Дипіридодіазепінони як інгібітори зворотної транскриптази
Номер патенту: 78774
Опубліковано: 25.04.2007
Автори: Дезіель Робер, Йоакім Крістіан, Огілві Уільям В., О'меара Джеффрі, Сімоно Брюно












Формула / Реферат
1. Сполука формули:
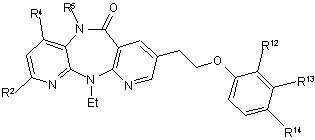 , (I)
, (I)
де
R2 означає H, галоген, С1-С4алкіл, ОС1-С4алкіл, NНС1-С4алкіл або N(С1-С4 алкіл)2;
R4 означає H або СН3;
R5 означає H або СН3, за умови, що й R4 й R5 не мають однакового значення;
R12 означає Н, галоген, С1-С4алкіл, СF3 або NO2;
R13 означає Н, С1-С4алкіл, галоген, ОН або NH2, за умови, що R12 й R13 обидва не означають Н; і
R14 означає COOR14a, де R14a означає Н або С1-С6алкіл; або R14 означає С2-C4aлкeнiл-COOR14a, де R14a має вказані вище значення; або R14 означає С1-С4алкіл-СООR14a, де R14a має вказані вище значення;
або її сіль або проліки.
2. Сполука за п. 1, де R2 означає Н, галоген, С1-С4алкіл, ОС1-С4алкіл або N(С1-С4алкіл)2 й R4 й R5 не мають однакових значень.
3. Сполука за п. 2, де R2 означає Н, Сl, F, С1-С4алкіл, ОС1-С4алкіл або N(С1-С4алкіл)2.
4. Сполука за п. 3, де R2 означає Н, Сl, F, СН3, ОМе або OEt.
5. Сполука за п. 4, де R2 означає H.
6. Сполука за п. 1, де R4 означає Н.
7. Сполука за п. 1, де R5 означає СН3.
8. Сполука за п. 1, де R12 означає галоген, С1-С4алкіл, СF3 або NO2.
9. Сполука за п. 8, де R12 означає Вr, Сl, СН3 або СН3СН2.
10. Сполука за п. 9, де R12 означає СН3 або СН3СН2.
11. Сполука за п. 1, де R13 означає Н, СН3 галоген, ОН або NH2.
12. Сполука за п. 11, де R13 означає Н, СН3 або ОН.
13. Сполука за п. 12, де R13 означає Н.
14. Сполука за п. 1, де R14 означає СООН, СООМе, С2-С4алкеніл-СООН або С1-С4алкіл-СООН.
15. Сполука за п. 14, де R14 означає СООН, СН=СН-СООН, СН2СООН або СН2СН2СООН.
16. Сполука за п. 15, де R14 означає СООН.
17. Сполука формули (І) за п. 1:
, (I)
де
R2 означає H, Сl, F, СН3, ОМе або OEt; R4 означає H; R5 означає СН3; R12 означає Вr, Сl, СН3 або СН2СН3; R13 означає Н, СН3 або ОН; і R14 означає СООН, СН=СН-СООН, СН2СООН або СН2СН2СООН; або її сіль або проліки.
18. Сполука за п. 17, де R2 означає Н; R4 означає Н; R5 означає СН3; R12 означає СН3 або СН3СН2; R13 означає Н; і R14 означає СООН; або її сіль або проліки.
19. Фармацевтична композиція, призначена для лікування або запобігання ВІЛ-інфекції, яка містить сполуку формули І за п. 1 або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій.
Текст
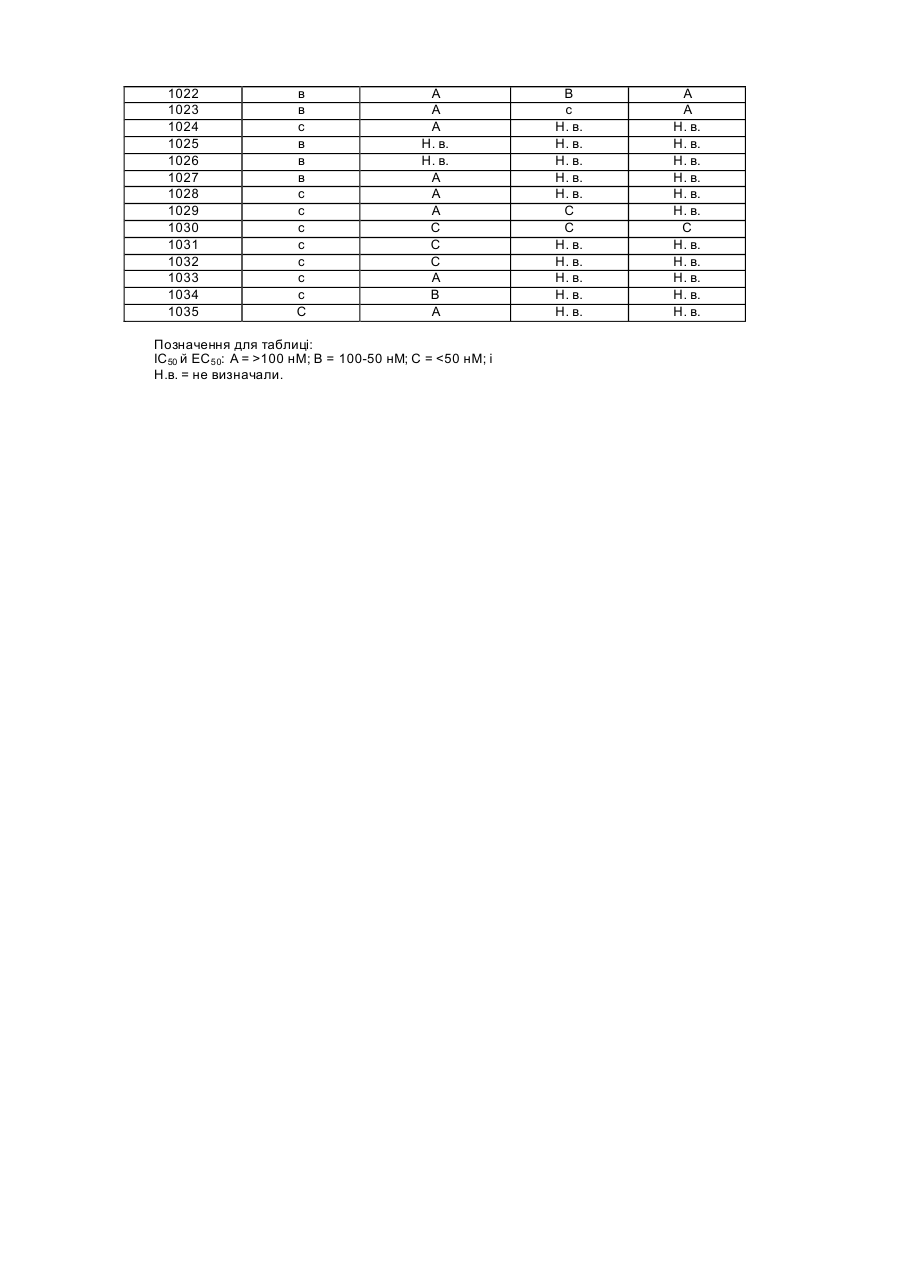
Винахід стосується нових сполук й їх фармацевтично прийнятних солей, їх застосування індивідуально або в сполученні з іншими терапевтичними агентами для лікування або профілактики ВІЛ-інфекції й фармацевтичних композицій, які містять такі сполуки. Захворювання, відоме як синдром набутого імунодефіциту (СНІД) викликається вірусом імунодефіциту людини (ВІЛ), насамперед штамом, відомим як ВІЛ-1. Для реплікації ВІЛ у клітині-хазяїні інформація, яка міститься у вірусному геномі, повинна бути інтегрована в ДНК клітини-хазяїна. Однак ВІЛ являє собою ретровірус, тобто вірус, у якого генетична інформація зчитується із РНК. Таким чином, цикл реплікації ВІЛ включає стадію транскрипції вірусного геному (РНК) з утворенням ДНК, цей процес є зворотним по відношенню до нормального ходу подій. Транскрипція вірусної РНК із утворенням ДНК відбувається за допомогою ферменту, який тому називається зворотною транскриптазою (ЗТ). Віріон ВІЛ містить копію ЗТ разом з вірусною РНК. Відомо, що зворотна транскриптаза має три ферментативні функції: вона діє як РНК-залежна ДНКполімераза, як рибонуклеаза й ДНК-залежна ДНК-полімераза. Як РНК-залежна ДНК-полімераза, ЗТ здійснює транскрипцію копії одноланюгової ДНК вірусної РНК. Як рибонуклеаза ЗТ розщеплює вихідну вір усну РНК і вивільняє ДНК отриману безпосередньо на матриці вихідної РНК. Нарешті, як ДНК-залежна ДНК-полімераза ЗТ створює другий комплементарний ланцюг ДНК, використовуючи перший ланцюг ДНК як матрицю. Два ланцюги утворюють дволанцюгову ДНК, яка інтегрується в геном клітин-хазяїв за допомогою іншого ферменту, який називається інтегразою. Сполуки, які мають здатність інгібувати ферментативні функції зворотної транскриптази ВІЛ-1, можуть інгібувати реплікацію ВІЛ-1 в інфікованих клітинах. Такі сполуки можна застосовувати для запобігання або лікування інфекції, викликаної ВІЛ-1, у хворих людей, як це встановлено при використанні відомих інгібіторів ЗТ, таких як 3'-азидо-3'-дезокситимідин (AZT), 2',3'-дидезоксіінозин (ddl), 2',3'-дидезоксицитидин (ddC), d4T, ЗТС, невірапін, делавірдин, ефавіренц й абакавір, які є основними лікарськими засобами, дозволеними для застосування при лікуванні СНІДу. Як і при будь-якій антивірусній терапії застосування інгібіторів ЗТ для лікування СНІДу в остаточному підсумку може приводити до зниження чутливості вірусу до конкретного лікарського засобу. Стійкість (знижена чутливість) до таких лікарських засобів є результатом мутацій, які відбуваються в сегменті зворотної транскриптази гена рої. Дотепер визначені характеристики декількох мутантних штамів ВІЛ і встановлено, що стійкість до відомих терапевтичних агентів зумовлена мутаціями в гені ЗТ. Одними з мутантів, які найбільш часто зустрічаються в клінічних умовах, є: мутант Y181C, у якому залишок тирозину (Y) у кодоні 181 замінений на залишок цистеїну (С), і K103N, у якому залишок лізину (К) у положенні 103 замінений на залишок аспарагіну (N). Інші мутанти, які виникають із зростаючою частотою, при лікуванні з використанням відомих антивірусних лікарських засобів, являють собою мутанти з однією мутацією V106 A, G190A, Y188C й P236L; і мутанти із двома мутаціями K103N/Y181C, K103N/P225H, K103N/V108I й K103N/L100I. Триваюче застосування антивірусних сполук для запобігання ВІЛ-інфекції безперечно буде приводити до збільшення випадків виникнення нових стійких штамів ВІЛ. Таким чином, існує необхідність в створенні нових інгібіторів ЗТ, які мають різні параметри ефективності відносно різних мутантів. Сполуки, які мають трициклічні структури, які є інгібіторами ВІЛ-1, описані в [U.S. No. 5366972]. Інші інгібітори зворотної транскриптази ВІЛ-1 описані в [Hargrave й ін., J. Med. Chem., 34, (1991), стор. 2231]. В [U.S. No. 5705499] описані 8-арилалкіл- і 8-арилгетероалкіл-5,11-дигідро-6Н-дипіридо[3,2-В:2',3'Е][1,4]діазепіни як інгібітори ЗТ. Встановлено, що вказані як приклади сполуки мають певну активність у відношенні ЗТ дикого типу й мутантного ВІЛ-1, насамперед Y181C, але при цьому вони менш ефективні відносно інших мутантів, які несуть одну мутацію, таких як K103N. В [WO 01/96338A1 й US 6420359] описані діазепінові структури, які містять хінолінові й хінолін-1Ч-оксидні замісники, як інгібітори ЗТ. Наведені як приклади сполуки мають активність відносно штамів ВІЛ дикого типу (WT), м утантів з однією й двома мутаціями. У винаході запропоновані заміщені похідні бензойної кислоти, які є ефективними інгібіторами ЗТ ВІЛ-1 дикого типу (WT) і мутантних штамів із двома мутаціями, насамперед мутантного штаму із двома мутаціями K103N/Y181C. Першим об'єктом винаходу є сполука формули І: де R2 означає Н, галоген, С1-С4алкіл, О С1-С4 алкіл, N С1-С4алкіл або N(С1-С4алкіл)2; R4 означає Н або СН3; R5 означає Н або СН3; R12 