Процесс (В) можно провести в инертном органическом растворителе, таком как спирт, например, метанол или эфир, например, тетрагидрофуран и при температуре в пределах 0-150°С. Соединения формулы (IV) можно получить реакцией гидразина формулы (II) с соответствующе замещенным соединением циклогексанона. Если Z - ацилокси или сульфонилокси, его можно получить из соединения (IV), в котором Z - гидрокси, с использованием стандартных процедур. Подходящие ацилирующие и сульфонилирующие агенты, которые могут использоваться в способе (С), включают хлориды карбоновой и сульфокислоты (например, ацетилхлорид 5 41254 как ацетил, трифтороацетил, бензоил, метоксикарбонил, т-бутоксикарбонил, бензилоксикарбонил или фталоил; и аралкильные группы, такие как бензил, дифенилметил или трифенилметил. Если R2 и R3, оба, представляют водород, атом азота, предпочтительно, защищен как фталимид. Защитные группы должны легко удаляться в конце последовательности реакции. L-защиту можно снять обычными способами, например, фталоиловую группу можно убрать реакцией с гидразином; ацильную группу, такую как бензоил, можно расщепить гидролизом, и аралкильную группу, такую как бензил можно расщепить гидрогенолизом. Если соединение формулы (I) получается в виде смеси энантиомеров, их можно разделить обычными методами, например, реакцией смеси с подходящей оптически активной кислотой, такой как д-винокаменная кислота, л-яблочная кислота, л-миндальная, л-гулоновая или 2,3,4,6/ди-О-изопропилиден-кето-L-гулоновая кислоты, чтобы получить две стереоизомерные соли, которые можно разделить, например, кристаллизацией. Иначе смеси энантиомеров можно разделить хроматографией, например, на хиральной ВЭЖХ колонне. Как было выявлено, соединения формулы (I) являются антагонистами и частичными антагонистами на рецепторах, подобных 5-НТ1, и найдут применение в лечении и/или профилактике мигрени и других состояний, связанных с головными болями. Для использования в медицине соединения настоящего изобретения обычно назначаются как стандартные фармацевтические композиции. Поэтому далее настоящее изобретение предусматривает фармацевтические композиции, включающие новое соединение формулы (I) или его физиологически приемлемую соль и физиологически приемлемый носитель. Соединения настоящего изобретения формулы (I) могут назначаться орально, парентерально, и приемом за щеку, под язык, в нос, через прямую кишку, через кожу в виде соответствующих фармацевтических композиций. Соединения формулы (I) и их физиологически приемлемые соли, которые активны при оральном приеме, могут быть в форме жидкостей, например, сиропов, суспензий или эмульсий, или таблеток, капсул и лепешек. Жидкая лекарственная форма может обычно состоять из суспензии или раствора соединения или его физиологически приемлемой соли в подходящем жидком носителе, например, водном растворителе (вода, этанол, глицерин), или неводном растворителе, таком как полиэтиленгликоль или масло, форма может также содержать суспендирующий агент, консервант, вкусовой или цветовой агент. Композицию в форме таблетки можно приготовить с использованием любого подходящего фармацевтического носителя, применяемого в приготовлении твердых форм. Примеры таких носителей включают стеарат магния, крахмал, лактозу, сахарозу и целлюлозу. Композицию в форме капсулы можно получить с использованием традиционных процедур капсулирования. Например, гранулы, содержащие ак тивный ингредиент, можно приготовить с использованием стандартных носителей и затем заполнением в твердую желатиновую капсулу; дисперсию или суспензию можно приготовить с применением подходящих фармацевтических носителей, например, водных смол, целлюлоз, силикатов или масел и заполнением дисперсии или суспензии в мягкую желатиновую капсулу. Обычные парентеральные композиции состоят из раствора или суспензии соединения или физиологически приемлемой соли в стерильном водном носителе или парентерально приемлемом масле, например, полиэтиленгликоле, поливинилпирролидоне, лецитине, арахисовом масле или кунжутном масле. В другом случае раствор может быть лиофилизирован и затем восстановлен подходящим растворителем перед приемом. Композиции для носового применения могут иметь форму аэрозолей, капель, гелей и порошков, форма аэрозоля обычно включает раствор или мелкую суспензию активного вещества в физиологически приемлемом водном или неводном растворителе и предназначена для одноразового или многоразового использования в стерильной форме в герметичном контейнере, который может иметь форму патрона или заполняться в емкость с распылителем. Герметичный контейнер может быть универсальным дозатором в виде одноразового ингалятора или в виде аэрозольного дозатора с измерительным клапаном, который используется, пока не закончится лекарство, а потом выбрасывается. Если дозирующая форма включает аэрозольный дозатор, она содержит пропеллант, который может быть сжатым газом, воздухом или органическим пропеллантом, таким как фторохлороуглеводород. Аэрозольная дозирующая форма может также быть насосом-распылителем. Композиции, предназначенные для применения за щеку или под язык, включают таблетки, лепешки и пастилки, в которых активный ингредиент формуется с носителем, таким как сахар и аравийская камедь, трагакант или желатин и глицерин. Композиции для ректального применения обычно в форме шариков, содержащих обычную суппозитарную основу – масло какао. Композиции, приемлемые для трансдермального применения, включают жидкие мази, гели и пластыри. Предпочтительна форма дозировки - единичная доза (таблетка, капсула или ампула). Каждая единица дозировки содержит для орального применения от 1 до 250 мг (а для парентерального - от 0,1 до 25 мг) соединения формулы (I) или его физиологически приемлемой соли, рассчитанного каксвободное основание. Физиологически приемлемые соединения изобретения обычно назначаются в ежедневном режиме (для взрослого пациента) с оральной дозой между 1 и 500 мг, предпочтительно, между 10 и 400 мг, при внутривенном, подкожном применении - 10-250 мг, внутримышечная доза составляет 0,1-100 