3-b-d-рибофуранозилтіазоло[4,5-d]піримідинові нуклеозиди та їх застосування
Номер патенту: 79764
Опубліковано: 25.07.2007
Автори: Аверетт Деврон Р., Веббер Стефен І., Леннокс Джозеф Р., Руден Ерік Дж.





Формула / Реферат
(21) 20040604959
(57)
1. Сполуки, представлені Формулою І:
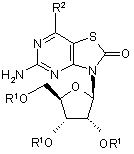 , (I)
, (I)
де:
R1 являє собою незалежно Н, -C(O)R3 або рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4, де R3 являє собою заміщений або незаміщений алкіл, а R4 являє собою Н або заміщений або незаміщений алкіл;
R2 являє собою Н, OR5 або N(R6)2, де R5 являє собою незалежно Н або алкіл та R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце; та
якщо R2 являє собою -ОН, тоді принаймні одна з R1-груп являє собою рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4;
або фармацевтично прийнятні солі.
2. Сполука або фармацевтично прийнятна сіль за п. 1, де принаймні одна з R1-груп являє собою рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 є незалежно вибраним з Н або алкілу та R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце.
3. Сполука або фармацевтично прийнятна сіль за п. 2, де принаймні одна з R1-груп являє собою L-амінокислотну групу -C(O)CHNH2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 являє собою заміщений алкіл та R6 являє собою незалежно Н або заміщений або незаміщений алкіл.
4. Сполука або фармацевтично прийнятна сіль за п. 3, де принаймні одна з R1-груп являє собою L-амінокислотну групу -C(O)CHNH2R4, де R4 являє собою -СН(СН3)2, та де R1-групи, що залишилися, являють собою Н; а R2 являє собою ОН.
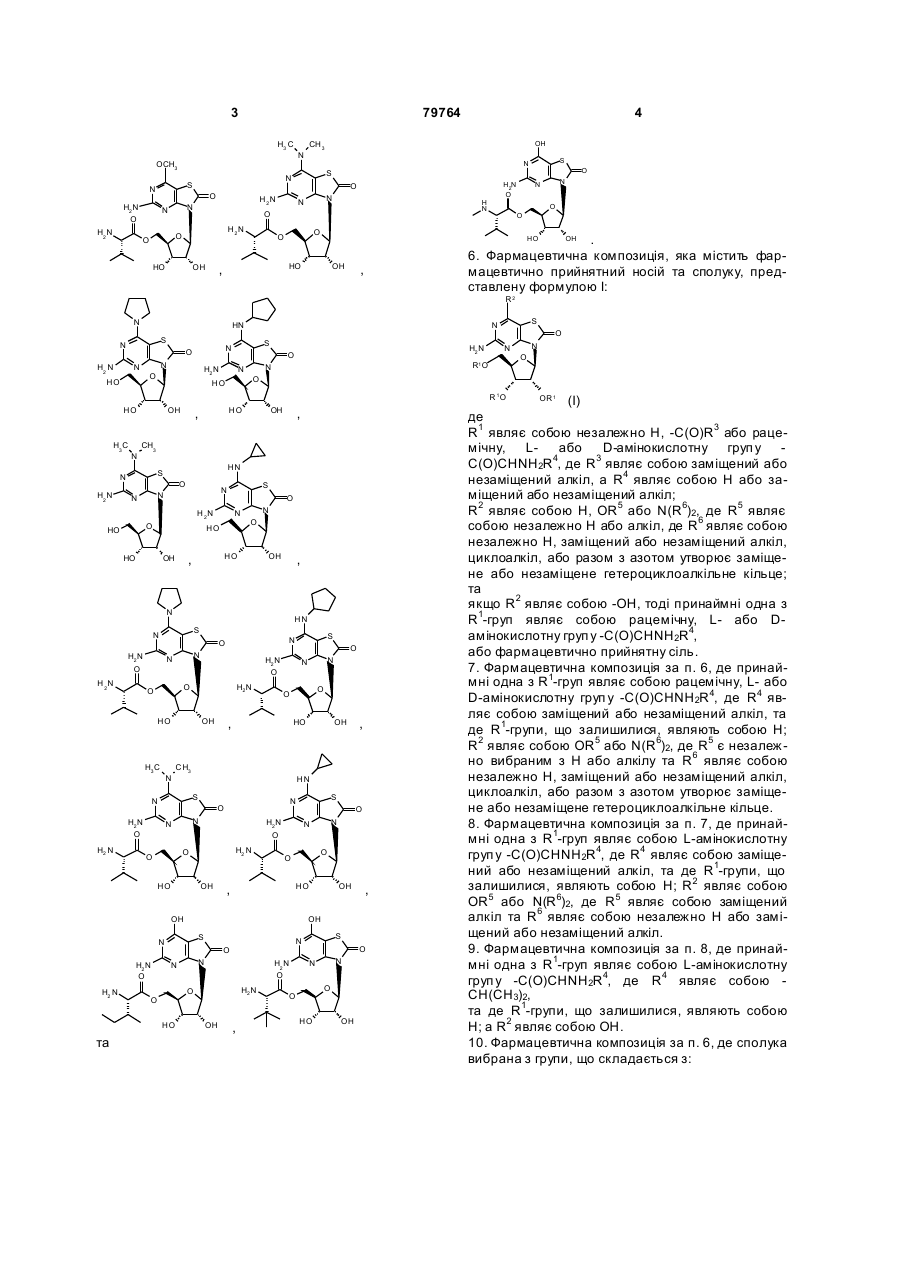
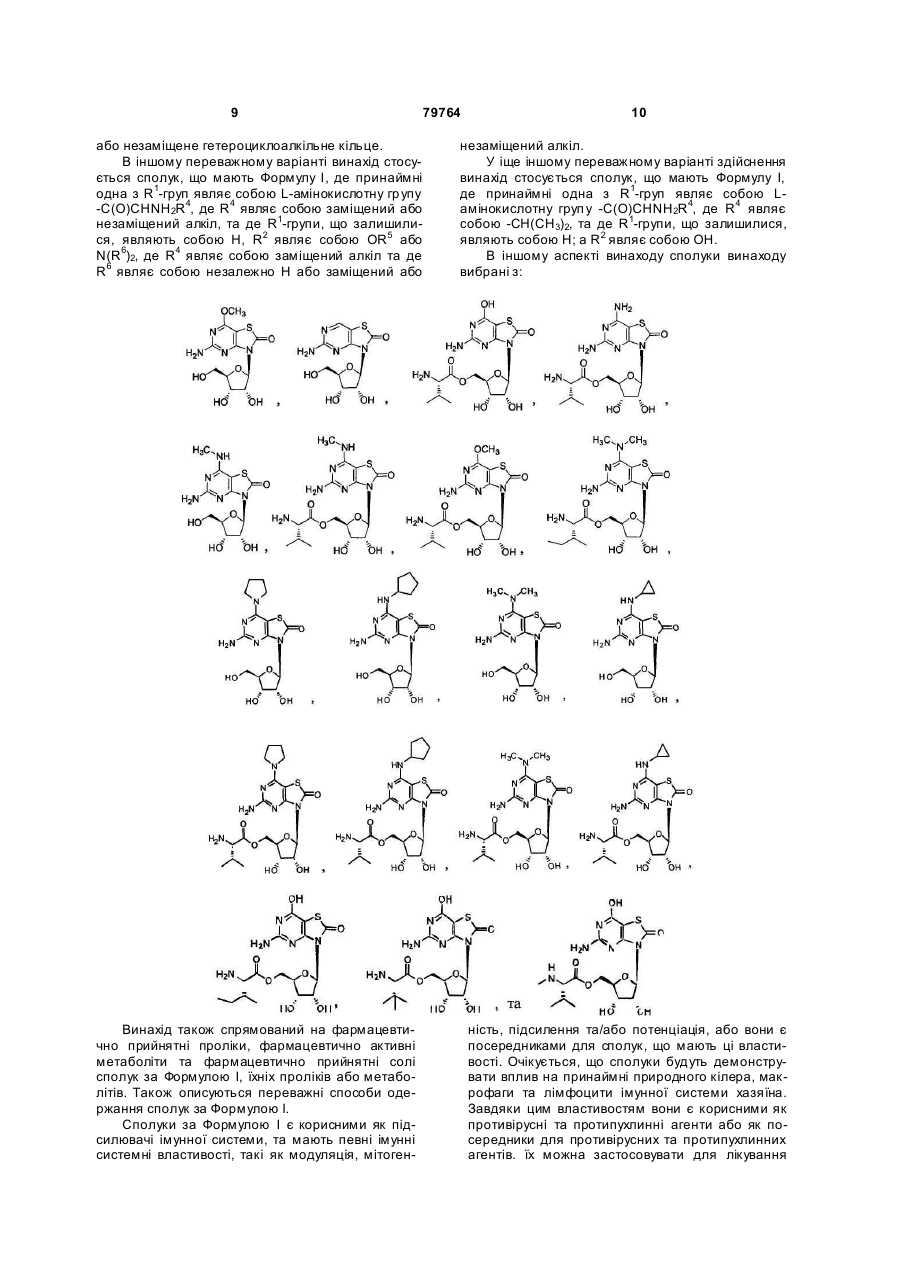
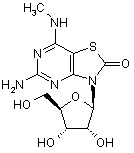
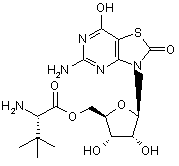
5. Сполука або фармацевтично прийнятна сіль за п. 1, вибрана з групи, яка
складається з:
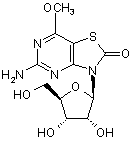 ,
, 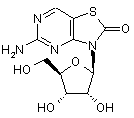 ,
,
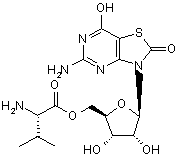 ,
, 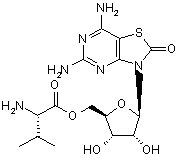 ,
,
 ,
, 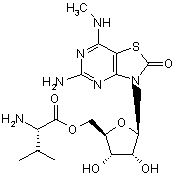 ,
,
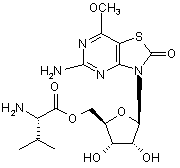 ,
, 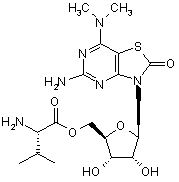 ,
