Похідне ізоксазоліну та спосіб його одержання
Номер патенту: 94395
Опубліковано: 10.05.2011
Автори: Хео Тае-Хо, Лі Джа Хоон, Чой Хьєонг Воок, Лі Кью-Воонг, Парк Кі-Соок, Шин Хьюн-Ік












Формула / Реферат
1. Сполука формули (1):
 ,
,
де
R1 являє собою ізохінолініл, хінолініл або нафтил, і
R2 являє собою метил, етил, пропіл або бутил.
2. Сполука за п. 1, в якій:
R1 являє собою ізохінолініл, і
R2 являє собою ізопропіл.
3. Сполука за п. 2, яка має кристалічну форму, що демонструє наступний патерн рентгеноструктурного аналізу:
D(X)
Відносна інтенсивність (I/I0)
Кут 2θ
9,665
0,555
9,15
7,284
0,397
12,15
5,825
0,260
15,21
5,563
0,228
15,93
5,372
0,302
16,5
4,840
1,000
18,33
4,695
0,477
18,9
4,341
0,454
20,46
3,663
0,230
24,3
3,414
0,219
26,1
4. Сполука за п. 1, яка призначена для застосування у виробництві лікарського засобу для лікування запалення або запобігання апоптозу.
5. Сполука за п. 4, яка відрізняється тим, що лікарський засіб застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця.
6. Фармацевтична композиція для лікування запалення або запобігання апоптозу, яка відрізняється тим, що містить сполуку формули (1) відповідно до п. 1 і фармацевтично прийнятні носії.
7. Фармацевтична композиція за п. 6, яка відрізняється тим, що композицію застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця.
8. Спосіб лікування запалення або запобігання апоптозу у суб'єкта, який відрізняється тим, що здійснюють введення суб'єкту терапевтично ефективної кількості сполуки формули (1) за п. 1.
9. Спосіб за п. 8, який відрізняється тим, що спосіб застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця.
10. Сполука за формулою
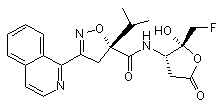 .
.
11. Сполука за п. 10, яка призначена для застосування у виробництві лікарського засобу для лікування запалення або запобігання апоптозу.
12. Сполука за п. 11, яка відрізняється тим, що лікарський засіб застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця.
13. Сполука за п. 11, яка відрізняється тим, що лікарський засіб застосовують при пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки.
14. Фармацевтична композиція, яка містить сполуку за п. 10 та один або більше фармацевтично прийнятних носіїв.
15. Спосіб одержання сполуки за п. 10, при якому здійснюють наступні етапи:
а) зняття захисту зі сполук формули (14) з отриманням суміші сполук формули (15) і формули (16); і
b) обробка суміші сполук формули (15) і формули (16) каталітичною кількістю основи разом із затравкою сполуки формули (1) з перетворенням як сполуки формули (15), так і сполуки формули (16) у сполуки формули (1),
де сполука формули (15) знаходиться в рівновазі із сполуками формули (16), формули (17) та формули (1), і сполука формули (16) знаходиться в рівновазі із сполуками формули (18) та формули (19),
 .
.
Текст
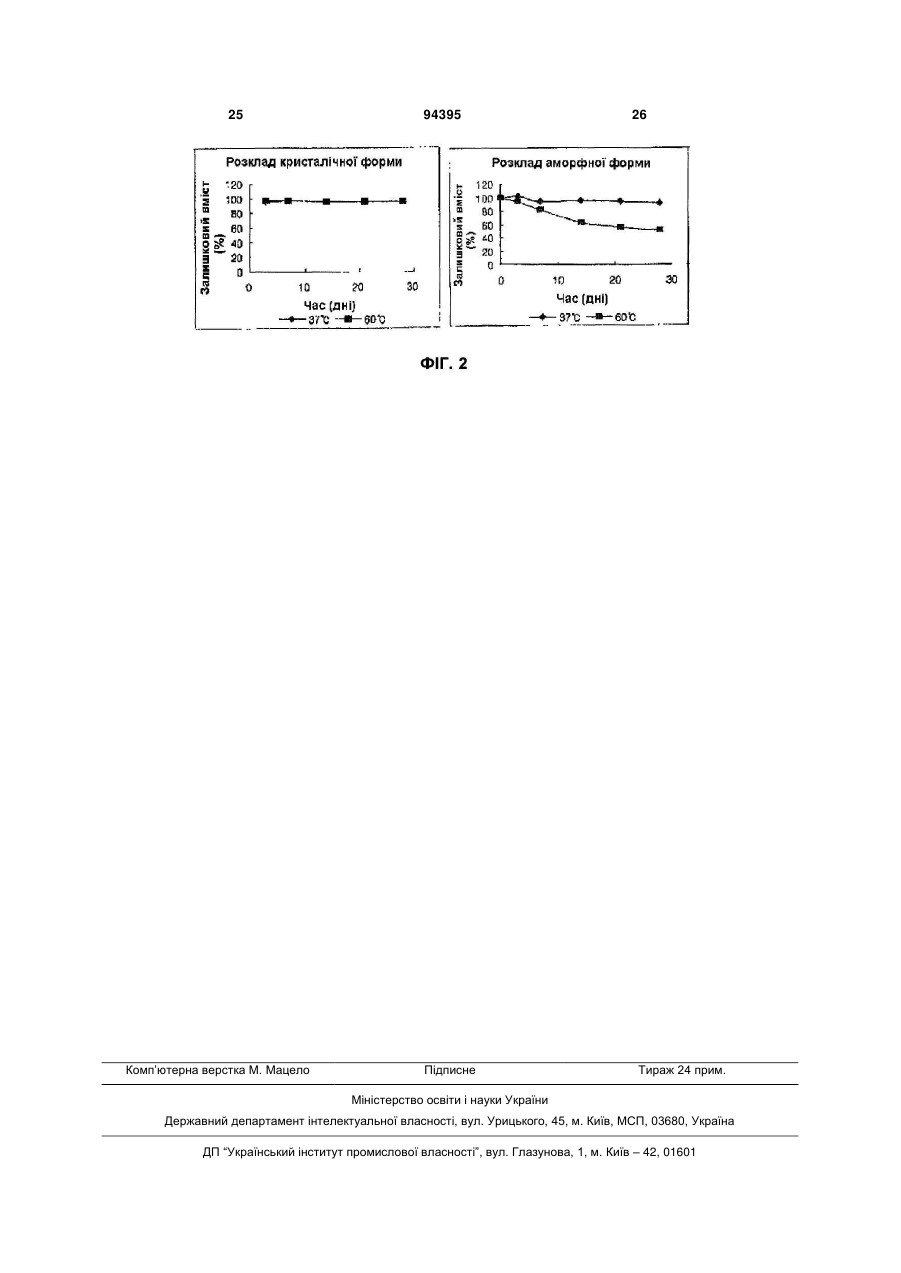
1. Сполука формули (1): (19) (21) a200709610 (22) 21.02.2006 (24) 10.05.2011 (86) PCT/KR2006/000576, 21.02.2006 (31) 10-2005-0016203 (32) 26.02.2005 (33) KR (46) 10.05.2011, Бюл.№ 9, 2011 р. (72) ШИН ХЬЮН ІК, KR, ЧОЙ ХЬЄОНГ ВООК, KR, ХЕО ТАЕ ХО, KR, ЛІ КЬЮ ВООНГ, KR, ЛІ ДЖА ХООН, KR, ПАРК КІ СООК, KR (73) ЕЛДЖІ ЛАЙФ САЄНСЕЗ ЛТД., KR (56) WO 0121600 A1, 29.03.2001 KR 1999-79267 A, 05.11.1999 KANEMASA S. ET AL: 'Lewis Acid Coordinated Nitrile Oxide and Nitrile Imine 1,3-Dipoles. syn-Selective Cycloadditions to 2-(1-Hydroxyalkyl)acrylates' BULL. CHEM. SOC. JPN. vol. 66, 1993, pages 2685 - 2693 ROBINSON R.R. ET AL.: 'Synthesis of a Peptidyl Difluoro Ketone Bearing the Aspartic Acid Site Chain: 3 94395 клітини серця внаслідок ішемічних захворювань серця. 8. Спосіб лікування запалення або запобігання апоптозу у суб'єкта, який відрізняється тим, що здійснюють введення суб'єкту терапевтично ефективної кількості сполуки формули (1) за п. 1. 9. Спосіб за п. 8, який відрізняється тим, що спосіб застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця. 10. Сполука за формулою N O H HO N печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки, сепсисі, відторгненні трансплантованого органа, ревматоїдному артриті або некрозі клітини серця внаслідок ішемічних захворювань серця. 13. Сполука за п. 11, яка відрізняється тим, що лікарський засіб застосовують при пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворюваннях, гострому гепатиті, швидкоплинній печінковій недостатності, цирозі печінки. 14. Фармацевтична композиція, яка містить сполуку за п. 10 та один або більше фармацевтично прийнятних носіїв. 15. Спосіб одержання сполуки за п. 10, при якому здійснюють наступні етапи: а) зняття захисту зі сполук формули (14) з отриманням суміші сполук формули (15) і формули (16); і b) обробка суміші сполук формули (15) і формули (16) каталітичною кількістю основи разом із затравкою сполуки формули (1) з перетворенням як сполуки формули (15), так і сполуки формули (16) у сполуки формули (1), де сполука формули (15) знаходиться в рівновазі із сполуками формули (16), формули (17) та формули (1), і сполука формули (16) знаходиться в рівновазі із сполуками формули (18) та формули (19), F O O N O . 11. Сполука за п. 10, яка призначена для застосування у виробництві лікарського засобу для лікування запалення або запобігання апоптозу. 12. Сполука за п. 11, яка відрізняється тим, що лікарський засіб застосовують при деменції, мозковому інсульті, пошкодженні мозку внаслідок СНІДу, діабеті, виразці шлунка, пошкодженні мозку вірусом гепатиту, спричинених вірусом гепатиту N 4 O H N F HO O N N МеО ОМе H F N CO2H N F O N N N O CO2H O F O N CO2H основа 16 O H N F HO N O H N O O N F HO O O O 1, осадження . O H N + 15 14 O 18 O H N F HO O O O H N N 17 O O O O N N N O 19 5 Опис винаходу Даний винахід стосується похідного ізоксазоліну, що містить фрагмент циклічного гемікеталю карбонової кислоти для застосування як інгібітора каспази, способу його одержання, фармацевтичної композиції, що його містить, та їх застосування. Рівень техніки Інгібітор каспази означає сполуку, яка може інгібувати активність каспази, таким чином контролюючи запалення або апоптоз, спричинений дією каспази. Відомо, що серед інгібіторів каспази необоротний інгібітор демонструє більш високу ефективність інгібування, оскільки він необоротно інактивує фермент для керування апоптозом (Wu J. et al., Methods: A Companion to Methods in Enzymology 1999, 17, 320). Наступні сполуки відомі як необоротні інгібітори і містять спільний фрагмент 3аміно-5-фтор-4-оксопентанової кислоти. Захворювання, які можна лікувати або полегшувати введенням згаданих вище сполук, являють собою ревматоїдний артрит, запальне захворювання кишечнику, захворювання "трансплантат проти хазяїна", сепсис, остеоартрит, остеопороз, гостру і хронічну