Спосіб одержання хіральних 8-(3-амінопіперидин-1-іл)ксантинів
Номер патенту: 100221
Опубліковано: 10.12.2012
Автори: Пахур Торстен, Пфренгле Вальдемар, Дуран Аділь, Нікола Томас




Формула / Реферат
1. Спосіб одержання сполуки загальної формули (І)
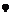 (I)
(I)
або її енантіомера або солі,
де
R1 являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну, нафтиридинілметильну або фенантридинілметильну групу, у якій ароматичний або гетероароматичний фрагмент в кожному випадку однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де
Ra являє собою атом водню, ціаногрупу, метильну, трифторметильну, етильну, фенільну групу, метокси-, дифторметокси-, трифторметокси- або етоксигрупу, або
два радикали Ra, за умови, що вони зв'язані із сусідніми атомами вуглецю, можуть являти собою також групу -О-СН2-О- або -О-СН2-СН2-О-,
R2 являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а
R3 являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2-хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2-(трифторметил)бензильну або 2-ціанбензильну групу,
який включає стадії синтезу, на яких:
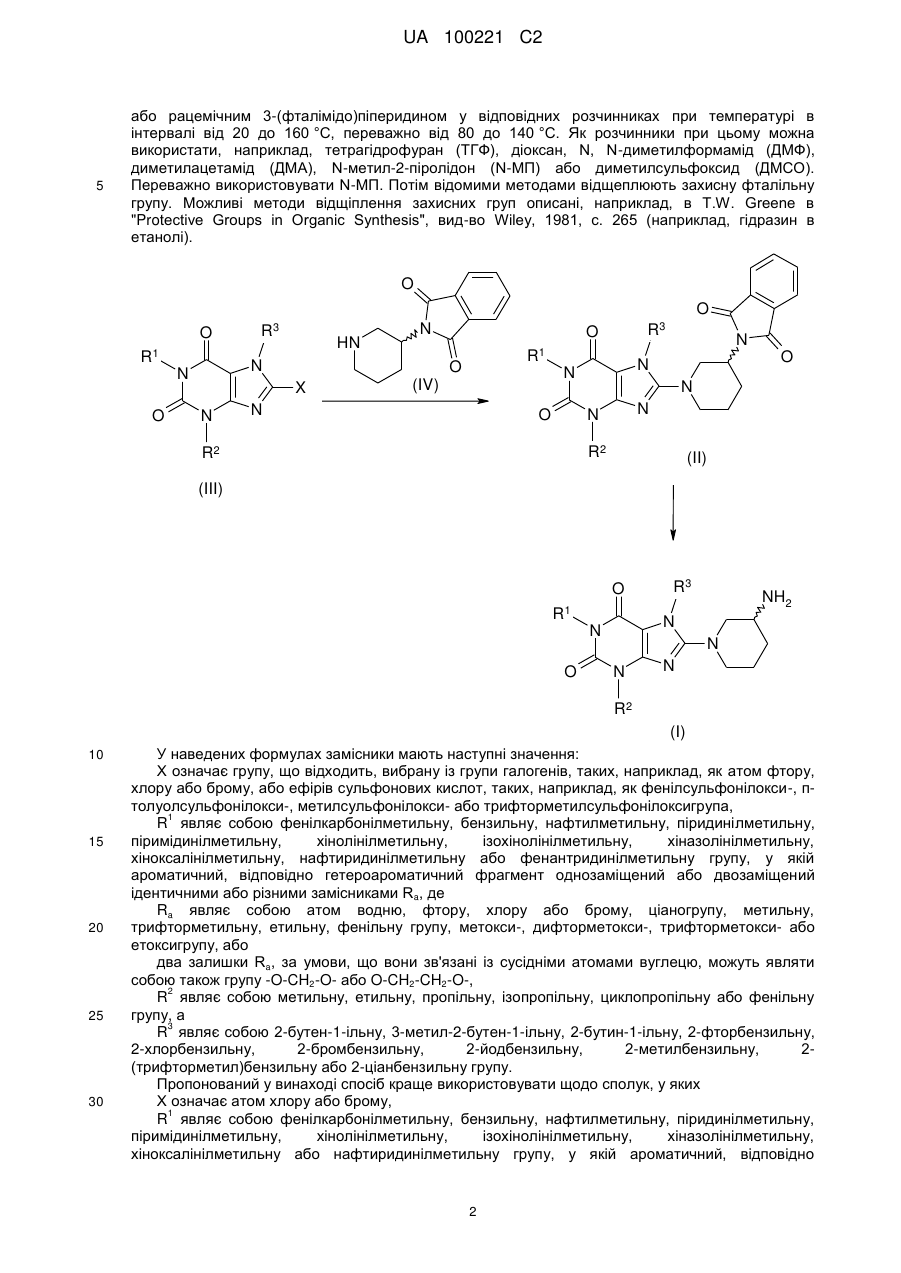
а) піддають взаємодії сполуку загальної формули (III)
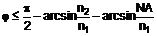 , (III)
, (III)
у якій X означає відхідну групу, вибрану із групи галогенів або ефірів сульфонових кислот, a кожний з R1-R3 мають зазначені вище значення,
з 3-(фталімідо)піперидином або його енантіомером,
б) видаляють захисну групу з одержаної таким шляхом сполуки загальної формули (II)
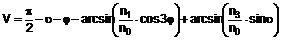 , (II)
, (II)
у якій кожен R1-R3 має зазначені вище значення, та
в) одержану сполуку при необхідності перетворюють на її фізіологічно сумісну сіль.
2. Спосіб за п. 1, в якому
X означає атом хлору або брому,
R1 являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну або нафтиридинілметильну групу, у якій ароматичний або гетероароматичний фрагмент однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де
Ra являє собою атом водню, ціаногрупу, метильну, етильну групу, метокси- або етоксигрупу,
R2 являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а
R3 являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2-хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2-(трифторметил)бензильну або 2-ціанобензильну групу.
3. Спосіб за п. 2, в якому
X означає атом хлору або брому,
R1 являє собою ціанобензильну, (ціанопіридиніл)метильну, хінолінілметильну, (метилхінолініл)метильну, ізохінолінілметильну, (метилізохінолініл)метильну, хіназолінілметильну, (метилхіназолініл)метильну, хіноксазинілметильну, (метилхіноксалініл)метильну, (диметилхіноксалініл)метильну або нафтиридинілметильну групу,
R2 являє собою метильну, циклопропільну або фенільну групу, а
R3 являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-хлорбензильну, 2-бромбензильну або 2-ціанобензильну групу.
4. Спосіб за п. 3, в якому
X означає атом брому,
R1 являє собою (4-метилхіназолін-2-іл)метильну, (3-метилізохінолін-1-іл)метильну або (3-ціанопіридин-2-іл)метильну групу,
R2 являє собою метильну групу, а
R3 являє собою 2-бутин-1-ільну групу.
5. Спосіб за будь-яким з пп. 1-4, де на стадії а) як реагент використовують (R)-3-(фталімідо)піперидин.
6. Спосіб за п. 5, в якому
X означає атом брому,
R1 являє собою (4-метилхіназолін-2-іл)метильну групу,
R2 являє собою метильну групу, та
R3 являє собою 2-бутин-1-ільну групу.
7. Спосіб за п. 6, в якому додатково проводять кристалізацію або перекристалізацію сполуки формули (І) з метанольного або етанольного розчину.
8. Спосіб за п. 7, в якому розчин додатково містить трет-бутилметиловий ефір.
9. Спосіб за п. 6, в якому додатково проводять кристалізацію сполуки формули (І) з етанолу.
