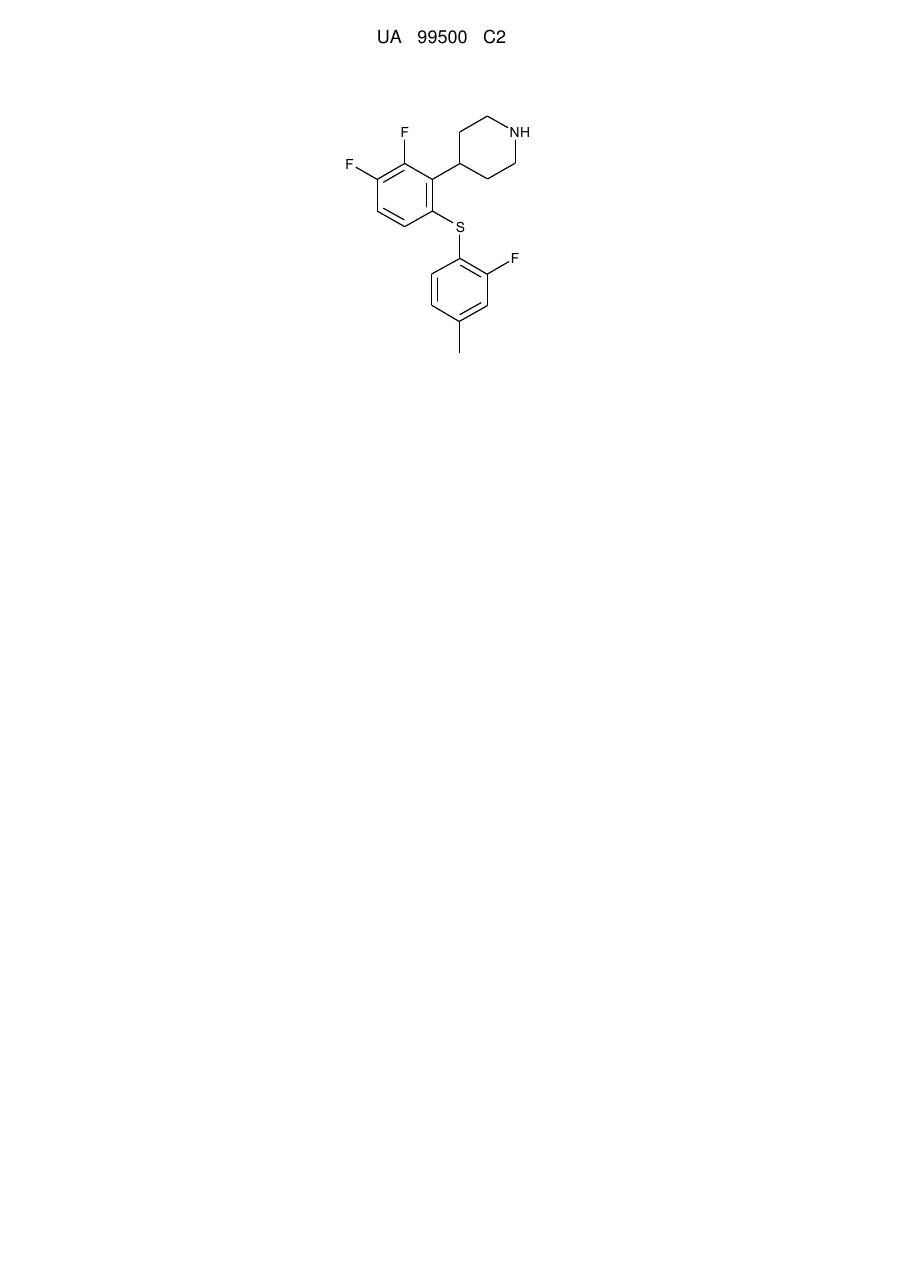
4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин
Номер патенту: 99500
Опубліковано: 27.08.2012
Автори: Андерсон Ніл, Йорґенсен Мортен, Фалт Андре, Банґ-Андерсен Бенні, Стенсбьол Тіне Брайан







Формула / Реферат
1. 4-[2,3-Дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин у відповідності зі структурною формулою
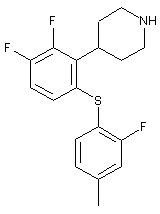
і його фармацевтично прийнятні солі.
2. Фармацевтична композиція, яка містить сполуку за п. 1 і принаймні один фармацевтично прийнятний носій або розріджувач.
3. Спосіб лікування болю, який передбачає введення терапевтично ефективної кількості сполуки у відповідності з п. 1 пацієнтові, що потребує цього.
4. Сполука за п. 1 для використання при лікуванні болю.
5. Застосування сполуки за п. 1 для виготовлення лікарського засобу для лікування болю.
Текст
Реферат: Запропонована сполука, 4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин, у відповідності із структурною формулою (формула І) і її фармацевтично прийнятні солі для лікування захворювань, пов'язаних із розладами ЦНС, таких як: великий депресивний розлад, дистимічний розлад; розлад настрою, пов'язаний з загальним медичним станом; атипова депресія; сезонний афективний розлад; меланхолія; стійка до лікування депресія; часткова відповідь; депресія, пов'язана з біполярним розладом, болем, хворобою Альцгеймера, психозом, хворобою Паркінсона, хворобою дифузних тілець Леві, хворобою Хантінгтона, розсіяним склерозом або тривожним станом; генералізований тривожний розлад; соціальний тривожний розлад; панічні атаки; фобія; соціофобія; обсесивно-компульсивний розлад; посттравматичний стресовий розлад; гострий стрес; синдром дефіциту уваги з гіперактивністю і біль. UA 99500 C2 (12) UA 99500 C2 NH F F S F UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 Галузь техніки, до якої відноситься винахід Даний винахід відноситься до сполуки 4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидину, фармацевтичних композицій, що містять вищезазначену сполуку, і до терапевтичного застосування вищезазначеної сполуки. Попередній рівень техніки Біль і, зокрема, хронічний біль та депресія часто є супутніми захворюваннями, з цієї причини забезпечення сполуками, які є ефективними при обох захворюваннях, було б корисним для пацієнта. Селективним інгібіторам зворотного захоплення серотоніну (5-гідрокситриптаміну, 5-НТ), (SSRI), протягом багатьох років лікарі віддавали перевагу для лікування багатьох захворювань центральної нервової системи (ЦНС), таких як депресія і тривога, оскільки вони ефективні і мають профіль безпеки, який є переважним в порівнянні з лікарськими препаратами попереднього покоління, що діють на ЦНС, тобто, так званими, трициклічними сполуками. Проте, застосування SSRI ускладнювалося наявністю значної частини пацієнтів із відсутньою клінічною відповіддю, тобто пацієнтів, які не відповідали на лікування, що проводилося, або відповідали не в повному обсязі. Більш того, як правило, SSRI починав демонструвати свою дію тільки після декількох тижнів лікування. Нарешті, хоча SSRI, як правило, викликають менше побічних реакцій, ніж трициклічні сполуки, введення SSRI часто викликає побічні реакції, такі як, наприклад, порушення сну. Відомо, що комбінування інгібування переносника серотоніну (SERT) і активності одного або декількох рецепторів серотоніну може приводити до більшого зростання рівня 5-НТ в порівнянні з тим, що дають SSRI, і що це пов'язано з швидшим початком дії і збільшенням ефективності в порівнянні з SSRI. Так, наприклад, повідомлялося, що комбінування піндололу, який є частковим агоністом рецептора 5-НТ1A, з інгібітором зворотного захоплення серотоніну (SRI) забезпечує більш швидкий початок дії [Psych. Res., 125, 81-86, 2004]. Також було виявлено, що комбінування SRI з антагоністом рецептора 5-НТ2С або із зворотним агоністом (сполуками, що мають негативну дію на рецептор 5-НТ2C) забезпечує значне збільшення рівня 5-НТ в термінальних ділянках в порівнянні з тільки SRI, як це вимірили в експериментах з мікродіалізу [WO 01/41701]. Оскільки вважають, що терапевтична ефективність SRJ пов'язана із збільшенням рівня 5-НТ, поєднання цих типів активності припускає швидший початок терапевтичного ефекту в клініці і посилення або потенціювання терапевтичного ефекту інгібітора зворотного захоплення серотоніну (SRI). Сприйняття болю може бути складнішим, ніж безпосередня передача сигналів від пошкодженої частини тіла до специфічних рецепторів в мозку, де сприйманий біль пропорційний пошкодженню. Швидше, пошкодження периферичної тканини і пошкодження нервів може викликати зміни в пов'язаних з центральною нервовою системою нервових структурах, залучених в сприйняття болю, впливаючи на подальшу чутливість до болю. Ця нейропластичність може приводити до пов'язаної з центральною нервовою системою сенсибілізації у відповідь на тривалий больовий подразник, яка може виявлятися сама по собі, як, наприклад, хронічний біль, тобто, при якому сприйняття болю залишається навіть після того, як тривале подразнення припинилося, або як гіпералгезія, тобто підвищена реакція на подразнення, яке зазвичай є безболісним. Одним з багатьох загадкових і драматичних прикладів цього є "фантомний біль в кінцівці", тобто відчуття болю, який існував в кінцівці до її ампутації. Нещодавній огляд стосовно пов'язаних з центральною нервовою системою нейропластичності і болю див. у Melzack et al. Ann. N. Y. Acad. Sci, 933, 157-174, 2001. Пов'язаний з центральною нервовою системою компонент хронічного болю може пояснити, чому хронічний біль, такий як, наприклад, невропатичний біль, часто погано реагує на класичні анальгетики, такі як нестероїдні протизапальні лікарські засоби (NSAIDS) і опіоїдні анальгетики. Трициклічні антидепресанти (ТСА), типовим представником яких служить амітриптилін, стали стандартом для лікування невропатичного болю, і ефект, як вважають, досягається за допомогою сумісної інгібуючої дії на SERT і переносник норадреналіну (NAT)) [Clin Ther., 26, 951-979, 2004]. Пізніше, так звані, антидепресанти подвійної дії, що мають інгібуючий ефект зворотного захоплення як 5-НТ, так і NA, використовували в клінічній практиці для лікування невропатичного болю [Human Psychopharm., 19, S21-S25, 2004]. Прикладами антидепресантів подвійної дії є венлафаксин і дулоксетин, і цей клас антидепресантів часто відносять до SSRI. Дані щодо застосування SSRI для лікування невропатичного болю є недостатніми, але, взагалі, припускають обмежений ефект [Bas. Clin. Pharmacol, 96, 399-409, 2005]. Фактично висунута гіпотеза, що SSRI мають тільки слабку антиноцицептивність самі по собі, але що інгібування SERT збільшує антиноцицептивний ефект інгібування зворотного захоплення норадреналіну. Це зауваження спирається на огляд по дослідженню 22 тварин і п'ятьох людей, 1 UA 99500 C2 5 10 15 20 25 30 який показує, що селективні інгібітори зворотного захоплення норадреналіну (SNRI) мають набагато кращий антиноцицептивний ефект в порівнянні з інгібіторами зворотного захоплення норадреналіну (NRI), які, в свою чергу, є набагато кращими за SSRI [Pain Med. 4, 310-316, 2000]. Таким