мієлогенну лейкемію, менінгіт, сальпінгіт, септичний шок, холангіт, коліт, енцефаліт, ендокардит, гломерулонефрит, гепатит, міокардит, панкреатит, цукровий діабет І типу, розсіяний склероз, хворобу Альцгеймера, хворобу Паркінсона, цироз печінки, і т. п. З іншого боку, для фрагменту 3-аміно-5-фтор4-оксопентанової кислоти інгібітора каспази, такий спосіб одержання, як показано на наступній Схемі реакції 1 (Revesz et al., Tetrahedron Lett, 1994 , 35, 9693), відомий з рівня техніки: Короткий опис винаходу Винахідники продовжили дослідження сполуки, яка може використовуватися як ефективний інгібітор каспази, і способу її одержання. В результаті, ними було виявлено, що похідне ізоксазоліну, яке містить фрагмент циклічного гемікеталю карбонової кислоти за даним винаходом, володіє високою активністю з інгібування каспази і може бути одержане з високою чистотою з використанням індукованого кристалізацією динамічного перетворення, для здійснення даного винаходу. Таким чином, одним об'єктом даного винаходу є сполука наступної формули (1) із структурою ізоксазоліну 94395 6 та фрагментами гемікеталю циклічної карбонової кислоти, а також новий спосіб її ефективного одержання. Іншим об'єктом даного винаходу є амінне похідне наступної формули (4) - проміжна сполука для одержання сполуки наступної формули (1), а також спосіб її одержання. Іншим об'єктом даного винаходу є фармацевтична композиція для лікування запалення або запобігання апоптозу, яка містить сполуку наступної формули (1) і фармацевтично прийнятні носії. Іншим об'єктом даного винаходу є спосіб лікування запалення або запобігання апоптозу у суб'єкта, який включає введення терапевтично ефективної кількості сполуки наступної формули (1) суб'єкту. Іншим об'єктом даного винаходу є застосування сполуки наступної формули (1) для виготовлення лікарського засобу для лікування запалення або запобігання апоптозу. Іншим об'єктом даного винаходу є кристалічна форма сполуки наступної формули (1), що має високу стабільність. Короткий опис фігур Фіг. 1 представляє спектр рентгеноструктурного аналізу кристалічної форми сполуки формули 1 2 (1) (R = ізохінолініл, R = ізопропіл) у відповідності до даного винаходу. Фіг. 2 представляє графік, де показані результати дослідження стабільності кристалічної форми 1 та аморфної форми сполуки формули (1) (R = 2 ізохінолініл, R = ізопропіл) у відповідності до даного винаходу. Розкриття винаходу Деякі важливі терміни, використані в даному винаході, можуть бути визначені наступним чином. У формулах і схемах реакції, використаних в даному винаході, алкіл означає лінійну або розгалужену алкільну групу, що складається з 1-8 атомів вуглецю, або циклоалкільну групу, що складається з 3-10 атомів вуглецю. Крім того, "арил" включає всі ароматичні групи, гетероароматичні групи, а також їх частково відновлені похідні. Вказана ароматична група позначає 5-15-членні ненасичені вуглеводи в простій або конденсованій циклічній формі, і вказана гетероароматична група означає ароматичні групи, що містять 1-5 гетероатомів, вибраних з групи, що складається з кисню, сірки та азоту. Додатково, один або більше атомів водню вказаного алкілу і вказаного арилу можуть бути заміщені іншим замісником. Приклади замісників, що можуть вибиратися, включають ацил, аміно, карбоалкокси, карбокси, карбоксиаміно, ціано, галоген, гідрокси, нітро, тіол, алкіл, циклоалкіл, алкокси, арилокси, сульфокси, гуанідо, і т. п. Даний винахід стосується похідного ізоксазоліну, що містить фрагмент гемікеталю циклічної карбонової кислоти наступної формули (1) для застосування як інгібітора каспази: 7 94395 8 1 R являє собою ізохінолініл, хінолініл або нафтил, і 2 R являє собою метил, етил, пропіл або бутил. 1 Більш переважно, R являє собою ізохінолініл, 2 і R являє собою ізопропіл. 1 Сполука формули (1) де R являє собою ізохі2 нолініл і R являє собою ізопропіл може існувати в кристалічній формі, яка демонструє наступний патерн рентгеноструктурного аналізу. 1 де R являє собою алкіл або арил, і 2 R являє собою алкіл. Переважно, D(X) 9,665 7,284 5,825 5,563 5,372 4,840 4,695 4,341 3,663 3,414 Відносна інтенсивність (І/Іо) 0,555 0,397 0,260 0,228 0,302 1,000 0,477 0,454 0,230 0,219 Даний винахід також стосується способу одержання сполуки формули (1), який включає: (a) активацію сполуки наступної формули (2), наступну реакцію її із сполукою наступної формули (4) з утворенням сполуки наступної формули (13); (b) гідроліз сполуки наступної формули (13) з утворенням сполуки наступної формули (14); (c) зняття захисту із сполуки наступної формули (14); і (d) здійснення індукованого кристалізацією динамічного перетворення; Кут 2 9,15 12,15 15,21 15,93 16,5 18,33 18,9 20,46 24,3 26,1 де 1 R являє собою алкіл або арил, 2 R являє собою алкіл, 3 3' кожен з R і R являє собою алкіл, 3 3’ або R і R разом з атомом кисню, до якого вони приєднані, утворюють гетероцикл, і 4 R являє собою алкіл. Переважно, 1 R являє собою ізохінолініл, хінолініл або нафтил, 2 R являє собою метил, етил, пропіл або бутил, 3 3’ кожен з R і R являє собою метил, етил або пропіл, 3 3’ або R і R разом з атомом кисню, до якого вони приєднані, утворюють діоксолан або діоксан, і 4 R являє собою метил, етил, пропіл або бутил. Кожна стадія згаданого вище способу одержання сполуки формули (1) може бути описана більш детально наступним чином. Як реактив для активації сполуки формули (2) на згаданій вище стадії (а) переважно використовують сполуку, вибрану з групи, що складається з оксалілхлориду, триметилацетилхлориду, фосфорилтрихлориду і тіонілхлориду. Також, реакцію на стадії (а) переважно здійснюють в присутності основи, вибраної з групи, що складається з триетиламіну, три(н-бутил)аміну, діізопропілетиламіну, піридину, 4диметиламінопіридину і 4-(4-метил-піперидин-І-іл)піридину, і, переважно, основа використовується в кількості 1,0-10,0 еквівалентів по відношенню до сполуки формули (2). Крім того, реакцію на стадії (а) переважно здійснюють в одному або більше розчинниках, 9 вибраних з групи, що складається з дихлорметану, хлороформу, тетрагідрофурану, диметоксиетану, діоксану та етилацетату. З іншого боку, сполука формули (4) на стадії (а) переважно використовується в кількості 1,0-3,0 еквівалентів по відношенню до сполуки формули (2). Гідроліз на стадії (b) переважно здійснюють в присутності основи, вибраної з групи, що складається з гідроксиду літію (переважно, безводного або кристалічного моногідрату), гідроксиду натрію, гідроксиду калію та гідроксиду кальцію, а також основу переважно використовують в кількості 0,110,0 еквівалентів по відношенню до сполуки формули (13). Крім того, реакцію на стадії (b) переважно здійснюють в одному або більше розчинників, вибраних з групи, що