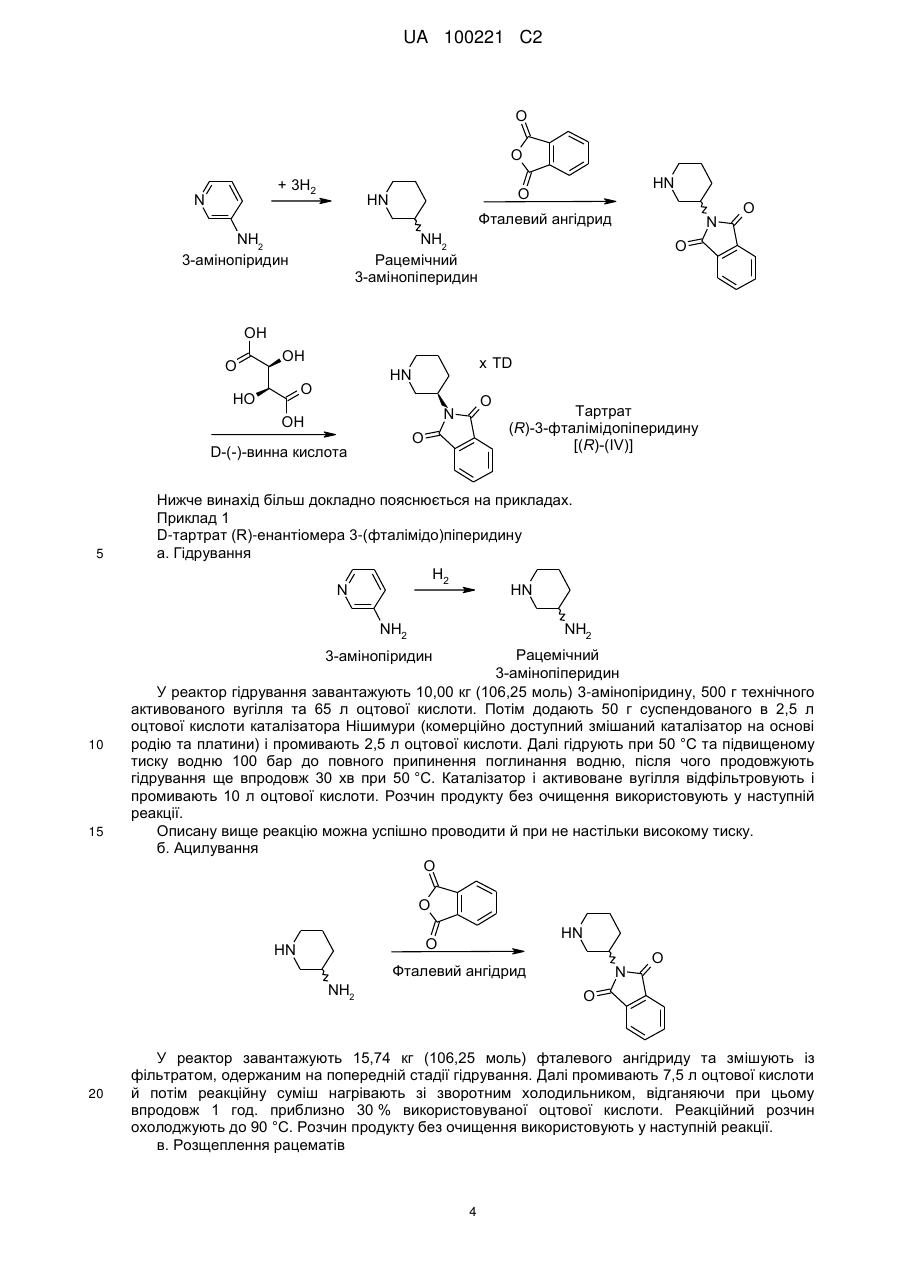
10. Спосіб одержання (R)-3-(фталімідо)піперидину, який включає стадії на яких:
а) піддають взаємодії рацемічний 3-амінопіперидин у відповідних розчинниках із фталевим ангідридом, і
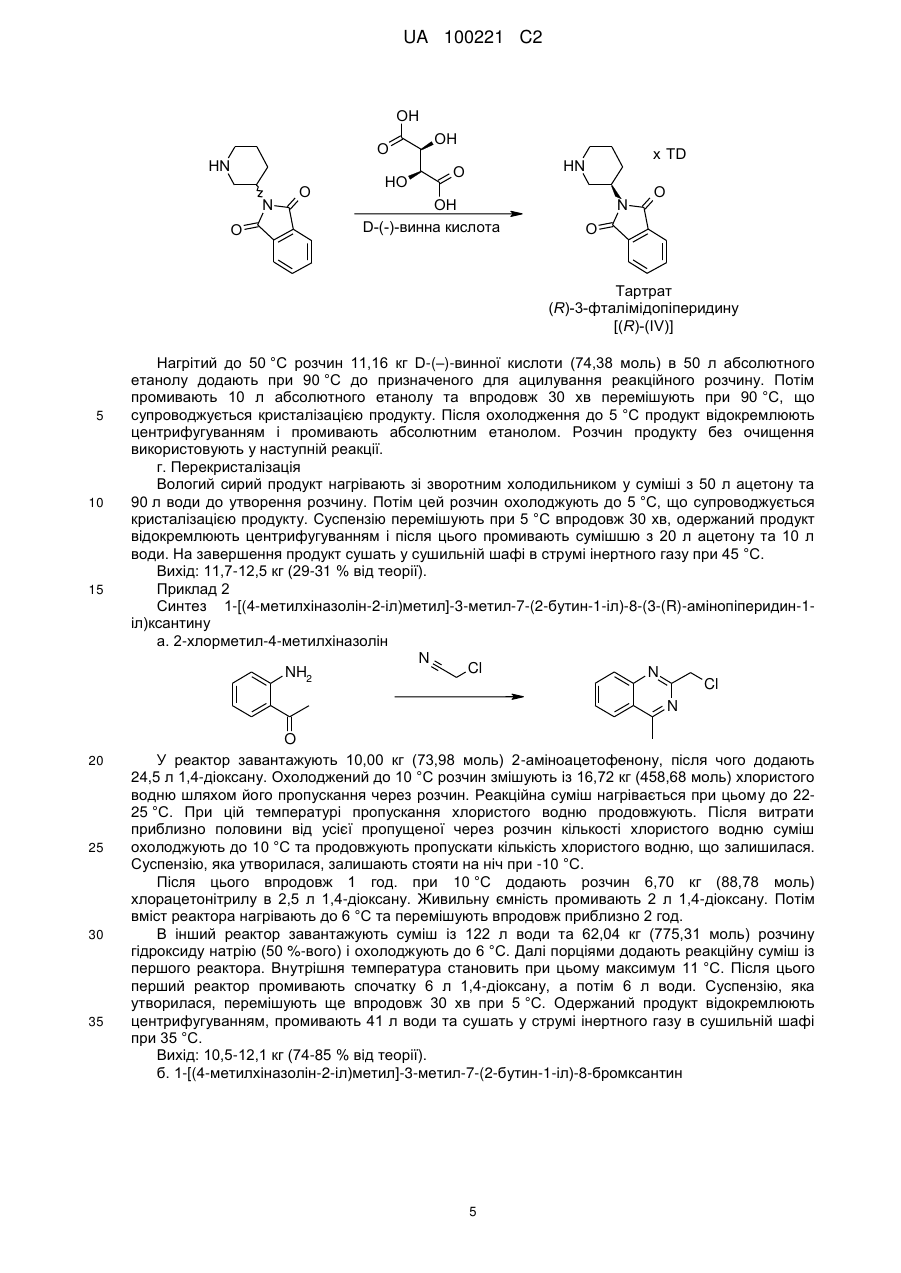
б) видаляють (R)-3-(фталімідо)піперидин із розчину одержаного таким чином рацемічного 3-(фталімідо)піперидину шляхом додавання D-винної кислоти та відокремлюють тартрат, що випав у осад.