означає Н, галоген, С1-С4алкіл, CF3 або NO2; R13 означає Н, С1-С4алкіл, галоген, ОН або NH2, за умови, що R12 й R13 обидва не означають Н; і R14 означає COOR14a, де R14a означає Н або СгС6алкіл; або R14 означає С2-С4алкеніл-СОOR14а, де R14a має вказані вище значення; або R14 означає С 1-С4алкіл- СОOR14а, де R14a має вказані вище значення; або її сіль або проліки. Другим об'єктом винаходу є фармацевтична композиція, призначена для лікування або запобігання ВІЛінфекції, яка містить представлену вище в даному описі сполуку формули І, або її фармацевтично прийнятну сіль, і фармацевтично прийнятний носій. Третім об'єктом винаходу є спосіб лікування або запобігання ВІЛ-інфекції, який полягає в тому, що пацієнтові вводять інгібуючу ВІЛ кількість сполуки формули І або її фармацевтично прийнятної солі, або фармацевтичної композиції, які вказані в даному описі. Четвертим об'єктом винаходу є спосіб лікування або запобігання ВІЛ-інфекції, який полягає в тому, що вводять фармацевтичну композицію, яка містить описану вище сполуку формули І у сполученні з антиретровірусним лікарським засобом. П'ятим об'єктом винаходу є спосіб лікування або запобігання перинатальної передачі ВІЛ від матері до дитини, який полягає в тому, що матері перед пологами вводять вказану в даному описі сполуку формули І або фармацевтичну композицію, як вони описані вище. Докладний опис винаходу Визначення Якщо не вказане інше, то в описі використовуються наступні визначення: У контексті даного опису поняття " С1-С2 галкіл", " С1-С4 алкіл" й " С1-С6 алкіл" індивідуально або в сполученні з іншим радикалом означають ациклічні алкільні радикали, які містять аж до двох, чотирьох або шести атомів вуглецю відповідно. Прикладами таких радикалів є метил, етил, пропіл, бутил, 1-метилетил, 1метилпропіл, 2-метилпропіл й 1,1-диметилетил. У контексті даного опису поняття "С2-С4алкеніл" індивідуально або в сполученні з іншим радикалом означає ненасичений ациклічний радикал, який містить від двох до чотирьох атомів вуглецю. У контексті даного опису поняття "галоген" означає атом галогену й включає фтор, хлор, бром і йод. У контексті даного опису поняття "фармацевтично прийнятна сіль" означає солі, які отримані з фармацевтично прийнятних основ й є нетоксичними. Прикладами придатних основ можуть служити холін, етаноламін й етилендіамін. Мається на увазі, що під обсяг винаходу підпадають також Na+-, K+- і Са++-солі [див. також Birge S.M. і ін., Pharmaceutical salts, J. Pharm. Sci., 66, (1977), стор. 1-19, включено в даний опис як посилання]. У контексті даного опису поняття "проліки" означає фармакологічно прийнятні похідні, з яких у результаті біотрансформації утворюється діючий лікарський засіб, який стосується сполук формули І. Прикладами таких похідних є (але не обмежуючись ними) складні ефіри й аміди [див. Goodman й Gilman, The Pharmacological Basis of Therapeutics, 9-е вид., McGraw-Hill, Int. Ed., "Biotransformation of Drugs, 1995, стор. 11-16, включене в даний опис як посилання]. Докладний опис переважних варіантів здійснення винаходу Переважно R2 означає Н, галоген, С1-С4 алкіл, О-С1С4 алкіл або N(С1-С4)2. Переважно R2 означає Н, СІ, F, С1-С4алкіл, О-С1-С4 алкіл або N-(C1-С4алкіл)2. Більш переважно R2 означає Н, СІ, F, СН3, ОМе або OEt. Найбільш переважно R2 означає Н. Переважно R4 й R5 обидва мають однакове значення. Більш переважно R4 означає Н. Більш переважно R5 означає СН3. Переважно R12 означає галоген, С1-С4 алкіл, CF3 або NO2. Більш переважно, R12 означає Br, Cl, СН3 або СН3СН2. Найбільш переважно R12 означає СН3 або СН 3СН2. Переважно R13 означає Н, СН3, галоген, ОН або NH2. Більш переважно R1 означає Н, СН3 або ОН. Найбільш переважно R13 означає Н. Переважно R14 означає СООН, СООМе, С 2-С4алкенілСООН або С 1-С4алкілСООН. Більш переважно R14 означає СООН, СН=СН-СООН, СН2СООН або СН 2СН2СООН. Найбільш переважно R14 означає СООН. Сполуки формули І є ефективними інгібіторами ВІЛ дикого типу, а також мають здатність інгібувати мутантний фермент, який несе дві мутації K1O3N/Y181C. Сполуки формули І мають інгібуючу активність у відношенні зворотної транскриптази ВІЛ-1. При введенні у вигляді придатних лікарських засобів їх можна застосовувати для лікування СНІД, стану, який передує синдрому набутого імунодефіциту (ARC) і споріднених захворювань, зв'язаних з ВІЛ-1-інфекцією. Таким чином, наступним об'єктом винаходу є спосіб лікування інфекції, яка викликається ВІЛ-1, який полягає в тому, що людині, інфікованій ВІЛ-1, вводять терапевтично ефективну кількість описаної вище нової сполуки формули І. Як для лікування, так і для профілактики, сполуку можна застосовувати також для запобігання перинатальної передачі ВІЛ-1 від матері до дитини шляхом введення сполуки матері перед пологами. Сполуки формули І можна вводити у вигляді однократної дози або у вигляді розділених доз оральним або парентеральним шляхом. Придатна оральна доза сполуки формули І може становити від приблизно 0,5 мг до 3 г на день. Переважно оральна доза сполуки формули І становить від приблизно 100 до 800 мг на день для пацієнта вагою 70 кг. У випадку композицій для парентерального введення придатна стандартна доза може становити від 0,1 до 250 мг сполуки, переважно від 1 до 200 мг. Однак слід мати на увазі, введена доза може змінюватися для різних пацієнтів й доза для кожного конкретного пацієнта залежить від рішення лікаря, який як критерій при призначенні відповідної дози повинен враховувати вагу і стан пацієнта, а також реакцію пацієнта на лікарський засіб. Якщо сполуки, запропоновані в даному винаході, вводять оральним шляхом, то їх можна вводити у формі фармацевтичних композицій, які містять лікарський засіб у сполученні із сумісним фармацевтичним носієм. Такий носій може являти собою інертний органічний або неорганічний носій, придатний для орального введення. Прикладами таких носіїв є вода, желатин, тальк, крохмаль, стеарат магнію, аравійська камедь, рослинні олії, поліалкіленгліколі, вазелін і т.п. Сполуки формули І можна застосовувати в сполученні з антиретровірусним лікарським засобом, відомим фа хівцеві в даній галузі, як комбінований лікарський засіб для одночасного, окремого або послідовного введення для лікування або запобігання ВІЛ-інфекції в індивідуума. Приклади антиретровірусних лікарських засобів, які можна застосовувати при комбінованому лікуванні з використанням сполук формули І, включають (але не обмежуючись ними) нуклеозидні/нуклеотидні інгібітори зворотної транскриптази (такі як АЗТ і тенофовір), ненуклеозидні інгібітори зворотної транскриптази (такі як невірапін), інгібітори протеази (такі як ритановір), інгібітори вірусного злиття (такі як Т-20), антагоністи CCR5 (такі як SCH-351125), антагоністи CXCR4 (такі як AMD-3100), інгібітори інтегрази (такі як L-870,810), інгібітори тирозинамінотрансферази (ТАТ), інші лікарські засоби, які знаходяться на стадії дослідження, (такі як PRO-542, BMS-806, ТМС-114 або АІ-183), протигрибкові або антибактеріальні агенти (такі як флуконазол), і імуномодулятори (такі як левамізол). Крім того, сполуку формули І можна застосовувати в сполученні з іншою сполукою формули І. Фармацевтичні композиції можна готувати стандартним способом і кінцеві лікарські форми можуть являти собою тверді лікарські форми, наприклад, таблетки, драже, капсули й т.