Бензоксазинонові сполуки, спосіб інгібування зворотної транскриптази, попередження або лікування, комбінації та фармацевтичні композиції
Номер патенту: 42699
Опубліковано: 15.11.2001
Автори: Тран Лікхан О., Янг Стівен Д., Пейн Лінда С., Брітчер Сюзн Ф., Ламма Вілльям К. (молодший)






Формула / Реферат
1. Бензоксазиноновые соединения формулы (I):
в которой:
Х представляет галоген;
X1 представляет тригалогенметил или пентагалогенэтил;
Z представляет О;
R представляет
(a) C1-8 алкил, незамещённый или замещённый заместителем А, и А представляет галоген, С3-6 циклоалкил, CN, гидрокси, C1-4 алкокси, C2-4 алкинил-С1-4 алкокси, арилокси, C1-4 алкилкарбонил, нитро, ди(С1-2 алкил)амино, C1-4 алкиламино-С1-2 алкил, гетероцикл, или арилтио;
(b) С2-4 алкенил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А;
(c) С2-5 алкинил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А; или
(d) С3-4 циклоалкил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А;
или их фармацевтически приемлемые соли, обладающие способностью ингибировать; ВИЧ обратную транскриптазу и полезные для предотвращения или лечения ВИЧ инфекции или для лечения СПИДа или СПИД ассоциированных состояний.
2. Соединения по п. 1, которые представляют собой:
(-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
(-)-6-хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
(+/-)-6-хлор-4-(2-цианофенил)этинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
(+/-)-4-(1-хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он, или
(+/-)-4-(2-[диметиламинометил]этинил)-4-трифторметил-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он,
или их фармацевтически приемлемые соли.
3. Соединение по п. 1, которым является:
(-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он или его фармацевтически приемлемые соли.
4. Фармацевтическая композиция, полезная для ингибирования ВИЧ обратной транскриптазы, для предотвращения или лечения инфекции ВИЧ, или для лечения СПИДа, или СПИД ассоциированных состояний, которая включает эффективное количество активного агента и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента она содержит соединение формулы (II):
в которой:
Х представляет галоген;
X1 представляет тригалогенметил или пентагалогенэтил;
Z представляет О или S;
R представляет
(a) C1-8 алкил, незамещённый или замещённый заместителем А, и А представляет галоген, С3-6 циклоалкил, CN, гидрокси, С1-4 алкокси, С2-4 алкинил-С1-4 алкокси, арилокси, С1-4 алкилкарбонил, нитро, ди(С1-2 алкил)амино. С1-4 алкиламино-С1-2 алкил, гетероцикл, или арилтио;
(b) С2-4 алкенил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А;
(c) С2-5 алкинил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А; или
(d) С3-4 циклоалкил, незамещённый или замещённый
(і) А, или
(іі) арилом, незамещённым или замещённым А;
или его фармацевтически приемлемую соль.
5. Способ ингибирования ВИЧ обратной транскриптазы, который включает введение млекопитающему эффективного количества активного агента, отличающийся тем, что в качестве активного агента используют соединение, определённое в любом из пп. 1-3, или соединение формулы (II), определённое в п. 4, или его фармацевтически приемлемую соль.
6. Способ предотвращения или лечения инфекции ВИЧ, или лечения СПИДа, или СПИД ассоциированных состояний, который включает введение млекопитающему эффективного количества активного агента, отличающийся тем, что в качестве активного агента используют соединение, определённое в любом из пп. 1-3, или соединение формулы (II), определённое в п.4, или его фармацевтически приемлемую соль.
7. Фармацевтическая композиция, полезная для ингибирования ВИЧ обратной транскриптазы, для предотвращения или лечения инфекции ВИЧ, или для лечения СПИДа, или СПИД ассоциированных состояний, которая включает эффективное количество активного агента и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента она содержит соединение, определённое в любом из пп. 1-3, или его фармацевтически приемлемую соль.
8. Комбинация, полезная для ингибирования ВИЧ обратной транскриптазы, для предотвращения или лечения инфекции ВИЧ, или для лечения СПИДа, или СПИД ассоциированных состояний, которая предусматривает эффективное количество каждого из соединений формулы (I), определённых в п. 1, или соединений формулы (II), определённых в п. 4, или их фармацевтически приемлемых солей и нуклеозидного аналога, обладающего активностью против ВИЧ обратной транскриптазы.
9. Комбинация по п. 8, в которой соединение представляет (-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (L-743726) или его фармацевтически приемлемую соль.
10. Комбинация антивирусных соединений против СПИДа, которая предусматривает эффективное количество каждого из соединений (-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она (L-743726) или его фармацевтически приемлемой соли, N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'(третбутилкарбоксамидо)пиперазинил))пентанамида (L-735524), или его фармацевтически приемлемой соли, и необязательно одного, или более ВИЧ ингибиторов, выбранных из группы, состоящей из 3-([4,7-дихлор-1,3-бензоксазол-2-ил)метил]амино)-5-этил-6-метилпиридин-2(1Н)-она (L-697661), зидовудина (AZT), дидезоксиинозина (ddl) и дидезоксицитидина (ddC), или их фармацевтически приемлемых солей.
11. Комбинация антивирусных соединений против СПИДа, которая предусматривает эффективное количество каждого из соединений (-)-6-хлор-4-цикпопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-она (L-743726) или его фармацевтически приемлемой соли и одного, или более ВИЧ ингибиторов, выбранных из группы, состоящей из 3-([4,7-дихлор-1,3-бензоксазол-2-ил)метил]амино)-5-этил-6-метилпиридин-2(1Н)-она (L-697661), зидовудина (AZT), дидезоксиинозина (ddl) и дидезоксицитидина (ddC), или их фармацевтически приемлемых солей.
Текст
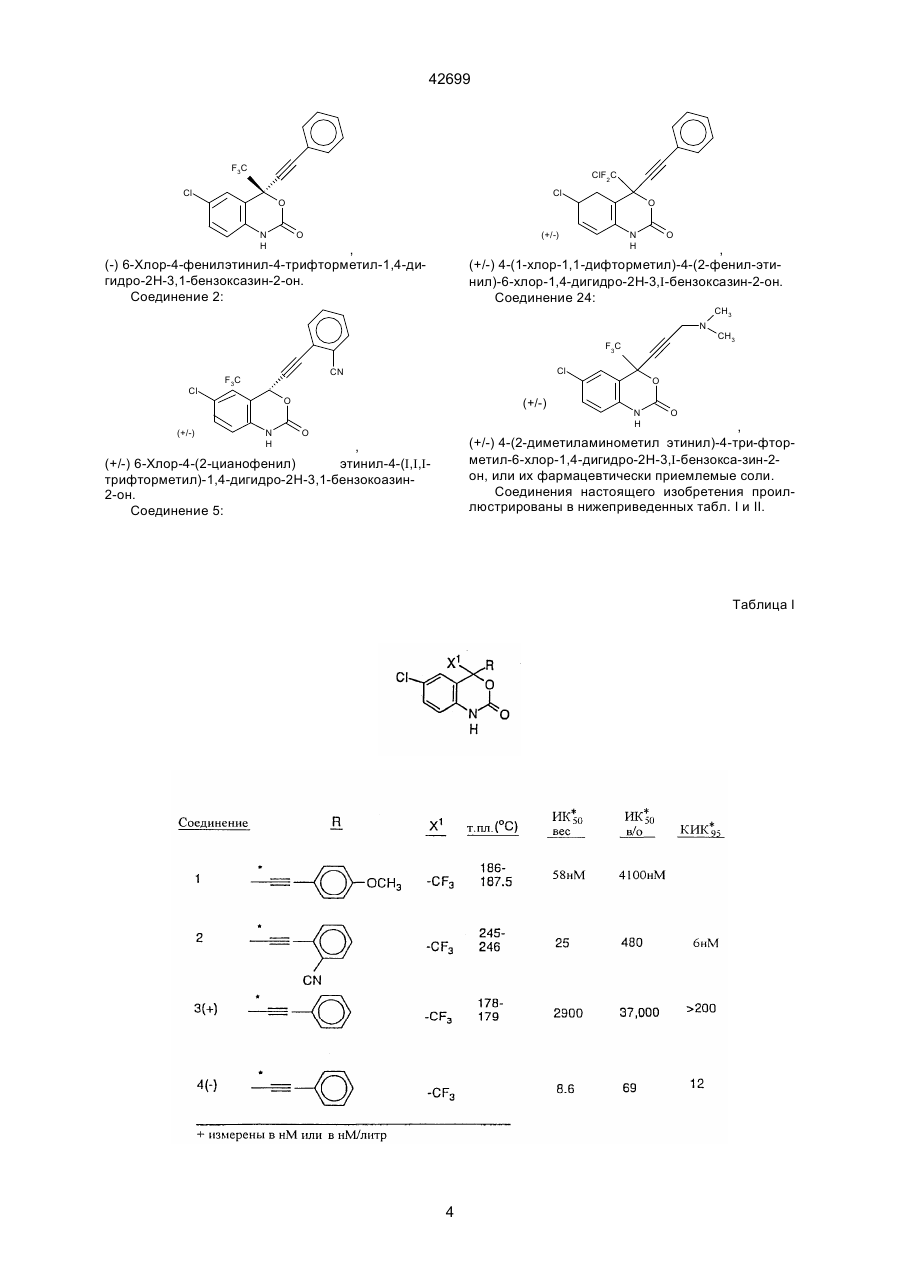
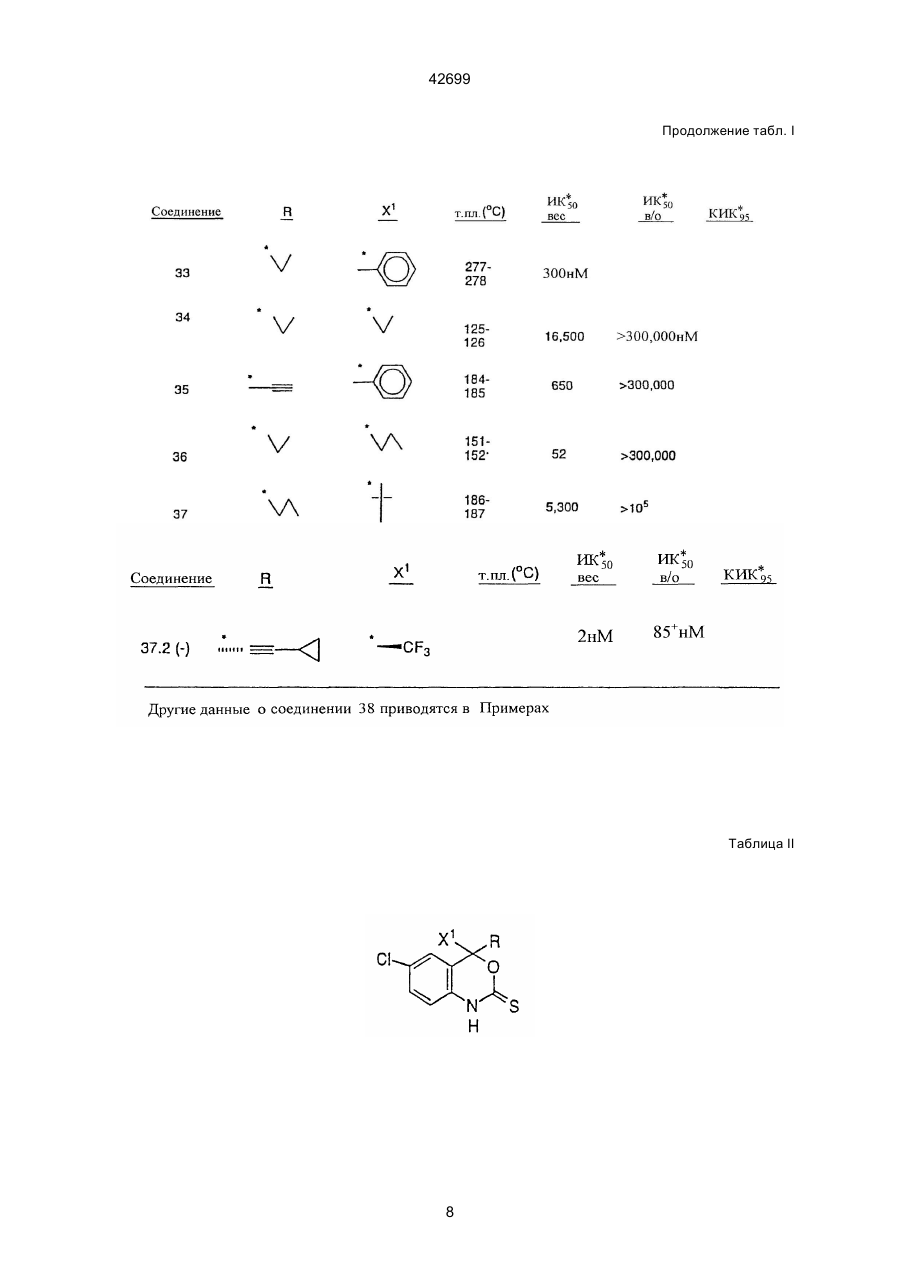
1. Бензоксазиноновые соединения формулы (I): C2 (54) БЕНЗОКСАЗИНОНОВІ СПОЛУКИ, ФАРМАЦЕВТИЧНА КОМПОЗИЦІЯ (ВАРІАНТИ), КОМБІНАЦІЯ СПОЛУК (ВАРІАНТИ), СПОСІБ ІНГІБУВАННЯ ВІЛ ЗВОРОТНОЇ ТРАНСКРИПТАЗИ, СПОСІБ ЗАПОБІГАННЯ АБО ЛІКУВАННЯ ВІЛ-ІНФЕКЦІЇ в которой: Х представляет галоген; X1 представляет тригалогенметил или пентагалогенэтил; Z представляет О или S; R представляет 42699 (a) C1-8 алкил, незамещенный или замещенный заместителем А, и А представляет галоген, С3-6 циклоалкил, CN, гидрокси, С1-4 алкокси, С2-4 алкинил-С1-4 алкокси, арилокси, С1-4 алкилкарбонил, нитро, ди(С1-2 алкил)амино, С1-4 алкиламино-С1-2 алкил, гетероцикл, или арилтио; (b) С2-4 алкенил, незамещенный или замещенный (і) А, или (іі) арилом, незамещенным или замещенным А; (c) С2-5 алкинил, незамещенный или замещенный (і) А, или (іі) арилом, незамещенным или замещенным А; или (d) С3-4 циклоалкил, незамещенный или замещенный (і) А, или (іі) арилом, незамещенным или замещенным А; или его фармацевтически приемлемую соль. 5. Способ ингибирования ВИЧ обратной транскриптазы, который включает введение млекопитающему эффективного количества активного агента, отличающийся тем, что в качестве активного агента используют соединение, определенное в любом из пп. 1-3, или соединение формулы (II), определенное в п. 4, или его фармацевтически приемлемую соль. 6. Способ предотвращения или лечения инфекции ВИЧ, или лечения СПИДа, или СПИД ассоциированных состояний, который включает введение млекопитающему эффективного количества активного агента, отличающийся тем, что в качестве активного агента используют соединение, определенное в любом из пп. 1-3, или соединение формулы (II), определенное в п. 4, или его фармацевтически приемлемую соль. 7. Фармацевтическая композиция, полезная для ингибирования ВИЧ обратной транскриптазы, для предотвращения или лечения инфекции ВИЧ, или для лечения СПИДа, или СПИД ассоциированных состояний, которая включает эффективное количество активного агента и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента она содержит соединение, определенное в любом из пп. 1-3, или его фармацевтически приемлемую соль. 8. Комбинация, полезная для ингибирования ВИЧ обратной транскриптазы, для предотвращения или лечения инфекции ВИЧ, или для лечения СПИДа, или СПИД ассоциированных состояний, которая предусматривает эффективное количество каждого из соединений формулы (I), определенных в п. 1, или соединений формулы (II), определенных в п. 4, или их фармацевтически приемлемых солей и нуклеозидного аналога, обладающего активностью против ВИЧ обратной транскриптазы. 9. Комбинация по п. 8, в которой соединение представляет (-)-6-хлор-4-циклопропилэтинил-4трифторметил-1,4-дигидро-2Н-3,1-бензоксазин2-он (L-743726) или его фармацевтически приемлемую соль. 10. Комбинация антивирусных соединений против СПИДа, которая предусматривает эффективное количество каждого из соединений (-)-6-хлор-4циклопропилэтинил-4-трифторметил-1,4-дигидро2Н-3,1-бензоксазин-2-она (L-743726) или его фармацевтически приемлемой соли, N-(2(R)-гидрокси1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси-5(1-(4-(3-пиридилметил)-2(S)-N'(третбутилкарбоксамидо)пиперазинил))пентанамида (L-735524), или его фармацевтически приемлемой соли, и необязательно одного, или более ВИЧ ингибиторов, выбранных из группы, состоящей из 3-([4,7-дихлор1,3-бензоксазол-2-ил)метил]амино)-5-этил-6-метилпиридин-2(1Н)-она (L-697661), зидовудина (AZT), дидезоксиинозина (ddl) и дидезоксицитидина (ddC), или их фармацевтически приемлемых солей. 11. Комбинация антивирусных соединений против СПИДа, которая предусматривает эффективное количество каждого из соединений (-)-6-хлор-4цикпопропилэтинил-4-трифторметил-1,4-дигидро2Н-3,1-бензоксазин-2-она (L-743726) или его фармацевтически приемлемой соли и одного, или более ВИЧ ингибиторов, выбранных из группы, состоящей из 3-([4,7-дихлор-1,3-бензоксазол-2ил)метил]амино)-5-этил-6-метилпиридин-2(1Н)она (L-697661), зидовудина (AZT), дидезоксиинозина (ddl) и дидезоксицитидина (ddC), или их фармацевтически приемлемых солей. Настоящая заявка является родственной заявкам фирмы Мерк, имеющим ссылочные №№ 18429, 184291A и 18727. Эта заявка является частичным продолжением заявки фирмы Мерк 18793, поданной 7 августа 1992 г., U.S.S.N. 07/926607. Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), является этиологическим фактором, ответственным за комплексное заболевание, выражающееся прогрессирующей деструкцией иммунной системы (синдром приобретенного иммунодефицита, СПИД) и дегенерацией центральной и периферической нервной системы. Этот вирус был ранее известен как LAY, НТLY-III, или ARY. Общей характерной особенностью репликации ретровирусов является обратная транскрипция РНК-генома, осуществляемая с помощью обратной транскриптазы, кодируемой вирусным геномом, в результате чего происходит синтез ДНК-копий ВИЧ-последовательностей, который является необходимой стадией для репликации вируса. Известно, что некоторые соединения являются ингибиторами обратной транскриптазы, и в качестве аффективных средств используются для лечения СПИДа и аналогичных заболеваний, например, азидотимидин или AZT. Путем секвенирования нуклеотидной последовательности ВИЧ было обнаружено, что в одной открытой рамке считывания присутствует pol-ген (Rather L и др., Nature, 313, 277 (1985)). На основании гомологии аминокислотных последовательностей было обнаружено, что роI-последовательность кодирует обратную транскриптазу, эндонуклеазу и ВИЧ-протеазу (Тоh Н. и др., ЕMВО J. 4, 2 42699 1267 (1985), Power M., и др., Science 231, 1567 (1986), Pearl L.Н и др., Nature 329, 351 (1987)). Авторами настоящей заявки было продемонстрировано, что соединения настоящего изобретения являются ингибиторами обратной транскриптазы ВИЧ. Преимущество соединений настоящего изобретения заключается в том, что они ингибируют резистентную обратную транскриптазу ВИЧ. В настоящей заявке раскрываются соединения формулы I, определенной ниже. Эти соединения могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ (и ее резистентных разновидностей); в целях предупреждения инфицирования вирусом ВИЧ; а также в целях лечения ВИЧ-инфекций, СПИДа и/или ARC (СПИД-ассоциированный синдром); причем указанные соединения могут быть использованы как таковые, или в виде фармацевтически приемлемых солей (если это целесообразно), либо в качестве ингредиентов фармацевтических композиций, используемых отдельно или в комбинации с другими противовирусными средствами, противоинфекционными средствами, иммуномодуляторами, антибиотиками или вакцинами. В настоящей заявке раскрываются также способы лечения СПИДа, способы предупреждения инфицирования вирусом ВИЧ, а также способы лечения ВИЧ-инфекций. Настоящее изобретение относится к соединениям формулы I, к их комбинациям, или к их фармацевтически приемлемым солям, которые могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ и ее резистентных разновидностей, а также в целях лечения ВИЧ-инфекций и вызываемого этими инфекциями синдрома приобретенного иммунодефицита (СПИД). Соединения формулы I имеют следующую структуру: (d) С3-4-циклоалкил, незамещенный или замещенный (I) А, или (II) арилом, незамещенным или замещенным А, или их фармацевтически приемлемые соли. Настоящее соединение также относится к фармацевтической Композиции, предназначенной для ингибирования обратной транскрипции ВИЧгенома, и содержащей эффективное количество соединения формулы II или его фармацевтически приемлемой соли: , и фармацевтически приемлемый носитель, где: Х представляет собой галоид; Х1 представляет собой тригалоидметил, пентагалоидэтил, С2-5-алкил, С2-5-алкинил, C3-5-циклоалкил, или арил; Z представляет собой О или S; R представляет собой: (А) C1-8-алкил, незамещенный или замещенный, А, а А представляет собой галоид, C3-6-циклоалкилом, СN, гидрокси, С1-4-алкокси, С2-4-алкинил-С1-4-алокси, арилокси, С1-4-алкилкарбонил, нитро, ди(C1-2-aлкил)амино С1-4-алкиламино-С1-2-алкил, гетероциклом, или арилтио, (в) С2-4-алкенил, незамещенный или замещенный: (I) А, или (II) арилом, незамещенным или замещенным А, (с) С2-5-алкинил, незамещенный или замещенный (I) А, или (II) арилом, незамещенным или замещенным, А, или (d) С3-4-циклоалкил, незамещенный или замещенный (I) А, или (II) арилом, незамещенным или замещенным А. Предпочтительными являются соединения 37.2, 4, 2, 5 и 24 табл. 2 (по нисходящей степени предпочтительности). Указанные соединения имеют следующую структуру. Соединение 37.2: где: Х представляет собой галоид; Х1 представляет собой тригалоидметил или пентагалоидэтил; Z представляет собой О; R представляет собой: (а) C1-8-алкил, незамещенный или замещенный, А, где А является галоидом, C3-6-циклоалкилом, СN, гидрокси, С1-4-алкокси, С2-4-алкинил-С1-4-алокси, арилокси, С1-4-алкилкарбонил, нитро, ди(C1-2-aлкил)амино С1-4-алкиламино-С1-2алкил, гетероциклом, или арилтио; (в) С2-4-алкенил, незамещенный или замещенный, (I) А, или (II) арилом, незамещенным или замещенным А, (c) С2-5-алкинил, незамещенный или замещенный (I) А, или (II) арилом, незамещенным или замещенным А, или Cl F3C O (-) N H O , (-) 6-Хлор-4-циклопропилэтинил-4-трифторметил1,4-дигидро-2Н-3,I-бензоксазин-2-он (наиболее предпочтительное соединение). Соединение 4: 3 42699 F3C ClF2C Cl Cl O N H O O N H (+/-) , (-)6-Хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он. Соединение 2: O , (+/-) 4-(1-хлор-1,1-дифторметил)-4-(2-фенил-этинил)-6-хлор-1,4-дигидро-2Н-3,I-бензоксазин-2-он. Соединение 24: CH3 N F3C Cl (+/-) Cl CN F3C O N H (+/-) CH3 O N H O , (+/-) 4-(2-диметиламинометил этинил)-4-три-фторметил-6-хлор-1,4-дигидро-2Н-3,I-бензокса-зин-2он, или их фармацевтически приемлемые соли. Соединения настоящего изобретения проиллюстрированы в нижеприведенных табл. I и II. O , (+/-) 6-Хлор-4-(2-цианофенил) этинил-4-(I,I,Iтрифторметил)-1,4-дигидро-2Н-3,1-бензокоазин2-он. Соединение 5: Таблица I 4 42699 Продолжение табл. I 5 42699 Продолжение табл. I 6 42699 Продолжение табл. I 7 42699 Продолжение табл. I Таблица II 8 42699 Продолжение табл. II Соединения настоящего изобретения могут иметь асимметрические центры, и могут существовать (за исключением тех случаев, о которых упоминается особо) в виде рацематов, рацемических смесей, или отдельных диастереомеров или энантиомеров, при этом, следует отметить, что все изомерные формы входят в объем настоящего изобретения. Символ (+/-) означает (+)оптические изомеры, (-)оптические изомеры, или их смеси. Если какая-либо переменная (например, R) встречается более, чем один раз в формуле I или в любой составной части этой формулы, то определение указанной переменной в данном конкретном случае не зависит от ее определения в любых других случаях. Кроме того, комбинации заместителей и/или переменных являются допустимыми, если только такие комбинации позволяют получить стабильные соединения. Используемый в настоящем описании термин "алкил" (если это не оговорено особо) означает прямые или разветвленные насыщенные алифатические углеводородные группы, имеющие определенное количество атомов углерода. Термин "алкенил" относится к прямым или разветвленным алкильным группам, имеющим, по крайней мере, одну углерод-углеродную двойную связь. Термин "алкинил" относится к прямым или разветвленным алкильным группам, имеющим, по крайней мере, одну углерод-углеродную тройную связь. Термин "галоид" или "галоин" означает фтор, хлор, бром и йод. Используемый в настоящем описании термин "арил" (если это не оговорено особо) означает фенил, нафтил, тетрагидронафтил, бифенил, фенатрил, антрил или аценафтил. Используемый в настоящем описании термин (если это не оговорено особо) "гетероцикл" или "гетероциклический" означает стабильное 5-7членное моноциклическое кольцо или стабильное 8-II-членное бициклическое гетероциклическое кольцо, которое является ненасыщенным или частично насыщенным, и которое состоит из атомов углерода и 1-4 гетероатомов, выбранных из N, О и S, причем, гетероатомы азота и серы могут быть, но необязательно, окислены, а в любой бициклической группе, любое из вышеуказанных гетероциклических колец является конденсированным с бензольным кольцом. Гетероциклическое кольцо может быть присоединено посредством 9 42699 любого гетероатома или атома углерода, что приводит к образованию стабильной структуры. Примерами таких гетероциклических элементов являются пиперидинил, пиперазинил, 2-оксипиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксоазецинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклинидил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, бензофуранил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинилсульфоксид, тиамофолинилсульфон, и оксадиазолил. Соединения настоящего изобретения могут быть синтезированы методами, описанными ниже. предпочтительными аминозащитними группами являются т-бутоксикарбонильная, ацетатная или изовалероильная группы. Затем соединение 2 подвергают реакции с алкиллитием, а предпочтительно с н-бутиллитием. В этой стадии металлирования могут быть также использованы и другие металлорганические соединения. Затем, после реакции с CF3СООЕ и последующего гашения реакции получают соединение 3. Синтез третичного карбинола 4 осуществляют с помощью реакции присоединения Гриньяра, которой подвергают полученный кетон 3. В качестве реагента Гриньяра должна быть использована соль двухвалентного катиона, например Mg++ или Zn++. Моновалентные катионы, например, такие, как Li++ или Na++, для данной реакции не подходят. Подходящими растворителями являются, но не ограничиваются, ими, ТГФ или простой эфир. Температурные условия предусматривают широкий диапазон реакционных температур, который составляет от около 0°С и примерно до комнатной температуры. Реакцию замыкания кольца осуществляют с использованием конденсирующих агентов, таких, как 1,1-карбонилдиимидазол, фосген, диметилкарбонат, дифенилкарбонат, или ди-(паранитрофенил)карбонат, и в результате этой реакции получают соединения настоящего изобретения 5. Циклизация может быть осуществлена с любым из указанных агентов, а также с другими агентами широкого ряда. Нижеприведенная схема IA представляет собой конкретный вариант схемы I. Эта схема иллюстрирует синтез соединения L-741, 211, которое представляет собой рацемат соединения 37.2, описанный ниже в примере 6. Синтез безоксазинов настоящего изобретения может быть осуществлен общим методом, согласно которому, конечная стадия представляет собой реакцию циклизации бензольного кольца (см. схему І). Сначала аминогруппу парахлоранилина защищают, например, пивалоилхлоридом, в результате чего получают соединение 2. Другими менее Схема II иллюстрирует один из методов дериватизации ацетиленовых заместителей в 4-по 10 42699 ложении бензоксазинового ядра. В соответствии с этой схемой, соединение 6 сначала подвергают металлизации, а затем добавляют цинковую соль. Для получения соединения 7 осуществляют реакцию Хека, в которой используют катализатор, тетракис(трифенилфосфин)-палладий (0) в сочетании с Cul. при получении соединения L-743.726. Схема IVA и пример 6. Схема III иллюстрирует замещение 4-ацетиленовой группы N-содержащим гетероциклом. Реакция Манниха представляет собой реакцию конденсации формальдегида с гетероциклом, например, пирролидоном. Замещение на конечном атоме углерода осуществляют в присутствии Cul в качестве катализатора. Схему IV иллюстрирует разделение оптических изомеров соединений формулы I или формулы II. На этой схеме разделяющим агентом является (-)камфановая кислота. При этом, могут быть использованы другие разделяющие агенты широкого диапазона, например, хлорид 0-метилминдальной кислоты или реагент Mosher. Впрочем, процедуры разделения таких изомеров хорошо известны любому специалисту. Схема IVA была специально разработана для разделения соединения L-741.211, образующегося Циклопропилацетилен получают по схеме V в соответствии с известными процедурами, описанными, например, C.E. Hudson и др., J. Am. Chem. Soc. 94, 1158 (1972), и W. Schoberth и др., Synthesis 703(1972). 