Аналоги 15-дезоксиспергуаліну, спосіб їх одержання, фармацевтична композиція та проміжні сполуки
Номер патенту: 37217
Опубліковано: 15.05.2001
Автори: Рено Патріція, Самрес Сос, Дютартре Патрік, ЛЕБРЕТОН Люк, Деррепас Філіпп




Формула / Реферат
І. Аналоги 15-дезоксиспергуалина общей формулы (І):
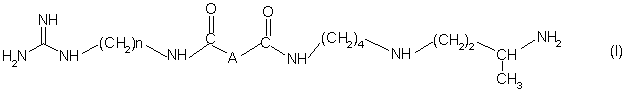 в которой:
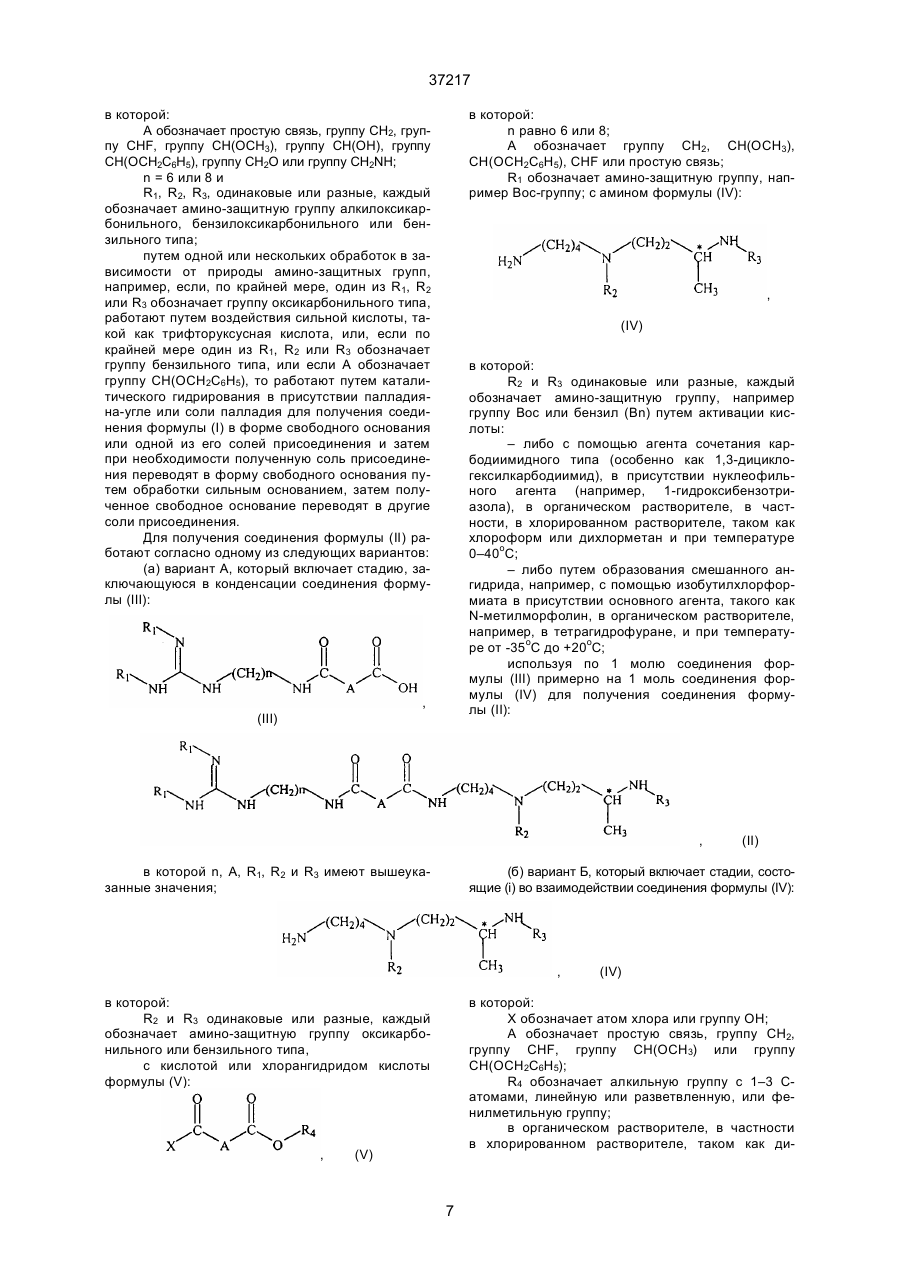
в которой:
А обозначает простую связь, группу –CH2-, группу –СН2О-, группу –CH2NH-, группу -СH(ОН)-, группу -СНF - или группу -СН(ОСНз)-;
"п" равно 6 или 8 и
их соли присоединения.
2. Соединения по п.І формулы І, в которой А обозначает группу СН2 или группу СН2О, и его соли присоединения.
3. Соединения по п.І формулы І, в которой С обозначает углерод конфигурации (R) или (R, S) и его соли присоединения.
4. Соединение по п.І, представляющее собой 2-{[[4-[/3-(амино)бутил/амино]бутил]амино]карбонилокси}-N-[6-/(аминоиминометил)-амино/гексил]ацетамид–трис (трифторацетат).
5. Соединение по п. І, представляющее собой 2-{[[4-[/3(R)-(амино)бутил/амино]бутил]амино]карбонилокси}-N-[6-/(аминоиминометил)амино/гексил]ацетамид–трис (трифторацетат).
6. Способ получения соединения формулы (I) или одной из его солей присоединения по п.1, отличающийся тем, что он включает удаление защитных групп из соединения формулы (II):
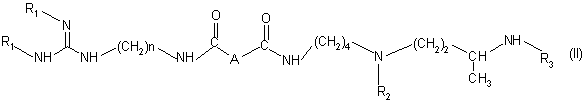
в которой:
А обозначает простую связь, группу –CH2-, группу -CHF, - группу CH(OCH3), группу CH(OH), группу CH(OCH2C6H5), группу СН2О или группу CH2NH;
"п" равно 6 или 8;
R1, R2, R3, одинаковые или разные, каждый обозначает амино- защитную группу алкилоксикарбонильного, бензилоксикар-бонильного или бензильного типа;
причем вышеуказанное удаление защитных групп включает в частности:
(а) обработку вышеуказанного соединения формулы (II) с помощью сильной кислоты, если, по крайней мере, один из R1, R2 или R3, обозначает группу оксикарбонильного типа; и/или
(б) каталитическое гидрирование вышеуказанного соединения формулы (II), если, по крайней мере, один из R1, R2 или R3, обозначает группу бензильного типа или если А обозначает группу CH(OCH2C6H5),для получения соответствующего соединения формулы (I) в виде свободного основания или одной из его солей присоединения.
7. Способ по п. 6, отличающийся тем, что перед снятием защитных групп соединение формулы (II) получают согласно одному из вариантов:
(а) вариант A, в котором (i) осуществляют конденсацию соединения формулы (III):
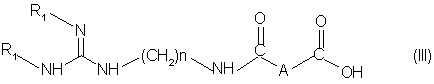
в которой:
"п" равно 6 или 8;
А обозначает группу OH2, CH(OCH3), CH(ΟCH2-C6Η5), CHF или простую связь;
R1 обозначает амино-защитную группу, с амином формулы (ІV):
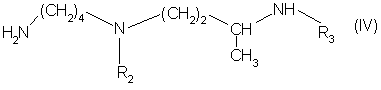
в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу, путём активации кислоти:
- либо с помощью агента сочетания карбодиимидного типа в присутствии нуклеофильного агента, в органическом растворителе и при температуре 0-40°С;
- либо за счет образования смешанного ангидрида, особенно с помощью изобутилхлорформиата в присутствии основного агента, в органическом растворителе и при температуре от -35°С до +20°С;
причем используют по I молю соединения формулы (III) примерно на I моль соединения формулы (ІV) для получения соединения формулы (II):
в которой "п", А, R1, R2, и R3, имеют вышеуказанные значения;
(іі) удаляют защитные группы из соединения формулы (II), получаемого на стадии (і) посредством одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получения соединения формулы (І), в которой:
А обозначает простую связь, СH2, СHF, СHОСН3 или СН(ОH);
"п" равно 6 или 8;
(iii) и при необходішостп получают соединение фортлулы (І) в виде свободного основания путем обработки сильным основанием, затем полученное свободное основание переводят в другие соли присоединения;
(б) вариант Б, в κοτοром
(i) осуществляет взаимодействие соединения пформулы (ІV):
в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу оксикарбонильного или бензильного типа;
с кислотой или хлорангидридом кислоты формулы (V):
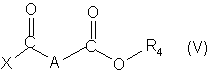
которой:
Χ обозначает атом хлора или группу ОН;
А обозначает простую связь, группу СН2, группу СНF, группу
CH(OCH3) или группу CH(OCH2C6H5);
R4 обозначает алкильную группу с 1-3 С-атомами, линейную или разветвлённую, или фенилметильную группу;
в органическом растворителе в присутствии активатора карбоксильной группы и нуклеофильного агента, когда X обозначает группу ОН, или в присутствии третичного амина, когда Х обозначает атом хлора при температуре 0-40°С, причем используют по I молю соединения формулы (IV) примерно на I моль соединения формулы (V)
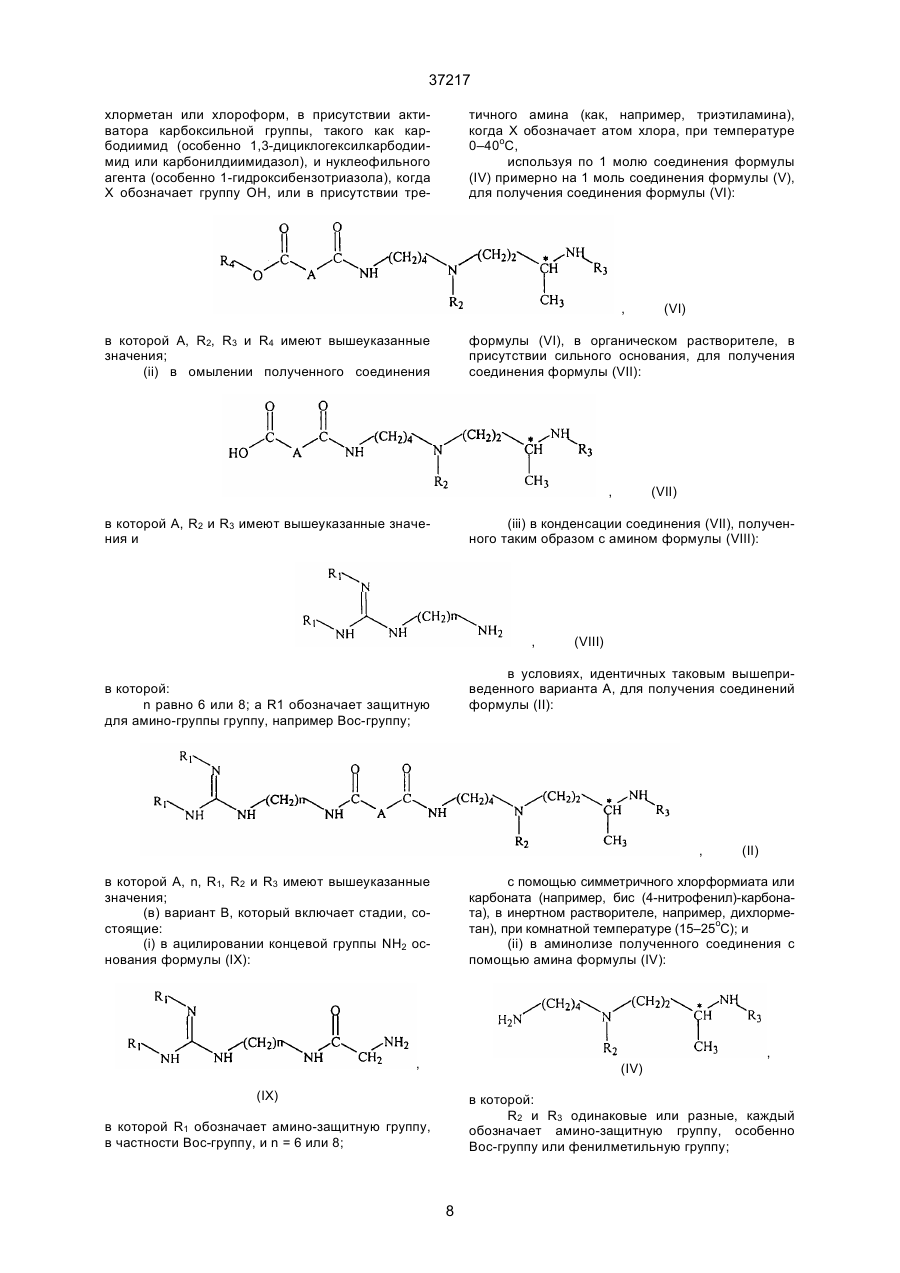
для получения соединения формулы (VI):
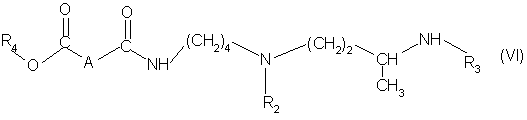
в которой А, R2, R3 и R4 имеют вышеуказанные значеная;
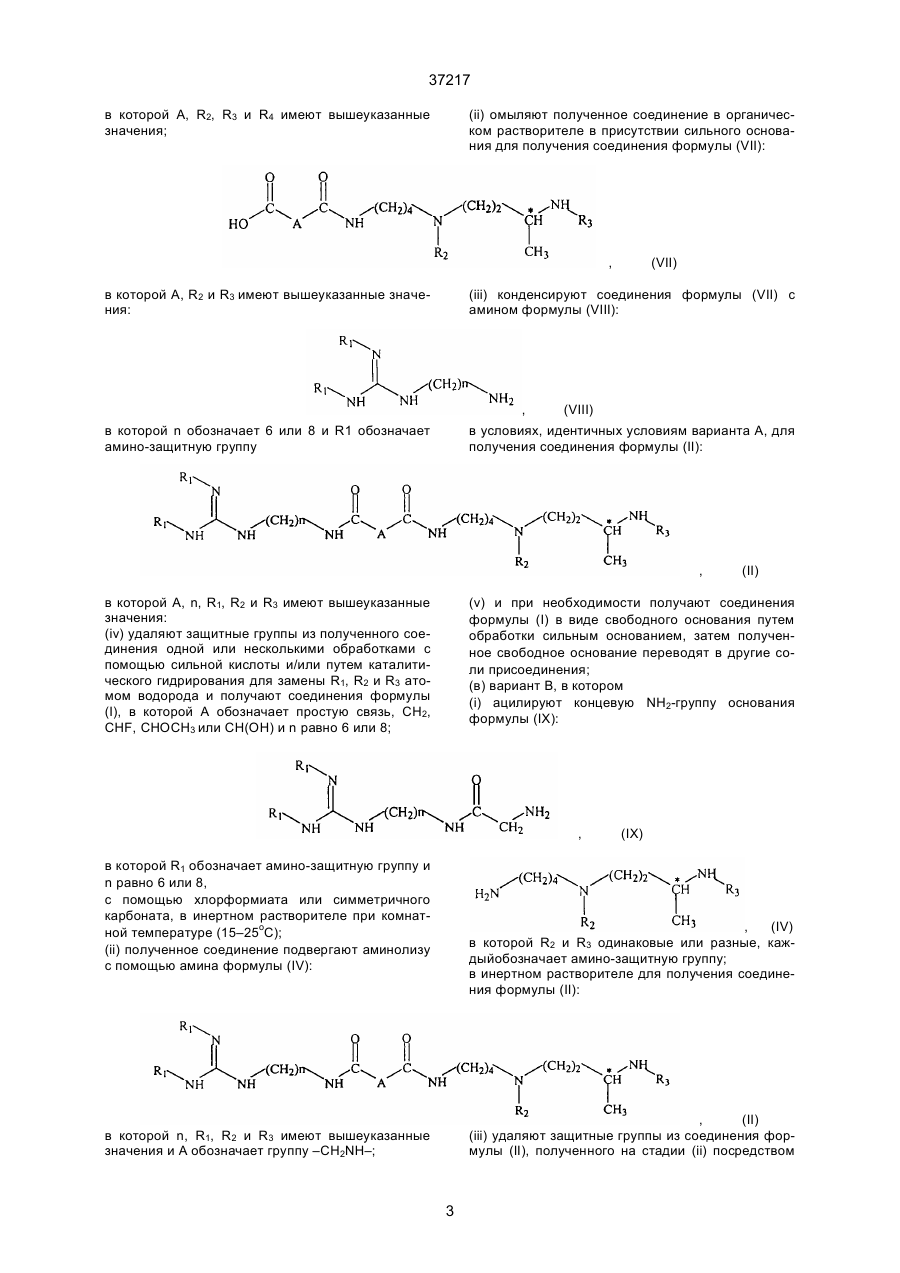
(іі) омыляют полученное соединение в органическом растворителе в присутствии сильного основания для получения соединения формулы (VII):
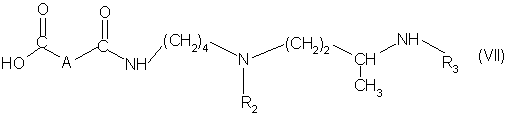
в которой А, R2 и R3 имеют вышеуказанные значения:
(iii) конденсируют соединения формулы (VII) с амином формулы (VIII):
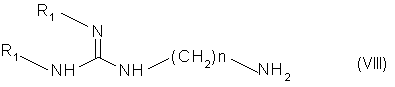
в которой "п" обозначает 6 или 8 и R1 обозначает амино-защитную группу в условиях, идентичных условиям варианта А для получения соединения формулы (II):
в которой А, "п", R1, R2, и R3 имеют вышеуказанные значения:
(iV) удаляют защитные группы из полученного соединения одной или несколькими обработками с помощью сильной кислоты и/или путём каталитического гидрирования для замены R1, R2, и R3 атомом водорода и получают соединения формулы (І), в которой А обозначает простую связь, CH2, СНF, CHOCH3 или СН(ОН) и "п" равно 6 или 8;
(\/) и при необходімости получают соединения формулы (I) в
виде свободного основания путём обработки сильным основанием, затем полученное свободное основание переводят в другие соли присоединения;
(в) вариант В, в котором
(і) ацилируют концевую NН2-группу основания формулы (IX):
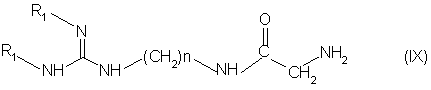
в которой R1 обозначает амино-защитную группу и "п" равно 6 или 8,
с помощью хлорформиата или симетричного карбоната, в инертном растворителе при комнатной температуре (15-25°С);
(іі) полученное соединение подвергают аминолизу с помощью амина формулы (IV):
в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу;
в инертном растворителе для получения соединения форгмулы (ІІ):
в которой "п", R1, R2 и R3 имеют вышеуказанные значения и А обозначает группу –CH2NH-;
(iii) удаляют защитные группы из соединения формулы (П), полученного на стадии (ii) посредством одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получают соединения формулы (I), в которой A обозначает группу СН2NH и "п" равно 6 или 8 , и
(iV), если необходимо, получают соединение формулы (I) в форме свободного основания путем обработки сильным основанием и полученное свободное основание переводят в другие соли присоединения; и
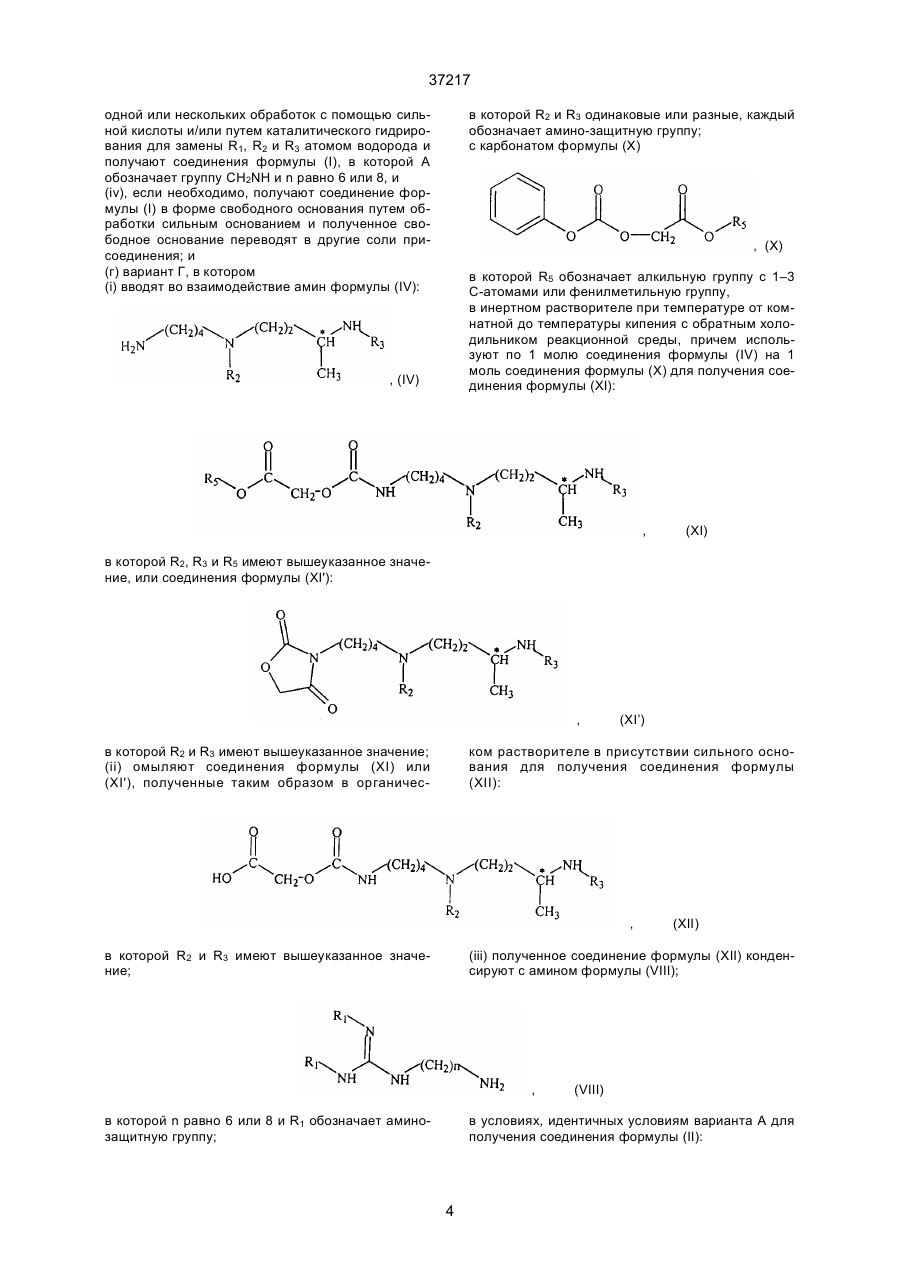
(г) вариант Г, в котором
(i) вводят во взаимодействие амин форцулы (iV):
в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу;
с карбонатом формулы (X)
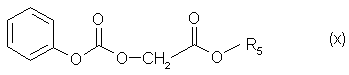
в которой R5 обозначает алкильную группу с 1-3 С-атомами или фенилметильную группу,
в инертном растворителе при температуре от комнатной до температуры кипения с обратным холодильником реакционной среды, причем используют по 1 молю соединения формулы (IV) на 1 моль соединения формулы (X) для получения соединения формулы (XI):
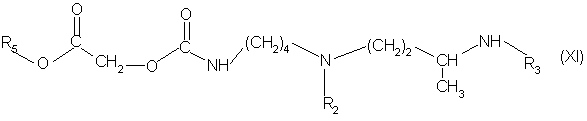
в которой R2, R3 и R5 имеют вышеуказанное значение, или соединения формулы (XI*):
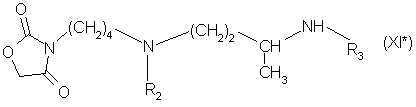
в которой R2 и R3 имеют вышеуказанное значение;
(ii) омыляют соединения формулы (ХI) или (XI*), полученные такім образом в органическом растворителе в присутствии сильного основания для получения соединения формулы (XII):
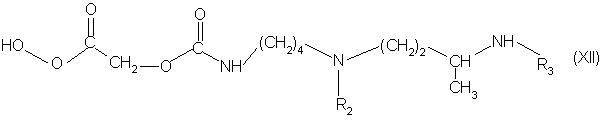 в которой R2 и R3 имеют вышеуказанное значение;
в которой R2 и R3 имеют вышеуказанное значение;
(iii) полученное соединение формулы (XII) конденсируют с амином формулы (VIII);
в которой "п" равно 6 или 8 и R1 обозначает амино-защитную группу;
в условиях, идентичных условиям варианта А для получения соединения формулы (II):
в которой "п", R1, R2 и R3 имеют вышеуказанные значения и А обозначает группу CН2О;
(iv) удаляют защитные группы из полученного соединения формулы (II) посредством одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гндрирования для замены R1, R2 и R3 атомом водорода и получают соединения формулы (I), в которой А обозначает группу CH2O и "п" равно 6 или 8; и
(v) если необходимо получают соединения формулы (I) в форме свободного основания обработкой сильным основанием, затем полученное свободное основание переводят в другие соли присоединения.
8. Фармацевтическая композиция, обладающая иммунодепрес-сивной активностью, содержащая активное начало и фармацевти-ческие добавки, отличающаяся тем, что в качестве активного начала она содержит, по крайней мере, одно соединение: формулы (І) в эффективном количестве.
9. Соединения формулы (II):
в которой:
А обозначает простую связь, группу СН2, группу CHF, группу
СН(ОН), группу CH(OCH3), группу CH(OCH2C6H5), группу СН2О
или группу CН2NН;
"п" равно 6 или 8;
R1, R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу алкилоксикарбонильного, бензилоксикарбонильного или бензильного типа в качестве промежуточного агента в синтезе соединений формулы (І).
Текст
1. Аналоги 15-дезоксиспергуалина общей формулы (I): C2 4. Соединение по п. 1, представляющие собой 2{[[4-[/3-(амино)бутил/амино]бутил]амино]карбонилокси}-N-[6-/(аминоиминометил)-амино/гексил]ацетамид-трис(трифторацетат). 5. Соединение по п. 1, представляющее собой 2{[[4-[/3-(R)-(амино)бутил/амино]бутил]амино]карбонилокси}-N-[6-/(аминоиминометил)амино/гексил] ацетамид-трис(трифторацетат). 6. Способ получения соединения формулы (I) или одной из его солей присоединения по п. 1, отличающийся тем, что он включает удаление защитных групп из соединения формулы (II): в которой: А обозначает простую связь, группу –СН2–, группу –СНF–, группу СН(OCH3), группу СН(ОН), группу СН(ОСН2С6Н5), группу СН2О или группу СH2NH; n равно 6 или 8; R1, R2, R3, одинаковые или разные, каждый обозначает аминозащитную группу алкилоксикарбонильного, бензилоксикарбонильного или бензильного типа; Ю (ІІ) причем вышеуказанное удаление защитных групп включает в частности: (а) обработку вышеуказанного соединения формулы (II) с помощью сильной кислоты, если, по крайней мере, один из R1, R2 или R3 обозначает группу оксикарбонильного типа; и/или (б) каталитическое гидрирование вышеуказанного соединения формулы (II), если, по крайней мере, один из R1, R2 или R3 обозначает группу (19) , UA (11) в которой: А обозначает простую связь, группу –СН2–, группу –СН2О–, группу –СН2NH–, группу –СН(ОН)–, группу –СHF– или группу –СН(ОСН3)–; n равно 6 или 8, и их соли присоединения. 2. Соединения по п. 1 формулы (I), в которой А обозначает группу СН2 или группу СН2О, и его соли присоединения. 3. Соединения по п. 1 формулы (I), в которой *C обозначает углерод конфигурации (R) или (R, S) и его соли присоединения. (І) (13) , 37217 (21) 95028166 (22) 17.02.1995 (24) 15.05.2001 (31) 9402125, 9406706 (32) 24.02.1994, 01.06.1994 (33) FR, FR (46) 15.05.2001, Бюл. № 4, 2001 р. 37217 бензильного типа или если А обозначает группу СН(ОСН2С6Н5), для получения соответствующего соединения формулы (I) в виде свободного основания или одной из его солей присоединения. 