Макролідні антибіотики або їх солі, спосіб їх одержання (варіанти), композиція для лікування тварин і людей, композиція і спосіб боротьби з шкідниками сільського господарства
Номер патенту: 27261
Опубліковано: 15.09.2000
Автори: Флеттон Річард Алан, НОБЛ Девід, Сазерленд Дерек Рональд, Ремсей Майкл Вінсент Джон, Вард Джон Беррі, Нобл Хейзел Мері, ПОРТЕР Ніл






















Формула / Реферат
(57) 1. Макролидные антибиотики общей формулы (lІ)
где R1 - метил, этил или изопропил,
R2 - атом водорода или группа OR5, где OR5 - гидроксильная или замещенная гидроксильная группа, содержащая до 25 атомов углерода, и R3 - атом водорода, или R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют > С=О группу, и
OR4 представляет группу OR5, как указано выше, причем, когда R2 представляет гидроксильную группу, то OR4 представляет замещенную гидроксильнуюгруппу иную, чем метоксигруппа, или их соли
2. Антибиотики по п. 1, где R1 – изопропильная группа
3. Антибиотики по п. 1, где R1 – иэопропильная группа, R2 - атом водорода или этокси-, н-пролокси-, ацетокси- или пропионокси группа и R3 - атом водорода или R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют группу > СО, и группа OR4 - гидроксильная, ацетокси- или метилоксикарбонилокси группа
4. Антибиотики по п. 1, где R1 – изопропильная группа, R2 - атом водорода, R3 - атом водорода и OR4 - гидроксильная группа или
R1 - иэопропильная группа, R2 - пропионоксигруппа, R3 - атом, водорода и OR4 – гидроксипьная группа, или
R1 - изопропильная группа, R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют группу > С=О, и OR4 - гидроксильная группа, или R1 - изопропильная группа, R2 – этоксигруппа, R3 - атом водорода и OR4 - гидроксильная группа, или
R1 - изопропипьная группа, R2 - н-пропоксигруппа, R3 - атом водорода и OR4 - гидроксильная группа, или R1 - метильная группа, R2 - ацетоксигруппа, R3 - атом водорода и OR4 - гидроксильная группа, или
R1 - этильная группа, R2 - ацетокси группа, R3 - атом водорода и OR4 - гидроксипькая группа, или
R1 - изопропильная группа, R2 - а цетокси группа, R3 - атом водорода и OR4 – гидроксильная группа.
5. Способ получения макролидных антибиотиков общей формулы (II)
где R1 - метил, этил или изопропил,
R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют > С=О группу, и OR4 представляет гидроксильную группу или защищенную гидроксильную группу OCOR5, где R6 =C1-C4 алкил, возможно, замещенный одним, двумя или тремя атомами галогена или OCО2R7, где R7 – С1-С4-алкил или бензил, отличающийся тем, что соединение общей формулы (ІІІ)
где R1 - метил, этил или изопропил,
OR4 представляет замещенную гидроксильную группу OCОR6, где R6 – С1-С4-алкил, возможно, замещенный одним, двумя или тремя атомами галогена, или OCO2R7, где R7 – C1-C4-алкил или бензил,
OR6 - гидроксильная группа,
подвергают окислению действием окислителя в среде органического растворителя или смеси органического растворителя с водой при температуре от -80 до 50°С и выделяют макролидный антибиотик формулы (ІІ) и, при необходимости получения антибиотика формулы (II), в котором OR4 - гидроксильная группа, с последующим снятием защиты с группы OR4
6. Способ получения макролидных антибиотиков общей формулы (II)
где R1 - метил, этил или изопропил,
R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют > С=О группу, и
OR4 представляет гидроксильную группу или метоксильную группу, отличающийся тем, что производят культивирование штаммов Steptomyces thermoarchaensis NCIB 12015 или NCIB 12111, или NCIB 12112, или NCIB 12113, или NCIB 12114 на жидкой питательной среде, содержащей источники углерода, азота и минеральные соли, при 20-50°С, предпочтительно 25—40°С более предпочтительно 34°С, при аэрации и рН 5,5-8,5, предпочтительно 5 5-7,5 в течение 2-10 суток
7. Композиция для лечения животных и людей, содержащая активное вещество и носитель и/или наполнитель, отличающаяся тем что в качестве активного вещества она содержит по крайней мере, один макролидный антибиотик формулы (II)
где R1 - метил, этил или изопропил,
R2 - атом водорода или группа OR5, где OR5 - гидроксильная или замещенная гидроксильная группа, содержащая до 25 атомов углерода, и R3 - атом водорода, или R2 и R3 вместе с атомом углерода, к которому они присоединяются, представляют > С=О группу, и
OR4 представляет группу OR5, как указано выше, причем когда R2 представляет гидроксильную группу, то OR4 представляет замещенную гидроксильную группу иную, чем метоксигруппа, или его соль в эффективном количестве
8. Композиция для борьбы с вредителями сельского хозяйства, содержащая активное вещество и носитель и/или наполнитель, отличающаяся тем, что в качестве активного вещества она содержит, по крайней мере, один макролидный антибиотик формулы (II)
где R1 - метил, этил или изопропил,
R2 - атом водорода или группа ОR5, где OR5 - гидроксильная или замещенная гидроксильная группа, содержащая до 25 атомов углерода, и R3 -атом водорода или R2 и R3 вместе с атомом углерода, к которому они присоединяются, предcтавляют > С=О группу и
OR4 представляет группу OR1 как указано выше, причем, когда R7 представляет гидроксильную группу, то OR4 представляет замещенную гидроксильную группу иную чем метоксигруппа, или его соль в эффективном количестве
9. Способ борьбы с вредителями сельского хозяйства, садоводства или лесоводства, а также в местах обитания вредителей путем обработки их или растений или мест, где они обитают, активным веществом, отличающийся тем, что в качестве активного вещества используют, по крайней мере, один макролидный антибиотик формулы (ІІ)
где R1 - метил этил или изопропил,
R2 - атом водорода ипи группа ОR5, где OR5 - гидроксильная или замещенная гидроксильная группа, содержащая до 25 атомов углерода, и R3 - атом водорода или R2 и R1 вместе с атомом углерода к которому они присоединяются, представляют > С=О группу, и
OR4 представляет группу OR как указано выше, причем, когда R2 представляет гидроксильную группу то OR4 представляет замещенную гидроксильную группу иную чем метоксигруппа или его соль в эффективном количестве.
Текст
Настоящее изобретение относится к новым соединениям антибиотика и способам их получения. В заявке на выдачу патента Великобритании № 8522699 описано получение анти биотика S541, которые можно выделить из продуктов ферментации вида Streptomyces. Антибиотики S541 пред- ставляют собой группу родственных соединений, имеющих неполную формулу (I) (I) Итак, как описано в данном описании, выявлена дополнительная группа соединений, обладающих антибиотической активностью, которые можно получить посредством химической модификации антибиотиков S541 или путем выделения их из культуры микроорганизмов рода Streptomyces. Новые соединения предлагаемого изобретения обладают антибиотической активностью и/или применяются как промежуточные продукты при по лучении других активных соединений и/или при выделении или очистке соединений антибиотиков S541. Соединения настоящего изобретения представляют собой 23-кетон, 23-деокси, и 23-гидроксил или замещенные гидроксильные аналоги анти биотиков S541, которые имеют гидроксильную или замещенную гидроксильную гр уппу в положении 5. Таким образом, в одном варианте настояще го изобретения предлагаются, в частности, соединения формулы (II) (II) где R1 представляет метильную, этильную или изопропильную гр уппу R2 представляет атом водорода или груп пу ОR5 (где ОR5 является гидроксильной группой, или замещенной гидроксильной группой, содержащей до 25 ато мов углерода) и R3 представляет атом водорода, или R2 и R3 вместе с ато мом углерода, к которому они присоединяются, представляют >> С=0 груп пу; и ОR4 представляет группу ОR 5, имеющую вышеуказанные значения, а соли указанных соединений, при условии, что в случае, когда R 2 представляет гидроксильную гр уппу, то ОR4 является помимо метокси группы замещенной гидроксильной группой. В случае, когда соединения формулы (1) прихо дится применять в качестве промежуточных продуктов, одна или обе из групп R2 или -ОR4 часто становятся защищен ной гидроксильной группой и предлагаемое изобретение, в частности, со держит такие защи щенные соединения. Когда груп пы R2 или ОR4 в сое динениях формулы (1) замещены гидроксильными группами, они могут быть одинаковыми или отличаться друг от др уга и могут представлять ацилоксигруппы [например, группу формулы -ОСОR5, -ОСО2R5 или -ОСSOR5 (где R5 является группой алифа тического, аромоалифа тического или ароматического ряда, например, алкильной, алкенильной, алкинильной, циклоалкильной, аралкильной или арильной группой)], формилоксигруппу, груп пу –ОR7 (где R7 имеет вышеуказанные для R5 значения), группу -OSO2R8 (где R8 является С 1–4 алкильной или С6–10 арильной группой), силилоксигруппу, циклическую или ациклическую ацетальокси группу или группу ОСО (СН2)nCO2R9 (где R9 является атомом водорода или группой, которая имеет вышеуказанные для R5 значения, а n равно 0, 1 или 2). В случае, когда R5 и R7 являются алкильными группами, они могут быть, например, С1–8 алкильной груп пой, т.е. метилом, этилом, н-пропилом, изопропилом, н-бутилом, изобутилом, трет-бутилом или н-гептилом, алкильные группы которых могут быть также замещены. В случае, когда R5 является замещенной алкильной группой, она может быть замеще на, например, одной или несколькими, например, двумя или тремя атомами галогена (атомами хлора или брома), или карбокси, С1–4 алкокси (например, метокси, этокси), фенокси или силилоксигруппой. Если R7 представляет собой замещенную алкильную гр уппу, она может быть замеще на С 3–7 циклоалкилом, например, циклопропильной группой. Когда R5 или R7 являются алкенильной или алкильной группами, они могут быть представлены, например, С2–8 алкенилом, например, аллилом, или С 2–8 алкинильными группами. В случае, когда R5 или R7 представляют собой циклоалкильные группы, они могут быть, например, С3–12 циклоалкилом, таким как С3–7 циклоалкилом. Таким образом, R5 может представлять, например, циклопропильную, циклобутильную, циклопентильную или циклогексильную гр уппу. R 7 может быть циклопентильной группой. Если R5 и R7 представляют аралкильные группы, они предпочти тельно имеют от 1 до 6 атомов углерода в алкильной части, а арильная группа (группы) может быть карбоциклической или гетероциклической и содержать предпочти тельно 4–15 атомов углерода, например, фе нил. Примеры таких групп содержат фен-С1–6 алкил, например, бензильные или фенэтильные группы. Когда R5 или R7 являются арильными группами, они могут быть карбоциклическими или гетероциклическими и включать предпочти тельно 4–15 атомов углерода, и могут представлять, например, фенильную груп пу. В том случае, когда R2 или -ОR4 являются группой -OSO2R8 они могут быть, например, метилсульфонилокси или п-толуолсуль фонилокси группу. Если R2 или -ОR4 представляют собой циклическую ацетальоксигруппу, они могут со держать, например, 5–7 членные циклы и могут быть, например, тетрагидропиранилокси груп пой. Когда R2 или -ОR4 представляют силиоксигруппу или R5 со держит силилокси-заместитель, силильная груп па может нести три гр уппы, которые могут быть одинаковыми или отличаются друг от др уга, выбранными из алкильной, алкенильной, алкокси, циклоалкильной, аралкильной, арильной и арилокси групп. Такие груп пы могут иметь вышеуказанные для R5 значения и, в частности, включать метильную, трет-бутильную и фенильную груп пы. Конкретными примерами таких силилоксигрупп являются тримети лосилилокси и трет-бутилдиметилсилилокси. В случае, когда R2 или ОR4 представляют группу ОСО(СН 2)nCO2R9 они могут быть, например, группой ОСОСО2R9 или ОСОСН 2СН2СО2R9, где R 9 представляет атом водорода или С1–4 алкильную (например, метильную или этильную) гр уппу. Соединения формулы (II), со держащие кислотную груп пу, могут образовывать соли с соответствующи ми основаниями. Примеры таких солей включают соли щелочного металла, например соли натрия и калия. К важной группе соединений формулы (II) относится группа, в которой R1 представляет собой метильную, этильную или изопропильную группу, R2 и ОR4, которые могут быть одинаковыми или разными, каждый представляет группу ОR5 (где ОR5 является гидроксильной группой или замещенной гидроксильной группой, включающей до 25 атомов углерода) и R3 представляет атом водорода и ее соли в том случае, когда R 2 представляет гидроксильную группу, ОR4 является помимо метоксильной замещенной гидроксильной группой. К другой важной группе соединений формулы (II) относится груп па, в которой R1 представляет со бой метильную, этильную или изопропильную груп пу, R2 и R3 вместе с атомом углерода, к которому они присоединяются, образуют груп пу >С=0 и ОR4 представляет группу ОR5 (где ОR5 является гидроксильной груп пой или замещенной гидроксильной группой, имеющей до 25 атомов углерода) и ее соли. К еще важной группе соединений формулы (II) относится группа, к которой R 1 представляет собой метильную, этильную, или изопропильную гр уп пу, R2 и R3 является каждый атом водорода и ОR4 представляет группу ОR5 (где ОR5 представляет гидроксильную или замещенную гидроксильную груп пу, содержащую до 25 ато мов углерода) и ее соли. В указанных соединениях формулы (II) R 1 является предпочтительно изопропильной группой. R2 является предпочтительно атомом водорода или гидроксильной группой, или группой формулы ОСОR5 (где R5 является С 1–8 алкильной группой (при желании замещенную С 1–4 а лк окси гр уп п ой ), или фе н -С 1–6 алк и льн ой гр уп по й ), -ОСО2R5 (где R5 – С1–8 алкильная группа, замещенная при желании одним или тремя атомами галогена, например, трихлорэтил), – ОСОСО2Н, –ОR7 (где R7 является С 1–8 алкил, С3–7 циклоалкил, алкильная или циклопропилметильная группа) или R 2 и R3 вместе с атомом углерода, к которому они присоединяются, образуют >С=0 груп пу. В частности, R2 предпочтительно является атомом водорода или этокси, н-пропокси, п-бутокси циклопропилметокси, ацетокси, фе нацетокси, пропионокси, изобутирионокси или циклопропанкарбонилокси группой или R2 и R3 вместе с атомом углерода, к которому они присоединяются образуют >С=0 гр уппу. Группа –ОR4 в соединениях фор мулы (II) является предпочти тельно гидроксильной, метоксильной, ацетоксильной или метилкарбонилокси груп пой. Важными активными соединениями предлагаемого изобретения являются соединения формулы (II), в которых R1 является метильной, этильной или, в частности, изопропильной группой, R2 представляет атом водорода или этокси, н-пропокси, н-бутокси ацетокси, или пропионокси группу и R3 является атомом водорода или R2 и R3 вместе с атомом углерода, к которому они присоединяются,образуют гр уппу >С=0, и ОR4 представляет собой гидроксильную, аце токсильную или метоксикарбонилокси груп пу. Особенно важными активными соединениями предлагаемого изобретения являются соединения формулы (II), в которых: R1 является изопропильной группой, R2 является атомом водорода, R3 является атомом водорода и 4 ОR представляет гидроксильную гр уппу; R1 является изопропильной группой, R2 является пропионоксигруппой, R3 является атомом водорода и ОR4 представляет гидроксильную гр уппу; R1 является изопропильной группой, R2 и R3 вместе с атомом углерода, к которому они присоединяются представляют груп пу >С=0 и –ОR4 является гидроксильной группой; R1 является изопропильной группой, R2 является этоксигруп пой, R3 представляет атом водорода и – ОR является гидроксильной группой; и R1 является изопропильной группой, R2 является н-пропоксигруп пой, R3 представляет атом водорода 4 и ОR является гидроксильной группой. R1 является метильной группой, R2 представляет ацетоксигруппу, R 3 является атомом водорода и 4 ОR является гидроксильной группой. R1 является этильной группой, R2 представляет ацетоксигруппу, R3 является атомом водорода и ОR4 представляет собой гидроксильную груп пу. R1 является изопропильной группой, R2 представляет ацетоксигруп пу, R3 является атомом водорода и ОR4 является гидроксильной группой. R1 является изопропильной