11 42699 Соединения настоящего изобретения могут быть использованы для получения препаратов в целях проведения анализов на поиск противовирусных соединений. Например, соединения настоящего изобретения могут быть использованы для выделения ферментов-мутантов, которые являются прекрасным средством для скрининга наиболее сильных противовирусных соединений. Кроме того, соединения настоящего изобретения могут быть использованы для установления или идентификации сайга связывания других противовирусных агентов с обратной транскриптазой ВИЧ, например, путем конкурентного ингибирования. Таким образом, соединения настоящего изобретения представляют собой коммерческие продукты, которые могут поставляться для вышеуказанных исследований. Соединения настоящего изобретения могут быть использованы в целях ингибирования обратной транскриптазы ВИЧ, в целях предупреждения и лечения инфекций, вызванных вирусом иммунодефицита человека (ВИЧ, а также в целях лечения патологических состоянии, обусловленных инфицированном вирусом ВИЧ, например, таких, как СПИД. Однако, настоящее изобретение не ограничивается лишь лечением или предупреждением СПИДа или ВИЧ-инфекций, в объем настоящего изобретения также входит лечение широкого ряда состояний, обусловленных ВИЧ-инфекциями, например, таких, как СПИД, АRС (СПИД-ассоциированный комплекс или предСПИД) (как симптомные так и бессимптомные), а также фактическое или возможное заражение вирусом ВИЧ. Например, соединения настоящего изобретения могут быть использованы для лечения ВИЧ-инфекций по подозрению заражения вирусом ВИЧ, например, после переливания крови, обменного вливания физиологических жидкостей, укусов, случайных уколов иглой, или хирургических операций. Основным преимуществом соединений настоящего изобретения является их способность к высокоэффективному ингибированию обратной транскриптазы ВИЧ, которая является резистентною по отношению к другим противовирусным агентам, таким, как L-697.661 (3-(/(4,7-дихлор-1,3бензоксазол-2-ил)етил/-амино)-5-этил-6-метилпиридин-2(1Н)-он), или _L-696.229 (3-/2-(1,3-бензоксазол-2-ил) этил/-5-этил-6-метил-пиридин-2(1Н)он), или АZТ. Для этих целей, соединения настоящего изобретения могут быть введены перорально, парентерально (подкожно, внутривенно, внутримышечно, внутригрудинно, или путем вливаний), путем ингаляций, или ректально, в виде унифицированных лекарственных препаратов, содержащих соответствующие нетоксичные фармацевтически приемлемые носители, адьюванты и наполнители. Поэтому, в другом своем варианте, настоящее изобретение относится к способу лечения и к фармацевтическим композициям, предназначенным для лечения ВИЧ-инфекций и СПИДа. Этот способ лечения предусматривает введение пациенту, нуждающемуся в таком лечении, фармацевтическую композицию, содержащую фармацевтический носитель и терапевтически эффективное количество соединения настоящего изобретения. Указанные фармацевтические композиции могут быть изготовлены в виде суспензий или таблеток для перорального введения, препаратов для ингаляций через нос, стерильных растворов для инъекций, например, в виде стерильных водных или масляных суспензии для инъекций, или в виде суппозиториев. Композиции в виде пероральных суспензий могут быть получены в соответствии с хорошо известной техников, обычно применяемой в фармацевтической практике для изготовления подобных препаратов, причем, эти суспензии могут содержать микрокристаллическую целлюлозу в качестве наполнителя, альгиновую кислоту или альгинат натрия, в качестве суспендирующего агента, метилцеллюлозу для повышения вязкости, и подслащивающие/ароматизирующие агенты. Хорошо известные специалистам. Композиции, изготовленные в виде таблеток немедленного действия, могут содержать микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния, лактозу, и/или другие наполнители, связующие, носители, дезинтеграторы, разбавители, и замасливающие агенты, хорошо известные специалистам. Композиции, предназначенные для введения через нос с помощью аэрозолей или ингаляций, могут быть получены в соответствии с традиционной фармацевтической практикой в виде растворов в физиологических растворителях с использованием бензилового спирта или других подходящих консервантов, стимуляторов абсорбции (для повышения биологической доступности), фторуглеродов, и/или других солюбилизирующих или диспергирующих агентов, хорошо известных специалистам. Инъецируемые растворы или суспензии могут быть получены традиционными методами с использованием подходящих нетоксичных парентерально приемлемых разбавителей или растворителей, таких, как маннит, 1,3-бутандиол, вода, раствор Рингеран или изотонический раствор хлорида натрия, либо подходящих диспергирующих или смачивающих и суспендирущих агентов, таких, как стерильное мягкое жирное масло, включая синтетические моно- или диглицериды, и жирные кислоты, такие, как олеиновая кислота. Композиции в виде суппозиториев для ректального введения могут быть получены путем смешивания активного ингредиента с подходящими нераздраживающим носителем, таким, как какао, масло, синтетические сложные эфиры глицерина, или полиэтиленгликоли, которые являются твердыми при обычной температуре, но при введении в прямую кишку расплавляются и/или растворяются с высвобождением лекарственного средства. Доза соединений настоящего изобретения, предназначенных для перорального введения человеку, составляет в диапазоне от 1 до 100 мг/кг веса тела в виде дробных доз. Предпочтительно, если диапазон доз для перорального введения составляет от 0,1 до 10 мг/кг, либо от 0,1 до 20 мг/кг веса тела в дробных дозах. При комбинированной терапии с нуклеозидными аналогами, предпочтительная доза соединений настоящего изобретения для перорального введения в дробных дозах составляет от 0,1 до 20 мг/кг, а 12 42699 предпочтительная доза нуклеозидных аналогов для перорального введения в дробных дозах составляет от 50 мг до 5 г/кг веса тела. При этом, следует ответить, что конкретная доза и частота ее введения каждому конкретному пациенту могут варьироваться в зависимости от различных факторов, например, таких, как активность конкретно используемого соединения, возраст, вес тела, общее состояние здоровья, пол, и режим питания пациента, способ и время введения, скорость высвобождения лекарственного средства, комбинация лекарственных средств, тяжесть состояния пациента, и организм-хозяин, подвергающийся терапии. Настоящее изобретение также относится к комбинациям соединений-ингибиторов обратной транскриптазы ВИЧ с одним или несколькими агентами, используемыми для лечения СПИДа. Например, соединения настоящего изобретения могут быть с успехом введены в комбинации с эффективными количествами агентов против вируса, вызывающего СПИД, иммуномодуляторов, антиинфекционных средств, или вакцин, например, указанных в нижеследующей табл. III, причем, соединения настоящего изобретения могут быть введены до введения и/или после введения вышеуказанных других активных агентов. Таблица III Противовирусные средства Название лекарственного средства АL-721 Рекомбинантный интерферонбета человека Производитель Ethigen (Los Angeles, CA) Triton Biosciences (Almeda, CA) Ацеманнан Carrington Labs (Irving, TX) Цитовен Syntex Ганцикловир (Palo Alto, CA) Дидегидродезокситимидин d4T Дидезоксиинозин dd1 Bristol-Myers (New York, NY) Bristol-Myers (New York, NY) EL10 Elan Corp, PLC (Gainesville, CA) Тринатрийфосфоноформат Дидезоксицитидин; ddC Новапрен Октапептидная последовательность пептида Т Зидовудин; AZT Astra Pharm. Products, Inc. (Westborough, MA) Hoffman-La Roche (Nutley, NJ) Novaferon Labs, Inc. (Akron, OH) Diapren, Inc. (Roseville, MN, marketer) Peninsula Labs (Belmont, CA) Burroughs Wellcome (Rsch. Triangle Park, NC) Анзамицин LM 427 Adria Laboratories (Dublin, OH) Erbamont (Stamford, CT) Декстрансульфат Ueno Fine Chem Ind. Ltd. Виразол, Рибавирин Viratek/ICN (Costa Mesa, CA) Альфа-интерферон Ацикловир Burroughs Wellcome (Rsch. Triangle Park, NC) Burroughs Wellcome Антитело, которое нейтрализует Advanced Biotherapy Concepts рН-лабильный аберрантный aинтерферон в иммуноадсорбц. ко- (Rockville, MD) лонке 13 Показания ARC, РGL, ВИЧ-инфицирование, СПИД СПИД, саркома Капоши, ARC (СПИД-ассоциированный комплекс) ARC (см. также иммуномодуляторы) Подозрение на CMV (цитомегаловирус) Периферическая CMV-инфекция, ретинит СПИД, ARC СПИД, ARC ВИЧ-инфекция (см. также иммуномодуляторы) Ретинит, вызванный СМVинфекцией, ВИЧ-инфекция, другие СМV-инфекция СПИД, ARC ВИЧ-ингибитор СПИД СПИД, ао (аденовирус АРС, СПИД у детей, саркома Капоши, бессимптомные ВИЧ-инфекции, ВИЧ-заболевания в нетяжелой форме, поражения нервной системы, в комбинации с другими терапевтическими средствами ARC СПИД, ARC, бессимптомная ВИЧинфекция Бессимптомная ВИЧ-инфекция, Ласса (арбовирус), ARC Саркома Капоши, ВИЧ в комбинации с ретровирусной инфекцией СПИД, ARC, бессимптомная