Сполука (варіанти), фармацевтична композиція, спосіб лікування (варіанти)
Номер патенту: 72945
Опубліковано: 16.05.2005
Автори: Волпоул Крістофер, Браун Вілл'ям, Плобек Ніклас











Формула / Реферат
1. Сполука формули I
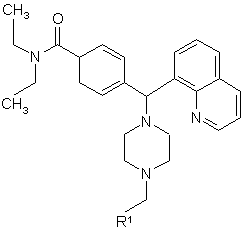 ,I
,I
де
R1 вибрано з групи, яка складається з
(і) фенілу;
(іі) піридинілу
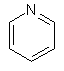 ;
;
(ііі) тіофенілу
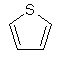 ;
;
(iv) фуранілу
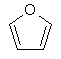 ;
;
(v) імідазолілу
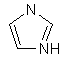 та
та
(vi) триазолілу
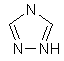 ,
,
де кожне фенільне кільце і гетероароматичне кільце може бути як варіант і незалежно, крім того, заміщеним 1, 2 або 3 замісниками, вибраними з лінійних та розгалужених С1-С6 алкілу, NО2, CF3, С1-С6 алкоксилу, хлору, флуору, брому та йоду; а також її фармацевтично прийнятні солі та ізомери.
2. Сполука за п. 1, яка відрізняється тим, що як варіант замісник(и) на ароматичному та гетероароматичному кільці(ях) вибрано з групи, яка складається з NO2, ізобутилу, CF3, метоксилу, метилу або хлору.
3. Сполука за п. 1 або 2, яку вибрано з групи, до складу якої входять
дигідрохлорид 4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил]-N,N-діетилбензамід (сполука 2);
N,N-діетил-4-[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 5);
дигідрохлорид 4-[{4-[4-(трет-бутил)бензил]-1-піперазиніл}(8-хінолініл)метил]-N,N-діетилбензамід (сполука 8);
дигідрохлорид N,N-діетил-4-[[4-(4-нітробензил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 9);
дигідрохлорид 4-[{4-[2,4-bis(трифлуорметил)бензил]-1-піперазиніл}(8-хінолініл)метил]-N,N-діетилбензамід (сполука 10);
дигідрохлорид N,N-діетил-4-[[4-(4-метоксибензил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 11);
дигідрохлорид 4-[[4-(2,4-дихлорбензил)-1-піперазиніл](8-хінолініл)метил]-N,N-діетилбензамід (сполука 12);
дигідрохлорид N,N-діетил-4-[[4-(2-піридинілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 13);
N,N-діетил-4-[[4-(3-тієнілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 14);
N,N-діетил-4-[[4-(2-фуранілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 15);
N,N-діетил-4-[[4-(3-фуранілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 16);
дигідрохлорид N,N-діетил-4-[[4-(2-тіофенілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 17);
дигідрохлорид N,N-діетил-4-[[4-(2-імідазолілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 18);
дигідрохлорид N,N-діетил-4-[[4-(4-імідазолілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 19) та
дигідрохлорид N,N-діетил-4-[[4-(3-триазолілметил)-1-піперазиніл](8-хінолініл)метил]бензамід (сполука 20).
4. Сполука за будь-яким з пп.1-3, яка відрізняється тим, що є (+)-енантіомером.
5. Сполука за будь-яким з пунктів 1-3, яка відрізняється тим, що є (-)-енантіомером.
6. Сполука за будь-яким з попередніх пунктів, яка відрізняється тим, що присутня у формі своїх солей гідрохлориду, сульфату, тартрату або цитрату.
7. Сполука за п. 4, яку вибрано групи, до складу якої входять
(+)4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил]-N,N-діетилбензамід та
(+)4-[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метил]-N,N-діетилбензамід.
8. Сполука за п. 5, яку вибрано групи, до складу якої входять
(-)4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил]-N,N-діетилбензамід та
(-)4-[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метил]-N,N-діетилбензамід.
9. Сполука за будь-яким з пп.1-8 для використання у терапії.
10. Сполука за п. 9, яка відрізняється тим, що терапія охоплює вгамування болю.
11. Сполука за п. 9, яка відрізняється тим, що терапію призначають для шлунково-кишкових розладів.
12. Сполука за п. 9, яка відрізняється тим, що терапію призначають для пошкоджень спинного мозку.
13. Сполука за п. 9, яка відрізняється тим, що терапію призначають для розладів симпатичної нервової системи.
14. Застосування сполуки формули І за п.1 у виготовленні медикаменту для використання при вгамуванні болю.
15. Застосування сполуки формули І за п.1 у виготовленні медикаменту для використання у лікуванні шлунково-кишкових розладів.
16. Застосування сполуки формули І за п.1 у виготовленні медикаменту для використання у лікуванні пошкоджень спинного мозку.
17. Фармацевтична композиція, яка містить сполуку формули І за п. 1 як активний інгредієнт разом з фармакологічно і фармацевтично прийнятним носієм.
18. Спосіб лікування болю, який полягає у тому, що ефективну кількість сполуки формули І за п. 1 уводять пацієнту для вгамування болю.
19. Спосіб лікування шлунково-кишкових розладів, який полягає у тому, що ефективну кількість сполуки формули І за п. 1 уводять пацієнту, який страждає від шлунково-кишкового розладу.
20. Спосіб лікування пошкоджень спинного мозку, який полягає у тому, що ефективну кількість сполуки формули І за п. 1 уводять пацієнту, який страждає від зазначеного пошкодження спинного мозку.
Текст
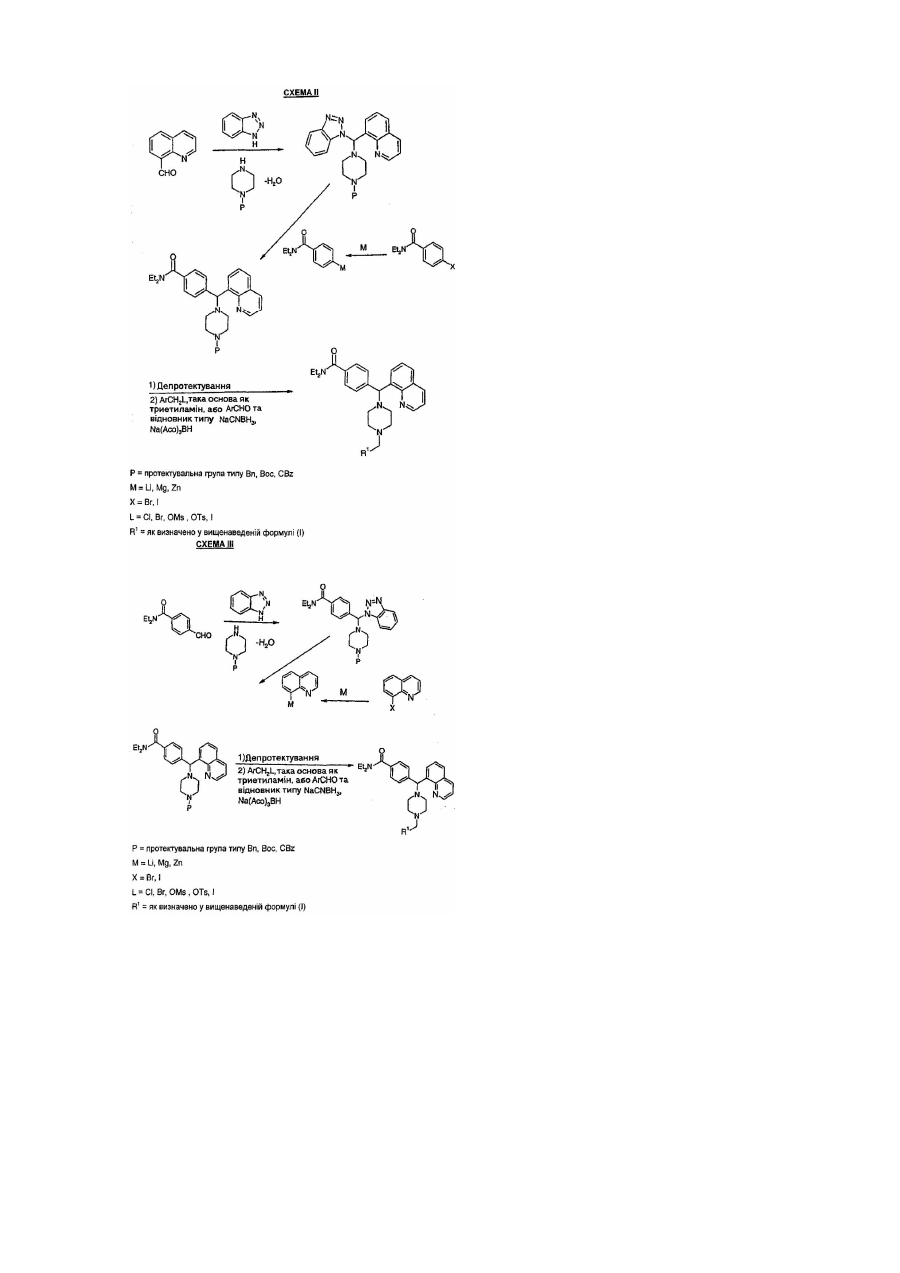
Винахід стосується нових сполук, способу їх одержання, їх використання, а також фармацевтичних композицій, які містять нові сполуки. Нові сполуки придатні для використання у терапії, і, зокрема, для вгамування болю. d-рецептор було визначено як такий, що відіграє роль у здійсненні багатьох фізичних функцій, наприклад, відповідає за системи циркуляції та болю. Тому, ліганди для d-рецептору можуть виявитися потенційно корисними як аналгетики, та/або як гіпотензивні засоби. Ліганди для d-рецептору також було виявлено як такі, що мають імуномодуляторну активність. Зараз добре визначено щонайменше три різні популяції опіоїдних рецепторів (m, d і к), і всі вони наявні у центральній і периферійній нервових системах багатьох видів, включно з людиною. Аналгезію спостерігали у багатьох моделях тварин, коли було активовано один або більше таких рецепторів. За деяким винятком, наявні зараз селективні опіоїдні d ліганди є пептидними за своєю природою і не придатні для уведення системними шляхами. Одним прикладом непептидного d-агоністу є SNC80 [Bilsky E.J. et al., Journal of Рhаrmасоlоgу and ЕхреrіmеnіаІ Therapeutics, 273(1), pp.359-366 (1995]. Однак, все ще існує потреба у селективних d-агоністах, які мають не лише удосконалену селективність, але й удосконалений профіль побічної дії. Отже, проблема даного винаходу полягала у тому, щоб винайти нові аналгетики, що мають удосконалену аналгезивну дію, з удосконаленим профілем побічної дії, на відміну від наявних зараз N-агоністів, і удосконалену системну