,
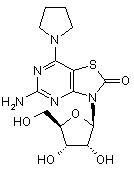 ,
, 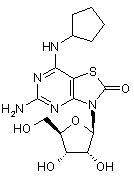 ,
,
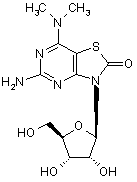 ,
, 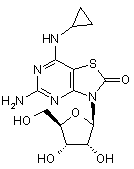 ,
,
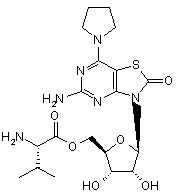 ,
, 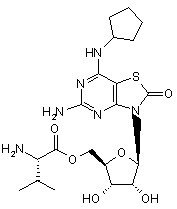 ,
,
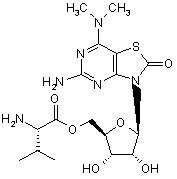 ,
, 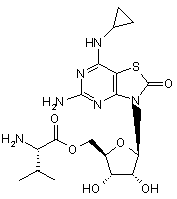 ,
,
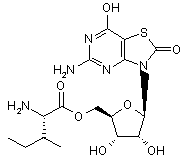 ,
, 
та
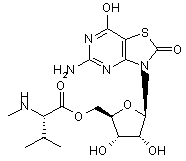 .
.
6. Фармацевтична композиція, яка містить фармацевтично прийнятний носій та сполуку, представлену формулою І:
(I)
де
R1 являє собою незалежно Н, -C(O)R3 або рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4, де R3 являє собою заміщений або незаміщений алкіл, а R4 являє собою Н або заміщений або незаміщений алкіл;
R2 являє собою Н, OR5 або N(R6)2, де R5 являє собою незалежно Н або алкіл, де R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце; та
якщо R2 являє собою -ОН, тоді принаймні одна з R1-груп являє собою рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4,
або фармацевтично прийнятну сіль.