ВИЧинфекция, в комбинации с AZT СПИД, ARC 42699 Продолжение табл. III L-697.661 Merck (Rahway, NJ) L-696.229 Merck (Rahway, NJ) L-735.524 Merck (Rahway, NJ) As-101 Бропиримин Ацеманнан CL246.738 Иммуномодуляторы Wyeth-Ayerst Labs. (Philadelphia, PA) Upjohn (Kalamazoo, MI) Carrington Labs, Inc. (Irving, TX) American Cyanamid (Pearl River, NY) Lederle Labs (Wayne, NJ) EL10 Elan Corp, PLC (Gainesville, CA) Гамма-интерферон Genentech (S. San Francisco, CA) Гранулоцитарно-макрофагальный колониестимулирующий фактор Гранулоцитарно-макрофагальный колониестимулирующий фактор Гранулоцитарно-макрофагальный колониестимулирующий фактор Частица ВИЧ-ядра иммуностимулянт Интерлейкин-2 1L-2 Иммуноглобулин (человека) для внутривенного введения IMPEG-1 IMPEG-2 Иммунотиолдиэтилдитиокарбамат Альфа-2-интерферон Метионин-Энкефалин МТР-РЕ (мурамилтрипептид) Гранулоцитарный колониестимулирующий фактор Рекомб. растворимый чел. CD4(rCD4) Рекомб. растворимый чел. rCD4 Интерферон a-2а SK&F106528 Растворимый Т4 Тимопентин Genetics Institute (Cambridge, MA) Sandoz (East Hanover, NJ) Hoeschst-Roussel (Somerville, NJ) Immunex (Seattle, WA) Schering-Plough (Madison, NJ) Rorer (Ft. Washington, PA) Hoffman-La Roche (Nutley, NJ) Immunex Gutter Biological (Berkeley, CA) Imreg (New Orleans, LA) Imreg (New Orleans, LA) Merieux Institute (Miami, FL) Schering Plough (Madison, NJ) TNI Pharmaceutical (Chicago, IL) Giba-Geigy Corp. (Summit, NJ) СПИД, ARC, бессимптомная ВИЧинфекция, также в комбинации с AZT СПИД, ARC, бессимптомная ВИЧинфекция, также в комбинации с AZT СПИД, ARC, бессимптомная ВИЧинфекция, также в комбинации с AZT СПИД Прогрессирующий СПИД СПИД, ARC (см. также противовирусные средства) СПИД, саркома Капоши ВИЧ-инфекция (см. также противовирусные средства) ARC, в комбинации с TNF (фактор некроза опухоли) СПИД СПИД СПИД Сероположительная реакция на ВИЧ СПИД, ARC, ВИЧ, в комбинации с AZT СПИД у детей, в комбинации с AZT СПИД, саркома Капоши, PGL СПИД, саркома Капоши, PGL СПИД, ARC Саркома Капоши, в комбинации с AZT: СПИД СПИД, ARC Саркома Капоши Amgen (Thousand Oaks, CA) СПИД, в комбинации с AZT Biogen (Cambridge, MA) СПИД, ARC Genetech (S. San Francisco, CA) Hoffman-La Roche (Nutley, NJ) Smith, Kline & French Laborat. (Philadelphia, PA) Immunobiology Research Institute (Annandale, NJ) Фактор некроза опухоли TNF Genentech (S. San Francisco, CA) Клиндамицин с примахином Противоинфекционные средства Upjohn (Kalamazoo, MI) Флуконазол Pfizer (New York, NY) Таблетки нистатина Squibb Corp. (Princeton, NJ) Орнидил Эфлорнитин Merrell Dow (Cincinnati, OH) 14 Саркома Капоши, СПИД, ARC, в комбинации с AZT. ВИЧ-инфекция ВИЧ-инфекция ARC, в комбинации с гаммаинтерфероном РСР (пневмоцистная пневмония) Криптококковый менингит, кандидоз Предупреждение кандидоза слизистой оболочки рта РСР 42699 Продолжение табл. III Пентамидин Изетионат (IM&IV) Пиритрексим Пентамидина изетионат для ингаляций Спирамицин LyphoMed (Rosemont, IL) Burroughs Wellcome (Rsch. Triangle Park, NC) Fisons Corporation (Bedford, MA) Rhone-Poulenc Pharmaceuticals (Princeton, NJ) Интраконазол-Р51211 Janssen Pharm. (Piscataway, NJ) Триметрексат Warner-Lambert Другие средства Рекомбинантный чел. эритропоэтин Мегестрола ацетат Полное энтеральное питание Ortho Pharm. Corp. (Raritan, NJ) Bristol-Myers (New York, NJ) Norwich Eaton Pharmaceuticals (Norwich, NY) Лечение РСР Лечение РСР Профилактика РСР Криптоспоридальная диарея Гистоплазмоз; криптококковый менингит РСР Тяжелая форма анемии, связанной с АТ-терапией Лечение СПИД-ассоциированной анорексии Диарея и СПИД-ассоциированная малабсорбция ридин-2(1Н)-он; соединение L-696229 представляет собой 3-[2-(1,3-бекзоксазол-2-ил)-этил]-5-этил6-метил-пиридин-2(1Н)-он. Синтез соединений L-697661 и L-696229 описан в ЕРО 484071 и ЕPO 462800 (эти два патента вводятся в настоящее описание в виде ссылки). Предпочтительными комбинациями является одновременное, периодическое или поочередное введение L-743726 в сочетании с ингибитором ВИЧ-протеазы или без него. Необязательным третьим компонентом в данной комбинации является нуклеозидный ингибитор обратной транскриптазы ВИЧ, такой, как АZТ, ddC или dd1. Предпочтительным ингибитором ВИЧ является соединение L-735524. Другим предпочтительным ингибитором обратной транскриптазы ВИЧ является L-697661. Указанные комбинации могут обладать синергическим действием, ограничивающим размножение ВИЧ. При этом, предпочтительными являются следующие комбинации: (1) L-743.726 с L-735.524 и необязательно с любым из L-697.661, АZT, dd1 или ddC; (2) L-743.726 с любым из L-697.661, АZT, dd1 или ddC. В объем настоящего изобретения входят также фармацевтически приемлемые соли указанных комбинаций. Пример 1 (+/-) 4-(1,1,1-Трифторметил)-4-(1-бутен-4-ил)6-хлор-1,4-дигидро-2Н-3,1-бензокоазин-2-он (соединение 15). Стадия А: N-(4-Хлорфенил)-2,2-диметилпропанамид. В 5-литровую трехгорлую круглодонную колбу, снабженную головной мешалкой, добавляли 4-хлоранилин (127,57 г, 1 М), 1200 мл CНCI3, и 1200 мл насыщенного водного раствора Nа2СО3. Капельную воронку подсоединяли к колбе и загружали 2,2-диметилпропаноилхлорид (129 мл, 1,05 М). После этого к энергично перемешанной смеси в течение часа по капле добавляли хлорангидрид. Затем полученную смесь перемешивали при комнатной температуре еще 23 часа. Некоторая часть продукта выделялась в виде белых кристаллов. Эти кристаллы собирали путем фильтрации. Фильтрат переносили в делительную воронку, а слои отделяли. После этого слой хлороформа промывали водой и солевым раствором. После Следует отметить, что объем настоящего изобретения не ограничивается лишь комбинациями соединений настоящего изобретения со средствами против СПИДа, иммуномодуляторами, противоинфекционными средствами или вакцинами, перечисленными в вышепривиденной таблице. В объем настоящего изобретения могут быть включены, в принципе, любые комбинации с любой фармацевтической композицией, используемой для лечения СПИДа. Например, соединение формулы I или формулы II могут быть с успехом введены в комбинации с нуклеозидным аналогом, обладающим биологической активностью против обратной транскриптазы ВИЧ. Подходящими нуклеозидными аналогами обычно являются терминаторы цепи, например, AZT, ddC, dd1, D4T, HEPT, и 3'-фтор-2',3'-дидезокситимидин. AZT может быть синтезирован по методам J.P. Horwitz и др., J. Org. Chem. 29, 2076 (1964); R. P. Glinski и др. J. Org. Chem. 38, 4299 (1973); C. K. Chu и др., Tetrahedron Letters 29, 5349 (1988). Применение АZT в качестве терапевтического средства для лечения СПИДа раскрывается в патенте США 4724232. Соединение ddC может быть синтезировано по методам J.P. Horwitz и др., J. Org. Сhem. 32, 817 (1967); P. Marumoto, Сhem. Pharm. Bull. 22,, 128 (1974); и T.-S. Lin и др., J. Med. Chem. 30, 440 (1987). D4T может быть синтезирован методами Herdewijn, P. и др., J. Med. Chem. 30, 1270 (1987). HEPT может быть синтезирован методами Miyasaka Т. и др. J. Med. Chem. 32, 2507 (1989); и A. Rosowsky, J. Med. Chem. 24, 1177 (1981). Синтез ddC, dd1. и AZT также описан в ЕРО 484071. Соединение 3'-фтор-2',3'-дидезокситимидин может быть синтезирован в соответствии с процедурами, описанным Herdewijn, Р. и др., J. Med. Chem. 30, 1270 (1987). Соединение L-735524 представляет собой N-(2(R)-гидрокси-1(s)-инданил)-2(R)-фенилметил-4-(s)-гидрокси-5-(1-(4-(3-пиридилмэтил)-2(s)N-(т-бутилкарбоксамидо)-пиперазинил))-пентанамид, или его фармацевтически приемлемая соль. Соединение L-697661 или '661' представляет собой 3-([4,7-дихлор-1,3-бензоксазол-2-ил) метил]-амино)-5-этил-этил-6-метил-пи 15 42699 смесь оставляли на 30 мин, а затем охлаждали до 0°С в ледяной бане. К тщательно перемешанному раствору в течение 30 минут по капле добавляли раствор 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанона (5,00 г, в тетрагидрофуране (35 мл). Охлаждающую баню оставляли для испарения, а смесь перемешивали в течение 20 часов при комнатной температуре. Затем полученную реакционную смесь разводили етилацетатом и 10%-ным водным раствором лимонной кислоты. После этого смесь перемешивали в течение 4 часов. Слои отделяли, а органическую фазу промывали водным раствором гидрокарбоната натрия и солевым раствором. После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме, и хроматографировании на силикагеле (300 г) (элюент: 15% EtОАс в гексане) получали 4,80 г (+/-) 2(2-амино-5-хлорфенил)-1,1,1-трифтор-5-гексаен-2ола в виде желтого твердого вещества. Стадия D: (+/-)4-(1,1,1-трифторметил)-4-(1-бутен-4-ил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксалин-2-он. В 200-миллилитровую круглодонную колбу, снабженную стержнем для перемешивания, отверстием для впуска аргона, и парциальным конденсатором горячего орошения добавляли (+/-) 2-(2-амино-5-хлорфенил)-1,1,1-трифтор-5-гексен2-ол (4,80 г, 17,16 мМ), 1,1’-карбонилдиимидазол (13,91 г, 85,81 мМ) и безводный тетрагидрофуран (75 мл). Затем эту смесь нагревали при 60°С в течение 18 часов. Охлажденную реакционную смесь разводили этилацетатом, а затем промывали водой (3х200 мл) и солевым раствором (250 мл). После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме, и перекристаллизации из кипящего ЕtОАс/-гексана получали 3,22 г (+/-) 4-(1,1,1-трифторметил)-4-(1-бутен-4-ил)-6хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-она в виде белого кристаллического твердого вещества, т.лл. 165-166°С. 1Н-ЯМР (СDСІ3): 1,99 (м, 1Н), 2,09-2,40 (м, 3Н), 5,00 (д, 1Н, J=1,4 Гц), 5,03 (дд, 1H, J=1,4, 7,9). 5,78 (м, 1Н), 6,85 (д, 1H, J=8,6 Гц). 7,21 (шир.с., 1Н), 7,35 (дд, J=2,2, 8,6 Гц), 9,63 (шир.