группой, R2 представляет н-бутоксигруппу, R3 является атомом водорода 4 и ОR является гидроксильной группой; Как указыва лось ранее, соединения предлагаемого изобретения могут применяться качестве антибиотиков и/или как промежуточные соединения для получения других активных соединений и/или при выделении и очистке соединений анти биотиков S541. В том случае, когда предлагаемые соединения приходится использовать как полуп родук ты, R2 и/или –ОR4 могут быть защи щенными гидроксильными группами. Ясно, что та кая группа должна иметь минимальное количество дополнительных функциональных групп, чтобы избежать дополнительных реакционных цепей и должна быть такова, чтобы стало возможным избирательно восста навливать из нее гидроксильную гр уппу. Примеры защитных гидроксильных гр упп известны и описаны, например, в монографии "Защитные группы в органическом синтезе" ("Protective Groupe in Organiс Synthesis" by Theodora W. Greene. Wiley–Interscience, New York 1981) и "Защи тные группы в органической химии" ("Protective Groups in Organie Chemistry" b y I.F.W. VoOmie Plenum Press, London, 1973). Примеры R2 и ОR4 защи щенных гидроксильных групп включают феноксиацетокси, силилоксиацетокси (например, триметил-силилоксиацетокси и трет-бутилдиметилсилилоксиацетокси), и силилокси, например, триметилсилилокси и трет-бутилдиметилсилилокси. Предлагаемые соединения, содержащие такие груп пы будут, гла вным образом, использоваться в качестве промежуточных соединений. Другие группы, например, ацетокси, могут выступать как защи щенные гидроксильные группы, но, кроме того, могут присутствовать в целевых активных соединениях. Другие активные соединения настояще го изобретения, кото рые также пригодны в качестве полупродуктов, являются соединения формулы (II), в которой R2 представляет группу –ОСОСО 2Н. В частности, указанные соединения находят использование при выделении и очистке соединений антибиотиков S541. Особенно значительным соединением в этом отношении является соединение формулы (II), в котором R1 представляет изопропильную гр уппу, R 2 является груп пой –ОСОСО2Н, R3 является атомом водорода и ОR4 представляет гидроксильную гр уп пу, которая может в основном использоваться при выделении и очистке соединения антибиоти ков S541, приводимого ниже в формуле (V), где R1а является изопропильной группой, а R представляет атом водорода. Соединение формулы (V) в неочищенном виде может быть превраще но в соответствующее соединение формулы (II), в которой R2 является группой –ОСОСО2Н и выделено, например, в кристаллической форме. Соединение формулы (V) может быть затем регенеpирова но в почти очищенном виде из последнего соединения описываемыми в данном описании способами, технология выделения и очистки которых образует до полнительные признаки настояще го изобретения. Заявленные соединения обладают антибиотической активностью, например, антигельминтной активностью, например, против нематодов, и в частности, анти-эндопаразитарной и анти-эктопаразитарной активностью. Эктопаразиты и эндопаразиты заражают людей и целый ряд животных, и особенно имеют широкое распространение среди сельскохозяйственных животных та ких как свиней, овец, крупного рога того скота, коз и домашней птицы, лошадей и домашних животных, например, собак и кошек. Паразитарная инфекция домашнего скота, приводящая к анемии, недостаточности питания и потере веса, является основной причиной экономических потерь во всем мире. К представителям эндопаразитов, заражающих указанных животных и/или людей относятся Ancylostoma, Ascaridia, Ascaris, Aspicularis, Brugia, Bunostomum, Capillaria, Chabertia, Cooperia, Diotyocaulus, Dirofilaria, Dracunculus, Enterobius, Haemonchus, Heterakis, Loa, Necator, Nematodirus, Nematospiroides (Heligomoroides), Nippostrongylus, Oesophagostomum, Onchocerca, Ostertagia, Oxyuris, Parasoaris, Strongulus, Stronyloides, Syphacia, Toxascaris, Toxocara,Trichonema, Trichostrongylus, Trichinella, Trichuris, Uncinaria и Wuchareria. Примерами эктопаразитов, заражающи х животных и/или людей являются членистоногие эктопаразиты, например, жалящие насекомые, падальная муха, блохи, вши, клещи, сосущие насекомые, искодовые клещи и др угие двукрылые насекомые – вредители. К примерам такого рода эктопаразитов, заражающи х животных и/или людей, относятся Ambylomma, Boophilus, Chorioptes, Culliphore, Demodex, Demallenia, Dermatobia, Gastrophilus, Haematobia, Haematopinus, Haemophysalis, Hyalomma, Hyperderma, Ixodes, Linognathus, Licilia, Melophagus, Oestrus, Otobius, Otodectes, Psorergates, Psoroptes, Rhipicephalus, Sarooptes, Stomoxys и Tabanus. Обнаружено, что предлагаемые соединения эффективно действуют как in vitro так и in vivo против широкого ряда эндопаразитов и эктопаразитов. В частности, обнаружено, что соединения настоящего изобретения активно действуют против нематодов, таких как Haemonchus contortus, Ostertagia circumcinota, Trichostrongylus colubiformis, Dictyocaulus viviparis, Cooperia oncophera, Ostertagia ostertagi, Nematospiroides dubius и Nippostrongylus braziliensis, и паразитарных клещей, как например, Sarcoptes sp. и Psoroptes sp. Таким образом, предлагаемые соединения находят применение при лечении животных и людей с эндопаразитарной и/или эктопаразитарной инфекциями. 4 Виды паразитов меняются в соответствии с хозяином (животным) и доминирующим местонахождением инфекции. Следовательно, например, возбудители Haemonchus contortus, Ostertagia circumcinota и Trichostrongylus colubiformis обычно поражают овец и преимущественно локализуются в желудке и тонкой кишке, в то время как Dictyocaulus viviparus, Cooperia oncophora и Ostertagia ostertagi обычно поражают крупный рогатый скот и локализованы, главным образом, в легких, кишечнике, или желудке, соответственно. Ан тибиотическая активность предлагаемых соединений может быть продемонстрирована, на-пример, своей активностью in vitro против сво бодно живущих нематодов, например, Caenorhabiditis elegans. Кроме того, заявленные соединения имеют применение в качестве противогрибковых ве ществ, например, против штаммов Candida как например, Candida albicans и Candida glabrata и против дрожжей, таких как Saccharomyces carlsbergensis. Cоединения настоящего изобретения, кроме того, имеют применение в борьбе с вредителями–насекомыми, клещами и нематодами в сельском хозяйстве, садоводстве, лесоводстве, здравоохранении и хранении продуктов. Ими можно успешно обрабаты вать вредите лей почвы и сельскохо зяйственных культур, включая хлебные злаки (например, пшеницу, ячмень, маис, рис), овощи (например, сою), фрукты (например, яблоки, виноград и цитрусовые), наряду с корнеплодами (например, сахарную свеклу, картофель), характерными примерами таких вредите лей являются фр уктовые клещи и тля, например, Aphis fabae, Aulacorthum ciroumflexum, Myzus persicae, Nephotettix cincticeps, Nilparvata lugens, Panonychus ulmi, Phorodon humuli, Phyllocoptruta cleivora, Tetranychus urticae и представители рода Trialeuroides нематоды, например, представители рода Aphelencoides, Globodera, Heterodera, Meloidogyne и Panagrellus чешуй чатые, например, Heliothis, Plutella и Spodoptera; долгоносик–каландрино, как например, Anthonomus grandis и Sitophilus granarius; хруща ки мучные, например, Tribilium castaneum; мухи, например, Musca domestica; муравьи Рихтера; моли-минеры/узкокрылые; Pear psylla; трипсы табачные; таракановые, например, Blatella germanica и Periplaneta americana; москитные, нак например Aedes aegypti. В соответствии с настоящим изобретением предложены соединения формулы (II), как указывалось выше, которые могут быть использованы в качестве антибиотиков. В частности, их можно использовать при лечении животных и людей, пораженных эндопаразитарной, эктопаразитарной и/или грибковой инфекциями и качестве пестицидов для борьбы с вредителями–насекомыми, клеща ми и нематодами, в сельском хозяйстве, са доводстве, или лесном хозяйстве. И х можно применять, кроме того обычно и в качестве пестицидов в других обстоятельствах, например, в хранилищах, зданиях или других общественных местах или местах ло кализации сельскохо зяйственных вредите лей. По-существу предлагаемые соединения могут быть применены либо к хозяину (животному или человеку, либо к культур ным расте ниям или другой растительности), либо к самим вредителям или месту и х расположения. Заявленные соединения могут быть сформулированы для приема в любой удобной форме при использовании в вете ринарии или практической медицине. Таким образом, настоящее изобретение содержит в пределах своего объема фармацевтические композиции, содержащие соединение согласно заявленному изо-брете нию, пригодное для применения ветеринарии или терапии. Такие композиции могут быть представлены при использовании в традиционной форме с помощью одного или нескольких подходящих носителей или наполнителей. Предлагаемые композиции содержат составы в форме, особенно пригодной для парентерального (в том числе, введение внутрь молочной железы), орального, ректального, местного применения в виде имплантанта, для применения при глазных, ушных или заболеваниях мочеполовой системы, подхо дящи ми способами и агента ми для приготовления состава соединений настоящего изобретения, пригодного для использования в ветеринарии или терапии, являются способы, которые описаны в патенте Южной Африки № 85/7049 для соединений антибиотиков S541. Заявленные соединения можно применять в комбинации с другими фармацевти чески активными ингредиента ми. Суммарная суточная доза предлагаемых соединений, применяемая как в вете ринарии, так и в терапии, будет соответственно в интервале 1–2000 мг/кг веса тела, предпочтительно 50–1000 мг/кг, причем указанные дозировки можно принимать в дробных дозах, например, 1–4 раза в день. Заявленные соединения можно составлять в любой удобной форме, пригодной для заземления или садоводства. Таким образом, настоящее изобретение включает в пределах своего объема составы, содержащие соединение, предназначенное для применения в земледелии или садоводстве. Та кие рецептуры включают сухие или жидкие формы, например, дусты, включая пылевидные субстраты или концентраты, порошки, в том числе растворимые или смачиваемые порошки, гранулы, включая микрогранулы и диспергируемые гранулы, таблетки, текучие вещества, эм ульсии, например, разбавленные эмульсии или эмульгируемые концентраты, пропиточные составы, например, составы для пропитки корней и семян, протравливатели семян, семенные гранулы, масляные концентраты, масляные растворы, инъекции, например, инъекции в ствол, растворы или опрыскивания, дымы и туманы. Подхо дящие способы и агенты для составления рецептуры предлагаемых соединений для применения в садоводстве или земледелии включают те, кото рые раскрыты в патенте Южной Африки № 85/7049 для антибиотиков S541. При составлении рецептуры концентрация активного вещества обычно составляет от 0,01 до 99 вес. % и более предпочтительно от 0,01–40 мас. %. Промышленные (товарные) продук ты обычно приготовлены в виде концентрированных композиций, которые при использовании разбавляют до требуе мой концентрации, например, от 0,001 до 0,0001 мас. %. При использовании в ветеринарии или в садоводстве и земледелии, и в случае, когда предлагаемые соединения являются продуктами, образуе мыми посредством ферментации, желательно использовать весь бродильный бульон в качестве источника активных соединений. Кроме того, может быть пригодным использование обезво женного бульона (содержащего мицелий) или мицелия, отделенного из бульона и пастеризованного, или более предпочтительно высушенного. При желании, указанный бульон или мицелий могут быть сформулированы в композиции, как указывалось выше, включающие традиционные инертные носители, наполните ли или разбавители. Ан тибиотические соединения предлагаемого изобретения могут вводиться и применяться в комбинации с другими активными ингредиентами. В частности, предлагаемые соединения антибиотика могут использоваться вместе с соединениями антибиотиков S541 или с другими антибиотическими соединениями настояще го изобретения. Это может возникнуть, например, в случае, когда неочищенные продукты брожения взаимодействуют согласно способу настояще го изобретения без предшествующе го или последующе го отделения; указанное может быть предпочтительным, например, при использовании соединений в земледелии, где важно обеспечить (сохранить) низкую себестоимость продук ции. Заявленные соединения можно получить способами, которые описаны ниже. В некото рых из указанных способов может быть необхо димым защитить гидроксильную груп пу в положении – 5 или – 23 в целевом материале до осуществления описываемого взаимодействия. В та ких случаях может быть необходимо впоследствии разрушить защи ту той же самой гидроксильной группы, как только возникло взаимодействие, чтобы получить требуемое соединение настоящего изобретения. Общепринятые способы защи ты и снятия защи ты можно использовать, например, как описано в вы шеуказанных монографиях Greene и MсOmie. Таким образом, например, ацильная группа, такая как ацетильная группа может быть отщеплена в результате гидролиза основанием, например, используя гидроокись натрия или калия в водном растворе спирта или с помощью гидролиза кислотой, например, используя концентрированную серную кислоту в метаноле. Аце тальные группы, такие как тетрагидроспиранил могут быть отщеплены, например, при гидролизе кислотой (используя кислоту, как, например, уксусную или трифторуксусную кислоту или разбавленную минеральную кислоту). Силильные группы могут быть отщеплены ионами фторида (например, из тетраалкиламмонийфторида, та кого как тетра-н-бути ламмонийфто рид), фтористым водородом в водном растворе ацетонитрила или кислоты такой как п-толуолсуль фокислоты (например, в метаноле). Арилметильные груп пы могут быть отщеплены при обработке кислотой по Льюису (например, эфирата фто ристого бора) в присутствии тиола (например, этантиола) в соответствующем растворите ле, как например, дихлормета не при комнатной температуре. Избирательное отщепление защитной группы в положении-5 5,23-дисилильных соединений настоящего изобретения может быть осуществлено с использованием тетра-н-бутиламмонийфторида, в то время как избирательное отщеп ление защитной группы в положении-5, 5,23-диацетоксисоединения может быть выполнено при использовании гидроокиси натрия в водном растворе метанола. Предлагаемые соединения, в которых R2 и/или группа -ОR4 являются замещенной гидроксильной груп пой обычно может быть получена путем взаимодействия соединений анти биотиков S541 (например, соединений формулы (V), см. далее) или 5 или 23-0-однозамещенных производных с реагентами, функционирующи ми для образования замещенной гидроксильной группы. В общем, гидроксильная группа в положении 5 является более химически активна, чем гидроксильная группа в положении 23. Гидроксильные груп пы в положении-5 более легко отщепляются, чем гидроксильные группы в положении 23. Вообще, 5однозамещенные соединения настоящего изобретения можно получить путем взаимодействия 5, 23-незамещенного оксисоединения антибиоти ков S541 с ограниченным количеством реагента при мягких условиях, в то время как 5,23-дизамещенные соединения образуются при использовании больше го количества реагента и в менее мягких условиях и/или в присутствии катализатора. 23-однозамещен ные соединения предлагаемого изобрете ния можно получить, во-первых, посредством образования 5,23-замещенного соединения и селективного отщеп ления защитной группы в положении-5. 