Інгібітори iap
Номер патенту: 114417
Опубліковано: 12.06.2017
Автори: Коен Фредерік, Газзард Льюїс Дж., Флайгер Джон А., Цуй Вікі Сяо-Вей

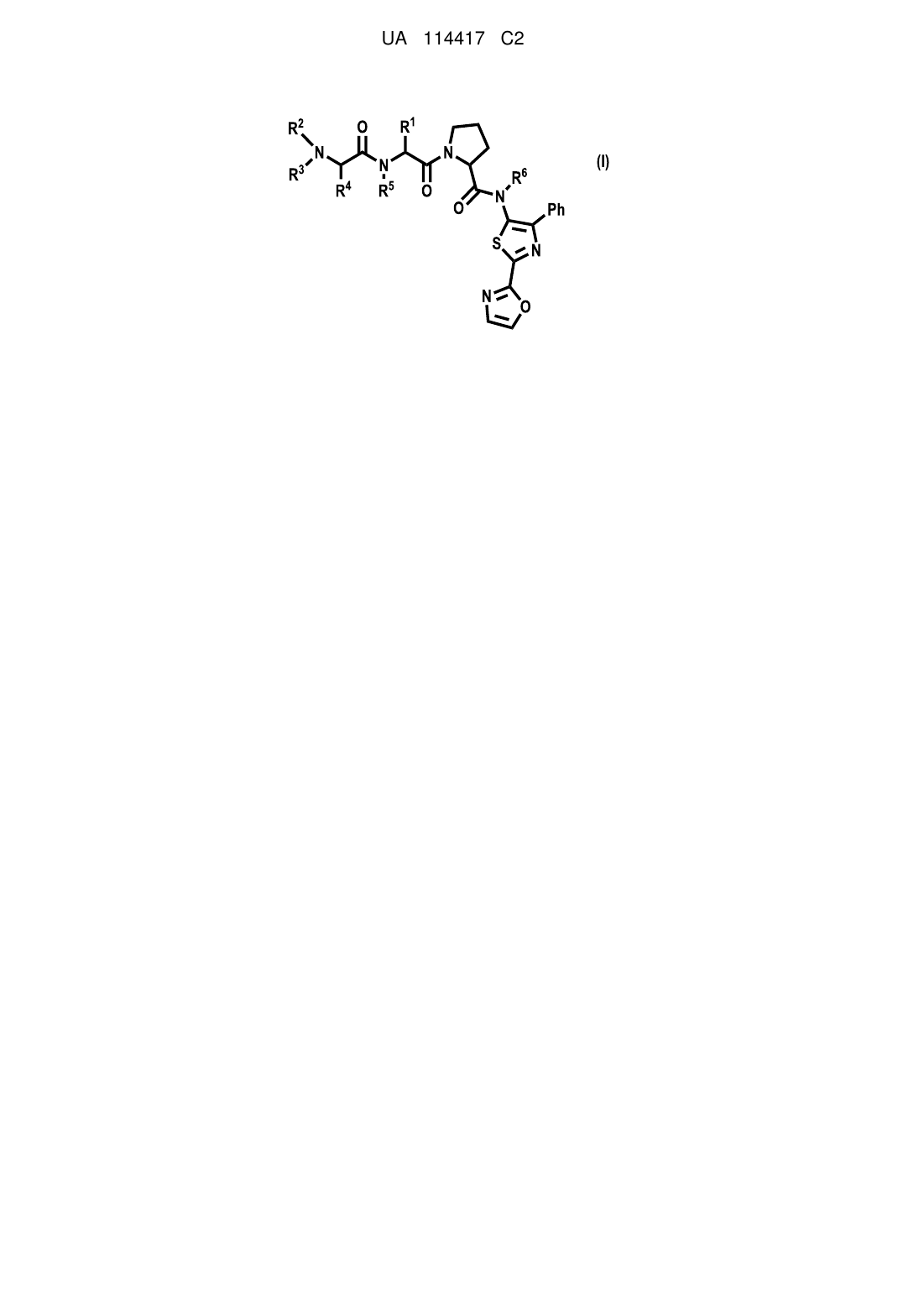





Формула / Реферат
1. Сполука формули І
 , (І)
, (І)
у якій
Ph являє собою феніл;
R1 являє собою С3-7-циклоалкіл;
кожний R2, R3, R4, R5 і R6 незалежно в кожному випадку являє собою Η або С1-6-алкіл; або
її фармацевтично прийнятна сіль.
2. Сполука за п. 1, що являє собою (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогексил-2-((S)-2-метиламінопропіоніламіно)ацетил]-піролідин-2-карбонової кислоти (Іа), або її фармацевтично прийнятна сіль.
3. Сполука за п. 1, яка відрізняється тим, що кожний R2-R6 незалежно являє собою Η або метил.
4. Сполука за п. 1, яка відрізняється тим, що R1 являє собою циклогексил.
5. Сполука за п. 3, яка відрізняється тим, що R1 являє собою циклогексил.
6. Сполука за п. 3, яка відрізняється тим, що один з R2 і R3 являє собою Н, а інший являє собою метил; або R4 являє собою метил; або кожний R5 і R6 являє собою Н.
7. Спосіб лікування захворювання або стану, пов'язаного з надлишковою експресією ІАР, у ссавця, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 1.
8. Спосіб лікування раку, який включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 1.
9. Спосіб лікування захворювання або стану, пов'язаного з надлишковою експресією ІАР, у ссавця, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 2.
10. Спосіб лікування раку, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 2.
11. Фармацевтична композиція, що містить сполуку за п. 1 і щонайменше один фармацевтично прийнятний носій, розріджувач або допоміжну речовину.
12. Фармацевтична композиція, що містить сполуку за п. 2 і щонайменше один фармацевтично прийнятний носій, розріджувач або допоміжну речовину.
Текст
Реферат: Запропоновані нові інгібітори ІАР, які є прийнятними терапевтичними агентами для лікування злоякісних утворень і мають загальну формулу (І): R 3 R O 2 N 1 N N R R 4 R 5 R O 6 N O Ph S N N O , (I) 1 2 3 4 5 6 при цьому R , R , R , R , R і R такі, як описано в цій заявці. UA 114417 C2 (12) UA 114417 C2 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 60 [0001] Даний винахід відноситься до органічних сполук, прийнятних для лікування і/або профілактики ссавця, зокрема до інгібіторів білків IAP, що прийнятні для лікування ракових захворювань. [0002] Апоптоз, або запрограмована загибель клітин, являє собою механізм, що регулюється генетично і біохімічно, який відіграє важливу роль у розвитку і гомеостазі у безхребетних, а також у хребетних. Порушення апоптозу, які призводять до передчасної загибелі клітин, зв'язують із рядом відхилень розвитку. Недостатність апоптозу, яка приводить до зниження загибелі клітин, зв'язують з раком і хронічними вірусними інфекціями (Thompson et al., (1995) Science 267, 1456-1462). [0003] Одними із ключових молекулярних ефекторів при апоптозі є каспази (аспартатспецифічні протеази, що містять цистеїн). Каспази є високоактивними протеазами, які розщеплюють білки після залишків аспарагінової кислоти і в активованій формі руйнують життєво важливі клітинні білки усередині клітини. Оскільки каспази є настільки високоактивними протеазами, для запобігання передчасної загибелі клітин необхідний точний контроль цього сімейства білків. У цілому, каспази синтезуються у вигляді великих неактивних зимогенів, для активації яких потрібний протеолітичний процесінг. Зазначений протеолітичний процесінг є тільки одним зі шляхів регуляції каспаз. Другий механізм опосередкований сімейством білків, що зв'язують та інгібують каспази. [0004] Сімейством молекул, що інгібують каспази, є інгібітори апоптозу (IAP) (Deveraux et al., J Clin Immunol (1999), 19:388-398). Спочатку IAP були відкриті в бакуловірусі, що було обумовлено їхньою функціональною здатністю заміняти білок Р35, антиапоптозний ген (Crook et al. (1993) J Virology 67, 2168-2174). Описи IAP наведені для різних організмів від Drosophila до людини. Незалежно від походження в структурах IAP містяться від одного до трьох доменів IAP бакуловірусу (BIR), що повторюються, і більшість із них також містять карбокситермінальний мотив RING finger. Домен BIR, як такий, являє собою цинк-зв'язувальний домен, що складається з приблизно з 70 залишків, що містить 4 альфа-спіралі і 3 бета-тяжі і залишки цистеїну і гістидину, який координує з іоном цинку (Hinds et al., (1999) Nat. Struct. Biol. 6, 648-651). Вважають, що домен BIR викликає антиапоптичну дію шляхом інгібування каспаз і, тим самим, інгібування апоптозу. Наприклад, людський IAP, пов'язаний з Х-хромосомою (XIAP), інгібує каспазу 3, каспазу 7, а також опосередковану Apaf-1-цитохромом С активацію каспази 9 (Deveraux et al., (1998) EMBO J. 17, 2215-2223). Домен BIR2 XIAP інгібує каспази 3 і 7, тоді як домен BIR3 XIAP відповідає за інгібування активності каспази 9. XIAP повсюдно експресуються в більшості тканин дорослих і плоду (Liston et al, Nature, 1996, 379(6563):349), їхня підвищена експресія відбувається в ряді пухлинних клітинних ліній, що належать до групи клітинних ліній NCI 60 (Fong et al, Genomics, 2000, 70:113; Tamm et al, Clin. Cancer Res. 2000, 6(5):1796). Було показано, що підвищена експресія XIAP у пухлинних клітинах забезпечує захист від ряду проапоптотичних стимулів і промотує резистентність до хіміотерапії (Lacasse et al, Oncogene, 1998, 17(25):3247). З цим узгоджується спостережувана сильна кореляція між рівнем білків XIAP і виживаністю пацієнтів з гострою мієлогенною лейкемією (Tamm et al, вище). Було показано, що понижувальна регуляція експресії XIAP античутливими олігонуклеотидами підвищує чутливість пухлинних клітин до загибелі, що викликається широким діапазоном проапоптотичних агентів in vitro і in vivo (Sasaki et al, Cancer Res., 2000, 60(20):5659; Lin et al, Biochem J., 2001, 353:299; Hu et al, Clin. Cancer Res., 2003, 9(7):2826). Також було показано, що Smac/ DIABLO-похідні пептиди підвищують чутливість ряду різних пухлинних клітинних ліній до апоптозу, викликаному різними проапоптотичними лікарськими засобами (Arnt et al, J. Biol. Chem., 2002, 277(46):44236; Fulda et al, Nature Med., 2002, 8(8):808; Guo et al, Blood, 2002, 99(9):3419; Vucic et al, J. Biol. Chem.,2002, 277(14):12275; Yang et al, Cancer Res., 2003, 63(4):831). [0005] IAP меланоми (ML-IAP) являє собою IAP, який не піддається виявленню в більшості нормальних тканин дорослої людини, але при меланомі відбувається його сильна підвищувальна регуляція (Vucic et al., (2000) Current Bio 10:1359-1366). Визначення структури білка показало значну гомологію доменів BIR і RING finger ML-IAP з відповідними доменами, що містяться в людських XIAP, C-IAP1 і C-IAP2. Вважають, що домен BIR ML-IAP найбільш схожий з BIR2 і BIR3 XIAP, C-IAP1 і C-IAP2 і відповідає за інгібування апоптозу, що визначали за допомогою делеційного аналізу. Крім того, Вучіч зі співавторами (Vucic et al.) показали, що MLIAP може інгібувати апоптоз, викликаний хіміотерапевтичними агентами. Агенти, такі як адріаміцин і 4-третинний бутилфенол (4-ТВР), тестували на системі клітинних культур меланоми, що мають підвищену експресію ML-IAP, і ефективність хіміотерапевтичних агентів відносно знищення клітин була значно знижена у порівнянні з нормальним контролем меланоцитів. Механізм, по якому ML-IAP здійснює антиапоптотичну активність, почасти опосередкований інгібуванням каспази 3 і 9. ML-IAP не інгібує ефективно каспази 1, 2, 6 або 8. 1 UA 114417 C2 5 10 15 20 25 30 35 40 45 [0006] Оскільки апоптоз є жорстко контрольованим процесом з безліччю взаємозалежних факторів, виявлення факту регуляції самих IAP не було несподіваним. У плодових мушок Drosophila білки Reaper (rpr), Head Involution Defective (hid) і GRIM вступають у фізичну взаємодію та інгібують антиапоптотичну активність сімейства IAP у Drosophila. У ссавців дія білків SMAC/DIABLO блокує IAP і забезпечує проходження апоптозу. Було показано, що при нормальному плині апоптозу відбувається процесінг SMAC з переведенням в активну форму і його вивільнення з мітохондрій у цитоплазму, де відбувається його фізичне зв'язування з IAP, що запобігає зв'язуванню IAP з каспазою. Зазначено інгібування IAP залишає каспазу в активній формі і, таким чином, відбувається апоптоз. Цікаво відзначити, що гомологія послідовностей в інгібіторах IAP підтверджує наявність мотиву з чотирьох амінокислот в N-кінці процесованих активних білків. Вважають, що зазначений тетрапептид зв'язується з гідрофобною "кишенею" домена BIR і руйнує домен BIR, що зв'язується з каспазами (Chai et al., (2000) Nature 406:855862, Liu et al., (2000) Nature 408:1004-1008, Wu et al., (2000) Nature 408 1008-1012). КОРОТКИЙ ОПИС ВИНАХОДУ [0007] Відповідно до одного з аспектів цього винаходу запропоновані нові інгібітори білків IAP, що мають загальну формулу (I) де [0008] R1 являє собою С3-7 циклоалкіл, [0009] Ph являє собою феніл, [0010] кожний R2, R3, R4, R5 і R6 у кожному випадку незалежно являє собою Н або С1-6 алкіл; або [0011] їх фармацевтично прийнятні солі. [0012] Формула I включає всі стереоізомери. [0013] Згідно з іншим аспектом цього винаходу запропоновані композиції, що містять сполуки формули I і носій, розріджувач або допоміжну речовину. [0014] Згідно з іншим аспектом цього винаходу запропонований спосіб ініціювання апоптозу в клітині, що включає введення в зазначену клітину сполуки формули I. [0015] Згідно з іншим аспектом цього винаходу запропонований спосіб збільшення чутливості клітини до апоптотичного сигналу, що включає введення в зазначену клітину сполуки формули I. [0016] Згідно з іншим аспектом цього винаходу запропонований спосіб інгібування зв'язування білка IAP з білком каспазою, що включає приведення зазначеного білка IAP у контакт зі сполукою формули I. [0017] Згідно з іншим аспектом цього винаходу запропонований спосіб лікування захворювання або стану, пов'язаного з підвищеною експресією білка IAP у ссавця, що включає введення зазначеному ссавцеві ефективної кількості сполуки формули I. [0018] Згідно з іншим аспектом цього винаходу запропонований спосіб лікування раку. КОРОТКИЙ ОПИС КРЕСЛЕНЬ [0019] На фігурі 1 показана ефективність Ia, що використовується окремо, а також у комбінації з апомабом, разом з даними ефективності відомої в рівні техніки сполуки III і його комбінації з апомабом у моделі ксенотрансплантата клітин аденокарциноми легенів Calu-6. Ia вводили п.