с., 1H). Пример 2 (+/-) 6-Хлор-4-этинил-4-(1,1,1-трифторметил)1,4-дигидро-2Н-3,1-бензоксаяин-2-он (соединение 26). Стадия А: 2-(2-Амино-5-хлорфенил)-1,1,1-трифтор-3-бутин-2-ол. 500-миллилитровую трехгорлую круглодонную колбу, снабженную капельной воронкой, отверстием для впуска аргона, стержнем для перемешивания, и цифровым термометром, загружали этинилмагнийбромидом (0,5 М в гексане, 268 мл, 134 мМ), а затем охлаждали до -78°С. Затем по капле в течение 15 минут добавляли раствор 1-(2амино-5-хлорфенил)-2,2,2-трифторэтанона (6,0 г, 26,8 мМ) в 50 мл ТГФ, поддерживая при этом температуру £ -55°С. После этого реакционную смесь перемешивали в течение 16 часов, а затем медленно нагревали до комнатной температуры. Темно-красный реакционный раствор гасили при -5°С путем добавления по капле насыщенного водного раствора хлорида аммония (60 мл). После экстрагирования этилацетатом, раствор промывали 10%-ной лимонной кислотой, насыщенным бикар осушки сульфатом магния, фильтрации, и удаления растворителя в вакууме получали дополнительный продукт. Две части этого продукта объединяли и перекристаллизовывали из кипящей смеси EtOAc/-гекоана, в результате чего получали 185,6 г N-(4-xлopфенил)-2,2-диметилпропанамида в виде белого кристаллического твердого вещества. Стадия В: 1-(2-Амино-5-хлорфенил)-2,2,2-трифторэтанон. В 3-литровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную головной мешалкой и отверстием для впуска аргона, и в 500-миллилитровую капельную воронку, осушенную в сушильном шкафу, добавляли N-(4-хлорфенил)-2,2-диметилпропанамид (100 г, 472 мМ) и 1 л безводного тетрагидрофурана. Этот раствор охлаждали в ледяной бане до 0°С, а в капельную воронку загружали н-бутиллитий (387 мл 2,5 М-раотвора в гексане, 968 мМ). После этого, медленно, в течение часа н-бутилдитиевый раствор по капле добавляли к амидному раствору, поддерживая при этом температуре ниже +5°С. После этого полученный раствор хранили при 0°С в течение часа, и в этот период времени образовывался оранжевый осадок. К полученной смеси по капле в течение 1 часа добавляли этил 1,1,1трифторацетат (115 мл, 968 мМ). Образовавшийся прозрачный раствор выдерживали еще 30 минут. Реакционную смесь гасили 5-ным водной соляной кислотой. После этого смесь разводили этилацетатом (1 л), а слои отделяли. Органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 160 г желтого маслообразного вещества. Это вещество суспендировали в 1 л 3 н. водного раствора соляной кислоты, и раствор нагревали с обратным холодильником в течение 24 часов. Затем охлажденный раствор разводили этилацетатом (1 л), и полученную смесь подщелачивали путем добавления концентрированной NН4ОН. Слои отделяли, а органическую фазу промывали солевым раствором и осушили сульфатом магния, после чего фильтровали, концентрировали в вакууме, и хроматографировали на силикагеле (1,5 кг), элюируя 15-ным EtOАс в гексане. Отхромато-графированный материал перекристаллизовывали из кипящего гексана, и получали 57 г (54%) чистого 1-(2-амино-5-хлорфенил)-2,2,2-трифторэтанола в виде ярко-желтых кристаллов, т.пл. 91-92°C. 1 Н-ЯМР(CDCH3): d 6,46 (шир.с.,2Н), 6,69 (д, 1H, J=9,2 Гц), 7,32 (дд, 1H, J=2,4, 9,2 Гц), 7,70 (д, 1Н, J=2,4 Гц). Стадия С: (+/-) 2-(2-Амино-5-хлорфенил)-1,1,1трифтор-5-гексан-2-ол. В 300-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную стержнем для перемешивания, отверстием для впуска аргона, капельной воронкой и конденсатором горячего орошения, добавляли магниевую стружку (3,03 г, 125 мМ) и 75 мл безводного тетрагидрофурана. К этой тщательно перемешанной смеси добавляли 4-бромо-1-бутен (12,0 мл, 118,21 мМ), таким образом, чтобы осуществлялось мягкое нагревание с обратным холодильником. После завершения добавления, 16 42699 бонатом натрия, водой и солевым раствором. В результате этой процедуры получали 8,5 г неочищенного продукта, который осушали сульфатом натрия, фильтровали, и выпаривали. После очистки с помощью флешхроматографии (элюент: 1520% этилацетат/гексан) получали чистый 2-(2амино-5-хлорфенил)-1,1,1-трифтор-3-бутин-2-ол (5 г светло-коричневого маслообразного вещества, выход 75%). Стадия В: (+/-) 6-Хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-он. Тетрагидрофурановый раствор 2-(2-амино-5хлорфенил)-1,1,1-трифтор-3-бутин-2-ола (5,0 г, 20,0 мМ в 225 мМ тетрагидрофурана) обрабатывали 1,1'-карбонилдиимидазолом (13,0 г, 80,0 мМ) и нагревали в масляной бане в течение 17 часов при 60°С. После удаления тетрагидрофурана в вакууме, остаток растворяли в этилацетате, а затем промывали 10% лимонной кислотой, бикарбонатом натрия, водой и солевым раствором После осушки сульфатом натрия, фильтрации и выпаривания в вакууме, образовавшийся неочищенный продукт выделяли (3,6 г) и перекристаллизовывали из этилацетата/гексана. Таким образом, получали (+/-) 6-хлор-4-этинил-4-(1,1,1-трифторметил)1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белого кристаллического твердого вещества (3,22 г, 1 выход 58,4%), т.пл. 226-227°С. Н-ЯМР (CDCI3+следовые количества ДМСО): d 3,16 (с, 1Н), 6,98 (д, J=3,3 Гц, 1H), 7,35 (м, 1Н), 7,51 (c, 1H), 10,66 (c, 1H). Пример 3 (+/-) 6-Хлор-4-(1,1,1-трифторметил)-4-/(3-(1-пирролидинил))-1-пропинил/-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 7). Диоксановый раствор (+/-) 6-хлор-4-этинил-4(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-она (150 мг, 0,544 мМ), пирролидина (52,2 мкл, 0,626 мМ), параформальдегида (20,5 мг, 0,681 мМ), уксусной кислоты (31,1 мкл, 0,544 мм), и хлорида меди (I) (20,5 мг, 0,207 мм в 3,5 мл диоксана) нагревали до 50°С в масляной бане, приблизительно в течение 2 часов. Затем реакционную смесь гасили в 2 н. соляной кислоте и экстрагировали этилацетатом. После этого водный слой нейтрализовали путем добавления твердого карбоната калия, и три раза экстрагировали этилацетатом. Объединенные экстракты промывали водой и солевым раствором, после чего осушали сульфатом натрия, и получали 140 мг неочищенного продукта. Этот неочищенный продукт очищали с помощью хроматографии на силикагеле и перекристаллизовывали из этилацетата/гексана, в результате чего получали кристаллический (+/-) 6хлор-4-(1,1,1-трифторметил)-4-/(3-(1-пирролидинил))-1-пропинил/-1,4-дигидро-2Н-3,1-бензоксазин2-он (89 мг, выход 46), т.пл. 160-161°С (при разлож.). 1 H-ЯМP (СDСІ3): d 1,85-1,89 (м, 4Н), 2,68-2,71 (м, 4Н), 3,67 (c, 1H), 6,88 (д, J=8,55 Гц, 1H), 7,40 (дд, J=2,19, 8,54 Гц, 1H), 7,55 (c, 1H), 9,45 (c, 1H). (+/-) 6-Хлор-4-(2-цианофенил)этинил-4-(1,1,1трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2он (соединение 2). Раствор 6-хлор-4-этинил-4-(1,1,1-трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2-она (138 мг, 0,5 мМ) в 3 мл безводного тетрагидрофу рана перемешивали при -78°С. Затем к этому раствору добавляли 0,4 мл (1,0 мМ) н-бутиллития (2,5 М в гексане). Анион выдерживали в течение 10 минут при -78°С после чего добавляли 1 млраствор ZnCl2 (1 M в эфире). Полученную реакционную смесь оставляли для перемешивания при -78°С на 15 минут, после чего ледяную баню удаляли, и смесь медленно нагревали до 0°С в течение 30 минут. Затем к этой реакционной смеси добавляли раствор 2-иодобензонитрила (149 мг, 0,65 мМ) в 2 мл ТГФ, после чего добавляли 56 мг (0,05 мМ) тетракис(трифенилфосфин) палладия (0). Полученную реакционную смесь оставляли для нагревания до комнатной температуры и продолжали перемешивание в течение 15 часов. Затем эту реакционную смесь гасили путем добавления 10 мл 2 н. соляной кислоты, экстрагировали этилацетатом (2х200 мл), а объединенные экстракты промывали водой, солевым раствором, и осушали сульфатом магния. Растворитель удаляли и получали 195 мг маслообразного вещества, которое очищали с помощью флеш-хроматографии на силикагеле (элюент: 20% ЕtОАс в гексане). В результате этой процедуры получали 60 мг непрореагировавшего исходного материала и 35 мг объединенного продукта. Этот объединенный продукт растирали с эфиром, и получали 25 мг (+/-) 6-хлор-4-(2-цианофенил)этинил-4-(1,1,1трифторметил)-1,4-дигидро-2Н-3,1-бензоксазин-2она, т.пл. 245-246°С. PAB-MС (М++): 377 m/е. 1НЯМР (CDCI3): d 6,82-6,85 (д, J=8,5 Гц, 1Н), 7,407,44 (дд, J=2,1, 8,5 Гц, 1н), 7,56-7,79 (м, 5Н), 8,00 (c, 1H). Пример 4 (+/-) 4-(1-Хлор-1,1-дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2он (соединение 5). Стадия А: 1-(2-Амино-5-хлорфенил-)-2-хлор2,2-дифторэтанон. В 300-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную магнитным стержнем для перемешивания, отверстием для впуска азота, и в 100-миллилитровую капельную воронку, осушенную в сушильном шкафу, добавляли N-(4-хлорфенил)-2,2-диметилпропанамид (10 г, 47,2 мМ) и 100 мл безводного тетрагидрофурана. Этот раствор охлаждали в ледяной бане до 0°С, а в капельную воронку загружали н-бутиллитий (38,7 мл 2,5 М-раствора в гексане, 96,8 мМ). Затем н-бутиллитиевый раствор медленно, по капле и в течение 1 часа добавляли к амидному раствору, поддерживания при этом температуру ниже +5°С. Полученный раствор хранили в течение часа при 0°С. В течение этого времени образовывался оранжевый осадок. После этого к смеси, по капле в течение 15 минут добавляли этил 1-хлор-1,1-дифторацетат (10,2 мл, 96,8 мМ). Подученный прозрачный раствор выдерживали еще в течение 30 минут. Реакцию завершали путем добавления 5%-ного водного раствора соляной кислоты. Смесь разводили этилацетатом (1 л), а слои отделяли. Органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали и концентрировали в вакууме о получением 160 г желтого маслообразного вещества. Это вещество суспендировали в 200 мл 3 н. водного раствора соляной 17 42699 ля (элюент: 20% этилацетат в гексане). Прохроматографированный материал перекристаллизовывали из кипящего этилацетата/гексана, в результате чего получали 507 мг (58) (+/-) 4-(1-хлор-1,1дифторметил)-4-(2-фенилэтинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-она в виде белых игольчатых кристаллов, т.пл. 154-155°С. 1 Н-ЯМР (CDCI3): d 6,89 (д, 1Н), J=8,4 Гц), 7,357,48 (М, 4Н), 7,56 (м, 2Н), 7,64 (шир.c. 1Н), 9,19 (шир.с. 1Н). Пример 5 (-)6-Хлор-4-фенилэтинил-4-трифторметил-1,4дигидро-2Н-3,1-бензоксазин-2-он (соединение 4). Стадия А: 2-(2-Амино-5-хлорфенил)-4-фенил1,1,1-трифтор-3-бутин-2-ол. Раствор литиофенилацетилида, полученного из 4,83 мл фенилацетилена (0,044 М) и 17,2 мл 2,5 н. растворы н-бутиллития в гексане (0,043 М) в 50 мл ТГФ, при -78°С и в течение 5 минут обрабатывали 11,4 граммами (0,044 М) этерата магнийбромида. Полученную смесь оставляли для нагревания до -20°С, а затем перемешивали в атмосфере аргона в течение 30 минут. После этого смесь охлаждали до -60°С, и добавляли раствор, содержащий 2,5 г (0,011 М) 1-(2-амино-5хлор)-2,2,2-трифторметилэтанона, предварительно объединенного с эквивалентом (2,8 г, 0,011 М) этерата магнийбромида в 25 мл ТГФ. Полученную реакционную смесь оставляли для перемешивания на 1 час при 15°С, после чего охлаждали до 0°С и обрабатывали по капле смесью насыщенного водного раствора хлорида аммония (30 мл) и воды (30 мл). Полученную смесь экстрагировали двумя порциями этилового эфира, а объединенные органические фазы промывали солевым растворов (100 мл) и осушали сульфатом магния. В результате удаления осушающего агента и растворителей оставалось 6 г маслообразного вещества, которое хроматографировали с помощью флеш-хроматографии на силикагеле (элюент: 20% ЕtОАс в гексане). Таким образом, получали 2-(2-амино-5-хлорфенил)-4-фенил-1,1,1-трифтор-3-бутин-2-ол. 1 Н-ЯМР (CDCI3): d 4,63 (шир.