ефективність. Аналгетики, які було визначено, і, які присутні у рівні техніки, мають багато недоліків у тому, що мають погану фармакокінетику і не є аналгезивними, якщо уводити їх системними шляхами. Також, було задокументовано, що бажані сполуки d-агоністу, описані у рівні техніки, виявляють значну конвульсивну дію, якщо уводити їх системно. Ми зараз виявили, що певні сполуки, які недостатньо описані, але включені до WO 98/28270, виявляють надзвичайно удосконалені властивості d-агоністу та потенційність in vivo на відміну від сполук, описаних в WO98/28270, якщо уводити їх системно. Сполуки даного винаходу виявляють значні та несподівано збільшені рівні агонізму g-рецептору та метаболічної стабільності. Нові сполуки згідно з винаходом визначено за формулою І де R1 вибрано з групи, яка складається з (і) фенілу; (іі) піридинілу (ііі) тіофенілу (iv) уранілу (ν) імідазолілу (vi) триазолілу де кожне фенільне кільце R1 і гетероароматичне кільце R1 може бути, як варіант і незалежно крім того заміщеним 1, 2 або 3 замісниками, вибраними з лінійних та розгалужених С 1-С6алкілу, NO2, CF3, С1С6алкоксилу, хлору, флуору, брому та йоду. Замісники на фенільному кільці та гетероароматичному кільці можуть знаходитися у будь-якій позиції на зазначених кільцевих системах; В рамках винаходу також передбачено фармацевтично прийнятні солі сполук формули І, їх ізомери. У бажаному втіленні винаходу, сполуки формули І присутні як (+)-енатіомер або як (-)-енантіомер. Під терміном "ізомери" розуміють сполуки формули І, які відрізняються позицією своїх функціональних груп та/або орієнтуванням. Під терміном "орієнтування" розуміють стереоізомери, діастереоізомери, регіоізомери та енантіомери. Нові сполуки згідно з винаходом придатні для використання у терапії, зокрема, для угамування різних станів болю, наприклад, хронічного, невропатологічного, різкого, болю, викликаного раком, болю, викликаного ревматичним артритом, мігренню, вісцерального болю, та інших видів болю. Однак, цей список не можна вважати обмеженим. Сполуки згідно з винаходом придатні для використання як імуномодулятори, зокрема, для лікування автоімунних захворювань, наприклад, артриту, трансплантатів шкіри, трансплантатів органів та подібних хірургічних потреб, для лікування колагенових захворювань, алергій різного виду, для використання як антиракових та антивірусних засобів. Сполуки згідно з винаходом придатні для використання у станах захворювання, де присутня або включена у таку парадигму дегенерація або дисфункція опіоїдних рецепторів. Це може включати використання мічених ізотопами сполук згідно з винаходом при застосуванні в діагностиці та візуалізації, наприклад, при застосуванні позитронної емісійної томографії (ПЕТ) Сполуки згідно з винаходом придатні для використання у лікуванні діареї, депресії, збудження, неутримання сечі, різних психічних захворювань, кашлю, набряку легенів різних шлунково-кишкових розладів, пошкодження спинного мозку та залежності від ліків, а також для лікування зловживання алкоголем, нікотином, опіоїдними та іншими ліками, і для лікування розладів симпатичної нервової системи, наприклад, гіпертонії. Сполуки згідно з винаходом придатні для використання як аналгезивний засіб для використання під час загальної та місцевої анестезій. Комбінації засобів з різними властивостями часто використовують для досягнення рівноваги ефектів, необхідних для підтримання стану анестезії (наприклад, амнезії, аналгезії, релаксації м'язів та седативного ефекту). Також до цієї комбінації входять анестезуючі засоби для вдихання, гіпнотичні засоби, анксіолітики, нервово-м'язові блокатори та опіоїдні засоби. Також, в рамки винаходу входить використання будь-якої зі сполук згідно з вищенаведеної формулою І, у виготовленні медикаменту для лікування будь-якого з вищеописаних станів. Ще одним аспектом винаходу є спосіб лікування пацієнта, який страждає від будь-якого з вищеописаних станів, причому пацієнту, який потребує такого лікування, уводять ефективну кількість сполуки згідно з вищенаведеною формулою І. Також в рамках винаходу передбачено будь-які нові проміжні сполуки (інтермедіати), як описано на схемі І, придатні для синтезу сполук вищенаведеної формули І Способи одержання І. Сполуки згідно з винаходом можна одержати за допомогою будь-якого з способів, описаних на схемах І, II, III та IV.. Ці відомі способи описано в J. March, Advanced Organic Chemistry, 4lh Edition, John Wiley and sons (1992); Katritsky, A.R., Lan, X. Chem. Soc. Rev., pp. 363-373 (1994),m уведено сюди посиланням. R1=як визначено у вищенаведеній формулі (І) Приклади Тепер винахід буде описано детальніше з посиланнями на приклади, які не повинні виходити за рамки даного винаходу. Приклад 1 Одержання дигідрохлориду 4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил-N,N-діетилбензаміду (сполука 2) Названу сполуку 2 одержали синтетичним способом, описаним на нижченаведеній Схемі 1. (і) Одержання N,N-діетил-4-формілбензаміду (сполука 1). 4-Формілбензойну кислоту (11,2г, 74,6ммоль) та триетиламін (10,4мл, 75ммоль) розчинили в тетрагідрофурані (ТГФ) (100мл) і охолодили до -10°С. і-Бутилхлорформат (10,3мл, 78ммоль) додали і перемішування продовжували протягом 10хв. при -10°С, перед тим як додали діетиламін (9,7мл, 94ммоль), і розчину дали можливість досягти темп. 25°С. Після концентрації, водної обробки та хроматографії на діоксиді силіцію (0-100% EtOAc в гептані) в загальному було отримано 7,4г (50%) сполуки І. (іі) Одержання N,N-діетил-4-[гідрокси(8-хінолініл)метил]бензаміду (сполука II) 8-Бромхінолін (3,0г, 14,4ммоль) розчинили в сухому ТГФ (150мл) і охолодили до -78°С під атмосферою азоту. s-BuLi (11,1мл, 1,3Μ в пентані, 14,4ммоль) додавали краплями протягом 5хв. [Preparation and reactions with 8-lithioquinoline: Suggs, J. Org: Chem. 1980, 45, 1514]. Через ще 5хв. додали розчинений в ТГФ (5мл) N,Nдіетил-4-формілбензамід (3,5г, 17.0ммоль). Розчин перемішували 1 годину, тоді додали NH4CI (aq.). Після концентрації, водної обробки і хроматографії на діоксиді силіцію, (0-100% EtOAc в гептані), в загальному отримали 3,5г (70%) сполуки II. Mac-спектрометрія (МС): 334, 262, 234, 215, 204, 178, 156, 129. Альтернативний шлях для отримання сполуки II з N,N-діетил-4-йодбензаміду (сполука IV) Сполуку IV (0,67г, 2.2ммоль) розчинили у сухому ТГФ (25мл) і охолодили до -78°С під атмосферою азоту. n-BuLi (1,3мл, 1.6Μ в гексані, 2.2ммоль) додавали краплями протягом 5хв. Через ще 10хв. додали розчинений в ТГФ (1мл) 8-формілхінолін (0,17г, 1.1ммоль) (8-формілхінолін отримали з 8-метилхіноліну окисленням за допомоги діоксиду селену при 150-155°С протягом 12 годин [Kingsbury, J. Med. Chem. 1993, 3308.]