п., або рідкі лікарські форми, наприклад, розчини, суспензії, емульсії й т.п. Фармацевтичні композиції можна піддавати стандартній фармацевтичній обробці, такій як стерилізація. Крім того, фармацевтичні композиції можуть містити стандартні допоміжні речовини, такі як консерванти, стабілізатори, емульгатори, коригенти, змочувальні агенти, буфери, солі для зміни осмотичного тиску й т.п. Тверді носії, які можна застосовувати, включають, наприклад, крохмаль, лактозу, маніт, метилцелюлозу, мікрокристалічну целюлозу, тальк, вторинний кислий фосфа т кальцію й високомолекулярні полімери (такі як поліетиленгліколь). Для парентерального застосування сполуку формули І можна вводити у вигляді водного або неводного розчину, суспензії або емульсії у фармацевтично прийнятному маслі або суміші рідин, які можуть містити бактеріостатичні агенти, антиоксиданти, консерванти, буфери або інші розчинені речовини, які надають розчину ізотонічність із кров'ю, загусники, суспендувальні агенти або інші фармацевтично прийнятні добавки. Такі добавки включають, наприклад, тартратний, цитратний й ацетатний буфери, етанол, пропіленгліколь, поліетиленгліколь, комплексоутворюючі агенти (такі як ЕДТК), антиоксиданти (такі як бісульфіт натрію, метабісульфіт натрію й аскорбінова кислота), високомолекулярні полімери (такі як рідкі поліетиленоксиди) для регулювання в'язкості й поліетиленові похідні ангідридів сорбіту. За необхідності можна додавати також консерванти, такі як бензойна кислота, метил- або пропілпарабен, бензалконійхлорид й інші похідні четвертинного амонію. Сполуки, запропоновані у винаході, можна вводити також у вигляді розчинів для назального введення й вони можуть містити крім сполук, запропонованих у даному винаході, придатні буфери, агенти для регулювання тонічності, протимикробні консерванти, антиоксиданти й агенти, які підвищують в'язкість, у водному носії. Прикладами агентів, які підвищують в'язкість, є полівініловий спирт, похідні целюлози, полівінілпіролідон, полісорбати або гліцерин. Протимікробні консерванти, які можна додавати, включають бензалконійхлорид, тимерозал, хлорбутанол або фенілетиловий спирт. Крім того, сполуки, запропоновані у винаході, можна вводити у вигляді супозиторію. Метод і синтез Фахівець в галузі синтетичної органічної хімії на основі своїх знань може одержувати сполуки, запропоновані у винаході. Приклади реакційних схем наведені нижче на схемах 1-4. Замісники R 2, R4, R5, R12, R13, R13 й R14 мають значення, вказані в описі. Загалом метод полягає в тому, що шля хом ароматичного заміщення (SNAR) сполуки 1(і) Et-NH2 одержують проміжну сполуку 1(іі). Потім шляхом галогенування положення 5 за допомогою бромувального агента (наприклад, NBS або брому) одержують 1(ііі). У результаті замикання кільця сполуки 1(ііі) за допомогою проведеної в присутності основи реакції SnAR утворюється трициклічний проміжний продукт l(iv). Шляхом введення замісника R2 за допомогою ароматичного заміщення хлору в положенні С-2 сполуки l(iv) одержують проміжну сполуку l(v). Послідовність реакцій на схемі 2 аналогічна до послідовності, описаної в J.M. Klunder й ін.; J. Med. Chem. 41, 1998, стор. 2960-2971 й в C.L. Cywin й ін.; J. Med. Chem., 41, 1998, стор. 2972-2984. Загалом, метод полягає в тому, що шля хом ароматичного заміщення (SNAR) сполуки 2(і) Et-NH2 одержують проміжний продукт 2(іі). Шляхом відновлення нітрогрупи (наприклад, за допомогою каталітичного гідрування) одержують сполуку 2(ііі). У результаті реакції конденсації, проведеної в присутності основи, сполуки 2(ііі), наприклад, з 5-бром-2хлор-3-піридинкарбонілхлоридом, одержують сполуку 2(iv). Після замикання кільця сполуки 2(iv), яке здійснюють шляхом реакції SnAR у присутності основи, одержують трициклічний проміжний продукт 2(v). Метальну гр уп у R5 у сполуці 2(vi) можна вводити за допомогою відомого в даній галузі алкілування з використанням, наприклад, метилйодиду. Схема 3: Одержання проміжних продуктів, де R2 означає С1-С4 алкіл Загалом метод полягає в тому, що шля хом реакції конденсації сполук 3(і) та 3(іі), яку проводять у присутності основи, одержують проміжний продукт З(ііі). Шляхом ароматичного заміщення (SNAR) сполуки З(ііі) Et-NH2 одержують проміжний продукт 3(iv). Замикання кільця сполуки 3(iv) проводять за допомогою SnARреакції в присутності основи, одержуючи трициклічний проміжний продукт, з якого шляхом алкілування одержують проміжний продукт 3(v). Загалом метод полягає в тому, що за допомогою реакції конденсації сполук 4(і) і 3(іі) у присутності основи одержують проміжний продукт 4(іі). Шляхом ароматичного заміщення (SNAR) сполуки 4(іі) Et-NH2 одержують проміжний продукт 4(ііі). Шляхом замикання кільця 4(ііі) за допомогою реакції SnAR у присутності основи одержують трициклічний проміжний продукт, який представляє собою сполуку l(v). Загалом метод полягає в тому, що бром-вмісне похідне 5(і), синтезоване відповідно до методу, представленому в описі, піддають реакції сполучення з реагентом, що представляє собою алілолово, в апротонному розчиннику (наприклад, ДМФ) у присутності каталізатора, у результаті чого утворюються С-8замісники 5(іі). Після окислення подвійного зв'язку (наприклад, шляхом озонолізу з одержанням озоніду), і наступного відновлення одержують С-8-гідроксиетильний замісник 5(ііі). За допомогою реакції типу реакції Міцунобу, нафтильні похідні 5(iv), 5(v) або 5(vi), де Y має значення, вказані для R14, за винятком СООН, конденсують зі сполукою 5(ііі), одержуючи сполуку формули І. В альтернативному варіанті, якщо Y означає попередник групи R14, наприклад, СООСН3, можна застосовувати реакцію типу реакції Міцунобу для конденсації сполуки 5(iv) або 5(v) зі сполукою 5(ііі), і після цього Y можна хімічним шляхом перетворювати в замісники R14, наприклад, шляхом омилення СООСН 3, з утворенням СООН, у результаті чого одержують сполуку формули І. Як вказано вище, сполука, запропонована у винаході, інгібує ферментативну активність ЗТ ВІЛ-1. Шляхом тестування цих сполук відповідно до описаного нижче методу встановлено, що вони інгібують РНК-залежну ДНК-полімеразну активність ЗТ ВІЛ-1. Встановлено (дані не представлені), що вони інгібують також ДНКзалежну ДНК-полімеразну активність ЗТ ВІЛ-1. За допомогою описаного нижче аналізу на основі зворотної транскриптази (ЗТ) сполуки можна тестувати відносно їх здатності інгібувати РНК-залежну ДНК-полімеразну активність ЗТ ВІЛ-1. Таким шляхом було проведене тестування деяких конкретних сполук, які описані в наведені нижче прикладах. Результати тестування наведені в таблиці 1 у вигляді значень ІС50 й ЕС 50. Приклади Нижче винахід більш докладно проілюстрований на прикладах, які не обмежують його обсяг. Всі реакції проводили в атмосфері азоту або аргону, якщо не вказане інше. Температури представлені в градусах Цельсію. Відсотки або співвідношення для розчинів являють собою об./об.