складається з метанолу, етанолу, н-пропанолу, ізопропанолу, тетрагідрофурану, диметоксиетану, діоксану і дихлорметану, або в змішаному розчиннику або розчиннику, вибраному із згаданої вище групи і води. Переважно, реакцію зняття захисту на стадії (с) здійснюють в присутності кислоти, вибраної з групи, що складається з хлористоводневої кислоти, сірчаної кислоти і трифтороцтової кислоти, і переважно, використовують кислоту в кількості 0,1-20,0 еквівалентів по відношенню до сполуки формули (14). Також, реакцію зняття захисту на стадії (с) переважно здійснюють в присутності або за відсутності розчинника, вибраного з дихлорметану або хлороформу. Індукована кристалізацією реакція динамічного перетворення на стадії (d) може бути здійснена шляхом додавання сполуки формули (1) як затравки або може бути здійснена в присутності затравки і каталітичної кількості основи, де основа переважно являє собою амін, вибраний з групи, що складається з триетиламіну, три(н-бутил)аміну, діізопропілетиламіну, діізопропіламіну, піридину, 4 94395 10 диметиламінопіридину, 4-(4-метил-піперидин-1-іл)піридину, оптично активного 1-фенілетиламіну та оптично активного 1-нафтилетиламіну. Переважно, на стадії (d) використовують вказаний амін в кількості 0,001-1,0 еквівалентів по відношенню до сполуки формули (14), і більш переважно використовувати 0,03-0,5 еквівалентів. Якщо кількість використаного аміну занадто маленька, швидкість реакції уповільнюється, а якщо кількість дуже велика, вихід сполуки формули (1) зменшується. Крім того, індуковану кристалізацією динамічну реакцію перетворення на стадії (d) переважно здійснюють в одному або більше розчинників, вибраних з групи, що складається з толуолу, бензолу, дихлорбензолу, тетрагідрофурану, диметоксиетану, діоксану, етилацетату, дихлорметану, ацетонітрилу, метил-трет-бутилового ефіру та діетилового ефіру. Спосіб одержання сполуки формули (1) у відповідності до даного винаходу нижче пояснюється більш детально з посиланням на Схему реакції 2. Похідне ізоксазоліну формули (2), що має високу оптичну активність, одержують у відповідності до способу, розкритого в PCT/KR2004/002060, поданій 17 серпня 2004 р. заявником даного винаходу, з наступним сполученням із сполукою формули (4) з утворенням сполуки формули (13). Далі естерну групу сполуки формули (13) піддають гідролізу з утворенням сполуки формули (14) і здійснюють реакцію зняття захисту з кетального фрагмента сполуки формули (14) з одержанням суміші сполук формули (15) і формули (16), яку ефективно перетворюють на сполуку формули (1) шляхом вибіркової динамічної кристалізації. Зокрема, якщо суміш сполук формули (15) і формули (16) розчинена в органічному розчиннику, і до розчину додають затравку сполуки формули (1), тільки сполука формули (15) в суміші перетворюється на сполуку формули (1), яку виділяють у вигляді твердої речовини. 11 94395 12 де 1 R являє собою алкіл або арил, 2 R являє собою алкіл, 3 3’ кожен з R і R являє собою алкіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють гетероцикл, і, 4 R являє собою алкіл. Також, якщо суміш сполук формули (15) і формули (16) обробляють каталітичною кількістю основи разом із затравкою, обидві сполуки - формули (15) та формули (16) - перетворюються на сполуку формули (1) з утворенням сполуки формули (1) з більш високим виходом (Схема реакції 3). Сполука формули (15) знаходиться в рівновазі із сполукою формули (16) внаслідок присутності основи в розчині. Також сполука формули (15) знаходиться в рівновазі із сполуками формули (17) і формули (1), а сполука формули (16) знаходиться в рівновазі із сполуками формули (18) і формули (19). Серед них, сполука формули (1), що володіє добрими кристалізаційними властивостями, вибірково осаджується таким чином, що рівновага всіх сполук зміщується на сполуку формули (1), що дає можливість вибіркового одержання з суміші сполук формули (15) і формули (16) тільки сполуки формули (1) з високим виходом. сполуки, використаної для одержання сполуки формули (1): (b) здійснення реакції сполучення вуглецьвуглець для сполуки формули (10) з утворенням сполуки наступної формули (11); (c) реакцію сполуки формули (11) з бензиламіном та наступне відновлення з утворенням сполуки наступної формули (12); і d) гідрогенізацію сполуки формули (12); де 3 3’ кожен з R і R являє собою алкіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють гетероцикл, і 4 R являє собою алкіл. Переважно, 3 3’ кожен з R і R являє собою метил, етил або пропіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють діоксолан або діоксан, і 4 R являє собою метил, етил, пропіл або бутил. Даний винахід також стосується способу одержання сполуки формули (4), який включає: (a) захист і зняття захисту із сполуки наступної формули (9) з утворенням сполуки наступної формули (10); 13 де 3 3’ кожен з R і R являє собою алкіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють гетероцикл, і 4 R являє собою алкіл. Переважно, 3 3’ кожен з R і R являє собою метил, етил або пропіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють діоксолан або діоксан, і 4 R являє собою метил, етил, пропіл або бутил. Далі наведений вище спосіб одержання сполуки формули (4) буде описаний більш конкретно. На стадії (а) сполуку формули (9) переважно захищають з використанням триметилортоформіату або триетилортоформіату, і реакцію зняття захисту здійснюють в присутності основи, вибраної з гідроксиду натрію, гідроксиду калію, карбонату натрію і гідрокарбонату натрію. Крім того, основу в згаданій вище реакції зняття захисту переважно використовують в кількості 1,0-2,0 еквівалентів по відношенню до сполуки формули (9). Також, реакцію захисту на стадії (а) переважно здійснюють в розчиннику метанолі або етанолі, і реакцію зняття захисту здійснюють в одному або більше розчинників, вибраних з групи, що складається з метанолу, етанолу, дихлорметану, хлороформу і води. Також, реакцію на стадії (b) переважно здійснюють, з використанням етилхлорформіату або метилхлорформіату в присутності основи, вибраної з н-бутиллітію, літію діізопропіламіну або літію гексаметилдисилазиду, і основу використовують в кількості 0,5- 3,0 еквівалентів по відношенню до сполуки формули (10), і вказаний етилхлорформіат або метилхлорформіат використовують в кількості 0,5-3,0 еквівалентів по відношенню до сполуки формули (10). Також, реакцію на стадії (b) переважно здійснюють в розчиннику, вибраному з групи, що складається з тетрагідрофурану, етилового ефіру та метил-трет-бутилового ефіру. Переважно, реакцію відновлення на стадії (с) здійснюють з використанням оцтової кислоти і боргідриду натрію, причому вказаний боргідрид на 94395 14 трію використовують в кількості 1,0-5,0 еквівалентів по відношенню