. Розчин перемішували 1 годину, тоді додали NH4Cl (aq.). Після концентрації, водної обробки та хроматографії на діоксиді силіцію (0-100% EtOAc в гептані) в загальному отримали 0,29г |78%) сполуки II. (iiі) Одержання 4-[хлор(8-хінолініл)метил]-N,N-діетилбензаміду (сполука III). Сполуку II (2,0г, 6,6ммоль) розчинили в сухому СН2СІ 2 (25мл) і додали SOCl2 (0,53мл, 7,3ммоль). Розчин перемішували при 25°С протягом 30хв. і розчинник випарювали у вакуумі. Сполуку III отримали у вигляді масла (-100%) і використали у наступній реакції, без подальшого очищення. МС: 348, 333, 233, 215, 204, 156. (iv) Одержання N,N-діетил-4-[1-піперазиніл(8-хінолініл)метил]бензаміду (сполука 1). Сирий продукт-сполуку III (~6,6ммоль) і піперазин (2,3г, 26ммоль) розчинили в сухому MeCN (50мл) і нагрівали під зворотним холодильником 12 годин. Розчинник було видалено у вакуумі, залишок розчинено в СН2СІ 2 і промито водою, а органічну фазу висушено (К2СО3) і випарено у вакуумі. Після хроматографії на діоксиді силіцію (0-20% МеОН в СН2СІ 2, 1% NH4OH), в загальному отримали 1,8г (68%, 2 етапи) сполуки 1. Подальше очищення можна було б досягти обернено-фазовою хроматографією (Li Chroprep RP-18,10-50% MeCN в воді, 0,1% ТФОК) для одержання 1,2г безкольорового продукту. Сіль дигідрохлориду отримали обробкою 2eq. НСІ в етері. Темп, плавл.: 180-90°С. ІЧ-спектр (KBr, Vмакс) 3297, 2982, 2716, 2474, 1611, 1434, 1380, 1288, 1098см-1. МС (амін): 402, 318, 246, 217, 109. 1 Н ЯМР (амін, CDCI 3): d 1.2, 1.1 (2s, 6Н), 2.94, 2.51 (2m, 8Н), 3.5-3.1 (m, 5H), 6.05 (s, 1H), 8.94-7.20 (m, 10H). Анал. (С25Н30N4Оx3.2CF3CO2H) С, N; Н: обчисл.. 4.36; виявлено, 3.90. (ν) Одержання дигідрохлориду 4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил-Ν,Ν-діетилбензаміду (названа сполука 2) Сполуку 1 (1,3г, 3,2ммоль) та триетиламін (0,90мл, 6,4ммоль) розчинили в MeCN (10мл). Бензилбромід (0,77мл, 6,4ммоль) додали з перемішуванням при 25°С. Через 4 години розчин було концентровано і очищена хроматографією на діоксиді силіцію (0-5% МеОН в СН2СІ 2, або обернено-фазовою хроматографією (LiChroprep RP-18, 20-80% MeCN у воді, 0,1% ТФОК). В загальному отримали 2,2г (72%) названої сполуки 2. Обробка 2eq. НСІ (aq.) і сушіння виморожуванням дало сіль дигідрохлориду (3,6г). ІЧ-Спектр (2Х НСІ, КВr): 2388,1606, 1434,1356, 1287 (см-1). ‘H ЯМР(вільний амін, CDCI 3) d=1.05 (m, 6Н), 2.5 (m, 8Н), 3.1-3.6 (m, 6Н), 6.04 (з, 1Н), 7.18-8.98 (m, 15H). Aнал..(C32H38Cl2N4O)C,H,N. Альтернативний спосіб отримання названої сполуки 2 з сполуки IIІ. Сирий продукт-сполуку III (~13,2ммоль), триетиламін (2,0мл, 14.5ммоль) та N-бензил-піперазин (2,6г, 14,5ммоль) розчинили в сухому MeCN (50мл) і нагрівали під зворотним холодильником 12 годин. Ще додали N-бензил-піперазин (0,5г, 2,8ммоль) і нагрівання продовжували протягом 12 годин. Розчинник видаляли у вакуумі, залишок розчиняли в СН2СІ 2 і промивали водою, а органічну фазу сушили (К2СО3) і випарювали у вакуумі. Після хроматографії на діоксиді силіцію (0-10% МеОН в СН2СІ 2), в загальному отримали 3.5г (53%) названої сполуки 2. Приклади 2 і 3 Виділення енантіомерів сполуки 2 (сполуки 3 і 4) Препаративне виділення цієї сполуки було проведено на колонці Chiralcel OD (50ммX50см), використовуючи Гексан/ЕtOН/Діетиламін 85:15:0.1 як рухому фазу. На колонці Chiralcel OD, (+)-ізомер було виявлено як такий, що елює першим. Приклад 2 (-)4-[(4-бензил-1-піперазиніл)(8-Хінолініл)метил-N,N-діетилбензамід (сполука 3) [а]D25: -130° (с 0.78, МеОН) ‘H ЯМР: (CD3OD): 6=1.05 (m, 6H), 3.0-3.6 (m, 14Н), 5.90 (s, 1H), 7.22-8.20 (m, 13H), 8.78 (m, 1H), 9.50 (m, 1H). АНАЛІЗ: Обчисл. w.3.1 Н2О, С: 61.85, Н: 7.17, N: 9.02. Виявлено С: 61.84, Н: 6.60, N: 8.89 Приклад 3 (+)4-[(4-бензил-1-піперазиніл)(8-хінолініл)метил-N,N-діетилбензамід (сполука 4). [a]D25: +130° (с 0.69, МеОН) ‘H ЯМР: (CD3OD): 6=1.05 (m, 6H), 3.0-3.6 (m, 14Н), 5.90 (s, 1H), 7.22-8.20 (m, 13Н), 8.78 (m, 1H), 9.50 (m, 1H). АНАЛІЗ: Обчисл. w.3.2 Н2О, С: 61.67, Н: 7.18, N: 8.99. Виявлено С: 61.70, Η: 6.46, Ν:8.84 Приклад 4 Одержання N,N-діетил-4-[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метилбензаміду (сполука 5) Названу сполуку 5 отримали, дотримуючись синтетичного способу, описаного нижче на схемі 2. До розчину сполуки 1 (0,80 г; 1,99ммоль) в СН2СІ 2 (20мл) додали Εt3Ν (0,83мл; 5,97ммоль), з наступним додаванням п-метилбензилброміду (773мг; 4,18ммоль). Реакційну суміш перемішували усю ніч і тоді концентрували під зниженим тиском. Очищення зворотною фазою, використовуючи 10%-30% СН3CN/H2O. (М+1) Обчислено: 507.70, (М+1) спостерігали: 507.20 ІЧ-Спектр (NaCI, вільний амін) 2969, 2807, 2360, 1628, 1455, 1425, 1286, 1134, 1095 (сm-1). ‘H ЯМР(СОСl3, вільний амін) d=1.0, 1.1 (2т, 6Н, амід-Ме), 2.31 (s, 3H, Аr-Ме), 2.5 (m, 8Н, піперазин-Н), 3.2, 3.5 (2m, амід-СН2), 3.49 (s, 2Н, ArCH2N), 6.03 (s, 1Н, Аr2СН), 7.06-7.68 (m, 11Н, Аr-Н), 8.01-8.12 (m, 2Н, Аr-Н), 8.93 (m, 1Н, Аr-Н). Анал. (С32Н38Сl2N4О) С, Н, N. Приклади 5 і 6 Виділення Енантіомерів сполуки 5 для отримання сполук 6 і 7 Препаративне виділення цієї сполуки було проведено на семі-препаративній колонці Ghiralcel AD (21ммX25см), використовуючи Гексан/ЕtOН/Діетиламін 80:20:0.1 як рухома фаза. На колонці Chiralcel AD, (-)ізомер було виявлено, як такий, що елює першим. Приклад 5 (-)4-[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метил-N,N-діетилбензамід (сполука 6) [a]D25:: -131°(c 1.0, MeOH) Приклад 6 (+)4[[4-(4-метилбензил)-1-піперазиніл](8-хінолініл)метил]-N,N-діетилбензамід (сполука 7) [a]D25: +124°(с 1.4, МеОН) Приклад 7 Одержання дигідрохлориду 4-[(4-[4-(mреm-бутил)бензил-1-піперазиніл](8-хінолініл)метил-N,Nдіетилбензаміду (сполука 8) Способом, аналогічним до одержання сполуки 2, отримали названу сполуку 8. Алкілування здійснювали з 4-mреm-бутилбензилброміду. МС (ES) 549.53 (МН+). ІЧ-Спектр (NaCI, вільний амін) 2963, 2807, 2360, 1631, 1456, 1425, 1285, 1135, 1094, 1001 (сm-1). ‘H ЯМР (CDCI 3, вільний амін) 5=1.0, 1.2 (2m, 6Н), 1.29 (s, 9Н), 2.50 (m, 8Н), 3.2, 3.5 (2m), 3.50 (s, 2Н), 6.04 (s, 1Н), 7.16-7.68 (m, 11Н), 7.98-8.10 (m, 2Н), 8.92 (m, 1Н). Анал. (C36H46Cl2 N4O) C, H, N. Приклад 8 Одержання дигідрохлориду N,N-діетил-4-[[4-(4-нітробензил)-1-піперазиніл](8-хінолініл)метилбензаміду (сполука 9) Способом, аналогічним до одержання вищезазначеної сполуки 2, отримали названу сполука 9. Алкілування здійснювали з 4-нітробензилброміду. МС (ES) 538.04 (МН+). ІЧ-Спектр (NaCI, вільний амін) 2969, 2809, 2360, 1626, 1518, 