11. Спосіб за п. 10, в якому на стадії б) як розчинник використовують етанол.
12. (R)-3-(фталімідо)піперидин.
13. D-(-)-тартрат (R)-3-(фталімідо)піперидину.
14. Спосіб одержання лікарського засобу, в якому одержують сполуку загальної формули (І), як визначено у пункті 1, або її енантіомер або сіль, шляхом здійснення способу за будь-яким з пп. 1-9, та об'єднують одержану сполуку загальної формули (І), як визначено у пункті 1, або її енантіомер або сіль, з одним або декількома інертними носіями і/або розріджувачами.
15. Сполука загальної формули (ІІ)
(ІI)
або її енантіомер, у якій кожен з R1-R3 має значення відповідно до визначення у будь-якому з пунктів 1-6.
16. Сполука наступної загальної формули
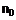 ,
,
у якій
R1 являє собою (4-метилхіназолін-2-іл)метильну, (3-метилізохінолін-1-іл)метильну або (3-ціанопіридин-2-іл)метильну групу,
R2 являє собою метильну групу, та
R3 являє собою 2-бутин-1-ільну групу.
17. Сполука за п. 16, у якій
R1 являє собою (4-метилхіназолін-2-іл)метильну групу,
R2 являє собою метильну групу, та
R3 являє собою 2-бутин-1-ільну групу.
18. Спосіб одержання лікарського засобу, в якому одержують сполуку наступної загальної формули
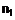
або її фізіологічно сумісну сіль,
у якій
R1 являє собою (4-метилхіназолін-2-іл)метильну групу,
R2 являє собою метильну групу, та
R3 являє собою 2-бутин-1-ільну групу,
шляхом здійснення способу за будь-яким з пп. 6, 7, 8 або 9, та об'єднують одержану сполуку з одним або декількома інертними носіями і/або розріджувачами.
19. Спосіб одержання сполуки наступної формули
,
де
R1 являє собою (4-метилхіназолін-2-іл)метильну групу,
R2 являє собою метильну групу, та
R3 являє собою 2-бутин-1-ільну групу,
причому в зазначеному способі видаляють фталільну захисну групу зі сполуки, вказаної в п. 17.
20. Спосіб за п. 19, в якому фталільну захисну групу видаляють в присутності етаноламіну, краще в толуолі або в суміші тетрагідрофурану та води.
21. Спосіб за п. 19 або 20, в якому додатково проводять кристалізацію незахищеної сполуки з етанолу або метанолу.
22. Спосіб за будь-яким з п. 19, 20 або 21, в якому незахищену сполуку кристалізують з етанолу.
23. Спосіб за п. 19, в якому додатково проводять кристалізацію незахищеної сполуки з етанолу.
Текст
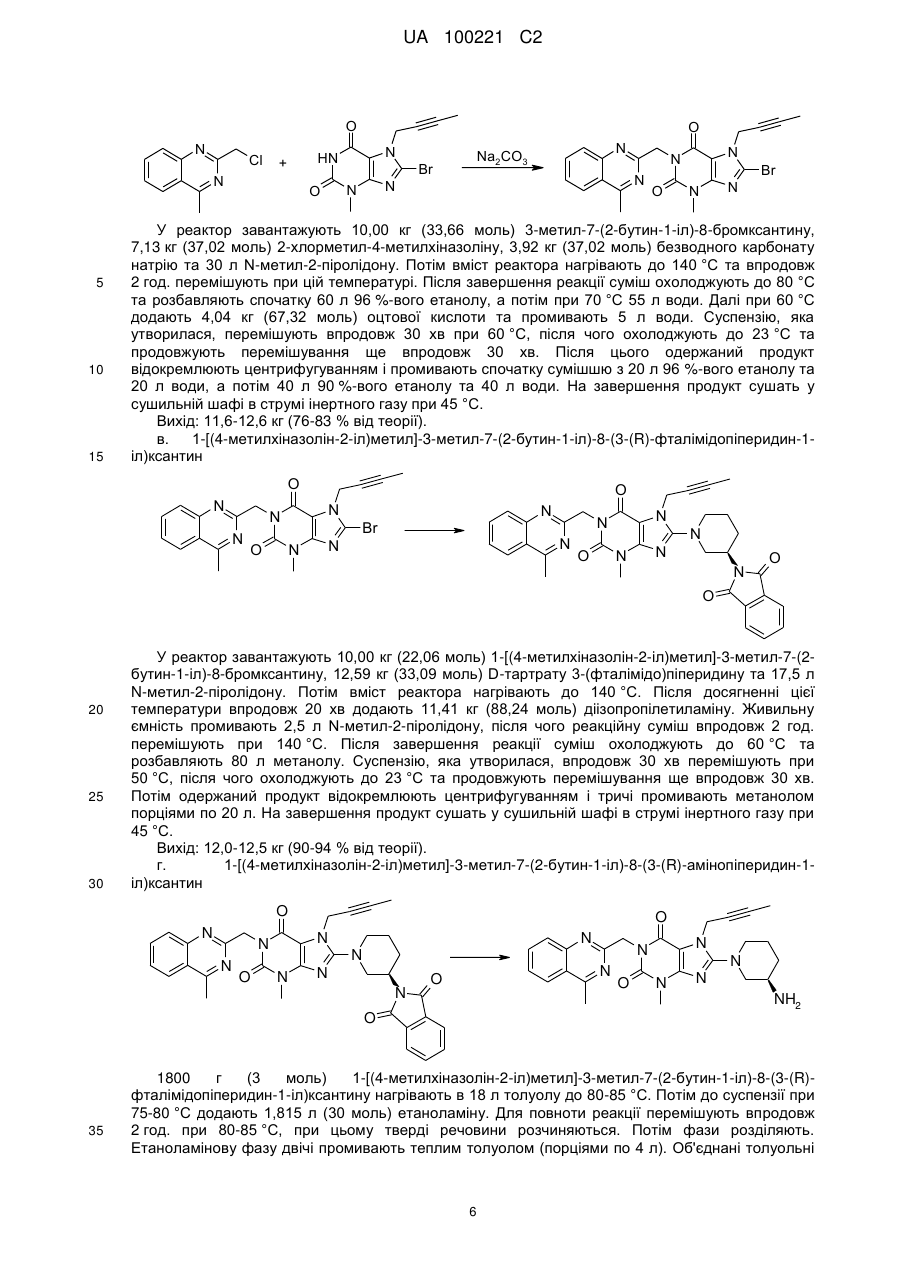
Реферат: У заявці описаний удосконалений спосіб одержання енантіомерно чистих 8-(3-амінопіперидин1-іл)ксантинів. UA 100221 C2 (12) UA 100221 C2 UA 100221 C2 Даний винахід стосується вдосконаленого способу одержання хіральних амінопіперидин-1-іл)ксантинів, їх енантіомерів та їх фізіологічно сумісних солей. 8-(3-амінопіперидин-1-іл)ксантини наступної загальної структурної формули R3 O R1 NH2 N N O 8-(3 N (I), N N R2 5 10 15 1 у якій R являє собою, наприклад, необов'язково заміщену арилметильну групу або 2 необов'язково заміщену гетероарилметильну групу, R являє собою, наприклад, алкільну групу, 3 а R являє собою, наприклад, необов'язково заміщену бензильну групу або прямоланцюгову або розгалужену алкенільну або алкінільну групу, вже відомі з WO 02/068420, WO 04/018468, WO 04/018467, WO 2004/041820 та WO 2004/046148, де описані сполуки з цінними фармакологічними властивостями, до яких належить насамперед пригнічуюча активність дипептидилпептидази-IV (DPP-IV) дія. Тому сполуки такого типу можуть застосовуватися для попередження або лікування захворювань або хворобливих станів, які пов'язані з підвищеною активністю DPP-IV або які можна запобігти або принаймні знизити їх гостроту за рахунок зменшення активності DPP-IV, насамперед цукрового діабету типу I або типу II, переддіабету або зниження толерантності до глюкози. В WO 04/018468 описаний спосіб одержання 8-(3-амінопіперидин-1-іл)ксантинів шляхом видалення захисної трет-бутилоксикарбонільної групи з відповідної похідної загальної формули (II) O R1 O O N N O H N R3 (II). N N N R2 20 25 30 35 40 При здійсненні цього способу, насамперед у промисловому масштабі, утворювалися важко відокремлювані домішки, обумовлені наявністю використовуваної захисної групи. Із цієї причини такий спосіб виявився не придатним для промислового одержання 8-(3-амінопіперидин-1іл)ксантинів, насамперед для одержання лікарських речовин з їх високими вимогами до чистоти. Ще один недолік цього відомого способу полягав у складності та високій вартості одержання енантіомерночистого попередника, тобто 3-(трет-бутилоксикарбоніламіно)піперидину. Щоб уникнути ж ризику побічних ефектів і з метою зниження дози, що вводиться в організм, до мінімуму для застосування у фармацевтиці кращі саме енантіомерночисті діючі речовини. Всі ці фактори спростовують можливість реалізації відомого способу для промислового одержання енантіомерночистих 8-(3-амінопіперидин-1-іл)ксантинів. З урахуванням описаних вище недоліків відомого способу в основу даного винаходу була покладена задача розробити спосіб, який дозволяв би з високою хімічною та оптичною чистотою без високих технічних витрат одержувати енантіомерночисті 8-(3-амінопіперидин-1іл)ксантини з використанням легко доступних вихідних речовин. Такий новий спосіб повинен бути також придатний для синтезу зазначених сполук у промисловому масштабі й тим самим для застосування в комерційних цілях. Зазначена задача вирішується за допомогою пропонованого у винаході способу одержання хіральних 8-(3-амінопіперидин-1-іл)ксантинів. До переваг пропонованого у винаході способу синтезу крім можливості його промислового здійснення й одержання цільових продуктів з високим виходом варто також віднести одержання цільових продуктів з винятково високим ступенем хімічної та оптичної чистоти. При здійсненні пропонованого у винаході способу відповідний попередник ксантинів формули (III) відповідно до наведеної нижче схеми 1 піддають взаємодії з енантіомерночистим 1 UA 100221 C2 5 або рацемічним 3-(фталімідо)піперидином у відповідних розчинниках при температурі в інтервалі від 20 до 160 °C, переважно від 80 до 140 °C. Як розчинники при цьому можна