чином, можна вважати, що сполуки, що інгібують SERT і рецептор 5-НТ2C, які також інгібують переносник норадреналіну, могли б бути сполуками, ефективними при лікуванні афективних розладів і болю. Міжнародна патентна заявка, опублікована як WO 2003/029232, розкриває, наприклад, сполуку 4-[2-(4-метилфенілсульфаніл)феніл]піперидин у вигляді вільної основи і відповідної солі НСl. Ця сполука, як повідомляють, є інгібітором SERT і рецептора 5-НТ2С і, як згадується, є корисною для лікування афективних розладів, наприклад, депресії і тривоги. Міжнародна патентна заявка, опублікована як WO 2004/087156, також розкриває ряд фенілсульфанілфенілпіперидинів з тим же самим фармакологічним профілем, що і у сполуки, розкритої в заявці WO 2003/029232. Суть винаходу Дані винахідники несподівано виявили, що сполука І, тобто 4-[2,3-дифтор-6-(2-фтор-4метилфенілсульфаніл)феніл]піперидин і його фармацевтично прийнятні кислотно-адитивні солі, є сильними інгібіторами SERT, що інгібують рецептори 5-НТ2А і 5-НТ2С і інгібують переносник норадреналіну (NAT). Таким чином, в одному варіанті здійснення цей винахід відноситься до 4[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]-піперидину і його фармацевтично прийнятних кислотно-адитивних солей. У одному варіанті здійснення цей винахід відноситься до способу лікування, який включає введення терапевтично ефективної кількості сполуки І пацієнтові, що потребує цього. У одному варіанті здійснення цей винахід відноситься до фармацевтичної композиції, яка містить сполуку І і принаймні один фармацевтично прийнятний носій або розріджувач. У одному варіанті здійснення цей винахід відноситься до сполуки І для використання в терапії. У одному варіанті здійснення цей винахід відноситься до сполуки І для використання при лікуванні певних захворювань. У одному варіанті здійснення цей винахід відноситься до застосування сполуки І для виготовлення лікарського засобу для лікування певних захворювань. Докладний опис винаходу Структурна формула 4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]-піперидину є наступною NH F F S F 35 40 45 і цей винахід відноситься до сполуки І, яку визначають як дану сполуку і її фармацевтично прийнятні кислотно-адитивні солі. У одному варіанті здійснення цього винаходу вказаними кислотно-адитивними солями є солі кислот, які є нетоксичними. Вказані солі включають солі, отримані з органічними кислотами, такими як малеїнова, фумарова, бензойна, аскорбінова, бурштинова, щавлева, бісметиленсаліцилова, метансульфонова, етандисульфонова, оцтова, пропіонова, винна, саліцилова, лимонна, глюконова, молочна, яблучна, малонова, мигдальна, корична, цитраконова, аспарагінова, стеаринова, пальмітинова, ітаконова, гліколева, п-амінобензойна, глутамінова, бензолсульфонова, теофіліноцтова кислоти, а також з 8-галогентеофілінами, наприклад, 8-бромтеофіліном. Вказані солі можуть також бути отримані з неорганічними 2 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 кислотами, такими як бромистоводнева, хлористоводнева, сірчана, сульфамінова, фосфорна і азотна кислоти. Пацієнти і медичні працівники часто віддають перевагу пероральним лікарським формам і, зокрема, пігулкам і капсулам, зважаючи на легкість введення і, як наслідок, краще дотримання призначеного режиму терапії. Для пігулок і капсул переважно, щоб активні інгредієнти були кристалічними. У одному варіанті здійснення сполука І є кристалічною. Кристалічні речовини, використовувані в цьому винаході, можуть існувати у вигляді сольватів, тобто кристалів, в яких молекули розчинника утворюють частину кристалічної структури. Сольвати можуть бути утворені з водою, в цьому випадку сольвати часто називають гідратами. Альтернативно, сольвати можуть бути утворені з іншими розчинниками, такими як етанол, ацетон або етилацетат. Точна кількість сольвату часто залежить від умов. Наприклад, гідрати зазвичай втрачають воду в міру збільшення температури або зменшення відносної вологості. Сполуки, які не змінюються або незначно змінюються при зміні умов, таких як, наприклад, вологість, в основному розглядаються як найбільш відповідні для фармацевтичних рецептур. Деякі сполуки є гігроскопічними, тобто вони абсорбують воду при дії на них вологості. Гігроскопічність в цілому розглядається як небажана властивість для сполук, які мають бути представлені у фармацевтичній препаративній формі, зокрема, в сухій препаративній формі, такій як пігулки або капсули. У одному варіанті здійснення цим винаходом пропонуються кристали з низькою гігроскопічністю. Для пероральних лікарських форм з використанням кристалічних активних інгредієнтів також сприятливо, якщо вказані кристали точно визначені. У цьому контексті термін "точно визначені", зокрема, означає, що стехіометрія точно визначена, тобто, що співвідношення між іонами, утворюючими сіль, представляє співвідношення між невеликими цілими числами, таке як 1:1, 1:2, 2:1, 1:1:1 тощо. У одному варіанті здійснення сполуки цього винаходу є точно визначеними кристалами. Розчинність активного інгредієнта також має значення для вибору лікарської форми, оскільки вона може безпосередньо впливати на біодоступність. Для пероральних лікарських форм в цілому вважається, що більш висока розчинність активного інгредієнта є переважною, оскільки вона збільшує біодоступність. Фармакологічний профіль сполуки І показаний в прикладах і може бути коротко описаний таким чином. Сполука І інгібує зворотне захоплення 5НТ і NA і інгібує рецептори 5-НТ2А і 5-НТ2C. Таким чином, сполука І може бути корисною для лікування афективних розладів, таких як депресія і тривожний стан, але її фармакологічний профіль може також зробити її корисною для лікування додаткових симптомів. Рецептори 5-НТ2А і 5-НТ2C розташовані, наприклад, на NA- і дофамінергічних (DA) нейронах, відповідно, де активація викликає тонізуючий інгібуючий вплив на виділення NA і DA, відповідно, і антагоністи рецепторів 5-НТ2А і 5-НТ2C впливатимуть на збільшення рівня NA і DA, відповідно. На цій підставі можна висунути гіпотезу про те, що антагоністи рецепторів 5-НТ2А і 5-НТ2С особливо добре підходять для лікування депресії, яка резистентна до лікування SRI (стійкої до лікування депресії (TRD) або резистентної депресії) [Psychopharmacol Bull, 39, 147-166, 2006]. Частина пацієнтів з депресією відповідатимуть на лікування, наприклад, SSRI в тому сенсі, що у них поліпшуватимуться показники клінічного прояву депресії за шкалами, такими як шкала оцінки депресії MADRD (Montgomery Aasberg Depression Rating scale) і шкала оцінки депресії HAMD (Hamilton Depression Rating Scale), але інші симптоми, такі як порушення сну і порушення когнітивних функцій, залишаться. У даному контексті ці пацієнти відносяться до пацієнтів з частковою відповіддю. Завдяки антагонізму сполуки І відносно рецепторів 5-НТ2А і 5-НТ2С, який, як вважають, відбивається у впливі на сон, сполука І може бути корисною в лікуванні пацієнтів з частковою відповіддю, або, перефразовуючи, лікування пацієнтів з депресією сполуками цього винаходу знижуватиме частку пацієнтів з частковою відповіддю. Порушення сну, мабуть, є загальним побічним ефектом більшості антидепресантів. Зокрема, SSRI, NRI і SNRI, як