7. Способ по п. 6, отличающийся тем, что перед снятием защитных групп соединение формулы (II) получают согласно одному из вариантов: (а) вариант А, в котором (i) осуществляют конденсацию соединения формулы (III): с амином формулы (IV): , (IV) в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу, путем активации кислоты: – либо с помощью агента сочетания карбодиимидного типа в присутствии нуклеофильного агента, в органическом растворителе и при температуре 0– 40оС; – либо за счет образования смешанного ангидрида, особенно с помощью изобутилхлорформиата в присутствии основного агента, в органическом растворителе и при температуре от -35оС до +20оС; причем используют по 1 молю соединения формулы (III) примерно на 1 моль соединения формулы (IV) для получения соединения формулы (II): , (ІІІ) в которой: n равно 6 или 8; А обозначает группу СН2, СН(ОСН3), СН(ОСН2– С6Н5), CHF или простую связь; R1 обозначает амино-защитную группу, , (II) нильного или бензильного типа; с кислотой или хлорангидридом кислоты формулы (V): в которой n, А, R1, R2 и R3 имеют вышеуказанные значения; (ii) удаляют защитные группы из соединения формулы (II), получаемого на стадии (i) посредством одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получения соединения формулы (I), в которой: А обозначает простую связь, СН2, CHF, CHOCH3 или СН(ОН); n равно 6 или 8; (iii) и при необходимости получают соединение формулы (I) в виде свободного основания путем обработки сильным основанием, затем полученное свободное основание переводят в другие соли присоединения; (б) вариант Б, в котором (i) осуществляют взаимодействие соединения формулы (IV): , (V) в которой: X обозначает атом хлора или группу ОН; А обозначает простую связь, группу СН2, группу СНF, группу СН(ОСН3) или группу СН(ОСН2С6Н5); R4 обозначает алкильную группу с 1–3 С-атомами, линейную или разветвленную, или фенилметильную группу; в органическом растворителе в присутствии активатора карбоксильной группы и нуклеофильного агента, когда Х обозначает группу ОН, или в присутствии третичного амина, когда Х обозначает атом хлора при температуре 0–40оС, причем используют по 1 молю соединения формулы (IV) примерно на 1 моль соединения формулы (V) для получения соединения формулы (VI): , (IV) в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу оксикарбо , 2 (VI) 37217 в которой А, R2, R3 и R4 имеют вышеуказанные значения; (ii) омыляют полученное соединение в органическом растворителе в присутствии сильного основания для получения соединения формулы (VII): , (VII) в которой А, R2 и R3 имеют вышеуказанные значения: (iii) конденсируют соединения формулы (VII) с амином формулы (VIII): в которой n обозначает 6 или 8 и R1 обозначает амино-защитную группу в условиях, идентичных условиям варианта А, для получения соединения формулы (II): , (VIII) , в которой А, n, R1, R2 и R3 имеют вышеуказанные значения: (iv) удаляют защитные группы из полученного соединения одной или несколькими обработками с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получают соединения формулы (I), в которой А обозначает простую связь, СН2, CHF, CHOCH3 или СН(ОН) и n равно 6 или 8; (II) (v) и при необходимости получают соединения формулы (I) в виде свободного основания путем обработки сильным основанием, затем полученное свободное основание переводят в другие соли присоединения; (в) вариант В, в котором (i) ацилируют концевую NH2-группу основания формулы (IX): , в которой R1 обозначает амино-защитную группу и n равно 6 или 8, с помощью хлорформиата или симметричного карбоната, в инертном растворителе при комнатной температуре (15–25оС); (ii) полученное соединение подвергают аминолизу с помощью амина формулы (IV): (IX) , (IV) в которой R2 и R3 одинаковые или разные, каждыйобозначает амино-защитную группу; в инертном растворителе для получения соединения формулы (II): , (II) (iii) удаляют защитные группы из соединения формулы (II), полученного на стадии (ii) посредством в которой n, R1, R2 и R3 имеют вышеуказанные значения и А обозначает группу –СН2NH–; 3 37217 одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получают соединения формулы (I), в которой А обозначает группу СН2NH и n равно 6 или 8, и (iv), если необходимо, получают соединение формулы (I) в форме свободного основания путем обработки сильным основанием и полученное свободное основание переводят в другие соли присоединения; и (г) вариант Г, в котором (i) вводят во взаимодействие амин формулы (IV): в которой R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу; с карбонатом формулы (X) , (X) в которой R5 обозначает алкильную группу с 1–3 С-атомами или фенилметильную группу, в инертном растворителе при температуре от комнатной до температуры кипения с обратным холодильником реакционной среды, причем используют по 1 молю соединения формулы (IV) на 1 моль соединения формулы (Х) для получения соединения формулы (XI): , (IV) , (XI) в которой R2, R3 и R5 имеют вышеуказанное значение, или соединения формулы (XI'): , в которой R2 и R3 имеют вышеуказанное значение; (ii) омыляют соединения формулы (XI) или (XI'), полученные таким образом в органичес (XI’) ком растворителе в присутствии сильного основания для получения соединения формулы (XII): , в которой R2 и R3 имеют вышеуказанное значение; (XII) (iii) полученное соединение формулы (XII) конденсируют с амином формулы (VIII); , в которой n равно 6 или 8 и R1 обозначает аминозащитную группу; (VIII) в условиях, идентичных условиям варианта А для получения соединения формулы (II): 4 37217 , в которой n, R1, R2 и R3 имеют вышеуказанные значения и А обозначает группу СН2О; (iv) удаляют защитные группы из полученного соединения формулы (II) посредством одной или нескольких обработок с помощью сильной кислоты и/или путем каталитического гидрирования для замены R1, R2 и R3 атомом водорода и получают соединения формулы (I), в которой А обозначает группу СН2О и n равно 6 или 8; и (v) если необходимо, получают соединения формулы (I) в форме свободного основания обработ (II) кой сильным основанием, затем полученное свободное основание переводят в другие соли присоединения. 8. Фармацевтическая композиция, обладающая иммунодепрессивной активностью, содержащая активное начало и фармацевтические добавки, отличающаяся тем, что в качестве активного начала она содержит, по крайней мере, одно соединение формулы (I) в эффективном количестве. 9. Соединение формулы (II): , в которой: А обозначает простую связь, группу СН2, группу CHF, группу СН(ОН), группу СН(ОСН3), группу СН(ОСН2С6Н5), группу СН2О или группу СН2NH; n равно 6 или 8; (II) R1, R2, R3 одинаковые или разные, каждый обозначает амино-защитную группу алкилоксикарбонильного, бензилоксикарбонильного или бензильного типа в качестве промежуточного агента в синтезе соединений формулы (I). ______________________________ 15-дезоксиспергуалина различными a- или wаминокислотами, или модифицируя цепь, несущую гуанидиновую функцию. Примеры таких модификаций изложены в европейских заявках на патент А–0181592 или А–0105193. В настоящем изобретении предлагаются новые продукты, общая структура которых остается аналогичной 15-дезоксиспергуалину, которые химически стабильны и обладают иммунодепрессивной активностью, выше таковой известных продуктов уровня техники. Продукты согласно изобретению отличаются в частности от известных продуктов уровня техники инверсией амидной связи, соединяющей гуанидино-гексильный остаток с центральной цепью молекулы, природой центральной цепи и введением разветвленной цепи, содержащей хиральный центр в спермидиновой части молекулы. Аналоги 15-дезоксиспергуалина, согласно изобретению, соответствуют соединениям формулы (I): Настоящее изобретение относится к новым соединениям структуры, аналогичной (близкой) 15-дезоксиспергуалину. Оно также относится к способу их получения и их применению в фармкомпозиции в качестве иммунодепрессивных агентов. Известно, что 15-дезоксиспергуалин, первоначальные исследования которого относились к противоопухолевой активности, обладает хорошей активностью в области иммунодепрессии. Эта активность констатируется в многочисленных публикациях, особенно: "Deoxyspergualin in lethal murine graft-versus-host disease", Transplantation, т. 51, 712–715, № 3 (март, 1991 г.) и "15deoxyspergualin: From Cytostasis to Immunosuppression", Behring Inst. Mitt., № 82, 231–239 (1988). Однако, 15-дезоксипергуалин не обладает химической стабильностью, поэтому пытались разработать более стабильные производные, например, заменяя a-гидроксиглициновый остаток , 5 (I) 37217 в которой: А обозначает простую связь, группу –СН2–, группу –СН2O–, группу –СН2NH–, группу –СН(ОН)– , группу –CHF или группу –СН(ОСН3); n равно 6 или 8 и их солям присоединения. В формуле (I) и в других формулах (т.е. (II), (IV), (VI), (XI), (XI') и (XII), которые следуют ниже, *C обозначает асимметрический атом углерода (другая номенклатура: "хиральный атом углерода"). Согласно изобретению, способ получения соединений формулы (I) и их солей присоединения включает удаление защитных групп из соединений формулы (II): , в которой: А и n имеют вышеуказанные значения и R1, R2 и R3, одинаковые или разные, каждый обозначает защитную группу аминной функции; путем одной или нескольких реакций, известных специалисту, для замены всех групп R1, R2 и R3 атомом водорода. Изобретение относится также к использованию соединений формулы (I) и их нетоксичных аддитивных солей в фармацевтических композициях, предназначенных для использования в терапии против иммунных нарушений или против малярии, или в качестве фармакологического реагента. Под аддитивными солями понимают соли присоединения кислоты, получаемые за счет взаимодействия неорганической или органической кислоты с соединением формулы (I). Предпочтительными неорганическими кислотами для солеобразования являются соляная, бромоводородная, серная и фосфорная кислоты. Предпочтительными для солеобразования органическими кислотами являются фумаровая, малеиновая, метансульфоновая, щавелевая, лимонная и трифторукусусная кислоты. Принимая во внимание наличие асимметрического атома углерода, вышеобозначенного как *C, и природу группы А, соединения формулы (I) могут содержать один или два хиральных атома углерода. Когда А обозначает –СН2–, СН2О– или – СН2NH–, настоящее изобретение охватывает из соединений формулы (I) рацематы, где *C имеет конфигурацию (R, S), и энантиомеры, где *C имеет конфигурацию (R) или (S). Когда A обозначает – СН(ОН)–, –СНF– или –СН(ОСН3)–, настоящее изобретение охватывает соединения формулы (I), которые содержат два хиральных положения, получаемые в виде эквимолекулярной смеси четырех диастереоизомеров, "полурацематов" (R, S)-A(R)-*C, (R, S)-A-(S)-*C, (R)-A-(S, R)-*C и (S)-A-(S, R)-*C и каждого из четырех диастереоизомеров. (II) Практически предпочитают, чтобы асимметрический атом углерода, обозначаемый как *C, имел конфигурацию (R, S) или (R). Соединения формулы (I) можно получать само по себе известными способами на основе классических реакций, таких как образование амидной связи, а также на основе методов химии пептидов. Способ получения, который предлагается согласно изобретению, включает, как указано выше, удаление защитных групп из соединения формулы (II). Практически защитные группы R1, R2, R3, которые нужно заменить на атом водорода, представляют собой группы, защищающие аминофункции и известны в области химии пептидов для временного блокирования неполностью замещенных "аминных" функций. Из групп, которые пригодны для этой цели, можно использовать: (а) группы оксикарбонильного типа, как например, алкоксикарбонильные и бензилоксикарбонильные группы: Вос = трет. – бутилоксикарбонил / или (1,1диметилэтокси) карбонил/ Fmoc = 9-флуоренилметоксикарбонил; Foc = фурфурилоксикарбонил; Z = бензилоксикарбонил; Z(n – Cl) = 4-хлорбензилоксикарбонил или Z(n – OMe) = 4-метоксибензилоксикарбонил; (б) группы бензильного типа, например: Bn = фенилметил. Из этих амино-защитных групп предпочтительными группами являются Вос и Вn. Практически, способ получения соединения формулы (I) или одной из его солей присоединения отличается тем, что он включает стадии, заключающиеся в удалении защитных групп из соединения формулы (II): , 6 (II) 37217 в которой: А обозначает простую связь, группу СН2, группу CHF, группу СН(ОСН3), группу СН(ОН), группу СН(ОСН2С6Н5), группу СН2О или группу СН2NH; n = 6 или 8 и R1, R2, R3, одинаковые или разные, каждый обозначает амино-защитную группу алкилоксикарбонильного, бензилоксикарбонильного или бензильного типа; путем одной или нескольких обработок в зависимости от пpиpоды амино-защитных групп, например, если, по крайней мере, один из R1, R2 или R3 обозначает группу оксикарбонильного типа, работают путем воздействия сильной кислоты, такой как трифторуксусная кислота, или, если по крайней мере один из R1, R2 или R3 обозначает группу бензильного типа, или если А обозначает группу СН(ОСН2С6Н5), то работают путем каталитического гидрирования в присутствии палладияна-угле или соли палладия для получения соединения формулы (I) в форме свободного основания или одной из его солей присоединения и затем при необходимости полученную соль присоединения переводят в форму свободного основания путем обработки сильным основанием, затем полученное свободное основание переводят в другие соли присоединения. Для получения соединения