с., 3Н), 6,69 (д, J=8,5 Гц, 1Н), 7,15 (д, J=2 Гц, 1Н), 7,17 (д, J=2 Гц, 1H), 7,35-7,44 (м, ЗH), 7,53-7,56 (м, 2H), 7,66 (д, J=2 Гц, 1Н). FАВ-MC: M+H=326 m/e. Стадия В: (+/-) 6-Хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 12). Раствор 2-(2-амино-5-хлорфенил)-4-фенил1,1,1-трифтор-3-бутин-2-ола (2,0 г, 6,1 мМ) и 11,0 г (12,0 мМ) 1,1-карбонилдиимидазола в 300 мл безводного тетрагидрофурана перемешивали в атмосфере аргона, при 55°C, в течение 24 часов. Растворитель удаляли на роторном испарителе, и полученный остаток распределяли между эфиром (200 мл) и водой (400 мл). Слои отделяли, а водный слой экстрагировали еще раз эфиром. После этого объединенные эфирные экстракты промывали 10%-ной лимонной кислотой (2х200 мл), а затем солевым раствором, и осушали сульфатом магния. После удаления осушающего агента и растворителя получали 1,5 г (70%) неочищенного целевого соединения в виде маслообразного вещества. В результате растирания с эфиром/гексаном получали 875 мг (+/-) 6-хлор-4фенилэтинил кислоты, и раствор нагревали с обратным холодильником в течение 24 часов. Охлажденный раствор разводили 500 миллилитрами этилацетата, и смесь подщелачивали путем добавления концентрированного NН4ОН. После этого слои разделяли, а органическую фазу промывали солевым раствором, осушали сульфатом магния, фильтровали, концентрировали в вакууме, и хроматографировали на силикагеле (350 г, элюент: 15% ЕtОАс в гексане). Отхроматографированный материал перекристаллизовывали из кипящего гексана, в результате чего получали 5,5 г чистого 1-(2-амино-5хлорфенил)-2-хлор-2,2-дифторэтанола в виде ярко-желтых кристаллов, т.пл. 55-56°С. 1Н-ЯМР (CDCI3): d 6,43 (шир.с., 2Н), 6,69 (д, 1Н, J=9,0 Гц), 7,31 (дд, 1H, J=2,4, 9,0 Гц), 7,80 (д, 1Н, J=2,4 Гц). Стадия В: (+/-) 2-(2-Амино-5-хлорфенил)-4фенил-1-хлор-1,1-дифтор-З-бутин-2-ол. В 100-миллилитровую трехгорлую круглодонную колбу, осушенную в сушильном шкафу и снабженную стержнем для перемешивания, отверстием для впуска аргона, парциальным конденсатором горячего орошения, и мембранной добавляли этинилбензол (2,13 г, 20,83 мМ), безводный тетрагидрофуран (50 мл) и этилмагнийбромид (6,94 мл 3,0 М раствора в эфире). Полученную смесь выдерживали в течение 2 часов при комнатной температуре, а затем, с помощью щирица добавляли раствор 1-(2-амино-5-хлорфенил)-2-хлор-2,2-дифторэтанона (1,00 г, 4,17 мМ) в тетрагидрофуране (6 мл). Полученный оранжевокрасный раствор перемешивали при комнатной температуре в течение 21,5 часа. Реакцию завершали путем добавления 1 н. соляной кислоты (50 мл), а затем смесь разводили этилацетатом. Полученный раствор подщелачивали путем добавления концентрированного NН4ОН, а слои отделяли. Органическую фазу промывали водой и солевым раствором. После осушки сульфатом магния, фильтрации, удаления растворителя в вакууме, и хроматографирования на силикагеле (элюент: 20% ЕОАс в гексане) получали (+/-) 2-(2амино-5-хлор-фенил)-4-фенил-1-хлор-1,1-дифтор3-бутин-2-ол (1,02 г) в виде беловатого твердого вещества. 1 Н-ЯМР (CDCI3): d 4,42 (шир.с., 2Н), 5,10 (шир.с., 1H), 6,65 (д, 1Н, J=8,5 Гц), 7,15 (дд, 1H, J=2,4, 8,5 Гц), 7,38 (м, 3Н), 7,55 (м, 2Н), 7,70 (д, J=2,4 Гц). Стадия С: (+/-) 4-(1-хлор-1,1-дифторфенил)-4(2-фенил-этинил)-6-хлор-1,4-дигидро-2Н-3,1-бензоксазин-2-он. В 100-миллилитровую круглодонную колбу, снабженную стержнем для перемешивания, парциальным конденсатором горячего орошения, и отверстием для впуска аргона, добавляли (+/-) 2(2-амино-5-хлорфенил)-4-фенил-1-хлор-1,1-дифтор-3-бутин-2-ол (0,81 г, 2,37 мМ), безводный тетрагидрофуран (25 мл), и 1,1'-карбонилдиимидазол (1,919 г, 11,84 мМ). Этот раствор нагревали в течение 20 часов при 60°С. Охлажденную реакционную смесь разводили этилацетатом, а затем промывали 0,5 н. соляной кислотой, водой, и солевым раствором. После осушки сульфатом магния, фильтрования и удаления растворителя в вакууме получали 890 мг маслообразного вещества. Это вещество хроматографировали на 80 г силикаге 18 42699 в CHCI3). Полученный раствор концентрировали в вакууме, а остаток растворяли в эфире. После промывания 0,1 н соляной кислотой и солевым раствором, эфирный раствор осушали сульфатом магния, фильтровали и выпаривали в вакууме с получением маслообразного твердого вещества, которое очишали с помощью хроматографии на двуокиси кремния (элюент: 5% 2-пропанол в гексане). После растирания о эфиром/гексаном получали (-) 6-хлор-4-фенил-этинил-4-трифторметил1,4-дигидро-2Н-3,1-бензо-ксазин-2-он в виде белых игольчатых кристаллов с т.пл. 178-179°С. [a]D20=-92,5° (СНСІ3, с=0,0012 г мл-1). 1 Н-ЯМР (CDCI3): d 6,87 (д, J=8,5 Гц, 1H), 7,377,50 (м, 4Н), 7,56-7,63 (м, 3Н), 8,60 (c, 1H). Стадия Е: (+) 6-Хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (соединение 3). В соответствии со способом, описанным в стадии D, (+)-6-хлор-4-фенилэтинил-4-трифтор-метил-1,4-дигидро-2Н-3,1-бензоксазин-2-он получали из некристаллического продукта стадии с (диастереомера I). Температура плавления полученного продукта составляла 178-179°С. [a]D20=+87,6° (СНСІ3, с=0,0050 г мл-1). 1 Н-ЯМР (CDCI3): d 6,87 (д, J=8,5 Гц, 1H), 7,377,50 (м, 4Н), 7,56-7,63 (м, 3Н), 8,60 (с, 1Н). Пример 6 (-) 6-Хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (L 743,726, соединение 37,2) и (+) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н3,1-бензоксазин-2-он (L 743,725). Стадия А: 2-(2-Амино-5-хлорфенил)-4-циклопропил-1,1,1-трифтор-3-бутин-2-ол. Раствор бромомагнийциклопропилацетилида получали из 23 г циклопропилацетилена (0,348 М) в 240 мл тетрагидрофурана путем добавления по капле в течение часа 116 мл 3,0 М раствора этилмагнийбромида в эфире (0,348 М). Полученный раствор поддерживали при 0°С в течение часа, а затем раствор поддерживали при 40°С в течение 3 часов. К полученному раствору, охлажденному до 0°С, порциями, в течение 5 минут добавляли 15,66 г твердого 1-(2-амино-5-хлорфенил)-2,2,2трифторметилтанола (0,0696 М). После этого реакционную смесь оставляли для перемешивания при 0°С в течение полутора часов. Реакцию завершали при 0°С путем добавления по капле насыщенного водного раствора хлорида аммония (700 мл). Полученную смесь дважды экстрагировали этилацетатом (400 мл), а объединенные органические фазы промывали солевым раствором и осушали сульфатом магния. После удаления осушающего агента и растворителей образовывалось желтое твердое вещество. Это вещество перекристаллизовывали из кипящего гексана (конечный объем=100 мл) и получали 14,67 г 2-(2-амино-5-хлорфенил)-4-циклопропил-1,1,1-трифтор-3бутин-2-ола, т.пл. 153-154°С. Второй сбор (2,1 г) продукта был получен из концентрированных маточных растворов. 1 Н-ЯМР (CDCI3): d 0,84 (м, 2Н), 0,90 (м, 2Н), 1,38 (м, 1Н), 4,50 (шир.с., 3Н), 6,69 (д, J=8,5 Гц, 1H), 7,13 (дд, J=2,5, 8,5 Гц, 1H), 7;55 (д. J=2,5 Гц, 1H). 4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин2-она в виде белого твердого вещества, которое размягчается при 137°С и становится прозрачным при 147°С. 1 Н-ЯМР (CDCI3): d 6,92 (д, J=8 Гц, 1H). 7,307,49 (м, 4Н), 7,58-7,65 (М, 3Н), 8,99 (с, 1Н). Стадия С: 6-Хлор-1-(1S)-камфаноил-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он. К раствору, перемешанному в атмосфере аргона и в ледяной бане, и содержащему (+/-) 6хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро2Н-3,1-бензоксазин-2-он (2,24 г, 6,37 мМ), 4-диметиламинопиридин (0,10 г, 0,8 мМ), и хлорид (-)-камфановой кислоты (2,07 г, 9,55 мМ) в 60 мл безводного дихлорметана, добавляли триэтиламин (2,22 мл, 15,9 мМ). Охлажденную баню удаляли, и реакционную смесь оставляли при комнатной температуре для продолжения реакции. После того, как реакция была полностью завершена, о чем свидетельствовала тонкослойная хроматография (SiO2, 4% EtOAc в CHCI3), раствор разводили 200 миллилитрам CHCI3, а затем промывали 10% лимонной кислотой (дважды), и солевым раствором. После осушки сульфатом магния, растворитель выпаривали на роторном испарителе, и образовавшийся пенообразный остаток подвергали флеш-хроматографии, элюируя хлороформом. Таким образом, получали 575 мг диастереомера (I) 6-хлор-(1S)-камфаноил-4-фенилэтинил-4-трифторметпл-1,4-дигидро-2Н-3,1-бензоксазин-2-она в виде маслообразного вещества. 1 Н-ЯМР (CDCI3): d 0,85 (с, 3Н), 1,08 (с, 3Н), 1,22 (с, 3Н), 1,73-1,85 (м, 1Н), 1,92-2,08 (м, 1Н), 2,50-2,67 (м, 2Н), 7,30-7,69 (м, 8Н). После этого получали 1,52 г смешанных фракций (диастереомеров I и II). Затем продолжали элюирование и получали 680 мг более медленно движущегося диастереомера II целевого соединения, которые растирали с эфиром/гексаном, и получали комкообразное вещество в виде белых игольчатых кристаллов, т.пл. 177-178,5°С. 1 Н-ЯМР (CDCI3): d 0,83 (c, 3H), 1,12 (с, 3H), 1,23 (c, 3H), 1,73-1,86 (м, 1Н), 1,93-2,06 (м, 1Н), 2,50-2,63 (м, 2Н), 7,38-7,51( м, 4Н), 7,49-7,62 (м, 2Н), 7,72 (д, J=9 Гц, 1H), 7,76 (д, J=2 Гц, 1H). 