7. Фармацевтична композиція за п. 6, де принаймні одна з R1-груп являє собою рацемічну, L- або D-амінокислотну групу -С(O)СНNН2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 є незалежно вибраним з Н або алкілу та R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце.
8. Фармацевтична композиція за п. 7, де принаймні одна з R1-груп являє собою L-амінокислотну групу -C(O)CHNH2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 являє собою заміщений алкіл та R6 являє собою незалежно Н або заміщений або незаміщений алкіл.
9. Фармацевтична композиція за п. 8, де принаймні одна з R1-груп являє собою L-амінокислотну групу -C(O)CHNH2R4, де R4 являє собою -СН(СН3)2,
та де R1-групи, що залишилися, являють собою Н; а R2 являє собою ОН.
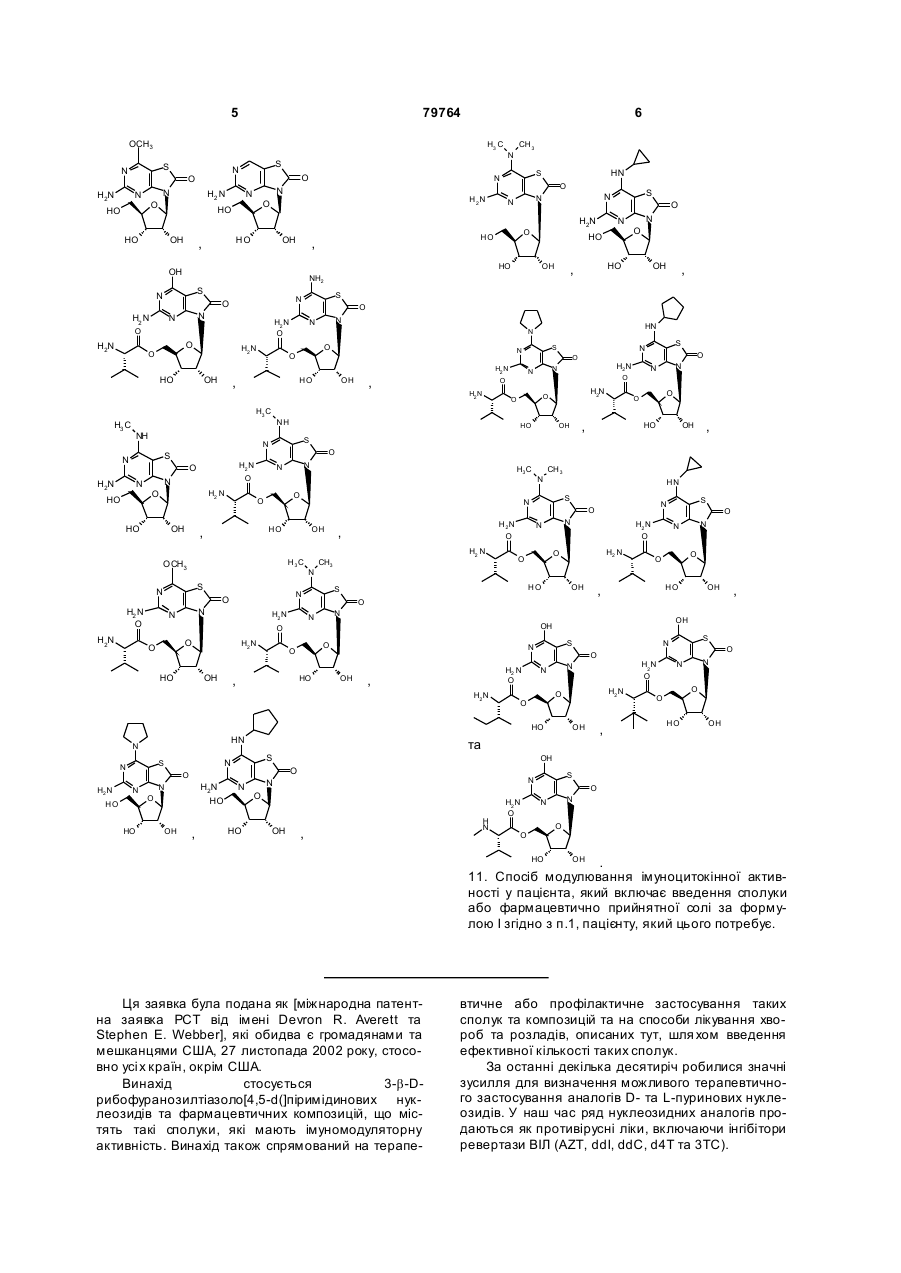
10. Фармацевтична композиція за п. 6, де сполука вибрана з групи, що складається з:
, ,
, ,
, ,
, ,
, 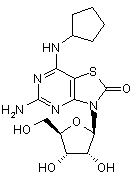 ,
,
, 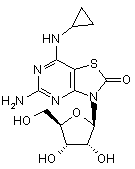 ,
,
, ,
, ,
,
та
.