о. III вводили в.в. Обрані дози являли собою максимальні стерпні дози для кожного лікарського засобу. [0020] На фігурі 2 показана ефективність Ia, що використовується окремо, а також у комбінації з апомабом, разом з даними ефективності відомої в рівні техніки сполуки III і його комбінації з апомабом у моделі ксенотрансплантата клітин колоректальної аденокарциноми 2 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 Colo205. Ia вводили п.о. III вводили в.в. Обрані дози являли собою максимальні стерпні дози для кожного лікарського засобу. [0021] Об'єкт, що описується у цій заявці у формі однини, відноситься до одного або більше зазначених об'єктів; наприклад, сполука відноситься до однієї або більше сполук або щонайменше до однієї сполуки. Таким чином, форму однини, "один або більше" і "щонайменше один" можна використовувати в цьому описі взаємозамінно. [0022] Використовувані в цьому описі як перехідна фраза або для опису пункту формули винаходу терміни "містити(ть)» і "що містить" слід розглядати як необмежуючі терміни. Тобто, ці терміни слід розглядати як синоніми з фразами "що має щонайменше" або "що включає щонайменше". При використанні в контексті опису способу термін "що включає" означає, що спосіб включає щонайменше зазначені стадії, але також може включати і додаткові стадії. При використанні в контексті опису сполуки або композиції термін "що містить" означає, що сполука або композиція включає всі зазначені відмітні ознаки або компоненти, але також може включати і додаткові відмітні ознаки або компоненти. [0023] Термін "незалежно" використовують у цьому описі для вказування на те, що змінну використовують у кожному випадку незалежно від наявності або відсутності в одній сполуці змінної, що має таке ж або відрізне визначення. Таким чином, у сполуці, у якій R" зустрічається двічі і визначений як "такий, що незалежно являє собою атом вуглецю або азоту", обоє R" можуть являти собою атоми вуглецю, обоє R" можуть являти собою атоми азоту або один R" може являти собою атом вуглецю, а інший – атом азоту. [0024] Терміни "можливий" або "можливо", використовувані в цьому описі, означають, що описувані з їхньою допомогою явища або умови можуть відбуватися, але не обов'язково, і в опис включені випадки, у яких явище або умова відбувається, і випадки, у яких вони відсутні. Наприклад, "можливо заміщений" означає, що можливо заміщений фрагмент може включати атом водню або замісник. [0025] Термін "приблизно" використовують у цьому описі в значенні приблизно, в районі, округлено або близько. Якщо термін "приблизно" використовують разом із числовим діапазоном, то він модифікує зазначений діапазон, розширюючи його границі вище і нижче числових значень, наведених для зазначеного діапазону. У цілому, термін "приблизно" використовують у цьому описі для модифікації числового значення в більшу і меншу сторону з погрішністю 20 % від зазначеного значення. [0026] При використанні в цьому описі зазначення числового діапазону для змінної означає можливість реалізації винаходу при використанні змінної, що має кожне зі значень усередині зазначеного діапазону. Таким чином, у випадку змінної, яка за визначенням може бути винятково дискретною, змінна може бути рівна будь-якому цілочисловому значенню усередині числового діапазону, включаючи кінці діапазону. Аналогічно, у випадку змінної, яка за визначенням є безперервною, змінна може бути рівна будь-якому фактичному значенню усередині числового діапазону, включаючи кінці діапазону. Наприклад, змінна, що описується як така, що має значення від 0 до 2, може становити 0, 1 або 2 у випадку змінних, які за визначенням є дискретними, і 0,0, 0,1, 0,01, 0,001 або мати будь-яке інше фактичне значення у випадку змінних, які за визначенням є безперервними. [0027] Сполуки формули I проявляють таутомерію. Таутомерні сполуки можуть існувати у вигляді двох або більше молекул, що взаємно перетворюються. Прототропні таутомери утворюються в результаті міграції ковалентно зв'язаного атома водню між двома атомами. Таутомери, у цілому, існують у рівновазі, і спроби виділити окремі таутомери звичайно приводять до одержання суміші, фізичні і хімічні властивості якої збігаються з властивостями суміші сполук. Положення рівноваги залежить від хімічних особливостей молекули. Наприклад, у безлічі аліфатичних альдегідів і кетонів, таких як ацетальдегід, переважає кето-форма, тоді як у фенолах переважає енольна форма. Розповсюджені прототропні таутомери включають системи кето/енол (-C(=O)-CH- Δ -C(-OH)=CH-), амід/імідинова кислота (-C(=O)-NH- Δ -C(OH)=N-) і амідинові таутомери (-C(=NR)-NH- Δ -C(-NHR)=N-). Два останні типи, зокрема поширені в гетероарильних і гетероциклічних кільцях. Цей винахід охоплює всі таутомерні форми сполук, описаних у цій заявці. [0028] "Алкіл" позначає розгалужену або нерозгалужену насичену аліфатичну вуглеводневу групу, що містить до 6 атомів вуглецю, якщо не зазначено інше, у тому числі у випадку використання як складової частини іншого терміну, наприклад, "алкіламіно". Приклади кращих алкільних груп включають метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, втор-бутил, третбутил, н-пентил, 2-метилбутил, 2,2-диметилпропіл, н-гексил, 2-метилпентил, 2,2-диметилбутил і т.д. Терміни "нижчий алкіл", "С1-С4 алкіл" і "алкіл, що містить від 1 до 4 атомів вуглецю" 3 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 60 являються синонімами, і їх використовують взаємозамінно для позначення метилу, етилу, 1пропілу, ізопропілу, циклопропілу, 1-бутилу, втор-бутилу або трет-бутилу. [0029] Циклоалкільні групи можуть являти собою моно-, бі- або трициклічні аліфатичні кільця, що містять від 3 до 7 атомів вуглецю. Кращі групи включають циклопропіл і циклогексил, найкращою є циклогексил. [0030] "Амінозахисна група" відноситься до похідного групи, що традиційно використовується для блокування або захисту аміногрупи, у результаті чого можна проводити взаємодії за участю інших функціональних груп сполуки. Приклади зазначених захисних груп включають карбамати, аміди, алкільні і арильні групи, іміни, а також численні N-гетероатомні похідні, які можна видаляти для регенерації цільової аміногрупи. Кращими амінозахисними групами є Вос, Fmoc і Cbz. Додаткові приклади зазначених груп наведені в T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, розділ 7; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. Mcomie, Ed., Plenum Press, New York, NY, 1973, розділ 5, і T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термін "захищений амін" відноситься до аміногрупи, заміщеної однією або більше із зазначених амінозахисних груп. Зазначені групи можна використовувати під час синтезу. [0031] "Карбоксизахисна група" відноситься до одному зі складноефірних похідних карбоксильної групи, традиційно застосовуваних для блокування або захисту карбоксильної групи, у результаті чого можна проводити взаємодії за участю інших функціональних груп сполуки. Приклади зазначених захисних груп карбонових кислот включають 4-нітробензил, 4метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгідрил, 4,4’диметоксибензгідрил, 2,2",4,4’-тетраметоксибензгідрид, алкіл, такий як трет-бутил або третаміл, тритил, 4-метокситритил, 4,4’-диметокситритил, 4,4",4’’-триметокситритил, 2-фенілпроп-2іл, триметилсиліл, трет-бутилдиметилсиліл, фенацил, 2,2,2-трихлоретил, бета(триметилсиліл)етил, бета-(ди(н-бутил)метилсиліл)етил, п-толуолсульфонілетил, 4нітробензилсульфонілетил, аліл, цинаміл, 1-(триметилсилілметил)проп-1-ен-3-іл і схожі фрагменти. Вид застосовуваної карбоксизахисної групи не важливий, оскільки похідні карбонових кислот є стабільними в умовах, у яких проводять наступну(і) взаємодію(ї) по інших положеннях молекули, і їх можна видаляти в прийнятний момент, не руйнуючи залишок молекули. Зокрема, важливо не піддавати молекулу із захищеними карбоксигрупами впливу сильних нуклеофільних основ, таких як гідроксид літію або NaOH, або впливу в умовах відновлення з використанням високоактивних гідридів металів, таких як LiAlH4. (Слід уникати використання таких жорстких умов і при видаленні амінозахисних груп і гідроксизахисних груп). Кращими захисними групами карбонових кислот є алкіл (наприклад, метил, етил, трет-бутил), аліл, бензил і п-нітробензильні групи. Для захисту карбокси-замісників також можна використовувати карбоксизахисні групи, схожі з тим, що застосовують в області цефалоспоринів, пеніцилінів і пептидів. Додаткові приклади зазначених груп наведені в T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, N.Y., 1991, розділ 5; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. Mcomie, Ed., Plenum Press, New York, N.Y., 1973, розділ 