мг, предпочтительно, 0,1-50 мг соединения формулы (I) или ее физиологически приемлемой соли, рассчитанной как свободное основание, причем соединение назначается от 1 до 4 раз в 6 41254 день на период продолжительной терапии, например, в течение недели или больше. Биологические данные Тестирование 5-НТ1-подобного рецептора Подкожная вена собаки Геликоиды подкожной вены собаки помещались при 37°С в модифицированный раствор Кребса с силой покоя 10 мN. Раствор также содержал 1 mмоль/л каждого из кетанзерин празозина, атропина и мепирамина, 6 mммоль/л кокаина и 200 mммоль/л аскорбата. На полиграфе с датчиками силы измерялись почти изомерные сокращения. Ткани дважды подвергались действию 5-окситриптамина (5-НТ) в количестве 2 mммоля/л с последующим промыванием. Определялась кривая эффекта кумулятивной концентрации, а затем кривая на 5-НТ в присутствии наивысшей используемой концентрации испытуемого соединения. Сокращения, вызванные испытуемым соединением, сравнивались с сокращениями, вызванными 5-НТ. Рассчитывалась активность испытуемого соединения как коэффициент максимального действия, вызванного соединением, над действием, вызванным 2 mмолями/л 5-НТ. Из соответствующей кривой действия был вычислен ЕС50 испытуемого соединения. Затем методом Марано и Кауманна (1976, J. Pharmacol. Exp. Ther. 198, 518525) были определены константы Кр соответствующей диссоциации равновесия. В этом тестировании соединения Примеров 2, 4, 5, 6, 9, 10, 11, 13, 17, 18, 21 и 24 имели ЕС50 в пределах от 0,1 до 15 mммолей. Основная артерия кролика Методы Эксперименты проводились на внутричерепных артериях кролика, на изолированной базилярной артерии таким же методом, как описан выше (Парзон и Уэлли, 1989, Eur. J. Pharmacol. 174, 189-196). Вкратце, кролики умерщвлялись большой дозой анестезирующего вещества (пентобарбитон натрия). Весь мозг быстро удалялся и погружался в холодный модифицированный раствор Кребса, и базилярная артерия удалялась с помощью препаровальной лупы. Раствор Кребса имел следующий состав (мМ) Na+ (120); К+ (5); Са2+ (2,25); Мg2+ (0,5); Cl- (98,5); SO42- (1); ЭДТК (0,04), уравновешенный 95% O2/5% CO2. Эндотелий удалялся мягким прикосновением к полости тонкой металлической проволоки. Артерии нарезались в кольцевые сегменты (шириной 4-5 мм) и погружались для записи изометрического напряжения в 50 мл ванночки для тканей с модифицированным раствором Кребса и добавлением (мМ) Na2+ (20); фумарата (10); пирувата (5); L-глютамата (5) и глюкозы (10). Затем артерии помещались в силу покоя 3-4 мN при 37°С ,и через раствор пузырьками пропускался 95% O2/5% CO2. После тестов на первоначальную реактивность деполяризующим раствором KCl (90 мМ) и на отсутствие расслабления, вызванного ацетилхолином, из-за предшествующего сокращения от 5-НТ (10 мМ), в присутствии аскорбата 200 мМ, кокаина 6 мМ, индометацина 2,8 мМ, кетанзерина 1 мМ и празозина 1 мМ были построены куму ля- тивные кривые концентрации - воздействия (2 нМ - 60 мМ) на 5-НТ. В этом тестировании соединения Примеров 2, 5, 6, 15, 17, 24, 25, 26, 28 и 29 имели ЕС50 в пределах 0,04-15. Пример 1 3-Амино-6-циано-1,2,3,4-тетрагирокарбазол гидрохлорид Раствор 4-аминоциклогексанол гидрохлорида (6,08 г, 0,04 М) в воде (60 мл) доводился до рН 8 водным раствором бикарбоната натрия. Добавлялся N-карбэтокси-фталимид (0,76 г, 0,04 М), а затем тетрагидрофуран (до получения гомогенного раствора). Чистый раствор перемешивался при комнатной температуре всю ночь. Во время этого выпадало в осадок вещество белого цвета. Тетрагидрофуран удалялся под вакуумом, и оставшийся водный раствор экстрагировался этилацетатом до полного очищения раствора. Экстракты этилацетата соединялись, промывались водой, высушивались (MgSO4) и концентрировались до получения 4-фталимидо циклогексанола в виде белого вещества, (7,1 г). Раствор 4-фталимидо циклогексанола (7,1 г, 0,029 М) в дихлорметане (250 мл) обрабатывался пиридиний хлорохроматом (8,6 г, 0,04 М), и полученная темная смесь перемешивалась при комнатной температуре всю ночь. Был добавлен диэтиловый эфир (50 мл), и смесь фильтровалась через кизельгур. Фильтрат концентрировался в вакууме, и остаток очищался колонной хроматографией (SiO2; CHCl3/EtOAc) для получения 4фталимидо циклогексанона в виде твердого вещества белого цвета (6,4 г). 4-Цианофенил гидразин гидрохлорид (4,41 г, 0,026 М) растворялся в уксусной кислоте (100 мл) и добавлялся ацетат натрия (2 г). Добавлялся 4фталимидо циклогексанон (6,4 г, 0,026 М), и смесь нагревалась с флегмацией всю ночь. Растворитель удалялся в вакууме, и остаток растирался с метанолом для получения 3-фталимидо-6-циано1,2,3,4-тетрагидрокарбазола в виде твердого вещества бежевого цвета (5,3 г). Суспензия указанного продукта (1 г) в этаноле (40 мл) обрабатывалась гидразином в воде (10 мл). Реакционная смесь перемешивалась при комнатной температуре всю ночь, в течение которой реактанты растворялись. Растворитель удалялся в вакууме, и остаток разделялся между водным карбонатом калия и этилацетатом. Раствор этилацетата промывался водой, высушивался и концентрировался в вакууме для получения 3-амино-6-циано-1,2,3,4-тетрагидрокарбазола в виде твердого вещества бежевого цвета (500 мг). Этот продукт был преобразован в соль гидрохлорида для получения искомого соединения, т. пл. 289°С (разл.). 