-співвідношення, якщо не вказане інше. Скорочення або символи, які використовуються в описі, мають наступні значення: ДЕАД: діетилазодикарбоксилат; ДІАД: діізопропілазодикарбоксилат; ДІЕА: діізопропіл етил амін; ДМАП: 4-(диметиламіно)піридин; ДМСО: диметилсульфоксид; ДМФ: диметилформамід; ДЦК: дициклогексилкарбодіімід; ES МС: мас-спектрометрія з використанням електронного пучка; Et: етил; EtOH: етанол; ЕtOАс: е тилацетат; Et2O: діетиловий ефір; ВЕРХ: рідинна хроматографія високого розділення; і-Рг: ізопропіл; Me: метил; МеОН: метанол; MeCN: ацетонітрил; NaHMDS: гексаметилдисилазид натрію; NEC: N-бромсукцинімід; Ph: феніл; ТБЕ: Трис-борат-ЕДТК; ТБТУ: те трафторборат 2-(1H-бензотриазол-1-ил)-N,N,N'N' -тетраметилуронію; ТФК: трифторогггова кислота; ТГФ: тетрагідрофуран; БУО: бляшкоутворюючі одиниці; ДЕПК: діетилпірокарбонат; ДТТ: ди тіотреїтол; ЕДТА: етилендіамінотетраацетат; УМФ: уридин-5'-монофосфат; УТФ: уридин-5'-трифосфат; МЕС: 2-(н-морфоліно)етансульфонова кислота; ДСН-ПААГ: гель-електрофорез у поліакриламідному гелі в присутності додецилсульфату натрію; ММСО: граничне значення пропускної молекулярної маси; Біс-трис-пропан: 1,3-біс{трис(гідроксиметил)метиламіно}пропан; GSH: відновлений глута тіон; ОБГ: н-октил-р-О-глюкозид. Синтез У наступних прикладах проілюстровані способи одержання сполук, запропонованих у винаході. Приклад 1 Стадія а: До розчину 2-хлор-3-нітропіридину 1а (51г, 325ммолів) у ТГФ (650мл) додавали 2М розчин етиламіну в ТГФ (365мл, 731ммоль). Реакційну суміш перемішували при кімнатній температурі протягом ночі. Реакційну суміш зливали у воду (~1,5 л) і утворений твердий продукт фільтрували й сушили при зниженому тиску, одержуючи сполуку 1b (52г). Стадія б: Розчин 2-(етиламіно)-3-нітропіридину 1Ь (52 г) в МеОН (600 мл) перемішували протягом ночі при кімнатній температурі в атмосфері водню (1 атм.) у присутності 20%-ного Pd(OH)2/C (10,4 г). Каталізатор видаляли шляхом фільтрації через діатомову землю. Фільтрат концентрували при зниженому тиску, одержуючи сполуку 1с у вигляді твердої речовини чорного кольору (39 г, ви хід 88% після стадій а й б). Стадія в: До охолодженого розчину 3-аміно-2-(етиламіно)піридину 1с (30,6 г, 223 ммоля) в MeCN (740 мл) додавали твердий NaHCO3 (56,3 г, 669 ммолів). Через 5 хв додавали неочищений 5-бром-2-хлор-3піридинкарбонілхлорид (отриманий з 5-бром-2-гідрокси-3-піридинкарбонової кислоти й SOCl2 (1екв., 223ммоля) [як описано в Т. W. Gero й ін. в Synth. Commun. 19, 1989, стор. 553-559 (документ включений у даний опис як посилання) за винятком водної обробки]. Через 2 год реакційну суміш зливали на лід/Н 2О (1,5л) і утворений твердий продукт фільтрували, промивали Н2О і потім гексаном. Після сушіння при зниженому тиску протягом ночі одержували сполуку Id у вигляді твердої речовини чорного кольору (54,9г, ви хід 69%). Стадія г: До розчину 2-хлор-М-{2-(етиламіно)-3-піридиніл}-5-бром-3- піридинкарбоксаміду 1d (54,9г, 154,4ммоля) у піридині (308мл) при 50°С додавали по краплях 1М розчин NaHMDS у ТГФ (355мл, 355ммолів). Через 10хв реакційної суміші давали охолонути до кімнатної температури й потім зливали на льодяну воду (2л). Утворений твердий продукт фільтрували, промивали водою й потім гексаном. Після сушіння при зниженому тиску протягом ночі одержували сполуку 1е (36г, вихід 75%) у вигляді твердої речовини темно-зеленого кольору. Стадія д: До розчину 8-бром-5,11-дигідро-11-етил-6Н-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону 1е (36,7г, 115ммолів) у ДМФ (380мл) додавали NaH (3,5г, 138ммолів) і суміш нагрівали до 50°С протягом 30 хв. Реакційну суміш охолоджували до кімнатної температури й обробляли МеІ (14,3мл, 230ммолів). Через 1,5 год реакційну суміш зливали на льодяну воду. Твердий продукт фільтрували, промивали водою й потім гексаном, одержуючи після сушіння сполуку 1f (37,9г, ви хід 99%) у вигляді твердої речовини темно-сірого кольору. Стадія e: Алілтрибутилолово (30,7мл, 99,0ммолів) і Pd(Ph3P)4 (5,20г, 4,50ммоля) додавали до дегазованого (шляхом пропускання N2 через розчин протягом 30хв) розчину 8-бром-5,11-дигідро-11-етил-5-метил-6Hдипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону 1f (30,0г, 90,0ммолів) у ДМФ (450мл) при кімнатній температурі. Суміш перемішували при 90°С протягом 1,5 год, потім охолоджували до кімнатної температури й концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (гексан:ЕtOАс, від 8:2 до 7:3), одержуючи сполуку 1g (22,19г, ви хід 84%). Стадія є: Струменем озонованого кисню барботували холодний (-78°С) розчин 5,11-дигідро-11-етил-5-метил-8-(2пропеніл)-6H-дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону 1g (22,19г, 75,4ммоля) в СН2Сl2 (150мл) і МеОН (150мл) протягом 2,5год. Потім розчин барботували струменем N2 протягом 15хв і після цього до розчину додавали твердий NaBH4 (4,99г, 132ммоля). Реакційної суміші давали нагрітися до кімнатної температури. Через 1год додавали водний насичений NH4Cl (200мл) і суміш перемішували при кімнатній температурі протягом 2год. Органічні розчинники видаляли при зниженому тиску. До залишку додавали воду (300мл) і СНСl3 (300мл). Фази розділяли й водний шар екстрагували СНСl3 (3 х 300мл). Об'єднані органічні шари сушили (MgSO4), фільтрували й концентрували при зниженому тиску. Залишок очищали експрес-хроматографією (ЕtOАс:СНСl3, 4:1) одержуючи сполуку lh (16,1 г, ви хід 72%) у вигляді твердої речовини білого кольору. Приклад 2 Стадія а: Охолоджений на льоді розчин EtNH2 (49,8г, 1,10моля) у толуолі (200мл) додавали протягом 15хв до охолодженого на льоді розчину 2,6-дихлор-3-нітропіридину 2а (100,0г, 0,52моля) у толуолі (225мл). Суміш перемішували при 0°С протягом 45хв. Додавали воду (500мл) і ЕtOАс (500 мл) і розділяли фази. Органічний шар послідовно промивали водою (200 мл) і соляним розчином (200 мл), сушили (MgSO4), фільтрували й концентрували при зниженому тиску. Твердий продукт, що залишився, перекристалізовували з МеОН, одержуючи сполуку 2b (83,7г, вихід 80%) у ви гляді голок жовтого кольору. Стадія б: Сполуку 2с одержували методом, аналогічним до методу, описаному в прикладі I, стадія б. Стадія в: Розчин 5-бром-2-хлор-3-піридинкарбонілхлориду (30,0г, 97,0ммолів) в MeCN (100мл) додавали через канюлю при кімнатній температурі до розчину 3-аміно-6-хлор-2-(етиламіно)піридину 2с (16,6г, 97,0ммолів) в MeCN (180мл), який містить твердий NaHCO3 (14,2г, 169ммоля). Суміш перемішували при кімнатній температурі протягом 1год. Додавали воду (200мл) і суміш перемішували протягом 10хв. Утворену суспензію фільтрували. Твердий продукт промивали водою, потім гексаном і сушили при зниженому тиску, одержуючи сполуку 2d (28,4г, ви хід 75%). Стадія г: 1М розчин NaHMDS у ТГФ (167,5мл, 167,5ммоля) повільно додавали до розчину 5-бром-2-хлор-N-{2(етиламіно)-6-хлор-3-піридиніл}-3-піридинкарбоксаміду 2d (28,4г, 72,8ммоля) у піридині (146мл), нагрітому до 50°С. Реакційну суміш перемішували при 50°С протягом 1,5 год. Потім суміш зливали на суміш води й льоду (1 л) і через 1 год фільтрували утворену суспензію. Твердий продукт промивали водою й сушили при зниженому тиску, одержуючи сполуку 2е (23,4г, вихід 91%). Стадія д: Твердий NaH (60%-на масляна дисперсія, 3,46г, 86,1ммоля) додавали протягом 30хв до розчину 8-бром-2хлор-5,11-дигідро-11-етил-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону 2е (23,4г, 66,3ммоля) у ДМФ (220мл) при 50°С. Суміш перемішували при 50°С протягом 