5, і T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981, розділ 5. Термін "захищений карбокси" відноситься до карбоксигрупи, заміщеної однією із зазначених вище карбоксизахисних груп. Зазначені групи можна використовувати під час синтезу. [0032] "Гідроксизахисна група" відноситься до похідного гідроксигрупи, традиційно використовуваного для блокування або захисту гідроксигруп, у результаті чого можна проводити взаємодії з використанням інших груп сполуки. Приклади зазначених захисних груп включають тетрагідропіранілокси, бензоїл, ацетокси, карбамоїлокси, бензил і прості силільні ефіри (наприклад, TBS, TBDPS). Додаткові приклади зазначених груп наведені в T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, розділи 2-3; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. Mcomie, Ed., Plenum Press, New York, NY, 1973, розділ 5, і T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термін "захищений гідрокси" відноситься до гідроксигрупи, заміщеної однією із зазначених вище гідроксизахисних груп. Зазначені групи можна використовувати під час синтезу. [0033] "Інгібітор" позначає сполуку, яка знижує або запобігає зв'язування білків IAP з білками каспазами або знижує або запобігає інгібування апоптозу білком IAP (наприклад, c-IAP1, c-IAP2, X-IAP або ML-IAP). Як альтернатива "інгібітор" позначає сполуку, яка запобігає зв'язування XIAP з каспазами або зв'язування ML-IAP з SMAC. 4 UA 114417 C2 5 10 15 20 25 30 35 40 45 "Фармацевтично прийнятні солі" включають солі приєднання кислот і основ. "Фармацевтично прийнятна сіль приєднання кислоти" відноситься до солей, які зберігають біологічну ефективність і властивості вільних основ, не є по біологічним або іншим причинам небажаними, отриманими з неорганічних кислот, таких як хлорводнева кислота, бромводнева кислота, сірчана кислота, азотна кислота, вугільна кислота, фосфорна кислота і т.д., і органічних кислот, які можуть бути обрані із класів аліфатичних, циклоаліфатичних, ароматичних, араліфатичних, гетероциклічних, карбонових і сульфо-органічних кислот, таких як мурашина кислота, оцтова кислота, пропанова кислота, гліколева кислота, глюконова кислота, молочна кислота, піровиноградна кислота, щавлева кислота, яблучна кислота, малеїнова кислота, малонова кислота, бурштинова кислота, фумарова кислота, винна кислота, лимонна кислота, аспарагінова кислота, аскорбінова кислота, глутамінова кислота, антранілова кислота, бензойна кислота, корична кислота, мигдальна кислота, ембонова кислота, фенілоцтова кислота, метансульфокислота, етансульфокислота, п-толуолсульфокислота, саліцилова кислота і т.д. "Фармацевтично прийнятні солі приєднання основ" включають солі, отримані з неорганічних основ, такі як солі натрію, калію, літію, амонію, кальцію, магнію, заліза, цинку, міді, марганцю, алюмінію і т.д. Особливо кращими є солі амонію, калію, натрію, кальцію і магнію. Солі, отримані з фармацевтично прийнятних органічних нетоксичних основ, включають солі первинних, вторинних і третинних амінів, заміщених амінів, включаючи природні заміщені аміни, циклічних амінів і основних іонообмінних смол, такі як солі ізопропіламіну, триметиламіну, диетиламіну, триетиламіну, трипропіламіну, етаноламіну, 2-диетиламіноетанолу, триметаміну, дициклогексиламіну, лізину, аргініну, гістидину, кофеїну, прокаїну, гідрабаміну, холіну, бетаїну, етилендиаміну, глюкозаміну, метилглюкаміну, теоброміну, пуринів, піперизину, піперидину, Nетилпіперидину, поліамінових смол і т.д. Особливо кращими органічними нетоксичними основами є ізопропіламін, диетиламін, етаноламін, триметамін, дициклогексиламін, холін і кофеїн. Передбачається, що формула I охоплює гідрати і сольвати сполук. [0034] У цьому винаході запропоновані нові сполуки, що мають загальну формулу I [0035] У конкретному варіанті реалізації R1 являє собою циклогексил. В іншому конкретному варіанті реалізації R1 являє собою циклопентил. У конкретному варіанті реалізації R1 орієнтований таким чином, що амінокислота або аналог амінокислоти, складовою частиною якого він є, має L-конфігурацію. [0036] R2 і R3 незалежно являють собою Н або С1-6 алкіл. В одному з варіантів реалізації R2 і R3 обоє являють собою Н. В іншому варіанті реалізації R2 являє собою метил, а R3 являє собою Н. [0037] R4 являє собою Н або С1-6 алкіл. У конкретному варіанті реалізації R4 являє собою Н або метил. В іншому варіанті реалізації R4 являє собою метил. В іншому варіанті реалізації R4 орієнтований таким чином, що амінокислота або аналог амінокислоти, складовою частиною якого він є, має L-конфігурацію. [0038] Кожний R5 і R6 незалежно являє собою Н або С1-6 алкіл. В одному з варіантів реалізації R5 і R6 являють собою Н або метил. В одному з варіантів реалізації R5 являє собою Н, а R6 являє собою метил. В іншому варіанті реалізації R5 являє собою метил, а R6 являє собою Н. В іншому варіанті реалізації R5 і R6 обоє являють собою метил. В іншому варіанті реалізації R5 і R6 обоє являють собою Н. [0039] Згідно з іншим аспектом цього винаходу сполука формули I являє собою (2-оксазол2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогексил-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти (Ia). 5 UA 114417 C2 5 10 [0040] Сполуки згідно з цим винаходом містять один або більше асиметричних атомів вуглецю. Відповідно, сполука може існувати у вигляді стереоізомерів, включаючи, диастереомери, енантіомери або їх суміші. У способах синтезу сполук як вихідних або проміжних речовин можна застосовувати рацемати, диастереомери або енантіомери. Диастереомерні сполуки можна розділяти за допомогою способів хроматографії або кристалізації. За аналогією, енантіомерні суміші можна розділяти за допомогою таких же або інших способів, відомих у даній галузі техніки. Кожний з асиметричних атомів вуглецю може мати R- або S-конфігурацію, і обидві зазначені конфігурації включені в обсяг цього винаходу. Краще сполуки згідно з цим винаходом мають наступну стереохімічну конфігурацію формули Ib, де R1, R2, R3, R4, R5 і R6 такі, як описано в цій заявці. 15 [0041] Сполуки формули II, де А являє собою можливо заміщений 5-членний гетероцикл, що містить від 1 до 4 гетероатомів, був запропонований в опублікованій заявці на патент США №20060014700. У деяких сполуках, запропонованих у зазначеній опублікованій заявці, А являє собою N-(4-фенілтіазол-5-іл). 20 25 [0042] Автори цієї заявки виявили, що сполуки згідно з цим винаходом, у яких А являє собою 2-(оксазол-2-іл)-4-фенілтіазол-5-іл, мають несподівано збільшену активність і пероральну біодоступність. Крім того, сполуки згідно з цим винаходом, у цілому, мають знижені побічні ефекти, включаючи поліпшену токсичність у легенях, наприклад, у порівнянні зі сполукою III, запропонованою в опублікованій заявці на патент США №20060014700. На фігурах 1 і 2 наведене порівняння активності в моделях ксенотрансплантатів пухлин після в.в. введення сполуки III і перорального введення сполуки Ia згідно з цим винаходом. 6 UA 114417 C2 5 10 СИНТЕЗ [0043] Сполуки згідно з цим винаходом одержували за допомогою стандартних способів органічного синтезу з комерційно доступних вихідних речовин і реактивів. Загальні способи синтезу описані в міжнародній заявці на патент WO 98/46576 і в патенті США №7244851, і в цю заявку шляхом посилань включені описи способів одержання, запропонованих у зазначених документах. Слід розуміти, що вибір способів синтезу, застосовуваних для одержання сполук згідно з цим винаходом, залежить від конкретних замісників, що містяться в сполуці, а також що у відповідності зі стандартною практикою органічного синтезу можуть вимагатися різні стадії введення і зняття захисту. Згідно із загальною схемою синтезу сполуки згідно з цим винаходом можна одержувати за допомогою традиційних способів хімії пептидів шляхом поєднування аналогів залишків амінокислот з використанням типових процедур амідного поєднування. На схемі 1 проводять поєднування амінозахищених залишків амінокислот і наступне зняття захисту з одержанням кінцевих сполук згідно з протоколами синтезу пептидів. Схема 1 15 20 25 30 [0044] Слід розуміти, що поєднування аналогів амінокислот можна проводити в будь-якому порядку, і їх можна одержувати з використанням твердофазної основи, що є традиційним у даній галузі техніки. [0045] Якщо сполуки згідно з цим винаходом містять замісники R2 або R3, відмінні від Н, їх також можна одержувати шляхом заміщення прийнятної проміжної кислоти, що містить групу, яка відходить, з використанням цільового аміну. Наприклад, Br-CH(R4)-C(O)-OH заміщають з використанням аміну R2-NH2 або R2-NH-R3 згідно зі схемою 2. [0046] Як альтернатива реакцію заміщення для введення замісників R2 або R3 можна проводити як кінцеву стадію способу одержання сполуки, як проілюстровано на схемі 3. Схема 3 7 UA 114417 C2 5 10 15 20 25 30 35 40 [0047] У конкретному варіанті реалізації проводять взаємодію 2-бромпропанової кислоти з відповідними амінами, розчиненими в ДМФ, при кип'ятінні до повного заміщення з утворенням N-заміщеного залишку аланіну. [0048] Сполуки з амінозаміщеним кільцем А, які виступають як проміжні речовини при одержанні сполук згідно з цим винаходом, є комерційно доступними або в іншому випадку їх можна одержувати з комерційно доступних реактивів за допомогою стандартних способів органічної хімії. 