1 Н ЯМР [250 МГЦ, CD3OD]. d 1,98-2,18 (1H, м), 2,25-2,40 (1H, м), 2,77 (1H, дд), 2,98 (2Н, м), 3,22 (1H, дд), 3,68 (1H, м), 7,34 (1H, д), 7,43 (1H, д), 7,82 (1H, с). Пример 2 3-Амино-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид Продукт Примера 1 (400 мг) растворялся в тетрагидрофуране, и добавлялся ди-т-бутил дикарбонат (500 мг). Смесь перемешивалась при ко 7 41254 (4,39 г, 18,1 мМ) в орошающем н-бутаноле в течение 20 минут с последующим охлаждением, фильтрацией и выпариванием фильтрата до сухости дала 3-фталимидо-6-бромо-1,2,3,4-тетрагидрокарбазол в виде твердого вещества оранжевого цвета (7,45 г). Этот продукт (0,33 г, 0,83 мМ) суспендировался в этаноле (13 мл) и обрабатывался гидратом гидразина (З мл), затем перемешивался при комнатной температуре всю ночь. Твердый осадок отфильтровывался, и фильтрат выпаривался до сухости и разделялся между К2СО3 (вод) и этилацетатом. После отделения органического слоя, промывания водой, высушивания (МgSО4) и выпаривания до сухости остаток растворялся в МеОН и обрабатывался газом НСl. Растворитель удалялся в вакууме, и остаток кристаллизовался из этанол/этилацетата для выхода искомого соединения в виде твердого вещества кремового цвета (0,15 г), т. пл. 308-310°С. 1 Н ЯМР [250 МГц, ДМСО-д6] d 1,91 (1H, м), 2,10-2,26 (1H, м), 2,63 (1H, дд), 2,84 (2Н, м), 3,04 (1H, дд), 3,50 (1H, м), 7,12 (1H, д), 7,24 (1H, д), 7,55 (1H, с), 8,15 (2Н, шир, с), 11,12 (1H, с). Пример 5 3-Амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол 4-Карбоксамидофенилгидразин гидрохлорид (2,87 г) и 4-фталимидоциклогексанон (3,00 г) смешивались в уксусной кислоте, и смесь нагревалась с флегмацией 2 часа. После охлаждения смесь нейтрализовалась с использованием водного раствора карбоната калия, и полученное желтое твердое вещество фильтровалось, промывалось водой и высушивалось. Очистка колонной хроматографией дала 3-фталимидо-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол (2,8 г). (SiO2, СНСl3/СН3OН). Указанный продукт (1,0 г) суспендировался в этаноле (10 мл) и добавлялся гидрат гидразина (5 мл). Получался чистый раствор, и смесь оставлялась перемешиваться всю ночь до получения осадка. Вся смесь выпаривалась до сухости, промывалась водным раствором К2СО3 и водой. Получалось в виде моногидрата искомое соединение 3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол, т. пл. 146-148°С. 1 Н ЯМР [250 МГц, ДМСО-д6] d 1,49-1,77 (1H, м), 1,83-2,03 (1H, м), 2,17-2,40 (1H, м), 2,62-2,80 (2Н, м), 2,90 (1H, дд), 1 сигнал невидим из-за воды на 3,1, 7,03 (1Н, шир. с), 7,18 (1H, д), 7,58 (1H, д), 7,83 (1H, шир. с), 7,98 (1H, с). Пример 6 (+)- и (-)-3-Амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид Способ 1 (±)-3-т-Бутилоксикарбониламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол разделялся на свои энантиомеры с использованием хиральной ВЭЖХ: (chiracel OD 4,6 мм колонна с промыванием гексаном/этанолом 85:15). (+)-Энантиомер собирался первым и имел т. пл. 150-152°С и [a]D25=+70,1 (в метаноле, 0,41% вес/об.). (-)-Энантиомер имел т. пл. 150-152°С и [a]D25=-79,4 (в метаноле, 0,40% вес/об.). (+)-Энантиомер был преобразован в родственный амингидрохлорид обработкой НСl газом в диоксане, чтобы получить мнатной температуре всю ночь. Растворитель удалялся в вакууме, и остаток очищался колонной хроматографией (SiO2; СНСl3/EtOAc) для получения 3-т-бутилокси-карбониламино-6-циано-1,2,3,4тетрагидрокарбазола (40 мг). Смесь нитрила вышеуказанного продукта (440 мг), водной перекиси водорода (30%, 0,5 мл) и гидроксида натрия (вод) (20%, 0,5 мл), в метаноле (25 мл) перемешивались при комнатной температуре всю ночь. Добавлялся метабисульфит натрия (100 мг), и растворитель удалялся в вакууме. Остаток растворялся в этилацетате, и слой этилацетата отделялся, высушивался и концентрировался в вакууме для получения тягучего твердого вещества, которое очищалось колонной хроматографией (SiO2; СНСl3/EtOAc) для получения 3-т-бутилокси-карбониламино-6-карбоксиамидо-1,2,3,4-тетрагидрокарбазола в виде вещества белого цвета (400 мг), т. пл. 270ºС (разл.). Вышеуказанный продукт (400 мг, 0,0012 М) растворялся в диоксане (100 мл), и через раствор в течение 20 минут пропускался газ HCl. В течение этого времени осаждалось белое вещество. Избыток хлороводорода удалялся из раствора пропусканием через него N2, и твердый продукт, 3амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид собирался фильтрацией, промывался диэтиловым эфиром и высушивался для получения искомого соединения в виде твердого вещества белого цвета (300 мг). т. пл. 270°С (разл.). 1 Н ЯМР [250 МГц, ДМСО-д6] d 1,96 (1H, м); 2,16-2,30 (1H, м), 2,74 (1H, дд), 2,85 (2Н, м), 3,12 (1H, дд), один сигнал затемнен Н2О на 3,6, 7,08 (1H, шир. с), 7,27 (1H, д), 7,61 (1H, д), 7,87 (1H, шир. с), 7,99 (1H, с), 8,39 (3Н, шир. с). Пример 3 3-Амино-6-метокси-1,2,3,4-тетрагидрокарбазол гидрохлорид Реакция 4-метоксифенил гидразин гидрохлорида (0,87 г, 5,0 мМ) с 4-фталимидо-циклогексаноном (1,22 г, 5,0 мМ) в этаноле (20 мл) нагревалась с флегмацией 2 часа с последующим охлаждением и удалением осажденного вещества фильтрацией, что дало 3-фталимидо-6-метокси1,2,3,4-тетрагидрокарбазол (1,62 г). Указанный продукт (1,57 г, 4,5 мМ) суспендировался в метаноле (100 мл) и обрабатывался гидразин гидратом (23 мл) с перемешиванием при комнатной температуре. После 30 минут растворитель удалялся в вакууме, и остаток делился между K2CO3 (вод) и EtOAc. Последний слой отделялся, промывался водой, высушивался (МgSО4) и выпаривался до сухости. Этот остаток растворялся в этаноле и обрабатывался эфирным НСl до помутнения, затем выстаивался всю ночь для получения искомого соединения (0,95 г) т. пл. > 250°С. 1 ЯМР [250 МГЦ, ДМСО-д6] d 1,81-2,02 (1H, м), 2,10-2,28 (1H, м), 2,65 (1H, дд), 2,82 (2Н, м), 3,02 (1H, дд), 1 сигнал затемнен Н2O на 3,5, 3,74 (3Н, с), 6,66 (1H, д), 6,84 (1H, д), 7,14 (1H, д), 8,16 (3Н, шир. с). Пример 4 3-Амино-6-бромо-1,2,3,4-тетрагидрокарбазол гидрохлорид Реакция 4-бромофенилгидразин гидрохлорида (4,0 г, 18,1 мМ) с 4-фталимидо циклогексаноном 8 41254 (+)-энантиомер 3-амино-6-карбоксамидо-1,2,3,4тетрагидрокарбазол гидрохлорида, т. пл. 248251°C, [a]D25=+26,2 (в метаноле, 0,50% вес/об.). (-)-Энантиомер 3-т-бутилоксикарбониламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола также был преобразован в (-)-энантиомер 3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид, т. пл. 248-251ºС, [a]D25=-28,6 (в метаноле, 0,50% вес/об.). Способ 2 (±)-6-Карбоксамидо-3-амино-1,2,3,4-тетрагидрокарбазол обрабатывался одним эквивалентом 2,3:4,6-ди-О-изопропилен-2-кето-L-гулоновой кислоты в метаноле, чтобы получить соль (+)-энантиомера в 38% выходе (относительно рацемата) и 84% энантиомерного избытка (эи). Этот материал рекристаллизовался дважды из метанола для получения соли (+)-энантиомера в 25% общем выходе (относительно