до сполуки формули (11), і вказану оцтову кислоту використовують в кількості 1,0-20,0 еквівалентів по відношенню до сполуки формули (11). Крім того, вказаний бензиламін на стадії (с) переважно використовують в кількості 1,0-10,0 еквівалентів по відношенню до сполуки формули (11). Додатково, реакцію на стадії (с) переважно здійснюють в присутності або за відсутності розчинника, вибраного з етилацетату, тетрагідрофурану, етилового ефіру і метил-трет-бутилового ефіру, і така реакція за бажанням може бути здійснена як однореакторна реакція. Реакцію на стадії (d) переважно здійснюють в присутності металевого каталізатора, більш переважно каталізатора з сімейства паладію або каталізатора з сімейства нікелю Ренея. Конкретно, каталізатор з сімейства паладію, що містить 1-20 % (мас.) паладію (Pd) або каталізатор з сімейства нікелю Ренея, що містить 1 % (мас.) або більше нікелю Ренея, нанесений на носій, вибраний з групи, що складається з вуглецю, кремнію діоксиду та оксиду алюмінію, і він може використовуватися в кількості 0,01-10 % (мас.) на базі металевого компоненту по відношенню до сполуки формули (12). Також, реакцію на стадії (d) переважно здійснюють в одному або більше розчинників, вибраних з групи, що складається з метанолу, етанолу, нпропанолу, ізопропанолу, тетрагідрофурану, диметоксиетану, діоксану, етилацетату і дихлорметану. Крім того, реакцію гідрогенізації на стадії (d) переважно здійснюють при температурі від 0 до 50 °С при тиску водню 1-100 атмосфер. Винахідниками розроблено новий спосіб одержання сполуки формули (4), тобто проміжної сполуки для одержання сполуки формули (1), з більш високим виходом, як проілюстровано на наступній Схемі реакції 4. ДЄ 3 3' кожен з R і R являє собою алкіл, або 3 3’ R і R разом з атомом кисню, до якого вони приєднані, утворюють гетероцикл, і 4 R являє собою алкіл. Даний винахід також стосується фармацевтичної композиції для лікування запалення або запобігання апоптозу, яка містить сполуку визначеної вище формули (1) і фармацевтично прийнятні носії, конкретно, фармацевтичної композиції для лікування деменції, мозкового інсульту, пошкодження мозку внаслідок СНІДу, діабету, виразки шлунку, ураження мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворювань, гострого гепатиту, швидкоплинної печінкової недо 15 статності, цирозу печінки, сепсису, відторгнення трансплантованого органу, ревматоїдного артриту або некрозу клітин серця внаслідок ішемічних захворювань серця. Даний винахід також стосується способу лікування запалення або запобігання апоптозу у суб'єкта, який включає введення терапевтично ефективної кількості сполуки визначеної вище формули (1) суб'єкту, конкретно, способу лікування деменції, мозкового інсульту, пошкодження мозку внаслідок СНІДу, діабету, виразки шлунку, ураження мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворювань, гострого гепатиту, швидкоплинної печінкової недостатності, цирозу печінки, сепсису, відторгнення трансплантованого органу, ревматоїдного артриту або некрозу клітин серця внаслідок ішемічних захворювань серця. Даний винахід також стосується застосування сполуки визначеної вище формули (1) для виробництва лікарського засобу для лікування запалення або запобігання апоптозу, особливо лікарського засобу для лікування деменції, мозкового інсульту, пошкодження мозку внаслідок СНІДу, діабету, виразки шлунку, ураження мозку вірусом гепатиту, спричинених вірусом гепатиту печінкових захворювань, гострого гепатиту, швидкоплинної печінкової недостатності, цирозу печінки, сепсису, відторгнення трансплантованого органу, ревматоїдного артриту, або некрозу клітин серця внаслідок ішемічних захворювань серця. Сполука формули (1) у відповідності до даного винаходу може бути введена в різноманітні лікарські форми з метою введення суб'єкту. Для виготовлення фармацевтичної композиції у відповідності до даного винаходу ефективну кількість сполуки формули (1) змішують з фармацевтично прийнятним носієм, який може бути вибраний з численних форм, в залежності від препарату, який виготовляють. Сполука формули (1) може бути введена в ін'єкційну лікарську форму для парентерального введення або для трансдермального або перорального введення в залежності від передбаченого застосування. Особливо переважним є виготовлення композиції у вигляді дозованої лікарської форми для легкого введення і однорідності дозування. У випадку препарату для перорального застосування може бути використаний будь-який звичайний фармацевтичний носій. Наприклад, вода, гліколі, масла, спирти і т. п. можуть бути застосовані для виготовлення рідких препаратів для перорального введення, таких як суспензії, сиропи, еліксири та розчини; або різні види крохмалю, цукрів, каолін, змащувачі, зв'язувальні агенти, дезинтегранти і т. п. можуть бути використані для твердих препаратів, таких як порошки, пігулки, капсули і таблетки. В наслідок легкості введення, таблетки і капсули являють собою найбільш переважні лікарські форми. Для таблеток і пілюль також бажано мати кишково-розчинне покриття. У випадку препарату для парентерального введення як носій звичайно використовують стерильну воду, однак можуть бути використані інші 94395 16 інгредієнти, такі як солюбілізувальні засоби. Лікарські форми для ін'єкцій, наприклад, стерильна водна або масляна суспензія для ін'єкцій, можуть бути виготовлені у відповідності до відомої методики з використанням придатного диспергувального агента, зволожувача або суспендувального агента. Розчинники, які можуть бути використані у виготовленні лікарських форм для ін'єкцій, включають воду, розчин Рингера та ізотонічний розчин NaCI; стерильне нелетке масло також може бути придатним чином використане як розчинник або середовище для суспендування. Будь-яке нелетке масло, що не володіє стимулювальною активністю, в тому числі моно- і дигліцериди, може бути використане для даної мети. Для ін'єкцій також може бути використана жирна кислота, така як олеїнова кислота. У випадку препарату для трансдермального введення, носій може містити агент для збільшення проникнення та/або відповідний зволожувач, необов'язково в поєднанні з відповідними добавками, які не спричиняють істотного подразнення шкіри. Вказані добавки можуть полегшувати введення крізь шкіру та/або можуть сприяти дії препарату бажаного складу. Такі трансдермальні лікарські форми вводять в різних формах, наприклад, трансдермального пластиру, точкового нанесення або мазі. Якщо сполуку формули (1) застосовують для клінічної мети, переважно її вводять суб'єктупацієнту в кількості, що варіює від 0,1 до 100 мг/кг маси тіла організму на добу. Загальна добова доза може бути введена за один раз або розділена на декілька прийомів. Однак, конкретна доза