повідомляється, викликають проблеми із засипанням і підтримкою сну, і про проблеми безсоння повідомляється часто [Int. Clin. Psychpharm., 21 (suppl 1), S25-S29, 2006]. Інші повідомляють, що такі сполуки викликають пригнічення фази швидкого сну, збільшують латентність сну, приводять до менш ефективного сну, збільшують число нічних пробуджень і фрагментують сон [Hum.Psychopharm.Clin.Exp., 20, 533-559, 2005]. Зазвичай припускають, що несприятливий вплив на сон викликається стимуляцією рецепторів 5-НT2А і 5-НT2С R.L. Fish повідомляє в Bioorg. Med. Chem. Lett., 15, 3665-3669, 2005, що певні 4-фторсульфонілпіперидини, які являють собою високоселективні антагоністи рецептора 5-НТ2А, є ефективними в збільшенні тривалості повільного сну і зменшенні числа пробуджень у щурів. Ці доклінічні спостереження підтверджуються клінічними 3 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 спостереженнями. Ритансерин, антагоніст рецептора 5-НТ2А, як було показано, збільшує загальний час сну, тривалість повільного сну, тривалість швидкого сну і покращує суб'єктивно сприйману якість сну у людини [Clin. Neurophys. 113, 429-434, 2002]. Нефазодон, сильнодіючий інгібітор рецепторів 5-НТ2А і слабкий інгібітор зворотного захоплення 5-НТ і NA, як було показано в клінічних дослідженнях, збільшує безперервність сну і загальний час швидкого сну і зменшує число пробуджень [Віоl. Psychiatry, 44, 3-14, 1998]. Аналогічно, тразадон, який являє собою антагоніст рецептора 5-НТ2А, і помірний інгібітор зворотного захоплення 5-НТ, як було показано, покращував клінічні показники за госпітальною шкалою оцінки тривоги (HAS) (розлади сну) і за шкалою HRSD (шкалою Гамільтона для оцінки ступеня депресії) (передчасне ранкове пробудження, недостатність міцного сну і ініціювання сну) [Psychiatr. Clin. Neurosci., 53, 193-194, 1999]. Sharpley в Neuropharmacology, 33, 467-471, 1994, повідомляє, що антагоністи рецепторів 5-НТ2А і, особливо, 5-НТ2C, покращують повільний сон. Вищезазначені результати і спостереження припускають, що виявлення сполук, які мають інгібуючу дію на зворотне захоплення 5-НТ і/або NA у поєднанні з антагоністичною активністю відносно рецепторів 5-НТ2А/C, могло б забезпечити сполуками, придатними для лікування афективних розладів, таких як, наприклад, депресія і тривожний стан, при відсутності побічного впливу на сон або із зниженим побічним впливом на нього. Біполярний розлад був раніше відомий як маніакально-депресивний синдром і характеризується епізодами манії і депресії, що повторюються час від часу. Основним завданням при лікуванні біполярного розладу (або депресії, пов'язаної з біполярним розладом) є уникнення маніакальних змін, тобто уникнення того, щоб у пацієнтів в стані депресії розвивалися маніакальні епізоди як наслідок лікування, направленого проти депресії. Для значної частини пацієнтів з біполярною депресією повідомлялося про появу манії внаслідок лікування після лікування антидепресантами [JClin. Psych., 67, suppl 11, 18-21, 2006]. Зазвичай маніакальні епізоди лікують антипсихотиками, такими як кветіапін або оланзапін, обидва з яких мають антагоністичний ефект відносно рецептора 5-НТ2А, або літієм. Сполука, що поєднує інгібування зворотного захоплення 5-НТ і NA з антагоністичною дією на рецептор 5-НТ2А, таким чином, представляється ідеальною сполукою для лікування біполярної депресії, що дозволяє уникнути маніакальних змін. Порушення сну і тривожність є відмітними ознаками посттравматичного стресового розладу (PTSD), таким чином, сполуки, що впливають на обидва ці симптоми були б добре відповідними для лікування цього захворювання. Меланхолія (ендогенна депресія) є особливим підтипом депресії, часто пов'язаним з тяжкою депресією; цей тип депресії також називають меланхолійною депресією. Меланхолія пов'язана з тривожністю, страхом перед майбутнім, безсонням і втратою апетиту. Сполуки, які інгібують зворотне захоплення як 5-НТ, так і NA, такі як, наприклад, венлафаксин, як було показано, є особливо ефективними в лікуванні пацієнтів з тяжкою депресією і меланхолією [Depres. Anxiety, 12, 50-54, 2000]. Розлад дефіциту уваги з гіперактивністю (ADHD) є найбільш звичайним з нейроповедінкових розладів. ADHD характеризується присутністю трьох ознак: соціальних і комунікативних порушень з обмеженими, повторюваними або стереотипними типами поведінки. ADHD зазвичай починається в дитинстві або в підлітковому віці, але симптоми можуть продовжитися і в зрілому віці. Атомоксетин є в даний час єдиним нестимулюючим лікарським засобом, дозволеним Комісією з контролю за продуктами харчування та ліками (FDA) для лікування ADHD [Drugs, 64, 205-222, 2004]. Атомоксетин є інгібітором зворотного захоплення NA, і це припускає, що сполуку І можна використовувати для лікування ADHD. На додаток до цього, сполуки, що є антагоністами рецепторів 5-НТ2А/C, можуть мати ефекти поліпшення сну, як обговорювалося вище, що є корисним в лікуванні ADHD. Фармакологічний профіль сполуки І і, зокрема, комбіноване сприяння 5-НТ- і NAнейротрансмісії за допомогою інгібуючого впливу на SERT і NAT і антагонізму відносно рецепторів 5-НТ2А і 5-НТ2С припускає, що сполука І може бути особливо корисною в лікуванні болю і, особливо, хронічного болю. Спеціальне згадування зроблене щодо застосування сполуки І для лікування болю і, особливо, хронічного болю, у пацієнтів, які також страждають від афективного розладу, такого як депресія і тривожність. Як показано в прикладах, сполука І, фактично, як було показано в експериментах на тваринах, демонструє виражений і дозо-залежний ефект при лікуванні невропатичного болю. У одному варіанті здійснення цей винахід відноситься до лікування захворювання, вибраного з великого депресивного розладу; дистимічного розладу; розладу настрою, пов'язаного із загальним медичним станом; атипової депресії; сезонного афективного розладу; меланхолії; стійкої до лікування депресії; при частковій відповіді; депресії, пов'язаної з 4 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 біполярним розладом, болем, хворобою Альцгеймера, психозом, хворобою Паркінсона, хворобою дифузних тілець Леві, хворобою Хантінгтона, розсіяним склерозом або тривожним станом; генералізованого тривожного розладу; соціального тривожного розладу; панічних атак; фобії; соціофобії; обсесивно-компульсивного розладу; посттравматичного стресового розладу; гострого стресу; синдрому дефіциту уваги з гіперактивністю (ADHD) і болю. У одному варіанті здійснення цей винахід відноситься до сполуки І для використання при лікуванні захворювань, вибраних з великого депресивного розладу; дистимічного розладу; розладу настрою, пов'язаного із загальним медичним станом; атипової депресії; сезонного афективного розладу; меланхолії; стійкої до лікування депресії; при частковій відповіді; депресії, пов'язаної з біполярним розладом, болем, хворобою Альцгеймера, психозом, хворобою Паркінсона, хворобою дифузних тілець Леві, хворобою Хантінгтона, розсіяним склерозом або тривожним станом; генералізованого тривожного розладу; соціального тривожного розладу; панічних атак; фобії; соціофобії; обсесивно-компульсивного розладу; посттравматичного стресового розладу; гострого