рацемата), и > 98% эи. Этот продукт преобразовывался в соль гидрохлорида сначала обработкой водным раствором щелочного металла, и осажденное свободное основание обрабатывалось 2 М вод. НСl в этаноле, чтобы получить (+)-энантиомер-3-амино-6-карбоксамидо1,2,3,4-тетрагидрокарбазол гидрохлорид. Пример 7 3-Амино-6-метил-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (2,16 г) с 4-толулгидразин гидрохлоридом (1,41 г) и последующее снятие защиты продукта способом, описанном в примере 3, дало свободное основание искомого соединения, которое было преобразовано в соль оксалата (0,23 г), т. пл. 272-5°С. Пример 8 3-Амино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (0,37 г) с 4-этокси-карбонилфенилгидразин гидрохлоридом (0,33 г), и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно было преобразовано в соль оксалата (0,11 г), т. пл. 230240°С разл. Пример 9 3-Амино-6-/N-метил-карбоксамидо/-1,2,3,4-тетрагидрокарбазол полуоксалат Реакция 4-фталимидоциклогексанона (1,20 г) с 4-/N-метилкарбоксамидо/-фенилгидразин гидрохлоридом (1,00 г), и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно было преобразовано в соль полуоксалата (0,22 г), т. пл. 227ºС разл. Пример 10 3-Амино-6-цианометил-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (1,05 г) с 4-циано-метилфенилгидразин гидрохлоридом (0,79 г), и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения, которое обрабатывалось щавелевой кислотой для получения соли оксалата (0,49 г), т. пл. 219-224°С разл. Пример 11 3-Амино-6-/N-метилсульфонамидометил/1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (0,42 г) с 4-/N-метилсульфонамидометил/ фенил гидразин гидрохлоридом (0,44 г), и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения. Оно обрабатывалось щавелевой кислотой, чтобы получить соль оксалата (0,15 г), т. пл. 218-222°С, разл. Пример 12 3-Амино-6-хлоро-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (6,7 г) с 4-хлорофенил гидразин гидрохлоридом (4,93 г), и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения, которое обрабатывалось щавелевой кислотой, чтобы получить соль оксалата (2,77 г), разл. 220°С. Пример 13 3-Амино-6-трифторометил-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (1,14 г) с 4-трифторометил фенил гидрохлоридом (1,0 г) и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения (0,40 г). Оно обрабатывалось щавелевой кислотой, т. пл. 212-213°С. Пример 14 3-Амино-6-н-бутилокси-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (1,12 г) с 4-н-бутилоксифенил гидразин гидрохлоридом (1,00 г), и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно обрабатывалось щавелевой кислотой, и получалась соль оксалата (0,47 г), т. пл. 227-229°С. Пример 15 3-Амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (1,00 г) с 4-сульфонамидо фенил гидразин гидрохлоридом (1,08 г), и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения. Оно было преобразовано в соль оксалата (0,090 г), разл. >200ºС. Пример 16 3-Амино-6-нитро-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидоциклогексанона (1,28 г) с 4-нитрофенил гидразин гидрохлоридом (1,00 г), и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения, которое было преобразовано в соль оксалата (0,25 г), т. пл. 275-277°С. Пример 17 3-Амино-6-/N,N-диметилкарбоксамидо/-1,2,3,4тетрагидрокарбазол полуоксалат 3-Амино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол (260 мг, 1,0 мМ) суспендировался в сухом ТГФ (5 мл) и добавлялся ди-трет бутил дикарбонат (320 мг, 1,5 мМ). После 10 минут раствор получался чистым. Смесь перемешивалась 20 ча 9 41254 сов, затем растворитель удалялся, и остаток растворялся в этилацетате, промывался водным раствором бикарбоната натрия и высушивался (MgSO4). После удаления этилацетата остаток растирался эфиром и гексаном, чтобы получить 3т-бутилокси-карбониламино-6-этоксикарбонил1,2,3,4-тетрагидрокарбазол (310 мг). Вышеуказанный продукт (556 мг, 1,55 мМ) суспендировался в этаноле (5 мл) и 2 М NаОН (3 мл) добавлялись к нему. Смесь нагревалась с флегмацией 1 час и выпаривалась до сухости. Остаток растворялся в воде и нейтрализовался уксусной кислотой, когда выпадал в осадок 3-тбутилоксикарбониламино-6-карбокси-1,2,3,4-тетрагидрокарбазол в виде твердого вещества белого цвета (425 мг). Раствор вышеуказанного продукта (400 мг, 1,2 мМ) в сухом ТГФ (8 мл) обрабатывался гексаметил фосфористым триамидом (198 мг, 1,2 мМ), и охлаждался до -10ºС. Газообразный диэтиламин пропускался через смесь 10 минут при этой температуре, затем по каплям добавлялся тетрахлорид углерода (185 мг, 1,2 мМ) в атмосфере азота. Смесь перемешивалась при комнатной температуре 1 час, затем ДМФ удалялся в вакууме. Остаток разделялся между этилацетатом и водой, и органический слой промывался насыщенным раствором бикарбоната натрия, затем рассолом и высушивался (MgSO4). Растворитель удалялся в вакууме, и остаточное масло растиралось эфиром и гексаном, и твердое вещество рекристаллизовалось из толуола, чтобы получить 3-т-бутилоксикарбониламино-6-/N,N-диметил карбоксамидо/-1,2,3,4-тетрагидрокарбазол (198 мг). Этот продукт растворялся в диоксане (5 мл), и через него пропускался газ НСl, чтобы осадить масло, растворитель удалялся в вакууме, и масло растворялось в воде и обрабатывалось раствором K2CO3, чтобы довести рН до 12. Затем свободное основание амина экстрагировалось этилацетатом, высушивалось (MgSO4) и выпаривалось до сухости. Полученное масло растворялось в метаноле и обрабатывалось щавелевой кислотой, чтобы получить искомое соединение в виде твердого вещества бледно-розового цвета (140 мг) т. пл.