2-(оксазол-2-іл)-4-фенілтіазол-5-амін можна одержувати шляхом конденсації гідрохлориду α-амінофенілацетонітрилу і оксазол-2-карбальдегіду в присутності сірки і ТЕА (схема 4). ЗАСТОСУВАННЯ [0049] Сполуки згідно з цим винаходом інгібують зв'язування білків IAP з каспазами, зокрема зв'язування X-IAP з каспазами 3 і 7. Сполуки також інгібують зв'язування ML-IAP з білком Smac. Відповідно, сполуки згідно з цим винаходом прийнятні для ініціювання апоптозу в клітинах або для збільшення чутливості клітин, зокрема ракових клітин, до апоптотичних сигналів. Сполуки згідно з цим винаходом прийнятні для ініціювання апоптозу в клітинах, у яких відбувається підвищена експресія білків IAP (наприклад, c-IAP1, c-IAP2, X-IAP або ML-IAP). Як альтернатива сполуки згідно з цим винаходом прийнятні для ініціювання апоптозу в клітинах, у яких у результаті порушення мітохондріального апоптотичного шляху відбувається інгібування вивільнення Smac з білків ML-IAP, наприклад, за рахунок підвищувальної регуляції Bcl-2 або понижувальної регуляції Bax/Bak. У більш широкому значенні сполуки можна застосовувати для лікування раку. [0050] Вони особливо прийнятні для лікування всіх типів раку, при яких не відбувається апоптоз. Приклади зазначених типів раку включають нейробластому, карциному кишечника, таку як карцинома прямої кишки, карцинома товстої кишки, сімейний аденоматозний поліпоз і вроджений неполіпозний колоректальний рак, карциному стравоходу, карциному губи, карциному гортані, карциному гіпофаринксу, карциному язика, карциному слинних залоз, карциному шлунку, аденокарциному, медулярну карциному щитовидної залози, папілярну карциному щитовидної залози, карциному нирки, карциному паренхіми нирок, карциному яєчників, карциному шийки матки, карциному тіла матки, карциному ендометрію, хоріонкарциному, карциному підшлункової залози, карциному простати, карциному яєчок, карциному грудей, карциному сечовивідних шляхів, меланому, пухлини мозку, такі як гліобластома, астроцитома, менінгіома, медулобластома, і периферичні нейроектодермальні пухлини, лімфому Ходжкіна, неХоджкінську лімфому, лімфому Беркітта, гостру лімфатичну лейкемію (ГЛЛ), хронічну лімфатичну лейкемію (ХЛЛ), гостру мієлоїдну лейкемію (ГМЛ), хронічну лімфатичну лейкемію (ХМЛ), Т-клітинну лейкемію/лімфому дорослих, печінковоклітинну карциному, карциному жовчного міхура, бронхіальну карциному, дрібноклітинну карциному легенів, недрібноклітинну карциному легенів, множинну мієлому, базаліому, тератому, ретинобластому, хоріоїдальну меланому, семіному, рабдоміосаркому, краніофарингеому, остеосаркому, хондросаркому, міосаркому, ліпосаркому, фібросаркому, 8 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 60 саркому Юінга і плазмоцитому. Можливе застосування для лікування солідних пухлин. Також можливе застосування для лікування раку грудей, аденокарциноми підшлункової залози або злоякісної меланоми. [0051] Сполуки згідно з цим винаходом прийнятні для збільшення чутливості клітин до апоптотичних сигналів. Відповідно, сполуки можна вводити до, під час або після проведення радіаційної терапії або цитостатичної або антинеопластичної хіміотерапії. Прийнятні сполуки для цитостатичної хіміотерапії включають, але не обмежуються ними, (i) антиметаболіти, такі як цитарабін, флударабін, 5-фтор-2’-деоксиуридин, гемцитабін, гідроксисечовина або метотрексат; (ii) агенти, що фрагментують ДНК, такі як блеоміцин, (iii) агенти, що забезпечують перехресну зшивку ДНК, такі як хлорамбуцил, цисплатин, циклофосфамід або азотистий іприт; (iv) інтеркалуючі агенти, такі як адріаміцин (доксорубіцин) або мітоксантрон; (v) інгібітори синтезу білків, такі як L-аспарагіназа, циклогексімід, пуроміцин або токсин дифтерії; (vi) "отрути" топоізомерази I, такі як камптотецин або топотекан; (vii) "отрути" топоізомерази II, такі як етопозид (VP-16) або теніпозид; (viii) агенти спрямованої дії в мікротрубочках, такі як колцемід, колхіцин, паклітаксел, вінбластин або вінкристин; (ix) інгібітори кіназ, такі як флавопіридол, стауроспорин, STI571 (CPG 57148B) або UCN-01 (7-гідроксистауроспорин); (х) допоміжні агенти для досліджень, такі як тіоплатин, PS-341, фенілбутират, ET-18-OCH3 або інгібітори фарнезилтрансферази (L-739749, L-744832); поліфеноли, такі як кверцетин, ресвератрол, піцеатанол, галат епігалокатехіну, теафлавіни, флаваноли, процианідини, бетулінову кислоту і її похідні; (xi) гормони, такі як глюкокортикоїди або фенретинід; (xii) антагоністи гормонів, такі як тамоксифен, фінастерид або антагоністи РФЛГ. У кращому варіанті реалізації сполуки згідно з цим винаходом вводять разом з цитостатичною сполукою, обраною із групи, що складається з цисплатину, доксорубіцину, паклітакселу, доцетакселу і мітоміцину С. Найкращою цитостатичною сполукою є доксорубіцин. Можна застосовувати комбінації з 5-FU, гемцитабіном, капецитабіном, винорелбіном, бевацизумабом або таксанами. [0052] Іншим класом сполук, які можна застосовувати в цьому винаході, є сполуки, які можуть збільшувати чутливість або ініціювати апоптоз шляхом зв'язування з рецепторіви смерті ("агоністи рецептора смерті"). Зазначені агоністи рецепторів смерті включають ліганди рецепторів смерті, такі як фактор некрозу пухлини а (ФНО-α), фактор некрозу пухлини β (ФНОβ, лімфотоксин-α), LT-β (лімфотоксин-β), TRAIL (Apo2L, ліганд DR4), ліганд CD95 (Fas, APO-1), ліганд TRAMP (DR3, Apo-3), ліганд DR6, а також фрагменти і похідні кожного із зазначених лігандів. Переважно лігандом рецептора смерті є ФНО-α. Ще краще лігандом рецептора смерті є Apo2L/TRAIL. Крім того, агоністи рецепторів смерті містять агоністичні антитіла до рецепторів смерті, такі як антитіло до CD95, антитіло до TRAIL-R1 (DR4), антитіло до TRAIL-R2 (DR5), антитіло до TRAIL-R3, антитіло до TRAIL-R4, антитіло до DR6, антитіло до ФНО-α і антитіло до TRAMP (DR3), а також фрагменти і похідні кожного із зазначених антитіл. [0053] Для збільшення чутливості клітин до апоптозу сполуки згідно з цим винаходом також можна застосовувати у комбінації з радіаційною терапією. Фраза "радіаційна терапія" відноситься до використання електромагнітного або корпускулярного випромінювання для лікування новотворів. Радіаційна терапія заснована на тому принципі, що високодозне опромінення, що доставляється до цільової ділянки, призводить до загибелі репродуктивних клітин пухлини і нормальних тканин. Схему дозування радіаційного випромінювання, у цілому, визначають із урахуванням дози випромінювання (рад), що поглинається, часу і фракціонування, і її точний вибір повинен здійснюватися онкологом. Кількість випромінювання, одержуваного пацієнтом, залежить від різних факторів, але двома найбільш важливими є розташування пухлини відносно інших життєво важливих структур або органів організму і ступінь поширення пухлини. Приклади радіотерапевтичних агентів запропоновані, але не обмежуються ними, у роботах з радіаційної терапії і відомі в даній галузі техніки (Hellman, Principles of Radiation Therapy, Cancer, in Principles I and Practice of Oncology, 24875 (Devita et al., 4th ed., vol 1, 1993)). Останні досягнення в галузі радіаційної терапії включають погоджене по трьом вимірам опромінення зовнішнім променем, радіаційну терапію з модульованою інтенсивністю (РТМІ), стереотаксичну радіохірургію і брахітерапію (інтерстиціальну радіаційну терапію), в останньому способі джерело випромінювання розміщають безпосередньо в пухлину у вигляді "зерен", що імплантуються. За допомогою зазначених сучасних режимів лікування відбувається доставка більш високих доз випромінювання до пухлини, що обумовлює підвищену ефективність у порівнянні зі стандартною терапією з використанням зовнішнього променя. [0054] Іонізуюче випромінювання з використанням радіонуклідів, що випускають бетачастинки, вважають найбільш прийнятним для застосування в радіаційній терапії внаслідок помірної лінійної передачі енергії (ЛПЕ) іонізуючої частинки (електрона) і помірного діапазону 9 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 60 впливу (як правило, на кілька міліметрів тканини). При використанні гамма-променів відбувається доставка менших дозувань на значно більш високі відстані. Альфа-частинки є іншим крайнім випадком, вони доставляють дуже високе дозування ЛПЕ, але мають сильно обмежений діапазон і, таким чином, повинні перебувати в безпосередньому контакті з клітинами оброблюваної тканини. Крім того, джерела альфа-випромінювання, у цілому, являють собою важкі метали, що обмежує можливі хімічні умови і являє підвищену загрозу, викликану можливістю витоку радіонукліду з ділянки, що зазнає лікування. Залежно від пухлини, що піддається лікуванню, усі види джерел випромінювання розглядають у рамках обсягу цього винаходу. [0055] Крім того, цей винахід охоплює різні типи неіонізуючого випромінювання, наприклад, ультрафіолетове (УФ) випромінювання, високоенергетичне видиме випромінювання, мікрохвильове випромінювання (при гіпертермічній терапії), інфрачервоне (ІК) випромінювання і лазери. У конкретному варіанті реалізації цього винаходу застосовують УФ випромінювання. [0056] Більш конкретно, сполуки згідно з цим винаходом можна використовувати в комбінованій терапії. "Комбінована терапія" включає введення суб'єктові сполук у комбінації з іншими біологічно активними інгредієнтами (включаючи, але не обмежуючись ним, другий і антинеопластичний агент, що відрізняється) і з проведенням нелікарської терапії (включаючи, але не обмежуючись ними, хірургію або радіаційне лікування). Наприклад, сполуки згідно з цим винаходом можна застосовувати в комбінації з іншими фармацевтично активними сполуками, краще зі сполуками, які здатні підсилювати дію сполук згідно з цим винаходом. Сполуки згідно з цим винаходом можна вводити одночасно (у складі одного препарату або різних препаратів) або