використати, наприклад, тетрагідрофуран (ТГФ), діоксан, N, N-диметилформамід (ДМФ), диметилацетамід (ДМА), N-метил-2-піролідон (N-МП) або диметилсульфоксид (ДМСО). Переважно використовувати N-МП. Потім відомими методами відщеплюють захисну фталільну групу. Можливі методи відщіплення захисних груп описані, наприклад, в T.W. Greene в "Protective Groups in Organic Synthesis", вид-во Wiley, 1981, с. 265 (наприклад, гідразин в етанолі). O O O R1 HN N N N O R3 N R1 O X R3 O N N N N O O N N (IV) N R2 R2 (II) (III) O R1 NH2 N N O R3 N N N R2 (I) 10 15 20 25 30 У наведених формулах замісники мають наступні значення: X означає групу, що відходить, вибрану із групи галогенів, таких, наприклад, як атом фтору, хлору або брому, або ефірів сульфонових кислот, таких, наприклад, як фенілсульфонілокси-, птолуолсульфонілокси-, метилсульфонілокси- або трифторметилсульфонілоксигрупа, 1 R являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну, нафтиридинілметильну або фенантридинілметильну групу, у якій ароматичний, відповідно гетероароматичний фрагмент однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де Ra являє собою атом водню, фтору, хлору або брому, ціаногрупу, метильну, трифторметильну, етильну, фенільну групу, метокси-, дифторметокси-, трифторметокси- або етоксигрупу, або два залишки Ra, за умови, що вони зв'язані із сусідніми атомами вуглецю, можуть являти собою також групу -O-CH2-O- або O-CH2-CH2-O-, 2 R являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2-хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2(трифторметил)бензильну або 2-ціанбензильну групу. Пропонований у винаході спосіб краще використовувати щодо сполук, у яких Х означає атом хлору або брому, 1 R являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну або нафтиридинілметильну групу, у якій ароматичний, відповідно 2 UA 100221 C2 5 10 15 20 25 30 35 40 гетероароматичний фрагмент однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де Ra являє собою атом водню, фтору або хлору, ціаногрупу, метильну, етильну групу, метокси- або етоксигрупу, 2 R являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2-хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2(трифторметил)бензильну або 2-ціанбензильну групу. Особливо краще використовувати пропонований у винаході спосіб щодо сполук, у яких X означає атом хлору або брому, 1 R являє собою ціанбензильну, (ціанпіридиніл)метильну, хінолінілметильну, (метилхінолініл)метильну, ізохінолінілметильну, (метилізохінолініл)метильну, хіназолінілметильну, (метилхіназолініл)метильну, хіноксазинілметильну, (метилхіноксалініл)метильну, (диметилхіноксалініл)метильну або нафтиридинілметильну групу, 2 R являє собою метильну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-хлорбензильну, 2-бромбензильну або 2-ціанбензильну групу, але насамперед використовувати для одержання сполук із групи, що включає 1-[(4метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1-іл)ксантин, 1-[(3метилізохінолін-1-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-((R)-3-амінопіперидин-1-іл)ксантин і 1-[(3ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1-іл)ксантин, з вихідних сполук, у яких Х означає бром. Як вихідний реагент в кожному випадку краще використовувати (R)-3-(фталімідо)піперидин. Сполуки формули (III) одержують відомими методами, описаними у вже зазначеній вище літературі. Ще одним об'єктом винаходу є спосіб одержання оптично активного 3(фталімідо)піперидину. При цьому спочатку 3-амінопіридин гідрують за відомою методикою, а потім одержаний таким шляхом рацемічний 3-амінопіперидин взаємодією із фталевим ангідридом переводять у відповідний фталімід. З розчину рацемічного сирого фталіміду формули (IV) за допомогою D-винної кислоти можна селективно осаджувати (R)-енантіомер. З такого ж маточного розчину, з якого осаджували сіль D-винної кислоти, можна далі додаванням L-винної кислоти простим шляхом одержувати відповідно (S)-енантіомер фталіміду (IV), а саме: без попереднього відділення надлишку D-винної кислоти, що ще міститься в маточному розчині. Подібне винятково просте розділення енантіомерів сполуки формули (IV) є для спеціаліста в даній галузі зовсім несподіваним. Крім цього немає необхідності в попередньому очищенні рацемічної основи, одержуваної в результаті реакції гідрування. Навіть при здійсненні такого способу в промисловому масштабі ніяких проблем не виникає. Крім цього непередбачувано чиста взаємодія 3-амінопіперидину із фталевим ангідридом вже сама по собі несподівана, оскільки відповідно до літературних джерел (див., наприклад, патент US 4005208, насамперед приклад 27) у цьому випадку передбачається одержання сумішей, які поряд з необхідним продуктом містять похідні, у яких кільцевий атом азоту ацилований. 3 UA 100221 C2 O O + 3H2 N HN O HN Фталевий ангідрид NH2 3-амінопіридин N NH2 Рацемічний 3-амінопіперидин O O OH O OH O HO O N OH O D-(-)-винна кислота 5 x TD HN Тартрат (R)-3-фталімідопіперидину [(R)-(IV)] Нижче винахід більшдокладно пояснюється на прикладах. Приклад 1 D-тартрат (R)-енантіомера 3-(фталімідо)піперидину а. Гідрування H2 N HN NH2 NH2 Рацемічний 3-амінопіперидин У реактор гідрування завантажують 10,00 кг (106,25 моль) 3-амінопіридину, 500 г технічного активованого вугілля та 65 л оцтової кислоти. Потім додають 50 г суспендованого в 2,5 л оцтової кислоти каталізатора Нішимури (комерційно доступний змішаний каталізатор на основі родію та платини) і промивають 2,5 л оцтової кислоти. Далі гідрують при 50 °C та підвищеному тиску водню 100 бар до повного припинення поглинання водню, після чого продовжують гідрування ще впродовж 30 хв при 50 °C. Каталізатор і активоване вугілля відфільтровують і промивають 10 л оцтової кислоти. Розчин продукту без очищення використовують у наступній реакції. Описану вище реакцію можна успішно проводити й при не настільки високому тиску. б. Ацилування O 3-амінопіридин 10 15 O HN O HN Фталевий ангідрид NH2 20 N O O У реактор завантажують 15,74 кг (106,25 моль) фталевого ангідриду та змішують із фільтратом, одержаним на попередній