= =190-195°С. Пример 18 3-Амино-6-/пиперидин-1-ил карбонил/-1,2,3,4тетрагидрокарбазол гидрохлорид Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4-тетрагидрокарбазола (175 мг) с пиперидином и продукт снятия защиты способом, описанным для примера 17, дали искомое соединение, т. пл. 246-249ºС (55 мг). Пример 19 3-Амино-6-/пирролидин-1-ил карбонил/-1,2,3,4тетрагидрокарбазол гидрохлорид Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4-тетрагидрокарбазола (140 мг) с пирролидином, и продукт со снятой защитой в последущем, как описано для примера 17, дали искомое соединение, т. пл. 201-212°C (8I мг). Пример 20 3-Амино-6-/N,N-диэтил карбоксамидо/-1,2,3,4тетрагидрокарбазол гидрохлорид Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4-тетрагидрокарбазола (105 мг) с ди этиламином и снятие защиты продукта, как описано в примере 17, дали искомое соединение, т. пл. 200-205°С (50 мг). Пример 21 3-Амино-6-/ацетамидо/-1,2,3,4-тетрагидрокарбазол оксалат Реакция 4-фталимидо циклогексанона (1,2 г) с 4-/ацетамидо/-фенил гидразин гидрохлоридом (1,0 г), и последующее снятие защиты продукта способом примера 3 дало свободное основание искомого соединения (570 мг). Часть этого продукта (50 мг) обрабатывалась щавелевой кислотой, в метаноле, чтобы получить соль оксалата, которая размягчается при >170°C (38 мг). Пример 22 3-Амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат 3-Фталимидо-6-нитро-1,2,3,4-тетрагидрокарбазол (4,00 г) растворялся в горячем этилацетате (130 мл). К охлажденному раствору добавлялся скелетный никелевый катализатор гидрирования, и смесь гидрировалась при первоначальном давлении в 39 ф/д2 (2,74 кг/см2) 4 часа. После отфильтровывания нерастворимых материалов фильтрат выпаривался до сухости и экстрагировался дважды в 20% водном метаноле, и экстракты соединялись и уменьшались в объеме, чтобы дать 3-фталимидо-6-амино-1,2,3,4-тетрагидрокарбазол (0,31 г). Указанный продукт (0,50 г) растворялся в свежедистиллированном пиридине (30 мл), и добавлялись метансульфонилхлорид (0,28 г) и 4-диметиламинопиридин (46 мг). Смесь нагревалась с перемешиванием при 50°С 5 часов, затем выпаривалась до сухости. Остаток растворялся в хлороформе, промывался водой, рассолом и водным бикарбонатом натрия, затем высушивался (MgSO4), и выпаривался до сухости, чтобы получить бледно-желтое вещество, которое рекристаллизовалось из водного этанола для выхода 3фталимидо-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазола (0,27 г). Указанное соединение суспендировалось в этаноле (15 мл) и добавлялся гидрат гидразина (2,72 г). После 25 минутного перемешивания при комнатной температуре смесь выпаривалась до сухости, разделялась между водой и этилацетатом, и водный слой повторно экстрагировался этилацетатом. Органические экстракты соединялись, промывались водой, высушивались (MgSO4) и выпаривались до получения бледно-желтого твердого вещества. Оно растворялось в метаноле и обрабатывалось щавелевой кислотой (89 мг). Добавление эфира привело к кристаллизации искомого соединения (50 мг), т. пл. 230-233°С. Пример 23 3-Амино-6-карбоксамидометил-1,2,3,4-тетрагидрокарбазол гидрохлорид 3-Амино-6-цианометил-1,2,3,4-тетрагидрокарбазол (2,5 г) и ди-т-бутил дикарбонат (3,63 г) перемешивались в ТГФ (56 мл) 2 часа. ТГФ выпаривался, и остаток разделялся между водным раствором бикарбоната натрия и этилацетатом. Водная фаза повторно экстрагировалась этилацетатом, и соединенные органические экстракты промывались водой, высушивались (MgSO4), и выпаривались до сухости, чтобы получить твердое ве 10 41254 щество, которое растиралось эфиром/гексаном (20%) для получения 3-т-бутилоксикарбониламино-6-цианометил-1,2,3,4-тетрагидрокарбазола в виде чисто-белого вещества (3,44 г). Продукт (7,0 г) растворялся в ДМСО (70 мл) и добавлялась перекись водорода (100 объемная, 3,5 мл). После перемешивания 1 час добавлялась еще перекись (8,5 мл), и смесь перемешивалась 2 часа при комнатной температуре. Добавлялся карбонат калия (0,84 г), и смесь перемешивалась всю ночь и еще 20 часов. Реакционная смесь выливалась в воду (500 мл), и полученное твердое вещество белого цвета отфильтровывалось и рекристаллизовалось из метанола, чтобы получить 3-т-бутилоксикарбониламино-6-карбоксамидо-метил-1,2,3,4-тетрагидрокарбазол (5,42 г). Указанный продукт (500 мг) растворялся в сухом диоксане (30 мл), и газ НСl пропускался через него 20 минут. Полученный раствор и отложившаяся смола выпаривались до сухости и обрабатывались водным раствором карбоната калия. Это экстрагировалось этилацетатом, и экстракты соединялись, высушивались (MgSO4) и выпаривались до сухости. Остаток растворялся в метаноле и обрабатывался избытком щавелевой кислоты. Добавление эфира давало кристаллизацию искомого соединения (250 мг), т. пл. 257-260°С. Пример 24 3-Метиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид 4-Цианофенил гидразин гидрохлорид (20,2 г) и 4-бензоилоксициклогексанон (25,9 г) растворялись в ледяной уксусной кислоте (400 мл), и смесь нагревалась с флегмацией 1,5 часа. После охлаждения смесь фильтровалась, и фильтрат выпаривался до сухости, и нейтрализовался водным раствором бикарбоната натрия, чтобы получить твердый осадок, который очищался хроматографией (SiO2; гексан/этилацетат) для выхода 3-бензоилокси-6-циано-1,2,3,4-тетрагидрокарбазола (18,0 г). Этот продукт (11,6 г) суспендировался в этаноле (230 мл) и обрабатывался 2,5% водным раствором гидроокиси калия (120 мл), и нагревался с флегмацией 1 час. Охлажденная смесь нейтрализовалась охлажденной уксусной кислотой и выпаривалась до твердого остатка, который промывался водой и высушивался, чтобы получить 3-гидрокси-6-циано-1,2,3,4-тетрагидрокарбазол (6,6 г). Указанный продукт (3,57 г) растворялся в сухом пиридине (35 мл), и смесь перемешивалась при 100°С 2 часа. После охлаждения раствор выливался в воду (500 мл). Экстрагировался этилацетатом, и последний экстракт промывался 2 М НСl, высушивался (MgSO4) и выпаривался до сухости. Очистка хроматографией (SiO2; гексан/этилацетат) дала 3-тосулокси-6-циано-1,2,3,4тетрагидрокарбазол (0,53 г). Этот продукт (0,40 г) растворялся в 33% метиламине в спирте (25 мл) и нагревался при 100°С в герметичном стальном сосуде 1,5 часа. После охлаждения смесь выпаривалась до сухости и очищалась хроматографией (SiО2; хлороформ/метанол), чтобы получить 3-метиламино-6-циано1,2,3,4-тетрагидрокарбазол (0,13 г). Указанный продукт (0,12 г) растворялся в ТГФ (10 мл) и реагировал с ди-трет-бутил дикарбонатом (0,36 г) в ТГФ (3 мл) при комнатной темпера туре всю ночь. Реакционная смесь выпаривалась до сухости, разделялась