послідовно з іншою лікарською терапією. У цілому, комбінована терапія припускає введення двох або більше лікарських засобів під час одного циклу або курсу терапії. Таким чином, відповідно до одного з аспектів цього винаходу запропоновані сполуки можна вводити в комбінації з одним або більше окремими агентами, які модулюють протеїнкінази, задіяні в різних хворобливих станах або мішені в спадних сигналах. Приклади зазначених кіназ можуть включати, але не обмежуються ними, серин/треонін-специфічні кінази, рецепторні тирозин-специфічні кінази і нерецепторні тирозин-специфічні кінази. Серин/треонін кінази включають протеїнкінази, що активуються мітогеном (МАРК), мейоз-специфічну кіназу (MEK), RAF і кіназу Aurora. Приклади сімейств рецепторних кіназ включають рецептор епідермального фактора росту (РЕФР) (наприклад, HER2/neu, HER3, HER4, ErbB, ErbB2, ErbB3, ErbB4, Xmrk, DER, Let23); рецептор фактора росту фібробластів (FGF) (наприклад, FGF-R1,GFFR2/BEK/CEK3, FGF-R3/CEK2, FGF-R4/TKF, KGF-R); рецептор фактора росту/розсіювання гепатоцитів (HGFR) (наприклад, MET, RON, SEA, SEX); інсуліновий рецептор (наприклад, IGFIR, PI3K, AKT, mTor); Eph (наприклад, CEK5, CEK8, EBK, ECK, EEK, EHK-1, EHK-2, ELK, EPH, ERK, HEK, MDK2, MDK5, SEK); Axl (наприклад, Mer/Nyk, Rse); RET; і рецептор фактора росту, виділеного із тромбоцитів (PDGFR) (наприклад, PDGFα-R, PDGβ-R, CSF1-R/FMS, SCF-R/C-KIT, VEGF-R/FLT, NEK/FLK1, FLT3/FLK2/STK-1). Сімейства нерецепторних протеїнкіназ включають, abl але не обмежуються ними, BCR-ABL (наприклад, p43 , ARG); BTK (наприклад, ITK/EMT, TEC); CSK, FAK, FPS, JAK, SRC, BMX, FER, CDK і SYK. Згідно з іншим аспектом винаходу запропоновані сполуки можна вводити у комбінації з одним або більше окремими агентами, які модулюють некіназні біологічні мішені або процеси. Зазначені мішені включають деацетилази гістонів (HDAC), метилтрансферазу ДНК (DNMT), білки теплового шоку (наприклад, HSP90), інгібітори Hedgehog і протеосоми. У кращому варіанті реалізації запропоновані сполуки можна поєднувати з антинеопластичними агентами (наприклад, з невеликими молекулами, моноклональними антитілами, античутливої РНК і гібридними білками), які інгібують одну або більше біологічних мішеней, такими як ериведж, золінза, тарцева, іреса, тайкерб, глівек, сутент, спріцел, нексавар, CNF2024, RG108, BMS387032, афінітак, авастин, герцептин, ербітукс, AG24322, PD325901, ZD6474, PD184322, обатодакс, ABT737 і AEE788. Також включені моноклональні антитіла, специфічні до кіназ і/або рецепторів, наприклад, зазначені в цій заявці або інші. Зазначені комбінації можуть підвищувати терапевтичну ефективність у порівнянні з ефективністю, що досягається при використанні кожного з агентів окремо, і можуть запобігати або відстрочувати появу резистентних мутантних змін. У конкретних кращих варіантах реалізації сполуки згідно з цим винаходом вводять у комбінації з хіміотерапевтичним агентом. Хіміотерапевтичні агенти охоплюють широкий діапазон способів лікування онкології. Зазначені агенти вводять на різних стадіях захворювання для зменшення розміру пухлини, знищення ракових клітин, що залишилися після хірургії, ініціювання ремісії, підтримки ремісії і/або ослаблення симптомів, що відносяться до раку або до способу його лікування. Приклади зазначених агентів (деякі з яких також обговорювалися вище) 10 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 включають, але не обмежуються ними, алкілюючі агенти, такі як похідні газоподібного іприту (мехлоретамін, циклофосфамід, хлорамбуцил, мелфалан, іфосфамід), етиленіміни (тіотепа, гексаметилмеланін), алкілсульфонати (бусульфан), гідразини і триазини (алтретамін, прокарбазин, дакарбазин і темозоломід), нітрозосечовини (кармустин, ломустин і стрептозоцин), іфосфамід і солі металів (карбоплатин, цисплатин і оксаліплатин); рослинні алкалоїди, такі як подофілотоксини (етопозид і теніпозид), таксани (паклітаксел і доцетаксел), алкалоїди вінку (вінкристин, вінбластин, віндесин і винорелбін) і аналоги камптотекану (іринотекан і топотекан); протипухлинні антибіотики, такі як хромоміцини (дактиноміцин і плікаміцин), антрацикліни (доксорубіцин, даунорубіцин, епірубіцин, мітоксантрон, валрубіцин та ідарубіцин) і допоміжні антибіотики, такі як мітоміцин, актиноміцин і блеоміцин; антиметаболіти, такі як антагоністи фолієвої кислоти (метотрексат, пеметрексед, ралтітрексед, аміноптерин), піримідинові антагоністи (5-фторурацил, флоксуридин, цитарабін, капецитабін і гемцитабін), пуринові антагоністи (6-меркаптопурин і 6-тіогуанін) та інгібітори аденозиндезамінази (кладрибін, флударабін, меркаптопурин, клофарабін, тіогуанін, неларабін і пентостатин); інгібітори топоізомерази, такі як інгібітори топоізомерази I (іринотекан, топотекан) та інгібітори топоізомерази II (амсакрин, етопозид, етопозиду фосфат, теніпозид); моноклональні антитіла (алемтузумаб, гемтузумаб озогаміцин, ритуксимаб, трастузумаб, ібритумомаб тіуксетан, цетоксимаб, панітумумаб, тоситумумаб, бевацизумаб); і допоміжні антинеопластичні засоби, такі як інгібітори рибонуклеотидредуктази (гідроксисечовина); інгібітори адренокортикальних стероїдів (мітотан); ферменти (аспарагіназа і пегаспаргаза); антимікротрубочкові агенти (естрамустин); і ретиноїди (бексаротен, ізотретіноїн, третіноїн (ATRA)) і т.д. [0057] Винахід також включає фармацевтичні композиції або лікарські засоби, що містять сполуки згідно з цим винаходом і терапевтично інертний носій, розріджувач або допоміжну речовину, а також способи застосування сполук згідно з цим винаходом для одержання зазначених композицій і лікарських засобів. Як правило, сполуки формули I, застосовувані в способах згідно з цим винаходом, вводять до складу шляхом перемішування при температурі навколишнього середовища і відповідному рН і при бажаному ступені чистоти з фізіологічно прийнятними носіями, тобто з носіями, які є нетоксичними для споживача в дозуваннях і концентраціях, застосовуваних у галенових лікарських формах. рН складу залежить, головним чином, від конкретного застосування і концентрації сполуки, але краще перебуває в діапазоні від приблизно 3 до приблизно 8. Прийнятним варіантом реалізації є сполука в ацетатному буфері з рН 5. [0058] Інгібуюча сполука для застосування згідно з цим винаходом краще є стабільним. Як правило, сполуку зберігають у вигляді твердої композиції, хоча ліофілізовані сполуки або водні розчини також є прийнятними. [0059] Композиції згідно з цим винаходом одержують, дозують і вводять відповідно до належної медичної практики. Важливі в цьому контексті фактори включають конкретне порушення, що зазнає лікування, конкретний ссавець, що зазнає лікування, клінічний стан конкретного пацієнта, причину порушення, ділянку доставки агенту, спосіб введення, схему введення та інші фактори, відомі практикуючим лікарям. "Ефективна кількість" сполуки, що вводиться, залежить від зазначених факторів і являє собою мінімальну кількість, необхідну для лікування зазначених захворювань, наприклад, для інгібування взаємодії IAP з каспазами, ініціювання апоптозу або збільшення чутливості злоякісних клітин до апоптотичного сигналу. Зазначена кількість краще є більш низькою у порівнянні з кількістю, яка є токсичною для нормальних клітин або для ссавця в цілому. [0060] У цілому, сполуки згідно з цим винаходом можна вводити один або кілька разів на день. Також їх можна вводити безупинно протягом дня без перерв у введенні. Таким чином, їх також можна вводити і з перервами. Зазначені варіанти також є доступними і у випадку застосування разом з іншими агентами або згідно з іншою схемою. Первісна фармацевтично ефективна кількість сполуки згідно з цим винаходом в дозі, що вводиться парентерально, перебуває в діапазоні приблизно 0,01-100 мг/кг, краще приблизно від 0,1 до 20 мг/кг маси тіла пацієнта на день, при цьому звичайно первісний діапазон кількості сполуки, що вводиться, становить від 0,3 до 15 мг/кг/день. Пероральні стандартні лікарські форми, такі як таблетки і капсули, переважно містять від приблизно 25 до приблизно 1000 мг сполуки згідно з цим винаходом. Пероральне введення є кращим. Можна використовувати схеми введення з інтервалами (режим on-off), які є традиційними для лікування раку, наприклад, щоденне пероральне введення протягом 1, 2, 3, 4 і т.д. тижнів, наступна перерва в лікуванні протягом 1, 2 і т.д. тижнів, і знову щоденне пероральне введення протягом 1, 2, 3, 4 і т.д. тижнів і т.