стресу; синдрому дефіциту уваги з гіперактивністю і болю. У одному варіанті здійснення цей винахід відноситься до застосування сполуки І для виготовлення лікарського засобу для лікування захворювання, вибраного з великого депресивного розладу; дистимічного розладу; розладу настрою, пов'язаного із загальним медичним станом; атипової депресії; сезонного афективного розладу; меланхолії; стійкої до лікування депресії; при частковій відповіді; депресії, пов'язаної з біполярним розладом, болем, хворобою Альцгеймера, психозом, хворобою Паркінсона, хворобою дифузних тілець Леві, хворобою Хантінгтона, розсіяним склерозом або тривожним станом; генералізованого тривожного розладу; соціального тривожного розладу; панічних атак; фобії; соціофобії; обсесивно-компульсивного розладу; посттравматичного стресового розладу; гострого стресу; синдрому дефіциту уваги з гіперактивністю і болю. У одному варіанті здійснення, вищезазначений біль є хронічним болем, який може бути далі вибраний з фантомного болю в кінцівці, невропатичного болю, діабетичної невропатії, постгерпетичної невралгії (PHN), синдрому каналу зап'ястка (CTS), викликаної HIV невропатії, комплексного місцевого больового синдрому (CPRS), тригемінальної невралгії/невралгії трійчастого нерва/tic douloureux (хворобливого тику), хірургічного втручання (наприклад, післяопераційні аналгетичні засоби), діабетичної васкулопатії, резистентності капілярів або діабетичних симптомів, пов'язаних з інсуліном, ангіозного болю, менструального болю, пов'язаного з раком болю, зубного болю, головного болю, мігрені, головного болю напруження, тригемінальної невралгії, синдрому скронево-нижньощелепного суглоба, міофасціального болю при пошкодженні м'язів, синдрому фіброміалгії, болю в кістках і суглобах (остеоартриту), ревматоїдного артриту, ревматоїдного артриту і набряку внаслідок травми, пов'язаної з опіками, болю при розтягуваннях або тріщинах в кістці або її переломах унаслідок остеоартриту, остеопорозу, метастазів в кістці або з невідомих причин, подагри, фіброзиту, міофасціального болю, компресійних синдромів верхньої апертури грудної клітки, болю у верхній частині спини або болю в нижній частині спини (де біль в спині відбувається із-за системного, місцевого або первинного захворювання хребта (радикулопатії)), болю в тазу, кардіального болю в грудях, болю в грудях, що не відносяться до кардіального, болю, пов'язаного з пошкодженням спинного мозку (SCI), центрального постінсультного болю, невропатії при раку, болю при СНІД, болю при серповидно-клітинній анемії та геріатричному болю. У варіанті здійснення винаходу сполуку згідно з винаходом вводять в кількості, рівній від приблизно 0,001 до приблизно 100 мг/кг маси тіла на добу. Типове дозування для перорального введення знаходиться в діапазоні від приблизно 0,001 до приблизно 100 мг/кг маси тіла на добу, переважно від приблизно 0,01 до приблизно 50 мг/кг маси тіла на добу, що вводиться у вигляді однієї або декількох доз, наприклад, від 1 до 3 доз. Точне дозування залежатиме від частоти і способу введення, статі, віку, маси і загального стану пацієнта, якого піддають лікуванню, природи і тяжкості захворювання, що лікується, і будь-яких супутніх захворювань, що підлягають лікуванню, та інших чинників, очевидних фахівцям в даній галузі. Типове дозування сполуки цього винаходу для перорального введення для дорослих знаходиться в діапазоні 1-100 мг/добу сполуки цього винаходу, наприклад, 1-30 мг/добу або 525 мг/добу. Це, як правило, досягається введенням 0,1-50 мг, наприклад, 1-25 мг, наприклад, 1,5, 10, 15,20 або 25 мг сполуки цього винаходу один раз або двічі на добу. "Терапевтично ефективна кількість" сполуки, як використано тут, означає кількість, достатню для одужання, полегшення або часткового припинення клінічних проявів даного захворювання і його ускладнень в результаті терапевтичного втручання, яке включає введення вказаної сполуки. Кількість, достатню для досягнення цього, визначають як "терапевтично ефективну 5 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 кількість". Термін також включає кількості, достатні для одужання, полегшення або часткового припинення клінічних проявів даного захворювання і його ускладнень при лікуванні, що включає введення вищезазначеної сполуки. Ефективні кількості, призначені для кожної мети, залежатимуть від тяжкості захворювання або пошкодження, а також від маси тіла і загального стану пацієнта. Зрозуміло, що визначення відповідних дозувань може проводитися в рамках рутинних експериментів шляхом побудови матриці значень і тестування різних точок цієї матриці, що знаходиться в межах звичайних знань навченого лікаря. Терміни "терапія" і "лікування", використовувані в цьому описі, означають комплекс заходів, направлених на ведення пацієнта і догляд за ним з метою боротьби з патологічним станом, таким як захворювання або розлад. Термін означає повний спектр лікувальних заходів по відношенню до даного патологічного стану, яким страждає пацієнт, таких як введення активної сполуки, щоб частково зняти симптоми або ускладнення, затримувати розвиток захворювання, розладу або патологічного стану, частково зняти або полегшити симптоми і ускладнення і/або вилікувати або усунути захворювання, розлад або патологічний стан, а також для профілактики вказаного патологічного стану, де вказану профілактику слід розуміти як систему ведення пацієнта і догляду за ним з метою боротьби із захворюванням, патологічним станом або розладом, і де вказана профілактика включає введення активних сполук для попередження початку появи симптомів або ускладнень. При цьому профілактичний (превентивний) і терапевтичний (лікувальний) підходи в лікуванні є двома окремими аспектами цього винаходу. Пацієнт, що підлягає лікуванню, є переважно ссавцем, зокрема людиною. Сполуки цього винаходу можуть вводитися самі по собі у вигляді чистої сполуки або у поєднанні з фармацевтично прийнятними носіями або ексципієнтами, у вигляді одиничної дози або множинних доз. Фармацевтичні композиції цього винаходу можуть бути виготовлені з використанням фармацевтично прийнятних носіїв або розріджувачів, а також будь-яких інших відомих ад'ювантів і ексципієнтів, відповідно до традиційних процедур, таких як ті, що описані в керівництві Remington: The Science and Practice of Pharmacy, 19 Edition, Gennaro, Ed., Mack Publishing Co, Easton, PA, 1995. Фармацевтичні композиції можна спеціально приготувати для введення будь-яким відповідним способом, таким як пероральний, ректальний, назальний, пульмональний, місцевий (включаючи букальний і під'язиковий), трансдермальний, інтрацистернальний, внутрішньочеревинний, вагінальний і парентеральний (включаючи підшкірний, внутрішньом'язовий, інтратекальний, внутрівенний і внутрішньошкірний) спосіб, при цьому пероральний спосіб є переважним. Слід враховувати, що переважний спосіб залежатиме від загального стану здоров'я і віку суб'єкта, якого треба лікувати, природи стану, який треба лікувати, і вибраного активного інгредієнта. Фармацевтичні композиції для перорального введення включають тверді лікарські форми, такі як капсули, пігулки, драже, пілюлі, пастили, порошки і гранули. При необхідності