1456, 1426, 1343, 1286, 1134,1095,1001 (0711). ‘H ЯМР(СDСl3, вільний амін) 6=1.0,1.2 (2m, 6Н), 2.50 (m, 8Н), 3.2,3.5 (2m), 3.60 (s, 2Н), 6.05 (s, 1Н), 7.188.16 (m, 13Н), 8.94 (m, 1Н). Aнал. (C32H37Сl2N5O3) C, H, N. Приклад 9 Одержання дигідрохлориду 4-[(4-[2,4-bis(трифлуорметил)бензил]-1-піперазиніл)(8-хінолінлметил]-N,Nдіетилбензаміду (сполука 10) 10. Дотримуючись способу, аналогічному до одержання вищезазначеної сполуки 2, отримали названу сполука Алкілування здійснювали з 2,4-bis(трифлуорметил)бензилброміду. МС (ES) 629.08 (МН+). ІЧ-спектр (NaCI, вільний амін) 2970, 2811, 2360, 1628, 1456, 1426, 1346, 1275, 1170, 1128 (сm-1) ‘H ЯМР (CDCl3, вільний амін) d=1.0,1.2 (2m, 6Н), 2.48 (m, 8Н), 3.2, 3.5 (2m), 3.71 (s, 2Н), 6.06 (s, 1Н), 7.208.14 (m, 12Н), 8.95 (m, 1Н). Анал.. (C34H36Cl2F6N4O) С, Η, Ν Приклад 10 Одержання дигідрохлориду N,N-діетил-4-[[4-(4-метоксибензил)-1-піперазиніл](8-хінолініл)метилбензаміду (сполука 11) Способом, аналогічним до одержання вищезазначеної сполуки 2, отримали названу сполуку 11. Алкілування здійснювали з 4-метоксибензилхлориду. МС (ES) 523.45 (МН+). 15 ІЧ-Спектр (NaCI, вільний амін) 2966, 2806, 2360, 1627, 1510, 1456, 1426, 1286, 1246, 1134,1095 (сm-1). Ή ЯМР(СDСl3, вільний амін) d=1.0, 1.2 (2m, 6Н), 2.48 (m, 8Н), 3.2, 3.5 (2m), 3.47 (s, 2Н), 3.78 (s, 3Н), 6.03 (s, 1Н), 6.80-7.68 (m, 11Н), 8.01-8.12 (m, 2Н), 8.93 (m, 1Н). Анал. (С33Н40СІ 2N4О2) С, Н, N. Приклад 11 Одержання дигідрохлориду 4-[[4-(2,4-дихлорбензил)-1-піперазиніл](8-хінолініл)метил]-N,N-діетилбензаміду (сполука 12) Дотримуючись способу, аналогічному до одержання вищезазначеної сполуки 2, отримали названу сполуку 12. Алкілування здійснювали з 2,4-дихлорбензилхлориду. МС (ES) 562.45 (МН+). ІЧ-Спектр (NaCI, вільний амін) 2968, 2810, 2360, 2341, 1627, 1470, 1426, 1285, 1134, 1095 (cm-1). ‘H ЯМР (CDCI 3, вільний амін) d=1.0,1.1 (2m, 6Н), 2.5 (m, 8Н), 3.2, 3.5 (2m), 3.58 (s, 2H), 6.05 (s, 1H), 7.147.70 (m, 10Н), 8.06 (m, 2H), 8.94 (m, 1H). Анал.. С, Η, Ν. Приклад 12 Одержання дигідрохлориду N,N-діетил-4-[[4-(2-піридинілметил)-1-піперазиніл[(8-хінолініл)метилбензамід (сполука 13) Сполуку 1 (80мг, 0,20ммоль) розчинили в МеОН (2мл) з 2-піридилкарбоксалдегідом (39мкл, 0,40ммоль) і НОАс (1мкл, 0,02ммоль). Ціаноборогідрид натрію (26мг, 0,40ммоль) додали і перемішування продовжували протягом 48 годин. Розчинник випарювали і залишок очищали хроматографії на діоксиді силіцію (0-10% МеОН in CH2Cl2). 38мг (39%) продукту отримали. MC(ES)494.19(MH+). ІЧ-Спектр (NaCI, вільний амін) 2968, 2809, 2360, 1626, 1455, 1428, 1286, 1134, 1094, 1001 (сm-1). ‘H ЯМР(СОСl3, вільний амін) d=1.0, 1.2 (2m, 6Н), 2.50 (m, 8Н), 3.2, 3.5 (2m), 3.69 (s, 2Н), 6.05 (s, 1Н), 7.127.70 (m, 10Н), 8.08 (m, 2Н), 8.54 (m, 1Н), 8.94 (m, 1Н). Анал.. (С33H37CI 2N5O) C, H, N. Приклад 13 Одержання N,N-діетил-4[[4-(3-тієнілметил)-1-піперазиніл](8-хінолініл)метил]бензаміду (сполука 14) Названу сполуку 14 отримали дотримуючись синтетичного способу з нижченаведеної схеми 3. До розчину сполуки 1 (500мг; 0,99ммоль) в метанолі (10мл) додали тіофен 3-карбоксалдегід (104мкл; 1,19ммоль), з наступним додаванням оцтової кислоти (0,1мл; 1%) і ціаноборогідрид натрію (186,6мг; 2,97ммоль). Реакційну суміш перемішували усю ніч, тоді додали гідроксид натрію 2N і суміш екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення оберненою фазою, з використанням 10%-30% CH3CN/H2O (ТФОК як буфер), дало 258мг бажаного продукту (сіль ТФОК). Чистота високоефективної рідинної хроматографії (ВЕРХ): >99% (215нм); >95% (254нм) (М+1) Обчислено: 499.25, (М+1 Спостерігали: 499.46 Анал..: Обчисл. для (C30H34N4OSX2.80C2HO2F3Χ1.80Н2О): С:50.28%; Н:4.79%; N: 6.59%;O: 15.80%;S: 3.77%;F: 18.77% виявлено: С: 50.28%; Н: 4.83%; N: 6.53% ‘H ЯМР: 8.95 (dd, IH, J=4.4, 2.0Гц), 8.38 (dd, IH, J=8.0, 2.0Гц), 8.00 (dd, IH, J=7.2, 1.6Гц), 7.84 (dd, IH, J=8.0, 1.6Гц), 7.52-7.62 (m, 5H), 7.45 (dd, IH, J=4.8, 2.8Гц), 7.20 (dd, 2H, J=8.8, 2.2Гц), 7.11 (dd, IH, J=4.8, 1.6Гц), 5.96(s, IH), 4.27 (s, 2H), 3.34-3.44 (m, 2H), 3.22-3.28 (m, 4H), 3.04-3.14 (m, 2H), 2.66-2.88 (m, 4H), 1.04-1.14 (m, 3H), 0.880.98(m, 3H) Приклад 14 Одержання N,N-діетил-4[[4-(2-фуранілметил)-1-піперазиніл](8-хінолініл)метил]бензаміду (сполука 15). Названу сполуку 15 отримали, дотримуючись синтетичного способу нижченаведеної Схеми 4. До розчину фурфурилового спирту при 0°С (0,19мл; 2.24ммоль) та триетиламіну (0,52мл; 3,73ммоль) в метиленхлориді (4мл) додали метансульфонілхлорид (0,17мл; 2,.24ммоль). Суміш перемішували 1 годину при 0°С, тоді додали сполуку 1 (300мг; 0,75ммоль). Реакційній суміші дали можливість нагрітися до кімнатної температури і перемішували усю ніч, тоді нагріли до 45°С і перемішували протягом півтори години. Реакційній суміші дали охолодитися до кімнатної температури і додали NaOH 2N, доки рівень рН був основним. Суміш екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення оберненою фазою, з використанням 10%-25% CH3CN/H2O (ТФОК як буфер), дало 197мг бажаного продукту (сіль ТФОК). Чистота ВЕРХ: >99% (215 нм, 254нм і 280нм) (М+1) Обчисл.: 483.63, (М+1) спостерігали: 483.30 ‘H ЯМР: 8.89 (dd, 1Η, J=4.4, 1.6Гц), 8.29 (dd, 1Η, J=8.0, 1.6Гц), 7.97 (dd, 1Η, J=7.2, 1.6Гц), 7.79 (d, 1H, J=7.2Гц), 7.61 (d, 2H, J=8.0Гц), 7.52-7.58 (m, 2H), 7.48 (dd, 1H, J=8.0, 4.4Гц), 7.19 (d, 2H, J=8.0Гц), 6.56 (d, 1H, J=3.2Гц), 6.40 (dd, 1H, J=3.2, 2.4Гц), 6.02 (s, 1H), 4.26 (s, 2Н), 3.34-3.44 (m, 2Н), 3.16-3.26 (m, 4Н), 3.04-3.14 (m, 2Н), 2.68-2.86 (m, 4Н), 1.06-1.14 (m, 3Н), 0.90-0.98 (m, 3Н) Приклад 15 Одержання N,N-дieтил-4-[[4-(3-фуранілметил)-1-піперазиніл](8-хінолініл)метил]бензаміду (сполука 16) Названу сполуку 16 отримали, дотримуючись синтетичного способу з нижченаведеної Схеми 5. До 0°С розчину 3-фуранметанолу (220мг; 2,24ммоль) та триетиламіну (0,52мл; 3,73ммоль) в метиленхлориді (4мл) додали метансульфонілхлорид (0,17мл; 2.24ммоль). Суміш перемішували 1 годину при 0°С, тоді додали сполуку 1 (300мл; 0,75ммоль). Реакційній суміші дали можливість нагрітися до кімнатної темп, і перемішували усю ніч, тоді нагрівали до 45°С і перемішували протягом 3.30 годин. Реакційній суміші дали можливість охолонути до кімнатної темп, і додавали NaOH 2N, доки рівень рН був основним. Суміш екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення оберненою фазою, використовуючи 10%-25% CH3CN/H2O (ТФОК як буфер) дало 293мг бажаного продукту ( сільТФОК). Чистота ВЕРХ: >98% (215нм та 280нм); >99% (254нм) (М+11 Обчисл.: 483.63, (М+1) спостережено: 483.34 Анал..: Обчисл. Для (С3ОН34О2X3.10C2HO2F3X1.70Н2О): С:50.17%; Η 4.71%; Ν:6.46%; 