11. Спосіб модулювання імуноцитокінної активності у пацієнта, який включає введення сполуки або фармацевтично прийнятної солі за формулою I згідно з п. 1, пацієнту, який цього потребує.
Начальник відділу
Л.С. Плюто
Виконавець
С.М. Томачинський
Текст
1. Сполуки, представлені Формулою І: 2 UA 1 OH , 3 79764 H3 C N OCH3 O H2 N O H2 N H 2N HO N N H N O O O N N O O . 6. Фармацевтична композиція, яка містить фармацевтично прийнятний носій та сполуку, представлену формулою І: HO HO , OH H2N O O O O O S N S H 2N N N OH CH 3 N S N 4 OH , OH R2 N N H2 N N N H2N O H2N O HO S N O S N HN S R1O N N O N R 1O H3 C N OH HO , OH CH3 HN S N H2 N , O S N N N O H 2N O HO N HO HO N O HO , OH OH , N HN S N H 2N N N H2 N HO H3C N OH H2 N O O , HO CH3 N O H2N N N O H2 N O O OH O O HO , OH H2N O N N H2N O H2N O HO , OH S N O O OH OH S N та , S N N HO H2 N OH HN O N N N S H2N O O H2 N O O O S N O H2N O , O O HO HO N O N N O O HO OH OR 1 (I) де R1 являє собою незалежно Н, -C(O)R3 або рацемічну, Lабо D-амінокислотну груп у C(O)CHNH2R4, де R3 являє собою заміщений або незаміщений алкіл, а R4 являє собою Н або заміщений або незаміщений алкіл; R2 являє собою Н, OR5 або N(R6)2, де R5 являє собою незалежно Н або алкіл, де R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце; та якщо R2 являє собою -ОН, тоді принаймні одна з R1-груп являє собою рацемічну, L- або Dамінокислотну груп у -C(O)CHNH 2R4, або фармацевтично прийнятну сіль. 7. Фармацевтична композиція за п. 6, де принаймні одна з R1-груп являє собою рацемічну, L- або D-амінокислотну груп у -С(O)СНNН2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 є незалежно вибраним з Н або алкілу та R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце. 8. Фармацевтична композиція за п. 7, де принаймні одна з R1-груп являє собою L-амінокислотну груп у -C(O)CHNH2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 являє собою заміщений алкіл та R6 являє собою незалежно Н або заміщений або незаміщений алкіл. 9. Фармацевтична композиція за п. 8, де принаймні одна з R1-груп являє собою L-амінокислотну груп у -C(O)CHNH2R4, де R4 являє собою СН(СН3)2, та де R1-групи, що залишилися, являють собою Н; а R2 являє собою ОН. 10. Фармацевтична композиція за п. 6, де сполука вибрана з групи, що складається з: 5 79764 6 OCH3 H3 C S N H2N N O N N O H2 N O HO S HO OH HO , H 2N O HO OH N H 2N N H2N HO OH HO H2 N O HO OH H2 N N H3C O OH S HO H2N H2N OH N N H2 N HN HO OH O O HO OH H2 N O , N HO HO H 2N N O HO OH , HO H2N O , OH O N N O O HO OH S N N O OH H2N O N N S OH O N , OH O O OH N S N S N HO , та O O O OH HO H2N H2 N O N N N HN S H2N O O O S N N O H2N N , CH 3 N S N , N OH O O O HO S H 2N O , CH3 N O O O O , OH N HO H 3C N O N N O O N H2N O O HO N , N H 2N , H2 N O S OCH3 H2 N O OH S N N N O N , O H2 N O NH H2N O OH HN S N HO N S HO , N O , NH N OH O N N O H3C H2N HO N O H2N O O N O O N S N O N O H3 C H2N NH2 S S N N N HO OH H2 N O O HO , HN S N N N CH 3 N , H N H2N O O O N N O HO OH . 