їх можна виготовити з покриттями. Рідкі лікарські форми для перорального введення включають розчини, емульсії, суспензії, сиропи і еліксири. Фармацевтичні композиції для парентерального введення включають стерильні водні і неводні відповідні розчини, дисперсії, суспензії або емульсії для ін'єкцій, так само як і стерильні порошки для виготовлення з них стерильних розчинів або дисперсій для ін'єкції перед використанням. Інші відповідні для введення форми включають супозиторії, спреї, мазі, креми, гелі, засоби для інгаляції, шкірні пластири, імплантати тощо. Зазвичай сполуки винаходу вводяться в дозованій лікарській формі, що містить вказані сполуки в кількості приблизно від 0,1 до 50 мг, наприклад 1 мг, 5 мг, 10 мг, 15 мг, 20 мг або 25 мг сполуки цього винаходу. Для парентеральних способів введення, таких як внутрівенне, інтратекальне, внутрішньом'язове і подібне введення, як правило, дози складають порядку приблизно половини дози, використовуваної для перорального введення. Для парентерального введення можна застосовувати розчини сполуки за винаходом в стерильному водному розчині, водному пропіленгліколі, водному вітаміні Е або кунжутній або арахісовій олії. Такі водні розчини треба відповідним чином забуферити, якщо це необхідно, і спочатку зробити рідкий розчинник ізотонічним за допомогою достатньої кількості сольового розчину або глюкози. Водні розчини є особливо придатними для внутрівенного, внутрішньом'язового, підшкірного і внутрішньочеревинного введення. Всі використовувані стерильні водні середовища легко одержати за загальноприйнятими способами, які відомі фахівцям в даній галузі. 6 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 60 Придатні фармацевтичні носії включають інертні тверді розріджувачі або наповнювачі, стерильний водний розчин і різні органічні розчинники. Прикладами твердих носіїв є лактоза, каолін, сахароза, циклодекстрин, тальк, желатин, агар, пектин, гуміарабік, стеарат магнію, стеаринова кислота і нижчі алкілові етери целюлози. Прикладами рідких носіїв є сироп, арахісова олія, оливкова олія, фосфоліпіди, жирні кислоти, аміни жирних кислот, поліоксіетилен і вода. Фармацевтичні композиції, одержані об'єднанням сполуки за винаходом і фармацевтично прийнятних носіїв, потім легко вводяться в різноманітних лікарських формах, відповідних до описаних способів введення. Склади за цим винаходом, придатні для перорального введення, можуть бути виготовлені у вигляді окремих одиниць, таких як капсули або пігулки, кожна з яких містить заздалегідь визначену кількість активного інгредієнта і які можуть включати підхожий ексципієнт. Крім того, придатні для перорального введення склади можуть бути у формі порошку або гранул, розчину або суспензії у водній або неводній рідині, або у формі рідкої емульсії типу "олія у воді" або "вода в олії". Якщо твердий носій використовують для перорального введення, тоді препарат може бути пігулкою, наприклад, у формі порошку або гранул, поміщених в тверду желатинову капсулу, або у формі пастилки або пігулки для розсмоктування. Кількість твердого носія може мінятися, але зазвичай вона становить від приблизно 25 мг до приблизно 1 г. Якщо використовують рідкий носій, препарат може бути у формі сиропу, емульсії, м'якої желатинової капсули або стерильної придатної для ін'єкції рідини, такої як водна або неводна рідка суспензія або розчин. Пігулки можна одержувати змішуванням активного інгредієнта із звичайними ад'ювантами і/або розріджувачами з подальшим пресуванням суміші в звичайній таблетувальній машині. Приклади ад'ювантів або розріджувачів включають: кукурудзяний крохмаль, картопляний крохмаль, тальк, стеарат магнію, желатин, лактозу, камедь тощо. Будь-які інші ад'юванти або добавки, зазвичай використовувані для таких цілей, такі як барвники, ароматизатори, консерванти тощо, можна використовувати, за умови, що вони є сумісними з активними інгредієнтами. Капсули, що містять сполуку за цим винаходом, можна одержати перемішуванням порошку, який містить вказану сполуку з мікрокристалічною целюлозою і стеаратом магнію, і поміщенням вказаного порошку в тверду желатинову капсулу. Необов'язково, вказану капсулу можна забарвити за допомогою відповідного пігменту. Як правило, капсули міститимуть 0,25-20 % сполуки за цим винаходом, наприклад, 0,5-1,0 %, 3,0-4,0 % або 14,0-16,0 % сполуки за цим винаходом. Ці концентрації можна використовувати для зручної доставки 1, 5, 10, 15, 20 і 25 мг сполуки цього винаходу в дозованій лікарській формі. Розчини для ін'єкцій можна одержати розчиненням активного інгредієнта і можливих добавок в частині розчинника для ін'єкції, переважно стерильній воді, доведенням розчину до бажаного об'єму, стерилізацією цього розчину і наповненням ним відповідних ампул або флаконів. Можна додавати будь-які відповідні добавки, загальноприйняті в даній галузі, такі як агенти ізотонічності, консерванти, антиоксиданти тощо. Сполука І може вводитися або сама по собі, або у комбінації з іншою терапевтично активною сполукою, при цьому дві такі сполуки можуть вводитися або одночасно, або послідовно. Приклади терапевтично активних сполук, які можна з перевагою комбінувати із сполукою І, включають седативні або снодійні засоби, такі як бензодіазепіни; протисудомні препарати, такі як ламотригін, вальпроєва кислота, топірамат, габапентин, карбамазепін; стабілізатори настрою, такі як літій; дофамінергічні лікарські засоби, такі як агоністи дофаміну і ліводопа; лікарські засоби, використовувані для лікування синдрому дефіциту уваги і гіперактивності (ADHD), такі як атомоксетин; психостимулятори, такі як модафініл, кетамін, метилфенідат і амфетамін; інші антидепресанти, такі як міртазапін, міансерин і бупропріон; гормони, такі як ТЗ, естроген, DHEA (дигідроепіандростерон) і тестостерон; атипові антипсихотичні засоби, такі як оланзапін і арипіпразол; типові антипсихотичні засоби, такі як галоперидол; лікарські засоби для лікування хвороби Альцгеймера, такі як інгібітори холінестерази і мемантин, фолат; S-аденозилметіонін; імуномодулятори, такі як інтерферони; опіати, такі як бупренорфіни; антагоністи рецептора 1 ангіотензину II (антагоністи ATI); інгібітори АСЕ; статини і антагоніст альфа 1-адренергічного рецептора, такий як празозин. Сполуку І, у вигляді вільної основи, можна отримати, наприклад, як викладено в WO 2004/087156. Солі можна отримати шляхом додавання відповідної кислоти з подальшим осадженням. Осадження можна викликати, наприклад, охолоджуванням, видаленням розчинника, додаванням іншого розчинника або комбінацією вказаного. Альтернативно, сполуку І можна отримати так, як розкрито в прикладах. 