формулы (II) работают согласно одному из следующих вариантов: (а) вариант А, который включает стадию, заключающуюся в конденсации соединения формулы (III): в которой: n равно 6 или 8; А обозначает группу СН2, СН(ОСН3), СН(ОСН2С6Н5), CHF или простую связь; R1 обозначает амино-защитную группу, например Вос-группу; с амином формулы (IV): , (IV) в которой: R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу, например группу Вос или бензил (Bn) путем активации кислоты: – либо с помощью агента сочетания карбодиимидного типа (особенно как 1,3-дициклогексилкарбодиимид), в присутствии нуклеофильного агента (например, 1-гидроксибензотриазола), в органическом растворителе, в частности, в хлорированном растворителе, таком как хлороформ или дихлорметан и при температуре 0–40оС; – либо путем образования смешанного ангидрида, например, с помощью изобутилхлорформиата в присутствии основного агента, такого как N-метилморфолин, в органическом растворителе, например, в тетрагидрофуране, и при температуре от -35оС до +20оС; используя по 1 молю соединения формулы (III) примерно на 1 моль соединения формулы (IV) для получения соединения формулы (II): , (III) , в которой n, A, R1, R2 и R3 имеют вышеуказанные значения; (б) вариант Б, который включает стадии, состоящие (i) во взаимодействии соединения формулы (IV): , в которой: R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу оксикарбонильного или бензильного типа, с кислотой или хлорангидридом кислоты формулы (V): , (II) (IV) в которой: Х обозначает атом хлора или группу ОН; А обозначает простую связь, группу СН2, группу CHF, группу СН(ОСН3) или группу СН(ОСН2С6Н5); R4 обозначает алкильную группу с 1–3 Сатомами, линейную или разветвленную, или фенилметильную группу; в органическом растворителе, в частности в хлорированном растворителе, таком как ди (V) 7 37217 хлорметан или хлороформ, в присутствии активатора карбоксильной группы, такого как карбодиимид (особенно 1,3-дициклогексилкарбодиимид или карбонилдиимидазол), и нуклеофильного агента (особенно 1-гидроксибензотриазола), когда Х обозначает группу ОН, или в присутствии тре тичного амина (как, например, триэтиламина), когда Х обозначает атом хлора, при температуре 0–40оС, используя по 1 молю соединения формулы (IV) примерно на 1 моль соединения формулы (V), для получения соединения формулы (VI): , в которой А, R2, R3 и R4 имеют вышеуказанные значения; (ii) в омылении полученного соединения (VI) формулы (VI), в органическом растворителе, в присутствии сильного основания, для получения соединения формулы (VII): , в которой А, R2 и R3 имеют вышеуказанные значения и (VII) (iii) в конденсации соединения (VII), полученного таким образом с амином формулы (VIII): , (VIII) в условиях, идентичных таковым вышеприведенного варианта А, для получения соединений формулы (II): в которой: n равно 6 или 8; а R1 обозначает защитную для амино-группы группу, например Вос-группу; , в которой А, n, R1, R2 и R3 имеют вышеуказанные значения; (в) вариант В, который включает стадии, состоящие: (i) в ацилировании концевой группы NH2 основания формулы (IX): (II) с помощью симметричного хлорформиата или карбоната (например, бис (4-нитрофенил)-карбоната), в инертном растворителе, например, дихлорметан), при комнатной температуре (15–25оС); и (ii) в аминолизе полученного соединения с помощью амина формулы (IV): , , (IV) (IX) в которой: R2 и R3 одинаковые или разные, каждый обозначает амино-защитную группу, особенно Вос-группу или фенилметильную группу; в которой R1 обозначает амино-защитную группу, в частности Вос-группу, и n = 6 или 8; 8 37217 в инертном растворителе, таком как дихлор метан, для получения соединения формулы (II): , (II) в которой n, R1, R2 и R3 имеют вышеуказанные значения и А обозначает группу –СН2–NH–; и (г) вариант Г, который включает стадии, состоящие: (i) во взаимодействии амина формулы (IV): , (X) в которой R5 обозначает алкильную группу с 1–3 С-атомами или фенилметильную группу, в инертном растворителе (особенно ароматическом растворителе, как, например, толуол), при температуре от комнатной до температуры рефлюкса реакционной смеси, используя по 1 молю соединения формулы (IV) примерно на 1 моль соединения формулы (X), для получения соединения формулы (XI): , (IV) в которой R2 и R3 одинаковые или разные, каждый обозначает защитную для амино-группы группу; с карбонатом формулы (Х): , в которой R2, R3 и R5 имеют вышеуказанное значение, (XI) или соединения формулы (XI'): , в которой R2 и R3 имеют вышеуказанное значение; (ii) в омылении полученного соединения формулы (XI) или (XI') в органическом раство (XI’) рителе в присутствии сильного основания для получения соединения формулы (XII): , в которой R2 и R3 имеют вышеуказанное значение и (iii) в конденсации соединения формулы (XII) c амином формулы (VIII): (XII) , 9 (VIII) 37217 в которой n равно 6 или 8 и R1 обозначают аминозащитную группу, особенно Вос-группу; в условиях, идентичных таковым, описанным для варианта А, приведенного выше, для получения соединения формулы (II): , (II) промывают с помощью 60 мл диэтилового эфира. Объединенные фильтраты концентрируют при пониженном давлении и полученный остаток обрабатывают с помощью 100 мл дихлорметана. Полученный раствор экстрагируют с помощью 50 мл 1М соляной кислоты; полученную водную кислую фазу промывают 2 раза 50 мл дихлорметана, затем подщелачивают путем медленного добавления 50 мл 5 н. раствора гидроксида натрия. Продукт экстрагируют из водной фазы 3 раза по 50 мл дихлорметана. Полученную органическую фазу сушат над карбонатом калия, затем концентрируют при пониженном давлении. Полученную желтую жидкость (7 г) затем перегоняют в вакууме, получая 6,02 г (выход=87%) целевого продукта. Т.кип. = 160–170оС /0,05 мм рт.ст. (0,05 мм рт.ст. соответствуют примерно 0,066 Па). Приготовление 2. 3-/N-(3-цианопропил)-N-(фенилметил)амино/-1- метилпропиловый эфир метансульфокислоты. Готовят раствор 9,11 г (37 × 10-3 моль) полученного согласно приготовлению 1 продукта в 150 мл безводного диэтилового эфира и охлаждают до 0оС. После этого добавляют 11,23 г (111 × 10-3 моль) триэтиламина, затем прикапывают 4,66 г (40 × 10-3 моль) метансульфонилхлорида. По окончании добавления перемешивают при 0oС в течение 1 часа, затем в течение 15 часов при комнатной температуре. Медленно добавляют 120 мл насыщенного раствора бикарбоната натрия, затем реакционную среду экстрагируют 3 раза по 50 мл диэтилового эфира. Органические фазы объединяют и сушат над карбонатом калия, после чего концентрируют при пониженном давлении. Таким образом получают 11,7 г (выход=98% в расчете на сырой продукт) целевого продукта в виде желтого вязкого масла. Продукт используют без дополнительной очистки в следующей стадии, однако его можно очищать путем хроматографии на диоксиде кремния и элюирования с помощью смеси н-гептана с диэтиловым эфиром (7:3 по объему). 1 Н-ЯМР-спектр (CDCl3): 1,33 (д., 3Н); 1,76 (м., 3Н); 1,86 (м., 1Н); 2,37 (т., 2Н); 2,53 (м., 4Н); 2,89 (с., 3Н); 3,48 (д., 1Н); 3,57 (д., 1Н); 4,84 (м., 1Н); 7,28 (м., 5Н). Приготовление 3. 3-/N-(3-цианопропил)-N-(фенилметил)-амино/-1- метил-1-азидопропан. Готовят раствор 10,59 г (33 × 10-3 моль) продукта из приготовления 2 в 70 мл диметилсульфоксида и добавляют 6,43 г (100 × 10-3 моль) азида натрия. Реакционную смесь перемешивают в течение 15 часов при 45–50оС, затем охлаждают; добавляют 100 мл воды, затем экстрагируют с по в которой n, R1, R2 и R3 имеют вышеуказанные значения, а А обозначает группу СН2О. Изобретение подробнее поясняется нижеследующими примерами и результатами фармакологических испытаний, получаемыми при использовании соединений согласно изобретению, по сравнению с результатами, получаемыми с известными продуктами уровня техники. Используемая в примерах номенклатура соответствует таковой, предлагаемой в Chemical Abstracts; согласно этой номенклатуре сложный моноэфир алкандикислоты с трет-бутанолом называется 1,1диметилэтиловый эфир алкандикислоты, а сложный диэфир типа трет.-бутил- и этил-алкандиоата называется здесь (1,1-диметилэтил)-этиловый эфир алкандикислоты. В экспериментальной части приготовления относятся к промежуточным продуктам, а примеры относятся к продуктам согласно изобретению. Когда соединения содержат в своей структуре асимметрический атом углерода, то отсутствие специального указания означает, что речь идет о практически эквимолекулярной смеси двух энантиомеров (рацемической смеси). Когда те же соединения обозначаются указанием (R) или (S) непосредственно после идентификации положения заместителя, то это означает, что содержащий этот заместитель углерод имеет (R) или (S)-конфигурацию, согласно правилам Cahn, Ingold и Prelog. Когда соединения содержат в своей структуре два центра асимметрии, то отсутствие специального указания означает, что речь идет о смеси четырех диастереоизомеров. Спектральные характеристики сигналов ядерного магнитного резонанса (ЯМР) даются для протона (1Н) или для изотопа 13 углерода (13С): указывается химсдвиг по отношению к сигналу ТМС, форма сигнала (с. для синглета; д. для дублета; т. для триплета; к. для квадруплета; м. для мультиплета; с.ш. – для сигнала уширенного) и число протонов, относящихся к сигналу. Для сведения 1Н-ЯМР-спектры снимают при 300 мГц. Приготовление 1. 4/N-(3-гидроксибутил)-N-(фенилметил)-амино/-бутаннитрил. 5 г (28 × 10-3 моль) 3-/N-(фенилметил)амино/-1-метилпропанола растворяют в 60 мл бутанола и добавляют 3,56 г (34 × 10-3 моль) карбоната натрия, 1,06 г (7 × 10-3 моль) иодида калия, затем 7,4 г (70 × 10-3 моль) 4-хлорбутиронитрила в виде раствора в 10 мл бутанола. Реакционную смесь при перемешивании кипятят с обратным холодильником в течение 20 часов. После охлаждения отфильтровывают и нерастворимые части 10 37217 1,05 г (8 × 10-3 моль) моноэтилового эфира малоновой кислоты растворяют в 15 мл безводного хлороформа, затем охлаждают до 0оС и добавляют 1,65 г (8 × 10-3 моль) 1,3-дициклогексилкарбодиимида и 0,108 г (0,8 × 10-3 моль) 1-гидроксибензотриазол-гидрата. После перемешивания в течение 0,5 часа при 0оС, добавляют раствор 1,5 г (4,19 × 10-3 моль) бис-(1,1-диметилэтилового эфира [[(6-аминогексил)имино]-метилен]-бис-карбаминовой кислоты в виде раствора в 5 мл безводного хлороформа. Затем оставляют при перемешивании в течение 48 часов при комнатной температуре, после чего реакционную среду концентрируют при пониженном давлении. Полученный сырой продукт очищают путем хроматографии на диоксиде кремния при среднем давлении и элюировании с помощью смеси гексана с этилацетатом (1:1 по объему). Таким образом получают 1,95 г (выход = 99%) целевого продукта в форме желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,30 (т., 3Н); 1,35– 1,65 (м., 26Н); 3,2–3,35 (м., 4Н); 3,4 (т.д., 2Н); 4,2 (к., 2Н); 7,1–7,2 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 7. 1-(1,1-Диметилэтиловый)-эфир 3-[/(1,1-диметилэтокси) карбонил/-амино]-12-оксо-2,4, 11-триаза-тетрадец-2-ендикислоты. 1,95 г (4,15 × 10-3 моль) полученного в приготовлении 6 выше продукта растворяют в 15 мл диметоксиэтана и добавляют 8,5 мл водного 1Н раствора NaOH. Перемешивают при комнатной температуре в течение 15 минут, затем добавляют 25 мл воды и 25 мл хлороформа и осторожно подкисляют до рН=2 с помощью 1Н водного раствора HCl. Экстрагируют несколько раз хлороформом, затем органическую фазу сушат и концентрируют при пониженном давлении. Таким образом получают 1,8 г (выход = 100%) целевого продукта в форме желтого цвета густого масла. 1 Н-ЯМР-спектр (CDCl3): 1,35–1,65 (м., 26Н); 3,3–3,45 (м., 6Н); 7,05 (с.ш., 1Н); 8,3 (т., 1Н); 9–11,5 (с.ш., 2Н). Приготовление 8. Бис(1,1-диметилэтиловый) эфир 3-{/(1,1диметилэтокси) карбонил/-амино}-23-метил-12,14диоксо-20- (фенилметил)-2,4,11,15,20,24-гексаазапентакоз-2-ен-дикислоты. Поступая аналогичным приготовлению 6 образом, но исходя из 6,21 г (14 × 10-3 моль), полученного в приготовлении 7 продукта и 4,7 г (13,5 × 10-3 моль) 1,1-диметилэтилового эфира 10-амино3-метил-6-(фенилметил)-2,6-диазадекановой кислоты и после очистки путем хроматографии на диоксиде кремния при элюировании смесью этилацетата с этанолом (9:1 по объему), получают 8,5 г (выход = 81%) целевого продукта в форме оранжевого масла. 1 Н-ЯМР-спектр (CDCl3): 1,01–1,03 (д., 3Н); 1,23–1,70 (м., 41Н); 2,4–2,7 (м., 4Н); 3,05–3,3 (м., 6Н); 3,35–3,45 (т.д., 2Н); 3,6–3,8 (м., 3Н); 5,1–5,4 (с.ш., 1Н); 7,1–7,4 (м., 5Н); 7,55 (с.