5,23-дизамещенные соединения данного изобретения, имеющие различные замести тели в положениях-5 и 23, можно получить путем взаимодействия соединения, кото рое однозамеще но в любом положении-5 или 23 с реагентом, пригодным для образования другой замещенной гидроксильной группы в другом положении. Согласно дополнительному варианту изобретения предлагается способ получения соединений формулы (II), в которой одна из групп R2 и ОR4 является замещен ной гидроксильной группой, а другая представляет гидроксильную или замещенную гидроксильную гр уппу, как указывалось выше, включающий взаимодействие соединений формулы (III): (III) (где R1 имеет выше указанные значения и одна из груп ОR4 -ОR5 представляет гидроксильную груп пу, в то время как другая является гидроксильной или замещенной гидроксильной группой), с реагентом, пригодным для превращения гидроксильной группы в замещенную гидроксильную груп пу, а при желании с последующим избирательным отщеп лением защитной группы соединения формулы (II), в которой R2 -ОR4 обе представляют замещенные гидроксильные группы для образования соединения формулы (II), где ОR4 является гидроксильной группой, а R2 представляет замещенную гидроксильную гр уппу. Вообще го воря, указанная реакция включает ацилирование, формилирование, введение сульфонильной группы, этерифи кацию, введение силильной группы и образование ацеталей. Таким образом, ацилирование может осуществляться с использованием ацилирующе го агента, например, кислоты формулы R5СООН или его реакционноспособного производного, например, галоидангидрида (такого как хлорангидрида), ангидрида или активи рованного сложного эфира, или реакционноспособного производного угольной кислоты R5ОСООН или тиоугольной кислоты R5ОСSOН. Аци лирование с использованием галоидангидридов и ангидридов, при желании может производиться в присутствии кислотного связующего агента такого, как третичный амин (например, триэтиламин, диметиланилина или пиридина), неорганических оснований (например, карбоната кальция или бикарбоната натрия) и оксирены такие как низшие 1,2-алкиленоксиды (например, этиленоксид или пропиленокси), которые связывают га логенводород, отщепляемый при ацилировании. Для селективного замещения положения-23, чтобы получить соединение формулы (II), в которой R2 представляет группу ОR5, где R5 является замещен ной гидроксильной группой и ОR4 является гидроксильной группой можно проводить реакции ацилирования с использованием высоко электрофильных хло рангидридов (например, окселилхлорида, метоксиацетилхлорида, хлорацетилхлорида или бромацетилхлорида), предпочтительно в присутствии кислотного поглотителя такого как карбоната кальция. Аци лирование с использованием кислот желательно проводить в присутствии конденсирующе го агента, например, карбодиимида такого как N,N-дициклогексилкарбодиимида или N-этил-N' g диметиламинопропилкарбодиимид; карбонильного соединения, например, карбонилдиимидазола; или изоксазолиевой соли, такой как N-этил-5-фенилизоксазолийперхлорат. Активи рованный сложный эфир может быть образован на месте, используя, например, 1-оксибензотриазол в присутствии конденсирующе го агента, как указывалось выше. При желании, акти визированный сложный эфир может быть брикетирован. Реакцию ацилирования можно проводить в водной или безводной реакционной среде, предпочтительно при температуре в интервале -20о – +100оС, например, -10о – +50оС. Формилирование можно проводить с применением активированного производного муравьиной кислоты, например, N-формид-имидазола или ангидрида муравьиной кислоты при стандартных условиях реакции. Введение сульфонильной группы может осуществляться с помощью реакционноспособного про-изводного сульфокислоты R8SO3H такого как сульфонилгалоид, например, хлорид R8SO2CL или ангидрид сульфокислоты. Сульфурилование предпочтительно проводить в присутствии подходящего кислотного связующего агента, как указывалось выше. Этерифи кацию можно осуществлять с использова нием реагента формулы R7Y (где R7 имеет вышеуказанные значения, а Y представляет ухо дящую груп пу та кую как атом хлора, брома или йода или гидрокарбонилсульфонилоксигруппу, та кую как метилсуль фонилокси или толилсуль фонилокси, или галоидалканоилокси группу, например, дихлорацетокси). Реакция может сопровождаться образованием алкоголята магния при использовании реагента Гриньяра, та кого как галоидметилмагний, например, йодметилмагний или галоид-триалкилсилилметилмагний, например, хлортриметилсилилметилмагний с последующей обработкой реагентом R7Y. При желании, реакция может проводиться в присутствии соли серебра, например, окиси серебра, перхлората серебра, карбоната серебра или салицилата серебра или их смесей, причем указанная система может быть особенно подходящей, когда этерифи кацию проводят в присутствии галоидалкила (йодметила). Этерифи кацию можно, соответственно, проводить в растворителе таком, как простой эфир, например, диэтиловом простом эфире. Образование ацеталей может осуществляться посредством реакции с циклическим или ациклическим простым виниловым эфиром. Указанный способ особенно пригоден для получения тетрагидропираниловых простых эфи ров с использованием в качестве реагента дигидропирана, или 1-алкоксиалкильных простых эфи ров, такого как 1-этоксиалкильный простой эфир, применяя в качестве реагента виниловый простой эфир. Указанную реакцию желательно проводить в присутствии сильно кислотного катализатора, например, минеральной кислоты, такой как серная кислота, или органической сульфокислоты, та кой как птолуолсульфо кислота, в негидроксильном, безводном растворите ле. Безводные раство рители, которые можно использовать в вышеуказанных реакциях включают кето ны (например, ацетон), амиды (например, N,N-димитилфор мамид, N,N-диметилацетамид или гексаметилфосфо рамид, простые эфиры (например, циклические простые эфиры, такие как тетрагидрофуран или диоксан, и ациклические простые эфи ры, например диметоксиэтан или диэтиловый простой эфир), нитрилы (например, ацетонитрил), углеводороды такие как галоидзамещенные углеводороды (например, мети ленхлорид) и сложные эфиры, например, этилацетат, а также смеси двух или более указанных растворите лей. Введение силильной группы может осуществляться путем взаимодействия с силилгалогенидом (например, хлоридом), преимущественно в присутствии основания, такого как имидазол-триэтиламина или пиридина с использованием растворите ля, например, диметилформамида. Во многих случаях, например, когда 5,23-дигидроксисоединение формулы (III) применяют в качестве целевого материала образуется смесь 5-однозамещенных и 5,23-двузамещенных производных. Однако их можно отделить с использованием общепринятых мето дов, например, хроматографией (хроматографией на силикагеле или препаративной тонкослойной хроматографией). В соответствии с еще одним способом настоящего изобретения, соединение формулы (II), в которой R2 и R3 представляют каждый атом водорода, может быть получено взаимодействием соединения формулы (III), где R1 имеет выше указанные значения и ОR4 является замещен ной гидроксильной группой и ОR5 является гидроксильной группой путем замеще ния 23-оксигруппы атомов водорода с последующим отщеплением защитной группы ОR4, где необхо димо соединение, в кото ром группа ОR4 является гидроксильной груп пой, и при желании, с последующим повторным замеще нием для получения соединения формулы (II), где ОR4 является замещенной гидроксильной группой. Таким образом, исходные материалы формулы (III), где ОR4 представляют замещен ную гидроксильную гр уппу, а ОR5 является гидроксильной группой может воздействовать с реагентом, служащим для замещения гидроксильной группы уходящей гр уппой для получения соединения формулы (IV) (IV) где R 1 и -ОR 4 имеют вы ше указанные зна чения, а L являе тся ато мом или гр уп пой, отщеп ляемой посредством реакции восста новле ния, напри мер, го молити ческого восста новления, та кого как гр уп па R11 OCSO [где R11 я вляется С1–6 алкилом, ари лом, нап ример, фе нилом, или (С1–6 а лкилом) ари лом, например, п -то лилокси тио карбонилокси гр уппой ]]. Пригодными реагентами, которые можно использовать для введения фрагмента L являются, например, арилга лотноформаты, такие как п-толилхлоротноформат. Указанную реакцию можно проводить в присутствии основания, например, амина, такого как пиридин, в растворите ле, такого как галогензамещенные углеводороды, например, дихлорметане. При желании, промежуточные соединения формулы (IV) можно выделить. Промежуточные соединения формулы (IV) можно затем восстановить с получением требуе мого соединения формулы (II) с помощью регенерирующего агента, такого как оловогидридалкил (например, три-нбутилолово гидрида) в присутствии инициатора радикалов, например, перекиси, азобисизобути ронитрила или под действием света. Реакцию можно также проводить в соответствующем растворителе, который может быть выбран из груп пы кетона, например, ацетона; простого эфи ра, например, диоксана, углево дорода, например, гексана или толуо ла; галогензамещенного углеводорода, например, трихлорбензола; или сложного эфи ра, например, этилацетата. Можно также использовать комбинации указанных растворителей либо самих, либо с водой. Указанную реакцию можно проводить при температуре от 0о до 200оС, предпочтительно в интервале 20–130оС. В соответствии с еще одним способом настоящего изобретения, соединение формулы (II), где R2 и 3 R вместе с атомом углерода, к которому они присоединяются представляют группу >С=0, может быть получено путем окисления соединения формулы (III), где ОR5 является гидроксильной груп пой, а ОR4 является замещенной гидроксильной группой с последующим отщеплением защитной группы ОR4, где необхо димо соединение, в котором ОR4 являлся гидроксильной группой, и при желании последующим повторном замещении для получения соединения формулы (II), где ОR4 является замещенной гидроксильной группой. Указанную реакцию можно проводить с помощью окислительного агента, выступающего для превраще ния вто ричной гидроксильной группой в оксогруппу, по лучая таким образом соединение формулы (II). Подхо дящие окислитель ные агенты включают бен зохи ноны в присутствии во ды, например, 2,3ди хлор-5,6-ди циан-1,4-бен зохи нон или 2,3,5 ,6-те трахлор, 1,4-бензохи нон; хром (IV) -окисли тель, например, дихромат пиридина или трио кись хрома в пиридине; марганце вый (IV) окислитель, нап ример, двуо кись марган ца в ди хлор мета не; N-га лосуцинимид, например, N-хлор суцинимид или N-бромсуцинимид; диалкилсуль фок сид, например, ди метилсуль фоксид, в при сутствии акти ва тора та кого как N,N' дициклогексилкарбодимида или ацилга лоида, нап ример, оксалилди хлорида; или комплекса-пи ридинсерный ангидрид. Указанную реакцию можно также прово дить в подхо дящем растворителе, кото рый может быть выбран из группы кето на, например, ацетона; простого эфи ра, например, диэтилового эфи ра, диоксана или тетрагидрофурана; углево дорода, например, гексана, галогензамещенного углеводорода, например, хлорофор ма или мети ленхлорида; или сложного эфи ра, например, этилацетата или замещенного амида, например, диметилформамида. В реакции можно также использовать комбинации указанных растворителей либо одних, ли бо с водой. Реакцию можно проводить при температуре от -80оС до +50оС. Согласно еще одному варианту настоящего изобретения предлагается способ получения соединений формулы (II), где R2 и R3 вместе с атомом углерода, к кото рому они присоединяются представляют груп пу >С=0, а ОR4 является гидроксильной или метоксильной группой, включающий стадию культивирования микроорганизма вида Streptomyces способного образовывать хо тя бы одно из предлагаемых соединений, и при желании выделять из него указанное соединение, а кроме того, если необхо димо модифи цировать груп пу OR4 спо собами, которые описаны выше. Предпочти тельными микроорганизмами, способными образовывать вышеуказанные вещества являются штаммы нового ви да рода Streptomyces, которые называются Streptomyces thermoarchaensis. Образец указанного микроорганизма, кото рый является почвенной культурой, помещен на хранение (10.09.84 г.) в постоянную коллекцию Национального собрания промышленных и морских бактерий Torry Research Station, Aberdeen, United Kingdom с присвоением ему регистрационного номера NCIB 12015. Мутанты штамма Streptomyces thermoarchaensis NCIB 12015 могут также быть преимущественно использованы, и в постоянную коллекцию культур На ционального собрания промышленных и морских бакте рий сданы в депозитарное хранение (26.06.85 г.) 4 штамма мутантов, которым были присвоены регистрационные номера NCIB 12111, NCIB 12112, NCIB 12113 и NCIB 12114. Получение предлагаемых соединений посредством ферментации подходяще го вида Streptomyces может быть достигнуто общеприняты ми методами, например путем выращи вания микроорганизма Streptomyces в присутствии способных к ассимиляции источников углерода, азота и минеральных солей. Способные к ассимиляции источники углерода, азота и минералов могут быть при наличии либо простых или комплексных питательных веществ. Источники углерода обычно включают глюкозу, мальтозу, крахмал, глицерин, кормовую патоку, декстрин, лактозу, са харозу, фрукто зу, карбоксильные кислоты, аминокислоты глицерида, спирты, алканы и растительные масла. Источники углерода обычно содержат 0,5–10 мас. % сбраживаемой среды. Источники азота обычно включают, бо бовосоевую муку, настойку пше ницы, раство ры перегонки, дрожжевые экстракты, муку из семян хлопка, пепто ны, муку из земляного ореха, со лодовый экстракт, черную патоку, казеин, смеси аминокислот, аммоний (газообразный или раствор), соли аммония и нитраты. Кроме того, можно использовать мочевину и другие амиды. Источники азота обычно содержат 0,1–10 мас. % сбраживаемой среды. Пита тель ные минераль ные со ли, ко то рые могут бы ть включе ны в к ультур ную сре ду, со держат обычно применяемые соли, име ющие спо собность отдавать ио ны натрия, ка лия, ам мония, же леза, магния, цин ка, никеля, ко бальта , марган ца, ва надия, хро ма, кальция, ме ди, мо либде на, бо ра, фосфа та , суль фа та , хло ри да и карбо ната . Культиви рование Streptomyces стрептомицеты обычно проводится при температуре в интервале, 20– 50оС, предпочтительно 25–40оС, более предпочти тельно около 34оС, причем желательно сопровождать ее аэрацией и взбалтыванием, например, встряхиванием или интенсивным перемешиванием. Среда может быть первоначально инокулирована небольшим количеством спорулированных вз весей микроорганизмов, но для избежания задержки роста можно приготовить растительный посевной материал микроорганизма путем инокуляции небольшого количества культуральной среды спорообразной формой указанного организма, при этом расти тельный инокулят, который получен, может быть перенесен в сбраживаемую среду, или, более предпочтительно в одну или более стадий посева, где возникает дополнительный рост перед переносом в основную ферментационную среду. Процесс ферментации обычно проводят при рН в интервале 5,5–8,5, предпочтительнее 5,5–7,5. Ферментацию можно проводить в течение 2–10 дней, например около 5 дней. В том случае, когда желательно отделить материал, содержащий соединения предлагаемого изобретения из всего бродильного бульона или выделить любое из отдельных соединений, указанное можно осуществлять посредством общепринятой методики выделения и отделения веществ. Предлагаемые соединения в основном содержатся в мицелиях клеток, но их также можно обнаружить в бродильном бульоне, и таким образом, техника выделения может также быть применена к бродильному бульону после очистки. Ясно, что мето дика выделения веществ может иметь широкое разнообразие. Предлагаемые соединения могут быть выделены и отделены посредством разнообразных методов фракционирования, например, элюированием, осаждением, фракционной кристаллизацией, экстракцией растворителей, кото рые можно комбинировать различными способами. Выявлено, что экстракция растворите лей и хроматография наиболее подхо дящи для выделения и отделения предлагаемых соединений. После фер ментации, мицелий может быть собран традиционными методами, например, фильтрацией или центрифугированием. После этого, например, соединения могут быть экстрагированы из мицелия соответствующи ми органическими растворителями, такими как кетоны, например, ацетоном, метилэтилкетоном или