д. У кращому варіанті реалізації сполуки згідно з цим винаходом вводять щодня в діапазоні від 11 UA 114417 C2 5 10 15 20 25 30 35 40 45 приблизно 25 до приблизно 3000 мг на день, ще краще від приблизно 300 до приблизно 1500 мг сполуки на день. [0061] Сполука згідно з цим винаходом можна вводити за допомогою будь-яких прийнятних способів, включаючи пероральне, місцеве, трансдермальне, парентеральне, підшкірне, інтраперитонеальне, внутрішньолегеневе та інтраназальне, а також при бажанні у випадку місцевого лікування внутрішньоосередкове введення. Парентеральні інфузії включають внутрішньом'язове, внутрішньовенне, внутрішньоартеріальне, інтраперитонеальне або підшкірне введення. Прикладом прийнятної пероральної лікарської форми є таблетка, що містить приблизно 25 мг, 50 мг, 100 мг, 250 мг або 500 мг сполуки згідно з цим винаходом разом з приблизно 90-30 мг безводної лактози, приблизно 5-40 мг кроскармелози натрію, приблизно 530 мг полівінілпіролідону (ПВП) K30 і приблизно 1-10 мг стеарату магнію. Спочатку перемішують один з одним порошкові інгредієнти, а потім їх перемішують з розчином ПВП. Отриману композицію можна сушити, гранулювати, змішувати зі стеаратом магнію і пресувати в таблетку за допомогою стандартного устаткування. Сполуку у вигляді аерозолю можна одержувати шляхом розчинення, наприклад, 5-400 мг сполуки згідно з цим винаходом в прийнятному буферному розчині, наприклад, у фосфатному буфері, і додавання агенту, що змінює тонічність, наприклад, солі, такої як хлорид натрію, при бажанні. Для видалення домішок і забруднюючих речовин розчин, як правило, фільтрують, наприклад, на фільтрі 0,2 мікрон. [0062] Більш конкретно, сполуки згідно з цим винаходом можна застосовувати згідно з описом, наведеним у міжнародній заявці на патент WO 98/46576 і патенті США №7244851, і наведені в них загальні рекомендації із застосування сполук включені в цю заявку шляхом посилань. ПРИКЛАДИ [0063] Винахід стане більш зрозумілим при розгляді наступних прикладів. Проте, їх не слід розглядати як обмежуючі обсяг винаходу. Далі наведені скорочення, використовувані в цьому описі: [0064] ACN: ацетонітрил; [0065] Chg: циклогексилгліцин; [0066] ДХМ: дихлорметан; [0067] DIPEA: диізопропілетиламін; [0068] DMAP: 4-диметиламінопіридин; [0069] ДМЕ: 1,2-диметоксиетан; [0070] ДМФ: диметилформамід; [0071] ДМСО: диметилсульфоксид; [0072] EDC: 1-етил-3-(3-диметиламінопропіл)карбодиімід; [0073] EEDQ: 2-етокси-1-етоксикарбоніл-1,2-дигідрохінолін; [0074] РХМС: рідинна хроматографія-мас-спектрометрія; [0075] HATU: гексафторфосфат O-(7-азобензотриазол-1-іл)-1,1,3,3-тетраметилуронію; [0076] HOBt: N-гідроксибензотриазол; [0077] HBTU: гексафторфосфат 2-(1Н-бензотриазол-1-іл)-1,1,3,3-тетраметилуронію; [0078] ВЕРХ: високоефективна рідинна хроматографія; [0079] NBS: N-бромсукцинімід; [0080] TASF: дифтортриметилсилікат трис(диметиламіно)сульфонію; [0081] ТЕА: триетиламін; [0082] ТФА: трифторацетат; [0083] ТГФ: тетрагідрофуран; [0084] Приклад порівняння 1 [0085] (2-феніл-2Н-піразол-3-іл)амід 1-[2-циклогексил-2-(2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти 50 12 UA 114417 C2 5 10 15 [0086] Розчин Boc-MeAla-Chg-Pro-OH (47,0 мг, 0,107 ммоль) і піридину (26 мкл, 0,32 ммоль) у безводному дихлорметані (300 мкл) прохолоджували до 0 °C и по краплях додавали розчин оксалілхлориду в дихлорметані (54 мкл, 2,0 М, 0,11 ммоль) протягом 10 хвилин. Суміш перемішували при 0 °C протягом 15 хвилин, потім при температурі навколишнього середовища протягом 45 хвилин і додавали розчин 5-аміно-1-фенілпіразолу (15,9 мг, 0,100 ммоль; № у каталозі TCI America А0174) і піридину (15,5 мкл, 0,191 ммоль) у дихлорметані (0,5 мл). Отриману суміш перемішували при температурі навколишнього середовища протягом 16 годин, розбавляли дихлорметаном до об'єму 20 мл і промивали 0,2н. водним гідроксидом натрію (20 мл). Органічну фазу сушили (MgSO4) і концентрували при зниженому тиску. Неочищений продукт очищали шляхом колонкової хроматографії (силікагель, 60 % етилацетат у гексанах, потім 100 % етилацетат) з одержанням жовтої оліїстої рідини: m/z 581 (М+Н+). Оліїсту рідину обробляли 5 % трифтороцтовою кислотою в дихлорметані (2 мл), через 18 годин розчинник видаляли у вакуумі. Отриману оліїсту рідину (29,3 мг, вихід 57 % за 2 стадії) додатково очищали шляхом звернено-фазової ВЕРХ з одержанням продукту (ТФА сіль, 9,6 мг, вихід 15 %): m/z 481 (М+Н+), 503 (M+Na+). Приклад порівняння 2 [0087] Спосіб поєднування з фтор ангідридом 20 25 30 35 [0088] Розчин Boc-MeAla-Chg-Pro-OH (2,3 ммоль) і піридину (6,9 мкмоль) у безводному ДХМ (23 мл) прохолоджували до 0 °C и по краплях додавали фторангідрид ціанурової кислоти (2,3 ммоль) протягом 30 секунд. Суміш перемішували при 0 °C протягом 15 хвилин, при КТ протягом 5 годин, а потім реакцію гасили водою. Суміш тричі екстрагували ДХМ (загальний об'єм 100 мл), об'єднані органічні фази промивали сольовим розчином і сушили (Na2SO4). Фільтрування і концентрування у вакуумі приводили до одержання фторангідриду пептиду у вигляді прозорої безбарвної оліїстої рідини, яку використовували безпосередньо без додаткового очищення. [0089] Розчин неочищеного фторангідриду (0,50 ммоль) і піридину (1,5 ммоль) у ДХМ (2,5 мл) додавали у твердий амін (14, 0,50 ммоль), отриману суміш перемішували при КТ або 50 °C (у герметичній посудині). Суміш виливали у водний NaHCO3, а потім тричі екстрагували дихлорметаном (загальний об'єм 100 мл). Об'єднані органічні фази промивали сольовим розчином, сушили (Na2SO4), фільтрували і концентрували у вакуумі. Неочищений амід пептиду використовували безпосередньо без додаткового очищення. [0090] Приклад порівняння 3 [0091] (4-феніл-[1,2,3]тіадіазол-5-іл)амід 1-[2-циклогексил-2-(2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти 13 UA 114417 C2 5 10 15 20 25 30 35 40 45 [0092] Стадія 1: У розчин Boc-L-Pro-OH (2 екв.), HOBt (1,9 екв.), EDC-HCl (1,9 екв.) і DIPEA (5 екв.) у ДМФ (10-15 об.) додавали 4-феніл-1,2,3-тіадіазол-5-амін (16). Реакція спочатку проходила з помірним виділенням тепла, суміш нагрівали до 75 °C и перемішували протягом ночі, прохолоджували до КТ, ДМФ частково видаляли у вакуумі. Розчин розбавляли EtOAc (1015 об.), після чого промивали 1М HCl (2х), NaHCO3 (1х) і сольовим розчином (1х) (1:1 водн./орг.). Органічний шар концентрували у вакуумі і отриману тверду речовину суспендували в киплячому MeCN (у мінімальному об'ємі, необхідному для простоти перемішування) протягом 30 хвилин, а потім прохолоджували до КТ. Вакуумне фільтрування приводило до одержання Вос-захищеного сполученого продукту у вигляді білуватої кристалічної твердої речовини з приблизно 77 % виходом. Вос-захищений продукт суспендували в 4М розчині HCl/діоксан (4-5 екв. кислоти) і MeCN (1 об.екв. відносно розчину в діоксані) і перемішували при КТ доти, поки за допомогою аналізу РХМС не підтверджували повне зняття захисту (приблизно 1 година). Реакційну суміш концентрували у вакуумі і отриману тверду речовину інтенсивно суспендували в киплячому MeCN (у мінімальному об'ємі, необхідному для простоти перемішування), прохолоджували до КТ, тверду речовину збирали шляхом вакуумного фільтрування і промивали холодним MeCN до зникнення фарбування осаду з одержанням солі HCl і (S)-N-(4феніл-1,2,3-тіадіазоліл-5-іл)піролідин-2-карбоксаміду (17) у вигляді білуватої твердої речовини приблизно з кількісним виходом. [0093] Стадія 2: У розчин 17 і DIPEA (5 екв.) у ДМФ (10-15 об.) додавали Boc-L-Chg (1,5 екв.), HOBt (1,4 екв.) і EDC-HCl (1,4 екв.). Реакційну суміш перемішували протягом приблизно 2 годин, потім розбавляли EtOAc (15 об.) і промивали 1М HCl (2х), NaHCO3 (1х) і сольовим розчином (1х) (1:1 водн/орг). Органічний екстракт сушили (Na2SO4), фільтрували і концентрували у вакуумі. Отриману тверду речовину суспендували в суміші EtOH/гексан (20:80) (у мінімальному об'ємі, необхідному для простоти перемішування) і фільтрували з одержанням Вос-захищеного сполученого продукту у вигляді білої пухкої твердої речовини з приблизно 80 % виходом. Восзахищену речовину розчиняли в 4М розчині HCl/діоксан (4-5 екв. кислоти) і MeCN (0,25 об.екв. відносно розчину в діоксані) і перемішували при КТ протягом приблизно 1 години. Реакційну суміш концентрували з толуолом (2х) досуха (у такому ж об'ємі, що й розчин, який застосовували для зняття захисту) з одержанням солі HCl і (S)-1-((S)-2-аміно-2циклогексилацетил)-N-(4-феніл-1,2,3-тіадіазол-5-іл)піролідин-2-карбоксаміду (18) у вигляді білої кристалічної твердої речовини приблизно з кількісним виходом. [0094] Стадія 3: У розчин 18 і DIPEA (5 екв.) у ДМФ (10-15 об.) додавали Boc-L-N-метил-Ala (1,5 екв.), HOBt (1,4 екв.) і EDC-HCl (1,4 екв.). Реакційну суміш перемішували протягом 1 години, розбавляли EtOAc (15 об.) і промивали 1М HCl (2х), NaHCO3 (1х) і сольовим розчином (1х) (1:1 водн/орг). Органічний екстракт сушили (Na2SO4), фільтрували і концентрували у вакуумі з одержанням Вос-захищеного сполученого продукту у вигляді бежевої твердої піни приблизно з 85 % виходом. Вос-захищений продукт розчиняли в 4М розчині HCl/діоксан (4-5 екв. кислоти) і MeCN (0,25 об.екв. відносно розчину в діоксані) з перемішували при КТ протягом приблизно 1 години. Реакційну суміш концентрували з толуолом (2х) досуха (у такому ж об'ємі, що й розчин, який застосовували для зняття захисту), отриману тверду речовину суспендували в розчині МТБЕ/EtOAc (70:30) (у мінімальному об'ємі, необхідному для простоти перемішування), фільтрували і збирали з одержанням неочищеного (S)-1-((S)-2-циклогексил-2-((S)-2(метиламіно)пропанамідо)ацетил)-N-(4-феніл-1,2,3-тіадіазол-5-іл)піролідин-2-карбоксаміду (d) у 14 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 55 вигляді білуватої вільно текучої твердої речовини. Неочищену сіль HCl суспендували в МеОН (4 об., мінімальна кількість) і розчиняли при перемішуванні при 65 °C. Теплий ізопропілацетат (6-8 об.) додавали двома частинами, підтримуючи температуру приблизно 60 °C, потім розчин залишали прохолоджуватися при перемішуванні. Швидко відбувалася кристалізація, суспензію перемішували при КТ протягом декількох годин, потім перемішували при 0 °C протягом ще години, після чого тверду речовину збирали шляхом вакуумного фільтрування. Неочищений продукт промивали МеОН/iPrOAc (1:4, 2 об.) і сушили з одержанням 19 у вигляді білої/білуватої кристалічної твердої речовини приблизно з 80 % виходом. [0095] Приклад порівняння 4 [0096] 5-аміно-2-(оксазол-2-іл)-4-фенілтіазол [0097] Суспензію гідрохлориду α-амінофенілацетонітрилу (1,52 г, 8,99 ммоль), порошкової сірки (289 мг, 9,01 ммоль) і оксазол-2-карбальдегіду (873 мг, 8,99 ммоль) в EtOH (18 мл) обробляли ТЕА (1,88 мл, 13,5 ммоль) і суміш перемішували при 