11. Спосіб модулювання імуноцитокінної активності у пацієнта, який включає введення сполуки або фармацевтично прийнятної солі за формулою I згідно з п.1, пацієнту, який цього потребує. Ця заявка була подана як [міжнародна патентна заявка РСТ від імені Devron R. Averett та Stephen E. Webber], які обидва є громадянами та мешканцями США, 27 листопада 2002 року, стосовно усі х країн, окрім США. Винахід стосується 3-b-Dрибофуранозилтіазоло[4,5-d(]піримідинових нуклеозидів та фармацевтичних композицій, що містять такі сполуки, які мають імуномодуляторну активність. Винахід також спрямований на терапе втичне або профілактичне застосування таких сполук та композицій та на способи лікування хвороб та розладів, описаних тут, шля хом введення ефективної кількості таких сполук. За останні декілька десятиріч робилися значні зусилля для визначення можливого терапевтичного застосування аналогів D- та L-пуринових нуклеозидів. У наш час ряд нуклеозидних аналогів продаються як противірусні ліки, включаючи інгібітори ревертази ВІЛ (AZT, ddI, ddC, d4T та 3ТС). 7 79764 Різноманітні аналоги D- та L-пуринових нуклеозидів також досліджували у пошуках імуномодуляторів. Було показано, наприклад, що аналоги гуанозину, що мають замісників на 7- та/або 8положеннях, стимулюють імунну систему. [Дивись Reitz et al, J. Med. Chem., 37, 3561-78 (1994); Michael et al., J. Med. Chem., 36, 3431-36 (1993)]. В іншому дослідженні, [патент США №5821236, автори Krenitsky et al.], описує 6-алкокси-похідні арабінофуранозилпуринових похідних, які є корисними для лікування пухлин. [У патенті США №5539098, автори Krenitsky et al.], також повідомляється про інгібітори вірусу вітряної віспи, включаючи 5'-О-пропріоніловий та 5'-О-бутириловий естери 2-аміно-6-метокси-9-(b-Dарабінофуранозил)-9Н-пурину. Було продемонстровано, що 7-деазагуанозин та аналоги мають противірусну активність у мишей проти різноманітних РНК-вірусів, навіть якщо сполука не має противірусних властивостей у клітинній культурі. 3Деазагуанінові нуклеозиди та нуклеотиди також продемонстрували суттєво широкий спектр противірусної активності проти певних ДНК- та РНКвірусів. [Revankar et al., J. Med. Chem., 27,1489-96 (1984)]. Певні 7- та 9-деазагуанінові С-нуклеозиди демонструють спроможність захищати від летального наслідку зараження вірусом [Semliki Forest. Girgis et al., J. Med. Chem., 33, 2750-55 (1990)]. Відібрані 6-сульфенамідта 6сульфінамідпуринові нуклеозиди [описані у патенті США №4328336, автори Robins et al.], як такі, що демонстрували значну протипухлинну активність. Певні піримідо[4,5-d]піримідинові нуклеозиди [описуються у патенті США №5041542, автори Robins et al.], як сполуки, ефективні у лікуванні від L1210 мишей BDF1. Ці певні нуклеозиди були запропоновані внаслідок їхньої ролі як імуномодуляторів. [Дивись Bonnet et al., J. Med. Chem., 36, 63553 (1993)]. Також Wang та інші [міжнародна патентна публікація WO 98/16184] повідомляють, що пуринові L-нуклеозидні сполуки та їх аналоги застосовувалися для лікування інфекції, інвазії, неоплазми, автоімунної хвороби або для модулювання аспектів імунної системи. Крім того, у [патентах США №5041426 та 4880784, автори Robins et al.], описуються 3-b-D-рибофуранозилтіазоло-[4,5d(]піримідини, що демонструють значну імунну активність, включаючи проліферацію клітин селезінки мишей, та активність in vivo щодо