7 UA 99500 C2 5 10 15 20 25 30 35 40 45 Всі посилання, включаючи публікації, патентні заявки і патенти, процитовані в цьому описі, включені в цей опис шляхом посилання в повному їх обсязі і в тому ступені, мовби для кожного посилання було вказано окремо і конкретно, що воно включено шляхом посилання і наведено в його повному обсязі в цьому описі (до максимального ступеня, що допускається законом), незалежно від будь-якого окремо наданого включення конкретних документів, зробленого будьде в цьому описі винаходу. Застосування форм однини і множини (застосування термінів з артиклями "a", "an" і "the") і подібних референтів в контексті опису цього винаходу слід тлумачити як такі, що охоплюють як форму однини, так і форму множини, якщо в цьому описі нема інших вказівок або якщо контекст явно не суперечить цьому. Наприклад, вираз "сполука" слід розуміти як вказівку на різноманітні сполуки цього винаходу або на конкретний описаний аспект, якщо нема інших вказівок. Якщо нема інших вказівок, всі точні значення, представлені в цьому описі, є репрезентативними для відповідних приблизних значень (наприклад, можна вважати, що всі приклади точних значень, представлені відносно конкретного чинника або вимірювання, також відносяться до відповідного приблизного значення, модифікованого за допомогою слова "приблизно", коли це доцільно). Передбачається, що в цьому описі винаходу опис будь-якого аспекту або аспекту цього винаходу, зроблений із застосуванням термінів, таких як "що містить", "що має", "що включає" або "що охоплює" щодо елементу або елементів, призначений для підтвердження аналогічного аспекту або аспекту цього винаходу, зробленого із застосуванням термінів "що складається з", "що в основному складається з" або "що по суті є" щодо даного конкретного елементу або елементів, якщо інше не вказано в даному описі або безсумнівно суперечить контексту (наприклад, композицію, описану в даному винаході, як таку, що містить конкретний елемент, слід розуміти також як опис композиції, що складається з даного елементу, якщо інше не вказано в даному описі або безсумнівно суперечить контексту). Приклади Якщо не вказано інше, рідинну хроматографію у поєднанні з мас-спектрометрією (LC/MS) проводили при наступних параметрах. LC/MS, загальна: система розчинників: А=вода/трифтороцтова кислота (100:0,05) і В=вода/ацетонітрил/трифтороцтова кислота (5:95:0,035) (ТРА=трифтороцтова кислота). Час утримування (RT) виражали в хвилинах. MS - установка фірми PESciex (API), обладнана АРРІджерелом, що працює в режимі визначення позитивних іонів. Спосіб: LC-система - це API 150EX і Shimadzu LC8/SLC-10A. Колонка: 30 × 4,6 мм Waters Symmetry C18 з 3,5 мкМ частинками, що працює при кімнатній температурі. Лінійний градієнт елюювання від 10 % В до 100 % В за 4 хвилини і з витратою потоку 2 мл/хв. Приклад 1. Фармакологічний профіль Величини інгібування, ІС50 (нМ), сполукою І зворотного захоплення в синаптосоми щурів: 3 [ Н]-серотонін: 2,4; 3 [ Н]-норадреналін: 12. Афінність (Кi нМ) сполуки І до серотонінових рецепторів людини обчислювали за рівнянням Ченга-Прусоффа: 5-НТ2А: 13; 5-НТ2с: 4,9. У функціональному аналізі сполука І, як було показано, була антагоністом рецептора 5-НТ2A з Kb близько 130 нМ, як було виміряно на флуоресцентному планшетному ридері з формуванням зображення (FLIPR). Аналогічно, сполука І є антагоністом рецептора 5-НТ2C з Kb близько 35 нМ. Приклад 2. Синтез сполуки І 8 UA 99500 C2 5 10 15 20 25 30 35 40 Стадія 1. 3,4-Дифторанізол (25,0 г) розчиняли в тетрагідрофурані (200 мл) і розчин охолоджували до -78 °C. Додавали н-бутиллітій (1,7 М в гексані, 102 мл) протягом 1 години при підтримці температури нижче -70 °C. Через 3 години при -78 °C додавали трет-бутиловий естер 4-оксо-піперидин-1-карбонової кислоти (31,2 г в 100 мл тетрагідрофурану) при такій швидкості, щоб температура підтримувалася нижче -65 °C. На наступний ранок неочищену суміш промивали насиченим водним хлоридом амонію (200 мл) і водою (100 мл). Органічний шар сушили над безводним сульфатом магнію і концентрували під вакуумом для отримання неочищеного продукту. Цю речовину очищали хроматографією на силікагелі (елюент: етилацетат/гептан, 1:1) з отриманням продукту (27,7 г; забруднений трет-бутиловим естером 4оксо-піперидин-1-карбонової кислоти). Стадія 2. Продукт попередньої стадії кип'ятили із зворотним холодильником в суміші 33 % бромистого водню в оцтовій кислоті (50 мл) і 48 % водного бромистого водню (50 мл) протягом ночі. На наступний ранок суміш охолоджували до кімнатної температури і осаджену тверду речовину (12,7 г) відфільтрували і використовували на наступній стадії. Стадія 3. Частину продукту з попередньої стадії (7,7 г) розчиняли в етанолі (150 мл). Додавали триетиламін (3,8 мл) з подальшим додаванням маленькими порціями протягом 5 хвилин ди-трет-бутил бікарбонату (5,8 г). Суміш перемішували протягом вихідних при кімнатній температурі. Осаджений продукт відфільтрували і фільтрат концентрували під вакуумом для отримання фракції другого неочищеного продукту. Цю речовину розподіляли між діетиловим етером (100 мл), водою (100 мл) і 10 % водним гідроксидом натрію (20 мл). Органічний шар промивали насиченим водним хлоридом натрію (100 мл) і сушили над сульфатом магнію. Фільтрування і концентрація під вакуумом давали другу партію продукту (загальний вихід 8,03 г). Стадія 4. Частину продукту з попередньої стадії (3,0 г) розчиняли в метиленхлориді (100 мл). Додавали (1,5-циклооктадієн)(піридин)(три-цикло-гексилфосфан)іридій(І) гексафторфосфат (каталізатор Крабтрі; 775 мг; 10 %) і цю суміш обробляли газоподібним воднем (3 бар), використовуючи струшувач Пара. Свіжий каталізатор додавали кілька разів протягом 24 годин (всього 30 %). Фільтрування дало білу тверду речовину, яку використовували на наступній стадії. Стадія 5. Неочищену суміш з попередньої стадії розчиняли в N,N-диметилформаміді (20 мл). Додавали етил-ді-ізо-пропіламін (основа Хуніга; 0,76 г) і 4-диметиламінопіридин (0,12 г), а потім 1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфонілфторид (NfF; 1,62 г). Через 1 годину леткі речовини видаляли під вакуумом і неочищений продукт очищали хроматографією на силікагелі (елюент: етилацетат/гептан, 1:4) для отримання бажаного продукту (2,04 г). Стадія 6. Продукт з попередньої стадії (2,04 г) додавали в колбу, що містить трет-бутоксид натрію (0,45 г) і сухий толуол (25 мл). Суміш дегазували аргоном, перш ніж додати її в колбу, що містить дегазовану суміш трис(дибензиліденацетон)дипаладію(0) (Pd 2dba3; 166 мг) і біс[(2дифенілфосфаніл)феніл]етеру (DPEphos; 195 мг) в сухому толуолі (10 мл). Нарешті додавали три-ізо-пропілсилантіол (0,78 мл) і цю суміш перемішували під аргоном при 100 °C протягом ночі. Після охолоджування до кімнатної температури неочищену суміш очищали 9 UA 99500 C2 5 10 15 20 хроматографією на силікагелі (елюент: етилацетат/гептан, 1:9) для отримання бажаного продукту (181 мг). Стадія 7. Продукт з попередньої стадії розчиняли в сухому толуолі (8 мл) під аргоном. Порцію цього вихідного розчину (1 мл) додавали в реакційну судину в Mettler-Toledo Bohdan block, використовуючи атмосферу аргону для виключення повітря. Додавали 2-фтор-1-йод-4-метилбензол (0,33 ммоль; отриманого з 2-фтор-4-метилфеніламіну, згідно загальній літературній методиці [S.E. Tunney and J.K. Stille, J. Org. Chem., 52, 748-53 (1987)]) у вигляді розчину в толуолі (1 мл), а потім 0,5 мл щойно приготованого розчину в толуолі суміші трис(дибензиліденацетон)дипаладію(0) (Рd2dba3) і біс[(2-дифенілфорсфаніл)феніл]етеру (DPEphos) (відповідно до 0,3 еквівалентів паладію і 0,6 еквівалентів DPEphos). Додавали трет-бутоксид калію (0,66 ммоль), а потім тетра-н-бутиламоній фторид (TBAF; 1,0 М в ТГФ; 80 мкл). Суміш перемішували при 100 °C протягом ночі під аргоном. На наступний ранок леткі компоненти видаляли, використовуючи апарат Genevac. Залишок розчиняли в метанолі (4 мл) і завантажували в VacMaster SCX-колонку (активовану 10 % оцтовою кислотою в метанолі). Продукт елюювали ацетонітрилом. Леткі компоненти видаляли під вакуумом. Залишок розчиняли в метанолі (1,5 мл) і додавали 4М НСl в діетиловому етері (1,5 мл). Суміш струшували при кімнатній температурі протягом вихідних, перш ніж всі леткі компоненти були видалені під вакуумом. Залишок розчиняли в диметилсульфоксиді (0,18 мл) і фільтрували. Додавали декілька крапель 20 % ацетонітрилу у воді і знову фільтрували. Продукт виділяли препаративною LC/MS, як описано, концентрували у вакуумі і цей продукт розчиняли в диметилсульфоксиді (0,78 мл) для отримання 10 мМ розчину. LC/MS-дані: метод 14, час утримування (УФ) 2,152 хв.; УФ-чистота 79,5 %; ELS-чистота 100 %; спостережувана маса 337,407. Приклад 3. Синтез сполуки І 25 30 35 40 Стадія 1. 