стадії гідрування. Далі промивають 7,5 л оцтової кислоти й потім реакційну суміш нагрівають зі зворотним холодильником, відганяючи при цьому впродовж 1 год. приблизно 30 % використовуваної оцтової кислоти. Реакційний розчин охолоджують до 90 °C. Розчин продукту без очищення використовують у наступній реакції. в. Розщеплення рацематів 4 UA 100221 C2 OH O HN N O O HO OH x TD HN O OH D-(-)-винна кислота N O O Тартрат (R)-3-фталімідопіперидину [(R)-(IV)] 5 10 15 20 25 30 35 Нагрітий до 50 °C розчин 11,16 кг D-(–)-винної кислоти (74,38 моль) в 50 л абсолютного етанолу додають при 90 °C до призначеного для ацилування реакційного розчину. Потім промивають 10 л абсолютного етанолу та впродовж 30 хв перемішують при 90 °C, що супроводжується кристалізацією продукту. Після охолодження до 5 °C продукт відокремлюють центрифугуванням і промивають абсолютним етанолом. Розчин продукту без очищення використовують у наступній реакції. г. Перекристалізація Вологий сирий продукт нагрівають зі зворотним холодильником у суміші з 50 л ацетону та 90 л води до утворення розчину. Потім цей розчин охолоджують до 5 °C, що супроводжується кристалізацією продукту. Суспензію перемішують при 5 °C впродовж 30 хв, одержаний продукт відокремлюють центрифугуванням і після цього промивають сумішшю з 20 л ацетону та 10 л води. На завершення продукт сушать у сушильній шафі в струмі інертного газу при 45 °C. Вихід: 11,7-12,5 кг (29-31 % від теорії). Приклад 2 Синтез 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1іл)ксантину а. 2-хлорметил-4-метилхіназолін N Cl NH2 N Cl N O У реактор завантажують 10,00 кг (73,98 моль) 2-аміноацетофенону, після чого додають 24,5 л 1,4-діоксану. Охолоджений до 10 °C розчин змішують із 16,72 кг (458,68 моль) хлористого водню шляхом його пропускання через розчин. Реакційна суміш нагрівається при цьому до 2225 °C. При цій температурі пропускання хлористого водню продовжують. Після витрати приблизно половини від усієї пропущеної через розчин кількості хлористого водню суміш охолоджують до 10 °C та продовжують пропускати кількість хлористого водню, що залишилася. Суспензію, яка утворилася, залишають стояти на ніч при -10 °C. Після цього впродовж 1 год. при 10 °C додають розчин 6,70 кг (88,78 моль) хлорацетонітрилу в 2,5 л 1,4-діоксану. Живильну ємність промивають 2 л 1,4-діоксану. Потім вміст реактора нагрівають до 6 °C та перемішують впродовж приблизно 2 год. В інший реактор завантажують суміш із 122 л води та 62,04 кг (775,31 моль) розчину гідроксиду натрію (50 %-вого) і охолоджують до 6 °C. Далі порціями додають реакційну суміш із першого реактора. Внутрішня температура становить при цьому максимум 11 °C. Після цього перший реактор промивають спочатку 6 л 1,4-діоксану, а потім 6 л води. Суспензію, яка утворилася, перемішують ще впродовж 30 хв при 5 °C. Одержаний продукт відокремлюють центрифугуванням, промивають 41 л води та сушать у струмі інертного газу в сушильній шафі при 35 °C. Вихід: 10,5-12,1 кг (74-85 % від теорії). б. 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-бромксантин 5 UA 100221 C2 O N Cl 10 15 N HN + N 5 O Br N N N N N O N Na2CO3 O Br N N У реактор завантажують 10,00 кг (33,66 моль) 3-метил-7-(2-бутин-1-іл)-8-бромксантину, 7,13 кг (37,02 моль) 2-хлорметил-4-метилхіназоліну, 3,92 кг (37,02 моль) безводного карбонату натрію та 30 л N-метил-2-піролідону. Потім вміст реактора нагрівають до 140 °C та впродовж 2 год. перемішують при цій температурі. Після завершення реакції суміш охолоджують до 80 °C та розбавляють спочатку 60 л 96 %-вого етанолу, а потім при 70 °C 55 л води. Далі при 60 °C додають 4,04 кг (67,32 моль) оцтової кислоти та промивають 5 л води. Суспензію, яка утворилася, перемішують впродовж 30 хв при 60 °C, після чого охолоджують до 23 °C та продовжують перемішування ще впродовж 30 хв. Після цього одержаний продукт відокремлюють центрифугуванням і промивають спочатку сумішшю з 20 л 96 %-вого етанолу та 20 л води, а потім 40 л 90 %-вого етанолу та 40 л води. На завершення продукт сушать у сушильній шафі в струмі інертного газу при 45 °C. Вихід: 11,6-12,6 кг (76-83 % від теорії). в. 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-фталімідопіперидин-1іл)ксантин O N O N N N O N Br N N N N N N N N O N O O 20 25 30 У реактор завантажують 10,00 кг (22,06 моль) 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2бутин-1-іл)-8-бромксантину, 12,59 кг (33,09 моль) D-тартрату 3-(фталімідо)піперидину та 17,5 л N-метил-2-піролідону. Потім вміст реактора нагрівають до 140 °C. Після досягненні цієї температури впродовж 20 хв додають 11,41 кг (88,24 моль) діізопропілетиламіну. Живильну ємність промивають 2,5 л N-метил-2-піролідону, після чого реакційну суміш впродовж 2 год. перемішують при 140 °C. Після завершення реакції суміш охолоджують до 60 °C та розбавляють 80 л метанолу. Суспензію, яка утворилася, впродовж 30 хв перемішують при 50 °C, після чого охолоджують до 23 °C та продовжують перемішування ще впродовж 30 хв. Потім одержаний продукт відокремлюють центрифугуванням і тричі промивають метанолом порціями по 20 л. На завершення продукт сушать у сушильній шафі в струмі інертного газу при 45 °C. Вихід: 12,0-12,5 кг (90-94 % від теорії). г. 