вірусу Semliki Forest. Одна можлива мішень імуномодуляції включає стимулювання або супресію лімфокінів Тh1 та Th2. Клітини типу І (Thl) виробляють інтерлейкін 2 (IL2), фактор некрозу пухлини (TNFa) та інтерферон гама (IFNg), та вони відповідають, перш за все, за клітина-опосередкований імунітет, такий як гіперчутливість уповільненого типу та противірусний імунітет. Клітини типу 2 (Th2) виробляють інтерлейкіни, IL-4, IL-5, IL-6, IL-9, IL-10 та IL-13, та вони, перш за все, сприяють гуморальним імунним реакціям, таким як реакції на алергени. [Дивись, наприклад, Mosmann, Annu. Rev. Immunol, 1, 145-73 (1989)]. Було продемонстровано, що D-гуанозинові аналоги неоднаково впливають на лімфокіни, IL-1, IL-6, INFa тa TNFa (не безпосередньо) in vitro 8 (Goodman, Int. J. Immunopharmacol, 10, 579-88 (1988); [патент США №4746651, автор Goodman)] та in vivo [Smee et alo., Antiviral Res., 15,229 (1991); Smee et al., Antimicrobial Agents and Chemotherapy, 33, 1487-92 (1989)]. Проте, спроможність аналогів D-гуанозину, таких як 7-тіо-8-оксогуанозин, модулювати цитокіни Типу 1 та Типу 2 безпосередньо у Т-клітинах була неефективною або її не було описано. Крім того, відомо, що пероральне введення багатьох пуринових нуклеозидних аналогів має деякі складнощі, які є наслідком поганої абсорбції, поганої розчинності або деградації у травному шляху, що спричиняється кислотними або лужними умовами або дією ензимів та/або комбінаціями цих явищ. Отже, залишається необхідність у пуринових нуклеозидних аналогах, удосконалених з точки зору доступності, толерантності для перорального введення, для модуляції аспектів імунної системи. Цей винахід звернувся до вирішення цієї потреби, відкривши 3-b-D-рибофуранозилтіазоло[4,5-d]піримідинові нуклеозиди, їхні фармацевтично прийнятні проліки, фармацевтично прийнятні метаболіти та фармацевтично прийнятні солі (такі сполуки, проліки, метаболіти та солі разом позначаються терміном "агенти"), описані нижче, які є корисними як імуномодулятори. У загальному аспекті винахід стосується сполук за Формулою І де R1 являє собою незалежно Н, -C(O)R3 або рацемічну, L- або D-амінокислотну групу C(O)CHNH2R4, де R3 являє собою заміщений або незаміщений алкіл, a R4 являє собою Η або заміщений або незаміщений алкіл; R2 являє собою Н, OR5 або N(R6)2, де R5 являє собою незалежно Η або алкіл та де R6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене або незаміщене гетероциклоалкільне кільце; та де, якщо R2 являє собою -ОН, тоді принаймні одна з R1-rpyn являє собою рацемічну, L- або Dамінокислотну груп у -С(О)СHNH2R4. У переважному варіанті здійснення винахід стосується сполук, що мають Формулу І, де принаймні одна з R1-rpyn являє собою рацемічну, Lабо D-амінокислотну груп у -C(O)CHNH 2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н; R2 являє собою OR5 або N(R6)2, де R5 є незалежно вибраним з Η або алкілу, та де R 6 являє собою незалежно Н, заміщений або незаміщений алкіл, циклоалкіл, або разом з азотом утворює заміщене 9 або незаміщене гетероциклоалкільне кільце. В іншому переважному варіанті винахід стосується сполук, що мають Формулу І, де принаймні одна з R1-груп являє собою