0:18.27%; F:20.39% виявлено: С:50.14%; Н:4.76%; N1:6.38% ‘H ЯМР: 8.93 (dd, 1Н, J=4.4, 2.0Гц), 8.36 (dd, 1Н, J=8.6, 2.0Гц), 8.00 (dd, 1H, J=7.4, 1.2Гц), 7.82 (dd, 1Н, J=7.6, 1.2Гц), 7.48-7.66 (m, 6Н), 7.19 (d, 2Н, J=8.0Гц), 6.46 (s, 5.97 (s, 1H), 4.13 (s, 2H), 3.32-3.44 (m, 2H), 3.203.28 (m, 4H), 3.04-3.14 (m, 2H), 2.66-2.86 (m, 4H), 1.04-1.14 (m, 3Н), 0.88-0.98 (m, 3Н) Приклад 16 Одержання дигідрохлориду N,N-діетил-4-[[4-(2-тіофенілметил)-1-піперазиніл](8-хінолініл)метил]бензаміду (сполука 17) Названу сполуку 17 отримали," дотримуючись синтетичного способу з нижченаведеної Схеми 6. До розчину сполуки 1 (0,99ммоль) в метиленхлориді (10мл) додали 2-тіофенкарбоксалдегід (190мкл; 1,98ммоль), з наступним додаванням оцтової кислоти (0.1мл; 1%). Суміш перемішували 30хв., тоді додали триацетоксиборогідрид натрію (0,63г; 2.97ммоль) і реакційну суміш перемішували усю ніч. Реакційну суміш нейтралізували гідроксидом 2N натрію і екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення оберненою фазою, використовуючи 10%-30% CH3CN/H2O (ТФОК як буфер) дало 15мг бажаного продукту (сіль ТФОК). Чистота ВЕРХ: >99% (215нм); >96% (254нм) (М+1) Обчисл.: 499.25, (М+1) спостережено: 499.33 Анал..: Обчисл. для (C30H34N4OSX2.50C2HO2F3X0.10Н2О): С:53.51%; Н:4.71%; N:7.13%; 0:12.42%; 5:4.08%; F:18.14% виявлено: С:53.49%; Н:4.63%; N.7.49% ‘H ЯМР: 8.91 (dd, 1Н, J=4.0, 1.6Гц), 8.30 (dd, 1Н, J=8.8, 1.6Гц), 7.96 (dd, 1H, J=7.4МГц), 7.81 (d, 1Н, J=7.2Гц), 7.62 (d, 2Н, J=8.0Гц), 7.46-7.58 (m, 3Н), 7.20 (d, 2H, J=8.0Гц), 7.14-7.22 (m, 1H), 7.00 (dd, 1H, J=5.2, 3.6Гц), 6.03 (s, 1H), 4.38 (s, 2H), 3.34-3.44 (m, 2H), 3.14-3.22 (m, 4Н), 3.06-3.12 (m, 2Н), 2.74-2.88 (m, 4Н), 1.04-1.14 (m, 3Н), 0.88-0.98 (m, 3Н) Приклад 17 Одержання дигідрохлориду N,N-діетил-4-[[4-(2-імідазолілметил)-1-піперазиніл](8-хінолініл)метилбензаміду (сполука 18) Названу сполуку 18 отримали, дотримуючись синтетичного способу з нижченаведеної Схеми 7. До розчину сполуки 1 (0,99ммоль) в метанолі (10мл) додали 2-імідазолкарбоксалдегід (114мг; 1.19ммоль), з наступним додаванням оцтової кислоти (0,5мл; 5%). Суміш перемішували 3 години, тоді додали ціаноборогідрид натрію (186,6мг; 2,97ммоль) і реакційну суміш перемішували усю ніч. Реакційну суміш нейтралізували гідроксидом 2N натрію і екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення оберненою фазою, використовуючи 10%-30% CH3CN/H2O (ТФОК як буфер) дало сіль ТФОК. Сіль НСІ отримали, використовуючи НСІ/етер. Вихід: 60.3мг бажаного продукту (сіль НСІ). Чистота ВЕРХ: >95% (215 нм); >93% (254нм) (М+1) Обчисл.: 483.29, (М+1) спостережено: 483.19 ‘H ЯМР: 9.12-9.22 (m, 1Н), 8.54-8.62 (m, 1Н), 8.08-8.16 (m, 1Н), 7.98-8.04 (m, 1Н), 7.60-7.86 (m, 4Н), 7.387.46 (m, 2Н), 7.22-7.32 (m, 2Н), 6.32 (s, 1H), 4.11 (s, 2H), 2.94-3.40 (m, 12Н), 0.88-1.12 (m, 6Н). Приклад 18 Одержання дигідрохлориду N,N-діетил-4-[[4-(4-імідазолілметил)-1-піперазиніл](8-хінолінл)метил]бензаміду (сполука 19) Названу сполуку 19 отримали, дотримуючись синтетичного способу з нижченаведеної Схеми 8. До розчину 434 кімнатної темп. (400мг; 0,99ммоль) та 4-імідазол карбоксалдегіду (95,5мг; 0,99ммоль) в метиленхлориді (Юмл) додали оцтову кислоту (0.1мл). Суміш перемішували протягом 5 годин, тоді додали триацетоксиборогідрид натрію (632мг; 2,98ммоль). Реакційну суміш перемішували усю ніч і нейтралізували гідроксидом 2N натрію. Суміш екстрагували метиленхлоридом (3Х). Поєднані екстракти метиленхлориду сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення обернено фазовою хроматографією, використовуючи 15% CH3CN/H2O (ТФОК як буфер) дало 103мг бажаного продукту (сіль ТФОК). Чистота ВЕРХ: >99% (215нм, 254нм та 280нм) (М+1) Обчисл.: 483.28, (М+1) спостережено: 482.96 Анал..: Обчисл. для (C29H34N6OX3.80C2HO2F3Χ0.80Н2О): С:47.25%; Н:4.27%; Ν:9.03%;' 0:16.17%; F:23.28% виявлено: С:47.31%; Н:4.40%; N:8.87 ‘H ЯМР: 8.99 (dd, IH, J=4.4, 1.2Гц), 8.76 (d, IH, J=1.2Гц), 8.39 (dd, IH, J=8.8, 1.2Гц),7.93 (dd, IH, 3=7.2, 1.6Гц), 7.86 (dd, 1H, J=8.0, 1.6Гц), 7.71 (d, 2H, J=8.8Гц), 7.60 (dd, 1H, J=8.8, 4.4Гц), 7.56 (dd, IH, J=8.0, 7.2Гц), 7.40 (s, 1H), 7.27 (d, 2H, J=8.8Гц), 6.12 (s, 1H) 3.74(s, 2H), 3.38 (q, 2H, J=6.4Гц), 3.10-3.25 (m, 6H), 3.06 (q, 2H, J=7.2Гц), 2.75-2.90 (m,2H),1.08 (t, 3H, J=6.4H4), 0.92 (t, 3H, J=7.2Гц) Приклад 19 Одержання дигідрохлориду N,N-діетил-4-[[4-(3-триазолілметил)-1-піперазиніл](8-хінолінл)метилбензаміду (сполука 20) Названу сполуку 20 отримали, дотримуючись синтетичного способу з нижченаведеної Схеми 9. До розчину 434 кімнатної темп. (200мг; 0,50ммоль) в диметилформаміді (10мл) додали карбонат калію (275мг; 1,99ммоль), з наступним додаванням N-формамід-2-(хлорметил)ацетамідину (170мг; 1,24ммоль). Реакційну суміш нагрівали до 60°С і перемішували протягом 2 днів, тоді темп, підвищували до 140°С і перемішували протягом 3 годин. Реакційній суміші дали можливість охолодитися до кімнатної темп, і додали води. Суміш екстрагували етилацетатом (3Х). Поєднані екстракти етилацетату сушили над Na2SO4, фільтрували і концентрували під зниженим тиском. Очищення обернено-фазовою хроматографією, використовуючи 20% CH3CN/H2O (ТФОК як буфер) дало 21мг бажаного продукту (сіль ТФОК). Чистота ВЕРХ: >99% (215нм, 254нм та 280нм) (М+1) Обчисл.: 484.28, (М+1) спостережено: 483.92 Анал..: Обчисл. для (С28Н33N7ОX3.30C2HO2F3Χ3.30Н2О): С:45.20%; Н:4.70%; N:10.66%;O:18.97%;F:20.46% виявлено: С:45.12%; Н:4.60%; N: 10.84 ‘H ЯМР: 8.94 (dd, ІН, J=4.4, 1.6Гц), 8.38 (s, 1Н), 8.33 (dd, 1Н, J=8.0, 1.2Гц), 7.93 (d, 1H, J=7.2Гц), 7.85 (d, 1H, J=7.2Гц), 7.65 (d, 2H, J=8.8Гц), 7.51-7.58 (m, 2H), 7.23 (d, 2H, J=8.8Гц), 6.15 (s, 1H), 4.21 (s, 2H), 3.40-3.50 (m, 2H), 3.10-3.30 (m,-8H), 2.90-3.10 (m, 2H), 0.90-1.30 (m, 6H) Фармацевтичні композиції Нові сполуки згідно з винаходом можна уводити перорально, внутрішньом'язово, підшкірно, місцево, через ніс, інтраперитонально, через грудну клітку, внутрішньовенно, епідурально, інтратекально, внутрішньоцереброшлуночково (інтрацеребровентикулярно), та ін'єкцією в суглоби. Бажаним шляхом уведення є пероральний, внутрішньовенний або внутрішньом'язовий. Доза залежатиме від шляху уведення, складності захворювання, віку і маси пацієнта та інших факторів, які звичайно враховує обслуговуючий лікар, при визначенні індивідуального режиму та рівня дози, як найбільш характерних для кожного пацієнта. Для одержання фармацевтичних композицій зі сполук згідно з винаходом, інертні, фармацевтично прийнятні носії можуть бути або у рідкій, або у твердій формі. До препаратів у твердій формі належать порошки, таблетки, дисперговані гранули, капсули, крохмальні