ш., 1Н); 7,75 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 9. Хлоргидрат бис-(1,1-диметилэтилового) эфира 3-[/(1,1-диметилэтокси)- карбонил/-амино]23-метил-12,14-диоксо-2,4,11,15,20,24-гексаазапентакоз-2-ен-дикислоты. мощью 100 мл диэтилового эфира. Водную декантированную фазу снова экстрагируют 3 раза по 30 мл диэтилового эфира и объединенные органические фазы промывают 50 мл водного насыщенного раствора хлорида натрия, затем сушат над сульфатом магния. После удаления растворителя при пониженном давлении остаточную жидкость очищают путем хроматографии на диоксиде кремния и элюирования смесью н-гептана с диэтиловым эфиром (7:3 по объему). Таким образом получают 7,8 г (выход=78%) целевого продукта в форме бесцветного вязкого масла. 1 Н-ЯМР-спектр (CDCl3): 1,11 (д., 3Н); 1,58 (м., 2Н); 1,77 (м., 2Н); 2,34 (т., 2Н); 2,52 (м., 4Н); 3,50 (м., 3Н); 7,26 (м., 5Н). Приготовление 4. 1,1-Диметилэтиловый эфир 9-циано-3-метил-6-(фенилметил)- 2,6-диазанонановой кислоты. В колбу для гидpиpования емкостью 250 мл вводят 3,00 г (11 × 10-3 моль) продукта, полученного согласно приготовлению 3, и 2,85 г (13 × 10-3 моль) ди-трет.-бутил-дикарбоната (продукт структуры: O/CO2C(CH3)3/) в виде раствора в 30 мл безводного этилацетата и добавляют 0,3 г 10%-ного палладия-на-угле. Смесь гидрируют при перемешивании при комнатной температуре и под давлением 2 × 105 Па в течение 15 часов. Затем катализатор удаляют путем отфильтровывания, после чего фильтрат концентрируют при пониженном давлении. Остаточный продукт очищают путем хроматографии на силикагеле, элюируя смесью н-гептата с диэтиловым эфиром (45:55 по объему). Таким образом получают 3,47 г (выход = 91%) целевого продукта в форме бесцветного вязкого масла. 1 Н-ЯМР-спектр (CDCl3): 1,06 (д., 3Н); 1,44 (с., 9Н); 1,45–1,80 (м., 4Н); 2,33 (т., 1Н); 2,48 (м., 3Н); 3,50 (к., 2Н); 3,70 (с.ш., 1Н); 5,08 (с.ш., 1Н); 7,27 (м., 5Н). Приготовление 5. 1,1-Диметилэтиловый эфир 10-амино-3-метил-6-(фенилметил)- 2,6-диазадекановой кислоты. В аппарат для гидрирования вводят суспензию 2 г никеля Ренея в 180 мл безводного этанола. Эту суспензию насыщают путем барботирования в течение 10 минут газообразным аммиаком, затем добавляют 2,95 г (8 × 10-3 моль), полученного в приготовлении 4 продукта в виде раствора в 20 мл безводного этанола. Затем реакционную смесь подвергают гидрированию под давлением 106 Па в течение 15 часов при 40оС. После охлаждения катализатор удаляют путем отфильтровывания и фильтрат концентрируют при пониженном давлении. Остаточное масло очищают путем хроматографии на силикагеле, элюируя смесью метанола с 32% аммиака (100:1 по объему). Таким образом получают 2,7 г (выход = 91%) целевого продукта в форме бесцветного вязкого масла. 1 Н-ЯМР-спектр (CDCl3): 1,04 (д., 3Н); 1,31 (с., 2Н); 1,44 (с., 9Н); 1,30–1,75 (м., 6Н); 2,63 (т., 2Н); 2,25–2,70 (м., 4Н); 3,43 (д., 1Н); 3,60 (д., 1Н); 3,60– 3,75 (м., 1Н); 5,74 (с.ш., 1Н); 7,30 (м., 5Н). Приготовление 6. 1-(1,1-Диметилэтил)-14-этиловый эфир 3[[(1,1-диметилэтокси) карбонил]-амино]-12-оксо2,4, 11-триазатетрадец-2-ен-дикислоты. 11 37217 1 8,5 г (10,9 × 10-3 моль), полученного в приготовлении 8, продукта растворяют в 100 мл этанола и добавляют 0,57 г хлорида палладия и 0,7 мл концентрированной соляной кислоты. Смесь затем гидрируют при атмосферном давлении в течение 8 часов. Катализатор отфильтровывают, споласкивают его небольшим количеством этанола, затем полученный фильтрат концентрируют при пониженном давлении. Таким образом получают 7,8 г (выход = 98%) целевого продукта в форме бесцветного масла. 1 Н-ЯМР-спектр (CDCl3): 1,20 (д., 3Н); 1,25– 1,9 (м., 41Н); 2,55–2,80 (м., 2Н); 2,80–3,55 (м., 10Н); 3,75 (с.ш., 1Н); 4,9 (д., 1Н); 7,9 (м., 1Н); 8,23 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 1. Трис-(трифторацетат) N-{4-[/3-(амино)бутил/-амино]- бутил}-N'-[6-/(аминоиминометил)амино/-гексил] -пропандиамида. 7,8 г (10,8 × 10-3 моль), полученного согласно предыдущему приготовлению 9, продукта растворяют в 40 мл дихлорметана и добавляют 40 мл трифторуксусной кислоты. Реакционную смесь перемешивают в течение 5 часов при комнатной температуре, затем концентрируют при пониженном давлении. Остаточное мало очищают путем хроматографии на привитом диоксиде кремния RP 18 (гранулометрия 5–20 мкм) при среднем давлении и элюировании смесью воды с ацетонитрилом и трифторуксусной кислотой (8:1:1 по объему). Таким образом полученные чистые фракции объединяют и лиофилизируют. После этого лиофилизат обрабатывают с помощью 100 мл воды и раствор промывают два раза 100 мл этилацетата, затем водную фазу снова лиофилизируют. Эту операцию повторяют два раза для удаления трифторуксусной кислоты. Таким образом получают 4,5 г (выход = 57%) целевого продукта в виде аморфного твердого вещества. 1 Н-ЯМР-спектр (DMCO-d6): 1,15–1,20 (д., 3Н); 1,25–1,5 (м., 8Н); 1,5–1,85 (м., 4Н); 1,85–2,0 (м., 2Н); 2,85–3,2 (м., 12Н); 3,25–3,35 (м., 1Н); 6,8– 7,55 (с.ш., 3Н); 7,6 (т., 1Н); 7,09–8,1 (м., 5Н); 8,5– 8,76 (с.ш., 3Н); 13 С-ЯМР-спектр (D2O): 18,01; 13,71; 26,21; 26,24; 26,35; 28,56; 28,84; 31,22; 39,51; 40,31; 41,88; 44,30; 44,60; 46,10; 48,14; 157,55; 170,01; 170,34. Приготовление 10. 1,1-Диметилэтиловый эфир 3-метил-10-/2,4диоксо-оксазолидин-3-ил/-6-(фенилметил)-2,6диазадекановой кислоты. Готовят раствор из 3 г (8,6 × 10-3 моль) 1,1диметилэтилового эфира 10-амино-3-метил-6-фенилметил-2,6-диазадекановой кислоты в 50 мл безводного толуола и добавляют раствор 1,8 г (8,6 × 10-3 моль) метилового эфира феноксикарбонилоксиуксусной кислоты в 10 мл безводного толуола, затем 1,08 г (10,7 × 10-3 моль) триэтиламина. Доводят до температуры 60оС и выдерживают при этой температуре при перемешивании в течение 48 часов. Реакционную среду концентрируют затем при пониженном давлении и остаток, полученный таким образом, очищают на диоксиде кремния, элюируя смесью метилциклогексана с этилацетатом (1:1 по объему). Таким образом получают 3,1 г (выход = 82%) продукта в виде вязкого желтого масла. Н-ЯМР-спектр (CDCl3): 1,0–1,1 (д., 3Н); 1,4– 1,7 (м., 15Н); 2,35–2,50 (м., 3Н); 2,50–2,65 (м., 1Н); 3,40–3,60 (м., 4Н); 3,60–3,75 (м., 1Н); 4,65 (с., 2Н); 5,35 (с.ш., 1Н); 7,2–7,3 (м., 5Н). Приготовление 11. 1-(1,1-Диметилэтиловый) эфир 3-метил-6(фенилметил)-13-окса- 12-оксо-2,6,11-триазапентадекандикислоты. 3,1 г (7,1 × 10-3 моль), полученного согласно приготовлению 10 выше продукта растворяют в 20 мл диметоксиэтана, затем добавляют 21 мл водного 1Н. раствора NaOH. Перемешивают при комнатной температуре в течение 4-х часов, затем добавляют 75 мл воды и 75 мл дихлорметана. Подкисляют до рН=2 с помощью 1Н. раствора соляной кислоты и экстрагируют дихлорметаном. Органические фазы промывают один раз раствором хлорида натрия, сушат над сульфатом магния, затем концентрируют при пониженном давлении. Таким образом получают 3 г (выход = 93%) целевого продукта в виде густого желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,15 (д., 3Н); 1,2–2,2 (м., 15Н); 2,35–2,55 (т., 2Н); 2,7–2,9 (т., 2Н); 2,9–3,3 (м., 4Н); 3,5–3,75 (м., 1Н); 4,0–4,3 (м., 2Н); 4,9 (д., 1Н); 5,1 (д., 1Н); 7,4–7,8 (м., 5Н); 12,3 (с.ш., 1Н). Приготовление 12. Бис(1,1-диметилэтиловый) эфир 3-[/(1,1диметилэтокси)карбонил/-амино]-24-метил-21(фенилметил)-14-окса-12, 15-диоксо- 2,4,11,16,21, 25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной приготовлению 6, но используя дихлорметан в качестве растворителя и исходя из 3 г (6,67 × 10-3 моль) полученной согласно приготовлению 11 кислоты и 2,61 г (6,65 × 10-3 моль) бис (1,1-диметилэтилового) эфира [/(6-аминогексил)-имино/метилен]-бис-карбаминовой кислоты, после очистки путем хроматографии на диоксиде кремния и элюирования с помощью смеси метилциклогексана с этилацетатом (3:7 по объему) получают 4 г (выход = 76%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 0,9–1,1 (д., 3Н); 1,25–1,75 (м., 41Н); 2,3–2,5 (м., 3Н); 2,6 (м., 1Н); 3,1–3,2 (с.ш., 2Н); 3,2–3,35 (к., 2Н); 3,35–3,45 (т., 2Н); 3,5 (к., 2Н); 3,7 (м., 1Н); 4,5 (с., 2Н); 5,3–5,6 (с.ш., 2Н); 6,3 (с.ш., 1Н); 7,2–7,3 (м., 5Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 13. 2-{[[4-[N'-[3-(амино)-бутил]-N'-(фенилметил)амино]- бутил]-амино]-карбонилокси}-N-{6-[(аминоиминометил)-амино]гексил}-ацетамид-трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, но исходя из 4 г полученного выше согласно приготовлению 12 продукта и после очистки путем хроматографии на колонке с привитым диоксидом кремния RP 18 и элюирования с помощью смеси воды с ацетонитрилом и трифторуксусной кислотой (7,5:2:0,5 по объему), получают 4,2 г (выход = 99%) целевого продукта в виде густого масла. 1 Н-ЯМР-спектр (СDCl3): 1,1–1,2 (д., 3Н); 1,2– 1,6 (м., 10Н); 1,65–2,2 (м., 4Н); 2,9-3,2 (м., 12Н); 3,2–3,35 (м., 1Н); 4,35 (с., 2Н); 6,8–7,3 (с.ш., 1Н); 7,35 (т., 1Н); 7,45–7,65 (м., 5Н); 7,85–8,1 (м., 3Н); 9,8 (с., 1Н). 12 37217 (м., 4Н); 3,39 (т.д., 4Н); 3,65–3,8 (м., 1Н); 4,8–4,9 (м., 1Н); 7,05 (с.ш., 1Н); 7,55 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 3. N-[4-[[3-(амино)-бутил]-амино]-бутил]-N'-[8[(аминоиминометил)-амино]-октил]-пропандиамидтрис (трифторацетат). Следуя методике, аналогичный таковой примера 1, но исходя из 2,15 г (3,01 × 10-3 моль) соединения, полученного согласно приготовлению 15, и после очистки на силикагеле RP 18 и элюирования с помощью смеси воды с ацетоном и трифторуксусной кислотой (7,5:2:0,5 по объему), получают 2,05 (выход = 90%) целевого продукта в виде аморфного твердого вещества. 1 Н-ЯМР-спектр (DMCO-d6): 1,18 (д., 3Н); 1,2– 1,65 (м., 16Н); 1,65–1,8 (м., 1Н); 1,8–2 (м., 1Н); 2,8– 3,1 (м., 12Н); 3,25–3,3 (м., 1Н); 6,8–7,5 (с.ш., 3Н); 7,55 (т., 1Н); 7,85–8,1 (м., 5Н); 8,45–8,65 (м., 3Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,69; 26,24; 26,51; 26,69; 28,60; 28,92; 28,96; 32,21; 39,49; 40,46; 41,97; 44,31; 44,59; 46,10; 48,14; 157,89; 169,96; 170,34. Приготовление 16. Бис(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)-карбонил]-амино]-23-метил-20-(фенилметил)-13-(фенилметокси)-12,14-диоксо-2,4,11, 15,20,24-гексаазапентакоз-2-ен-дикислоты. 2,78 г (5,05 × 10-3 моль) 1-(1,1-диметилэтилового) эфира 3-[[(1,1-диметилэтокси)-карбонил]амино]-13- (фенилметокси)-12-оксо-2,4,11-триазатетрадец-2-ен-дикислоты растворяют в 50 мл безводного тетрагидрофурана (ТГФ). Раствор охлаждают до -25 оС и добавляют 1,02 г (10,1 × 10-3 моль) N-метилморфолина и 0,69 г (5,05 × 10-3 моль) изобутилхлорформиата. Немедленно выпадает белого цвета осадок. Перемешивают в течение 0,5 часа, затем добавляют 1,76 г (5,05 × 10-3 моль), полученного согласно приготовлению 5 соединения, в виде раствора в 10 мл ТГФ. Перемешивают в течение 1 часа, затем концентрируют при пониженном давлении. Продукт очищают путем хроматографии на силикагеле и элюирования смесью метилциклогексана с этилацетатом (7,5:2,5 по объему). Таким образом получают 3,76 г (выход = 86%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,04 (дд., 3Н); 1,2– 1,8 (м., 41Н); 2,3–2,7 (м., 4Н); 3,1–3,3 (м., 4Н); 3,3– 3,5 (м., 3Н); 3,5–3,75 (м., 2Н); 4,28 (с., 1Н); 4,79 (с., 2Н); 5,3–5,5 (с.ш., 1Н); 6,95 (с.ш., 1Н); 7,2–7,4 (м., 10Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 17. Бис(1,1-диметилэтиловый) эфир 3-{[(1,1диметилэтокси)- карбонил]-амино}-13-гидрокси-23метил-12, 14-диоксо- 2,4,11,15,20,24-гексаазапентакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, но исходя из 3,76 г (4,35 × 10-3 моль) продукта, полученного согласно приготовлению 16, получают 3,05 г (выход количественный) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,15 (д., 3Н); 1,25– 1,75 (м., 42Н); 2,55–2,7 (м., 4Н); 3,1–3,35 (м., 4Н); 3,35–3,45 (тд., 2Н); 3,6–3,8 (м., 1Н); 4,42 (с., 1Н); 4,75–4,85 (м., 1Н); 7,2–7,45 (м., 3Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 2. 2-{[[4-[[3-(Амино)-бутил]-амино]-бутил]-амино]-карбонилокси}-N-{6-[(аминоиминометил)-амино]- гексил}-ацетамид-трис(трифторацетат). 4,2 г (5,05 × 10-3 моль) продукта, полученного в приготовлении 13 выше, растворяют в 275 мл метанола, затем добавляют 0,25 мл концентрированной соляной кислоты, после чего добавляют 0,25 г хлорида палладия. Смесь затем гидрируют под атмосферном давлении и при комнатной температуре в течение 14 часов. Реакционную среду отфильтровывают, затем фильтрат концентрируют при пониженном давлении. Полученное масло обрабатывают с помощью 100 мл воды и 1 мл трифторуксусной кислоты, затем раствор промывают 3 раза по 75 мл этилацетата и лиофилизируют. Таким образом получают 3,9 г (выход = 99%) целевого продукта в виде аморфного твердого вещества. 