метилизобутилкетоном; углеводородом, например, гексаном; галогензамещенным углеводоро дом, например, хлороформом, карбонтетрахлоридом, или метиленхлоридом; спирта ми, например, мета нолом или этанолом; или сложными эфирами, например, метилацетатом, или этиленацета том, ясно, что если мицелий содержит значительное количество воды, предпочтительно использовать водорастворимые растворители. Для дости жения опти мального извле чения сое динений обычно желате льно прово дить бо лее одной экстракции. П редпочти тель но первую экстракцию вы полнять с и спль зова нием смеши вающе го ся с во дой раство рите ля, нап ример, мета нола или аце то на. Ан ти био ти ки можно извле чь как неочи щен ный экстракт п утем уда ле ния раство рите ля . Экстракты ра ство рите ля могут да лее бы ть эк странирова ны, если желате льно, после уменьше ния объема раство рите ля, нап ример, вы парива нием. На этой ста дии пре дпочти те льно использова ть несмеши вающий ся с во дой раство рите ль, нап ример, гек сан, хло ро форм, ме ти лен хлорид или эти ла цета т, или и х смеси , при этом для дости жения хо роше го раз деле ния соеди нений анти био ти ка вво дится до ста точное коли чество во ды. Пр и уда ле нии во донераствори мой фа зы получают про дук т, со держащий одно или бо лее сое ди нений предлагае мого изобре тения, воз можно вместе с сое ди нениями анти био ти ка S541 . Очистку и/или отделение предлагаемых соединений можно осуществлять традиционными методами, например, хроматографией (включая высокоэффективную жидкостную хроматографию на соответствующей подложке), такой как двуокись кремния, нефункциональная макросетчатая адсорбционная смола, например, поперечносши тые полистирольные смолы, такие как смолы Amberlite XAD-2, XAD-4 или XAD-1180 (Rohm & Haus Ltd) или смола S112 (Kastell Ltd) или поперечносшитый декстран, смеши вающий ся с органическим раство рите лем, например, Sephadex LH20 (Pharmacia UK Ltd) или, в случае высокоэффективной жидкостной хроматографии та кой на подложках с обратной фазой, та ких как углеводород, нанесенный на двуо кись кремния, например, С18 сши тая двуо кись кремния. Подложка может быть в ви де слоя с насадкой размещен ного в колонне. В случае использования нефункциональных макросетчаты х смол, например XAD-1180 или S112, для элюирования можно использовать смеси органических раство рителей, например, ацето нитрила с во дой. Раствор указанных соединений в соответствующем растворителе обычно загружают в хроматографи ческую колонку из двуокиси кремния или Sephadex при желании после первого уменьше ния объема растворителя. Колонку могут по выбору промыть, а затем вымыть растворителем подходящей полярности. В случае, колонок из Sephadex и двуокиси кремния, в качестве растворителей можно использовать спирты, например метанол; углеводороды, например, гексан; ацетонитрил; галогензамещен ные углеводороды, например, хлороформ или метиленхлорид; или сложные эфиры, например, этилацетаты. Элюирование и разделение/очистку заявленных соединений можно регулировать общепринятыми способами, например, тонкослойной хроматографией и высокоэффективной жидкостной хроматографией или используя свойства соединений, описываемых далее. Предлагаемые соединения могут первоначально быть очище ны хроматографией на двуо киси кремния, предпочтительно используя элюат, та кой как хлороформ: эти лацетат, при желании с последующей высокоэффективной жидкостной хроматографией. Полученный таким образом очищенный продукт может быть затем подвергнут хроматографии на сефе дексовых колонках, предпочти тельно с использованием элюата, например, ацето нитрила, а затем заявленные соединения могут быть вы делены жидкостной хроматографией с высокой характе ристикой. Используя подхо дящие комбинации вышеуказанных методик, соединения формулы (II), как уточнено сейчас, вы делены как твердые вещества в почти чистом виде. Ясно, что последовательность, при которой осуществляют выше указанные стадии очистки, выбор тех, которые используют и степень достигаемой очистки могут очень отличаться. Указанные соединения могут применяться, однако, как указывалось выше, при степени чистоты, соответствующей их предназначению. При использованию их в медицине, желательно иметь степень чистоты хо тя бы 90%, предпочтительно более, чем 95%. Для ветеринарии и другого применения достаточна более низкая степень чистоты, например, 50% или ниже. Промежуточные соединения антибиотиков S541 формулы (III), где ОR5 является гидроксильной группой и OR4 представляет гидроксильную или метоксильную гр уп пу могут также быть получены с использованием способов ферментации и выделения, например, как описано в патенте Южной Африки № 85/7049. Другие промежуточные соединения формулы (III) можно получить из указанных соединений, используя описанные выше способы для получения соединений формулы (II). Настоящее изобрете ние далее представлено следующими препаратами и примерами. Все температуры приведены в оС. Соединения обозначены со ссылкой на источник "факторы", которые представляют собой перечисленные ниже соединения формулы (V) (V) Фактор A B C D E F R –H –CH3 –H –H –CH3 –CH3 R1 –CH(CH3)2 –CH3 –CH3 –CH2CH3 –CH2CH3 –CH(CH3)2 Факторы А, В, С, D, E, F можно получить способом, который описан в патенте Южной Африки № 85/7049. Пример 1. 5-Феноксиацетокси и 5,23-дифе ноксиацетокси фактор А. Фактор А (2,0 г) в растворе дихлорметана (25 мл) и пиридина (0,35 мл) при 0 оС обрабатывают раствором феноксиацетилхлоридом (0,6 мл) в дихлорметане. Через 18 часов при температуре 3оС полученный раствор обрабатывают пиридином (1,0 мл) и феноксиацетилхлоридом (1,0 мл) в дихлорметане (5 мл). Раствор тща тельно перемешивают при температуре 0–5оС в течение 30 минут, прежде чем вылить в воду со льдом. Простой эфир (100 мл) вводят в раствор, и смесь перемешивают в течение 20 мин. Водный слой экстрагируют простым эфиром. Эфирный слой объединяют, последовательно промывают во дой и соляным раствором, высушивают и выпаривают. Остаток очищают хроматографией на колонке из двуокиси кремния смесью дихлорметана с ацето ном (40:1) с выходом названных соединений в виде смеси (1,8 г моноацил:диацил = 6:1), кото рую разделяют высокоэффективной препаративной жидкостной хроматографией с обратной фазой, получая 5-феноксиацетокси фактор А, d (CDCl3) со держит 6,8–7,4 (м; 5Н) и 4,66 (с; 2Н) м/з включает 746, 728, 710, 594 и 576, и 5,23-дифеноксиацетокси фактор А, d (CDCl3) содержит 6,8– 7,4 (м, 10Н), 4,60 (с, 2Н) и 4,70 (с, 2Н). Пример 2. 5-Феноксиацетокси-23-(4-метилфенокситиокарбонилокси) фактор А 5-феноксиацетокси фактор А (747 мг) в дихлорметане (10 мл) при 0оС в атмосфере азота обрабатывают пиридином (0,81 мл), а затем 4-метилфенилхлортиоформато (0,75 г) в дихлорметане (2 мл). Полученный темный раствор тщательно перемешивают в те чение 15 минут при температуре 0оС, и затем в течение еще 22 часов без охлаждения. Полученную смесь выливают в хо лодную во ду и со ляной раствор (рассол) и экстрагируют простым эфиром. Соединившиеся слои простого эфи ра промывают во дой и соляным раствором, высуши вают и выпаривают. Остаток очищают хроматографией на колонке двуокиси кремния и препаративной жидкостной хроматографией с обратной фазой с получением названного соединения (430 мл), d (CDCl3) содержит 3,34 (м; 1Н), 3,58 (м; 1Н), 3,97 (д. 10; 1Н), 4,72 (с. 2Н), 5,4 (м, 1Н), 5,59 (д 6; 1Н) и 6,9 до 7,4 (м; 9Н), м/з включает 728, 616, 576, 466, 464, 448, 354, 297, 247, 219 и 151. Пример 3. 5-Трет-бутилдиметилсилилоксиацетокси фактор А. Фактор А (2,144 г) в безводном простом эфире (25 мл) и пиридине (2,5 мл) при 0оС в атмосфе ре азота обрабатывают с прибавлением по капле трет-бутилдиметилсилилоксиацетилхлорида (1,2 г) в простом эфи ре (10 мл). Полученную смесь перемешивают в те чение 90 минут при температуре 0оС прежде, чем обработать по капле опять кислотным хлоридом (1,10 г) в простом эфире (10 мл). Полученную смесь тщательно перемешивают при температуре 0оС в течение 60 минут и выливают в холодную во ду и простой эфир. Водный слой промывают простым эфиром. Соединившиеся органические слои промывают водой и соляным раствором, высушивают и выпаривают. Полученный остаток очищают посредством хроматографии на двуо киси кремния, используя смесь дихлорметана с ацето ном (25:1) в качестве элюата с получением названного соединения, d (CDCl3) включают 0,09 (о; 6Н), 0,78 (д 6; 3Н), 0,90 (с, 9Н), 0,93 (д 6, 3Н), 0,97 (д 6, 3Н), 1,03 (д 6, 3Н), 1,51 (с; 3Н), 1,59 (с; 3Н), 1,74 (с; 3Н), 3,32 (м; 1Н), 3,52 (д 10; 1Н), 3,64 (м; 1Н), 3,74 (д 10; 1Н), 3,82 (м; 1Н), 4,32 (с, 2Н) 5,57 (д 5; 1Н), м/з содержит 784, 766, 748, 595, 577, 484, 466, 354, 314, 297, 255, 247, 237, 219 и 151. Пример 4. 5-Трет-бутилдиметилсилилокси. Фактор А. Фактор А (250 г) и имидазол (163 мг) в сухом диметилформамиде (10 мл) обрабатывают трет-бутилдиметилсилилхлоридом (197 мг). Полученный раствор тщательно перемешивают в те чение 2 часов и выливают в хо лодную воду. Указанную смесь тща тельно экстрагируют простым эфиром, и соединившиеся экстракты эфи ра высушивают и выпаривают. Остаток очищают хроматографией на двуо киси кремния, используя смесь дихлорметана с ацето ном (10:1) в качестве элюата с получением названного соединения (235 мг), d (CDCl3) содержит 0,13 (с, 6Н), 0,80 (д 6; 3Н) 0,92 (с; 9Н), 0,96 (д 6, 3Н), 1,00 (д6; 3Н), 1,03 (д6; 3Н), 1,53 (с; 3Н), 1,60 (с; 3Н), 1,80 (с; 3Н), 3,37 (м; 1Н), 3,56 (д 10; 1Н), 3,64 (м; 1Н), 3,75 (д. 10; 1Н) и 4,43 (д 5; 1Н), м/з содержит 726, 708, 691, 633, 466, 448, 354, 314, 297, 265, 247, 219 и 151. Пример 5. 5-Ацетокси и 5,23-диацетокси. Фактор А. Фактор А (3,0 г) и пириди на (20 мл) при тем пературе -5оС обраба тывают кислотным ангидридом (8 мл) и полученный раствор оста вляют при температуре 3оС в те че ние 20 часов. К раство ру до бавляют бен зол (100 мл) и полученную смесь сгущают в ва кууме. Оста точное масло хро мато графи руют на двуо киси кремния, используя в качестве элюа та смесь ди хлор мета на с ацето ном (40:1), по лучая 5ацета т Факто ра А (2 ,06 г), со держащий 5 ,23-диа цета т (10%). По лученные соединения отделяют пре паративной жидкостной хро мато графией с обратной фа зой с получением 5-ацеток си Фактора А (79% выхо да), lmax (EtOH) 244,5 нм (Е11; 4,62), d (CDCl3 ) содер жит 2,14 (с; 3Н), м/з со держит 654, 594 и 576, и 5,23-диа цетокси Факто ра А (6,5% вы хо да, d (CDCl3) со держит 2,01 (с; 3Н) и 2,13 (с; 3Н ), м/з со держит 696 и 636. Пример 6. 5,23-Диацетокси Фактор А. Раствор Фактора А (600 кг) в сухом пиридине (1,0 мл) обрабатывают из быточным количеством кислотного ангидрида (0,50 мл) и несколькими кристаллами 4-N,N-димети ламинопиридина. Через 24 часа при комнатной температуре раствор вы ливают в простой эфир и за тем поочередно промывают ор ганическую фа зу с помощью 2N соляной кислотой, насыщен ным раствором бикарбоната натрия и, наконец соляным раствором. Выпаривание просушенного органического слоя обеспечивает камедь, кото рую очищают хроматографией через колонку Merck Kieselgel 60 с двуо кисью кремния (45 г) размером 230–400 меш. Элюирование колонны смесью дихлорметана с эфиром (9:1) дало названное соединение в ви де бесцветной пены (560 мг), [a]D25+160o (с 0 ,48, CHCl 3). Соединение примера 7 получено аналогичным способом: Пример 7. 5,23-Диацетокси Фактор С. Т. пл. 211–213о, [a]D22+200o (с, 0,40, CHCl 3), lmaxEtOH 238,5 (30,400) и 245 нм (emax 32,500); n max 3440 (OH) и 1718 см -1 (ацетокси и сложный эфир); d(CDCl3) содержит 4,90 (м,1Н), 4,04 (д 6Гц, 1Н), 3,96 (д 10Гц, 1Н), 3,58 (м, 1Н), 3,32 (м, 1Н), 2,14 (с, 3Н), 1,75 (с, 3Н), 1,67 (д 6Гц, 3Н), 1,60 (с, 3Н), 1,52 (с, 3Н), 0,99 (д 6Гц, 3Н), и 0,71 (д 7Гц, 3Н), м/з = 668 (М+), из Фактора С (312 мг). Реакционную смесь выливают в этил, а не в простой эфир и в заключении кристаллизуют на порошкование диизопропиловым эфиром. Пример 8. 23-Аце токси Фактор А. К перемешанному и охлажденному (0–5о) раствору соединения примера 6 (530 мг) в мета ноле (10 мл) вводят по каплям водный раствор (1 мл) гидроокиси натрия (30 кг). Через 1,3 часа смесь выливают в этилацетат и органическую фазу затем промывают поочередно с помощью 2N соляной кислотой; водой и в конце соляным раствором. Выпаривание просушенной орга нической фазы дает желтую смолу, которая в дихлорметане вводится в хро матографи ческую колонку Merck Kieselgel 60, с добавлением в тот же самый растворитель двуо киси кремния (50 г), размером частиц 230–400 меш. Элюирование смесью дихлормета на с простым эфиром (9:1) дало названное соединение, которое получено в виде почти бесцветного твердого вещества путем выпаривания раствора н-пентана. Указанное вещество (330 г) имеет [a]D23+166о (c, 0,64, СНСl3), lmax EtOH 239 (26,600) и 245 нм (emax 29,300); n max (CHBr3) 3540, 3460 (OH) и 1712 cм -1 (сложного эфи ра); d(CDCl3) содержит 4,89 (м, 1Н), 4,27 (t 6Гц, 1Н), 3,93 (д 6Гц, 1Н), 3,91 (д 10Гц, 1Н), 3,55 (м, 1Н), 3,25 (м 1Н), 2,01 (с, 3Н), 1,85 (с, 3Н), 1,59 (с, 3Н), 1,51 (с, 3Н), 1,03 (д 6Гц 3Н), 0,98 (д 6Гц, 3Н), 0,94 (д 6Гц, 3Н), и 0,69 (д 7Гц, 3Н), м/з = 654 (М+). Соединение примера 9 получено аналогичным путем: Пример 9. 23-Ацетокси Фактор С. [a]D23+139o (с 0,52, СН2Сl2); lmax EtOH 244,5 нм (emax 29,400); n max (CHBr3) 3550, 3480 (ОН); 1716 (сложный эфир и ацетат) и 1255 см -1 (ацетат); d(CDCl3) содержит 4,91 (к, 3Гц, 1Н); 2,03 (с, 3Н), и 1,64 (л, 7Гц, 3Н) и 1,60 (с, 3Н), м/з = 626 (М+), из соединения примера 7 в ви де белого аморфного твер дого ве щества. Пример 10. 5-Ацетокси-23-кето Фактор А Раствор оксалилхлорида (1,96 мл) в сухом дихлормета не (25 мл) при температуре -70о в атмосфере азота обработан по каплям раствором диметилсульфоксида (3,19 мл) в сухом дихлорметале (15 мл) и затем по каплям раствором 5-ацетокси Фактора А (4,91 г) в сухом дихлорметане (30 мл). Полученный раствор тща тельно перемешивают в те чение 1,5 часа при температуре -70оС, прежде чем обработать по каплям раствором триэтиламина (12,6 мл) в сухом дихлорметане (40 мл). Реакционную смесь перемешивают в течение 1,25 часа без охлаждения и выливают в смесь (1:1) холодной воды и просто го эфира. Водный слой экстрагируют простым эфиром. Соединившиеся органические слои промывают во дой, соляным раствором, высушивают и выпаривают. Остаточную пену хро матографируют на двуокиси кремния, используя смесь дихлормета на с ацетоном (50:1) с получением названного соединения (3,4 г), d(CDCl3) содержит 3,33 (м, 1Н), 3,49 (м, 1Н), 3,70 (д 10, 1Н), и 5,52 (д 5, 1Н), м/з содержит 652, 634, 609, 591, 482, 263, 235 и 151. Пример 11. 23-Кето Фактор А Соединение примера 10 (276 мг) в мета ноле (5 мл) при температуре 0 оС обрабатывают по капле раствором 2N-натрий-гидроксида (0,42 мл) в метаноле (1,0 мл). Перед тем, как вылить раствор в хо лодную воду, его вы держивают при температуре 5оС в течение 5 часов. Смесь экстраги руют простым эфиром и этилацетатом. Соединившиеся органические слои промывают соляным раствором, высушивают и выпаривают с по лучением твердого ве щества, которое очищают препаративной тонкослойной ххроматографией, используя в качестве растворителя смесь дихлорметана с ацетоном (10:1) для получения названного соединения (140 мг), d(CDCl3) содержит 3,28 (м, 1Н), 3,48 (м, 1Н), 3,70 (д 10; 1Н), и 4,28 (тр 7 1Н), м/з включает 592, 549, 482, 370, 263, 235 и 151. Пример 12. 