3,4-дифторфенол (100 г) розчиняли в 3,4-дигідро-2H-пірані (DHP; 280 мл). Додавали 0,5 мл концентрованого водного хлористого водню і суміш перемішували протягом ночі при кімнатній температурі. Неочищену суміш екстрагували насиченим водним гідрокарбонатом натрію (200 мл) і діетиловим етером (400 мл), і органічний шар промивали насиченим водним хлоридом натрію (200 мл), і сушили над сульфатом магнію. Фільтрування і концентрація під вакуумом давали бажану сполуку (169 г) у вигляді блідо-жовтої оливи. Стадія 2. Через розчин продукту з попередньої стадії (інше завантаження; 214,2 г) в тетрагідрофурані (2 л) продували азот і охолоджували до -35 °C. Протягом 70 хвилин додавали розчин н-бутиллітію (10 М в гексані; 120 мл) і одержану суміш перемішували при -35 °C протягом 260 хвилин. Потім по краплях протягом 70 хвилин додавали етиловий естер 4-оксипіперидин-1-карбонової кислоти (205,4 г) при підтримці температури нижче -30 °C, перш ніж суміш перемішували протягом ночі при кімнатній температурі. На наступний ранок суміш охолоджували до 0 °C і додавали 2 М водний гідрохлорид (200 мл). Суміш перемішували при 10 UA 99500 C2 5 10 15 20 25 30 35 40 45 50 55 кімнатній температурі протягом 3 годин. Неочищену суміш розподіляли між водою (500 мл) і етилацетатом (200 мл). Водний шар екстрагували етилацетатом (200 мл). Об'єднані органічні шари промивали 15 % водним хлоридом натрію (3 × 200 мл) і концентрували під вакуумом спільно з толуолом (3 × 250 мл) для отримання жовтої оливи (442,4 г). Стадії 3+4. Продукт з попередньої стадії додавали до триетилсилану (160 мл) і суміш нагрівали до 60 °C. Додавали трифтороцтову кислоту (TFA; 250 мл), за якою слідувало додавання додаткової кількості триетилсилану (50 мл). Через 90 хвилин додавали активоване вугілля (25 г) і суміш перемішували при 70 °C протягом 0,5 години. Додавали етанол (500 мл) і суміш перемішували протягом ночі при кімнатній температурі. На наступний ранок суміш нагрівали при кип'ятінні із зворотним холодильником протягом 1 години, перш ніж відфільтрувати в теплому вигляді. Фільтрат концентрували під вакуумом. Залишок розчиняли в етанолі (100 мл) при 0 °C протягом 2,5 години. Осаджену тверду речовину (7,7 г) зібрали шляхом фільтрування. Фільтрат перемішували в етилацетаті (50 мл) і гептані (300 мл) для одержання другої порції цього продукту у вигляді твердої не зовсім білої речовини (153,8 г), яку виділяли фільтруванням. Об'єднані фракції продукту розчиняли в суміші тетрагідрофуран/етанол (1:3; 1,5 л) і обробляли Pd/C (5,4 г) і газоподібним воднем (3 бар) при кімнатній температурі, використовуючи струшувач Пара. Каталізатор відфільтрували і фільтрат концентрували під вакуумом, отримуючи тверду речовину, яку перемішували в гептані (300 мл) і потім виділяли фільтруванням, отримуючи білу тверду речовину (144,6 г). Стадія 5. Суспензію продукту з попередньої стадії (інше завантаження; 175 г) в ацетонітрилі (1,5 л) і триетиламіні (255 мл) обробляли 1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфонілфторидом (NfF; 142,6 мл) при кімнатній температурі. Через 25 хвилин суміш концентрували під вакуумом для отримання неочищеного нонафлату (405,2 г). Стадія 6. Продукт з попередньої стадії розчиняли в толуолі (3,4 л). До цього розчину додавали карбонат калію (168,6 г), етиловий естер 3-меркапто-пропіонової кислоти (85,4 г), трис(дибензиліденацетон)дипаладій(0) (Pd2dba3; 2,84 г) і біс(2-дифенілфосфаніл)етер (DPEphos; 4,1 г). Суміш дегазували азотом, перш ніж кип'ятити із зворотним холодильником протягом ночі. Суміш охолоджували до 0 °C і осаджену тверду речовину відфільтрували і промивали толуолом (100 мл). Об'єднані фільтрати використовували на наступній стадії. Стадія 7. Продукт з попередньої стадії додавали до охолодженої на льоду суспензії третбутоксиду калію (95,4 г) в толуолі (2,8 л) протягом ~2 годин. Потім додавали 1-бром-2-фтор-4метилбензол (121 г), трис(дибензиліденацетон)дипаладій(0) (Pd2dba3; 1,7 г) і біс(2дифенілфосфаніл)етер (DPEphos; 2,48 г) і цю суміш кип'ятили із зворотним холодильником протягом ~1 години. Неочищену суміш охолоджували до кімнатної температури і фільтрували через силікагель і концентрували під вакуумом для отримання неочищеного продукту (240 г). Стадія 8. Продукт з попередньої стадії розчиняли в 33 % бромистому водні в оцтовій кислоті (368 мл; 3 еквіваленти HBr) і розчин перемішували при 110 °C протягом ~4 годин. Потім додавали додаткову кількість 33 % бромистого водню в оцтовій кислоті (~0,5 еквівалента HBr) і суміш перемішували при 110 °C протягом 45 хвилин, перш ніж охолодити її до кімнатної температури. На наступний ранок розчин охолоджували на льодяній бані і додавали діетиловий етер (2,25 л). Через 1,5 години осаджену тверду речовину зібрали фільтруванням для отримання бажаного продукту у вигляді гідробромідної солі (185 г). Приклад 4. Вплив на невропатичний біль Існують декілька добре обґрунтованих тваринних моделей невропатичного болю, доступних для оцінки антиноцицептивного потенціалу лікарських засобів. Серед найчастіше використовуваних моделей знаходиться модель постійного пошкодження стискуванням (наприклад, Bennett and Xie, Pain, 1988), модель із застосуванням капсаїцину (Gilchrist et al, Pain 1996) і модель із застосуванням формаліну [Neuropharm., 48, 252-263, 2005; Pain, 51,5-17, 1992]. Для демонстрації ефективності проти невропатичного болю сполуку І тестували на моделі невропатичного болю із застосуванням формаліну. У цій моделі миші отримували ін'єкцію формаліну (4,5 %, 20 мкл) в підошовну поверхню лівої задньої лапи і потім поміщалися в індивідуальні скляні стакани (ємністю 2 л) для спостереження. Подразнення, викликане формаліновою ін'єкцією, виявляє характерну двофазну поведінкову реакцію, яка кількісно виражається в кількості часу, що витрачається на зализування пошкодженої лапи. Перша фаза (0-10 хвилин) відображає безпосереднє хімічне подразнення і ноцицепцію, тоді як друга фаза (20-30 хвилин), як вважають, відображає біль невропатичного походження. Дві фази розділено періодом спокою, під час якого поведінка повертається до нормальної. Вимірювання кількості часу, витраченого на зализування пошкодженої лапи в двох фазах, дає оцінку ефективності досліджуваної сполуки відносно зниження реакції на біль невропатичного походження. 