1 Н-ЯМР-спектр (DMCO-d6): 1,2 (д., 3Н); 1,25– 1,70 (м., 12Н); 1,70–1,9 (м., 1Н); 1,9–2,05 (м., 1Н); 2,85–3,15 (м., 11Н); 4,3 (с., 2Н); 4,3 (с., 2Н); 6,8–7,5 (м., 5Н); 7,6–7,75 (т., 1Н); 7,85–7,95 (т., 1Н); 7,95– 8,20 (с.ш., 3Н); 8,70–8,90 (с.ш., 2Н); 13 С-ЯМР-спектр (D2O): 18,02; 23,65; 26,19; 26,28; 26,76; 28,56; 28,93; 31,22; 39,77; 40,59; 41,88; 44,61; 46,11; 48,22; 63,76; 157,54; 158,21; 171,45. Приготовление 14. Бис(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)карбонил]-амино]-25-метил-22фенил-метил-14,16-диоксо- 2,4,13,17,22,26-гексаазагептакоз-2-ен-дикислоты. Следуя методике, идентичной таковой приготовления 6, но исходя из 2,1 г (4,45 × 10-3 моль) 1-(1,1-диметилэтилового) эфира 3-[[(1,1-диметилэтокси)-карбонил]-амино]-14-оксо -2,4,13-триазагексадец-2-ен-дикислоты и 1,55 г (4,45 × 10-3 моль) соединения, полученного в приготовлении 5, после очистки путем хроматографии на силикагеле и элюирования с помощью смеси этилацетата с этанолом и гидроксидом аммония (7:3:0,1 по объему), получают 3,26 г (выход = 93%) целевого продукта в виде аморфного твердого вещества желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,02 (д., 3Н); 1,2–1,8 (м., 45Н); 2,55–2,8 (м., 4Н); 3,1–3,3 (м., 6Н); 3,38 (т.д., 2Н); 3,5–3,7 (м., 1Н); 3,7–3,9 (м., 2Н); 5–5,3 (с.ш., 1Н); 7,1–7,45 (м., 5Н); 7,5–7,6 (с.ш., 1Н); 7,7– 7,8 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 15. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-25-метил-14,16диоксо-2,4,13,17,22,26-гексаазагептакоз-2-ен-дикислоты. 3,2 г (4,06 × 10-3 моль), полученного согласно приготовлению 14, соединения растворяют в 75 мл этанола и добавляют 0,25 г 5%-ного палладияна-угле. Смесь подвергают гидрированию при комнатной температуре и при атмосферном давлении в течение 12 часов. Катализатор отфильтровывают и фильтрат концентрируют при пониженном давлении. Таким образом получают 2,15 г (выход = 74%) целевого продукта в виде аморфного твердого вещества. 1 Н-ЯМР-спектр (CDCl3): 1,15 (д., 3Н); 1,25– 1,8 (м., 45Н); 2,55–2,7 (м., 4Н); 3,11 (с., 2Н); 3,2–3,3 13 37217 Следуя методике, аналогичной таковой приготовления 16, исходя из 2,55 г (4,4 × 10-3 моль) продукта, полученного согласно приготовлению 19, и 1,54 (4,4 × 10-3 моль) соединения, полученного согласно приготовлению 5, получают 3,5 г (выход = 89%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,04 (дд., 3Н); 1,2– 1,7 (м., 45Н); 2,3–2,65 (м., 4Н); 3,1–3,3 (м., 4Н); 3,39 (тд., 2Н); 3,44 (д., 1Н); 3,57 (д., 1Н); 3,6–3,75 (м., 1Н); 4,28 (с., 1Н); 4,79 (с., 2Н); 5,35–5,5 (с.ш., 1Н); 6,85-7,1 (с.ш., 2Н); 7,2–7,4 (м., 10Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 21. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-15-гидрокси-25метил- 14,16-диоксо-2,4,13,17,22,26-гексаазагептакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, исходя из 3,5 г (3,92 × 10-3 моль) соединения, полученного согласно приготовлению 20, получают 2,4 г (выход = 85%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,15 (д., 3Н); 1,2–1,8 (м., 46Н); 2,55–2,75 (м., 4Н); 3,2–3,45 (м., 6Н); 3,65–3,8 (м., 1Н); 4,42 (с., 1Н); 4,75–4,9 (д., 1Н); 7,2 (т., 1Н); 7,4 (т., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 5. N-{4-[[3-(амино)-бутил]амино]бутил}-2- гидрокси-N'-[8-[(аминоимино-метил)-амино]-октил]пропандиамид- трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 2,4 г (3,29 × 10-3 моль) соединения, полученного согласно приготовлению 21, после очистки путем хроматографии на силикагеле RP 18, элюируя смесью воды с ацетонитрилом и трифторуксусной кислотой (8:1,5:0,5), получают 2,27 г (выход = 89,5%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DМCO-d6): 1,15–1,65 (м., 19Н); 1,65–1,85 (м., 1Н); 1,85–2 (м., 1Н); 2,85–3,2 (м., 10Н); 3,2–3,35 (м., 1Н); 4,31 (с., 1Н); 6,7–7,5 (с.ш., 3Н); 7,59 (т., 1Н); 7,8–8,05 (м., 6Н); 8,45–8,7 (м., 2Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,65; 26,32; 26,49; 26,61; 28,60; 28,95; 29,04; 31,21; 39,20; 40,15; 41,97; 44,59; 46,09; 48,12; 73,14; 157,56; 171,15; 171,53. Пример 4. N-{4-[[3-(амино)-бутил]-амино]-бутил}-N'- [6[(аминоиминометил)-амино]-гексил]-2-гидроксипропандиамид-трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 3,03 г (4,32 × 10-3 моль) соединения, полученного согласно приготовлению 17, и после очистки путем хроматографии на силикагеле RP 18 и элюирования смесью воды с ацетонитрилом и трифторуксусной кислотой (8:1:1 по объему), получают 1,89 г (выход = 60%) аморфного белого твердого вещества. 1 Н-ЯМР-спектр (DMCO-d6): 1,18 (д., 3Н); 1,2– 1,65 (м., 12Н); 1,65–1,85 (м., 1Н); 1,85–2 (м., 1Н); 2,8–3,15 (м., 10Н); 3,2–3,35 (м., 1Н); 4,31 (с., 1Н); 6,7–7,4 (м., 3Н); 7,6 (т., 1Н); 7,85–8,05 (м., 5Н); 8,45–8,65 (м., 3Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,66; 26,19; 26,28; 26,31; 28,55; 28,94; 31,21; 39,22; 40,00; 41,87; 44,59; 46,09; 48,12; 73,13; 157,23; 171,20; 171,53. Приготовление 18. 1-(1,1-Диметилэтил)-16-этиловый эфир 3[[(1,1-диметилэтокси)- карбонил/-амино]-15-фенилметокси-14-оксо-2,4,13-триазагексадец-2-ен-дикислоты. 2,6 г (10,9 × 10-3 моль) этилового эфира 2фенилметоксипропан-дикислоты растворяют в 50 мл дихлорметана и смесь охлаждают до 0оС. Добавляют 4,34 г (22 × 10-3 моль) N,N'-дициклогексилкарбодиимида и 0,57 г (4 × 10-3 моль) 1-гидроксибензотриазола и выдерживают при перемешивании в течение 0,5 часа, после чего добавляют 4,21 г (10,9 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[(8-амино-октил)-имино]-метилен] бис-карбаминовой кислоты в виде раствора в 15 мл дихлорметана и оставляют при перемешивании и при комнатной температуре в течение 48 часов. Реакционную смесь концентрируют при пониженном давлении, затем остаток очищают путем хроматографии на диоксиде кремния, элюируя смесью метилциклогексана с этилацетатом (7:3 по объему). Таким образом получают 2,7 г (выход = 40,8%) целевого продукта в виде масла бледно-желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,25–1,6 (м., 33Н); 3,25 (тд., 2Н); 3,39 (тд., 2Н); 4,25 (к., 2Н); 4,44 (с., 1Н); 4,54 (д., 1Н); 4,70 (д., 1Н); 6,62 (т., 1Н); 7,3– 7,45 (м., 5Н); 8,25 (т. 1Н); 11,5 (с., 1Н). Приготовление 19. 1-(1,1-Диметилэтиловый) эфир 3-[[(1,1диметилэтокси)-карбонил]-амино]-15-фенилметокси)-14-оксо-2,4,13-триазагексадец-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 7, исходя из 2,7 г (4,45 × 10-3 моль) соединения, полученного согласно приготовлению 18, получают 2,55 г (выход = 99%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,2–1,7 (м., 3 ОН); 3,2–3,4 (м., 4Н); 4,41 (с., 1Н); 4,71 (д., 1Н); 5,11 (д. 1Н); 6,99 (т., 1Н); 7,35–7,5 (м., 5Н); 8,3 (т., 1Н); 11,3–11,8 (с.ш., 1Н). Приготовление 20. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)карбонил]-амино]-15-фенилметокси-25-метил-22-фенилметил-14,16-диоксо- 2,4, 13,17,22,26-гексаазагептакоз-2-ен-дикислоты. Приготовление 22. Бис-(1,1-диметилэтиловый)-эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-13-метокси-23метил-20-фенилметил-12,14-оксо-2,4,11,15,20,24гексаазапентакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 16, исходя из 3 г (6,3 × 10-3 моль) 1-(1,1диметилэтилового) эфира 3-[[(1,1-диметилэтокси)карбонил]-амино]-13- метокси-12-оксо-2,4,11-триазатетрадец-2-ен-дикислоты и 2,2 г (6,3 × 10-3 моль) соединения, полученного согласно приготовлению 5, получают 3,6 г (выход = 68%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,05 (дд., 3Н); 1,2– 1,8 (м., 41Н); 2,3–2,7 (м., 4Н); 3,1–3,8 (м., 12Н); 4,1 (с., 1Н); 5,5 (м., 1Н); 6,9 (м., 2Н); 7,29 (м., 5Н); 8,29 (т., 1Н); 11,5 (с., 1Н). 14 37217 Приготовление 26. Бис-(1,1-диметилэтиловый)-эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-15-метокси-25метил-22-фенилметил-14,16-диоксо- 2,4,13,17,22, 26-гексаазагептакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 16, исходя из 4 г (8 × 10-3 моль) соединения, полученного согласно приготовлению 25, получают 4,8 г (выход = 72%) целевого продукта в виде светло-желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,05 (дд., 3Н); 1,2– 1,8 (м., 45Н); 2,3–2,7 (м., 4Н); 3,1–3,3 (м., 4Н); 3,3– 3,6 (м., 7Н); 3,6–3,75 (м., 1Н); 4,09 (с., 1Н); 5,35– 5,55 (с.ш., 1Н); 6,75–7 (м., 2Н); 7,2–7,35 (м., 5Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 27. Бис-(1,1-диметилэтиловый)-эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-15-метокси-25метил-14,16-диоксо- 2,4,13,17,22,26-гексаазагептакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 23, исходя из 4,8 г (5,76 × 10-3 моль) соединения, полученного согласно приготовлению 26, получают 4,15 г (выход = 97%) целевого продукта в виде масла. 1 Н-ЯМР-спектр (CDCl3): 1,15 (д., 3Н); 1,25– 1,8 (м., 45Н); 1,9–2,1 (с.ш., 1Н); 2,6–2,75 (м., 4Н); 3,2–3,35 (м., 4Н); 3,35–3,45 (м., 2Н); 3,57 (с., 3Н); 3,65–3,8 (м., 1Н); 4,12 (с., 1Н); 4,8–4,9 (с.ш., 1Н); 6,85 (т., 1Н); 7,2–7,35 (м., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 7. N-{4-[[3-(амино)-бутил]-амино]-бутил}-2-метокси-N'[8-[(аминоиминометил)-амино]-октил]пропандиамид-трис (трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 4,15 г (5,58 × 10-3 моль) соединения, полученного согласно приготовлению 27, получают 3,8 г (выход = 86,5%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DМСО-d6): 1,18 (д., 3Н); 1,2– 1,35 (м., 8Н); 1,35–1,65 (м., 8Н); 1,65–1,85 (м., 1Н); 1,85–2 (м., 1Н); 2,8–3,2 (м., 10Н); 3,25–3,4 (м., 4Н); 4,08 (с., 1Н); 6,6–7,5 (с.ш., 3Н); 7,62 (т., 1Н); 7,9–8,1 (м., 6Н); 8,5–8,7 (с.ш., 2Н). 13 С-ЯМР-(D2O): 18,01; 23,67; 26,33; 26,50; 26,63; 28,60; 28,94; 28,93; 29,01; 31,21; 39,18; 40,14; 41,97; 44,59; 46,09; 48,11; 58,68; 82,44; 157,54; 169,60; 169,97. Приготовление 28. 1-(1,1-диметилэтил)-14-этиловый эфир 13фтор-6-фенилметил-12-оксо-2,6,11-триазатетрадекандикислоты. Готовят раствор из 1,3 г (8,7 × 10-3 моль) этилового эфира 2-фторпропандикислоты в 30 мл дихлорметана и 3 мл диметилформамида и добавляют 2,2 г (13,5 × 10-3 моль) 1,1-карбонилдиимидазола. В течение 3-х часов при комнатной температуре перемешивают, затем добавляют 3 г (8,7 × 10-3 моль) соединения, полученного согласно приготовлению 5, в виде раствора в 10 мл дихлорметана. Выдерживают при перемешивании и при комнатной температуре в течение 48 часов. Реакционную среду концентрируют при пониженном давлении и очищают путем хроматографии на силикагеле, элюируя смесью этилацетата с циклогексаном (6:4 по объему). Таким образом полу Приготовление 23. Хлоргидрат бис-(1,1-диметилэтилового) эфира 3-[[(1,1-диметилэтокси)- карбонил]-амино]13-метокси-23-метил-12,14-диоксо-2,4,11,15,20, 24-гексаазапентакоз-2-ен-дикислоты. Готовят раствор из 3,3 г (4,1 × 10-3 моль) соединения, полученного согласно приготовлению 22, в 100 мл этанола и добавляют 0,2 мл концентрированной соляной кислоты и 300 мг 10%-ного палладия-на-угле. Смесь гидрируют при комнатной температуре и при атмосферном давлении в течение 2-х часов. Катализатор отфильтровывают и фильтрат концентрируют при пониженном давлении. Таким образом получают 2,92 г (выход = 94,8%) целевого продукта в виде твердого белого вещества. 1 Н-ЯМР-спектр (CDCl3): 1,1–2,3 (м., 44Н); 2,8–3,6 (м., 11Н); 3,4–3,6 (с., 3Н); 4,4 (с., 1Н); 4,9 (м., 1Н); 7,6 (м., 1Н); 7,9 (т., 1Н); 9,3 (м., 1Н); 9,9 (м., 1Н); 11,4 (с., 1Н). Пример 6. N-{4-[[3-(амино)-бутил]-амино]-бутил}-N'-[6[(аминоиминометил)-амино]-гексил]-2-метоксипропандиамид-трис (трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 2,7 г (3,6 × 10-3 моль) соединения, полученного согласно приготовлению 23, и после очистки путем хроматографии на силикагеле RP 18 и элюирования смесью воды с ацетонитрилом и трифторуксусной кислотой (8:1,5:0,5 по объему), получают 2,11 г (выход = 78%) целевого продукта в виде аморфного твердого вещества. 1 Н-ЯМР-спектр (DMCO-d6): 1,2 (д., 3Н); 1,4 (м., 12Н); 1,75–1,95 (2м., 1Н); 3,0 (м., 11Н); 3,3 (с., 3Н); 4,1 (с., 1Н); 7,1 (с.ш., 3Н); 7,6 (т., 1Н); 8,0 (м., 4Н); 8,6 (м., 2Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,68; 26,18; 26,29; 26,32; 28,55; 28,92; 31,21; 39,21; 39,99; 41,87; 44,60; 46,10; 48,11; 58,65; 82,43; 157,54; 165,65; 169,96. Приготовление 24. 1-(1,1-Диметилэтил)-16-метиловый эфир 3[[(1,1-диметилэтокси)- карбонил]-амино]-15-метокси-14-оксо- 2,4,13-триазагексадец-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 18, исходя из 3,5 г (23,65 × 10-3 моль) метилового эфира 2-метокси-пропандикислоты и 7 г (18,1 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[(8-аминооктил)-имино]- метилен]-бискарбаминовой кислоты, получают 8,6 г (выход = 95%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,2–1,65 (м., 30Н); 3,2– 3,35 (м., 2Н); 3,38 (тд., 2Н); 3,47 (с., 3Н); 3,82 (с., 3Н); 4,30 (с., 1Н); 6,6 (т., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 25. 