облатки та супозиторії. Твердим носієм може бути одна або більше речовин, які також можуть діяти як розріджувачі, ароматизатори, солюбілізатори, змащувальні, суспендувальні засоби, зв'язувальні засоби, або дезинтегратори таблеток; ним також може бути капсулюючий матеріал. В порошках носієм є тонкоподрібнена тверда речовина у суміші з тонкоподрібненим активним компонентом. В таблетках активний компонент змішано з носієм, що має необхідні зв'язувальні властивості у відповідних пропорціях, і спресовано у бажану форму і розмір. Для одержання супозиторних композицій, спочатку розплавляють низькоплавкий віск, як наприклад, суміш гліцеридів жирної кислоти і масла какао, а активний інгредієнт диспергують у ньому, наприклад, перемішуванням. Розплавлену однорідну суміш тоді виливають у форми відповідного розміру і залишають охолоджуватися і твердіти. Придатними носіями є карбонат магнію, стеарат магнію, тальк, лактоза, сахароза, пектин, декстрин, крохмаль, трагакант, метилцелюлоза, натрій-карбоксиметилцелюлоза, низькоплавкий віск, масло какао, та подібні їм. Фармацевтично прийнятними солями є ацетат, бензолсульфонат, бензоат, гідрокарбонат, гідротартрат, бромід, ацетат кальцію, камзилат, карбонат, хлорид, цитрат, дигідрохлорид, едетат, едизилат, естолат, езилат, фумарат, глюкаптат, глюконат, глютамат, гліколіларазанілат, гексилрезорцинат, гідрабамін, гідробромід, гідрохлорид, гідроксинафтоат, йодид, ізетіонат, лактат, лактобіонат, малат, малеат, манделат, мезилат, метилбромід, метилнітрат, метилсульфат, мукат, напсилат, нітрат, памоат (ембонат), пантотенат, фосфат/дифосфат, полігалактуронат, саліцилат, стеарат, субацетат, сукцинат, сульфат, танат, тартрат, теоклат, триетіодид, бензатин, хлорпрокаїн, холін, діетаноламін, етилендіамін, меглумін, прокаїн, солі алюмінію, кальцію, літію, магнію, калію, натрію та цинку. Бажаними фармацевтично прийнятними солями є гідрохлориди, і гідротартрати. Солі гідрохлориду є бажанішими. Названа композиція повинна включати композицію активного компоненту з капсулюючим матеріалом у вигляді носія, що забезпечує капсулу, в якій активний компонент (з іншими носіями або без них) оточений носієм, який таким чином пов'язаний з ним. Подібним чином, включені і крохмальні облатки. Таблетки, порошки, крохмальні облатки і капсули можуть бути використані як тверді дозовані форми, придатні для перорального уведення. Рідина з композицій включає розчини, суспензії та емульсії. Стерильну воду або водопропіленгіколеві розчини активних сполук можна визнати як приклад рідких препаратів, придатних для парентерального уведення. Рідкі композиції також можуть бути сформовані у розчині у водному розчині поліетиленгліколю. Водні розчини для перорального уведення можна виготовити розчиненням активного компоненту у воді і додаванням відповідних барвників, коригентів, стабілізаторів, та згущувачів, як бажаних елементів. Водні суспензії для перорального застосування можна виготовити розподіленням тонкоподрібненого активного компонента у воді разом з клейким матеріалом, як наприклад, натуральна синтетична гума, смола, метилцелюлоза, натрій-карбоксиметилцелюлоза, та інші суспендувальні засоби, відомі у рівні фармацевтичного складу. Бажано, щоб фармацевтичні композиції були у формі одиничної (разової) дози. У такій формі, композицію розподіляють на одиничні дози, які містять відповідну кількість активного компонента. Одинична доза може бути упакованим препаратом, упаковкою, що містить роздільні кількості препаратів, наприклад, упаковані таблетки, капсули, та порошки в пузирчиках або ампулах. Одинична доза сама також може бути капсулою, крохмальною облаткою, або таблеткою, або нею може бути відповідна кількість будь-якої з цих упакованих форм. БІОЛОГІЧНА ОЦІНКА Модель In vitro Клітинна культура Клітини 293S людини, що експресують рецептори m, d і к людини і резистентні до неоміцину було вирощено у суспензіях при 37°С і 5% СО2 у вібраційних колбах, що містять вільний від кальцію DMEM10% FBS, 5% BCS, 0,1% Pluronic F-68, і 600мкг/мл генетицину. Одержання мембран Клітини гранулювали і повторно суспендували в лізісному буфері (50мМ Трис, рН 7,0, 2.5 мМ EDTA, з PMSF, який додають як безпосередньо перед використанням до 0,1мМ з 0,1Μ вихідного розчину в етанолі), інкубували на льоді протягом 15хв., потім гомогенізували з політроном протягом 30с Суспензію центрифугували (прокручували) при 1000г. (макс.) протягом 10хв. при 4°С. Супернатант (надосадковий шар) було збережено на льоді, а гранули повторно суспендували і центрифугували як раніше. Супернатанти з двох центрифугувань було поєднано і центрифуговано при 46000г (макс.) протягом 30хв. Гранули було повторно суспендовано у холодному буфері Трис (50мМ Трис/СІ, рН 7,0) і центрифуговано знову. Отримані гранули було повторно суспендовано в буфері для мембран (50мМ Трис, 0,32Μ сахароза, рН 7,0). Аліквоти (1мл) у поліпропіленових трубках було заморожено у сухому льоді/етанолі і збережено при -70°С до використання. Концентрації білків було визначено модифікованим аналізом Лоурі за допомогою SDS. Аналіз зв'язування Мембрани розморожували при 37°С, охолоджували на льоді, пропускали 3 рази через голку 25 розміру, і розбавляли буфером для зв'язування (50мМ Трис, 3мМ мrСІ 2, 1мг/мл BSA (Бідма А-7888), рН 7,4, які зберігали при 4°С, після фільтрування через 0,22м фільтр, і до яких тільки що було додано 5мкг/мл апротиніну, 10мкМ бестатину, 10мкМ дипротину А, без DTT). Аліквоти по 100мкл (на мкг білку, див. Таблицю 1) було додано у охолоджені льодом пропіленові туби 12x75мм, що містять 100мкл відповідного радіоліганду (див. Таблиця 1) і 100мкл тестованих при різних концентраціях пептидів. Загальне (33) та неспецифічне (НС) зв'язування визначали у відсутності і присутності 10мкМ налоксану, відповідно. Туби струшували та інкубували при 25°С протягом 60-75хв., після чого вміст швиденько фільтрували у вакуумі та промивали приблизно 12м/тубу льодяним буфером для промивання (50мМ Трис, рН 7,0, 3мМ МgСІ 2) через фільтри TF/B (Whatman), попередньо просочених протягом щонайменше 2 годин у 0,1% поліетиленіміні. Радіоактивність (сірм), що залишилась на фільтрах, було визначено бета-лічильником, після промочування фільтрів протягом щонайменше 12 годин в мінісклянках, що містять 6-7мл сцинтиляційної рідини. Якщо аналіз проведено у планшетах з 96 глибокими комірками, фільтрування здійснювали на 96-місцевих попередньо просочених РЕІ уніфільтрах, які було промито 3x1мл промивочним буфером, і висушено у сушильній шафі при 55°С протягом 2 годин. Фільтр-плати було