1 год, потім давали нагрітися до кімнатної температури. Суміш зливали на воду (1 л) і утворену суспензію фільтрували. Твердий продукт послідовно промивали водою й гексаном, потім сушили при зниженому тиску, одержуючи сполуку 2f (23,0 г, ви хід 94%). Стадія e: Алілтрибутилолово (21,3мл, 68,7ммоля) і Pd(Ph3P)4 (3,61г, 3,12ммоля) додавали до дегазованого (шляхом пропускання N2 через розчин протягом 30хв) розчину 8-бром-2-хлор-5,11-дигідро-11-етил-5-метил-6Hдипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону 2f (23,0г, 62,5ммоля) у ДМФ (312мл). Суміш нагрівали до 90°С протягом 2 год. Суміш концентрували при зниженому тиску. Залишок очищали за допомогою експресхроматографії (гексан: ЕtOАс, 7:3) одержуючи сполуку 2g (13,4 г, ви хід 65%). Стадія є: Озонований кисень вводили в холодний (-78°С) розчин 2-хлор-5,11-дигідро-11-етил-5-метил-8-(2пропеніл)-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону 2g (13,4г, 40,7ммоля) в МеОН (102мл) і СН2Сl2 (102мл) до повного поглинання алкену. Розчин барботували азотом для видалення надлишку О3. Потім невеликими порціями додавали твердий NAВН4 (2,69г, 71,1ммоля) і суміші давали повільно нагрітися до кімнатній температурі. Через 1год додавали водний насичений NH 4Cl (150мл) і суміш перемішували протягом 20хв. Органічні розчинники видаляли при зниженому тиску. До водного розчину додавали воду (100мл). Розчин екстрагували СНСl3 (3 х 200мл). Об'єднані органічні шари сушили (MgSO 4), фільтрували й концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (ЕtOАс:СНСl3, 4:1), одержуючи сполуку 2h (10,4г, ви хід 77%). Приклад 3: Стадія а: До суміші концентрованої сірчаної кислоти (600мл) і димлячої азотної кислоти (90%, 400мл) у льодяній бані (внутрішню температуру підтримували на рівні 5-10°С) додавали по краплях 2,6-дифторпіридин 3а (200г, 1,74моля). Утворену суміш перемішували протягом ночі при кімнатній температурі. Суміш повільно зливали на 3 кг льоду й екстрагували Еt2О (2 х 2л). Об'єднані органічні шари промивали водним 1,5н. NaOH (2x1л), потім водним насиченим NaHCO3 (400мл) або до досягнення значення рН приблизно 8-9. Органічні шари сушили над MgSO4, фільтрували й концентрували при зниженому тиску до постійної маси (для видалення 2,6дифторпіридину, який не прореагував: 10-12%). У результаті одержували сполуку 3b у вигляді рідини жовтого кольору (207,3г, вихід 74%). Стадія б: До розчину 2,6-дифтор-З-нітропіридину 3b (45,7г, 285ммолів) у ТГФ (500мл) при -40°С додавали по краплях розчин етиламіну (25,7г, 570ммолів) у ТГФ (250мл). Через 30хв, реакційну суміш концентрували при зниженому тиску й залишок розчиняли в EtOAc. Органічну фазу промивали соляним розчином, сушили (MgSO4), фільтрували й концентрували. Утворений твердий продукт очищали за допомогою експресхроматографії (15% ЕtOАс у гексані), одержуючи сполуку 3с (43, 2г, ви хід 82%) у вигляді твердої речовини жовтого кольору. Стадія в: Розчин 2-етиламіно-6-фтор-3-нітропіридину 3с (43,2г, 230ммолів) у ТГФ (1л) перемішували протягом ночі при кімнатній температурі в атмосфері водню (1атм.) у присутності 20%-ного Pd(OH)2/C (4,35г). Каталізатор видаляли фільтрацією через діатомову землю. Фільтрат концентрували при зниженому тиску, одержуючи сполуку 3d (36,3г, ви хід 95%) у вигляді твердої речовини чорного кольору. Стадія г: До охолодженого розчину (4°С) 3-аміно-2-етиламіно-6-фторпіридину 3d(31,0г, 200ммолів) у MeCN (160мл) додавали твердий NaHCO3 (50,4г, 600ммолів). Через 15хв додавали розчин 5-бром-2-хлор-3піридинкарбонілхлориду (1екв., 200ммолів) у MeCN (155мл). Після витримування протягом 60хв при кімнатній температурі реакційну суміш зливали на воду (1,2л) і перемішували протягом 30 хв. Утворений твердий продукт фільтрували, сушили протягом ночі при зниженому тиску при 50°С. У результаті одержували сполуку 3е (73,7г, вихід 99%) у вигляді твердої речовини чорного кольору. Стадія д: До розчину 2-хлор-N-{2-(етиламіно)-6-фтор-3-піридиніл}-5-бром-3-піридинкарбоксаміду 3е (73,5г, 216ммолів) у піридині (435мл) при 50°С додавали по краплях 1М розчин NaHMDS у ТГФ (520мл, 520ммолів). Через 10хв реакційної суміші давали нагрітися до кімнатної температури, після чого зливали на льодяну воду (2л). Утворений твердий продукт фільтрували, промивали водою і потім гексаном. Твердий продукт сушили при зниженому тиску, одержуючи сполуку 3f (50,6г, вихід 69%) у вигляді твердої речовини темно-зеленого кольору. Стадія e: До розчину 8-бром-5,11-дигідро-11-етил-2-фтор-6H-дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону 3f (44г, 130,5ммоля) у ДМФ (520мл) додавали NaH (4,28г, 178ммолів) і суміш нагрівали до 50°С протягом 30хв. Реакційну суміш охолоджували до кімнатної температури й обробляли МеІ (24,4мл, 522ммоля). Через 1,5год реакційну суміш зливали на льодяну воду. Твердий продукт фільтрували, промивали водою і потім гексаном, сушили при зниженому тиску, одержуючи сполуку 3g (43,2г, вихід 94%) у вигляді твердої речовини темносірого кольору. Стадія є: Алілтрибутилолово (32,0мл, 103,4ммоля) і Pd(Ph3P)4 (5,43г, 4,70ммоля) додавали до дегазованого (шляхом пропускання N2 через розчин протягом 45хв) розчину 8-бром-5,11-дигідро-11-етил-2-фтор-5-метил6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону 3g (33,0г, 94,0ммоля) у ДМФ (470мл). Через 1, 2, 3, 4 і 5 год додавали додаткові кількості Pd(Ph3)4 (1,09г, 0,94ммоля) для завершення реакції. Суміш нагрівали до 90°С протягом 6 год. Суміш концентрували при зниженому тиску. Залишок очищали за допомогою експресхроматографії (гексан:EtOAc, від 8:2 до 7:3), одержуючи сполуку 3h (22,4г, ви хід 76%). Стадія є: Струменем озонованого кисню барботували холодний (-78°С) розчин 5,11-дигідро-11-етил-2-фтор-5метил-8-(2-пропешл)-6H-дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону 3h (22,38г, 71,6ммоля) у СН 2Сl2 (100мл) і МеОН (100мл) протягом 3год. Після цього розчин барботували протягом 15хв струменем N2 і потім до розчину додавали твердий NaBH4 (5,05г, 133ммоля). Реакційній суміші давали нагрітися до кімнатної температури. Через 1 год до реакційної суміші додавали додаткову порцію NaBH4 (1,62г, 43,0ммоля). Ще через 1 год додавали водний насичений NH 4Cl (150мл) і суміш перемішували при кімнатній температурі протягом 30хв. Органічні розчинники видаляли при зниженому тиску. Додавали воду (200мл) і суміш екстрагували СНСl3 (3 х 300мл). Об'єднані органічні шари сушили (MgSO4), фільтрували і концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (ЕtOАс:СНСl3, 4:1), одержуючи сполуку 3І (19,7г, вихід 72%) у вигляді твердої речовини білого кольору. Приклад 4 (сполуки 1003 і 1031) Стадія а: Розчин 1,6М н-BuLi у гексані (6,22 мл, 9,95 ммоля) швидко додавали до холодного (-78°С) розчину сполуки 4а (0,87г, 4,33ммоля) у ТГФ (20мл). Суміш перемішували при -78°С протягом 10хв, давали нагрітися до 0°С, витримували при 0°С протягом 1 год. У реакційну суміш вводили струмінь СО2 протягом 10хв і розчин підкисляли шляхом додавання водного 1,0н. розчину НСд. Суміш екстрагували ЕtOАс. Органічний шар сушили (MgSO4), фільтрували і концентрували при зниженому тиску. Залишок розчиняли в Еt2О (20мл) і обробляли надлишковою кількістю розчину CH2N2 у Et2O (приблизно 0,6М, 10мл) протягом 