50 °C протягом 60 хвилин. Охолоджену суміш обробляли водним гідроксиламіном (1,00 мл, 50 мас. %, 15 ммоль) при КТ протягом ночі, фільтрували і концентрували у вакуумі. Залишок розділяли в EtOAc і водному NaHCO3, виділену органічну фазу промивали сольовим розчином, сушили (Na2SO4), фільтрували і концентрували у вакуумі з одержанням темно-коричневої оліїстої рідини. Неочищену оліїсту рідину попередньо наносили в SiO2 і очищали шляхом автоматизованої флеш-хроматографії, елюируя з градієнтом 5-70 % сумішами етилацетату в гексанах, з одержанням 5-аміно-2-(оксазол-2-іл)-4-фенілтіазолу (20, 159 мг, 7,3 %). Приклад 1 [0098] (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогексил-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти (Ia) [0099] Стадія 1: У розчин Boc-L-проліну (4,5 г, 0,02 ммоль) і піридину (8,45 мл, 0,104 ммоль) у ДХМ (20 мл), охолоджений на крижаній лазні, по краплям додавали фторангідрид ціанурової кислоти (5,35 мл, 0,0627 ммоль). Після завершення додавання реакційна суміш набувала молочно-білий колір. Розчин перемішували при 0 °C протягом 10 хвилин, потім нагрівали до КТ і перемішували протягом 4 годин. Реакцію гасили водою і суміш тричі екстрагували ДХМ. Об'єднані органічні екстракти промивали сольовим розчином, сушили і концентрували у вакуумі з одержанням трет-бутил-2-(фторкарбоніл)піролідин-1-карбоксилату. Неочищений фторангідрид негайно використовували в наступній реакції поєднування. [00100] Свіжоотриманий фторангідрид кислоти (4,55 г, 0,02 ммоль) розчиняли в MeCN (20 мл) і обробляли 20 (1,7 г, 0,007 ммоль) і піридином (2,82 мл, 0,035 ммоль). Реакційну суміш нагрівали до 50 °C протягом ночі. Реакцію гасили нас. NaHCO3 і тричі екстрагували EtOAc. Об'єднані органічні шари промивали сольовим розчином, сушили і концентрували. Неочищений продукт очищали шляхом хроматографії на SiO2, шляхом елюювання з градієнтом сумішами EtOAc/гексан (від 50 до 80 % EtOAc), з одержанням (S)-трет-бутил-2-(2-(оксазол-2-іл)-4фенілтіазол-5-ілкарбамоїл)-піролідин-1-карбоксилату (21). [00101] Видалення Вос-захисної групи і наступне поєднування з BocNH-Chg-OH і H(Me)NAla-OH проводили згідно зі способами, описаними для стадій 2 і 3 у прикладі порівняння 3. Очищення продукту комбінації Chg проводили шляхом хроматографії SiO2, елюируя з градієнтом сумішами EtOAc/гексан (від 50 до 80 % EtOAc). Неочищений Вос-захищений трипептидний продукт очищали шляхом ISCO (50-80 % EtOAc/гексан). Після видалення Восгрупи кінцевий продукт очищали шляхом препаративної ВЕРХ з одержанням чистого Ia: (М+Н)+ = мс 565,3. Приклад 2 [00102] Інші сполуки згідно з цим винаходом, наприклад, [00103] 1. (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклопропіл-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти; [00104] 2. (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклопентил-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти; [00105] 3. (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогептил-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти; [00106] 4. (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогексил-2-((S)-2етиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти; [00107] 5. (2-оксазол-2-іл-4-фенілтіазол-5-іл)-N-метиламід (S)-1-[(S)-2-циклогексил-2-((S)-2метиламінопропіоніламіно)ацетил]піролідин-2-карбонової кислоти; і [00108] 6. (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2-циклогексил-2-((S)-2метиламінопропіоніламіно)-2-метилацетил]піролідин-2-карбонової кислоти, 15 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 [00109] можна одержувати аналогічно сполуці згідно з прикладом 1 з використанням відповідних аналогічних вихідних речовин у відповідності зі способом згідно з прикладом 1. Приклад 3 [00110] Дослідження інгібування IAP [00111] У наступних експериментах використовували химерний домен BIR, що називається MLXBIR3SG, у якому 11 з 110 залишків відповідали тим, що містяться в XIAP-BIR3, а залишок відповідав ML-IAP-BIR. Було показано, що химерний білок MLXBIR3SG значно краще зв'язує та інгібує каспазу 9 у порівнянні з природними доменами BIR, при цьому афінність зв'язування з пептидами на основі Smac і "дозрілим" Smax схожа з показником для природнього ML-IAP-BIR. Поліпшене інгібування каспази-9 химерним доменом BIR в MLXBIR3SG зв'язують з поліпшеним інгібуванням апоптозу, викликаного доксорубіцином, при трансфіціюванні клітин MCF7. [00112] Послідовність MLXBIR3SG: [00113] MGSSHHHHHHSSGLVPRGSHMLETEEEEEEGAGATLSRGPAFPGMGSEELRLASFYDWPLTAEVP PELLAAAGFFHTGHQDKVRCFFCYGGLQSWKRGDDPWTEHAKWFPGCQFLLRSKGQEYINNIHLTH SL (№ послідовності: 1) [00114] Дослідження пептидного зв'язування РПЕФ-ВР [00115] Проводили конкурентні експерименти в рамках дослідження резонансного переносу енергії флуоресценції з тимчасовим дозволом на багатоцільовому аналізаторі Wallac Victor2 (Perkin Elmer Life and Analytical Sciences, Inc.) згідно зі способами, описаними в Kolb et al (Journal of Biomolecular Screening, 1996, 1(4):203). Готували коктейль реагентів, що містить 300 нМ his-мічений MLXBIR3SG; 200 нМ біотинільований пептид SMAC (AVPI); 5 мкг/мл анти-his алофікоцианін (XL665) (Cisbio International); і 200 нг/мл стрептавідин-європій (Perkin Elmer), у буфері для реагентів (50 мМ Tris [рН 7,2], 120 нМ NaCl, 0,1 % бичачих глобулінів, 5 мМ DTT і 0,05 % октилглюкозид). (Як альтернативу зазначений коктейль можна одержували з використанням міченого європієм анти-His (Perkin Elmer) і стрептавідин-алофікоцианіну (Perkin Elmer) у концентраціях 6,5 нМ і 25 нМ, відповідно). Коктейль реагентів інкубували при кімнатній температурі протягом 30 хвилин. Після завершення інкубації коктейль додавали до сполукиантагоніста в концентрації, отриманій шляхом послідовних розведень 1:3 (від початкової концентрації 50 мкМ), в 384-ямкові чорні планшети FIA (Greiner Bio-One, Inc.). Після 90хвилинної інкубації при кімнатній температурі аналізували флуоресценцію з використанням фільтрів для довжин хвиль порушення європію (340 нм) і випущення європію (615 нм) і алофікоцианіну (665 нм). Дані антагоністів розраховували як відношення сигналу випущення алофікоцианіну при 665 нм до сигналу випущення європію при 615 нм (для спрощення обробки даних значення цього відношення множили на 10000). Будували графіки залежності отриманих значень від концентрації антагоніста, і отримані значення підставляли в 4-параметрове рівняння з використанням програмного забезпечення Kaleidograph (Synergy Software, Reading, PA). Дані активності антагоністів визначали за значеннями IC50. Було виявлено, що сполуки згідно з цим винаходом мали інгібуючу активність у відношенні IPA, що продемонстрували в зазначеному дослідженні. [00116] Поляризаційне флуоресцентне дослідження зв'язування пептидів [00117] Експерименти з використанням поляризації проводили на Analyst HT 96-384 (Molecular Devices Corp.) згідно зі способом, наведеним в Keating, S.M., Marsters, J, Beresini, M., Ladner, C., Zioncheck, K., Clark, K., Arellano, F., and Bodary., S.(2000), Proceedings of SPIE: In Vitro Diagnostic Instrumentation (Cohn, G.E., Ed.) pp 128-137, Bellingham, WA. Зразки для визначення афінності по поляризації флуоресценції одержували шляхом додавання MLXBIR3SG у кінцевій концентрації, отриманій шляхом послідовного розведення 1:2, починаючи з 5 мкМ, у поляризаційному буфері (50 мМ Tris [рН 7,2], 120 мМ NaCl, 1 % бичачих глобулінів, 5 мМ DTT і 0,05 % октилглюкозид) у карбоксифлуоресцеїн-сполучений AVPdi-Phe-NH2 (AVP-diPhe-FAM) у кінцевій концентрації 5 нМ. 16 UA 114417 C2 5 10 15 20 25 30 35 40 45 50 Зонд AVP-diPhe-FAM [00118] Реакційні суміші аналізували після 10-хвилинної інкубації при кімнатній температурі з використанням стандартних обмежуючих фільтрів для фторофору флуоресцеїну (λвозб = 485 нм; λісп = 530 нм) в 96-ямкових чорних планшетах НЕ96 (Molecular Devices Corp.). Будували графіки залежності значень флуоресценції від концентрації білка, значення IC50 одержували шляхом підстановки даних в 4-параметрове рівняння з використанням програмного забезпечення Kaleidograph (Synergy Software, Reading, PA). Проводили конкурентні експерименти шляхом додавання 30 нМ MLXBIR3SG у лунки, що містять 5 нМ зонд AVP-diPheFAM, а також сполуки-антагоністи в концентрації, отриманій шляхом послідовного розведення 1:3, починаючи з 300 мкМ концентрації, у поляризаційному буфері. Зразки аналізували після 10хвилинної інкубації. Будували графіки залежності значень поляризації флуоресценції від концентрації антагоніста, значення IC50 одержували шляхом підстановки даних в 4параметрове рівняння з використанням програмного забезпечення Kaleidograph (Synergy Software, Reading, PA). Константи інгібування (Ki) антагоністів визначали за значеннями IC50. Було виявлено, що сполуки згідно з цим винаходом мають інгібуючу активність у відношенні IAP, що продемонстрували в зазначеному дослідженні. Наприклад, значення Ki для сполуки Ia становило 0,014 (ML-IAP-BIR). [00119] Приклад 4 [00120] Дослідження ксенотрансплантатів пухлин (фіг.1 і 2) [00121] Усі процедури із застосуванням тварин проводили згідно з рекомендаціями інституціонального комітету по утриманню і використанню тваринних компанії Genentech. Ракові клітини, такі як клітини раку грудей людини MDA-MB-231, колоректального раку Colo205, або НМРЛ Calu6, одержували в Американській колекції типових культур (Manassas, VA). Клітини повторно суспендували в HBSS (Colo205), або ж клітинну суспензію перемішували у відношенні 1:1 з матригелем (BD Biosciences; MDA-MB-231, Calu-6). Потім клітини (1,5 × 107 у випадку MDA-MB-231; 5,0 × 107 у випадку Colo205, Calu6) підшкірно імплантували в правий бік самок бестимусних мишей (Charles River Laboratories, Hollister, CA) віком 6-8 тижнів. Розраховували об'єм пухлин з використанням значень середнього діаметра, виміряних за допомогою штангенциркуля, за формулою v=0,5 ( a ( b2, де a і b являють собою найбільший і найменший взаємно перпендикулярні діаметри пухлини, відповідно. По десять мишей з відповідним середнім об'ємом пухлини випадковим чином розподіляли в кожну із шести груп. На момент початку дослідження (день 0) середній об'єм пухлини ± стандартна помилка середнього (СПС) для всіх шести груп становив 168±3 мм3. Мишей спостерігали щодня під час дослідження, об'єм пухлин і масу тіла вимірювали два рази на тиждень. Пригнічення росту пухлини у відсотках розраховували за формулою %TGI=100 ( (1 – об'єм пухлинидоза/об'єм пухлининосій). [00122] У цьому дослідженні з використанням клітин раку грудей людини MDA-MB-213• X1 Сполука Ia згідно з цим винаходом мала значення МЕД 3,4 мг/кг (в.