23-Кето Фактор А 0,4 мл спорообразной суспензии Strepto-myces thermoarchaensis NCIB 12015 в 10% глицерине используют для инокуляции в 250 мл колбе Erlenmyer, содержащей 50 мл культур ной среды, состоящей из Dглюкозы 15,0 gL-1, глицерина 15,0 gL-1, соевого пептона 15,0 gL-1, NaCl 3,0 gL-1 и CaCO3 1,0 gL-1, добавлено до нормы дистиллированной водой. Перед помещением в автоклав, рН до ведена до 7,0 водным раствором NaOH. Колбу инкубируют при температуре 28о в течение 2 дней на круговой качалке, работающей при скорости 250 оборотов в минуту с 50 мл в диа метре движением по орбите, 4-х мл порции используют затем для инокуляции каждой из четырех 2-х ли тровых фла конов с ши роким горлышком, содержащая каждая по 200 мл одной и той же среды перед инкубированием в те х же самих условиях в течение 2 дней. Содержимое указанных 4 фла конов далее используют для инокуляции 70 литрового ферментатора, содержащего 40 литров той же культуральной среды с добавлением пропиленгликоля 2000 (0,06%). Полипропиленгликоль 2000 вводится, как необхо димо, в течение всего процесса фермента ции для регулирования пенообразованием. Ферментацию проводят при температуре 28о с энергичным перемешиванием и аэрацией, достаточной для поддержания уровня растворенного кислорода больше 30% насыщения. Через 24 часа процесса брожения 800 мл, и 9-литровые порции переносят в 70 литровый ферментатор, содержащий 40 литров культуральной среды, и 700-литровый ферментатор, содержащий 450 литров культуральной среды, соответственно. Оба из указанных ферментаторов содержат культуральную среду, состоящую из D-глюкозы 2,5 gL -1 Malt декстрина (MD30E) 25,0 gL-1, Arkasoy 50 12,5 gL-1, Beet Molasses 1,5 gL-1, K2HPO4 0,125 gL-1, CaCO3 (Ar) 1,25 gL-1 и силикона 1520 (Dow Corning) 0,6 gL-1 приведенную к норме дистиллированной водой. Перед стерилизацией рН доводят до 6,5 водным раствором Н2SO4. Указанные процессы ферментации выполняют при температуре 34оС при энергичном размеши вании и аэрации, достаточной для сохранения уровня растворенного кислорода больше, чем 30% насыще ния. При необхо димости добавляют антивоспламените ли из полипропиленгликоля 2000. Через 24 часа, рН в каждом из ферментаторов доводят до 7,2 путем введения водного раствора Н 2SO4. Продукты ферментации собирают и объединяют через 4 дня. Ми целий (10,4 кг) из собранной жидкой среды (423 л) выбирают в центрифуге Sharples PS16AY. Мицелий интенсивно перемешивают в метаноле (50 л) в течение 40 минут и затем отфильтровывают. Остаток повторно суспендируют в метаноле (15 мл) и вновь фильтруют. Соединившиеся фильтраты (55 л) затем смешивают с во дой (27 л) и 60–80 петролейным эфиром (предел кипения) 60–80о) (30 л) и перемеши вают в течение 20 минут. Полученные фазы отделяют в центрифуге Westfalia MEM1256 а метанольную фа зу (75 л) смешивают снова с водой (38 л) и 60–80 петролейным эфиром (30 л). Через 20 минут фазы вновь отделяют в центрифуге, причем в фа зу петролейного эфи ра, чтобы разрушить полученную эмуль сию вводят ацетон (2,0 л). Ме танольную фа зу (110 л) смешивают с водой (38 л) и 60–80 петролейным эфиром (30 л) в третий раз, фазы отделяют как раньше. Аце тон (3 л) добавляют снова к петролейной эфирной фазе для разруше ния полученной эмульсии. Три фазы гексана объединяют (90 л) и концентрируют при низком давлении (температура выпарки 25о). Концентрат (9,8 л) вы сушивают суль фатом натрия (3 кг) и выпаривают до масла. Получен ное масло раство ряют в ди хлорметане (0,5 л) и фильтр уют че рез Dicalite 478. Раствор (0,9 л) загружают в хро матографи ческую ко лонну (150 х10 см) из двуо киси кремния (Merck) при за грузке 6 л/час, промывают ди хлор мета ном (4 л) и элюир уют смесью хло рофор ма с этила цетатом (3:1). Фракции, элюи р ующие между 14,6 и 33,3 л, концентрируют до твер дого ве ще ства и растворяют в смеси хло рофор ма с аце то ном (3:1). Полученный раствор повторно хроматографируют на колонке двуокиси кремния с тем же самым растворителем. Фракции, элюирующие между 14,5 и 31,5 л, высуши вают до твердого вещества и растворяют в смеси хлорофрма с этилацетатом (3:1). Полученный раствор вновь хроматографи руют на двуо киси кремния при тех же условиях, что и раньше, и затем фракции элюирования меду 14 и 31 л высуши вают до твердого вещества. Твердое вещество растворяют в 70%-ацетонитриле в во де (1,23 л) с доста точным количеством метанола, добавляемого для получения светлого раствора. Указанный раствор хроматографи руют в 5 мл порциях на колонке Spherisorb ODS2. Содержимое колонки элюируют с по мощью 70% ацето нитрила при скорости течения, которая возрастает от 20 мл/мин до 34 мл/мин через 21 минуту. Фракции из каждой порции, которые элюированы между 12,4 и 16, были собраны вместе и разбавлены равным объемом воды. Полученный раствор затем загружают в колонку Montedison S112 на полистироле с сетчатой структурой (2 л). Колонку промывают 35% раствором ацетонитрила и элюируют ацетоном. Фракции, элюирующие между 0,5 и 1,25 л, высуши вают до твердого вещества. Твердое вещество растворяют в ацетонитриле (20 мл) и хроматографи руют на колонке из Sephadex LH20 в том же растворителе. Фракции, элюирующие между 1,08 и 1,26 л, собирают и высушивают до твердого вещества. Твердое вещество растворяют в 60% ацетонитриле (10 мл) с достаточным количеством метанола, добавляемого для получения чистого раствора. Раствор хроматографи руют в 2 мл порциях еще раз на колонке из Spherisorb ODS2 и элюируют с помощью 60% ацетонитрила при 25 мл/мин. Фракции, элюирующие между 0,95 л и 1,08 л, со бирают вместе и вы сушивают до твердого ве щества. Полученное твердое вещество повторно растворяют в хло роформе (5 мл) и хроматографи руют на колонке из силикагеля Merck 60. Раствор элюируют хло роформом при 10 мл/мин, и фракции, элюирующие между 400 мл и 790 мл, высушивают с получением названного соединения (33 мг) в виде твердого вещества. ЯМР-спектр соответствует содержащемуся 23-кето Фактору А. Э.И. – масс-спектроскопия дала ион молекулы при 610 и характеристические фрагменты при 592, 549, 498, 482, 370, 263 и 151. Образец представлен, что бы сравнить 23-кето Фактор А, полученный при высокоэффективной жидкостной хроматографией с аналогичным кето ном, образуемым при химическом окислении Факто ра А. Пример 13. 5-феноксиацетокси-23-деокси Фактор А Соединение примера 2 (350 мг) и азобисизобутиронитрила (25 мг) в сухом толуо ле (20 мл) и атмосфе ре азота при температуре 120о обработано по каплям раствором три-н-бутилолово гидрида (0,5 мл) в сухом толуо ле (10 мл). Раствор нагревают с обратным холодильником в течение 90 мин., охлаждают и выпаривают. Оста ток хроматографируют че рез двуокись кремния, используя смесь дихлорметана с ацетоном (40:1) в качестве растворителя с выхо дом названного соединения (280 мг), d(CDCl3) содержит 3,32 (м, 1Н), 3,42 (д 10; 1Н), 3,57 (м; 1Н), 4,71 (с; 2Н), 5,59 (д 6; 1Н), и 6,8–7,4 (м; 5Н), м/з содержит 730, 712, 578, 560, 468, 450, 314, 299, 249, 248, 221 и 151. Пример 14. 23-деокси Фактор А Соединение примера 13 (240 мг) вве дено в неочищенный раствор аммония в метаноле (10 мл) при температуре -5о. Полученный раствор перемеши вают при температуре от 0о до 10о в те чение 2 часов перед его выпариванием до сухого состояния. Оста ток хроматографируют че рез двуокись кремния, используя смесь дихлорметана с ацетоном (20:1) с получением названного соединения (180 мг), d(CDCl3) содержит 3,27 (м; 1Н), 3,42 (д 9; 1Н), 3,54 (м; 1Н), и 4,29 (т6; 1Н), м/з содержит 596, 578, 560, 468, 450, 356, 314, 299, 249, 248, 221 и 151. Пример 15. 5-Аце токси-23-метилсуль фонилокси Фактор А Раствор 5-ацетокси Фактор А (3,46 г) в пиридине (30 мл) охлаждают в ледяной бане и обрабатывают метансульфо ниевым ангидридом (2,2 г). Через 30 минут смесь подогревают до температуры окружающей среды и еще че рез 60 минут разделяют между этилацета том и 2N-соляной кислотой. Ор ганическую фазу отделяют и промывают последовательно 2N-соляной кислотой, насыщен ным водным раствором бикарбоната натрия и в заключении насыщенным соляным раствором. Орга нический раствор высушивают (Na2SO4) и растворитель выпаривают с вы ходом пены, которую хроматографируют через колонку на двуокиси кремния (Merck Art 9385), добавленного в смесь гексана (пределы кипения 60–80о) с ацетилацетатом (3:1) и элюируют в том же растворителе. Соответствующие фракции основного компонента соединяют, и растворитель выпаривают с получением названного соединения в виде пены (2,08 г), [a]D22+154o (o=0,56; CHCl3), l maxCHCl3 + 247 нм (e29070); n max(CHBr3) 3550, 3470 (OH) и 1735, 1715 (сложный эфир); d(CDCl3) содержит 4,90 (м, 1Н), 3,05 (с, 3Н), и 2,16 (с, 3Н). Пример 16. 23-Tозилокси Фактор В Фактор В (250 кг), тозиловый ангидрид (204 мг) и несколько кристаллов 4-N,N-диметиламинпиридина перемеши вают в сухом пиридине (0,5 мл) в течение 24 часов. Смесь выливают в простой эфир, и органическую фазу промывают последовательно 2М со ляной кислотой, насыщенным водным раствором бикарбоната натрия и, наконец, соляным раствором. Выпаривание высушенного (Na2SO4) эфир ного слоя дает неочищенный продукт, в который в дихлормета не вводят в хроматографи ческую колонну на силикагеле (50 г, Merck Kieselgel 60, 203–400 меш), включенного в тот же растворитель. Осуществляют элюирование из смеси дихлормета на с простым эфиром (9:1) основного компонента, который добавочно очищают препаративной высокоэффективной жидкостной хроматографией. Названное соединение получено в виде смолы [a]D23+173o (0,84, CHCl3); lmax (EtOH) 235 (30,000) и 244,5 нм (emax 30,800); n max (Cand 1708 см -1 (эфир); d(CDCl3) содержит 7,82 (д, 10 Гц, 2Н), 7,29 (д, 10 Гц, 2Н), 4,83 (к, 3 Гц, 1Н), 3,50 (с, 3Н) и 2,43 (с, 3Н); м/з = 752 (М+). Пример 17. 5-Ацетокси-23-н-бутокси Фактор А Карбонат серебра (лг) добавляют к раствору 5-ацетокси Фактора А (325 мг) в сухом простом эфире с последующим введением иодобутана (0,5 мл) и перхлората серебра (550 мг). Полученную смесь перемешивают при комнатной температуре в течение 20 часов, когда вводят соллидин (0,5 мл). После перемешивания в течение еще 20 минут, смесь фильтруют и фильтрат промывают поочередно с помощью 2N соляной кислоты, насыщенного водного раствора бикарбоната натрия и воды. Высушенную органическую фазу выпаривают почти досуха и масло очищают хроматографией на колонке из Merck Kieselgel 60 с размерами пор 230–400 меш. Элюирование из колонки смесью гексана с этилацетатом (3:1) дает названное соединение в виде бесцветной пены (276 мг) [a]D22+160o (c 0,94, CHCl 3) l max (EtOH) 245 нм (emax). Пример 18. Фактор А-23-гемиоксалат Фактор А, (лг, ка 70% очистки) и карбонат кальция (1,5 г) перемешивают дихлорметаном (20 мл) при температуре 21оС и к полученной суспензии добавляют в избытке однократно оксалилхлорид (1,0 мл). Через 4–5 минут, указанную смесь обрабатывают водой (15 мл) и еще через 5 минут 2 М со ляной кислотой (10 мл) и этилацета том (70 мл). Органическую фа зу отделяют, промывают во дой и соляным раствором и далее обрабатывают обесцвечивающим углем. Через 5–10 минут образованный органический раствор отфильтровывают через фазоразделительную бумагу и фильтрат десорбируют из растворителя. Остаток в простом эфире (50 мл) очищают п утем фильтрования через "Hyflo" и, уда ляя растворитель, получают бледно-желтую пе ну. Порошкование указанного вспененного материала диизопропиловым эфиром (ка 7 мл) дает названное соединение в виде кристаллического твердого вещества (750 мг) после промывания твердого вещества диизопропиловым эфи ром и н-пента ном. n max (CHBr3) 3360–3600 (ОН), 1805 (кислотный мономер) и 1720 см-1 (кислотный димер и сложные эфиры): d(СDCl3) содержит 4,30 (д, 5Гц, 1Н) и 5,14 (м, 1Н). Пример 19. Фактор А. Раствор 23-гемиоксалат Фактора А (500 мг) в дихлорметане (30 мл) энергично перемешивают с водным раствором гидроокиси натрия (1,54 г в 30 мл Н2О) в течение 2 часов при температуре 21о. Органическую фа зу собирают, а водную фа зу промывают небольшим количеством дихлорметана. Органические фазы соединяют, промывают с помощью 2М НСl, затем высушивают (Na2SO 4) и де сорбируют из растворителя с получением названного соединения (430 кг) в виде вспененного вещества. Указанное соединение продемонстрировано в качестве Фактора А при сравнении получения высокоэффективной жидкостной хроматографией с аналогичным образцом Факто ра А. Пример 20. Фактор А, 5-ге мисукцинат. Раствор Фактор А (306 мг) и сукцинового ангидрида (60 мг) в сухом пиридине (0,5 мл) перемешивают при температуре 22оС в течение 24 часов. Смесь разбавляют простым эфиром и промывают органическую фа зу с помощью 2N соляной кислоты и соляного раствора. При выпаривании высушенного эфирного слоя получают вспененное вещество, из кото рого выделяют посредством высокоэффективной жидкостной препаративной хроматографии с обратной фазой названное соединение. Полученный продукт в эфи ре осаждается в виде белого аморфного твердого вещества (86 мг) при введении дополнительно н-пента на [a]D22+125o (с. 0,44, CHCl 3) l maxEtOH 245 нм (emax 30,700), n max (CНBr3) 3490 (ОН), 3,300–2,200 (СО2Н), 1730 (кислотный карбонил) и 1710 см-1 (сложный спирт); d(CDCl3) со держит 2,74 (с, 4Н), м/з = 712 (M+). Пример 21. 5-Аце токси, 23-этилоксалилокси Фактор А. Раствор 5-ацетокси Фактор А (320 мг) и избыточный этилоксалилхлорид (0,5 мл) в сухом дихлорметане (6 мл) смешивают в присутствии карбоната кальция (300) мг в те чение 1 часа. Образованную смесь, разбавленную простым эфиром, выливают в на сыщенный бикарбонат натрия, и затем перед выливанием в этилацетат перемешивают в те чение 20 минут. Вы париванием высушенной (Na2SO4) органической фазы получают смолу (325 мг), кото рая в дихлорметане вводится в хроматографическую колонку из Marck Kieselgel 60, причем двуокись кремния (25 г) с размером частиц 70–230 меш добавлена в тот же растворитель. Элюирова ние из колонки дихлорметаном и затем смесью дихлормета на с простым эфиром (95:5) дает названное соединение (265 мг) в виде белого вспененного вещества [a]D23+157o (c 0,41, СНСl3); lmax EtOH 245 нм (emax 32,800); n max (CHBr3) 3530, 3480, (ОН), 1760 и 1735 см -1 (сложные эфиры); d(CDCl3) содержит 5,4–5,6 (м, 2Н); 5,05 (м, 1Н), 4,32 (к, 7 Гц, 2Н), 2,13 (с, 3Н) и 1,35 (t, 7 Гц, 3Н). Пример 22. 23-Ме тилоксалилокси Фактор А. Раствор 5-ацетокси, 23-этилоксалилокси – Фактора А (50 мг) в метаноле (1 мл), содержащий концентрированную сер ную кислоту (0,01 мл) выдерживают при температуре 21оС в течение 17 часов. Затем смесь выливают в эти лацетат и органическая фаза действует в качестве нейтрального продукта. Указанный продукт очищают хро матографией через колонку из Marck Kieselgel 60, двуо киси кремния (15 г) с размером частиц 70–230 меш, использующих в качестве элюирующи х растворителей первоначально дихлорметан, а затем смеси дихлорметана с эфиром (9:1). Названное соединение получено в виде бесцветной смолы (25 мг), [a]D23+147o (c, 0,28 СНСl3), l maxEtOH 245 нм (emax 26,000); n max (CHBr3) 3550, 3480 (OH), 1763, 1737 и 1710 см -1 (сложные эфи ры); d(CDCl3) содержит 4,29 (br, т., 7 Гц, 1Н), 5,09 (м, 1Н) и 3,88 (с, 3Н). Пример 23. 5-Аце токси-23-циклопропилкарбонилокси – Фактор А. Раствор 5-ацетокси – Фактора А (140 мг) в пи ридине (1 мл) обрабатывают хлоридом циклопропанкарбоксиловой кислоты (0,08 мл). Через 3 часа реакционную смесь разделяют между этилацетатом и 2N соляной кислоты. Органическую фазу отделяют и промывают последовательно с помощью 2N соляной кислоты и насыщенным водным раствором бикарбоната натрия. Органический раствор высуши вают (Na2SO4) и растворитель упаривают с получением масла, которое хроматографируют в колонке из двуокиси кремния (Marck Art 9385; 80 мл), введенного в смесь гексана (пределы кипения 60–80оС) с этилацетатом (3:1) и элюируемого таким же растворителем. Соответствующие фракции основного компонента объединяют и выпаривают растворитель с получением названного соединения в виде вспененного вещества (70 мг), [a]D22+143o (с=0,54; СНСl3); lmaxCНCl3 250,2 (e = 20310); n max (CHBr3) 3550, 3470 (ОН) и 1733, 1712 (сложный эфир); d(CDCl3) содержит 2,16 (с, 3Н), 3,33 (м, 1Н), 3,88 (д, J 10 Гц, 1Н), 5,01 (м, 1Н), и 5,5 – 5,5 (м, 2Н). Соединения примера 24 получают аналогичным путем. Пример 24. 5-Аце токси-23-циклопентилкарбонилокси – Фактор А. 5-Ацетокси – Фактора А (0,28 г) и циклопентилкарбонилхлорида (0,2 мл) дают названное соединение в виде вспененного вещества (0,17 г), [a]D22+149 (c=0,475; CHCl3); l max CHCl3 250,2 нм (e = 19900); n max (CHBr3) 3480 (OH). Пример 25. 