для введення індивідуальному пацієнту може варіювати в залежності від сполуки, яку застосовують, маси тіла, статі, стану здоров'я або харчування суб'єкта-пацієнта, часу або способу введення, швидкості екскреції, співвідношення змішування для агента, тяжкості захворювання, яке підлягає лікуванню, і т. п. Нижче даний винахід буде описаний більш детально з посиланням на наступні Приклади, але контекст даного винаходу не повинен бути обмежений таким чином в будь-якій формі. Препаративний приклад 1 1-Фтор-4-триметилсиланіл-3-бутин-2-он (9) 49,1 г (499 ммоль) триметилсилілацетилену розчиняють в 250 мл безводного тетрагідрофурану та внутрішню температуру знижують приблизно до -55 °С, після чого додають 210 мл (525 ммоль) 2,5 М н-BuLi в н-гексані протягом приблизно 25 хв, підтримуючи внутрішню температуру нижче -30 °С. Після перемішування протягом приблизно 40 хв до реакційної суміші додають 52,9 г (499 ммоль) етилфторацетату протягом 5 хв, підтримуючи внутрішню температуру нижче -25 °С, і потім додають 74,4 г (524 ммоль) BF-OEt протягом 15 хв, підтримуючи внутрішню температуру в інтервалі від -55 °С до -65 °С. Після закінчення додавання реакційну суміш перемішують при 20 °С, витримуючи при цій температурі протягом 2 год, та додають 250 мл 10 % водного розчину амонію хлориду для закінчення реакції. Органічну фракцію відокремлюють, і водну фракцію екстрагують 200 мл етилацетату. 17 Об'єднані органічні фракції промивають 250 мл розсілу та упарюють при зниженому тиску. Залишок дистилюють під вакуумом при тиску 10 мбар і температурі 68 °С з одержанням сполуки формули (9) (67,3 г, 85 %) у вигляді прозорого масла 1 Н ЯМР (500 МГц, CDCI3): 4,90 (д, J = 47,1 Гц, 2Н), 0,26 (с, 9Н). 13 С ЯМР (125 МГц, CDCI3): 181,0 (д, J = 21,5 Гц), 104,0, 98,1, 84,8 (д, J = 187 Гц). Препаративний приклад 2 4-Фтор-3,33 3’ диметокси-1-бутин (10, R , R = метил) 33,6 г (316 ммоль) триметилортоформіату та 6,0 г (31,5 ммоль) n-TsOH-H2O разом з 50,0 г (316 ммоль) сполуки формули (9), одержаної в Препаративному прикладі 1, вміщують у 260 мл метанолу та перемішують при температурі кипіння із зворотним холодильником (внутрішня температура 60-64 °С) протягом приблизно 6 год. Реакційну суміш упарюють при зниженому тиску для видалення приблизно 130 мл розчинника та розбавляють 260 мл метиленхлориду. Додають 130 мл 10 % водного розчину натрію гідрокарбонату і фракції розділяють та водну фракцію екстрагують 130 мл метиленхлориду. Об'єднані органічні фракції упарюють при зниженому тиску з одержанням 4-фтор3,3-диметокси-1-триметилсилілбутина (59,0 г, 92 %) як проміжної сполуки, прекурсора цільової сполуки (10). Цю сполуку використовують в наступній реакції без додаткового очищення. 1 Н ЯМР (500 МГц, CDCI3): 4,38 (д, J = 47,1 Гц, 2Н), 3,40 (с, 6Н), 0,20 (с, 9Н). 4-Фтор-3,3-диметокси-1-триметилсилілбутин 59,0 г (289 ммоль), прекурсор цільової сполуки (10), одержаний вище, розчиняють в 280 мл метиленхлориду, обробляють 59 мг (0,183 ммоль) тетра-н-бутиламонію броміду і 347 мл (347 ммоль) 1 н водного розчину натрію гідроксиду та перемішують протягом приблизно 2 год. Органічну фракцію відокремлюють і водну фракцію екстрагують 110 мл метиленхлориду. Об'єднані органічні фракції промивають 110 мл розсілу та упарюють при зниже3 ному тиску з одержанням цільової сполуки (10, R , 3’ R = метил; 40,9 г, кількісний вихід). Одержану сполуку застосовують в наступній реакції без додаткового очищення. 1 Н ЯМР (500 МГц, CDCI3): 4,42 (д, J = 47,1 Гц, 2Н), 3,42 (с, 6Н), 2,64 (с, 1Н) 13 С ЯМР (125 МГц, CDCI3): 96,1 (д, J = 20,3 Гц), 82,9 (д, J = 180 Гц), 77,5, 75,5, 51,0 Препаративний приклад 3 Етил 5-фтор-4,4-диметокси-2-пентиноат (11, 3 3’ 4 R , R = метил, R = етил) Розчин 40,9 г (405 ммоль) діізопропіламіну в 270 мл тетрагідрофурану охолоджують до 0 °С та додають 112 г (405 ммоль) 2,5 М н-BuLi в н-гексані протягом приблизно 1 год, підтримуючи внутрішню температуру нижче 14 °С. Реакційну суміш перемішують при 0 °С, витримуючи при цій температурі протягом приблизно 30 хв, та температуру доводять до -78 °С. Розчин 41,0 г (311 ммоль) сполуки, одержаної в Препаративному прикладі 2 вище (10, 3 3’ R , R = метил) в 160 мл тетрагідрофурану додають до реакційної суміші протягом приблизно 2 год, підтримуючи внутрішню температуру нижче 40 °С, після чого додають 60,4 г (557 ммоль) етил 94395 18 хлорформіату протягом приблизно 1 год, підтримуючи внутрішню температуру нижче -40 °С. Реакційну суміш додатково перемішують при 0 °С, витримуючи при цій температурі протягом приблизно 2 год. До реакційної суміші додають 250 мл 10 % водного розчину амонію хлориду для закінчення реакції, та органічну фракцію відокремлюють. Водну фракцію екстрагують 100 мл етилацетату та об'єднані органічні фракції промивають 100 мл розсілу та упарюють при зниженому тиску з одер3 3’ жанням сирої цільової сполуки (11, R , R =метил, 4 R = етил; 95,0 г, обчислений вихід 70 %). Одержану сполуку застосовують в наступній реакції без додаткового очищення 1 Н ЯМР (500 МГц, CDCI3): 4,45 (д, J = 46,5 Гц, 2Н), 4,25 (к, J = 7,1 Гц, 2Н), 3,43 (с, 6Н), 1,31 (т, J = 7,3 Гц, 3Н) Препаративний приклад 4 Етил 3-(бензиламіно)-5-фтор-4,43 3’ 4 диметоксипентаноат (12, R , R = метил, R = етил) 88 г (431 ммоль) сирої сполуки, одержаної в 3 3’ Препаративному прикладі 3 вище (11, R , R = 4 метил, R = етил), розчиняють в 430 мл метилтрет-бутилового ефіру (МТВЕ) і температуру доводять до 0 °С. До реакційної суміші додають 31,4 г (293 ммоль) бензиламіну, перемішують при температурі 20 °С, витримуючи при цій температурі протягом приблизно 1 год, і розбавляють 450 мл метил-трет-бутилового ефіру. Температуру реакційної суміші знову доводять до 0 °С, до реакційної суміші додають 33 г (873 ммоль) NaBH4, після чого додають 259 г (4320 ммоль) оцтової кислоти протягом приблизно 30 хв. Реакційну суміш витримують при 0 °С та повільно додають 880 мл (2640 ммоль) 3 н водного розчину натрію гідроксиду повільно додають протягом приблизно 2 год. Органічну фракцію відокремлюють та відокремлену органічну фракцію промивають 880 мл 10 % водного розчину амонію хлориду, після чого додають 880 мл 1 н водної хлористоводневої кислоти. Водну фракцію відокремлюють, промивають 400 мл метил-трет-бутилового ефіру, підлужують за допомогою 246 мл 10 н водного розчину натрію гідроксиду, та екстрагують 2 х 700 мл метил-третбутилового ефіру. Об'єднані органічні фракції промивають 400 мл розсілу та упарюють при зниженому тиску з одержанням цільової сполуки [12, 3 3’ 4 R і R = метил, R = етил; 60,0 г, 44 % та 65 % від кількості сполуки формули (10)]. Одержану сполуку застосовують в наступній реакції без додаткового очищення. 