10хв. С уміш концентрували при зниженому тиску і залишок очищали за допомогою експрес-хроматографіі (гексан: ЕtOАс, від 4:1 до 7:3), одержуючи 4b (0,13г, ви хід 17%). Стадія б: Розчин ДІАД (86мкл, 0,44ммоля) у ТГФ (2,0мл) додавали протягом 30хв до розчину, який містить сполуку 1h (100мг, 0,33ммоля), сполуку 4b (60,0мг, 0,33ммоля) і PPh3 (114мг, 0,44ммоля) у ТГФ (10мл), при 25°С. Суміш перемішували протягом 1год і потім концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (гексан:EtOAc, від 7:3 до 1:1), одержуючи сполуку 1031 (128 мг, вихід 83%). Стадія в: Водний 1,0н. розчин LiOH (1,52мл, 1,52ммоля) додавали до розчину сполуки 1031 (100мг, 0,22ммоля) у ТГФ (6мл) і МеОН (2мл). Реакційну суміш перемішували при 25°С протягом 24год, потім нагрівали до температури дефлегмації протягом 1год. Розчин підкисляли шляхом додавання водного 1,0н. розчину НСl, потім екстрагували ЕtOАс. Ор ганічний шар промивали водою (2 x) і соляним розчином, сушили (MgSO 4), фільтрували і концентрували при зниженому тиску. Залишок розтирали з Et2O/гексаном, одержуючи сполуку 1003 (80мг, вихід 83%) у вигляді твердої речовини білого кольору. Суміш сполуки 1003 (38,0 мг, 0,085 ммоля) і водного 0,02н. розчину NaOH (4,3 мл, 0,085ммоля) у MeCN (3мл) опромінювали ультразвуком. Утворений розчин заморожували і ліофілізували, одержуючи відповідну натрієву сіль (37мг, вихід 98%) у вигляді твердої речовини білого кольору. Приклад 5 (сполуки 1019, 1020 і 1028) Стадія а: Розчин ДІАД (86мкл, 0,44ммоля) у ТГФ (2,0мл) додавали протягом 2год до розчину, який містить сполуку 3і (106мг, 0,34ммоля), сполуку 5а (56,0мг, 0,34ммоля) і PPh3 (114мг, 0,44ммоля) у ТГФ (7мл), при 25°С. Суміш перемішували протягом 1год і потім концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (гексан: ЕtOАс, від 7:3 до 1:1), одержуючи сполуку 5b (115мг, вихід 73%) у вигляді твердої речовини білого кольору. Стадія б: Водний 1,0н. розчин LiOH (1,0мл, 1,0ммоль) додавали до розчину сполуки 5b (100мг, 0,21ммоля) у МеОН (6мл). Реакційну суміш перемішували при 25°С протягом 24год. Розчин підкисляли шляхом додавання водного 1,0н. розчину НСl і потім екстрагували ЕtOАс. Органічний шар промивали водою і соляним розчином, сушили (MgSO4), фільтрували і концентрували при зниженому тиску. Залишок очищали за допомогою експресхроматографії (гексан: ЕtOАс:АсОН, 50:50:1), одержуючи спочатку сполуку 1019 (25мг, вихід 25%) у вигляді твердої речовини білого кольору, а потім сполуку 1020 (48мг, вихід 50%) у вигляді твердої речовини білого кольору. Відповідні натрієві солі одержували шляхом обробки водним 0,02н. розчином NaOH. Стадія в: 1,0М розчин диметиламіну в ТГФ (5,0мл, 5,0ммолів) і водний 1,0н. розчин LiOH (1,0мл, 1,0ммоль) додавали до розчину сполуки 5b (50,0мг, 0,11ммоля) у ізо-РгОН (3мл). Реакційну суміш нагрівали до температури дефлегмації протягом 48год. Додавали водний 1,0н. розчин НСl (2мл) і суміш екстрагували ЕtOАс. Органічний шар промивали водою і соляним розчином, сушили (MgSO4), фільтрували і концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (гексан: ЕtOАс:АсОН, 50:50:1), одержуючи сполуку 1028 (18мг, ви хід 35%) у вигляді твердої речовини білого кольору. Відповідну натрієву сіль одержували шляхом обробки водним 0,02н. розчином NaOH. Приклад 6 (сполука 1014) Стадія а: До розчину кислоти 6а (1,00г, 5,55ммоля) у СН 2СІ2 (50мл) додавали оксалілхлорид (0,73мл, 8,3ммоля) і ДМФ (100мкл). Реакційну суміш перемішували протягом 90хв, потім додавали ЕtOН (15мл) і реакційну суміш перемішували ще протягом 1год. Реакційну суміш концентрували при зниженому тиску, залишок розбавляли ЕtOАс і промивали послідовно водою, соляним розчином, сушили (MgSO4), фільтр ували і концентрували, одержуючи складний ефір 6b, який використовували без додаткового очищення. Стадія б: До розчину складного ефіру 6b у СН 2Сl2 (50мл) додавали 1М розчин ВВг3 у СН2СІ2 (7,2мл, 7,20ммоля). Після витримування протягом 3год при кімнатній температурі реакційну суміш о холоджували до 0°С і додавали EtOH (5мл). Реакційну суміш перемішували протягом 30хв при кімнатній температурі, потім концентрували при зниженому тиску. Залишок розбавляли ЕtOАс і послідовно промивали насиченим водним розчином NaHCO3, водою і соляним розчином, сушили (MgSO4), фільтр ували і концентрували досуха. Залишок очищали за допомогою експрес-хроматографії (гексан: ЕtOАс; 70:30), одержуючи фенол 6с (802мг, вихід 74% після двох стадій) у ви гляді прозорої смоли. Стадія в: Розчин ДІАД (87 мкл, 0,44 ммоля) у ТГФ (2,0 мл) додавали протягом 2 год до розчину, який містить сполуку lh (100 мг, 0,33 ммоля), Ph3P (104 мг, 0,44ммоля) і фенол 6с (65 мг, 0,34 ммоля) у ТГФ (7,0 мл), при кімнатній температурі. Суміш перемішували протягом 4 год і потім концентрували при зниженому тиску. Залишок очищали за допомогою експрес-хроматографії (гексан:ЕЮАс; від 30:70 до 50:50), одержуючи сполуку 6d (46 мг, вихід 29%) у вигляді піни білого кольору. Стадія г: До розчину складного ефіру 6d (44мг, 0,09ммоля) у суміші ТГФ (3мл) і МеОН (1мл) додавали водний 1н. розчин LiOH (1,0мл, 1,0ммоль). Після витримування протягом 4год при кімнатній температурі додавали 1н. НСl (2мл). Суміш екстрагували ЕtOАс. Органічний шар промивали водою і соляним розчином, сушили (MgSO 4), фільтрували і концентрували досуха, одержуючи сполуку 1014 (39мг, вихід 93%) у вигляді твердої речовини білого кольору. Відповідну натрієву сіль одержували шляхом обробки 1екв. водного гідроксиду натрію і утворений розчин ліофілізували, одержуючи пухнасту тверду речовину білого кольору. Аналіз зворотної транскриптази (ЗТ) Теоретичні основи аналізу До переліку ферментів, які кодуються вірусом імунодефіциту людини (ВІЛ-1), входить зворотна транскриптаза (1), яка одержала таку назву тому, що вона транскрибує копію ДНК на матриці РНК. Цю активність можна кількісно вимірювати за допомогою безклітинного ферментативного аналізу, який заснований на встановленому факті, що зворотна транскриптаза має здатність використовувати синтетичну матричну poly r(C), примовану біотинільованим олігонуклеотидом d(G), для транскрипції радіоактивноміченого ланцюга ДНК із використанням як субстрату ЗН-дГТФ. В описаному нижче методі аналізу використовується фермент дикого типу (який є домінуючою формою ферменту, виявленою в пацієнтів, інфікованих ВІЛ-1), однак у методі можна застосовувати також мутантні ЗТ-ферменти (наприклад, Y181C, отриманий шляхом сайтнаправленого мутагенезу, у якому залишок тирозину в кодоні 181 замінений на залишок цистеїну) з використанням аналогічних умов проведення аналізу. Цей аналіз дозволяє оцінювати ефективність сполук відносно їх здатності інгібувати мутантні ферменти. Матеріали: а) Одержання ферменту Деякі мутанти ЗТ ВІЛ-1 клону ВН10 ПІВ були отримані від Dr. С.