обчислено лічильником TopCount (Packard) після додавання 50мкл/комірку сцинтиляційної рідини MS-20. Аналіз даних Специфічне зв'язування (СЗ) було обчислено як TB-NS, і СЗ у присутності різних тестованих пептидів було виражено як процент від контрольного СЗ. Значення ІК50 і коефіцієнту Хілла (nн) для лігандів при витісненні специфічно зв'язаного радіоліганду було обчислено за логіт-перетворенням графіків або програмами підгонки кривих, наприклад, Ligand, GraphPad PrisM, SigMaPlot, або ReceptorFit. Значення К, було обчислено за рівнянням Ченга-Пруссофа. Значення ± стандартна похибка величин ІК50, К, та nн повідомлено для лігандів, тестованих за щонайменше трьома кривими заміщення. Біологічні дані зафіксовано у нижченаведеній таблиці 1. Таблиця 1 Підсумок біологічних даних HDELTA Приклад № 12 0.692 13 1.033 14 0 181 15 0.787 ΙΑ 1.509 17 1.091 18 1.54 19 18.751 HDELTA Мозок щурів Мозок з мишей МLM RLM % % % 100000% 100000% ЕС50 ЕС50 ЕС50 10000%rem. 10000%rem. ЕМАХ ЕМАХ ЕМАХ rem. rem. 0.76 97.73 20.99 106.43 27.14 91.34 0 53.5 21.5 66 1.44 101.18 17.7 111.96 25.77 112.68 1.667 71.667 10 62.667 0.76 88.65 14.26 102.02 20.49 106.48 0 49 13.5 84 S 0.79 88.99 14.16 108.81 16.01 109.85 0 68 10 86 2.39 99.36 30.83 100.5 24.2 98.41 0.5 46.5 9.5 64.5 3.03 95.66 49.47 105.91 75.1 92.47 0 21.5 5.5 75 5.85 93.82 452.31 111.0! 429.56 108.41 85.24 97.88 2807.47 56.35 1365.82 48.68 Експерименти на насичення рецепторів Значення радіоліганду Кd було визначено на основі проведення аналізу зв'язування на мембранах клітин з прийнятними радіолігандами при концентраціях в межах 0,2-5 разів оціненого Кd (до 10 разів, якщо кількості необхідного радіоліганду є допустимими). Специфічне зв'язування радіолігандів було виражено як пмоль/мг мембранного білку. Значення Κd і Вмакс з окремих експериментів було отримано з нелінійних пригінок специфічно зв'язаного (Б) проти нМ вільного радіоліганду (F) від індивідуалу згідно з одномісцевою моделлю. ВИЗНАЧЕННЯ МЕХАНО-АЛОДИНІЇ, ВИКОРИСТОВУЮЧИ ТЕСТУВАННЯ ВОН-ФРЕЯ Тестування проводили між 08:00 і 16:00, застосовуючи спосіб, описаний Chaplan et al. (1994). Щурів помістили у клітки з плексигласу на сітчасте дно з дроту, яке дає доступ до лапи, і залишили там для звикання на 10-15хв. Зоною тестування було вибрано середину підошви лівої задньої лапи, уникаючи менш чутливих подушечок ступні. До лап торкалися набороом з 8 волосинок Вон Фрея з логарифмічно зростаючою жорсткістю (0,41, 0,69,1,20, 2,04, 3,63, 5,50, 8,51, і 15,14г.; Stoelting, III, USA). Волосинку Вон Фрея підкладали з під-низу сітчастої підлоги, перпендикулярно до поверхні підошви, з достатньою силою, щоб спричинити таким чином легкий вигин лапи, і тримали приблизно 6-8 сек. Позитивну реакцію було помічено, коли лапа різко відсмикувалася. Відсмикування відразу після усування волосинки, також приймалося за позитивну реакцію. Рухомість вважали сумнівною реакцією, і у таких випадках подразнення повторювали знову. ПРОТОКОЛ ТЕСТУВАННЯ Тварин тестували в день 1 після операції для FCA-лікувальної групи. 50% поріг відсмикування визначали, використовуючи понижуючий спосіб Діксона (1980). Тестування почали з волосинки 2,04г, у середині набору. Стимули завжди представляли у послідовному напрямку, при зростанні чи зменшенні. У відсутність реакції відсмикування лапи на спочатку вибрану волосинку, спостерігали сильне збуджування; якщо лапа відсмикувалася, спостерігали наступне слабше збудження. Для обчислення оптимального порогу на основі цього способу необхідно 6 реакцій у безпосередній близькості до 50% порогу, і обчислювання 6 реакцій почалося тоді, коли відбулася перша зміна реакції, наприклад, поріг було вперше перейдено. У випадках, коли пороги виходили за межі стимулів, значення 15,14 (нормальна чутливість) або 0,41 (максимальна алодинічна). задавалися відповідно. Отриману картину позитивної та негативної реакцій було занесено у таблицю, використовуючи умовні позначення, X=ніякого відсмикування; 0=відсмикування, а 50% поріг відсмикування було уведено, використовуючи формулу: 50%г поріг=I0(xf+kd )/10,000 де Xf=значення останньої використаної волосинки Вон Фрея (у логарифмічних одиницях); k=табличне значення (з Chaplan et ai. (1994)) для зразка позитивної/негативної реакцій; і d=слабка різниця між стимулами (у логарифмічних одиницях). Тут d=0,224. Пороги Вон Фрея було перетворено у процент максимально можливої дії (% МРЕ), згідно з Chaplan et al. 1994. Наступне рівняння було використано для обчислення % МРЕ:% МРЕ=Поріг обробки ліками (г) - поріг алодинії (r) X 100 поріг для контролю (г) - поріг алодинії (г) УВЕДЕННЯ ТЕСТ-РЕЧОВИНИ Щурам робили ін'єкції (підшкірно, інтраперитонально, внутрішньовенно або. перорально) з тест-речовиною до початку тестування за Вон Фреєм, час між уведенням тест-сполуки і тестом Вон Фрея змінювали, залежно від природи тест-сполуки. ТЕСТ ВРИТИНГА Оцтова кислота призвела би до абдомінальних скорочень при інтраперитональному уведенні мишам, витягуючи таким чином їх тіло у типовій картині. Коли уводять аналгезивні ліки, цю описану зміну спостерігали рідше, і ліки вибирали як потенційно гарний засіб. Повним і типовим рефлекс Вритинга вважають лише тоді, коли присутні наступні елементи: тварина не рухається; нижча частина спини послаблена; видно підошву обох лап. (і) Одержання розчинів Оцтова кислота (АсОН): 120мкл Оцтової кислоти додають до 19,88мл дистильованої води для того, щоб отримати кінцевий об'єм 20мл з кінцевою концентрацією 0,6% АсОН. Розчин тоді змішують (перемішують мішалкою) і він готовий для ін'єкцій. Сполука (лікувальний засіб): Кожну сполуку виготовляють і розчиняють у найпридатнішому носії згідно зі стандартними способами. (іі) Уведення розчинів Сполуку (лікувальний засіб) уводять перорально, інтраперитонально (і.р.), підшкірно (s.c.) або внутрішньовенно (i.v.)) при 10мл/кг (враховуючи середню масу тіла мишей) за 20, 30 або 40хв. (згідно з класом сполук і їх характеристиками) перед тестуванням. Коли сполуку уводять центральним шляхом: інтравентркулярно (i.v.) або інтратекально (i.t.) уводять об'єм 5мкл. АсОН уводять інтраперитонально (і.р.) у двох місцях при концентрації 10мл/кг (враховуючи середню масу тіла мишей) відразу до тестування. (ііі) Тестування За твариною (мишею) спостерігали протягом 20хв. і записували кількість випадків (рефлекс Вритинга), і компілювали під кінець експерименту. Мишей тримали в індивідуальних клітках типу "коробка для взуття" з контактною підкладкою. Всього спостерігали за 4 мишами протягом того самого часу: одна контрольна і три для доз лікувального засобу.