1,52 г изомерной смеси, полученной в результате флеш-хроматографии, растворяли в 75 мл эфира, и полученный раствор разводили 50 миллилитрами гексана, а затем в этот раствор в качестве затравки добавляли кристалл изомера II. После медленной кристаллизации получали еще 385 мг изомера II, который перекристаллизовывали из эфира/гексана, и получали диастереомерный материал со степенью чистоты >96% (ЖХBPанализ). Стадия D: (-) 6-Хлор-4-фенилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он. Кристаллический диастереомер (II) 6-хлор-1(1)-камфаноил-4-фенилэтинил-4-трифторметил1,2-дигидро-4(Н)-3,1-бензоксазин-2-она (53 мг, 0,10 мМ) растворяли в 8 миллилитрах 2-пропанола при 45°С и в атмосфере аргона. К этому раствору добавляли 0,27 мл 10% водного раствора К2СО3. После 10-минутного перемешивания, исходный материал был полностью израсходован, о чем свидетельствовала ТСХ (SiО2, 4%-ный EtOАс 19 42699 Стадия D: (-) 6-Хлор-4-циклопропилэтинил-4трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2он (L-743,726) (соединение 37,2). В атмосфере аргона, 6-хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифтор-метил-1,2-дигидро-4(Н)-3,1-бензоксазин-2-он (7,50 г, 0,01512 М) растворяли в 150 мл н-бутанола при 60°С. К этому раствору добавляли 10 мл 1 н. соляной кислоты. После этого раствор выдерживали при 60°С в течение 72 часов. Затем смесь нейтрализовали путем добавления водного раствора гидрокарбоната натрия, и инбутанол удаляли в вакууме. Образовавшийся остаток растворяли в 150 мл тетрагидрофурана и обрабатывали при комнатной температуре в течение 3 часов 50 миллилитрами 2 н. LiОН. Полученную смесь разводили этилацетатом и промывали двумя порциями воды и одной порцией солевого раствора. После осушки сульфатом магния, фильтрации, и удаления растворителя в вакууме получали белое твердое вещество. Это вещество перекристаллизовывали из горячего гексана, и получали (-) 6-хлор-4-циклопропилэтинил4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин2-он (3,43 г) в виде белых кристаллов, т.пл. 131132°C. [a]20D=-84,7° (СНСІ3, с=0,005 г мл-1). 1 Н-ЯМР (CDCI3): d 0,85 (м, 2Н), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J=8,5 Гц, 1H), 7,37 (дд, J=2,5, 8,5 Гц, 1H), 7,43 (д, J=2,5 Гц, 1H), 8,87 (шиpгc. 1H). Стадия Е: (+) 6-Хлор-4-циклопропилэтинил-4трифторметил-1,4-дигидро-2Н-3,1-бензокоазин-2он (L-743,725). Маточные растворы, полученные в вышеописанной стадии С, очищали с помощью колоночной хроматографии на силикагеле (элюент: 10% етилацетат в гексане). Чистый, нежелательный диастереомер (бесцветная пена) гидролизовали в соответствии с вышеописанной стадией D. Таким образом, был получен энантиомерный бензоксазинон, (+) 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белых кристаллов, т.пл. 131-132°C. [a]20D =+84,4° (CHCI3, с=0,005 г мл-1). 1 Н-ЯМР (CDCI3) d: 0,85 (м, 2Н), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J=8,5 Гц, 1H), 7,37 (дд, J=2,5, 8,5 Гц, 1H), 7,49 (д, J=2,5 Гц, 1H), 8,87 (шир.с., 1Н). Анализ обратной транскриптазы. В это анализе измеряли включение с помощью рекомбинантной обратной транскритазы ВИЧ (ВИЧ-RТR) (или другой RT) меченного тритием дезоксигуанозин-монофосфата в осажденную кислотой кДНК при определенных значениях Кm dGTP и роІуr(С) олигоd(G)12-18. Это включение ингибируется ингибитором настоящего изобретения. Этот анализ проводили в 55 мМ Триса (рH 8,2), 30 мМ КСІ, 30 мм MgСІ2, 1 мМ дитиотреитола, 20 мкг rC:dG12-18 (Pharmacia)/мл, 8 мМ/3Н/dGTP (New England Nuclear) 0,01% Тритона Х-100, 50 мМ этиленгликоль-бис (b-амино-этил-эфир)N,N,N,N-тетрауксусной кислоты (EGTA), 1 мг альбумина бычьей сыворотки/мл. После 60-минутного инкубирования при 37°С, осаждаемый кислотой материал собирали на стекловолоконные фильтры с помощью полуавтоматического харвестера клеток. Бактериальные клеточные экстракты, содержащие RТ, разводили до концентраций в пределах линейного диапазона анализа, после чего определяли активность в присутствии или отсут Стадия В: (+/-) 6-Хлор-4-циклопропилэтинил-4трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2он (L-741,211). Раствор 2-(2-амино-5-хлорфенил)-4-циклопро-пил-1,1,1-трифтор-3-бутин-2-ола (15,00 г, 0,0518 М) и 41,98 г (0,259 М (1,1'-карбонилдиимидазола в 250 мл безводного тетрагидрофурана перемешивали в атмосфере аргона, в течение 24 часов, при 55°С. Растворитель удаляли на роторном испарителе, и образовавшийся остаток распределяли между 500 мл этилацетата и 400 мл воды. Затем слои отделяли, а водную фазу еще раз экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали 2%-ным водным раствором соляной кислоты (2х200 мл), насыщенным водным раствором гидрокарбоната натрия, и солевым раствором. После осушки сульфатом магния, фильтрации, и удаления растворителя в вакууме получали 16,42 г целевого соединения в виде твердого вещества. Это вещество перекристаллизовывали из этилацета/-гексана, в результате чего получали аналитически чистый (+/-) 6-хлор-циклопропилэтинил-4-трифтор-метил1,4-дигидро-2Н-3,1-бензоксазин-2-он в виде белых кристаллов (12,97 г). т.пл. 178-180°С. 1 Н-ЯМР (CDCI3): d 0,85 (м, 2Н), 0,94 (м, 2Н), 1,40 (м, 1Н), 6,81 (д, J=8,5 Гц, 1Н), 7,37 (дд, J=2,5, 8,5 Гц, 1Н), 7,49 (д, J=2,5 Гц, 1Н), 8,87 (шир.с. 1Н). Стадия С: 6-Хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н3,1-бензоксазин-2-он. К раствору, перемешанному в атмосфере аргона и в ледяной бане, и содержащему (+/-) 6хлор-4-циклопропилэтинил-4-трифторметил-1,4дегидро-2Н-3,1-бензоксазин-2-он (12,97 г, 0,041 М), 4-диметиламинопиридин (1,02 г, 0,0083 М), и (-)-хлорид (-)-камфановой кислоты (14,22 г, 0,06556 М) в 350 мл безводного дихлорметана, добавляли триэтиламин (22,84 мл, 0,164 М). Охлаждающую баню удаляли, в реакционную смесь оставляли при комнатной температуре для продолжения реакции. Через 75 минут, реакция была полностью завершена, о чем свидетельствовала тонкослойная хроматография (SiO2, 4% ЕtОАc в CHCI3). После этого, раствор разводили 500 миллилитрами хлороформа, а затем промывали 2 раза 10% лимонной кислотой, один раз водой, и один раз солевым раствором. После осушки сульфатом магния, фильтрации, и удаления растворителя в вакууме образовывалась бесцветная пена. Это пенообразное вещество растирали с 200 миллилитрами кипящего гексана. После охлаждения до комнатной температуры осаждалось нужное соединение, а именно диастереомерный имид камфаната. Это твердое вещество собирали на фритте, промывали слегка охлажденным гексаном, и осушали в вакууме, в результате чего получали 6-хлор-1-(1S)-камфаноил-4-циклопропилэтинил-4-трифтор-метил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (7,79 г) в виде белых кристаллов, т.пл. 164-165°С. ЖХВР-чистота: 99,2 при 254 нм. 1 Н-ЯМР (CDCI3): 0,77 (с, 3Н), 0,86-0,96 (м, 4Н), 1,08 с, 3Н), 1,19 (с, 3Н), 1,44 (м, 1Н), 1,76 (м, 1Н), 1,95 (м, 1Н), 2;51 (м, 2Н), 7,42 (дд, J=2,4, 9,0 Гц, 1H), 7,63 (м, 2Н). 20 42699 Labs) и 1:100 пенициллин-стрептомицин (Gibco Labs). Смесь инкубировали в течение ночи при 37°С в атмосфере 5% CO2. В. Обработка ингибиторами. Получали матрикс (в наномолярных концентрациях) для парных комбинаций (см. таблицу S). На день 1, аликвоты 125 мкл ингибиторов добавляли к равным объемам ВИЧ-инфицированных МТ-4-клеток (50000 кл. на лунку) в 96-луночном планшете для микротитрования. Инкубирование продолжали в течение 3 дней при 37°С в 5% СО2атмосфере. С. Оценка размножения вируса. С использованием многоканальной пипетки, осажденные клетки ресуспендировали, и 125 мкл собирали в отдельный планшет для микротитрования. Супернатант анализировали на ВИЧ-антиген р24 (после 24 ч.). Концентрацию ВИЧ-р24-антигена измеряли с помощью иммуноанализа, описанного ниже. Аликвоты р24-антигена, предназначенного для измерения, добавляли в микролунки, покрытыемоноклональным антителом, специфичным к антигену, ВИЧ-ядру. На этой стадии лунки промывали (а также на других последующих соответствующих стадиях). После этого добавляли биотинидированное ВИЧ-специфическое антитело, а затем конъюгированную со стрептавидином пероксидазу хрена. После добавления перекиси водорода и тетраметилбензилинового субстрата наблюдалась цветовая реакция. Интенсивность цвета была пропорциональна концентрации ВИЧ-р24-антигена. Вычисление степени синергизма. Было обнаружено, что парные комбинации ингибиторов (см. таблицу S) ингибируют размножение вируса в более высокой степени, чем каждый ингибитор, взятый отдельно, либо по сравнению с суммарным ингибированием, наблюдаемым для каждого ингибитора. Так, например, было установлено, что парная комбинация 726 и AZT дает значительно более эффективное ингибирование распространения вируса, чем одно соединение 726 или AZT, либо, чем суммарное ингибирование соединения 726 и AZT. Эти данные обрабатывали следующим образом: отношения фракционных ингибирующих концентраций (FIС) вычисляли методом, описанным ЕІіоn и др., J. Biol. Chem., 208, 477 (1954). Для различных пар комбинации определяли минимальную сумму FICS, которая представляет максимальный синергизм. Альтернативно, вычисляли среднюю сумму FІСS, которая представляет средний синергизм. См. таблицу S. Эти результаты иллюстрируют значительную степень синергизма в ингибировании распространения вируса. Чем меньше число, тем выше синергизм. Таблица S ствии ингибитора. В качестве контроля служил также очищенный гетеродимер ВИЧ-1-RT, продуцированный в E.coli. Результаты анализа выражали как концентрацию ингибитора (IC50 наномоль/литр) дающего 50%-ное ингибирование. В этом анализе, для оценки двойного мутанта (dm) использовали A17-RT. A17-RT является резистентной к различным аминопиридонам, как описано Nunberg J.Н и др. J. Virol 65, 4887 (1991). Результаты выражали как IC50 dm в нМ/л. Анализ на размножение клеток. Ингибирование размноженного в клеточной культуре вируса ВИЧ оценивали в соответствии с описанием Nunberg J.Н и др., J. Virol 65, 4887 (1991). В этом анализе, МТ-4-Т-лимфоидные клетки инфицировали вирусом ВИЧ-1 (дикого типа, если это не оговорено особо) посредством предварительно детерминированного инокулята, после чего культуры инкубировали в течение 24 часов. По истечении этого времени, £ 1% клеток, проанализированных с помощью непрямой иммуносоресценции, показали положительную реакцию. Затем клетки тщательно промывали и распределяли по 96-луночным планшетам. После этого, в лунки, добавляли серийные двухкратное разведения ингибитора, и продолжали культивировать еще 3 дня. Через 4 дня после инфицирования, в контрольных культурах было инфицировано 100 клеток. Аккумуляция ВИЧ (после 24 ч.) непосредственно коррелировала с размножением вируса. Ингибирующую концентрацию определяли как концентрацию ингибитора (в наномолях на литр), способствующую снижению распространения инфекции, по крайней мере, на 95% или СІС95. Суммарные результаты для соединения 37,2. А. Анализ обратной транскритазы и размножения клеток Дик. типа К103N* Y181C DM RT-2 IC50 0,002b 0,030 0,008 0,085 80,8 (мкМ) CIC95 не
ДивитисяДодаткова інформація
Назва патенту англійськоюBenzoxazinones, pharmaceutical composition (variants), combination of compounds (variants), method for inhibition of hiv reverse transcriptase, method for preventing or treating hiv infections
Автори англійськоюYoung Steven D., Britcher Susan F., Payne Linda S., Tran Lekhanh O., Lumma Jr. William C.