1 Н ЯМР (400 МГц, CDCI3): 7,35-7,21 (м, 5Н), 4,53 (2дд, J = 46,8, 10,4 Гц, 2Н), 4,13 (к, J = 7,2 Гц, 2Н), 3,80 (2д, J = 12,8 Гц, 2Н), 3,53 (дд, J = 8,4, 4,0 Гц, 1Н), 3,30 (с, 3Н), 3,22 (с, 3Н), 2,79 (дд, J = 15,6, 3,6 Гц, 1Н), 2,40 (ддд, J = 15,6, 8,0, 1,6 Гц, 1Н), 1,25 (т, J = 7,2 Гц, 3Н). Приклад 1 Етил 3-аміно-5-фтор-4,4-диметоксипентаноат 3 3’ 4 (4, R , R = метил, R = етил) Сполуку, одержану в Препаративному прикла3 3’ 4 ді 4 вище (12, R , R = метил, R = етил) (18,3 г, 58,5 ммоль) розчиняють в 180 мл етанолу та здійснюють дебензилювання з використанням каталі 19 затора 5 % паладію на активованому вугіллі (5 % 2 Pd/C) при тиску водню 50 фунт/дюйм протягом приблизно 4 год. Реакційну суміш фільтрують крізь 5,0 г броунмілериту, промивають 90 мл етанолу і фільтрат упарюють при зниженому тиску з одержанням цільової сполуки (4, 12,8 г, 98 %). Одержану сполуку застосовують на наступній стадії без додаткового очищення. 1 Н ЯМР (500 МГц, CDCI3): 4,53 (2дд, J = 46,5, 10,4 Гц, 2Н), 4,14 (к, J = 7,3 Гц, 2Н), 3,57 (дд, J = 11,0, 1,9 Гц, 1Н), 3,29 (д, J = 11,7 Гц, 6Н), 2,73 (дд, J = 16,5, 2,5 Гц, 1Н), 2,36 (ддд, J = 16,5, 10,4, 2,5 Гц, 1Н), 1,25 (т, J = 7,3 Гц, 3Н). Препаративний приклад 5 5-Фтор-3-[((Р)-5-ізопропіл-3-(І-ізохінолініл)-4,5дигідро-ізоксазол-5-карбоніл)-аміно]-4,4диметокси-пентанової кислоти етиловий естер (13, 1 2 3 3' R = І-ізохінолініл, R = ізопропіл, R , R = метил, 4 R = етил) (5R)-5-Ізопропіл-3-(1-ізохінолініл)-4,5-дигідро1 5-ізоксазолкарбонову кислоту (2, R = 12 ізохінолініл, R = ізопропіл) (15,5 г, 54,5 ммоль)розчиняють в 150 мл метиленхлориду, температуру доводять до 0 °С, після чого додають 7,1 мл (81,7 ммоль) оксалілхлориду та 0,2 мл (2,6 ммоль) ДМФА, підтримуючи внутрішню температуру нижче 12 °С. Реакційну суміш перемішують при 20 °С, витримуючи при цій температурі протягом приблизно 2 год, та упарюють при зниженому тиску. Реакційну суміш розчиняють в 150 мл метиленхлориду, температуру доводять до 0 °С, додають триетиламін та повільно додають розчин 12,8 г (57,4 ммоль) сполуки, одержаної в Прикладі 1 (4, 3 3' 4 R , R = метил, R = етил), в 30 мл метиленхлориду протягом 20 хв. Реакційну суміш перемішують при 25 °С, витримуючи при цій температурі протягом 1,5 год і додають суміш 120 мл 10 % водного розчину натрію гідрокарбонату та 60 мл 1 н водного розчину натрію гідроксиду для завершення реакції. Органічну фракцію відокремлюють, і водну фракцію екстрагують 3 150 мл метиленхлориду. Об'єднані органічні фракції упарюють при зниже1 ному тиску з одержанням цільової сполуки (13, R 2 3 3’ 4 = 1-ізохінолініл, R = ізопропіл, R , R = метил, R = етил; 30,1 г, кількісний вихід). Одержану сполуку застосовують на наступній стадії без додаткового очищення. 1 Н ЯМР (500 МГц, CDCI3): 9,12 (к, 1Н), 8,53 (м, 1Н), 7,85-7,25 (м, 4Н), 4,80 (м, 1Н), 4,54-4,34 (м, 2Н), 4,14 (к, J = 7,4 Гц, 2Н), 3,99 (2d, J = 18,4 Гц, 1Н), 3,81 (м, 1Н), 3,78 (2d, J = 8,6 Гц, 1Н), 3,33 (д, 3Н), 3,20 (д, 3Н), 2,75 (м, 3Н), 2,53 (м, 1Н), 2,39 (гептет, J = 6,7 Гц, 1Н), 1,27 (т, J = 7,4 Гц, 1,5Н), 1,07 (м, 6Н), 0,97 (т, J = 7,4 Гц, 1,5Н). Препаративний приклад 6 5-Фтор-3-[((R)-5-ізопропіл-3-(І-ізохінолініл)-4,5дигідро-ізоксазол-5-карбоніл)-аміно]-4,41 диметокси-пентанова кислота (14, R = І2 3 3’ ізохінолініл, R = ізопропіл, R , R = метил) Сполуку, одержану в Препаративному прикла1 2 ді 5 вище (13, R =1- ізохінолініл, R = ізопропіл, 3 3' 4 R , R = метил, R = етил) (30,1 г, 61,6 ммоль), разом з 7,76 г (185 ммоль) літію гідроксиду моногідрату розчиняють в розчиннику, що являє собою суміш 168 мл тетрагідрофурану та 42 мл води, і 94395 20 перемішують при приблизно 40 °С, витримуючи при цій температурі протягом 4 год. Реакційну суміш упарюють при зниженому тиску для видалення тетрагідрофурану з розчинника, додають 180 мл 1 н водного розчину натрію гідроксиду і суміш промивають 2120 мл толуолу. Водну фракцію підкислюють 66 мл 6 н водної хлористоводневої кислоти, екстрагують 3150 мл метиленхлориду та об'єднані органічні фракції упарюють при зниже1 ному тиску з одержанням цільової сполуки (14, R 2 3 3’ = 1-ізохінолініл, R = ізопропіл, R , R = метил; 25,4 г, 89 %). Одержану сполуку застосовують на наступній стадії без додаткового очищення. 1 Н ЯМР (400 МГц, CDCI3): 9,10-8,92 (м, 1Н), 8,52 (м, 1Н), 7,86-7,13 (м, 4Н), 4,77 (м, 1Н), 4,544,34 (м, 2Н), 3,95 (2d, J = 8,0 Гц, 1Н), 3,75 (2d, J = 18,4 Гц, 1Н), 3,35-3,16 (2d, 6H), 2,78 (2дд, J = 16,0, 4,4 Гц 1Н), 2,54 (м, 1Н), 2,39 (м, 1Н), 2,35 (с, 1Н), 1,06 (м, 6Н). Приклад 2 (45,55)-5-Фторметил-5-гідрокси-4-({[(5Р)-5ізопропіл-3-(І-ізохінолініл)-4,5-дигідро-5ізоксазоліл]карбоніл}аміно)-2-дигідрофуранон (1, 1 2 R = 1-ізохінолініл, R = ізопропіл) Сполуку, одержану в Препаративному прикла1 2 ді 6 вище (14, R = 1-ізохінолініл, R = ізопропіл, 3 3’ R , R = метил) (17,0 г, 36,9 ммоль) та 6,6 мл (110 ммоль) оцтової кислоти розчиняють в 123 мл (738 ммоль) 6 н водної кислоти хлористоводневої та перемішують протягом приблизно 4 год. Внутрішню температуру реакційної суміші доводять до 0 °С та додають 150 мл етилацетату. Повільно додають 220 мл (660 ммоль) 3 н водного розчину натрію гідроксиду для доведення рН приблизно до 3. Органічну фракцію відокремлюють, і водну фракцію екстрагують 2150 мл етилацетату. Об'єднані органічні фракції промивають 100 мл розсілу та упарюють при зниженому тиску. Залишок розбавляють 50 мл толуолу та знову упарюють при зниженому тиску з одержанням суміші сполук форму1 2 ли (15) та формули (16) (R = 1-ізохінолініл, R = ізопропіл) (15,4 г, кількісний вихід, хімічна чистота 87,0 %). 1 Н ЯМР (500 МГц, ДMCO-d6): 8,99 (м, 1Н), 8,65 (м, 1Н), 8,19-7,78 (м, 4Н), 5,15 (м, 1,5Н), 4,77 (м, 1Н), 4,42 (м, 0,5Н), 3,91 (2d, J = 17,6 Гц, 1Н), 3,74 (м, 1Н), 2,99 (м, 0,2Н), 2,82 (м, 1Н), 2,63 (м, 0,8Н), 2,33 (м, 1Н), 0,97 (м, 6Н). До 146 мл толуолу додають 14,6 г (35,2 ммоль) суміші сполук формули (15) та формули 1 2 (16) (R = 1-ізохінолініл, R = ізопропіл) (хімічна чистота: 87,0 %), і суміш нагрівають до 100 °С, витримуючи до повного розчинення. Далі додають 1 14 мг затравки цільової сполуки (1, R = 12 ізохінолініл, R = ізопропіл), температуру повільно знижують до 20 °С і реакційну суміш перемішують з утворенням твердої речовини. Додають 0,25 мл (1,8 ммоль) діізопропіламіну та перемішують при 20 °С, витримуючи при цій температурі протягом приблизно 2 тижнів, до підтвердження за допомогою ВЕРХ співвідношення між сполукою формули 1 (15) та сполукою формули (16) (R = 1-ізохінолініл, 2 R = ізопропіл) 92,8:7,2. Реакційну суміш упарюють при зниженому тиску для видалення толуолу, додають 88 мл етилацетату і суміш нагрівають до 65 21 94395 °С, витримуючи до повного розчинення. Далі додають 88 мл н-гексану додають, температуру повільно знижують та перемішують при приблизно 20 °С, витримуючи при цій температурі протягом 2 днів. Одержану тверду речовину відфільтровують і промивають сумішшю 15 мл етилацетату та 15 мл н-гексану. Після висушування твердої речовини азотом одержують цільову сполуку у вигляді білої 1 2 твердої речовини (1, R = 1-ізохінолініл, R = ізопропіл) з виходом 54,7 % від кількості сполуки формули (2) (8,0 г, хімічна чистота 98,6 %). Дані твердофазного ЯМР для кристалічної форми одержані з використанням VACP MAS (крос-поляризація з обертанням зразка під магічним кутом із змінною амплітудою) при швидкості обертання 9 кН. 1 Н ЯМР (CDCI3): 9,02 (шс, 1Н), 8,54 (д, J = 5,5 Гц, 1Н), 7,85 (д, J = 7,95 Гц, 1Н), 7,70 (м, 3Н), 7,60 (шс, 1Н), 4,86 (шс, 1Н), 4,2-5,2 (шс, 2Н), 4,05 (b, J = 19,0 Гц, 1Н), 3,78 (ш, J = 19,0 Гц, 1Н), 2,7-3,1 (шм, 2Н), 2,40 (м, 1Н). 