между 2 М раствора бикарбоната натрия и этилацетатом, и органический слой высушивался и выпаривался до получения вещества белого цвета. Оно растиралось эфиром/гексаном, чтобы получить 3-т-бутилоксикарбонил-метил амино-6-циано-1,2,3,4-тетрагидрокарбазол (0,14 г). Этот продукт растворялся в метаноле (15 мл) и обрабатывался смесью 20% водного гидроксида натрия (0,20 мл) и 30% перекисью водорода (0,20 мл), и вся смесь перемешивалась при комнатной температуре всю ночь. Добавлялся метабисульфит натрия (38 мг), и раствор выпаривался до сухости и хроматографировался (SiО2; хлороформ) 10% NН4ОН в метаноле), чтобы получить 3метиламино-6-карбоксамид-1,2,3,4-тетрагидрокарбазол (0,12 г). Указанное соединение (0,11 г) растворялось в метаноле (10 мл) и обрабатывалось 3 М хлористоводородной кислоты при комнатной температуре. Смесь выпаривалась до сухости, азеотропировалась этанолом, чтобы получить твердое вещество, которое рекристаллизовалось из метанола (эфира для получения искомого соединения, т. пл. 327-328°С /80 мг). 1 Н ЯМР [250 МГц, МеОН-д4] д 1,98-2,20 (1H, м), 2,29-2,49 (1H, м), 2,75-2,90 (5Н, с+м), 2,90-3,09 (2Н, м), 3,52-3,69 (1H, м), 7,31 (1H, д), 7,63 (1H, д), 8,05 (1H, с). Пример 25 3-Этиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат 1,4-Циклогександион моно-2',2'-диметил триметилен кетал (2,00 г) смешивался с безводным этиламином (10,0 г) и бензолом (10,0 мл), и смесь охлаждалась до 5°С. По каплям добавлялся раствор титаний тетрахлорида (0,95 г) в бензоле (10 мл), затем смесь перемешивалась при комнатной температуре 1 час. Смесь фильтровалась и выпаривалась до сухости, чтобы получить масло, которое растворялось в этаноле (30 мл). К этому раствору добавлялся катализатор палладий-на-углероде (100 мг), и смесь гидрировалась при 50 ф/д2 (3,5 кг/см2) давлении всю ночь. Катализатор отфильтровывался, и этанол выпаривался для получения 4-этиламино-циклогексанон 2',2'диметил триметилен кетала в виде масла (2,0 г). Это соединение (0,80 г) растворялось в муравьиной кислоте (20 мл), и раствор нагревался до 90ºС 1 час. Муравьиная кислота выпаривалась, и остаток разделялся между хлороформом и 1M хлористоводородной кислоты. Водный слой выпаривался до сухости, чтобы дать выход 4-этиламиноциклогексанону (0,40 г). Смесь указанного продукта (0,40 г) и 4-карбоксамидо-фенил гидразин гидрохлорида (0,60 г) в ледяной уксусной кислоте (20 мл) нагревалась с флегмацией 1 час. Кислота выпаривалась в вакууме до масла, которое очищалось хроматографией (SiO2; СНСl3/10% NH3 в МеОН), чтобы получить масло (0,50 г). Часть этого продукта (150 мг) растворялась в метаноле и обрабатывалась щавелевой кислотой. Раствор обрабатывался эфиром для получения искомого соединения в виде кристаллического твердого вещества, т. пл. 165170°С (100 мг). 11 41254 1 Н ЯМР [250 МГц, ДМСО-д6] д 1,25 (3Н, т), 1,81-2,05 (1H, м), 2,20-2,38 (1H, м), 2,61-2,79 (1H, м), 2,79-2,94 (2Н, м), 2,98-3,28 (3Н, дд+с), 3,41-3,60 (1H, м), 7,08 (1H, шир. с), 11,12 (1H, с). Пример 26 3-н-Пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат Пропиламин (1,81 г) растворялся в метаноле (12,5 мл), и с охлаждением добавлялось 1,5 М НСl в метаноле (5,6 мл). После 1 минуты добавлялся 1,4-циклогександион моно-2',2'-диметил триметилен кетал (1,0 г), а через еще 10 минут - цианоборогидрид натрия (0,23 г). Смесь перемешивалась при комнатной температуре 3 дня. Полученная смесь фильтровалась, и фильтрат выпаривался и обрабатывался 1M НСl (10 мл) с охлаждением. Остаток вываривался для образования раствора, который промывался эфиром, доводился основанием до рН 12 с помощью водного гидроксида натрия и экстрагировался дихлорметаном. Этот экстракт промывался насыщенным водным раствором бикарбоната натрия, высушивался (MgSO4), и выпаривался до сухости. Хроматография (SiO2; хлороформ/метанол/аммиак) дала 4-н-пропиламино циклогексанон 2',2'-диметил триметилен кетал (0,72 г). Этот продукт (0,66 г) гидролизовался до кетона, который реагировал с 4-карбоксамидофенил гидразин гидрохлоридом и преобразовывался в соль оксалата, как описано в Примере 25, чтобы дать выход искомому соединению (0,44 г), т. пл. >168°С разл. Пример 27 3-i-Пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалата Реакция изопропиламина (9,54 г) с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом (2,0 г) способом, описанным для Примера 25, дала 4-i-пропиламино циклогексанон 2',2'-диметил триметилен кетал (2,38 г). Этот продукт (0,66 г) гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом (0,45 г), и смесь обрабатывалась, как описано выше, чтобы получить свободное основание искомого соединения (0,34 г). Оно преобразовывалось в оксалат, т. пл. >235°С. разл. Пример 28 3-Диметиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат Диметиламин (10,0 г) реагировал с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом (2,0 г) способом, описанным для Примера 25, чтобы получить 4-диметиламино-циклогексанон2',2'-диметил триметилен кетал (0,72 г). Этот продукт (0,72 г) гидролизовался и реагировал с 4-карбоксамидо-фенил гидразин гидрохлоридом (0,47 г), и продукт преобразовывался в соль оксалата, как описано выше, чтобы получить искомое соединение (0,20 г), т. пл. 99-101°C. 1 Н ЯМР [250 МГц, ДМСО-д6] д 1,83-2,05 (1H, м), 2,27-2,40 (1H, м), 2,72-3,00 (9Н, 2м+с), 2,37-3,22 (1H, дд), 3,50-3,68 (1H, м), 7,05 (1H, шир. с), 7,27 (1Н, д), 7,60 (1H, д), 7,81 (1H, шир. с), 8,00 (1H, с), 11,11 (1H, с). Пример 29 3-Бензиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат Реакция бензиламина (0,59 г) с 1,4-цикло- гександион-моно-2',2'-диметил триметил кеталом (1,0 г) и последующее восстановление имина цианоборогидридом натрия способом, описанным для Примера 26, дала 4-бензиламино-циклогексаном 2',2'-диметил триметилен кетал (0,54 г). Этот продукт (0,52 г) реагировал с 4-карбоксамидофенил гидразин гидрохлоридом (0,34 г), и продукт обрабатывался щавелевой кислотой, чтобы получить искомое соединение, т. пл. >190ºС, разл. (0,11 г). Пример 30 3-Пирролидинил-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат Реакция пирролидина (15,6 