23-Циклопропилкарбонилокси – Фактор А. Перемешанный раствор 5-ацетокси-23-циклопропилкарбонилокси – Фактора А (230 мг) в метаноле (15 мл) охлаждают в ледяной бане и обрабатывают 3% водным раствором гидроокиси натрия (0,5 мл). Через 5 часов, в течение которых реакционная смесь нагревалась до комнатной температуры, раствор разделяют между этилацетатом и 2N соляной кислотой. Ор ганическую фазу отделяют, промывают 2N соляной кислотой (х 2), вы сушивают (Na2SO4) и вы паривают растворитель. Остаток хроматографи руют на колонке из двуокиси кремния (Marck Art 9385; 150 мл), введенного в смесь гексана (пределы кипения 60–80оС) с этилацетатом и элюируемого тем же растворителем. Соответствующие фракции основного компонента объединяют и выпаривают растворитель с выхо дом названного сложного эфи ра в виде вспененного вещества (170 мг), [a]D22+146o (c=0,46; CHCl 3); l max CHCl3 250,2 нм (e = 20090); n max (СНBr3) 3550, 3480 (ОН) и 1709 (сложный эфир); d(СНСl3) содержит 3,26 (м; 1Н), 3,88 (д, J 10 Гц, 1Н), 4,29 (т, J 7Гц, 1Н), и 5,01 (м; 1Н). Соединения примера 26 получают аналогичным способом. Пример 26. 23-Циклопентилкарбонилокси – Фактор А. Отщепление защитной группы 5-ацетокси-23-циклопентилкарбонилокси – Факто ра А (0,25 г) дают названный сложный эфир в виде вспененного вещества (0,215 г) [a]D20+141o (c=0,63; СНCl3); lmaxCHCl3 250,2 нм (e = 19990); n max (CHBr3) 3550, 3480 (ОН), 1710 (сложный эфир); d(CDCl3) содержит 2,74 (квинтет, J 7 Гц, 1Н), 3,26 (м, 1Н), 3,91 (д, J 10 Гц, 1Н), 4,30 (т, J 7 Гц, 1Н), 4 ,95 (м, 1Н). Пример 17. 23-Циклопропилкарбонилокси-5-метоксикарбонилокси – Фактор А. Раствор 23-циклопропилкарбонилокси – Фактора А (0,1 г) в сухом дихлорметане (15 мл) охлаждают в ледяной бане и обрабатывают пиридином (0,2 мл) с последующей обработкой 1М раствором метилхлорформата в дихлорметане (0,3 мл). Через 20 минут раствор разбавляют дихлормета ном и промывают 2N соляной кислотой, затем высуши вают суль фатом натрия и выпаривают растворитель. Осадок хроматографи руют в колонке из двуокиси кремния (Merck Art 9385; 100 мл), входяще го в смесь гексана (60–80о С) с этилацетатом (3:1) и элюированным тем же самым растворителем. Соответствующие фракции основного компонента объединяют, и растворитель выпаривают с вы хо дом названного соединения в виде вспененного вещества (0,106 г) [a]D20+149o (c=0,63; CHCl 3) lmaxCHCl3 250,2 (e = 20470); n max (CHBr3) 3470 (OH), 1745 (карбонат), 1710 (сложный эфир), 998 (С–О); d(CDCl3) содержит 5,56 (с, 1Н), 5,01 (м, 1Н), 3,88 (д, J 10 Гц, 1Н), 3,83 (с, 3Н), 0,67 (д; J 7 Гц, 3Н). Пример 28. Фактор А, 5-ацетат, 23-изобутират. Раствор Факто ра А, 5-ацетат (131 мг), изобути рилхлорида (0,05 мл), пиридина (0,1 мл) и 4-диметиламинопиридина (25 мг) в сухом дихлормета не (10 мл) перемеши вают при тем пературе 20оС в те чение 16 часов. Раствор разбавляют ди хлорметаном (30 мл), промывают 2N со ляной кислотой и вы суши вают (сульфа том магния), и упаривают до суха. Оста ток очищают хроматографией на Kieselgel 60 (15 г). Элюирование из колонки смесью светлой нефти с этилацетатом (4:1) дают названное соединение (57 мг) в виде бесцветного вс 20 o пененного продукта, [a]D +159 (c. 0,61, СНСl 3), lmax (EtOH) 244 нм (e=30, 400), n max (СHBr3) 3480 (ОН), и 1713 см -1 (сложный эфир), d(СDCl3) содержит 5,5–5,6 (м, 2Н), 4,95 (м, 1Н), 3,98 (д, J 10 Гц, 1Н), 2,55 (септет J 7 Гц, 1Н), 2,17 (с, 3Н), 1,20 (д. J 7 Гц, 6Н) и 0,70 (д, J 7 Гц, 3Н). Пример 29. Фактор А, 5-23-ди-н-бутирата. Раствор Фактора А (306 мг), н-бутирового ангидрида (0,33 мл) и 4-диметиламинопиридина (244 мг) в пиридине (5 мл) перемешивают при температуре 20 оС в течение 18 часов, а затем выливают в смесь этилацетата и 2N соляной кислоты (50 мл каждого). Ор ганическую фа зу промывают 2N соляной кислотой, насыщенным бикарбонатом натрия (25 мл), и соляным раствором (25 мл), и высушивают (сульфа том магния) и упаривают досуха. Остаток очищают хроматографией на Kieselgel 60 (40 г). Элюирование из колонки смесью светлой нефти с эти лацетатом (5:1) дают названное соединение (200 мг) в виде бесцветного вспененного вещества, [a]D20+144o (c 1,13, CHCl3), lmax (EtOH), 245 нм (e=31,800), n max (CHBr3) 1720 см -1 (сложный эфир), d(CDCl3) содержит 5,5–5,6 (м, 2Н), 4,94 (м, 1Н), 3,92 (д, J 10 Гц, 1Н), 2,39 (т, J 7 Гц, 2Н), 2,28 (т, J 7 Гц 2Н), и 0,70 (д, J 7 Гц, 3Н), м/з = 752 (М+). Соединения примеров 30–32 получают аналогичным способом: Пример 30. Фактор А, 5-ацетат, 23-пивалоат (70 мг). [a ]D 20+159 o (c. 0,6 9, СНСl 3) l max (EtOH) 244 нм (e = 29 ,900), n max (CHBr) 3470 (OH) и 1730 и 1710 см-1 (сложный эфир ), d(CDCl 3) со держит 5,5– 5,6 (м, 2Н), 4 ,95 (м , 1Н), 3,9 1 (д, J 10 Гц , 1Н), 2 ,16 (с, 3Н), 1 ,22 (с, 9Н ), и 0,68 (д, J 7 Гц , 3 Н), из Факто ра А, 5-а це та та (131 мг) и пи ва лоилхло рида (0 ,06 мл). Пример 31. Фактор А, 5-ацетат, 23-бензоат (180 мг). [a]D20+153o (c, 0,59, CHCl3), lmax (EtOH) 236 нм (e = 36,200), n max (СHCl3) 3450 (ОН) и 1730 и 1707 см-1 (сложный эфир), d(CDCl3) содержит 8,21 (д, J 7 Гц, 2Н), 7,56 (т, J 7 Гц,1Н), 7,44 (т, J 7 Гц, 2Н), 5,5–5,6 (м, 2Н), 4,07 (д, J 10 Гц, 1Н), 2,17 (с, 3Н), и 0,75 (д, J 7 Гц, 3Н), из Факто ра А, 5-ацетат (327 мг) и ангидрид бензольной кислоты (339 мг). Пример 32. Фактор А, 5,23-дибензоат (370 мг) и 5-бензоат (200 мг). Из Фактора А (613 мг) и бензонхлорида (0,35 мл) начальные фракции дают названный дибензоат, [a]D20+86o (c 0,65, СHCl3), lmax (EtOH) 236 нм (а 46,200), n max (CHBr3) 3480 (OH), 1710 (сложные эфиры) и 1602 и 1585 см-1 (фе нил), d(CDCl3) содержит 8,2–8,0 (м, 4Н), 7,7–7,5 (м, 2Н), 7,46 (т, J 7 Гц, 4Н), 5,61 (с, 1Н), 4,07 (д, J 10 Гц, 1Н), и 0,76 (д, J 7 Гц, 3Н). Последующие фракции дают названный бензоат [a]D20+80o (c, 0,61, CHCl3), l max (EtOH) 237 нм (e = 39,200), n max (CHBr3) 3500 (ОН), 1712 (сложные эфиры), и 1601 и 1585 см-1 (фенил), d(CDCl3) содержит 8,09 (д, J 7 Гц, 2Н), 7,58 (т, J 7 Гц,1Н), 7,45 (т, J 7 Гц, 2Н), 5,60 (с, 1Н), 3,83 (м, 1Н), 3,76 (д, J 10 Гц, 1Н), и 0,81 (д, J 7 Гц, 3Н). Пример 33. Фактор А6 5-хло рацетат. Раствор Фактора А (123 мг) пиридина (0,1 мл) в сухом дихлорметане (5 мл) перемешивают при температуре 0оС и вводят в хлорацетилхлорид (0,3 мл 1М – раствор в ди хлорметане). Полученный раствор перемеши вают при 0оС температуре в те чение 15 минут и затем добавляют еще хло рацетилхлорид (0,1 мл 1М-раствор) и продолжают перемеши вание дополнительно в те чение 15 минут. Раствор разбавляют дихлорметаном (50 мг), затем промывают 2N со ляной кислотой (2х20 мл), и насыщенным бикарбонатом натрия (20 мл), и высуши вают (сульфатом магния), и упаривают до суха. Остаток (140 мг) очищают хроматографией в колонке из Kieselgel 60 (15 г). Элюирование из колонки смесью светлой нефти с этилацетатом (4:1) дают названное соединение (90 мг) в виде бесцветного вспененного вещества, [a]D20+143o (c, 1,11 CHCl3), l max (EtOH) нм (e=30,400), n max (CHBr3) 3500 (ОН) 1760 (хлорацетат) и 1710 см -1 (сложный эфир), d(CDCl3) содержит 5,5–5,6 (м, 2Н), 4,18 (с, 2Н), са. 3,81 (м, 1Н), 3,74 (д, J 10 Гц,1Н), и 0,80 (д, J 7 Гц, 3Н), м/з = 688 (М+, 35Cl). Соединение примеров 35, 45, 49 и 50 получают аналогичным образом. Пример 34. Фактор А, 5-хло рацетат, 23-кетон. Раствор Фактора А, 5-хлорацетата (276 мг) и дихромата пиридина (602 мг) в сухом N,N-диметилформамида (10 мл) перемешивают при температуре 20 оС в течение 48 часов и затем вливают в смесь этилацетата (50 мл) и 2N-хлористоводородной кислоты (25 мл). Органическую фазу промывают 2N-хлористоводородной кислотой и насыщенным бикарбонатом натрия, высушивают (суль фат магния) и выпаривают досуха. Остаток очищают хро матографией на колонке Kieselgel 60 (25 г). Элюирование колонки смесью петролейный эфир: этилацетат (4:1) дает названное соединение (85 мг) в качестве бесцветной пены, [a]D20+120o (c 1,05 CHCl3), lmax (EtOH) 245 нм (e=26,500), n max (CHBr3) 3480 (ОН), 1760 (хлорацетат) и 1715 см -1 (сложный эфир и кетон), d(CDCl3) включает 5,5–5,7 (м, 2Н), 4,17 (с, 2Н), 3,72 (д, J 10 Гц, 1Н), 2,50 (с, 2Н, и 0,85 (д, J 7 Гц, 3Н), м/з = 686 (М+, 35Сl). Пример 35. Фактор А, 5-бромацетат (447 мг). [a]D20+132o (с 0,77, СНCl3), lmax (EtOH) 245,5 нм (e = 31,500), n max (CHBr3) 3495 (ОН) 1730 (бром-ацетат) и 1712 см -1 (сложный эфир), d(CDCl3) включает 5,5–5,6 (м, 2Н), 3,97 (д, J 12 Гц, 1Н), 3,90 (д, J 12 Гц,1Н), 3,81 (м, 1Н), 3,75 (д, J 10 Гц, 1Н), и 0,81 (д, J 7 Гц, 3Н), из Фактора А (613 г) и бромангидрида бромуксусной кислоты (0,13 мл). Пример 36. Фактор А, 5,23-дихлорацетат. Смесь Фактора А, 5-дихлорацетата (365 мг), хлорангидрида хлоруксусной кислоты (0,21 мл) и карбоната кальция (265 мг) в сухом дихлорметане (15 мл) перемеши вают при температуре 20 о в течение 48 часов. Полученную суспензию разбавляют дихлорметаном (50 мл), затем промывают 2N-хлористоводородной кислотой и насыщенным бикарбонатом натрия, высушивают (сульфат магния) и выпаривают досуха. Оста ток очищают хроматографией на колонке Kiselgel 60 (30 г). Элюирование колонки смесью петролейный эфир: эти лацетат (4:1) дает названное соединение (167 мг) в качестве бесцветной пены, [a]D20+151o (c, 1,17, CHCl3), lmax (EtOH) 245,5 нм (e = 29,400), n max (CYBr 3) 3470 (OH) и 1725 см -1 (сложные эфиры, d(CНCl3) включает 5,5–5,6 (м, 2Н), 5,00 (м, 1Н), 4,17 (с, 2Н, 4,10 (д, J 15 Гц,1Н), 4,02 (д, J 15 Гц, 1Н), 3,92 (д, J 10 Гц, 1Н), и 0,73 (д, J 7 Гц, 3Н), м/з = 764 (М+, 35Cl). Соединение примеров 37, 38 и 46 получают аналогичным образом. Пример 37. Фактор А, 23-метоксиацетат (92 мг). [a]D20+157o (c, 0,75 CHCl 3), lmax (EtOH) 244 нм (e = 32300), n max (CHBr3) 3560 b 3480 (ОН), 1740 (метоксиацетат) и 1712 см -1 (сложный эфир, d(CDCl3) включает 5,01 (м, 1Н), 4,28 (т, J 7 Гц, 1Н), 4,01 (с, 2Н), 3,89 (д, J 10 Гц, 1Н), 3,45 (с, 3Н), и 0,71 (д, J 7 Гц, 3Н), из фактора А (184 мг) и метоксиацетилхлорида (0,3 мл). Пример 38. Фактор А, 23-феноксиацетат (112 мг). [a]D20+129o (c 0,75, CHCl3), lmax (EtOH) 244 нм (e = 25900), n max (CHBr3) 3550 и 3470 (ОН), и 1745 и 1708 см-1 (сложные эфиры), d(CDCl3) включает 7,30 (т, J 8 Гц, 2Н), 7,00 (т, J 8 Гц, 1Н), 6,94 (д, J 8 Гц, 2Н), 5,02 (м, 1Н), 4,68 (д, J 16 Гц, 1Н), 4,58 (д, J 16 Гц, 1Н), 4,30 (д, J 7 Гц, 1Н), 3,92 (д, J 10 Гц, 1Н), и 0,68 (д, J 7 Гц, 3Н), из факто ра А (184 мг) и фе ноксиацетилхлорида (0,38 мл). Пример 39. Фактор А, 5,23-ди-(2,2,2-трихлорэтил) карбонат (362 мг). [a]D22+110o (c 0,94, CHCl3), lmax (EtOH) 244 нм (e = 28000), n max (CHBr3) 3550 и 3460 (ОН), 1750 (карбонат), и 1710 см-1 (сложный эфир), d(CDCl3) включает 5,60 (с, 1Н), 4,89 и 4,75 (ABq, J 12 Гц, 2Н), 4,85 (м, 1Н), 4,81 и 4,71 (ABq, J 14 Гц, 2Н), и 0,79 (д, J 7 Гц, 3Н), из фактора А (306 мг) и 2,2,2-три хлорэтилхлорформата (0,14 мл). Пример 40. Фактор А, 23–(2,2,2-трихлорэтил) карбонат. Раствор Фактора А 5,23-ди-(2,2,2-трихлор-этил)карбоната (200 мг) в диоксане (10 мл) перемешивают при температуре приблизительно 20оС и добавляют 1М-гидроокись натрия (0,5 мл). Полученный раствор перемеши вают в течение 30 минут, затем прибавляют еще 1М-гидроокись натрия (1 мл) и перемеши вание продолжают в течение 1 часа. Раствор разбавляют этилацета том (50 мл), промывают 2N-хло ристоводородной кислотой, высуши вают (сульфат магния) и выпаривают досуха. Остаток хроматографируют на колонке Keselgel 60 (15 г). Элюирование колонки смесью петролейный эфир: эти лацетат (2:1) дает названное соединение (80 мг) в качестве бесцветной пены, [a]D20+142o (c, 0,92, CHCl3), lmax (EtOH) 245 нм (e=29200), n max (CHBr3) 3560 и 3500 (ОН), 1745 (карбонат) и 1710 см-1 (сложный эфир), d(CDCl3) включает 5,40 (с, 1Н), 4,80 и 4,70 (ABq, J 14 Гц, 2Н), 4,28 (м, 1Н), и 0,79 (д, J 7 Гц, 3Н). Пример 41. Фактор А, 5-ацетат, 23-/2,2,2-трихлорэтил/карбонат (385 мг). [a]D20+140o (c, 1,07, CHCl3), lmax (EtOH) 245,5 нм, (e = 29200), n max (CHBr3) 3300–3600 (ОН), 1738 (карбонат), и 1720 см-1 (сложный эфир), d(CDCl3) включает 5,53 (с, 1Н), 4,81 и 4,70 (ABq, J 14 Гц, 2Н), 4,84 (м, 1Н), 3,98 (д, J 10 Гц, 1Н), 2,16 (с, 3Н), и 0,79 (д, J 7 Гц, 3Н)), из фактора А, 5-ацетат (393 мг) и 2,2,2трихлорэтилхлорформата (2 мл 1М-раствора в дихлорметане). Пример 42. Фактор А. 5,23-ди-н-бутират Раствор Фактора А (306 мг), н-масляного ангидрида (0,33 мл) и 4-димети ламинопиридина (244 мг) в пиридине (5 мл) перемешивают при температуре 20оС в течение 18 часов и затем вливают в смесь этилацетата и 2N-хлористоводородной кислоты (50 мл каждого компонента). Органическую фа зу промывают 2Nхло ристоводородной кислотой, насыщенным бикарбонатом натрия (25 мл) и рассолом (25 мл), высушивают (сульфат магния) и выпаривают до суха. Остаток очищают хроматографией на колонке Kieselgel 60 (40 г). Элюирование колонки смесью петролейный эфир: этилацетат (5:1) дает названное соединение (200 мг) в качестве бесцветной пены, [a]D20+144o (c, 1,13 CHCl3), lmax (EtOH) 245 нм (e = 31800), n max (CHBr3) 1720 см-1 (сложные эфиры), d(CDCl3) включает 5,5–5,6 (м, 2Н), 4,94 (м, 1Н), 3,92 (д, J 10 Гц, 1Н), 2,39 (т, J 7 Гц, 2Н), 2,28 (т, J 7 Гц, 2Н), и 0,70 (д, J 7 Гц, 3Н), м/з = 752 (М+). Пример 43. Фактор А, 23-н-бути рат. Раствор Фактора А, 5,23-ди-н-бути рата (160 мг) в метаноле (10 мл) перемешивают при 0оС и прибавляют 1М-гидроокись натрия (0,25 мл). Полученный раствор перемешивают в течение 90 минут при температуре от 0 до 5оС, затем прибавляют еще (0,25 мл) 1М-гидроокись натрия и перемешивание продолжают в течение 4 часов. Раствор вливают в смесь этилацетата (50 мл) и 2N-хлористоводородной кислоты (25 мл), органическую фазу промывают водой, рассолом, высушивают (суль фат магния) и выпаривают досуха. Оста ток очищают хроматографией на колонке Kieselgel 60 (20 г). Элюирова ние колонки смесью петролейный эфир: этилацетат (2:1) дает названное соединение (68 мг) в качестве бесцветной пены, [a]D20+164o (c 1,03, CHCl3), lmax (EtOH) 244,5 нм (e = 29600), n max (CHBr3) 3550 и 3470 (ОН), и 1710 см -1 (сложные эфиры), d(CDCl3) включает 4,93 (м, 1Н), 4,28 (т, J 7 Гц,1Н), 3,91 (д, J 10 Гц, 1Н), 2,28 (т, J 7 Гц, 2Н), 1,65 (м, 2Н), 0,94 (т, J 7 Гц, 3Н), и 0,69 (д, J 7 Гц, 3Н), м/з = 682 (М+). Соединение примера 44 получают аналогичным образом. Пример 44. Фактор А, 23-метилкарбонат (155 мг). [a]D20+169o (c, 0,81, CHCl3), lmax (EtOH) 245 нм (26600), n max CHBr3) 3300–3610 (ОН), 1735 (карбонат), и 1710 см-1 (сложный эфир), d(CDCl3) включает 5,40 (с, Гц, 1Н), 4,28 (м, Гц, 1Н), 3,94 (д, J 10 Гц, Гц, 1Н), 3,77 (с, 3Н), и 0,76 (, J 7 Гц, 3Н), м/з = 670 (М+), из фактора А, 5-ацетата, 23-/2,2,2-трихлорэтил/карбоната (300 мг). Пример 45. 23-Деокси-Фактор А, 5-метилкарбонат (57 мг). [a]D20+152o (c 0,6, CHCl3), lmax (EtOH) 244,5 нм (e = 28200), n max (CHBr3) 3530 и 3460 (ОН), 1740 (карбонат) и 1707 см-1 (сложный эфир), d(CDCl3) включает 5,55 (с, Гц, 1Н), 3,82 (с, 3Н), 3,42 (д, J 10 Гц, Гц, 1Н), и 0,69 (д, J 4 Гц, 3Н), из 23-деокси-фактора А (90 мг) и метилхлорформата (0,3 мл 1М-раствора в дихлорметане). П ример 46 . Факто р А, 23 -хлорацета т (198 м г). [a]D20+162o (c, 1,04, CHCl3), lmax (EtOH) 245 нм (e = 28900), n max (СHBr3) 3320–3620 (ОН), 1748 (хлорацетат), и 110 см -1 (сложный эфир), d(CDCl3) включает 5,42 (с, Гц, 1Н), 4,28 (м, Гц, 1Н), 4,09 и 4,01 (ABq, J 15 Гц, 2Н), 3,91 (д, J 10 Гц, Гц, 1Н), и 0,73 (д, J 7 Гц, 3Н), м/з =688 (М+, 35Cl) из фактора А (306 мг) хлорангидрида хлоруксусной кислоты (0,2 мл). Пример 47. Фактор А, 5,23-дипропионат (387 мг). [a]D20+157o (c 0,96, CHCl 3), lmax (EtOH) 244,5 нм (e = 30200), n max (CНBr3) 3500 (ОН) и 1720 см-1 (сложные эфиры), d(CDCl 3) включает 5,52 (с, Гц, 1Н), 3,93 (д, J 10 Гц, Гц, 1Н), 2,43 (к, J 7 Гц, 2Н), 2,32 (к, J 7 Гц, 2Н), 1,18 (т, J 7 Гц, 3Н), 1,16 (т, J 7 Гц, 3Н) и 0,70 (д, J 7 Гц, 3Н), м/з = 724 (М+), из фактора А (613 мг) и пропионового ангидрида (0,5 мл). Пример 48. Фактор А, 23-пропионат (155 мг). [a]D20+168o (c 1,03, CHCl3), lmax (EtOH) 244,5 нм (e = 30600), n max (CHBr3) 3550 и 3480 (ОН), и 1710 см-1 (сложный эфир) d(CDCl3) включает 5,39 (с, 1Н), 4,27 (м, 1Н), 3,91 (д, J 10 Гц, 1Н), 2,31 (к, J 7 Гц, 2Н), 1,14 (т, J 7 Гц, 3Н) и 0,69 (д, J 7 Гц, 3Н), м/з = 668 (М+), из факто ра А, 5,23-дипропионата (327 мг), аналогичным образом для соединения примера 43. Пример 49. Фактор А, 5-метилкарбонат, 23-кетон (830 мг). [a]D20+132o (c 0,82, СHCl3), l max (EtOH) 245 нм (e = 29800), ИР и ЯМР спектры аналогичны тем, которые имеют соединение, описанное в примере 40, из факто ра А, 23-кетона (916 мг) и метилхлорфор мата (0,23 мл). Пример 50. Фактор А, 5-бензилкарбонат, 23-кетон (57 мг). [a]D20+99o (c 0,4 СHCl3), lmax (EtOH) 245,5 нм (e = 29600), n max (CHBr3) 3600= 3550 и 3480 (ОН), 1740 (карбонат) и 1715 см-1 (сложный эфир и кетон), d(CDCl3) включает 7,5–7,2 (м, 5Н), 5,57 (с, 1Н), 5,21 (с, 2Н), 3,70 (д, J 10 Гц, 1Н), 2,49 (с, 2Н), и 0,86 (д, J 6 Гц, 3Н), из фактора А, 23-кето на (92 мг) и бензилхлорформата (0,05 м). Приме р 5 1. 