-К. Shih (фірма Boehringer Ingelheim Pharmaceuticals Inc., США) у векторі рКК233-2 (фірма Pharmacia). Зокрема, клон ЗТ ВІЛ pKRT2, що несе тільки ген рбб ЗТ, яка знаходиться під контролем оперону lac/промотору trc, одержували від Dr. W. Summers (Єльський університет) (2). Шляхом сайтнаправленого мутагенезу в ген ЗТ дикого типу інтродукували різні амінокислотні заміни. Клони ЗТ субклонували в бактеріальному експресійному векторі рКК233-2. Отримані клони включали клон дикого типу, VallO6Ala, Tyrl81Cys, Tyrl88Cys, Tyrl88Leu, Glyl90Ala й Pro236Leu. Інші клони були отримані заявниками шляхом сайтнаправленого мутагенезу ЗТ рКК233-2 і включали LyslO3Asn, LyslO3Asn/Tyrl81Cys, Lysl03Asn/Leul00Ile, Lysl03Asn/Pro225His ftLyslO3Asn/VallO8Ile. б) Очищення ферменту Очищення рекомбінантної зворотної транскриптази здійснювали за допомогою комбінації описаних раніше методів (3). Для ініціації росту попередньої культури, яку вирощували при 37°С, використовували індивідуальну колонію із планшета, який містить свіжі трансформовані клітини лінії JM109. Цією попередньою культурою інокулювали два літри середовища для росту. Після досягнення ОГ 600 ~1,5 (5-6 год при 37°С), експресію гена ЗТ індукували за допомогою ІПТГ (кінцева концентрація імМ) і ферментацію здійснювали ще протягом декількох годин при 37°С. Після центрифугування відкидали супернатанти, пелетовані клітини пулювали й поміщали на зберігання при -80°С до здійснення очищення. Клітинам давали розмерзтися при 4°С протягом ночі й суспендували в буфері для лізису (MES 50мМ рН 6, ЕДТА імМ, 10 об.% гліцерину, 0,02% мас/об. ОБГ, 0,02% мас./об. азиду натрію). Додавали лізоцим і суміш інкубували на льоді протягом 40 хв. Після гомогенізації з використанням Dounce у присутності лізоциму й опромінення ультразвуком клітини центрифугували протягом 30 хв. Збирали супернатант (S1) і поміщали на зберігання при 4°С. Отриманий центрифугуванням пелет ресуспендували в буфері для екстракції (MES 50мМ рН 6, КРО 4 50мМ рН 6, КС1 100мМ, 10 об.% гліцерину, 0,02% мас/об. ОБГ, 0,02% мас/об, азиду натрію) і перемішували протягом 30хв при 4°С. Цю другу суміш знову центрифугували й збирали супернатант (S2). Описану вище процедуру повторювали ще два рази, одержуючи супернатанти S3 й S4, і протягом ночі проводили останню екстракцію (S5). До об'єднаних супернатантів додавали полімін (кінцева концентрація 0,005%) для видалення нуклеїнових кислот. Цей розчин перемішували протягом 75хв при 4°С і центрифугували протягом 1год. Супернатант (SS1) осаджували на льоді за допомогою 20% мас/об, сульфа ту амонію й перемішували протягом 1год при 4°С. Потім суміш центрифугували й утворений супернатант (SS2) осаджували шляхом додавання 40% мас/об, сульфату амонію (усього 60%), перемішували протягом 1год і знову центрифугували. Кінцевий дебрис (Р1) зберігали протягом ночі при 4°С перед проведенням очищення наступного дня. Всі стадії очищення здійснювали при 4°С, якщо не вказане інше. Дебрис (Р1) ресуспендували в буфері MES, який містить 50мМ рН 6, КРО4 10мМ рН 6, КСl 100мМ, 10 об.% гліцерину, 0,02% мас/об. ОБГ, 0,02% мас/об, азид натрію. Суспензію протягом ночі піддавали діалізу в протитечії того ж самого буфера з використанням діалізних трубок із ММСО 12-14кДа. Діалізат центрифугували й супернатант фільтрували через фільтри типу Millex-PF з розміром отворів 0,8 мкм. Профільтрований зразок завантажували в гідроксіапатитну колонку (об'єм шару 30-мл) і промивали тим же самим буфером. Зв'язаний фермент елюювали з використанням лінійного градієнта від 10 до 300 мМ КРО4 (200 мл) у вказаному вище буфері. Фракції, які містять гетеродимер р66/р51 (за даними аналізу за допомогою ДСН-ПААГ 8% і вестерн-блотингу) об'єднували для внесення в наступну колонку. Фракції, які містять ЗТ, розбавляли у два рази біс-Трис-буфером, який містить пропан 50 мМ рН 7,0, 0,02 %мас./об. ОБГ, 10 об.% гліцерину, 0,02% мас/об, азиду натрію й завантажували в Ні-Тгар гепарин-сефарозну колонку (об'єм шару 5мл) і промивали тим же самим буфером. Потім зв'язану ЗТ елюювали з використанням лінійного градієнта від 0 до ЇМ сульфату амонію (75мл) у тому ж самому буфері. Фракції, які містять ЗТ, об'єднували на основі даних аналізу, отриманих за допомогою ДСН-ПААГ і вестерн-блотингу. Концентрацію протеїну в такому пулі визначали за допомогою методу Брадфорда, використовуючи як стандарт БСА. Кінцевий ферментний препарат піддавали діалізу в протитечії буфера, який містить MES 50мМ рН 6, КРО4 300 мМ рН 6, КСl 175мМ, 10 об.% гліцерину, 0,02% мас/об, азиду натрію, розділяли на аліквоти і зберігали при -80°С. Процедура аналізу: Радіометричний ферментативний аналіз був адаптований до формату 96-лункового титраційного мікропланшета й заснований на використанні завантажених стрептавідином сцинтиляційних гранул. Метод аналізу коротко описаний нижче. Фермент ЗТ ВІЛ-1 піддавали розморожуванню й розбавляли відповідним чином Трис/НСІ-буфером, 50 мМ рН 7,8, який містить NaCl 60 мМ, гексагідрат MgCl2 2 мМ, ДТТ 6 мМ, GSH 2 мМ й 0,02% мас/об. Chaps, одержуючи » 3 н М фермент. До 30мкл розчину цього ферменту додавали 10мкл розчину інгібітора (від 50мкМ до 2,5нМ інгібітора в цьому ж самому буфері для аналізу, доповненому 15 об.% ДМСО). Перед здійсненням наступної стадії планшет піддавали попередній інкубації протягом 15хв при кімнатній температурі. На цій стадії попередньої інкубації найбільша й найменша концентрації інгібітора становили 12,5мкМ й 0,62нМ відповідно, а концентрація ДМСО становила 3,75об.%. Потім ініціювали ферментативну реакцію шляхом додавання 10мкл розчину субстрату. Кінцева реакційна суміш містила Трис/НСІ 50 мМ рН 7,8, NaCl 60 мМ, MgCl2·6H2O 2 мМ, ДТТ 6 мМ, GSH 2 мМ, Chaps 0,02% мас/об. ДМСО 3об.%, Poly rC 179 нМ, біотин дГ 15 18 н М, дГТФ 288 нМ, 3Н-дГТФ 71 нМ й 1-2 н М фермент. На цієї стадії інкубації найбільша й найменша концентрації інгібітора становили 10 мкМ й 0,5 нМ відповідно. Після додавання субстратів планшет заклеювали пластиковим покриттям й інкубували протягом 1 год при 37°С у сухій камері. Потім реакцію припиняли шляхом додавання 75 мкл 0,5М розчину ЕДТК, який містить 5 мг/мл завантажених стрептавідином сцинтиляційних гранул. Планшет струшували протягом 2 хв при середній швидкості й інкубували протягом 1 год при кімнатній температурі. Потім додавали 75 мкл 7М розчину хлориду цезію, планшет струшували протягом 2 хв при середній швидкості й знову інкубували протягом 1 год при кімнатній температурі. Потім планшет заклеювали пластиковим покриттям і здійснювали кількісну оцінку з використанням сцинтиляційного й люмінісцентного лічильника для мікропланшетів типу TopCount-NXT™ (фірма Packard). Радіоактивність у кожній лунці оцінювали протягом 60 с. У кожній серії для одержання екстремальних значень використали порожні й контрольні лунки. Відсоток інгібування розраховували в такий спосіб: cpm у лунці –cpm у чист. лунці %інгібув ання =(1-[----------------------------------------------------])(100 cpm у контр.