в. один раз на тиждень). Це значення в п'ять разів нижче кількості сполуки III, яку тестували в цьому ж дослідженні в однакових умовах (МЕД = 18,6 мг/кг). Значення AUC для Ia у чотири рази нижче значення AUC для сполуки III при забезпеченні однакової ефективності. [00123] Відмітні ознаки, описані в наведеному вище описі або в наступній формулі винаходу, виражені конкретно або у вигляді способів реалізації запропонованої функції або способу або процесу досягнення запропонованого результату, можна окремо або як яку-небудь комбінацію зазначених ознак застосовувати для реалізації цього винаходу в його різноманітних формах. [00124] Для ясності і простоти розуміння запропонований вище винахід був докладно описаний за допомогою ілюстрацій і прикладів. Фахівцям у даній галузі техніки повинно бути очевидним, що в рамках обсягу прикладеної формули винаходу можна реалізовувати зміни і модифікації. Таким чином, слід розуміти, що наведений вище опис є ілюстративним і 17 UA 114417 C2 5 10 необмежуючим. Таким чином, обсяг винаходу повинен визначатися не наведеним вище описом, але наступною формулою винаходу, що прикладається, а також повним обсягом еквівалентів, можливих для зазначеної формули винаходу. [00125] Джерела інформації: Патенти, опубліковані заявки на патенти і наукова література, посилання на які наведені в цьому описі, визначають рівень знань фахівців у даній галузі техніки включені в цю заявку у всій повноті шляхом посилань тією самою мірою, як і у випадку якби для кожної окремої роботи було конкретно зазначено, що вона включена за допомогою посилання. Будь-які протиріччя між якимнебудь посиланням, наведеним в цьому описі, і конкретними відомостями, наведеними в цьому описі, слід дозволяти на користь останніх. Аналогічно, будь-які протиріччя між визначенням слова або фрази, яким мається на увазі в рівні техніки, і конкретним визначенням слова або фрази, наведеним у цьому описі, слід дозволяти на користь останнього. ФОРМУЛА ВИНАХОДУ 15 1. Сполука формули І R 3 R O 2 N 1 N N R R 4 R 5 R O 6 N O Ph S N N O 20 25 30 35 40 , (І) у якій Ph являє собою феніл; 1 R являє собою С3-7-циклоалкіл; 2 3 4 5 6 кожний R , R , R , R і R незалежно в кожному випадку являє собою Η або С1-6-алкіл; або її фармацевтично прийнятна сіль. 2. Сполука за п. 1, що являє собою (2-оксазол-2-іл-4-фенілтіазол-5-іл)амід (S)-1-[(S)-2циклогексил-2-((S)-2-метиламінопропіоніламіно)ацетил]-піролідин-2-карбонової кислоти (Іа), або її фармацевтично прийнятна сіль. 2 6 3. Сполука за п. 1, яка відрізняється тим, що кожний R -R незалежно являє собою Η або метил. 1 4. Сполука за п. 1, яка відрізняється тим, що R являє собою циклогексил. 1 5. Сполука за п. 3, яка відрізняється тим, що R являє собою циклогексил. 2 3 6. Сполука за п. 3, яка відрізняється тим, що один з R і R являє собою Н, а інший являє 4 5 6 собою метил; або R являє собою метил; або кожний R і R являє собою Н. 7. Спосіб лікування захворювання або стану, пов'язаного з надлишковою експресією ІАР, у ссавця, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 1. 8. Спосіб лікування раку, який включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 1. 9. Спосіб лікування захворювання або стану, пов'язаного з надлишковою експресією ІАР, у ссавця, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 2. 10. Спосіб лікування раку, що включає введення зазначеному ссавцеві ефективної кількості сполуки за п. 2. 11. Фармацевтична композиція, що містить сполуку за п. 1 і щонайменше один фармацевтично прийнятний носій, розріджувач або допоміжну речовину. 12. Фармацевтична композиція, що містить сполуку за п. 2 і щонайменше один фармацевтично прийнятний носій, розріджувач або допоміжну речовину. 18 UA 114417 C2 19 UA 114417 C2 Комп’ютерна верстка М. Мацело Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 20
ДивитисяДодаткова інформація
Назва патенту англійськоюInhibitors of iap
Автори англійськоюCohen, Frederick, Gazzard, Lewis, Tsui, Vickie Hsiao-Wei, Flygare, John A.
Автори російськоюКоэн Фрэдэрик, Газзард Льюис Дж., Цуй Вики Сяо-Вэй, Флайгэр Джон А.
МПК / Мітки
МПК: A61K 31/417, C07D 417/14
Мітки: інгібітори
Код посилання
<a href="https://ua.patents.su/22-114417-ingibitori-iap.html" target="_blank" rel="follow" title="База патентів України">Інгібітори iap</a>
Заміщені 6-циклогексилалкілом заміщені 2-хінолінони та 2-хіноксалінони як інгібітори полі-(адф-рибоза)полімерази

Номер патенту: 91007
Опубліковано: 25.06.2010
Автори: Вутерс Вальтер Будевійн Леопольд, Сомерс Марія Вікторіна Франсіска, Мабір Домінік Жан-П'єр, ван Дун Якобус Альфонсус Йозефус
МПК: C07D 241/44, A61K 31/498, A61P 43/00, C07D 401/06
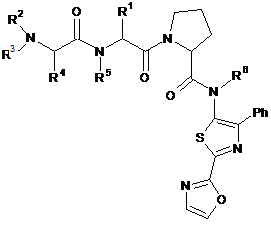
Мітки: полі-(адф-рибоза)полімерази, 6-циклогексилалкілом, інгібітори, 2-хінолінони, заміщені, 2-хіноксалінони
Формула / Реферат:
1. Сполука формули (І), (I)її N-оксидні форми, адитивні солі та стереохімічно ізомерні форми, деn означає 0 або 1;s означає 0 або 1;X являє собою -N= або -CR4=, де R4 являє собою водень, або, взятий разом з R1, може утворювати бівалентний радикал формули -СН=СН-СН=СН-;Y являє собою -N< або -СН<;Q являє собою -NH-, -О-,...
Тієнопіримідини як інгібітори фосфодіестерази, спосіб їх одержання та фармацевтичний препарат на їх основі

Номер патенту: 72256
Опубліковано: 15.02.2005
Автори: Крістадлер Марія, Клюхен Франц-Вернер, Джонас Рохус, Шеллінг П'єр
МПК: A61K 31/519, A61P 43/00, A61P 15/10, A61P 15/00, A61P 9/04, A61P 9/00, C07D 495/04
Мітки: спосіб, основі, одержання, тієнопіримідини, фосфодіестерази, препарат, інгібітори, фармацевтичний
Формула / Реферат:
1. Сполука формули I, Ів якійR1, R2 у кожному випадку незалежно один від одного являють собою Н, А, ОН, ОА або Hal,Х являє собою R4, R5 або R6, монозаміщений R7,R4 являє собою нерозгалужений або розгалужений алкілен з 1-10 С-атомами, в якому одна або дві CH2-групи можуть бути заміщені групами -СН=СН-,R5 являє собою циклоалкіл або циклоалкілалкілен, що містить 5-12 С-атомів,R6 являє собою феніл...
Інгібітори vla-4: omepupa-v

Номер патенту: 65623
Опубліковано: 15.04.2004
Автори: Лі Вен-Чернг, Джилл Алан
МПК: A61K 31/401, A61P 3/10, A61P 37/06, A61P 25/00, A61P 11/06, A61P 1/00, A61P 11/08, A61P 37/02, A61P 9/10, A61P 35/00, A61P 43/00, A61P 17/06, C07D 207/16, A61P 29/00
Мітки: інгібітори, omepupa-v, vla-4
Формула / Реферат:
1. Сполука, що інгібує клітинну адгезіюВІО 1591і її фармацевтично прийнятні похідні, проліки або солі.2. Сполука за п. 1, де проліки являють собою складний ефір.3. Сполука за п. 2, де проліки - складний ефір одержують взаємодією сполуки ВІО 1591 з С1-С10алкіловим спиртом з прямим або розгалуженим ланцюгом.4. Сполука за п. 2, де...
Похідні фталазіну як інгібітори parp

Номер патенту: 93351
Опубліковано: 10.02.2011
Автори: Мертенс Йозефус Каролюс, ван Дун Якобус Альфонсус Йозефус, Кенніс Людо Едмон Жозефін, Мевеллек Лоренс Анн, Сомерс Марія Вікторіна Франціска, Вутерс Вальтер Будевійн Леопольд
МПК: C07D 401/04, C07D 403/12, A61P 31/00, C07D 403/14, A61K 31/502
Мітки: фталазину, інгібітори, похідні
Формула / Реферат:
1. Сполука формули (І) (I)або її форми N-оксидів, фармацевтично прийнятні адитивні солі, стереохімічно ізомерні форми, де пунктирна лінія позначає необов'язковий зв'язок; n дорівнює 0, 1, 2 або 3, та, якщо n дорівнює 0, тоді мається на увазі прямий зв'язок; Q являє собою -С(=О)- або -CR3-, де...
Пуринові інгібітори циклінозалежної кінази 2 та ik-a

Номер патенту: 74142
Опубліковано: 15.11.2005
Автори: Заблоцкі Джеффері А., Блам Чері Лінн, Скоу Стівен Р., Ібрагім Прабха, Макмен Річард, Лам Роберт Т., Уік Майкл М.
МПК: A61P 19/06, A61P 3/10, A61K 31/52, A61P 13/12, A61P 31/10, A61P 35/00, C07D 473/40, C07D 473/16, A61P 25/00, A61P 9/00, A61P 43/00, A61P 19/02, C07D 473/00, A61P 29/00
Мітки: циклінозалежної, інгібітори, пуринові, кінази
Формула / Реферат:
1. Сполука, вибрана з групи, до складу якої входять:{2-[(2-аміноетил)аміно]-9-(метилетил)пурин-6-іл}[(4-хлорфеніл)метил]амін;{2-[(2-амінопропіл)аміно]-9-(метилетил)пурин-6-іл}[(4-хлорфеніл)метил]амін;2-[(2-аміноетил)(6-{[4-хлорфеніл)метил]аміно}-9-(метилетил)пурин-2-іл)аміно]етан-1-ол;2-[(2-гідроксіетил)(6-{[(4-хлорфеніл)метил]аміно}-9-(метилетил)пурин-2-іл)аміно]етан-1-ол.2. Сполука за п. 1, яка являє собою...
Попередній патент: Гербіцидна композиція, яка містить деякі піридинкарбонові кислоти і (2,4-дихлорфеноксі)оцтову кислоту
Наступний патент: Визначення контекстів для кодування даних коефіцієнтів перетворення при кодуванні відео
Випадковий патент: Модифікатор алюмінієвих сплавів