5-Аце ток си-23-н -буток си-фак тор А. Карбонат серебра (1 г) прибавляют к раствору 5-ацетокси факто ра А (325 мг) в сухом эфире с последующим йодбутаном (0,5 мл) и перхлоратом серебра (550 мг). Смесь перемешивают при комнатной температуре в течение 20 часов, после чего прибавляют коллидин (0,5 мл). После перемешивания в течение еще 20 минут смесь отфильтровывают и фильтрат промывают последовательно 2N хлористоводородной кислотой, насыщен ным водным раствором бикарбоната натрия и водой. Высушенную органическую фазу выпаривают почти досуха и масло очищают хро матографией на колонке Merck Kieselgel 60 с размером 230–400 меш (100 мл). Элюирование колонки смесью гексан: этилацетат (3:1) позволяет получить названное соединение в качестве бесцветной пены (276 мг, 78%), [a]D22+160o (c 0,94, СHCl3), l max (EtOH) 245 нм (emax 28300), d(CDCl3) включает 3,16 (м, 1Н), 3,43 (м, 1Н), 3,59 (м, 1Н). Пример 52. 5-Аце токси-23-циклопропилметокси – фактора А. [a]D21+174o (c 1,74, СHCl3), lmax (EtOH) 244 нм (emax 28360) n max (CHBr3) 3470 (ОН), 1732, 1710 (сложные эфиры) 998 см-1 (С–О) d(CDCl 3) включает 5,5–5,6 (м, 2Н), 3,96 (д, 10; 1Н), 3,49 (м, 1Н), 3,37 (дд, д. 6, 10, 1Н), 3,17 (дд. 6, 10, 1Н), 2,16 (с, 3Н), 0,76 (д 7, 3Н), 0,44 (м, 2Н), 0,22 (м, 2Н), из 5-ацетокси-фактора А и бромметилциклопропана, аналогичным образом для соединения примера 17. Пример 53. 5-т-Бутилдиметилсилилокси-23-метокси-фактор А. Раствор йодметилмагния в простом эфире (0,35 мл 3М-раствора) прибавляют при комнатной температуре под азотом к магнитно перемешанному раствору 5-т-бутилдиметилсилокси-факто ра А (239 мг) в сухом гексаметилфосфортриамиде (18 мл) с бурным выделением газа. Через 30 минут прибавляют йодметан (0,4 мл), и полученную смесь перемешивают в те чение 5 часов, разбавляя простым эфиром (100 мл) и промывая обильно водой. Эфирный раствор затем промывают рассолом (50 мл) и высушенную органическую фазу выпаривают. По лученную пену (280 мг) очищают хро матографией на колонке Merck Kieselgel 60 с размером 230–400 меш (80 мл). Элюирование смесью гексан: этилацетат (5:1) позволяют получить названное соединение в качестве бесцветной пены (46%), [a]D21+142o (c 1,13, СHCl3), lmax (EtOH) 244 нм (emax 27900), n max (CHBr3) 3460 (ОН), 1708 (широкий, сложный эфир) и 995 см-1 (С–О), d(CDCl3) включает 4,42 (м, 1Н), 3,92 (д 10, 1Н), 3,3–3,4 (м, 2Н), 3,32 (с, 3Н), 0,92 (с, 9Н), 0,76 (д 7, 3Н), 0,12 (с, 6Н). Соединение примера 54 получают аналогичным образом. Пример 54. 5-т-Бутилдиметилсилилокси-23-н-пропилокси–Фактор А. [a]D21 + 153o (c 1,16, СHCl3), lmax (EtOH) 244 нм (emax 29950), n max (CHBr3) 3460 (ОН), 1705 (сложный эфир) и 995 см -1 (С–О), d(CDCl3) включает 4,44 (м, 1Н), 3,96 (д 10, 1Н), 3,56 (м, 1Н), 3,45 (м, 1Н), 3,14 (м, 1Н), 0,93 (с, 9Н), 0,75 (д 7, 3Н), 0,13 (с, 6Н), из 5-т-б утилдиметилсилилокси-фактора А и 1-йодпропана. Пример 55. 5-Ацетокси–фактор D (923 мг). [a]D21 + 143,9o (c 0,9, СHCl3), lmax (EtOH) 239 (28700) и 245 нм (emax 31000), n max (CHBr3) 3490 (ОН) 1730 и 1710 (сложный эфир), d(CDCl3) 0,81 (д, 7 Гц, 3Н), 0,99 (д, 7 Гц, 3Н), 1,00 (т, 7 Гц, 3Н), 1,53 (с, 3Н), 1,59 (с, 3Н), 1,75 (с, 3Н), 2,16 (с, 3Н), 3,32 (м, 1Н), 3,65 (м, 1Н), 4,04 (д, 6 Гц, 1Н), и 5,53 (м, 2Н), м/з = 640 (М+), из фактора D )2,5 г) и уксусного ангидрида (0,47 мл), аналогичным образом для соответствующе го соединения примера 5. Приме р 5 6. 5 ,23-Диа цеток си-фак тор D (286 мг). Температура плавления 147–149o ромбы из пентана в простом эфире [a]D21+152,5o (c, 0,9, СHCl3), lmax (EtOH) 232,5 (21500), 238 (26600), и 244,5 нм (emax 28800), n max (CHBr3) 1720 см-1 (сложный эфир, d(СHCl3), 0,72 (д, 6 Гц, 3Н), 0,99 (д, 6 Гц, 3Н), 1,01 (т. 7 Гц, 3Н), 1,53 (с, 3Н), 1,60 (с, 3Н), 1,75 (с, 3Н), 2,02 (с, 3Н), 2,15 (с, 3Н), 3,31 (м, 1Н), 4,04 (д, 6 Гц, 1Н), 4,91 (м, 1Н), и 5,5–5,6 (м, 2Н), м/з = 682 (М+), из фактора D (439 мг) и уксусного ангидрида (0,25 мл), аналогичным образом для соединения примера 5. Пример 57. 23-Ацетокси–фактор D (127 мг) [a]D21+150o (c 0,5, СHCl3), lmax (EtOH) 238 (29200) и 244,5 нм (emax 15600), n max(CHBr3) 3300, 3590 (ОН) и 1710 см -1 (сложный эфир), d(CDCl3) 0,72 (д, 7 Гц, 3Н), 0,99 (д, 7 Гц, 3Н), 1,01 (т, 7 Гц, 3Н), 1,53 (с, 3Н), 1,60 (с, 3Н), 1,86 (с, 3Н), 3,26 (м, Н), 3,95 (д, 6 Гц, 1Н), 4,26 (т, 6 Гц, 1Н) и 4,91 (м, 1Н), м/з = 640 (М+), из 5,23-диацетокси–фактора D (207 мг), аналогичным образом для соединения примера 8. Пример 58. 5-Аце токси, 23-кето-фактор D (152 мг). Температура плавления 228–230о [a]D21+84o (c 0,6, CHCl3), lmax (EtOH) 244,5 нм (emax 31100), n max (CHBr3) 3500 (ОН), 1732 и 1714 см -1 (сложный эфир и кетон), d(CDCl3) 0,86 (д, 6 Гц, 3Н), 0,98 (д, 6 Гц, 3Н), 1,00 (т, Гц, 3Н), 1,49 (с, 3Н), 1,67 (с, 3Н), 1,74 (с, 3Н), 2,14 (с, 3Н), 3,33 (м, 1Н), 4,03 (д, 6 Гц, 1Н), и 5,5–5,6 (м, 2Н), м/з = 638 (М+). Из 5-ацетокси-фактора D (336 мг), аналогичным образом для соединения примера 10. Пример 59. 23-Кето–фактор D (59 мг). [a]D21+84o (c 0,4, СНСl3), lmax (EtOH) 244,5 нм (emax 28000), n max (CHBr3) 3550 и 3500 и 1712 см -1 (сложный эфир и кетон), d(CDCl3) 0,86 (д, 6 Гц, 3Н), 0,98 (д, 7 Гц, 3Н), 1,00 (т, 7 Гц, 3Н), 1,50 (с, 3Н), 1,69 (с, 3Н), 1,86 (с, 3Н), 3,27 (м, 1Н), 3,73 (д, 10 Гц, 1Н), 3,95 (д, 6 Гц, 1Н), и 4,27 (т, 6 Гц, 1Н), из 5-ацетокси-23-кетофактора D (96 мг), аналогичным образом для соединения примера 11. Приме р 60 . Фак тор А, 23-фе нилаце тат (240 мг). [a]D20+140o (c 0,92, CHCl3), l max (CHBr3) 3550 и 3470 (ОН) и 1730 см -1 (сложные эфиры), d(CDCl3) включает 7,32 (с, 5Н), 5,0 (м, 1Н), 4,29 (т, J 7 Гц, 1Н), 3,88 (д, J 10 Гц, 1Н), 3,62 (с, 2Н), и 0,54 (д, J 7 Гц, 3Н), получают способом, аналогичным для соединения примера 38 из факто ра А (306 мг) и фенилацетилхлорида (0,33 мл). Пример 61. 23-Этокси– фактор А. Раствор 5-ацетокси-23-этокси фактора А (806 мг) в метаноле (18 мл) охлаждают в ледяной бане, затем вводят 1N водный раствор гидроокиси натрия (1,3 мл) и полученный раствор светло-желтого цвета перемеши вают в ледяной бане в течение 1,25 часа. Раствор разбавляют этилацетатом (80 мл), затем последовательно промывают 1N хлористо водородной кислотой, водой и соляным раствором. Высушенную органическую фазу вы паривают и полученную в ре зультате это го смолу очи щают хро матографией на Merck Kieselgel 60 с размером частиц двуо киси кремния 230–400 меш. (200 мл). Элюирование из колонки 15% этилацетатом в дихлорметане дает названное соединение в ви де бесцветной пены (623 мг). [a]D21 + 178 o (c, 1,13, CHCl 3) lmax (EtOH) 244 нм (emax 29400), d(СDCl3) включает 4,29 (т, 7, 1Н), 3,65 (м, 1Н), 3,47 (м, 1Н), 3,26 (м, 2Н), 1,15 (т, 7, 3Н). Соединения примеров 123–126 получены аналогичным образом. Пример 62. 23-н-Бутокси–фактор А (61%) получают в виде бесцветной пены [a]D21+161o (c. 1,47 CHCl3) lmax (EtOH) 244 нм (e 33100), d(CDCl3) включает 4,30 (т. 7, 1Н), 3,60 (м, 1Н), 3,43 (м, 1Н), 3,17 (м, 1Н) из 5-ацетокси-23-н-бутокси Фактора А. Пример 63. 23-н-Пропокси Фактор А (83%) получают в виде бесцветной пены [a]D21+165o (c, 1,01, CHCl3) lmax (EtOH) 2,45 нм (e 30970), d(CDCl3) включает 4,29 (т, 7, Н), 3,55 (м, 1Н), 3,44 (м, 1Н), 3,13 (м, 1Н) из 5-ацетокси-23-н-пропокси Фактора А. Пример 64. 23-Метокси Фактор А (66%) получают в виде бесцветной пены [a]D21+175o (c, 1,01 CHCl 3) lmax (EtOH) 244 нм (e 19100) d(CDCl3) включает 4,29 (т, 7, 1Н), 3,40 (м, 1Н), 3,33 (с, 3Н) из 5-ацетокси-23метокси – Факто ра А. Пример 65. 23-Циклопентилокси Фактор А (75%) получают в ви де бесцветной пены [a]D21+160o (c, 1,65, CHCl3) lmax (EtOH) 244 нм (e19,800), d(CDCl3) вк лючает 4,29 (т, 7, 1Н), са 3,96 (невидимый м, 1Н), 3,95 (д, 5, 1Н), 3,91 (д, 10, 1Н), 3,45 (м, 1Н). 0,69 (д, 7, 3Н) из 5-ацетокси-23-циклопентилокси Фактора А. Пример 66. (а) 5-ацетокси-23-аллилокси – Фактор А. Салицилат се ребра (872 мг) до бавляют к раство ру 5-аце токси Фак тора А (207 мг) аллилйоди да (1,0 мл) в сухом простом эфи ре (25 мл), и получен ную смесь перемеши вают при комнатной температуре в те че ние 4 дней, после че го филь труют. Филь трат вы паривают с по луче нием желтого масла, которое очищают хро матографией на колонке Merck Kieselgel 60, с размером частиц двуо киси кремния 230–400 меш. Элюи рова ние смесью дихлорметан с эти лаце татом (19:1 ) дае т наз ванное соединение в ви де бесцветной пены (105 мг), [a]D21+152 o (c 1,00 , CHCl3) l max (EtOH) 245 нм (e 28400), d(СDCl 3) включает 3,54 (м, 1Н), 4,14 (м, 1Н). Аналогичным образом получают сле дующие соединения: (b) 5-ацетокси-23-н-пропилокси Фактор А получают из 5-ацетокси – Факто ра А и н-пропилйодида. При очистке хроматографией на колонке Merck Kieselgel 60, с размером частиц двуо киси кремния 230–400 меш, и элюировании смесью гексана с этилацетатом (3:1) получают названное соединение в виде бесцветной пены [a]D22+160o (c 0,75, CHCl 3) lmax (EtOH) 245 нм ( 27600), d(CDCl3) включает 3,33 (с, 3Н), 3,39 (м, 1Н). (с) 5-ацетокси-23-метокси-Фактора А получают из 5-ацетокси Фактора А и метилйодида. Названное соединение получают в виде бесцветной пены [a]D22+159o (c 0,98, CHCl3) lmax (EtOH) 245 нм (e 27600), d(CHCl3) включает 3,33 (с, Н), 3,39 (м, 1Н). Пример 67. 5-Ацетокси-23-этокси–Фактора А получают из 5-ацетокси–Фактора А и этилйодида. Названное соединение выхо дит в ви де бесцветной пены [a]D22+167o (c 1,02, CHCl3) lmax (EtOH) 245 нм ( 28070), d(СDCl3) включает 3,25 (м, 1Н), 3,46 (м, 1Н), 3,64 (м, 1Н), получаемое способом, аналогичным для соединения примера 17. (1) Активность в соотношении Caenorhabditis elegans. Cоединения по изобретению были испытаны на воздействие, оказываемое ими на свободно живущи х нематод Caenorhabditis elegans. Приготовили порции раствора или суспензии этих соединений в метаноле, а также серию растворов в 20% пропиленгликоле (400 мкг/мл и ниже). 10 мкл каждого из получивши хся растворов смешали с 20 мкл водного раствора, содержаще го 100–200 червей Caenorhabditis elegans и выдерживали при комнатной температуре в те чение 20 часов. В вышеприведенном испытании соединения, приведенные в примерах, в основном, были эффективны и после 20 часов привели к гибели или парализова ли более 98% червей при концентрациях менее 10 мг/мл. (2) Активность в отноше нии Nematospiroides dubius. Эффективность соединений, приготовленных в соответствии с изобрете нием, была испытана на мышах, зараженных глистами Nematos-piroides dubius. Мышам женской особи CR/H (18–22 г) ввели 100 L 3 личинок глистов Nematos-piroides dubius и оставили до полного развития инфекции (обычно на три недели). Затем мышам однократно перорально ввели соединение в соответствии с изобретением в количестве, равном одному из 3/4 уровней доз. Соединения вводили в виде раствора пропиленгликоля. После этого мышей оставили по меньшей мере на 3 дня (обычно 5 дней), затем мышей с развившейся в них инфекцией умертвили и уда лили тонкую кишку. Удаленную часть кишки разрезали ножницами с тупыми концами для обнажения слизистой оболочки тонкой кишки. Глисты взрослых особей собрали при помощи модифи цированного устройства Баэрмана. Время миграции соста вило 5 часов, причем во время этого периода мигрирующие глисты поддерживали при температуре 37оС. Спустя пять часов нейлоновую сетку, через которую мигрировали глисты, исследовали под увеличительным стеклом (Х2). Подсчитали количество глистов, уловленных сеткой и проникших че рез нее, получив общее содержание глистов у каждой мыши, которое сравнили с контрольными мышами. Таким образом, было обнаружено, что доза 10 мг/кг соединений в соответствии с изобретением значительно уменьшила содержание глистов у мышей, принимавших препарат. Например, каждое соединение из Примеров 6, 8, 11, 14, 23, 25, 26, 28, 38, 43, 45, 52, 60–63, 66(а), 66(b), и 67 дало снижение среднего процента содержания глистов у мышей, подвергнутых ле чению, в сравнении с контрольными мышами, больше чем на 80%. (3) Активность по отноше нию к множеству экто- и эндопаразитов в различных моделях с использованием животных. (а) Однократная доза (800 мг/кг) соединения из Примеров 14 и 61, введенная внутривенно или подкожно, полностью избавила от Сooperia oncophera телят, инфи цированных этими глистами. (b) Лечение кроликов соединением из Примера 14 в течение 5 дней убило 97% личинок Rhinocephalus appendiculatus. (с) Соединения из примеров 14 и 61 проявили полную эффективность при пероральном введении в дозе 200 мкг/кг лоша дям, инфицированным Parascaries. (d) Оба соединения из Примеров 14 и 61 при пероральном введении в дозе 200 мкг/кг были эффективны как ивермектин (Ivermectin) против личинок и взрослых особей Nematodirus batters и против взрослых особей Ostertagia circumcincta, Trichostrongylus axei, Haemonchus contortus, Nematodirus spathiger, Cooperia curticei и Trichostrongylus vitrinus (е) Соединения из Примеров 14 и 61 прояви ли полную эффективность при пероральном введении в дозе 150 мкг/кг собакам, инфи цированным Toxocara. (f) Вве денная подкожно доза (800 мкг/кг) соединений из Примеров 14 и 61 имела 98% эффективность при лечении свиней, инфи цированных Oesophagostomum. Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrolide antibiotics or salts thereof, method for obtaining thereof (variants), composition for animal and human treatment, composition and method for combating agricultural pests
Автори англійськоюWard John Berri, Noble Heizel Mary, PORTER Hil, Fletton Richard Alan, NOBLE David, Satherland Derek Ronald, Remsey Michael John
Назва патенту російськоюМакролидные антибиотики или их соли, способ их получения (варианти), композиция для лечения животных и людей, композиция и способ борьбы с вредителями сельского хозяйства
Автори російськоюВард Джон Берри, Нобл Хейзел Мери, ПОРТЕР Нил, Флеттон Ричард Алан, НОБЛ ДевИд, Сазерленд Дерек Рональд, Ремсей Майкл Винсент Джон
МПК / Мітки
МПК: A01P 5/00, A01N 43/32, A01P 7/02, A61K 31/365, A01P 3/00, C07D 493/22
Мітки: лікування, боротьби, людей, солі, сільського, макролідні, спосіб, шкідниками, одержання, господарства, тварин, антибіотики, композиція, варіанти
Код посилання
<a href="https://ua.patents.su/22-27261-makrolidni-antibiotiki-abo-kh-soli-sposib-kh-oderzhannya-varianti-kompoziciya-dlya-likuvannya-tvarin-i-lyudejj-kompoziciya-i-sposib-borotbi-z-shkidnikami-silskogo-gospodarstva.html" target="_blank" rel="follow" title="База патентів України">Макролідні антибіотики або їх солі, спосіб їх одержання (варіанти), композиція для лікування тварин і людей, композиція і спосіб боротьби з шкідниками сільського господарства</a>
Похідні піримідину, що проявляють гербіцидні властивості, спосіб їх одержання (варіанти), проміжні сполуки, гербіцидна композиція та спосіб боротьби із бур’янами

Номер патенту: 26408
Опубліковано: 30.08.1999
Автори: Ріо Йосіда, Масахіро Сато, НОРІХІРО Кавамуро, Масатосі Тамару, Фуміакі Такабе, Січехіко Татікава
МПК: C07C 249/00, A01P 13/00, A01N 43/54, C07D 239/28, C07C 251/40
Мітки: властивості, сполуки, варіанти, гербіцидна, боротьби, гербіцидні, проміжні, похідні, одержання, композиція, спосіб, проявляють, піримідину, бур'янами
Формула / Реферат:
1. Производные пиримидина общей формулыгде R - низшая алкилтиогруппа, низшая диалкиламиногруппа, имидазольная группа или группа формулы OR3, где R3 - атом водорода, низшая алкильная группа, которая может быть замещена цианогруппой, низшей алкоксильной группой, низшей алкилтиогруппой, фенильной группой, фенилтиогруппой, фенилсульфинильной группой, фенилсульфонильной группой, бензилоксильной группой, низшей алкилкарбонилоксильной...