Назва патенту російськоюБензоксазиноновые соединения, фармацевтическая композиция (варианты), комбинация соединений (варианты), способ ингибирования вич обратной транскриптазы, способ предотвращения или лечения вич-инфекции
Автори російськоюЯнг Стивен Д., Бритчер Сюзн Ф., Пейн Линда С., Тран Ликхан О., Ламма Вилльям К. (молодший)
МПК / Мітки
МПК: A61P 43/00, A61P 31/12, A61P 31/18, A61K 31/52, A61K 31/54, C07D 265/18, A61K 45/06, A61K 31/505, A61P 37/04
Мітки: сполуки, бензоксазинонові, транскриптази, інгібування, зворотної, спосіб, композиції, попередження, фармацевтичні, лікування, комбінації
Код посилання
<a href="https://ua.patents.su/22-42699-benzoksazinonovi-spoluki-sposib-ingibuvannya-zvorotno-transkriptazi-poperedzhennya-abo-likuvannya-kombinaci-ta-farmacevtichni-kompozici.html" target="_blank" rel="follow" title="База патентів України">Бензоксазинонові сполуки, спосіб інгібування зворотної транскриптази, попередження або лікування, комбінації та фармацевтичні композиції</a>
Інгібітор вірусу імунодефіциту людини (віл) та зворотної транскриптази

Номер патенту: 34946
Опубліковано: 15.03.2001
Автори: Даниленко Валентина Пилипівна, Гужова Світлана Василівна, Даниленко Георгій Іванович, Рибалко Світлана Леонтіївна, Пацковський Юрій Васильович, Максимов Юрій Миколайович
МПК: A61K 31/015, A61P 31/18, C07C 13/00, A61K 31/13
Мітки: транскриптази, інгібітор, віл, зворотної, людини, імунодефіциту, вірусу
Формула / Реферат:
Застосування як інгібітору вірусу імунодефіциту людини та зворотної транскриптази сполуки наступної структури:
Антрациклінові дисахариди, спосіб їх одержання та фармацевтичні композиції, які їх містять
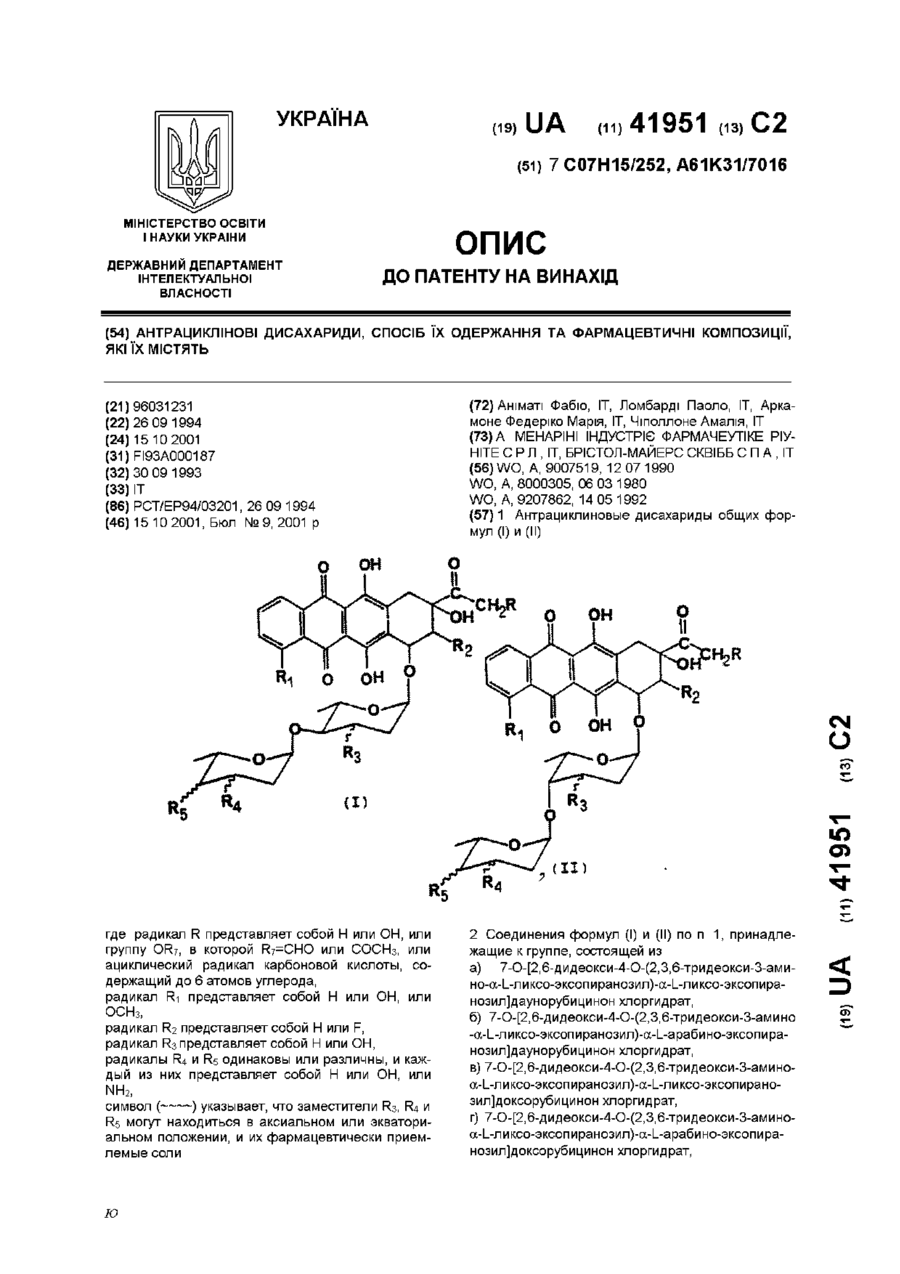
Номер патенту: 41951
Опубліковано: 15.10.2001
Автори: Аніматі Фабіо, Чіполлоне Амалія, Аркамоне Федеріко Марія, Ломбарді Паоло
МПК: A61P 35/00, A61K 31/70, A61K 31/7028, A61K 31/704, C07H 15/252, A61K 31/7034
Мітки: композиції, спосіб, фармацевтичні, одержання, дисахариди, містять, антрациклінові
Формула / Реферат:
1. Антрациклиновые дисахариды общих формул (I) и (II):где:радикал R представляет собой Н или ОН, или группу OR7, в которой R7=СНО или СОСН3, или ациклический радикал карбоновой кислоты, содержащий до 6 атомов углерода;радикал R1 представляет собой Н или ОН, или ОСН3;aрадикал R2 представляет собой Н или F;радикал R3 представляет собой Н или ОН;радикалы R4 и R5 одинаковы или различны, и каждый из...
Сполуки,які містять біциклічне конденсоване ядро, спосіб їх одержання, проміжні сполуки, спосіб їх одержання (варіанти) та фармацевтична композиція для інгібування перетворення ферменту ангіотензину та нейтраль

Номер патенту: 39924
Опубліковано: 16.07.2001
Автори: Кронентал Девід Р., Робл Джефрі А., Годфрей Джоллі Д.
МПК: A61K 31/54, C07K 5/078, A61K 31/535, C07D 498/04, C07D 513/04, A61K 38/00, C07C 327/00, A61P 43/00, C07C 319/00, A61K 31/553, A61K 31/55, A61K 31/542, A61P 9/04, A61P 9/12, A61K 31/554, C07C 323/59, C07C 317/50
Мітки: ангіотензину, інгібування, сполуки,які, спосіб, містять, біциклічне, фармацевтична, композиція, ферменту, варіанти, одержання, перетворення, нейтраль, проміжні, конденсоване, ядро, сполуки
Формула / Реферат:
1. Соединения, содержащие конденсированное бициклическое ядро формулы,или их фармацевтически приемлемая соль, где А представляетили,Χ представляет О или S,r1 и R12 независимо...
Сполуки, композиції та способи, які інгібують протеази, для лікування та профілактики імуномедіаторних запальних хвороб
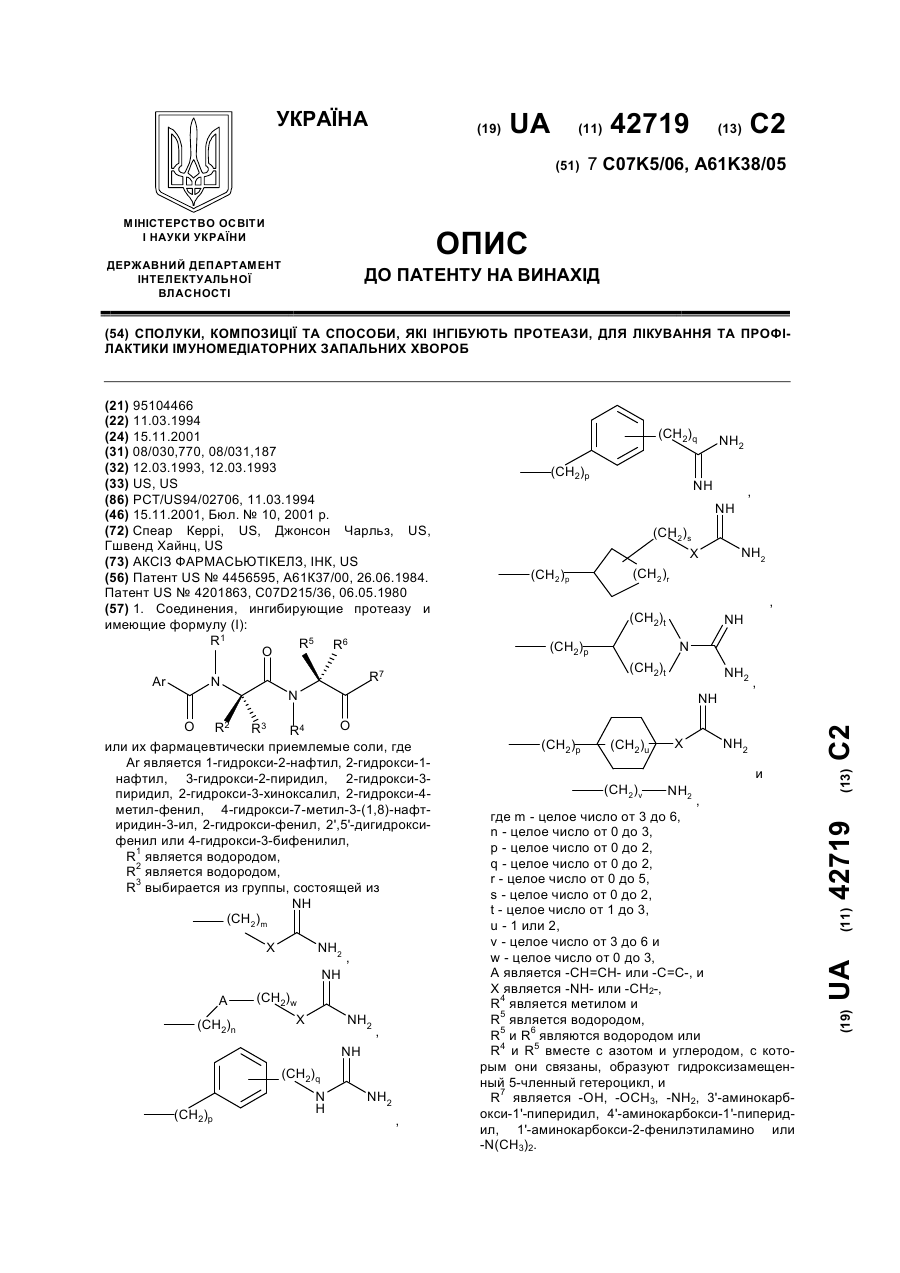
Номер патенту: 42719
Опубліковано: 15.11.2001
Автори: Гшвенд Хайнц, Джонсон Чарльз, Спеар Керрі
МПК: A61M 11/00, C07K 5/068, A61K 31/57, A61K 31/44, C07K 5/065, A61P 11/00, A61K 38/00, A61K 9/12, C07K 5/072, A61P 29/00, A61P 27/16, A61P 37/08, A61P 43/00, C07D 471/04
Мітки: хвороб, сполуки, інгібують, імуномедіаторних, способи, протеази, запальних, композиції, лікування, профілактики
Формула / Реферат:
1. Соединения, ингибирующие протеазу и имеющие формулу (I):или их фармацевтически приемлемые соли, гдеАr является 1-гидрокси-2-нафтил, 2-гидрокси-1-нафтил, 3-гидрокси-2-пиридил, 2-гидрокси-3-пиридил, 2-гидрокси-3-хиноксалил, 2-гидрокси-4-метил-фенил, 4-гидрокси-7-метил-3-(1,8)-нафтиридин-3-ил, 2-гидрокси-фенил, 2',5'-дигидрокси-фенил или 4-гидрокси-3-бифенилил,R1 является водородом,R2 является...
Комбінація для профілактики та лікування сніду, фармацевтична композиція на її основі, спосіб отримання фармацевтичної композиції

Номер патенту: 26587
Опубліковано: 11.10.1999
Автори: Дорсі Брюс Д, Холлоуей Катарін М, Вакка Джозеф П, ХАНГЕЙТ Рендалл, Гар Джеймс П
МПК: A61K 31/70, A61K 45/06
Мітки: сніду, комбінація, композиція, фармацевтична, композиції, основі, отримання, фармацевтично, лікування, профілактики, спосіб
Формула / Реферат:
1. Комбинация соединенияили его фармацевтически приемлемой соли и любого из соединений, выбранных из AZT, ddl или ddC.2. Комбинация по п.1, предназначенная для лечения СПИДа, профилактики заражения ВИЧ, лечения ВИЧ-инфицирования или для ингибиропания ВИЧ-протеазы.3. Фармацевтическая композиция, включающая активный ингредиент и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного...
Наступний патент: Друкарська секція для ротаційної друкарської машини
Випадковий патент: Спосіб подрібнення рослинної маси і пристрій для його здійснення