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1іл)ксантин O N N N O O N N N N N O N N N N O N N N NH2 O 35 1800 г (3 моль) 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)фталімідопіперидин-1-іл)ксантину нагрівають в 18 л толуолу до 80-85 °C. Потім до суспензії при 75-80 °C додають 1,815 л (30 моль) етаноламіну. Для повноти реакції перемішують впродовж 2 год. при 80-85 °C, при цьому тверді речовини розчиняються. Потім фази розділяють. Етаноламінову фазу двічі промивають теплим толуолом (порціями по 4 л). Об'єднані толуольні 6 UA 100221 C2 5 10 15 20 25 30 35 40 45 50 фази двічі промивають при 75-80 °C теплою водою порціями по 8 л. З толуольної фази під вакуумом відганяють 22 л толуолу. До суспензії, яка утворилася, при 40-50 °C додають 4 л третбутилметилового ефіру й потім охолоджують до 0-5 °C. Одержаний продукт відокремлюють шляхом фільтрації, промивають трет-бутилметиловим ефіром і відокремлюють вакуумфільтрацією через сухий вакуум-фільтр. Вологу сиру речовину потім нагрівають зі зворотним холодильником разом з 5-кратною кількістю абсолютного етанолу й гарячий розчин для очищення фільтрують через активоване вугілля. Після охолодження фільтрату до 20 °C та початку кристалізації розбавляють трет-бутилметиловим ефіром до подвоєння об'єму. Далі суспензію охолоджують до 2 °C, перемішують впродовж 2 год., фільтрують вакуум-фільтрацією та на завершення сушать у вакуумній сушильній шафі при 45 °C. Вихід: 1174 г (83,2 % від теорії). Інший варіант здійснення стадії г 1400 г (2,32 моль) 1-[(4-метилхіназолін-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)фталімідопіперидин-1-іл)ксантину додають в 4,9 л тетрагідрофурану, після чого нагрівають до 55-65 °C. Потім до суспензії додають 350 мл води та 1433 г (2,32 моль) етаноламіну. Для повноти реакції перемішують впродовж 3 год. при 60-63 °C. Далі додають 619 мл 45 %-вого розчину їдкого натру та 3,85 л води й перемішування продовжують ще впродовж 30 хв при 5565 °C. Потім до реакційної суміші додають 5,6 л толуолу, перемішують впродовж 15 хв, після чого фази розділяють. Органічну фазу промивають 2,8 л води при 55-65 °C і потім її відокремлюють. Органічну фазу відганяють під вакуумом у кількості 4,2 л. Потім при 65-75 °C додають 1,4 л метилциклогексану, що супроводжується кристалізацією продукту. Суспензію впродовж 8-16 год. перемішують при температурі в інтервалі від 15 до 25 °C, після чого охолоджують до 0-5 °C. Продукт відокремлюють шляхом фільтрації, промивають 4,2 л метилциклогексану, відфільтровують через сухий вакуум-фільтр і сушать у вакуумі при 35 °C. Після сушіння сиру речовину (991 г) нагрівають зі зворотним холодильником разом з 5-кратною кількістю метанолу, додають активоване вугілля та фільтрують. Об'єм фільтрату за рахунок відгону метанолу зменшують до 1,5 л. Після охолодження фільтрату до 45-55 °C його розбавляють трет-бутилметиловим ефіром до досягнення 4-кратного об'єму. Суспензію охолоджують до 0-5 °C, перемішують впродовж 2 год., відфільтровують вакуум-фільтрацією, промивають трет-бутилметиловим ефіром і на завершення сушать у вакуумній сушильній шафі при 35 °C. Вихід: 899 г (81,9 % від теорії). Приклад 3 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1-іл)ксантин а. 3-ціанo-2-(хлорметил)піридин 165,5 г (0,98 моль) 2-гідроксиметил-3-піридинкарбоксаміду нагрівають впродовж 1 год. разом з 270 мл оксихлориду фосфору до 90-100 °C. Потім реакційну суміш охолоджують до кімнатної температури та по краплях додають до приблизно 800 мл нагрітої до 50-60 °C води. Після гідролізу оксихлориду фосфору при охолодженні нейтралізують розчином їдкого натру, що супроводжується випаданням продукту в осад. Продукт відфільтровують, промивають 300 мл води та на завершення сушать при 35-40 °C. Вихід: 122,6 г (82 % від теорії). Інший варіант здійснення стадії а: 3-ціанo-2-(хлорметил)піридин 20,0 г (131,45 ммоль) 2-гідроксиметил-3-піридинкарбоксаміду суспендують в 110 мл ацетонітрилу та нагрівають до 78 °C. Потім впродовж 15 хв додають 60,65 г (395,52 ммоль) оксихлориду фосфору й впродовж 2 год. нагрівають до 81 °C. Після охолодження до 22 °C реакційну суміш домішують до 200 мл нагрітої до 40 °C води. Після додавання 100 мл толуолу при охолодженні нейтралізують розчином їдкого натру. Після розділення фаз органічну фазу промивають 100 мл води. Далі органічну фазу відокремлюють, а розчинник випарюють у вакуумі, одержуючи в результаті спочатку маслянистий залишок, який потім кристалізується при стоянні. Вихід: 16,66 г (83 % від теорії). б. 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-бромксантин N N O O N N N + N Br Br N N O N N O N N Cl 7 UA 100221 C2 5 10 У реактор завантажують 202 г (0,68 моль) 3-метил-7-(2-бутин-1-іл)-8-бромксантину, 188,5 г (1,36 моль) безводного карбонату калію та 1,68 л N-метил-2-піролідону й нагрівають до 70 °C. Потім по краплях додають 119 г (0,75 моль) 2-хлорметил-3-ціанопіридину в 240 мл N-метил-2піролідину (N-MП). Вміст реактора перемішують впродовж 19 год. при 70 °C. Після завершення реакції до реакційної суміші додають 2,8 л води та охолоджують до 25 °C. Одержаний продукт відфільтровують, промивають 2 л води та на завершення сушать у сушильній шафі в струмі інертного газу при 70 °C. Вихід: 257,5 г (91 % від теорії). в. 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-фталімідопіперидин-1іл)ксантин N N O O N N N O N N N Br N N N N N O N O O 15 20 25 У реактор завантажують 230 г (0,557 моль) 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2бутин-1-іл)-8-бромксантину, 318 г (0,835 моль) D-тартрату 3-(фталімідо)піперидину та 1,15 л Nметил-2-піролідону. Потім вміст реактора нагрівають до 140 °C. Після досягненні цієї температури впродовж 20 хв додають 478 мл (2,78 моль) діізопропілетиламіну й далі реакційну суміш впродовж 2 год. перемішують при 140 °C. Потім реакційну суміш охолоджують до 75 °C і розбавляють 720 мл метанолу. Після цього при температурі в інтервалі від 60 до 68 °C додають 2,7 л води та охолоджують до 25 °C. Одержаний продукт відфільтровують і промивають 2 л води. На завершення продукт сушать у сушильній шафі в струмі інертного газу при 70 °C. Потім одержаний таким шляхом сирий продукт розмішують в 1 л метанолу при температурі кипіння, фільтрують у гарячому стані, промивають 200 мл метанолу та на завершення сушать у струмі інертного газу при 70 °C. Вихід: 275 г (88 % від теорії). г. 