г) с 1,4-цикло- гександион моно-2',2'-диметил триметилен кеталом (2,0 г) способом, описанным для Примера 26, дала 4-пирролидинил-циклогексанон-2',2'-диметил триметилен кетал (1,74 г). Этот продукт (1,70 г) гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом (1,70 г), и продукт обрабатывался щавелевой кислотой, как описано выше, чтобы получить искомое соединение (32 мг), т. пл. >190°С разл. Пример 31 3-/N-Метилэтиламино/-6-карбоксамидо-1,2,3,4тетрагидрокарбазол оксалат Реакция N-метил этиламина (13,0 г) с 1,4циклогександион моно-2',2'-диметил триметилен кеталом (2,0 г) способом, описанным для Примера 25, дала 4-/N-метил этиламино/-циклогексанон2',2'-диметил триметилен кетал (1,71 г). Этот продукт (0,86 г) гидролизовался и реагировал с 4карбоксамидо-фенил гидразин гидрохлоридом (0,52 г) и обрабатывался, как описано выше, чтобы получить искомое соединение (76 мг), т. пл. >130°С разл. Пример 32 3-Амино-6-/2-карбоксамидоэтил/-1,2,3,4-тетрагидрокарбазол оксалат Смесь 4-нитрокоричной кислоты (22,5 г) и тионил хлорида (20,8 г) в бензоле (160 мл) нагревалась с флегмацией 4 часа. Полученная оранжевая смесь фильтровалась и выпаривалась для получения кислотного хлорида (22,9 г). Это растворялось в дихлорметане (1 л), пропускался аммиак с охлаждением до ниже 20°С и перемешиванием. Растворитель удалялся в вакууме, и остаток растворялся в горячем этилацетате, и раствор встряхивался с 1M раствора гидроксида натрия. Полученная органическая фаза высушивалась, фильтровалась и выпаривалась. Оставался осадок, который шламовался этилацетатом, чтобы получить 4-нитро циннамамид в виде кристаллического вещества (18,6 г). Этот продукт (18,6 г) суспендировался в этаноле (1 л) и гидрировался с использованием Рd-С катализатора (6,6 г) при давлении 50 фунт/д2 (3,5 кг/см2) в течение 1 часа. Полученная смесь фильтровалась и выпаривалась до сухости с получением 4-аминофенил пропионамида (17,1 г). Медленно добавлялась концентрированная хлористоводородная кислота (4 мл) к продукту 4аминофенил пропионамида (0,80 г) с поддержанием температуры ниже 5°С. К этому шламу добав 12 41254 лялся раствор нитрита натрия в (0,37 г) в воде (2 мл) в течение 15 минут по каплям с последующим перемешиванием еще в течение 15 минут. Полученный таким образом мутный раствор порциями добавлялся к охлажденному перемешанному раствору двухлористого олова (2,19 г). НСl (4 мл), и полученная смесь перемешивалась 1 час. После фильтрования раствор восстанавливался в объеме, до тех пор, пока не формировался неорганический осадок. Он отфильтровывался, и фильтрат выпаривался до сухости. Остаточная смола кристаллизовалась из уксусной кислоты для получения сырого 4-гидразинофенил пропионамид гидрохлорида (1,05 г). Смесь вышеуказанного продукта (1,05 г) и 4фталимидо-циклогексанона (1,18 г) в уксусной кислоте (40 мл) нагревалась с флегмацией 40 минут. Растворитель удалялся в вакууме, и остаток разделялся между водным раствором карбоната калия и этилацетатом. Органическая фаза высушивалась (MgSO4) и выпаривалась до сухости, и остаток хроматографировался (Sio2; CH2Cl2) МеОН, чтобы дать 3-фталимидо-6-карбоксамидоэтил-1,2,3,4-тетрагидрокарбазол (0,70 г). Этот продукт (0,70 г) растворялся в метаноле (50 мл), обрабатывался гидразин гидратом (1,0 мл) и нагревался с флегмацией 30 минут. Смесь выпаривалась до сухости, затем разделялась между этилацетатом и водным раствором карбоната калия. Органическая фаза высушивалась (MgSO4) и выпаривалась до сухости, и оста ток растворялся в этаноле и обрабатывался щавелевой кислотой (83 мг) в этаноле. Получалось твердое вещество, которое рекристаллизовалось из этанола, чтобы получить искомое соединение (110 мг), т. пл. 232-5°С. Лекарственные формы Пример А Таблетка для орального применения готовится соединением Мг/табл. Соединения формулы (I) 100 Лактоза 153 Крахмала 33 Crospovidone 12 Микрокристаллической целлюлозы 30 Стеарата магния 2 330 330 мг в 9 мм таблетке. Пример В Инъекционный раствор для парентерального назначения готовится из следующих компонентов. % вес/вес Соединения формулы (I) 0,50% (вес/об) 1M лимонной кислоты 30% (об/об) Гидроксида натрия (gs) до рН 3,2 Воды для инъекции ВР до 100 мл. Соединение формулы (I) растворяется в лимонной кислоте, и рН медленно доводится до 3,2 раствором гидроксида натрия. Затем раствор дополняется до 100 мл водой, стерилизуется фильтрацией и запаивается в ампулы разных размеров. __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 50 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 13
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61P 25/04, A61K 31/454, A61K 31/445, C07D 209/88, A61K 31/4427, A61K 31/40, C07D 403/04, A61P 25/06, A61P 43/00, A61K 31/403, C07D 401/04
Мітки: одержання, фармацевтична, композиція, 5-нт1-подібного, гідрат, похідне, спосіб, тетрагідрокарбазолу, сольват, рецептора, агоніст, сіль
Код посилання
<a href="https://ua.patents.su/13-41254-pokhidne-tetragidrokarbazolu-abo-jjogo-sil-solvat-abo-gidrat-yak-agonist-5-nt1-podibnogo-receptora-sposib-kh-oderzhannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Похідне тетрагідрокарбазолу або його сіль, сольват або гідрат як агоніст 5-нт1-подібного рецептора, спосіб їх одержання та фармацевтична композиція</a>
Фармацевтична композиція, яка містить алкілсульфонамід-агоніст 5нті- подібного рецептора для ректального введення

Номер патенту: 37206
Опубліковано: 15.05.2001
Автори: Річард Ізабель, Мерлет Надін, Тілеманс Ізабель
МПК: A61K 31/40, A61K 31/435
Мітки: подібного, містить, композиція, введення, ректального, яка, фармацевтична, 5нті, рецептора, алкілсульфонамід-агоніст
Формула / Реферат:
1. Фармацевтическая композиция для ректального введения в твердой дозированной форме для лечения и/или предупреждения головной боли, в частности, мигрени, включающая в качестве активного ингредиента алкилсульфонамидное соединение, являющееся агонистом 5НТ1 - подобного рецептора, отличающаяся тем, что она содержит упомянутое соединение в форме его свободного основания или его физиологически приемлемого сольвата и носитель или наполнитель в...