1-(1,1-Диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-15-метокси-14оксо- 2,4,13-триазагексадец-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 7, исходя из 8,6 г (16,7 × 10-3 моль) соединения, полученного согласно приготовлению 24, получают 8,2 г (выход = 98%) целевого продукта в виде аморфного твердого вещества желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,25–1,7 (м. 30Н); 3,2–3,45 (м., 4Н); 3,69 (с., 3Н); 4,26 (с., 1Н); 6,9–7,1 (с.ш., 1Н); 8,25–8,45 (с.ш., 1Н); 11,3–11,9 (с.ш., 1Н). 15 37217 Приготовление 32. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-15-фтор-25-метил-22-фенилметил-14,16-диоксо- 2,4,13,17,22,-26гексаазагептакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 16, исходя из 670 мг (1,5 × 10-3 моль) соединения, полученного согласно приготовлению 29, и 571 мг (1,5 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[(8-аминооктил)-имино]-метилен] бискарбаминовой кислоты, получают 1 г (выход = 83,3%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,06 (д., 3Н); 1,2–1,8 (м., 45Н); 2,5 (м., 4Н); 3,2–3,8 (м., 9Н); 5,1–5,2 (д., 1Н); 5,2 (м., 1Н); 6,9 (м., 2Н); 7,3 (м., 5Н); 8,23 (т., 1Н); 11,5 (с., 1Н). Приготовление 33. Хлоргидрат бис-(1,1-диметилэтилового)эфира 3-[[(1,1-диметилэтокси)- карбонил]-амино]15-фтор-25-метил-14,16-диоксо- 2,4,13,17,22,26гексаазагептакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 23, исходя из 1 г (1,2 × 10-3 моль) соединения, полученного согласно приготовлению 32, получают 746 мг (выход = 81%) целевого продукта в виде гигроскопичного твердого вещества белого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,1–2,1 (м., 48Н); 2,8–3,5 (м. 9Н); 3,6–3,8 (м., 2Н); 4,9 (с.ш., 1Н); 5,6 (д., 2Н); 7,9 (с.ш., 1Н); 8,5 (м., 1Н); 11,4 (м., 1Н). Пример 9. N-{4-[[3-(амино)бутил]-амино]бутил}-2-фторN'-[8[(аминоиминометил)-амино]октил]-пропандиамид-трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 740 мг (0,96 × 10-3 моль) соединения, полученного согласно приготовлению 33, получают 400 мг (выход = 54%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DMCO-d6): 1,1–1,6 (м., 19Н); 1,75–1,95 (2м., 2Н); 2,8–3,2 (м., 10Н); 3,3 (м., 1Н); 5,12–5,28 (д., 1Н); 7,1 (с.ш., 1Н); 7,6 (т., 1Н); 7,94 (с., 3Н); 8,3–8,6 (м., 4Н). 13 С-ЯМР-спектр (D2О): 18,01; 23,65; 26,19; 26,48; 26,57; 28,59; 28,88; 28,91; 28,94; 31,21; 39,30; 40,22; 41,97; 44,59; 46,10; 48,09; 87,27; 89,87; 156,81; 170,12; 170,14. Приготовление 34. 1-(1,1-диметилэтил)-13-этиловый эфир 3-метил-6-фенилметил-12-оксо-2,6,11-триазатридекандикислоты. 7 г (20 × 10-3 моль) соединения, полученного согласно приготовлению 5, растворяют в 100 мл безводного дихлорметана и добавляют 5,25 г (52 × 10-3 моль) триэтиламина, затем медленно добавляют 3,56 г (26 × 10-3 моль) этоксалилхлорида, охлаждая на водяной бане до 10оС. По окончании добавления перемешивают в течение 15 минут, затем концентрируют при пониженном давлении. Очищают путем хроматографии на силикагеле и элюирования смесью метилциклогексана с этилацетатом (4:6, затем 1:9 по объему). Таким образом получают 6,8 г (выход = 75,7%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (СDCl3): 1,0 (д., 3Н); 1,3–1,8 (м., 18Н); 2,3–2,65 (м., 4Н); 3,2–3,35 (м., 2Н); 3,4 чают 2,3 г (выход = 55,6%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,04 (д., 3Н); 1,2–1,7 (м., 18Н); 2,3–2,5 (м., 2Н); 2,5–2,65 (м., 2Н); 3,4–3,8 (м., 5Н); 4,25–4,4 (м., 2Н); 5,25 (д., 2Н); 6,7 (с.ш., 1Н); 7,3 (м., 5Н). Приготовление 29. 1-(1,1-Диметилэтиловый) эфир 13-фтор-6фенилметил-12-оксо-2,6,11-триазатетрадекандикислоты. Следуя методике, аналогичной таковой приготовления 7, исходя из 2,25 г (4,7 × 10-3 моль) продукта, полученного согласно приготовлению 28, получают 1,44 г (выход = 68%) целевого продукта в виде порошка белого цвета. Т.пл. = 50оС. 1 Н-ЯМР-спектр (CDCl3): 1,1 (д., 3Н); 1,4–2,1 (м., 15Н); 2,7–3,5 (м., 7Н); 4,0–4,3 (м., 2Н); 4,8 (м., 1Н); 5,4 (м., 1Н); 6,3–6,8 (м., 1Н); 7,4 (м., 5Н). Приготовление 30. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-13-фтор-23-метил-20-фенилметил-12,14-диоксо- 2,4,11,15,20,24гексаазапентакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 16, исходя из 670 мг (1,5 × 10-3 моль) соединения, полученного согласно приготовлению 29, и 530 мг (1,5 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[6-(аминогексил)имино]-метилен]бискарбаминовой кислоты, получают 570 мг (выход = 49%) целевого продукта в виде масла светло-желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,0–1,1 (д., 3Н); 1,2– 1,7 (м., 41Н); 2,3–2,7 (м., 4Н); 3,2–3,8 (м., 9Н); 5,07–5,2 (дд., 1Н); 6,8–7,1 (м., 2Н); 7,2–7,4 (м., 5Н); 8,29 (т., 1Н); 11,5 (с., 1Н). Приготовление 31. Хлоргидрат бис-(1,1-диметилэтилового)эфира 3-[[(1,1-диметилэтокси)- карбонил]-амино]13-фтор-23-метил-12,14,-диоксо- 2,4,11,15,20,24гексаазапентакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 23, исходя из 560 мг (0,7 × 10-3 моль) продукта, полученного согласно приготовлению 30, получают 484 мг (выход = 93%) целевого продукта в виде твердого вещества белого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,1–2,1 (м., 44Н); 3,0–3,8 (м., 11Н); 4,8 (м., 1Н); 5,4–5,6 (д., 1Н); 8 (м., 1Н); 8,5 (м., 1Н); 9,7 (м., 1Н); 10,7 (м., 1Н); 11,3 (с., 1Н). Пример 8. N-{4-[[3-(амино)-бутил]амино]бутил}-2-фторN'-[6- [(аминоиминометил)-амино]гексил]-пропандиамид-трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 460 мг (0,62 × 10-3 моль) соединения, полученного согласно приготовлению 31, получают 350 мг (выход = 67%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DMCO-d6): 1,1 (д., 3Н); 1,2– 1,6 (м., 12Н); 1,7–1,9 (2 м., 2Н); 2,8–3,2 (м., 10Н); 3,27 (м., 1Н); 5,1–5,28 (д., 1Н); 7,1 (м., 4Н); 7,6 (т., 1Н); 7,95 (с., 3Н); 8,35–8,65 (м., 4Н). 13 С-ЯМР-спектр (D2О): 17,89; 22,83; 25,67; 25,81; 25,86; 28,34; 28,68; 30,40; 37,87; 38,45; 40,64; 43,17; 44,38; 46,43; 86,46; 156,51; 170,12; 170,14. 16 37217 Следуя методике, аналогичной таковой приготовления 18, исходя из 2,73 г (6,47 × 10-3 моль) соединения, полученного согласно приготовлению 35, и 2,5 г (6,47 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[(8-аминооктил)имино]-метилен]-бискарбаминовой кислоты, получают 2 г (выход = 39%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,05 (д., 3Н); 1,1–1,8 (м., 45Н); 2,3–2,7 (м., 4Н); 3,15–3,30 (м., 4Н); 3,4 (тд., 2Н); 3,45 (д., 1Н); 3,6 (д., 1Н); 3,6–3,75 (м., 1Н); 5,3–5,5 (с.ш., 1Н); 7,2–7,35 (м., 5Н); 7,35–7,6 (м., 2Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 39. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-24-метил-14,15диоксо- 2,4,13,16,21,25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, исходя из 2 г (2,53 × 10-3 моль) соединения, полученного согласно приготовлению 38, получают 1,75 г (выход = 99%) целевого продукта в виде бесцветного масла. 1 Н-ЯМР-спектр (CDCl3): 1,2 (дд., 3Н); 1,2-1,8 (м., 45Н); 2,45–2,9 (м., 4Н); 3,1–3,8 (м., 8Н); 4,7– 4,85 (с.ш., 1Н); 7,4–7,5 (с.ш., 1Н); 7,7–7,85 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 11. N-{4-[[3-(амино)-бутил]-амино]-бутил}-N'-[8[(аминоиминометил) амино]-октил]-этандиамидтрис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 1,75 г (2,5 × 10-3 моль) соединения, полученного согласно приготовлению 39, получают 250 мг (выход = 13,5%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DMCO-d6): 1,2 (д., 3Н); 1,2– 1,6 (м., 16Н); 1,6–1,8 (м., 1Н); 1,8–2,0 (м., 1Н); 2,8– 3,2 (м., 10Н); 3,2–3,35 (м., 1Н); 6,6–7,4 (с.ш., 3Н); 7,55 (т., 1Н); 7,85–8,05 (с.ш., 4Н); 8,4–8,6 (с.ш., 2Н); 8,7 (т., 1Н); 8,77 (т., 1Н). 13 С-ЯМР-спектр (D2O): 15,00; 20,77; 23,12; 23,51; 23,74; 25,60; 25,91; 25,94; 25,98; 28,20; 36,53; 37,45; 38,97; 41,60; 43,09; 45,11; 157,45; 161,55; 161,88. Приготовление 40. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)карбонил]-амино]-24-метил-21фенилметил-12,15-диоксо- 2,4,11,14,16,21,25-гептаазагексакоз-2-ен-дикислоты. 2 г (4,8 × 10-3 моль) 1,1-диметилэтилового эфира 13-амино-3-[[(1,1-диметилэтокси)карбонил]амино]-12- оксо-2,4,11-триазатридец-2-еновой кислоты растворяют в 50 мл дихлорметана и добавляют порциями при комнатной температуре 1,6 г (5,3 × 10-3 моль) бис-(4-нитрофенил)-карбоната. Выдерживают при перемешивании в течение 5 часов при комнатной температуре, затем добавляют 1,68 г (4,8 × 10-3 моль) соединения, полученного согласно приготовлению 5, в виде раствора в 15 мл дихлорметана. Реакционную среду перемешивают при комнатной температуре в течение 16 часов, затем концентрируют при пониженном давлении. Очищают путем хроматографии на силикагеле, элюируя смесью этилацетата с циклогексаном (4:6 по объему), затем смесью этилацетата с этанолом (9:1 по объему). Таким образом получают (д., 1Н); 3,58 (д., 1Н); 3,6–3,8 (м., 1Н); 4,3 (к., 2Н); 5,2–5,4 (с.ш., 1Н); 7,2–7,5 (м., 6Н). Приготовление 35. 1-(1,1-диметилэтиловый) эфир 3-метил-6фенилметил-12-оксо-2,6, 11-триазатридекандикислоты. Следуя методике, аналогичной таковой приготовления 7, исходя из 6,8 г (15,14 × 10-3 моль) соединения, полученного согласно приготовлению 34, получают 6,4 г (выход количественный) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,1 (д., 3Н); 1,3–1,9 (м., 15Н); 2,5–3,45 (м., 8Н); 3,5–3,7 (м., 1Н); 5,3–5,6 (с.ш., 1Н); 7,25–7,6 (м., 5Н); 7,7–8 (с.ш., 1Н). Приготовление 36. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)карбонил]-амино]-22-метил-19фенилметил-12,13-диоксо- 2,4,11,14,19,-23-гексаазатетракоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 18, исходя из 4 г (9,5 × 10-3 моль) соединения, полученного согласно приготовлению 35, и 3,4 г (9,5 × 10-3 моль) бис-(1,1-диметилэтилового) эфира [[(6-аминогексил)имино]-метилен]-бискарбаминовой кислоты, получают 1,46 г (выход = 20%) целевого продукта в виде желтого масла. 1 Н-ЯМР-спектр (CDCl3): 1,05 (д., 3Н); 1,3–1,8 (м., 41Н); 2,3–2,6 (м., 4Н); 3,1–3,35 (м., 4Н); 3,40 (тд., 1Н); 3,45 (д. 1Н); 3,6 (д., 1Н); 3,6–3,8 (м., 1Н); 5,3–5,5 (с.ш., 1Н); 7,2–7,35 (м., 5Н); 7,4–7,6 (м., 2Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 37. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-22-метил-12,13диоксо- 2,4,14,19,23-гексаазатетракоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, исходя из 1,46 г соединения, полученного согласно приготовлению 36, получают 1,25 г (выход = 97,5%) целевого продукта в виде желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,2 (д., 3Н); 1,3-1,8 (м., 41Н); 2,6–2,8 (м., 4Н); 3,25–3,85 (м., 8Н); 4,85 (д., 1Н); 7,45 (т., 1Н); 7,85 (т., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 10. N-{4-[[3-(амино)бутил]амино]бутил}-N'-[6[(аминоиминометил)- амино]-гексил]-этандиамидтрис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 1,25 г соединения, полученного согласно приготовлению 37, получают 580 мг (выход = 43%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DMCO-d6): 1,2 (д., 3Н); 1,25– 1,65 (м., 12Н); 1,65–1,85 (м., 1Н); 1,85–2,0 (м., 1Н); 2,8–3,2 (м., 10Н); 3,2–3,4 (м., 1Н); 6,8–7,5 (с.ш., 3Н); 7,65 (т., 1Н); 7,9–8,1 (с.ш., 4Н); 8,5–8,7 (с.ш., 2Н); 8,71 (т., 1Н); 8,77 (т., 1Н). 13 С-ЯМР-спектр (D2O): 18,00; 23,77; 26,13; 26,23; 26,41; 28,54; 28,82; 31,21; 39,54; 40,31; 41,88; 44,61; 46,09; 48,12; 157,55; 161,65; 161,98. Приготовление 38. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)-карбонил]-амино]-24-метил-21-фенилметил-14,15-диоксо- 2,4,13,16,21,25-гексаазагексакоз-2-ен-дикислоты. 17 37217 на и кристаллизуют. Сырой продукт затем перекристаллизовывают из 350 мл циклогексана и получают 49,3 г (выход = 87%) целевого продукта в виде кристаллов белого цвета. Т.пл. = 60oC. (a)D22 = +12,1o (c = 1,00; CHCl3). Приготовление 43. 1,1-диметилэтиловый эфир [2-(метилсульфонилокси)-1(R)-метилэтил]-карбаминовой кислоты. 55 г (0,314 моль) продукта, полученного согласно приготовлению 42, растворяют в 600 мл дихлорметана и добавляют 90 г (0,89 моль) триэтиламина. Смесь охлаждают до -5оС и медленно добавляют раствор 51 г (0,445 моль) метансульфонилхлорида в 100 мл дихлорметана. Затем оставляют температуру повышаться до комнатной и выдерживают при перемешивании в течение 15 часов. Реакционную среду затем выливают в 200 мл воды со льдом. Органическую фазу промывают раствором хлорида натрия, затем сушат над сульфатом магния и концентрируют при пониженном давлении. Таким образом получают 79 г (выход количественный) сырого целевого продукта в виде оранжевого цвета кристаллов, которые используют также для следующей операции. Продукт может быть очищен путем перекристаллизации из гептана. Т.