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)-амінопіперидин-1іл)ксантин N N O O N N N O N N N N N N N O O N N N NH2 O 30 35 40 412,5 г (0,733 моль) 1-[(3-ціанопіридин-2-іл)метил]-3-метил-7-(2-бутин-1-іл)-8-(3-(R)фталімідопіперидин-1-іл)ксантину нагрівають в 4125 мл толуолу до 80 °C. Потім при 75-80 °C до суспензії додають 445 мл (7,33 моль) етаноламіну. Для повноти реакції впродовж 2 год. перемішують при 80-85 °C, що супроводжується розчиненням твердих речовин. Потім фази розділяють. Етаноламінову фазу двічі екстрагують теплим толуолом (порціями по 1 л). Об'єднані толуольні фази двічі промивають нагрітою до 75-80 °C водою порціями по 2 л. Толуольні фази сушать над сульфатом натрію, фільтрують і потім шляхом перегонки концентрують у вакуумі до об'єму біля 430 мл. Далі при 50-55 °C додають 1 л третбутилметилового ефіру та після цього охолоджують до 0-5 °C. Одержаний продукт відфільтровують, промивають трет-бутилметиловим ефіром і на завершення сушать у сушильній шафі при 60 °C. Вихід: 273 г (86 % від теорії). tпл 188±3 °C. Аналогічно прикладам 2 і 3 одержують також 1-[(3-метилізохінолін-1-іл)метил]-3-метил-7-(2бутин-1-іл)-8-((R)-3-амінопіперидин-1-іл)ксантин. 8 UA 100221 C2 ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання сполуки загальної формули (І) O R1 5 10 15 20 NH2 N N O R3 N N N R2 (I) або її енантіомера або солі, де 1 R являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну, нафтиридинілметильну або фенантридинілметильну групу, у якій ароматичний або гетероароматичний фрагмент в кожному випадку однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де Ra являє собою атом водню, ціаногрупу, метильну, трифторметильну, етильну, фенільну групу, метокси-, дифторметокси-, трифторметокси- або етоксигрупу, або два радикали Ra, за умови, що вони зв'язані із сусідніми атомами вуглецю, можуть являти собою також групу -О-СН2-О- або -О-СН2-СН2-О-, 2 R являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2(трифторметил)бензильну або 2-ціанобензильну групу, який включає стадії синтезу, на яких: а) піддають взаємодії сполуку загальної формули (III) O R1 N N O R3 X N N R2 25 , (III) у якій X означає відхідну групу, вибрану із групи галогенів або ефірів сульфонових кислот, a 1 3 кожний з R -R мають зазначені вище значення, з 3-(фталімідо)піперидином або його енантіомером, б) видаляють захисну групу з одержаної таким шляхом сполуки загальної формули (II) O O R1 N N N O R3 O N N N R2 30 35 , (II) 1 3 у якій кожен R -R має зазначені вище значення, та в) одержану сполуку при необхідності перетворюють на її фізіологічно сумісну сіль. 2. Спосіб за п. 1, в якому X означає атом хлору або брому, 1 R являє собою фенілкарбонілметильну, бензильну, нафтилметильну, піридинілметильну, піримідинілметильну, хінолінілметильну, ізохінолінілметильну, хіназолінілметильну, хіноксалінілметильну або нафтиридинілметильну групу, у якій ароматичний або 9 UA 100221 C2 5 10 15 20 25 30 35 40 45 гетероароматичний фрагмент однозаміщений або двозаміщений ідентичними або різними замісниками Ra, де Ra являє собою атом водню, ціаногрупу, метильну, етильну групу, метокси- або етоксигрупу, 2 R являє собою метильну, етильну, пропільну, ізопропільну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-фторбензильну, 2хлорбензильну, 2-бромбензильну, 2-йодбензильну, 2-метилбензильну, 2(трифторметил)бензильну або 2-ціанобензильну групу. 3. Спосіб за п. 2, в якому X означає атом хлору або брому, 1 R являє собою ціанобензильну, (ціанопіридиніл)метильну, хінолінілметильну, (метилхінолініл)метильну, ізохінолінілметильну, (метилізохінолініл)метильну, хіназолінілметильну, (метилхіназолініл)метильну, хіноксазинілметильну, (метилхіноксалініл)метильну, (диметилхіноксалініл)метильну або нафтиридинілметильну групу, 2 R являє собою метильну, циклопропільну або фенільну групу, а 3 R являє собою 2-бутен-1-ільну, 3-метил-2-бутен-1-ільну, 2-бутин-1-ільну, 2-хлорбензильну, 2бромбензильну або 2-ціанобензильну групу. 4. Спосіб за п. 3, в якому X означає атом брому, 1 R являє собою (4-метилхіназолін-2-іл)метильну, (3-метилізохінолін-1-іл)метильну або (3ціанопіридин-2-іл)метильну групу, 2 R являє собою метильну групу, а 3 R являє собою 2-бутин-1-ільну групу. 5. Спосіб за будь-яким з пп. 1-4, де на стадії а) як реагент використовують (R)-3(фталімідо)піперидин. 6. Спосіб за п. 5, в якому X означає атом брому, 1 R являє собою (4-метилхіназолін-2-іл)метильну групу, 2 R являє собою метильну групу, та 3 R являє собою 2-бутин-1-ільну групу. 7. Спосіб за п. 6, в якому додатково проводять кристалізацію або перекристалізацію сполуки формули (І) з метанольного або етанольного розчину. 8. Спосіб за п. 7, в якому розчин додатково містить трет-бутилметиловий ефір. 9. Спосіб за п. 6, в якому додатково проводять кристалізацію сполуки формули (І) з етанолу. 10. Спосіб одержання (R)-3-(фталімідо)піперидину, який включає стадії на яких: а) піддають взаємодії рацемічний 3-амінопіперидин у відповідних розчинниках із фталевим ангідридом, і б) видаляють (R)-3-(фталімідо)піперидин із розчину одержаного таким чином рацемічного 3(фталімідо)піперидину шляхом додавання D-винної кислоти та відокремлюють тартрат, що випав у осад. 11. Спосіб за п. 10, в якому на стадії б) як розчинник використовують етанол. 12. (R)-3-(фталімідо)піперидин. 13. D-(-)-тартрат (R)-3-(фталімідо)піперидину. 14. Спосіб одержання лікарського засобу, в якому одержують сполуку загальної формули (І), як визначено у пункті 1, або її енантіомер або сіль, шляхом здійснення способу за будь-яким з пп. 1-9, та об'єднують одержану сполуку загальної формули (І), як визначено у пункті 1, або її енантіомер або сіль, з одним або декількома інертними носіями і/або розріджувачами. 15. Сполука загальної формули (ІІ) O O R1 N N N O R3 O N N R2 N (ІI) 10 UA 100221 C2 1 3 або її енантіомер, у якій кожен з R -R має значення відповідно до визначення у будь-якому з пунктів 1-6. 16. Сполука наступної загальної формули O R1 N N O R3 N N O N N R2 O 5 10 15 , у якій 1 R являє собою (4-метилхіназолін-2-іл)метильну, (3-метилізохінолін-1-іл)метильну або (3ціанопіридин-2-іл)метильну групу, 2 R являє собою метильну групу, та 3 R являє собою 2-бутин-1-ільну групу. 17. Сполука за п. 16, у якій 1 R являє собою (4-метилхіназолін-2-іл)метильну групу, 2 R являє собою метильну групу, та 3 R являє собою 2-бутин-1-ільну групу. 