Похідне тетрагідробензимідазолу або його фармацевтично прийнятна сіль, що проявляють активність антагоніста 5-нт3-рецептора, та фармацевтична композиція на його основі

Номер патенту: 27290
Опубліковано: 15.09.2000
Автори: Акіра Матсухіса, Такесі Сузукі, Ісао Янагісава, Дзунйа Охморі, Токуо Коіде, Мітсуакі Охта, Кейдзі Міята
МПК: C07D 235/06, C07D 403/12, C07D 403/08, A61K 31/40, C01B 7/00, A61K 31/415
Мітки: фармацевтично, антагоніста, композиція, основі, похідне, фармацевтична, прийнятна, 5-нт3-рецептора, активність, сіль, проявляють, тетрагідробензимідазолу
Текст:
...%: C 71,77; H 6,13; N 15,13. Масс-спектр (Е1): m/z; 358 (M+). Примеp 17. Гидрохлорид N-[(4,5,6,7- тетрагидробензимидазол-5-ил)карбонил]-фенотиазина Физико-химические свойства: Т.пл. 268 – 270оС. Элементный анализ для C20H17N3О × HCl × 0,5 х х х H2O: Рассчитано, %: C 61,14; H 4,87; N 10,69; Cl 9,02. Найдено, %: C 61,15; H 4,64; N 10,60; Cl 8,59. Масс-спектр (Е1): m/z; 347 (M+, сво бодное соединение). Пример 18....
Похідне піколінової кислоти або його сіль, що має гербіцидну активність, спосіб його одержання, гербіцидна композиція, спосіб знищення бур’янів
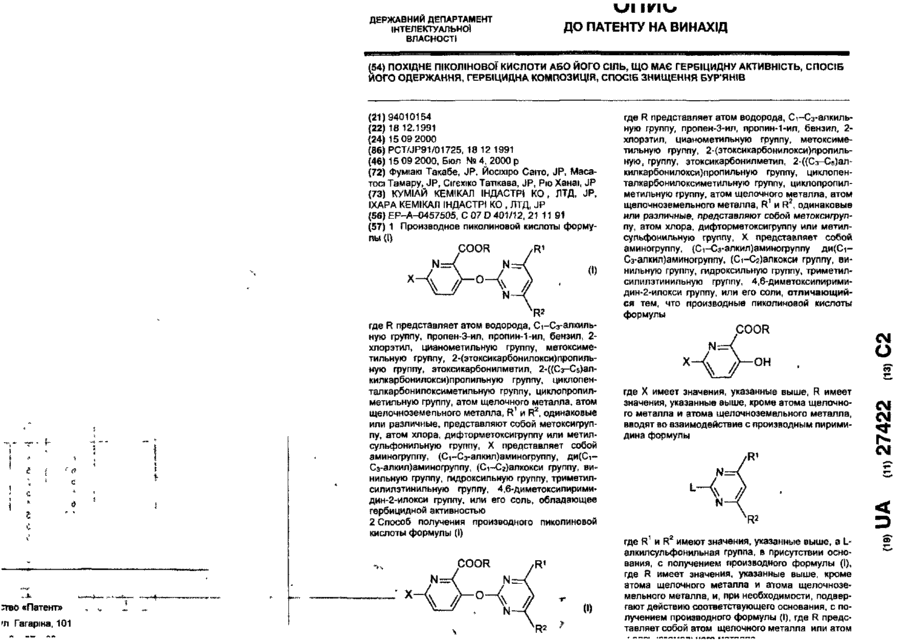
Номер патенту: 27422
Опубліковано: 15.09.2000
Автори: Масатосі Тамару, Фуміакі Такабе, Ріо Ханаі, Йосіхіро Саіто, Сігєхіко Татікава
МПК: A01P 13/00, A01N 43/54, C07D 401/12
Мітки: похідне, сіль, бур'янів, знищення, гербіцидну, кислоти, піколінової, спосіб, має, гербіцидна, композиція, одержання, активність
Текст:
...Процесс G COOR' осн, COOR 4 Основание Base [П - 4 ) 6 /где R имеет значение, определенное выше, R10 представляет алкильную группу, замещенную алкильную группу или 4,6-диметоксипиридинильную группу, и L2 представляет атом галогена, при условии, что L2 представляет атом галогена или алкилсульфонильную группу, когда R10 представляет 4,6-диметоксипиримидинильную группу/. Так, соединение формулы Н-8 может быть получено путем...
Похідне хіназоліну, спосіб його одержання, фармацевтична композиція на його основі

Номер патенту: 34426
Опубліковано: 15.03.2001
Автор: Баркер Ендрю Джон
МПК: C07D 403/04, C07D 491/056, C07D 491/04, C07D 239/94
Мітки: спосіб, похідне, фармацевтична, хіназоліну, основі, композиція, одержання
Текст:
...суль финильную груп пу,также можно использовать более слабый окислитель, например, метапериодат натрия или калия, обычно в полярном растворителе, таком как уксусная кислота или этанол. При необходимости получения соединения формулы I, содержащего /1-4С/алкилсульфонильную группу, его можно получить путем окисления соответствующего /1-4С/алкилсульфи нильного соединения, а также соответствующего /14С/алкилтиосоединения. (d) Для получе ния...
Похідні n-алкіленпіперидинів як антагоністи рецептора нейрокініну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 26894
Опубліковано: 29.12.1999
Автори: Мартінез Серж, ван Броєкк Дідьє, Емонд-Альт Ксавьє, Пруаєтто Венсанзо
МПК: A61P 43/00, C07D 401/06, C07D 211/54, C07D 211/46, A61K 31/4433, C07D 403/12, C07D 409/12, A61K 31/4465, A61K 31/454, A61K 31/445, C07D 211/58, A61K 31/4545, A61K 31/44, A61K 31/4427, A61K 31/4468, C07D 401/12
Мітки: похідні, спосіб, одержання, рецептора, сполуки, антагоністи, фармацевтична, проміжні, нейрокініну, n-алкіленпіперидинів, композиція
Текст:
...где Alk является (С,-Сэ)алкильной группой, R представляет собой атом водорода 40 или С^-С^алкил, Z представляет собой фенил, незамещенный или моно- или дизамещенный галогеном или (С^-Сзіалкоксигруппой, наф 26894 7 тил, замещенный галогеном, фенилметильной группой, замещенной на фениле (С,С4}алкоксигруппой, * обозначает хиральный центр, в виде рацемата или оптически чистых изомеров, или их соли с минеральными или органическими кислотами,...
Попередній патент: Друкарське видання
Наступний патент: Система зв’язку з багатостанційним доступом та кодовим розділенням сигналів з розширеним спектром (варіанти) і спосіб здійснення зв’язку у цій системі
Випадковий патент: Концентратор сонячного випромінювання