пл. = 76oC. (a)D23 = +29,7о (с = 1,02; CHCl3). Приготовление 44. 1,1-диметилэтиловый эфир (2-циано-1(R)метилэтил)-карбаминовой кислоты. 79 г (0,31 моль) соединения, полученного согласно приготовлению 43, растворяют в 600 мл диметилсульфоксида и добавляют 40,5 г (0,62 моль) цианида калия. Затем реакционную среду перемешивают при 50оС в течение 15 часов. После этого охлаждают и гидролизуют в 600 мл воды со льдом. Экстрагируют 4 раза 500 мл диэтилового эфира и органические фазы сушат над сульфатом магния. После концентрирования при пониженном давлении сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью метилциклогексана с этилацетатом (8:2 по объему). Таким образом получают 42 г масла, которое кристаллизуется. После перекристаллизации из смеси метилциклогексана с диизопропиловым эфиром получают 33 г (выход = 57%) целевого продукта в виде белых кристаллов. Т.пл. = 70оС. (a)D23 = +93,2о (с = 1,00; CHCl3). Приготовление 45. 1,1-диметилэтиловый эфир (3-амино-1(R)метилпропил)-карбаминовой кислоты. Готовят раствор из 23,64 г (0,128 моль) соединения, полученного согласно приготовлению 44, в 500 мл этанола и добавляют 10 мл 1н. раствора NaOH и 7 г никеля Ренея. Смесь перемешивают в течение 48 часов под давлением водорода 20 × 105. После отфильтровывания катализатора, фильтрат нейтрализуют путем добавления 1 н. соляной кислоты и концентрируют при пониженном давлении. Остаток очищают путем хроматографии на силикагеле, элюируя смесью метилциклогексана с этилацетатом и гидроксидом аммония (7,5:2:0,5 по объему). Таким образом получают 22,1 г (выход = 91,5%) чистого целевого продукта, который кристаллизуется. Т.пл. = 73оС. (a)D23,5 = +12,0о (с = 1,00; CHCl3). 2,59 г (выход = 68%) целевого продукта в виде твердого вещества желтого цвета. 1 Н-ЯМР-спектр (CDCl3): 1,0–1,1 (д., 3Н); 1,2– 1,8 (м., 41Н); 2,3–2,7 (2м., 4Н); 3,1–3,4 (м., 8Н); 3,5–3,6 (м., 1Н); 3,9 (м., 2Н); 5,2 (м., 1Н); 5,7 (м., 1Н); 6,4 (м., 1Н); 6,8 (м., 1Н); 7,3 (м., 5Н); 8,3 (т., 1Н); 11,45 (с., 1Н). Приготовление 41. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-24-метил-12,15диоксо- 2,4,11,14,16,21,25-гептаазагексакоз-2-ендикислоты хлоргидрат. Раствор 2,52 г (3,2 × 10-3 моль) соединения, полученного согласно приготовлению 40, в 40 мл этанола подвергают гидрированию при атмосферном давлении в присутствии 200 мг 10%-ного палладия-на-угле и 0,1 мл соляной кислоты. После протекания реакции в течение 5 часов, катализатор удаляют путем отфильтровывания и раствор концентрируют при пониженном давлении. Остаток обрабатывают водой и дихлорметаном и подкисляют до рН=2. После двух экстракций водой объединенные водные фазы лиофилизируют. Полученное аморфное твердое вещество (2,2 г) обрабатывают без всякой другой очистки в следующем приготовлении. 1 Н-ЯМР-спектр (CDCl3): 1,0–1,1 (д., 3Н); 1,15–1,7 (м., 39Н); 1,8 (м., 1Н); 2,1 (м., 1Н); 2,8–3,1 (м., 9Н); 3,2–3,4 (2м., 2Н); 3,6 (с., 2Н); 7,3 (м., 2Н); 8,2 (т., 2Н); 7,8 (т., 1Н); 9 (м., 1Н); 11 (с., 1Н). Пример 12. Трис-(трифторацетат)-N-{4-[[3-(амино)бутил]-амино]-бутил}-N'-{[[[6-[(аминоиминометил)амино]-гексил]-амино]-карбонил]-метил}-мочевины. Следуя методике, аналогичной таковой примера 1, исходя из 2,2 г соединения, полученного согласно приготовлению 41, получают 1 г (выход = 43%) целевого продукта в виде аморфного твердого вещества белого цвета. 1 Н-ЯМР-спектр (DMCO-d6): 1,1–1,65 (м., 15Н); 1,7 (м., 1Н); 1,9 (м., 1Н); 3,0 (м., 10Н); 3,3 (м., 1Н); 3,6 (с., 2Н); 4,5–5,5 (с.ш., 3Н); 6,0–6,4 (м., 2Н); 6,9–7,5 (м., 2Н); 7,6 (т., 1Н); 7,8 (т., 1Н); 8(с., 2Н); 8,6 (м., 2Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,69; 26,20; 26,31; 27,20; 28,56; 29,02; 31,22; 39,85; 39,95; 41,87; 43,98; 44,59; 46,10; 48,26; 157,90; 161,07; 173,72. Приготовление 42. 1,1-диметилэтиловый эфир (2-гидрокси1(R)-метилэтил)-карбаминовой кислоты. Готовят раствор из 24,3 г (0,323 моль) 2-(R)аминопропанола в 450 мл тетрагидрофурана и 8 мл воды. Добавляют 32,6 г (0,323 моль) триэтиламина, затем медленно приливают раствор 70,5 г (0,323 моль) ди-трет.-бутил-дикарбоната (т.е. O[CO2C(CH3)3]) в 150 мл ТГФ. Реакционную среду выдерживают при перемешивании в течение 1 часа при комнатной температуре, затем концентрируют при пониженном давлении. Маслянистый остаток обрабатывают с помощью 400 мл диэтилового эфира и промывают 2 раза по 100 мл 0,1 н. раствора соляной кислоты, содержащего хлорид натрия, затем раствором бикарбоната натрия. Органическую фазу сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток обрабатывают с помощью 200 мл циклогекса 18 37217 Приготовление 46. 1,1-диметилэтиловый эфир [3-(фенилметиламино)-1(R)-метилпропил]-карбаминовой кислоты. Готовят раствор из 22,1 г (0,117 моль) соединения, полученного согласно приготовлению 45, в 300 мл диэтилового эфира и добавляют 20 г молекулярных сит 3 Å, затем 12,47 г (0,117 моль) бензальдегида. Смесь перемешивают в течение 15 часов при комнатной температуре, затем удаляют молекулярные сита путем отфильтровывания и фильтрат концентрируют при пониженном давлении. Таким образом получают 32,6 г промежуточного имина, который растворяют в 350 мл этанола. К раствору добавляют порциями 6,7 г (0,177 моль) боргидрида натрия, поддерживая температуру около 10оС. Затем перемешивают 3 часа, после чего концентрируют при пониженном давлении. Остаток обрабатывают с помощью 500 мл диэтилового эфира и органическую фазу промывают водой, сушат над сульфатом магния и концентрируют при пониженном давлении. После перекристаллизации остатка из циклогексана получают 32,5 г (выход = 98%) целевого продукта в виде желтых кристаллов. Т.пл. = 79оС. (a)D23 = -5,2о (с = 2,00; CHCl3). Приготовление 47. 1,1-диметилэтиловый эфир 9-циано-3(R)-метил-6-фенилметил- 2,6-диазанонановой кислоты. Готовят раствор из 32 г (0,115 моль) соединения, полученного согласно приготовлению 46, и 18,2 г (0,175 моль) 4-хлорбутиронитрила в 300 мл бутанола. Добавляют 14,6 г (0,138 моль) карбоната натрия и 4,8 г иодида калия и кипятят с обратным холодильником при перемешивании в течение 15 часов. Концентрируют при пониженном давлении и остаток очищают путем хроматографии на силикагеле, элюируя смесью метилциклогексана с этилацетатом (8:2, затем 1:1 по объему). Таким образом получают 39 г (выход = 98%) целевого продукта в виде очень вязкого желтого масла. (a)D23 = -6,9о (с = 2,00; CHCl3). Приготовление 48. 1,1-диметилэтиловый эфир 10-амино-3(R)метил-6-фенилметил2,6-диазадекановой кислоты. Следуя методике, аналогичной таковой приготовления 45, работая под давлением водорода 3,5 × 105 Па и исходя из 32,8 г (95 × 10-3 моль) соединения, полученного согласно приготовлению 45, получают 33 г (выход = 99%) целевого продукта в виде вязкого бесцветного масла. (a)D23 = -1,9о (с = 1,00; CHCl3). 1Н-ЯМР-спектр (CDCl3): 1,04 (д., 3Н); 1,35– 1,8 (м., 17Н); 2,3–2,5 (м., 4Н); 2,64 (т., 2Н); 3,44 (д., 1Н); 3,61 (д., 1Н); 3,62–3,8 (м., 1Н); 5,6–5,8 (с.ш., 1Н); 7,2–7,35 (м., 5Н). Приготовление 49. 1,1-диметилэтиловый эфир 10-амино-3(S)метил-6-фенилметил-2,6-диазадекановой кислоты. Вследствие реакций, аналогичных таковым приготовлений 42–48, исходя из 2(S)-амино-пропанола, получают хиральное производное целевой конфигурации (S). (a)D23 = +1,6о (с = 1,20; CHCl3). Приготовление 50. 1,1-диметилэтиловый эфир 3-(R)-метил-10[2,4-диоксо-оксазолидин-3-ил]6-фенилметил2,6-диазадекановой кислоты. Следуя методике, аналогичной таковой приготовления 10, исходя из 1,8 г (5,15 × 10-3 моль) соединения, полученного согласно приготовлению 48, получают целевой продукт в виде белых кристаллов с выходом 90%. Т.пл. = 62оС. (a)D22 = -1о (с = 1,00; CHCl3). Приготовление 51. 1,1-диметилэтиловый эфир 3-(S)-метил-10[2,4-диоксо-оксазолидин-3-ил]6-фенилметил2,6-диазадекановой кислоты. Согласно способу, идентичному таковому приготовления 50, исходя из соединения приготовления 49, получают целевой продукт в виде бесцветного масла. (a)D22 = +0,5о (с = 1,00; СHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,05 (д., 3Н); 1,35– 1,65 (м., 15Н); 2,35–2,65 (м., 4Н); 3,4–3,75 (м., 5Н); 4,67 (с., 2Н); 5,2–5,4 (с.ш., 1Н); 7,2–7,4 (м., 5Н). Приготовление 52. 1-(1,1-диметилэтиловый) эфир 3-(R)-метил6-фенилметил-13-окса-12-оксо- 2,6,11-триазапентандикислоты. Следуя методике, аналогичной таковой приготовления 11, исходя из 2 г (4,6 10-3 моль) соединения, полученного согласно приготовлению 50, получают 2,1 г (выход = 99%) целевого продукта в виде аморфных белых кристаллов. (a)D22 = -3,9о (с = 1,00; CHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,17 (д., 3Н); 1,25– 2,1 (м., 15Н); 2,35–3,3 (м., 8Н); 3,45-3,7 (м., 1Н); 4– 4,35 (м., 2Н); 4,8–5,2 (м., 1Н); 7,15–7,8 (м., 6Н); 12,1–12,7 (с., 1Н). Приготовление 53. 1-(1,1-диметилэтиловый) эфир 3-(S)-метил6-фенилметил-13-окса-12-оксо- 2,6,11-триазапентадикислоты. Следуя методике, аналогичной таковой приготовления 52, исходя из соединения, полученного согласно приготовлению 51, получают целевой продукт в виде аморфного твердого вещества белого цвета. (a)D22 = +6,0о (с = 0,41; CHCl3). Приготовление 54. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-24(R)-метил-21фенилметил-14-окса- 12,15-диоксо- 2,4,11,16,21, 25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 16, исходя из 2,08 г (4,61 × 10-3 моль) соединения, полученного в приготовлении 52, получают 3,6 г (выход = 99%) целевого продукта в виде бесцветного масла. (a)D22 = -2,3о (с = 1,00; CHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,01 (д., 3Н); 1,3-1,8 (м., 41Н); 2,3–2,5 (м., 3Н); 2,5–2,65 (м., 1Н); 3,05– 3,2 (м., 2Н); 3,2–3,3 (м., 2Н); 3,35–3,5 (м., 3Н); 3,6 (д., 1Н) 3,65–3,8 (м., 1Н); 4,5 (с., 2Н); 5,35–5,6 (м., 2Н); 6,25–6,4 (с.ш., 1Н); 7,2–7,35 (м., 5Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 55. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-24(S)-метил-21 19 37217 14-диоксо-20-фенилметил- 2,4,11,15,20,24-гексаазапентакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 18, исходя из 4,88 г (11 × 10-3 моль) соединения, полученного согласно приготовлению 7, и 2,75 г (7,88 × 10-3 моль) соединения, полученного в приготовлении 48, получают 4,8 г (выход = 80%) целевого продукта в виде вязкого масла. (a)D23 = -4,1о (с = 1,00; CHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,02 (д., 3Н); 1,251,85 (м., 41Н); 2,25–2,7 (м., 4Н); 3,05–3,8 (м., 11Н); 5,2–5,4 (с.ш., 1Н); 7–7,15 (с.ш., 1Н); 7,15–7,4 (м., 6Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 59. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)карбонил]-амино]-23(R)-метил12,14-диоксо- 2,4,11,15,20,24-гексаазапентакоз-2ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, исходя из 4,9 г (6,32 × 10-3 моль) соединения, полученного согласно приготовлению 58, получают 4,32 г (выход = 99%) целевого продукта в маслянистой форме. (a)D22 = -2,8о (с = 1,00; CHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,2 (д., 3Н); 1,25-2,0 (м., 42Н); 2,6–2,8 (м., 4Н); 3,16 (с., 2Н); 3,2–3,55 (м., 6Н); 3,6–3,85 (м., 1Н); 4,75–4,95 (с.ш., 1Н); 7,1– 7,35 (с.ш., 1Н); 7,65–7,8 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Пример 15. N-{4-[[3(R)-(амино)-бутил]амино]бутил}-N'-[6[(аминоиминометил) -амино]-гексил]-пропандиамид-трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 4,3 г (6,27 × 10-3 моль) соединения, полученного согласно приготовлению 59, после очистки путем хроматографии на силикагеле RP 18 и элюирования смесью воды с ацетонитрилом и трифторуксусной кислотой (8:1:1) по объему), получают 2,1 г (выход = 46%) целевого продукта в виде прозрачного аморфного твердого вещества. (a)D23 = +1,1о (с = 1,00; СН3ОН). 1 Н-ЯМР-спектр (DMCO-d6): 1,2 (д., 3Н); 1,2– 1,65 (м., 12Н); 1,65–1,85 (м., 1Н); 1,85–2 (м., 1Н); 2,8–3,15 (м., 12Н); 3,2–3,35 (м., 1Н); 6,7–7,5 (с.ш., 3Н); 7,62 (т., 1Н); 7,7–8,2 (м., 6Н); 8,3–8,8 (с.ш., 2Н). 13 С–ЯМР-спектр (D2O): 18,02; 23,71; 26,23; 26,35; 28,56; 28,74; 28,84; 31,23; 39,50; 40,31; 41,88; 44,30; 44,61; 46,10; 48,15; 115,25; 119,11; 158,30; 170,02; 170,34. Иммунодепрессивная активность продуктов согласно изобретению демонстрируется с помощью теста реакции трансплантата против хозяина самцов мышей B6D2F1 (гибриды первой генерации C57B1/6 x DBA/2) иммуноугнетают за счет интраперитональной инъекции циклофосфамида. Спустя три дня (день 0 эксперимента: ДO) они получают внутривенно 4 х 107 селезеночных клеток мышей C57B1/6. Животных затем распределяют на партии минимально по 8 и проводят ежедневную обработку в дни Д1–Д5 и Д7–Д10 интраперитонально. Контрольная группа получает один эксципиент. За гибелью следят до Д60. Результаты, выражаемые в виде среднего значения выживания в виде количества дней на указанную дозу, фенилметил- 14-окса-12,15-диоксо- 2,4,11,16,21, 25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 54, исходя из 1,56 г соединения, полученного согласно приготовлению 53, получают целевой продукт в количестве 2,02 г (выход = 74%) в виде бесцветного масла. (a)D22 = +1,8о (с = 1,00; СHCl3). Приготовление 56. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)- карбонил]-амино]-24(R)-метил-14окса- 12,15-диоксо- 2,4,11,16,21,25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 15, исходя из 3,6 г (4,5 × 10-3 моль) соединения, полученного согласно приготовлению 54, получают 3,2 г целевого продукта (выход = 99%) в виде светло-желтого масла. (a)D22 = -4,4о (с = 1,00; CHCl3). 1 Н-ЯМР-спектр (CDCl3): 1,18 (д., 3Н); 1,251,95 (м., 42Н); 2,6–2,8 (м., 4Н); 3,1–3,45 (м., 6Н); 3,7–3,9 (м., 1Н); 4,54 (с., 2Н); 4,65–4,8 (с.ш., 1Н); 5,8–6,1 (с.ш., 1Н); 6,4–6,6 (с.ш., 1Н); 8,3 (т., 1Н); 11,5 (с., 1Н). Приготовление 57. Бис-(1,1-диметилэтиловый) эфир 3-[/(1,1диметилэтокси)- карбонил/-амино]-24(S)-метил-14окса-12,15-диоксо- 2,4,11,16,21,25-гексаазагексакоз-2-ен-дикислоты. Следуя методике, аналогичной таковой приготовления 56, исходя из 2,0 г (2,5 × 10-3 моль) соединения, полученного согласно приготовлению 55, получают целевой продукт в виде желтого масла. (a)D22 = +5,8о (с = 1,00; СHCl3). Пример 13. 2-{[[4-[[3(R)-(амино)бутил]амино]бутил]амино]карбонилокси}-N-[6-[(аминоиминометил)-амино]-гексил]-ацетамид- трис(трифторацетат). Следуя методике, аналогичной таковой примера 1, исходя из 3,2 г (4,56 × 10-3 моль) соединения, полученного согласно приготовлению 56, получают 2,48 г (выход = 73%) целевого продукта в виде аморфного твердого белого вещества. (a)D22 = +1,1о (с = 2,00; СН3ОН). 1 Н-ЯМР-спектр (DMCO-d6): 1,18 (д., 3Н); 1,25–1,65 (м., 12Н); 1,65–1,85 (м., 1Н); 1,85–2,0 (м., 1Н); 2,85–3,15 (м., 10Н); 3,2–3,35 (м., 1Н); 4,33 (с., 2Н); 6,8–7,3 (с.ш., 3Н); 7,32 (т., 1Н); 7,62 (т., 1Н); 7,86 (т. 1Н); 7,9–8,05 (с.ш., 4Н); 8,5–8,7 (с.ш., 2Н). 13 С-ЯМР-спектр (D2O): 18,01; 23,64; 26,19; 26,28; 26,76; 28,56; 28,93; 31,21; 39,77; 40,58; 41,87; 44,60; 46,10; 48,21; 63,75; 157,55; 157,89; 171,44. Пример 14. 2-{[[4-[[3(S)-(амино)-бутил]амино]бутил]амино]карбонилокси}-N-[6-[(аминоиминометил)-амино]-гексил]-ацетамид- трис(трифторацетат). Следуя методике примера 13 и исходя из 1,75 г (2,5 × 10-3 моль) соединения, полученного согласно приготовлению 57, получают 1,5 г (выход = 82%) целевого продукта в виде аморфного твердого вещества. (a)D22 = -0,95о (с = 2,00; СН3ОН). Приготовление 58. Бис-(1,1-диметилэтиловый) эфир 3-[[(1,1диметилэтокси)-карбонил]-амино]-23(R)-метил-12, 20 37217 представлены в таблице 1, где указанные величины являются показательными согласно тесту Логранка (вероятность ниже или равна 5%). В конце сравнения также в таблице 1 приводятся значения, полученные при применении известных продуктов уровня техники: 15-дезоксиспергуалин (15– DSG) (циклоспорин А), который представляет собой в настоящее время стандартный иммунодепрессант, используемый в терапии, и продукт примера 1, описанный в европейской заявке на патент А–0105193. Из этого сравнения следует, что продукты согласно изобретению в 100 раз более активны, чем известные продукты уровня техники. Особенно продукты согласно изобретению обладают значительной активностью, начиная с 0,3 мг/кг (наиболее слабая испытуемая доза), тогда как сравнительный продукт примера 1 европейской заявки на патент А–0105193 обладает значительной активностью только начиная с 1 мг/кг, а циклоспорин А – начиная с 25 мг/кг. Более того, соединения согласно изобретению обладают отчетливо более высокой стабильностью в растворе, чем известные продукты уровня техники, особенно 15-дезоксиспергуалин. Продукты согласно изобретению могут использоваться в терапии в качестве иммунодепрессивных лечебных или профилактических агентов, особенно для предупреждения отторжения аллогенных или ксеногенных васкуляризованных или нет органов; реакции трансплантата против хозяина в результате васкуляризованной или нет трансплантации (пересадки); для лечения аутоиммунных заболеваний, генетически определенных или при обретенных (как, например, диссеминированная эритематозная волчанка, бляшечный склероз, ревматоидный полиартрит); хронических воспалительных заболеваний таких, как, например, ревматизмы суставов; так же, как для лечения любых патологий, где появляющееся иммунное нарушение является причиной или фактором, ответственным за сохранение ухудшенного клинического состояния. Продукты согласно изобретению также можно вводить в дополнение к цитотоксическим противораковым лекарственным средствам, чтобы ограничивать их вторичные эффекты, и вводить дополнительно с продуктами, полученными методом биотехнологии, в частности с рекомбинантными цитокинами, моно- и поликлональными антителами, чтобы уменьшить появление защитных антител, продуцируемых пациентом. Продукты согласно изобретению могут быть использованы для лечебной обработки паразитов, в особенности в случае малярии. Продукты согласно изобретению можно вводить орально, путем инъекции (особенно внутримышечно или внутривенно), топически (особенно в виде крема для локального нанесения, в виде глазных капель), чрескожно, ректально в виде свечей или путем ингаляции. Продукты согласно изобретению также находят свое применение в качестве фармакологических реагентов, особенно при исследовании аутоиммунных заболеваний. В таблице 1 все указанные продукты примеров согласно изобретению находятся в форме трис(трифторацетата). Таблица 1 Пример A n Хиральность 1 2 3 4 5 6 7 8 9 10 11 12 13 15 15-DSG циклоспорин А Пр. 1 по ЕР– А–0105193 -CH2-CH2-O-CH2-CH(OH)-CH(OH)-CH(OCH)3-CH(OCH)3-CHF-CHFпростая связь простая связь -CH2-NH-CH2-O -CH2 6 6 8 6 8 6 8 6 8 6 8 6 6 6 рацемат рацемат рацемат смесь диастереоизомеров смесь диастереоизомеров смесь диастереоизомеров смесь диастереоизомеров смесь диастереоизомеров смесь диастереоизомеров рацемат рацемат рацемат хиральный (форма R) хиральный (форма R) Активности, Выживание, доза, (мг/кг) (дни) 60 53 57 60 60 58 60 38 60 56 60 43 36 1 21 3 0,3 3 3 3 3 3 0,3 3 1 0,3 1 25 32 37217 Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 22 (03122) 2 – 57 – 03
ДивитисяДодаткова інформація
Назва патенту англійськоюAnalogs of 15-desoxyspergualine, method for preparation thereof, pharmaceutical composition and intermediary compounds
Автори англійськоюReno Patricia, Lebreton Luc, Dutartre Patric, Derrepas Philippe, Samres Sos
Назва патенту російськоюАналоги 15-дезоксиспергуалина, способ их получения, фармацевтическая композиция и промежуточные соединения
Автори російськоюРено Патриция, Лебретон Люк, Дютартре Патрик, Деррепас Филипп, Самрес Сос
МПК / Мітки
МПК: A61K 31/17, A61K 31/16, A61P 33/02, A61P 37/06, C07C 279/12, C07C 277/00, C07C 279/24, A61K 31/27
Мітки: 15-дезоксиспергуаліну, проміжні, сполуки, композиція, аналоги, спосіб, фармацевтична, одержання
Код посилання
<a href="https://ua.patents.su/22-37217-analogi-15-dezoksispergualinu-sposib-kh-oderzhannya-farmacevtichna-kompoziciya-ta-promizhni-spoluki.html" target="_blank" rel="follow" title="База патентів України">Аналоги 15-дезоксиспергуаліну, спосіб їх одержання, фармацевтична композиція та проміжні сполуки</a>
Похідні n-алкіленпіперидинів як антагоністи рецептора нейрокініну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 26894
Опубліковано: 29.12.1999
Автори: Мартінез Серж, Емонд-Альт Ксавьє, ван Броєкк Дідьє, Пруаєтто Венсанзо
МПК: C07D 401/12, A61K 31/44, A61K 31/454, C07D 403/12, A61P 43/00, C07D 211/46, A61K 31/4427, C07D 401/06, A61K 31/4468, C07D 211/58, A61K 31/445, C07D 211/54, C07D 409/12, A61K 31/4465, A61K 31/4545, A61K 31/4433
Мітки: проміжні, одержання, похідні, нейрокініну, сполуки, фармацевтична, n-алкіленпіперидинів, композиція, рецептора, спосіб, антагоністи
Текст:
...где Alk является (С,-Сэ)алкильной группой, R представляет собой атом водорода 40 или С^-С^алкил, Z представляет собой фенил, незамещенный или моно- или дизамещенный галогеном или (С^-Сзіалкоксигруппой, наф 26894 7 тил, замещенный галогеном, фенилметильной группой, замещенной на фениле (С,С4}алкоксигруппой, * обозначает хиральный центр, в виде рацемата или оптически чистых изомеров, или их соли с минеральными или органическими кислотами,...
Заміщені фенілімідазолідини, спосіб їх одержання, фармацевтична композиція, проміжні сполуки

Номер патенту: 34428
Опубліковано: 15.03.2001
Автори: ГУБЕ Франсуа, ТЕТШ Жан-Жорж, ФІЛІБЕР Даніель, Гейар-Келлі Мартін
МПК: C07D 233/76, A61K 31/4166, A61P 13/02, C07D 233/88, C07D 233/86, A61P 15/00, C07D 233/84, A61K 31/415, C07D 233/80, C07D 233/74, A61P 35/00, A61P 43/00
Мітки: одержання, фенілімідазолідини, заміщені, проміжні, спосіб, композиція, сполуки, фармацевтична
Текст:
...(Г) в которой R"i, R^, - А"—В"— имеют значения, указанные выше для Ri, R2 и —А—В—, с учетом того, что когда —А"—В"— представляет собой группу —СО—N(RMt3)—. в которой R"'3 представляет собой атом водорода или линейный или разветвленный алкильнмй радикал, включающий не более 7 атомов yf лерода, Y является атомом кислорода, R"i представляет собой цианильный радикал, причем данный метод отличается тем, что продукт формулы (V). Hal 34428 в...
Аналоги вітаміну d3, спосіб їх одержання, фармацевтична композиція
Номер патенту: 27370
Опубліковано: 15.09.2000
Автори: Ускоковіч Мілан Радоє, Скалоне Мікельанджело, Окабе Масамі, Маклейн Джон Артур, Доран Томас І.
МПК: A61P 17/00, C07F 7/18, C07J 13/00, A61K 31/59, C07J 51/00, C07J 9/00, A61P 3/02, A61K 31/695, C07C 401/00
Мітки: аналоги, фармацевтична, спосіб, вітаміну, композиція, одержання
Текст:
...примерно 290-320 нм 5 Фармацевтическая композиция для лечения гиперпролиферативных заболеваний кожи, таких как псориаз, и для печения заболеваний сальных желез, таких как угри и себорейная экзема, включающая активное на 11 'по и фармацевтически пригодный носитель, отличающаяся тем, что в качестве активного начала фармацевтическая композиция содержит эффективное количество соединения формулы I CM ,16-тетраэн23-ин-3,25-диол [диацетат или...
Сірковмісні похідні імідазолу, що мають гіпотензивну активність, спосіб їх отримання, проміжні сполуки і фармацевтична композиція

Номер патенту: 26130
Опубліковано: 07.06.1999
Автори: Вевер Жан-Поль, Жукей Сімон, Фортен Мішель, Амон Жіль, Кай Жан-Клод, Корб'є Ален
МПК: A61K 31/415, C07D 401/12, A61K 31/4178, A61P 15/00, C07D 233/84, C07D 403/12, A61P 9/12, C07D 233/94, A61P 43/00, A61K 31/4164, A61P 9/10, C07D 233/90, A61P 21/00, A61P 9/00, A61P 13/02, C07D 403/10
Мітки: фармацевтична, сірковмісні, активність, проміжні, спосіб, композиція, сполуки, імідазолу, гіпотензивну, похідні, отримання, мають
Формула / Реферат:
1. Серосодержащие производные имидазола общей формулы (I)где R1 - алкил;R2 - водород, гидроксиметил, карбоксигруппа, группа формулы -S(=O)2-A, где A - низший алкил или фенил,- группа формулы -S(=O)2(CH2)k-B, где k = 1 и 2, B - низшая алкокси- или гидроксигруппа,- группа -S-D, где D - незамещенный фенил или замещенный галогеном, низшей алкоксигруппой низший алкил, возможно замещенный галогеном, и...
Похідні хінолон- і нафтиридонкарбонової кислоти у вигляді суміші ізомерів або окремих ізомерів, фармацевтична композиція, що має антибактеріальну активність та проміжні сполуки

Номер патенту: 35554
Опубліковано: 16.04.2001
Автори: ФІЛІПС Томас, ПЕТЕРСЕН Уве, БРЕММ Клаус Дітер, ХАЛЛЕР Інго, КРЕБС Андреас, ШЕНКЕ Томас, Ендерманн Райнер, МЕТЦГЕР Карл-Георг, ГРОХЕ Клаус
МПК: C07D 471/04, C07D 498/04, C07D 519/00
Мітки: суміші, композиція, хінолон, фармацевтична, окремих, похідні, антибактеріальну, має, сполуки, ізомерів, кислоти, нафтиридонкарбонової, проміжні, активність, вигляді
Формула / Реферат:
1. Производные хинолон- и нафтиридонкарбоновой кислоты общей формулы (I),(І)где:А = СН, CF, CCl, С-ОСН3, N,X1 = Н, Hal, NH2, СН3,R1 = С1-С3-алкил, циклопропил, фенил, который может быть от одного до трех раз замещен галогеном, илиА и R1 вместе означают С-О-СН2-СН(СН3)-,R2 = Н или С1-С3-алкил,В = остаток...
Попередній патент: Спосіб виготовлення тонкого листового полірованого скла
Наступний патент: Багатоцільовий коток
Випадковий патент: Модифікований спосіб препарування щитовидної залози у щурів