18. Спосіб одержання лікарського засобу, в якому одержують сполуку наступної загальної формули O R1 N N O R3 N N N NH2 R2 20 або її фізіологічно сумісну сіль, у якій 1 R являє собою (4-метилхіназолін-2-іл)метильну групу, 2 R являє собою метильну групу, та 3 R являє собою 2-бутин-1-ільну групу, шляхом здійснення способу за будь-яким з пп. 6, 7, 8 або 9, та об'єднують одержану сполуку з одним або декількома інертними носіями і/або розріджувачами. 19. Спосіб одержання сполуки наступної формули O R1 N N O 25 30 35 R3 N N R2 N NH2 , де 1 R являє собою (4-метилхіназолін-2-іл)метильну групу, 2 R являє собою метильну групу, та 3 R являє собою 2-бутин-1-ільну групу, причому в зазначеному способі видаляють фталільну захисну групу зі сполуки, вказаної в п. 17. 20. Спосіб за п. 19, в якому фталільну захисну групу видаляють в присутності етаноламіну, краще в толуолі або в суміші тетрагідрофурану та води. 21. Спосіб за п. 19 або 20, в якому додатково проводять кристалізацію незахищеної сполуки з етанолу або метанолу. 22. Спосіб за будь-яким з п. 19, 20 або 21, в якому незахищену сполуку кристалізують з етанолу. 23. Спосіб за п. 19, в якому додатково проводять кристалізацію незахищеної сполуки з етанолу. 11 UA 100221 C2 Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 12
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing chiral 8-(3-amino-piperidin-1-yl)-xanthines
Автори англійськоюPfrengle Waldemar, Pachur Thorsten, Nikolaus Thomas, Duran Adil
Назва патенту російськоюСпособ получения хиральных 8-(3-аминопиперидин-1-ил)ксантинов
Автори російськоюПфренгле Вальдемар, Пахур Торстен, Никола Томас, Дуран Адиль
МПК / Мітки
МПК: A61K 31/522, C07D 473/04
Мітки: хіральних, спосіб, 8-(3-амінопіперидин-1-іл)ксантинів, одержання
Код посилання
<a href="https://ua.patents.su/14-100221-sposib-oderzhannya-khiralnikh-8-3-aminopiperidin-1-ilksantiniv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання хіральних 8-(3-амінопіперидин-1-іл)ксантинів</a>
8-[3-амінопіперидин-1-іл]ксантини, спосіб їх одержання і їх застосування як інгібіторів dpp-iv

Номер патенту: 87835
Опубліковано: 25.08.2009
Автори: Екхардт Маттіас, Хіммельсбах Франк, Томас Лео, Лангкопф Ельке, Тадайон Мохаммед
МПК: A61P 3/10, A61K 31/437, C07D 473/04
Мітки: застосування, інгібіторів, дпп-iv, 8-[3-амінопіперидин-1-іл]ксантини, спосіб, одержання
Формула / Реферат:
1. Сполука загальної формули, (I)у якій R означає бензильну, 2-фторбензильну, 3-фторбензильну, 4-фторбензильну, 2,6-дифторбензильну, 3,4-дифторбензильну, 2-хлорбензильну, 3-хлорбензильну або 4-хлорбензильну групу, 2-трифторметилбензильну, 3-трифторметилбензильну або 4-трифторметилбензильну групу,3-трифторметоксибензильну або...
8-[3-амінопіперидин-1-іл]ксантини, спосіб їх одержання та їх застосування як лікарських засобів

Номер патенту: 84275
Опубліковано: 10.10.2008
Автори: Лангкопф Ельке, Марк Міхаель, Майер Роланд, Тадайон Мохаммад, Хіммельсбах Франк, Лотц Ральф Ріхард Херманн, Екхардт Маттіас
МПК: A61K 9/28, A61K 47/02, C07D 473/08, C07D 473/06, A61P 3/10, C07D 473/04, A61K 9/02, A61K 9/48, A61K 9/20
Мітки: лікарських, застосування, засобів, одержання, спосіб, 8-[3-амінопіперидин-1-іл]ксантини
Формула / Реферат:
1. Сполуки загальної формули І, (I)у якійR1 означає 4-метокси-1-нафтилметильну групу,2-хінолінілметильну, 4-хінолінілметильну або 6-хінолінілметильну групу,1-ізохінолінілметильну, 3-метил-1-ізохінолінілметильну, 4-метил-1-ізохінолінілметильну або 3-ізохінолінілметильну групу або 2-хіназолінілметильну, 4-метил-2-хіназолінілметильну або...
Комбінація та фармацевтична композиція, що містить 8-[3-амінопіперидин-1-іл]ксантини, спосіб її одержання та їх застосування як лікарського засобу

Номер патенту: 97468
Опубліковано: 27.02.2012
Автори: Марк Міхаель, Майер Роланд, Екхардт Маттіас, Лотц Ральф Ріхард Херманн, Хіммельсбах Франк, Тадайон Мохаммад, Лангкопф Ельке
МПК: A61K 31/4188, C07D 473/00
Мітки: композиція, 8-[3-амінопіперидин-1-іл]ксантини, засобу, одержання, застосування, фармацевтична, містить, комбінація, спосіб, лікарського
Формула / Реферат:
1. Комбінація, яка містить сполуку загальної формулив якійR1 означає 4-метокси-1-нафтилметильну групу, 2-хінолінілметильну, 4-хінолінілметильну або 6-хінолінілметильну групу, 1-ізохінолінілметильну, 3-метил-1-ізохінолінілметильну, 4-метил-1-ізохінолінілметильну або 3-ізохінолінілметильну групу або 2-хіназолінілметильну, 4-метил-2-хіназолінілметильну...
Спосіб одержання хіральних 1,4-дизаміщених піперазинів, проміжна сполука (варіанти)

Номер патенту: 79774
Опубліковано: 25.07.2007
Автори: Джирковскі Іво, Зелдіс Джозеф, Фейгельсон Грегг Брайан
МПК: C07D 405/12, C07D 213/75, C07B 53/00, C07D 487/10, C07D 295/08, C07D 319/00, C07C 229/12, C07D 295/10
Мітки: сполука, варіанти, 1,4-дизаміщених, спосіб, піперазинів, проміжна, хіральних, одержання
Формула / Реферат:
1. Спосіб стереоселективного одержання 1,4-дизаміщених піперазинів формули IX, (IX)де R являє собою С1-С3-алкільну групу;Аr являє собою дигідробензодіоксиніл, бензодіоксиніл або феніл, необов'язково заміщений до трикратно замісниками, незалежно один від одного вибраними з галогену, метокси, галогенметилу, дигалогенметилу і тригалогенметилу; і Aryl...
Спосіб одержання хіральних 4-аміно-4-арил-5,5,5-трифторопентан-2-онів

Номер патенту: 84821
Опубліковано: 25.11.2008
Автори: Сукач Володимир Андрійович, Піроженко Володимир Валентинович, Вовк Михайло Володимирович, Головач Наталія Михайлівна, Русанов Едуард Борисович
МПК: C07C 221/00, C07C 225/00, C07B 53/00
Мітки: спосіб, одержання, хіральних, 4-аміно-4-арил-5,5,5-трифторопентан-2-онів
Формула / Реферат:
Спосіб одержання хіральних 4-аміно-4-арил-5,5,5-трифторопентан-2-онів загальної формули І:, Іде Аr = Ph, 4-FC6H4, 3-MeC6H4, 4-MeC6H4, 4-МеОС6Н4,який відрізняється тим, що арилтрифторометилкетіміни піддають взаємодії з ацетоном в розчині диметилсульфоксиду при кімнатній температурі в присутності 10%-вої каталітичної кількості L- або D-проліну з наступним...
Попередній патент: Спосіб пакування, транспортування і зберігання свіжих продуктів
Наступний патент: Тканинозахисні пептиди і їх застосування
Випадковий патент: Генератор синусоїдальної напруги, синхронізований з мережею перемінного струму