ДивитисяДодаткова інформація
Назва патенту англійськоюA compound (variants), a pharmaceutical composition, a method for treatment (variants)
Автори англійськоюPlobeck Niklas
Назва патенту російськоюСоединение (варианты), фармацевтическая композиция, способ лечения (варианты)
Автори російськоюПлобек Никлас
МПК / Мітки
МПК: A61P 25/00, A61P 25/02, A61P 1/16, C07D 215/14, A61P 25/04, C07D 405/12, C07D 409/12, A61P 1/04, A61K 31/496, C07D 215/12, C07D 401/12, C07D 403/12, A61P 43/00
Мітки: фармацевтична, лікування, спосіб, варіанти, сполука, композиція
Код посилання
<a href="https://ua.patents.su/13-72945-spoluka-varianti-farmacevtichna-kompoziciya-sposib-likuvannya-varianti.html" target="_blank" rel="follow" title="База патентів України">Сполука (варіанти), фармацевтична композиція, спосіб лікування (варіанти)</a>
Похідне 3-арил-2-гідроксипропіонової кислоти, спосіб його отримання (варіанти), проміжна сполука, фармацевтична композиція (варіанти) та спосіб профілактики та/або лікування асоційованих з резистентністю до інс

Номер патенту: 71912
Опубліковано: 17.01.2005
Автор: Андерссон К'єлль
МПК: C07C 69/734, C07D 263/04, C07C 233/75, C07C 335/00, A61P 3/10, C07C 311/16, C07C 323/44, C07C 311/13, C07C 323/56, C07C 317/28, C07C 317/22, C07C 255/54, C07C 303/00, A61K 31/255, C07C 271/58, A61P 9/12, C07C 217/76, C07C 323/19, C07C 271/38, C07C 275/34, A61P 9/10, C07C 309/00, C07C 271/28, C07C 271/44, A61P 43/00, A61K 31/216, C07C 233/25, C07C 235/42, A61P 3/06, A61P 3/04, C07C 233/29, A61K 31/192, C07C 311/08
Мітки: 3-арил-2-гідроксипропіонової, резистентністю, похідне, інс, варіанти, отримання, проміжна, спосіб, асоційованих, сполука, кислоти, лікування, композиція, фармацевтична, профілактики
Формула / Реферат:
1. Похідне 3-арил-2-гідроксипропіонової кислоти формули (I) Iта його фармацевтично прийнятні солі, сольвати та кристалічні форми.2. Сполука за п. 1, яка відрізняється тим, що призначена для використання в терапії.3. Сполука за п. 1, яка відрізняється тим, що її використовують у виробництві лікувального засобу для профілактики та/або лікування асоційованих з резистентністю до інсуліну клінічних станів.4....
Гетероциклічна сполука, спосіб її одержання (варіанти), проміжні сполуки, фармацевтична композиція та спосіб лікування неврологічних захворювань або розладів (варіанти)

Номер патенту: 64006
Опубліковано: 16.02.2004
Автори: Палмер Майкл Джон, Кемп Марк Ян, Уайтс Мартін Джеймс, Сеннер Марк Аллен
МПК: C07D 401/14, A61K 31/4427, C07D 413/04, C07D 471/14, A61P 25/16, A61K 31/4709, A61P 43/00, C07D 417/04, A61P 37/00, A61P 25/00, A61P 25/28, A61P 25/14, A61P 25/02, A61P 21/00, A61P 27/16, C07D 417/14, A61K 31/454, C07D 413/14, C07D 401/04, A61P 19/00, A61K 31/4545, A61P 21/04, C07D 521/00, A61P 27/02
Мітки: неврологічних, сполука, проміжні, гетероциклічна, одержання, захворювань, розладів, лікування, сполуки, спосіб, варіанти, композиція, фармацевтична
Формула / Реферат:
1. Гетероциклічна похідна формули (І): , (I)або її фармацевтично прийнятна сіль, в якійA є нерозгалуженим C3-C5алкіленом, необов’язково, заміщеним C1-C6алкілом;X є O, S, NH або N(C1-C6алкіл);Y є О, S, NH або N(C1-C6алкіл);R є C-приєднаною 4-6-членною неароматичною гетероциклічною групою, що містить один атом азоту як гетероатом, згадана група, необов’язково, заміщена 1, 2 або 3 замісником(ами), кожний...
Похідні 4-діариламінопіперидину, їх використання, фармацевтична композиція на їх основі, спосіб лікування (варіанти), проміжна сполука

Номер патенту: 72287
Опубліковано: 15.02.2005
Автори: Браун Вілл'ям, Волпоул Крістофер
МПК: A61P 25/04, A61P 9/12, A61K 31/4468, A61P 29/00, A61P 1/00, A61P 25/06, A61P 25/24, A61K 31/4535, A61P 37/02, A61P 25/36, A61K 31/454, A61P 31/12, C07D 401/06, C07D 405/06, A61P 19/08, A61P 25/34, A61P 25/22, A61P 25/32, A61P 13/02, A61P 11/00, A61P 35/00, C07D 211/58, A61K 31/4545, A61P 1/12, A61P 25/02, A61K 31/4525, C07D 409/06, A61P 23/00
Мітки: спосіб, проміжна, сполука, основі, похідні, варіанти, 4-діариламінопіперидину, використання, лікування, фармацевтична, композиція
Формула / Реферат:
1. Сполука формули І, (І)деR1 вибрано з групи, яка складається з(і) фенілу;(іі) піридинілу;(ііі) тієнілу;(iv) фурилу
Сполука 2-феніл-1-[4-(2-аміноалкокси)-бензил]індолу як естрогенний засіб, спосіб її одержання (варіанти), фармацевтична композиція та спосіб лікування (варіанти)

Номер патенту: 48148
Опубліковано: 15.08.2002
Автори: Сантіллі Артур Аттіліо, Трен Беч Дінх, Міллєр Кріс Пол, Колліні Мішель Девід
МПК: C07D 209/32, C07D 405/12, C07D 209/12, C07D 487/08, C07D 209/42, A61K 31/40, A61K 31/55, C07D 401/12, C07D 405/04, C07D 209/08, A61K 31/445, A61K 31/495, A61P 43/00, A61K 31/403, A61K 31/4427, C07D 209/10, C07D 209/30, A61P 15/00, A61P 13/02, C07D 403/12, A61K 31/404
Мітки: лікування, 2-феніл-1-[4-(2-аміноалкокси)-бензил]індолу, фармацевтична, естрогенний, сполука, засіб, спосіб, одержання, композиція, варіанти
Формула / Реферат:
1. Соединение 2-фенил-1-[4-(2-аминоалкокси)-бензил]индола формулы I или II:,,где:R1 выбран из Н, ОН или его C1-C12 сложных эфиров (линейная цепь или разветвленная) или C1-C12алкиловых эфиров (линейная цепь или разветвленная, или циклическая), или галогенов; или C1-C4галогенированных простых эфиров, включая трифторметиловый эфир и трихлорметиловый эфир.R2, Rз, R4, R5 и R6, независимо, выбраны из Н, ОH или его...
N-6-заміщені 7-деазапурини (варіанти), спосіб їх одержання, фармацевтична композиція для лікування аденозинопосередкованих захворювань (варіанти)

Номер патенту: 72736
Опубліковано: 15.04.2005
Автори: Кастелано Арліндо Л., Уіттер Девід Дж., Маккіббен Брайан
МПК: A61K 31/505
Мітки: фармацевтична, n-6-заміщені, 7-деазапурини, спосіб, варіанти, захворювань, аденозинопосередкованих, одержання, лікування, композиція
Формула / Реферат:
1. N-6-заміщений 7-деазапурин формули (І),де: кожний з R1 і R2 незалежно являє собою атом водню, заміщений (С1-С30)алкіл з прямим ланцюгом, заміщений (С3-С10)алкіл з розгалуженим ланцюгом, заміщений (С4-С10)циклоалкіл, заміщений циклопропіл або заміщену або незаміщену арильну групу; де тільки один з R1 і R2 може бути воднем;де, коли...
Попередній патент: Мікропроцесорний пристрій з кодуванням
Наступний патент: Похідні хіназоліну як інгібітори васкулярного ендотеліального фактора росту (vegf)
Випадковий патент: Спосіб диференціальної діагностики деструктивної та недеструктивної форм гострого холециститу