1,08 (дд, J = 6,7, 4,9 Гц, 6Н) 13 С ЯМР (CDCI3): 173,8, 172,4, 160,2, 147,6, 141,7, 136,8, 130,7, 129,0, 127,4, 127,3, 126,8, 122,9, 92,3, 82,7 (д, J = 215 Гц), 48,9 (b), 44,6, 34,4, 33,9, 17,7, 16,3. 13 С ЯМР (тверда речовина): 176,4, 171,8, 160,3, 150,2, 139,5, 137,5, 132,3 (2С), 127,7 (3С), 123,0, 104,3, 94,1, 86,4, 48,8, 42,9, 32,7 (2С), 19,6, 15,4. Мас-спектр (співвідношення маси до заряду): 416,14 (М+1). 25 []D 3,2(c 10, ацетонітри ) , л Експериментальний приклад 1 Дослідження стабільності Як показано на фіг. 2, в результаті досліджень стабільності, проведених на аморфній формі і кри1 сталічній формі сполуки формули (1) (R = 12 ізохінолініл, R = ізопропіл), спостерігалося, що 50 % аморфної форми розкладалося після проходження 28 днів в суворих умовах (60 °С), але кількість кристалічної форми в таких же умовах (60 °С) не зменшувалася взагалі навіть після проходження 28 днів. Таким чином, слід розуміти, що кристалічна форма має в достатній мірі більш високу стабільність, в порівнянні з аморфною формою з точки зору застосування в композиції інгібітора або терапевтичного агента. Експериментальний Приклад 2 Вплив лікування на індукований ліпополісахаридом (ЛПС) гострий гепатит у мишей Стадія 1: Одержання зразка крові Мишей-самців (6 тижнів, Charles River Laboratory, Осака, Японія) Balb/c розводили при температурі 22 °С та відносній вологості 55 %, із зміною 22 ночі/дня через 12 годин. Харчування і воду надавали без обмежень. ЛПС (ліпополісахарид) і Dгалактозамін розчиняли в концентраціях 0,4 мг/мл і 280 мг/мл, відповідно, в апірогенному сольовому розчині, при співвідношенні змішування 1:1. Розчин вводили ін'єкційним способом мишам в кількості 5 мл/кг. Безпосередньо після ін'єкції ЛПС і Dгалактозаміну, вводили носій з розчиненою досліджуваною сполукою (суміш, що складається з РЕС400/етанолу/Твіну 80 у співвідношенні 15:7,5:2,5, розведена 1/5 сольовим розчином) або тільки носій ін'єкційним способом досліджуваній тварині. Зразки крові одержували з серця мишей через 8 годин після ін'єкції лікарського засобу. Стадія 2: Аналіз активності амінотрансферази плазми крові Активність АЛТ плазми крові в зразках крові, одержаних на Стадії 1, вимірювали з використанням набору для аналізу АЛТ (Asan pharmaceutical company) у відповідності до протоколу виробника. Спостерігалося, що введення ЛПС і Dгалактозаміну швидко підвищувало активність АЛТ в плазмі крові, і досліджуваний матеріал інгібував таку підвищену ферментну активність дозозалежним чином. На базі вказаних результатів значення ED50 для кожного зразка досліджуваного матеріалу обчислювали із застосуванням програмного забезпечення Prism (GraphPad Co.). Експериментальний приклад 3 Вплив лікування на індукований антитілом Fas гострий гепатит у мишей Мишей-самців (6 тижнів, Charles River Laboratory, Осака, Японія) Balb/c розводили при температурі 22 °С та відносній вологості 55%, із чергуванням ніч/день через 12 годин. Харчування і воду надавали без обмежень. Антитіло Fas розчиняли в концентраціях 30 мкг/мл в апірогенному сольовому розчині, і розчин вводили ін'єкційним способом мишам в кількості 5 мл/кг. Безпосередньо після ін'єкції антитіла Fas, вводили носій з розчиненою досліджуваною сполукою (суміш, що складається з РЕС400/етанолу/Твіну 80 у співвідношенні 15:7,5:2,5, розведена 1/5 сольовим розчином) або тільки носій ін'єкційним способом досліджуваній тварині. Зразки крові одержували з серця мишей через 8 годин після ін'єкції лікарського засобу. Значення ED50 для кожного зразка досліджуваного матеріалу обчислювали із застосуванням вказаного вище метода. Наступна табл. 1 демонструє результати дослідження фармакологічного впливу на моделі гострого гепатиту у відповідності до способу введення сполуки формули (1), одержаної в наведених вище Прикладах 2 і 3. Таблиця 1 Модель 1 Ліпополісахарид/D-глюкозамін Антитіло Fas Спосіб введення Внутрішньовенне введення Пероральне введення Внутрішньовенне введення Пероральне введення ED50 (мг/кг) 95% довірчий інтервал (мг/кг) 0,015 0,002-0,111 0,02 0,003-0,118 0,003 0,001-0,006 0,018 0,013-0,026 23 Промислова придатність Похідне ізоксазоліну, що містить циклічний гемікеталь карбонової кислоти формули (1) у відповідності до даного винаходу, володіє чудовою активністю з інгібування каспази і чудовою стабільністю. Крім того, спосіб у відповідності до даного винаходу дає тільки один діастереоізомер високої чистоти за допомогою індукованого кристалізацією динамічного перетворення. Крім того, якщо амінне похідне, що містить кеталь у відповідності до даного винаходу, застосовують як проміжну сполуку, фахівець в даній галузі може легко одержати похідне ізоксазоліну, що містить циклічний гемікеталь карбонової кислоти без додаткового очищення. Література: [Dementia: Arch Neurol 2003 Mar; 60(3): 369-76, Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Pompl PN, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R, Pasinetti GM.; Cerebral stroke: Proc Natl Acad Sci USA 2002 Nov 12; 99(23): 15188-93, Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA.; Brain impairment due to AIDS: J Neurosci 2002 May 15; 22(10): 4015-24, Caspase cascades in human immunodeficiency virus-associated neurodegeneration. Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA.; Diabetes: Diabetes 2002 Jun; 51(6): 1938-48, Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase3 activation pathway. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ.; Gastric ulcer: J Physiol Pharmacol 1998 Dec; 49(4): 489-500, Role of basic fibroblast growth factor in the suppression of apoptotic caspase-3 during chronic gastric ulcer healing. Slomiany BL, Piotrowski J, Slomiany A.; Cerebral injury by hepatitis: J Viral Hepat 2003 Mar; 10(2): 81-6, Cerebral dysfunction in chronic he 94395 24 patitis C infection. Forton DM, Taylor-Robinson SD, Thomas HC; Fulminant hepatic failure: Gastroenterology 2000 Aug; 119(2): 446-60, Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Streetz K, Leifeld L, Grundmann D, Ramakers J, Eckert K, Spengler U, Brenner D, Manns M, Trautwein C; Sepsis: Nat Immunol 2000 Dec; 1(6): 496-501, Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S, Roy S, Black C, Grimm E, Aspiotis R, Han Y, Nicholson DW, Karl IE.; Organ transplantation rejection: Xenotransplantation 2001 May; 8(2): 115-24, In vitro prevention of cell-mediated xenograft rejection via the Fas/FasLpathway in CrmA-transducted porcine kidney cells. Fujino M, Li XK, Suda T, Hashimoto M, Okabe K, Yaginuma H, Mikoshiba K, Guo L, Okuyama T, Enosawa S, Amemiya H, Amano T, Suzuki S.; Rheumatoid arthritis: Prog Med Chem 2002; 39: 1-72, Caspase inhibitors as antiinflammatory and antiapoptotic agents. Graczyk PP.; Ischemic cardiac diseases: Am J Physiol Heart Circ Physiol 2002 Sep; 283(3): H990-5, Hypoxiainduced cleavage of caspase-3 and DFF45/ICAD in human failed cardiomyocytes. Todor A, Sharov VG, Tanhehco EJ, Silverman N, Bernabei A, Sabbah HN.; Anti-inflammation: J Immunol 2003 Mar 15; 170(6): 3386-91, A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. Iwata A, Nishio K, Winn RK, Chi EY, Henderson WR Jr, Harlan JM.; Hepatocirrhosis: i) J Pharmacol Exp Ther. 2004 Mar; 308(3): 1191-6; The caspase inhibitor ldn-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. Canbay A., Fledstein A., Baskin-Bey E., Bronk FS. Gores GJ.; ii) Hepatology. 2004 Feb; 39(2): 273-8, Apoptosis: the nexus of liver injury and fibrosis. Canbay A, Friedman S, Gores GJ.; iii) Hepatology. 2003 Nov; 38(5): 1188-98, Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Canbay A, FeldsteinAE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ.]. 25 Комп’ютерна верстка М. Мацело 94395 Підписне 26 Тираж 24 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюIsoxazoline derivative and process for its preparation
Автори англійськоюShin Hyun Ik, Choi Hyeong Wook, Heo Tae Ho, Lee Kyu Woong, Lee Jae Hoon, Park Ki Sook
Назва патенту російськоюПроизводное изоксазолина и способ его получения
Автори російськоюШин Хьюн Ик, Чой Хьеонг Воок, Хео Тае Хо, Ли Кью Воонг, Ли Джа Хоон, Парк Ки Соок
МПК / Мітки
МПК: C07D 413/12
Мітки: ізоксазоліну, спосіб, похідне, одержання
Код посилання
<a href="https://ua.patents.su/13-94395-pokhidne-izoksazolinu-ta-sposib-jjogo-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Похідне ізоксазоліну та спосіб його одержання</a>
Спосіб одержання циталопраму та проміжні сполуки для його одержання

Номер патенту: 63034
Опубліковано: 15.01.2004
Автори: Петерсен Ханс, Сван Хенрік, Рок Майкл Харольд
МПК: C07D 307/81, A61K 31/343, C07D 307/87, A61P 29/00, A61P 25/24, C07B 61/00
Мітки: сполуки, одержання, спосіб, проміжні, циталопраму
Формула / Реферат:
1. Спосіб одержання циталопраму, під час якого здійснюють взаємодію сполуки формули IV:, IVу якій R являє собою галоген або групу СF3-(СF2)n-SO2-O-, де n являє собою ціле число в інтервалі 0-8, включно, із джерелом ціаніду в присутності паладієвого каталізатора і каталітичної кількості Сu+ або Zn2+, або з Zn(CN)2 у присутності паладієвого каталізатора, і виділяють відповідну 5-ціаносполуку, тобто циталопрам:, (I)у...
Похідне триазолу, його фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб його отримання та фармацевтична композиція
Номер патенту: 27672
Опубліковано: 15.09.2000
Автори: Стріт Леслі Дж., Матасса Віктор Г., Бейкер Реймонд
МПК: C07D 403/06, C07D 401/14, C07D 409/04
Мітки: 5-нt1-подібних, прийнятні, проліки, агоністами, фармацевтично, композиція, отримання, солі, триазолу, спосіб, рецепторів, похідне, фармацевтична
Текст:
...призначення або призначення шля хом ін'єкцій , включають водні розчини , ароматизо вані сиропи, во дні або масляні емульсії, ароматизовані емульсі ї, з такими маслами, як бавовняне масло, сезамове масло, кокосове масло, а також еліксири та подібні засоби . Відпо відні диспер гуючі та суспензуючі аген ти для водни х суспензій включають синте ти чні і натуральні смоли, такі як трагант, акація, альгінат, декстран, натрієва сіль...
Похідне хіназоліну, спосіб його одержання, фармацевтична композиція на його основі

Номер патенту: 34426
Опубліковано: 15.03.2001
Автор: Баркер Ендрю Джон
МПК: C07D 491/056, C07D 239/94, C07D 403/04, C07D 491/04
Мітки: фармацевтична, основі, композиція, похідне, хіназоліну, спосіб, одержання
Текст:
...суль финильную груп пу,также можно использовать более слабый окислитель, например, метапериодат натрия или калия, обычно в полярном растворителе, таком как уксусная кислота или этанол. При необходимости получения соединения формулы I, содержащего /1-4С/алкилсульфонильную группу, его можно получить путем окисления соответствующего /1-4С/алкилсульфи нильного соединения, а также соответствующего /14С/алкилтиосоединения. (d) Для получе ния...
Лікарський препарат іресси, який містить водорозчинне похідне целюлози (варіанти), та спосіб його одержання

Номер патенту: 76325
Опубліковано: 17.07.2006
Автори: Геллерт Пол Річард, Паркер Майкл Девіс, де Матас Марсель
МПК: A61K 47/38, A61K 31/537
Мітки: містить, варіанти, одержання, спосіб, целюлози, препарат, водорозчинне, лікарський, іресси, похідне
Формула / Реферат:
1. Фармацевтична композиція, яка містить 4-(3'-хлор-4'-фтораніліно)-7-метокси-6-(3-морфолінопропокси)хіназолін або його фармацевтично прийнятну сіль (засіб) і простий водорозчинний ефір целюлози або складний ефір простого водорозчинного ефіру целюлози.2. Фармацевтична композиція відповідно до пункту 1, яка містить засіб і простий водорозчинний ефір целюлози, де простий водорозчинний ефір целюлози вибраний з гідроксіетилцелюлози,...
Похідне піколінової кислоти або його сіль, що має гербіцидну активність, спосіб його одержання, гербіцидна композиція, спосіб знищення бур’янів
Номер патенту: 27422
Опубліковано: 15.09.2000
Автори: Сігєхіко Татікава, Ріо Ханаі, Масатосі Тамару, Фуміакі Такабе, Йосіхіро Саіто
МПК: A01N 43/54, C07D 401/12, A01P 13/00
Мітки: сіль, бур'янів, активність, знищення, має, піколінової, гербіцидну, гербіцидна, похідне, одержання, спосіб, кислоти, композиція
Текст:
...Процесс G COOR' осн, COOR 4 Основание Base [П - 4 ) 6 /где R имеет значение, определенное выше, R10 представляет алкильную группу, замещенную алкильную группу или 4,6-диметоксипиридинильную группу, и L2 представляет атом галогена, при условии, что L2 представляет атом галогена или алкилсульфонильную группу, когда R10 представляет 4,6-диметоксипиримидинильную группу/. Так, соединение формулы Н-8 может быть получено путем...
Попередній патент: Ліофілізований продукт з клітин бурих водоростей, спосіб його одержання та використання
Наступний патент: Курильний пристрій та тютюновий картридж для використання в наргіле або з іншим курильним пристроєм
Випадковий патент: Вакуумний вимикач