лунці – cpm у чист. лунці За допомогою описаного вище аналізу тестували сполуку, запропоновану у винаході, у відношенні інгібування ЗТ дикого типу (WT) і мутантних ферментів. Результати наведені в таблиці 4 у вигляді значень ІС5о (нМ). Для підтвердження здатності сполуки інгібувати реплікацію ВІЛ її тестували відповідно до описаного нижче методу аналізу людської Т-клітинної культури (фірма Syncytia). Твердофазний імуноферментний аналіз (ELISA) для оцінки активності в клітинній культурі Здатність сполуки, запропонованої у винаході, інгібувати реплікацію ВІЛ у клітинній культурі тестували у форматі 96-лункового планшета. Для розведення сполуки, а також середовища для росту клітин використовували повне середовище RPMI 1640, яке містить RPMI 1640 + 10% фетальної телячої сироватки, 10 мкг/мл гентаміцину й ЮмкМ (b-меркаптоетанолу. Клітинну лінію Т-лімфоцитів С8166 інфікували із множинністю зараження 0,001 вірусами, які кодують зворотну транскриптазу дикого типу й мутантну транскриптазу. Потім клітини інкубували протягом трьох днів у присутності серійних розведень сполуки, запропонованої у винаході. Об'єднували супернатант із восьми лунок-копій і визначали концентрацію позаклітинного р24 з використанням набору для аналізу антигену р24 ВІЛ-1, який надходить в продаж (фірма Beckman-Coulter®). Рівень інгібування (% інгібування) розраховували на основі наступного рівняння: p24 пг/мл інгібітора %інгібув ання =(1-[-----------------------------------])(100 з24 пг/мл контролю Результати наведені в таблиці 2 у вигляді ЕС 50 (н М). Процитована література (включена в опис як посилання) 1. Benn, S. І ін., Science 230:, 1985, стор. 949. 2. D'Aquila R. Т. і Summers W. С. J., Acq. Imm. Def. Syn. 2: 1989, стор. 579. 3. a) Warren Т. С і ін., Protein Expression & Purification 3:, 1992, стор. 479; 6) Kohlstaedt L. A. Science 256 (5065):, 1992, стор. 1783. Інгібування ЗТ дикого типу ймутантних штамів ЗТ сполукою формули I Сполука № 1001 1002 1003 1004 1005 1006 1007 1008 1009 1010 1011 1012 1013 1014 1015 1016 1017 1018 1019 1020 1021 іс50 (WT) (н М) С с с с с с с С с с с с с с с с с с с с с ІС50 K103N/ Y181C (нМ) А А А А А А В А С с А А А А В А А А А А А ЕС50 (WT) (нМ) С с с с с NT С С с с Н. в. Н. в. Н. в. С С NT С С с с Н. в. Таблиця 2 ЕС5о K103N/ Y181C (нМ) С С с А С NT В А С С Н. в. Н. в. Н. в. А С NT С С с с Н. в. 1022 1023 1024 1025 1026 1027 1028 1029 1030 1031 1032 1033 1034 1035 в в с в в в с с с с с с с С А А А Н. в. Н. в. А А А С С С А В А Позначення для таблиці: ІС50 й ЕС 50: А = >100 нМ; В = 100-50 нМ; С =
ДивитисяДодаткова інформація
Назва патенту англійськоюDipyridodiazepinones as inhibitors of reverse transcriptase
Назва патенту російськоюДипиридодиазепиноны как ингибиторы обратной транскриптазы
МПК / Мітки
МПК: A61K 31/55, A61K 31/551, A61P 43/00, A61P 31/18, C07D 471/14
Мітки: дипіридодіазепінони, інгібітори, транскриптази, зворотної
Код посилання
<a href="https://ua.patents.su/13-78774-dipiridodiazepinoni-yak-ingibitori-zvorotno-transkriptazi.html" target="_blank" rel="follow" title="База патентів України">Дипіридодіазепінони як інгібітори зворотної транскриптази</a>
Ненуклеозидні інгібітори зворотної транскриптази та фармацевтична композиція на їх основі

Номер патенту: 74834
Опубліковано: 15.02.2006
Автор: Сімоне Бруно
МПК: A61P 31/18, C07D 471/14, A61K 31/551, A61P 43/00
Мітки: ненуклеозидні, інгібітори, композиція, транскриптази, основі, фармацевтична, зворотної
Формула / Реферат:
1. Сполука загальної формули I:,деR2 вибирають із групи, яка включає H, F, Cl, C1-С4алкіл, C3-С4циклоалкіл і CF3;R4 означає H або Me;R5 означає H, Me або Et за умови, що R4 і R5 обидва не означають Me, і, якщо R4 означає Me, то R5 не означає Et;R11 означає Me, Et, циклопропіл, пропіл, ізопропіл або циклобутил; іQ вибирають...
Інгібітор вірусу імунодефіциту людини (віл) та зворотної транскриптази

Номер патенту: 34946
Опубліковано: 15.03.2001
Автори: Рибалко Світлана Леонтіївна, Гужова Світлана Василівна, Даниленко Валентина Пилипівна, Даниленко Георгій Іванович, Пацковський Юрій Васильович, Максимов Юрій Миколайович
МПК: A61P 31/18, C07C 13/00, A61K 31/13, A61K 31/015
Мітки: вірусу, зворотної, інгібітор, імунодефіциту, людини, транскриптази, віл
Формула / Реферат:
Застосування як інгібітору вірусу імунодефіциту людини та зворотної транскриптази сполуки наступної структури:
Бензоксазинонові сполуки, спосіб інгібування зворотної транскриптази, попередження або лікування, комбінації та фармацевтичні композиції

Номер патенту: 42699
Опубліковано: 15.11.2001
Автори: Ламма Вілльям К. (молодший), Тран Лікхан О., Пейн Лінда С., Янг Стівен Д., Брітчер Сюзн Ф.
МПК: A61K 31/505, A61K 31/54, A61P 31/12, A61P 31/18, C07D 265/18, A61K 31/52, A61K 45/06, A61P 37/04, A61P 43/00
Мітки: інгібування, фармацевтичні, спосіб, композиції, бензоксазинонові, комбінації, транскриптази, попередження, сполуки, зворотної, лікування
Формула / Реферат:
1. Бензоксазиноновые соединения формулы (I):в которой:Х представляет галоген;X1 представляет тригалогенметил или пентагалогенэтил;Z представляет О;R представляет(a) C1-8 алкил, незамещённый или замещённый заместителем А, и А представляет галоген, С3-6 циклоалкил, CN, гидрокси, C1-4 алкокси, C2-4 алкинил-С1-4 алкокси, арилокси, C1-4 алкилкарбонил, нитро, ди(С1-2 алкил)амино, C1-4 алкиламино-С1-2...
Спірогетероциклічні нітрили як інгібітори зворотної дії цистеїнових протеаз, фармацевтична композиція та спосіб лікування захворювань

Номер патенту: 73378
Опубліковано: 15.07.2005
Автори: Патель Уша Р., Янг Ерік Річард Роуш, Хікей Еужен Річард, Беккалі Юнес, Ворд Янси Девід, Сперо Деніс Мері, Ліу Веймін, Сан Санхінг, Томсон Девід С.
МПК: C07D 211/60, A61P 1/04, A61P 21/04, C07D 413/12, A61P 31/00, A61K 31/40, C07D 417/12, A61K 31/496, A61P 25/28, C07D 207/16, A61K 31/541, C07D 401/12, C07D 455/00, A61P 11/06, A61K 31/5377, A61P 9/10, A61K 31/454, C07D 211/66, C07D 405/12, A61P 19/10, A61K 31/517, A61K 31/4468, C07D 487/08, C07D 413/14, A61K 31/506, A61P 29/00, A61P 37/06, A61K 31/538, A61P 3/10, A61K 31/5365, A61P 37/00, A61K 31/55
Мітки: нітрили, протеаз, захворювань, спірогетероциклічні, фармацевтична, інгібітори, лікування, спосіб, цистеїнових, дії, зворотної, композиція
Формула / Реферат:
1. Сполука формули (Ia): , (Ia)у якій її фрагменти , і , позначені як A, B і C...
Засоби клітинної диференціації і інгібітори гістонової деацетилази та способи їх використання

Номер патенту: 74345
Опубліковано: 15.12.2005
Автори: Річон Вікторія М., Ріфкайнд Річард А., Бельведере Сандро, Бреслоу Рональд, Міллер Томас А., Гершелль Леланд, Маркс Поль А.
МПК: C07C 311/03, C07C 271/22, A61P 43/00, A61K 31/165, C07D 333/38, C07C 259/00, C07D 215/38, C07C 311/06, C07C 327/00, C07C 233/13, A61P 35/00, C07D 521/00, C07F 9/44, A61K 31/27, C07F 9/24, C07C 69/604, A61K 31/47, C07D 231/40, C07D 215/54, C07C 311/42, C07D 233/54, C07F 9/36, C07C 233/12, C07D 213/82, C07C 69/602, C07C 233/30, A61K 31/4406, C07D 277/46, C07D 213/75, C07C 237/22, C07C 69/716, C07D 215/40, C07C 323/67, C07C 237/52
Мітки: гістонової, інгібітори, засоби, деацетилази, диференціації, клітинної, способи, використання
Формула / Реферат:
1. Сполука, що має формулу:,де радикали R1 і R2 є однаковими чи різними, і обидва радикали є гідрофобними групами;радикал R3 є гідроксамовою кислотою, гідроксиламіно-, гідроксильною, аміно-, алкіламіно- чи алкілоксигрупою; і n є цілим числом від 3 до 10;або її фармацевтично прийнятна сіль.2. Сполука за п. 1, яка відрізняється тим, що кожен із радикалів R1 і R2 приєднаний безпосередньо чи через лінкер і є...
Попередній патент: Спосіб і пристрій для етерифікації жирних кислот
Наступний патент: Спосіб одержання вафельного виробу під кондитерський продукт, що містить жир
Випадковий патент: Спосіб заміщення дефектів м'яких тканин довгих пальців кисті при ушкодженні пальцьових артеріальних дуг