11 UA 99500 C2 Таблиця 1 нижче, показує кількість часу, витраченого на зализування пошкодженої лапи, в двох фазах, тобто, 0-5 хвилин і 20-30 хвилин після ін'єкції формаліну. Для кожної дози було узято вісім мишей і 12 мишей для групи, якій вводили наповнювач. Таблиця 1 0-5 хвилин (сек.) 20-30 хвилин (сек.) Наповнювач 51 66 0,25 мг/кг підшкірно 40 48 1 мг/кг підшкірно 40 21 2,5 мг/кг підшкірно 31 7 5 10 Дані таблиці 1 показують, що сполука І надає незначну дію в першій фазі, що відображає безпосереднє хімічне подразнення і ноцицепцію. Більш значним є те, що ці дані також показують чітке і дозо-залежне зменшення кількості часу, витраченого на зализування пошкодженої лапи під час другої фази, вказуючи на ефект сполуки цього винаходу при лікуванні невропатичного болю. ФОРМУЛА ВИНАХОДУ 15 1. 4-[2,3-Дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин у відповідності зі структурною формулою NH F F S F 20 і його фармацевтично прийнятні солі. 2. Фармацевтична композиція, яка містить сполуку за п. 1 і принаймні один фармацевтично прийнятний носій або розріджувач. 3. Спосіб лікування болю, який передбачає введення терапевтично ефективної кількості сполуки у відповідності з п. 1 пацієнтові, що потребує цього. 4. Сполука за п. 1 для використання при лікуванні болю. 5. Застосування сполуки за п. 1 для виготовлення лікарського засобу для лікування болю. 25 Комп’ютерна верстка Л. Ціхановська Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 12
ДивитисяДодаткова інформація
Назва патенту англійською4-[2,3-difluoro-6-(2-fluoro-4-methyl-phenylsulfanyl)-phenyl]-piperidine
Автори англійськоюBang-Andersen, Benny, Jorgensen Morten, Faldt, Andre, Andercon, Neil, Stensbol, Tine, Bryan
Назва патенту російською4-[2,3-дифтор-6-(2-фтор-4-метилфенилсульфанил)фенил]пиперидин
Автори російськоюБанг-Андерсен Бенни, Йоргенсен Мортен, Фалт Андре, Андерсон Нил, Стенсбьол Тине Брайан
МПК / Мітки
МПК: C07D 211/20, A61P 25/00, A61K 31/451
Мітки: 4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин
Код посилання
<a href="https://ua.patents.su/14-99500-4-23-diftor-6-2-ftor-4-metilfenilsulfanilfenilpiperidin.html" target="_blank" rel="follow" title="База патентів України">4-[2,3-дифтор-6-(2-фтор-4-метилфенілсульфаніл)феніл]піперидин</a>
Рідкі композиції солей 4-[2-(4-метилфенілсульфаніл)феніл]-піперидину

Номер патенту: 97660
Опубліковано: 12.03.2012
Автори: Лопес де Дієґо Хейді, Ласкоґен Ґудрун, Стенсбьол Тіне Брайан
МПК: A61K 9/08, A61K 31/451, A61P 25/00
Мітки: 4-[2-(4-метилфенілсульфаніл)феніл]-піперидину, солей, композиції, рідкі
Формула / Реферат:
1. Рідка фармацевтична композиція, яка містить сіль 4-[2-(4-метилфенілсульфаніл)феніл]піперидину, вибрану з солі приєднання DL-молочної кислоти, солі приєднання глутарової кислоти, солі приєднання L-аспарагінової кислоти і солі приєднання глутамінової кислоти.2. Рідка композиція за п. 1, яка відрізняється тим, що згаданою сіллю є сіль приєднання DL-молочної кислоти.3. Рідка композиція за п. 1, яка відрізняється тим, що згаданою...
Поліморфна форма 2-(r)-(1(r)-(3,5-біс(трифторметил)феніл)етокси)-3-(s)-(4-фтор)феніл-4-(3-(5-оксо-1н,4н-1,2,4-триазоло)метил)морфоліну як антагоніст рецептора тахікініну

Номер патенту: 57089
Опубліковано: 16.06.2003
Автори: Крокер Луіс, МакКолі Джеймс
МПК: A61P 29/00, A61P 25/04, A61P 1/08, A61P 43/00, A61P 11/06, A61K 31/5377, C07D 413/06, A61P 25/00
Мітки: 2-(r)-(1(r)-(3,5-біс(трифторметил)феніл)етокси)-3-(s)-(4-фтор)феніл-4-(3-(5-оксо-1н,4н-1,2,4-триазоло)метил)морфоліну, антагоніст, тахікініну, рецептора, поліморфна, форма
Формула / Реферат:
1. Поліморфна форма сполуки 2-(R)-(1-(R)-(3,5-біс(трифторметил)феніл)етокси)-3-(S)-(4-фтор)феніл-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфоліну.2. Поліморфна форма за пунктом 1, яка по суті відрізняється рентгенівською порошковою дифрактограмою з характерними відображеннями приблизно 12.0, 15.3, 16.6, 17.0, 17.6, 19.4. 20.0, 21.9, 23.6, 23.8 і 24.8° (2 тета).3. Фармацевтична композиція, що містить фармацевтично прийнятний...
Кристалічні форми 4-[2-(4-метилфенілсульфаніл)-феніл]піперидину з комбінованим інгібуванням зворотного захоплення серотоніну та норепінефрину для лікування невропатичного болю

Номер патенту: 95801
Опубліковано: 12.09.2011
Автори: Фалт Андре, Міллер Зілке, Стенсбьол Тіне Брайан, Лопес де Дієґо Хейді, Банґ-Андерсен Бенні
МПК: A61K 31/451, A61P 25/32, C07D 211/20, A61P 25/36, A61P 25/24, A61P 25/28, A61P 25/04, A61P 25/34, A61P 25/22
Мітки: 4-[2-(4-метилфенілсульфаніл)-феніл]піперидину, комбінованим, болю, невропатичного, зворотного, лікування, захоплення, інгібуванням, серотоніну, форми, норепінефрину, кристалічні
Формула / Реферат:
1. Кристалічна форма кислотно-адитивної солі 4-[2-(4-метилфенілсульфаніл)феніл]піперидину формули, де НХ являє собою кислоту, вибрану з кристалічної форми:адитивної солі НВr, яка характеризується піками на порошковій рентгенограмі (XRPD) при кутах 2q (±0,1°) 6,08, 14,81, 19,26 і 25,38,адитивної солі DL-молочної кислоти,...
Похідні 5-феніл-3(піперидин-4-іл)-1,3,4-оксадіазол-2(3н)ону та фармацевтична композиція

Номер патенту: 50748
Опубліковано: 15.11.2002
Автори: Соліньяк Аксель, Локхед Алістер, Неделек Ален, Галлі Фредерік, Де Круз Лоран, Жегам Самір
МПК: A61K 31/445, A61K 31/454, C07D 413/04, A61K 31/00, C07D 413/14, A61K 31/4523, A61P 43/00, A61K 31/4465
Мітки: композиція, 5-феніл-3(піперидин-4-іл)-1,3,4-оксадіазол-2(3н)ону, похідні, фармацевтична
Формула / Реферат:
1. Похідні 5-феніл-3-(піперидин-4-іл)-1,3,4-оксадіазол-2-(3Н)-ону у формі чистого оптичного ізомеру або суміші таких ізомерів, які відповідають загальній формулі (І)в якійR1 - (С1-С4)-алкіл, або (С3-С7)-циклоалкіл,Χ1 - атом гідрогену чи галогену або (С1-С4)-алкоксил, абоOR1 та Χ1 разом представляють групу формули-OCH2O-, -О(СН2)2-, -О(СН2)3-, -О(СН2)2О- або -О(СН2)3О-,Х2- атом...
Похідні n-[феніл(піперидин-2-іл)метил]бензаміду, спосіб їх отримання та застосування у терапії

Номер патенту: 78025
Опубліковано: 15.02.2007
Автори: Естенн-Буту Женев'єв, Дарґазанлі Жіад, Медеско Флоран, Роже П'єр, Веронік Корінн, Маґа Паскаль, МАРАБУ Бенуа, Севрен Мірей
МПК: A61P 25/24, A61P 25/22, A61P 25/02, C07D 211/26, A61P 11/16, C07D 211/94, A61K 31/452, A61P 25/28, A61P 25/32, A61P 25/04, A61P 25/18, A61P 1/00, A61K 31/445, A61P 29/00, A61P 25/00, A61P 25/06, A61K 31/4458, A61P 25/16, A61P 43/00
Мітки: n-[феніл(піперидин-2-іл)метил]бензаміду, спосіб, отримання, застосування, похідні, терапії
Формула / Реферат:
1. Сполука, у формі чистого оптичного ізомеру (1R,2R) або (1S,2S), або у формі трео-діастереоізомеру загальної формули (І) , (I)в якій А представляєгрупу загальної формули N-R1, в якій R1 представляє атом гідрогену, лінійний або розгалужений (С1-С7)-алкіл, як варіант, заміщений одним або більше атомами флуору, або (С4-С7)-циклоалкіл, або...
Попередній патент: Композиція рідкого пального, спосіб її одержання та використання у двигуні внутрішнього згорання
Наступний патент: Пристрій для переорієнтування картонних пакетів
Випадковий патент: Запобіжний кульковий патрон