Похідні n-заміщеного 4-феніл-4-піперидинкарбоксаміду та їх фармацевтично прийнятні солі, що проявляють анестезувальну та анальгезувальну дію, спосіб їх одержання та фармацевтична композиція

Номер патенту: 26403
Опубліковано: 30.08.1999
Автори: Аск Анна-Лена, Сандберг Руне
МПК: A61P 29/00, A61K 31/451, C07D 211/52, C07D 211/64, A61P 25/04, A61K 31/445
Мітки: 4-феніл-4-піперидинкарбоксаміду, n-заміщеного, одержання, анальгезувальну, анестезувальну, проявляють, дію, похідні, спосіб, прийнятні, солі, композиція, фармацевтична, фармацевтично
Формула / Реферат:
1. Производные N-замещенного 4-фенил-4-пиперидинкарбоксамида общей формулыгде R1 представляет собой алкильную группу с 2 - 6 атомами углерода или алкоксиалкильную группу R4O(CH2)m, в которой R4 представляет собой алкильную группу с 1 - 4 атомами углерода и m равно 2 - 4;R2 и R3 являются одинаковыми или различными и каждый представляет собой алкильную группу с числом атомов углерода до 6, или R2 и R3 образуют вместе цепь...
Спосіб одержання препарату для лікування паразитарних захворювань тварин
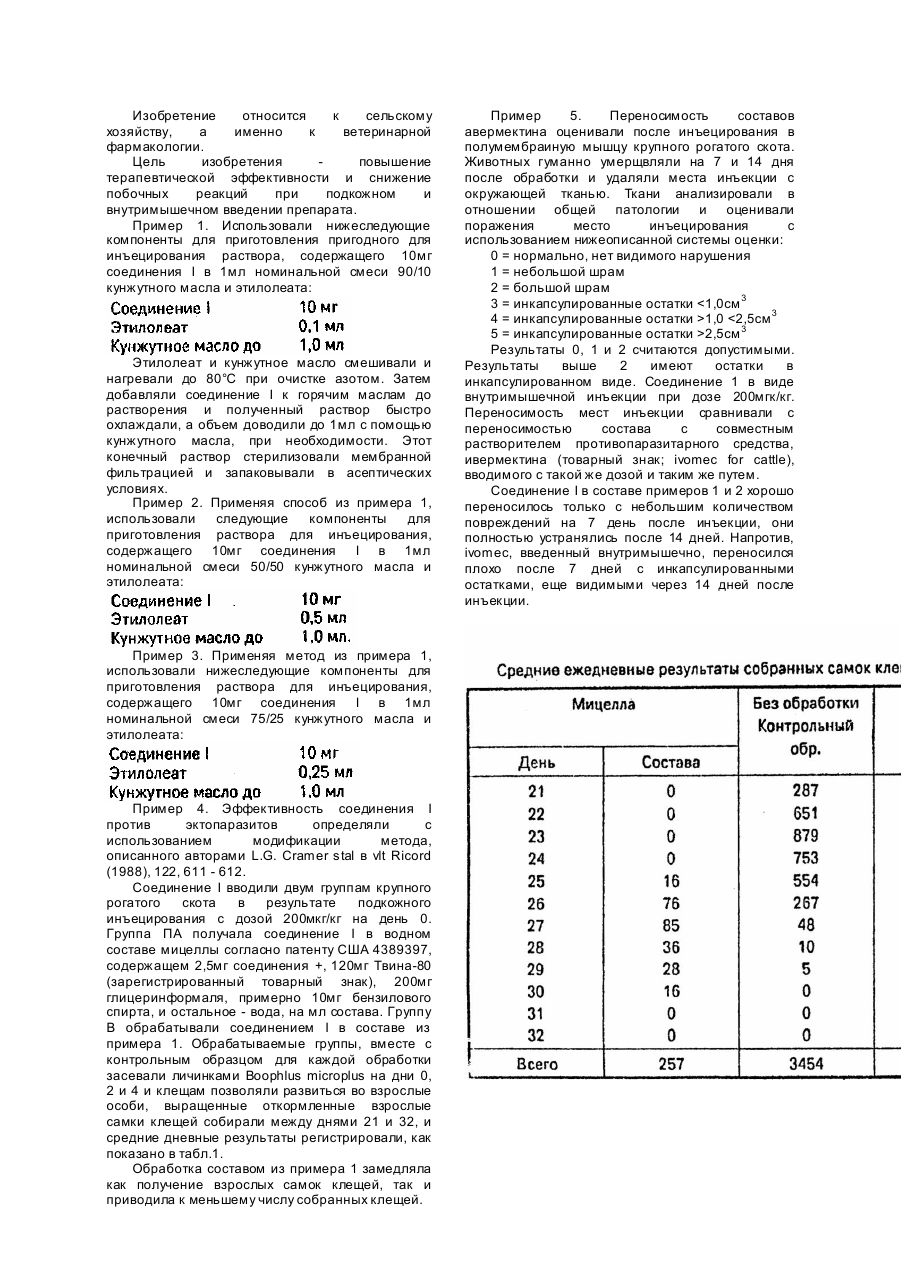
Номер патенту: 18250
Опубліковано: 25.12.1997
Автори: Едвард Девісон, Стефен Річард Вікс
МПК: A61K 31/70, A61K 47/44, A61P 33/00
Мітки: паразитарних, захворювань, спосіб, тварин, препарату, лікування, одержання
Формула / Реферат:
1. Способ получения препарата для лечения паразитарных заболеваний животных, включающий растворение вещества из группы авермектинов в растворителе, отличающийся тем, что в качестве авермектина используют 25-циклогексилавермектина В1, а в качестве растворителя берут состав, содержащий 50 - 90об.% кунжутного масла, этилолеат - остальное.2. Способ по п.1, отличающийся тем, что препарат содержит 10мг/мл 25-циклогексилавермектина В1.
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
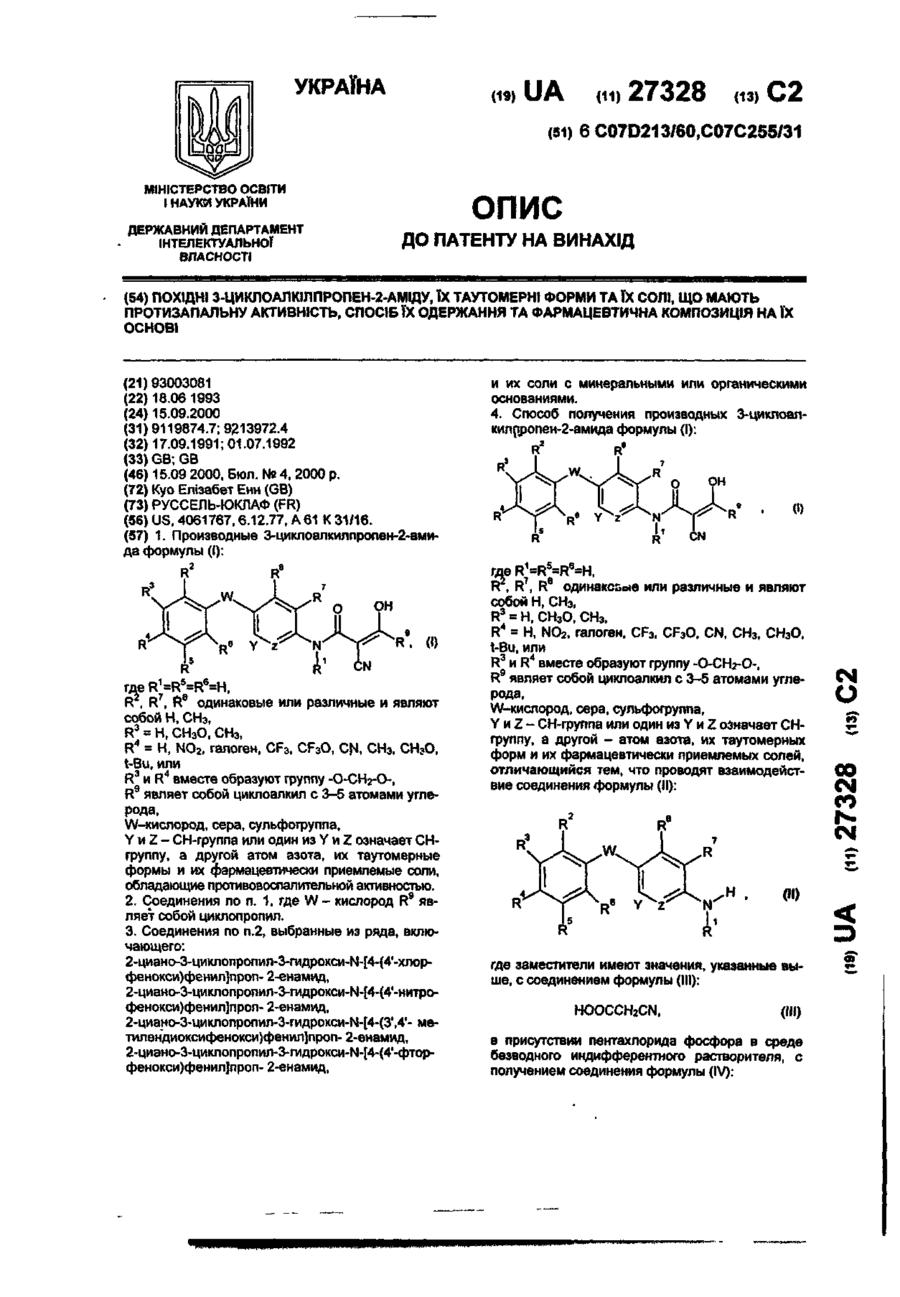
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: основі, похідні, 3-циклоалкілпропен-2-аміду, таутомерні, спосіб, мають, активність, протизапальну, форми, композиція, солі, фармацевтична, одержання
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...
Похідні 3,4-дигідроізохіноліну або їх фармацевтично прийнятні солі, спосіб їх одержання і фармацевтична композиція з інгібуючою трансмембранний перехід кальцію в клітинах активністю
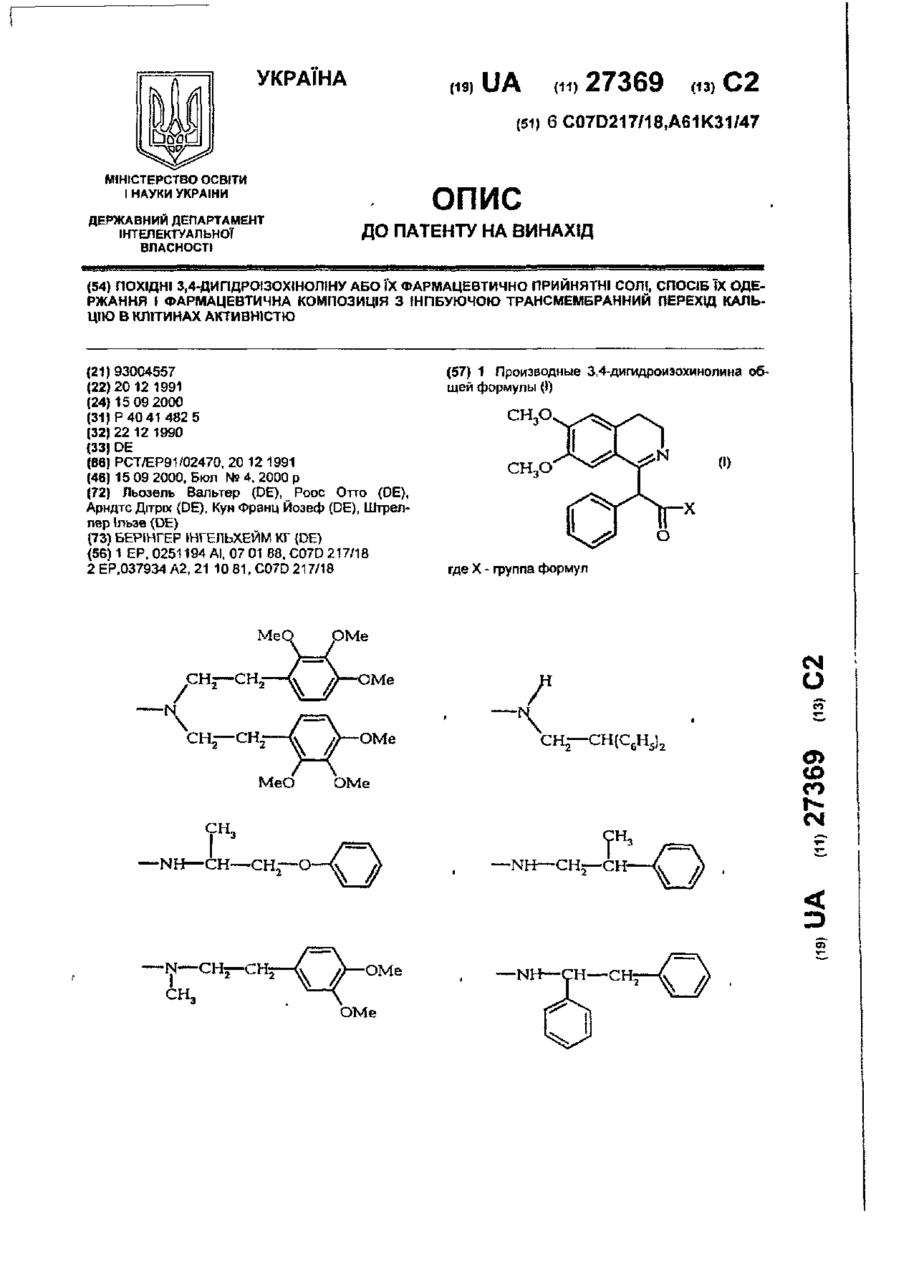
Номер патенту: 27369
Опубліковано: 15.09.2000
Автори: Арндтс Дітріх, Льозель Вальтер, Роос Отто, Штреллер Ільзе, Кун Франц-Йозеф
МПК: A61K 31/496, A61P 25/28, A61P 7/02, A61K 31/535, A61K 31/44, A61K 31/47, A61K 31/445, A61K 31/4725, A61P 9/00, A61K 31/475, A61K 31/54, A61K 31/4741, C07D 217/18, A61P 11/06, A61K 31/437, A61K 31/5377, A61K 31/4365, A61K 31/495, A61K 31/472, A61P 29/00, C07D 217/12, A61K 31/4745, A61K 31/435
Мітки: кальцію, солі, прийнятні, фармацевтично, трансмембранний, перехід, похідні, активністю, інгібуючою, клітинах, 3,4-дигідроізохіноліну, одержання, спосіб, фармацевтична, композиція
Текст:
...FMLP), или изопреналином) "перегрузку кальцием" и гибель клетки (значение KTso (= концентрация торможения) находится в области 1 мкмоль). Измеряли полумаксимальную концентрацию торможений соединения примера, которую ингибируют индуцируемый тринуклеотид ом FMLP (1 наномоль) переход кальция на клетках HL-60 (10 6 клеток в суспензии}, содержащих FLUO3. Она составляет 5,4 • 106 моль. На отдельных клетках HL-60 с использованием...
Попередній патент: Спосіб безокисної термічної обробки прокату з вуглецевих марок сталі та сплавів на основі титану
Наступний патент: Адаптогенний засіб “силлард”
Випадковий патент: Конвеєрний перевантажувальний агрегат