L-амінокислотну гр yпy -C(O)CHNH2R4, де R4 являє собою заміщений або незаміщений алкіл, та де R1-групи, що залишилися, являють собою Н, R2 являє собою OR5 або N(R6)2, де R4 являє собою заміщений алкіл та де R6 являє собою незалежно Η або заміщений або Винахід також спрямований на фармацевтично прийнятні проліки, фармацевтично активні метаболіти та фармацевтично прийнятні солі сполук за Формулою І, їхніх проліків або метаболітів. Також описуються переважні способи одержання сполук за Формулою І. Сполуки за Формулою І є корисними як підсилювачі імунної системи, та мають певні імунні системні властивості, такі як модуляція, мітоген 79764 10 незаміщений алкіл. У іще іншому переважному варіанті здійснення винахід стосується сполук, що мають Формулу І, де принаймні одна з R1-груп являє собою Lамінокислотну груп у -C(O)CHNH 2R4, де R4 являє собою -СН(СН 3)2, та де R1-групи, що залишилися, являють собою Н; a R2 являє собою ОН. В іншому аспекті винаходу сполуки винаходу вибрані з: ність, підсилення та/або потенціація, або вони є посередниками для сполук, що мають ці властивості. Очікується, що сполуки будуть демонструвати вплив на принаймні природного кілера, макрофаги та лімфоцити імунної системи хазяїна. Завдяки цим властивостям вони є корисними як противірусні та протипухлинні агенти або як посередники для противірусних та протипухлинних агентів. їх можна застосовувати для лікування 11 ураженого хазяїна, коли вони служать як активний початок придатних фармацевтичних композицій. В одному аспекті винаходу сполуки за Формулою І застосовуються для лікування повного ряду вірусних хвороб у ссавців шля хом введення ссавцю терапевтично ефективної кількості сполук. Вірусні хвороби, які, як передбачається, можна лікувати сполуками за Формулою І, включають гострі та хронічні інфекції, спричинені як РНК, так і ДНК-вірусами. Не обмежуючи в жодному разі ряд вірусних інфекцій, які можна лікувати, сполуки за Формулою І є особливо корисними у лікуванні інфекцій, спричинених аденовірусом, цитомегаловірусом, вірусом гепатиту A (HAV), вірусом гепатиту В (HBV), флавівірусами, включаючи вірус жовтої гарячки та вірус гепатиту С (HCV), вірусом простого герпесу типу 1 та 2, оперізувальним герпесом, вірусом герпесу 6 людини, вірусом імунодефіциту людини (ВІЛ), вірусом папіломи людини (HPV), вірусом грипу А, вір усом грипу В, кіром, вірусом парагрипу, поліовірусом, поксвірусом (включаючи вірус віспи людини та вірус віспи мавпи), риновірусом, респіраторносинцитіальним вірусом (RSV), численними родинами вірусів, які спричиняють геморагічні гарячки, включаючи Аренавіруси (LCM, вірус Junin, вірус Machup, вірус Guanarito та вірус гарячки Lassa), віруси Вunyа (вірус Hunta та вірус Рифт-Валлі) та Філовіруси (Ебола та Марбург), рядом вірусних енцефалітів, включаючи вірус енцефаліту За хідного Нілу, вір ус ЛаКросс, вірус каліфорнійського енцефаліту, вір ус енцефаліту венесуельського сапу, вір ус енцефаліту східного сапу, вірус енцефаліту західного сапу, вір ус японського енцефаліту, вір ус Kysanur Forest та віруси кліщового енцефаліту, такі як вірус конго-кримської геморагічної гарячки. В іншому аспекті винаходу сполуки за Формулою І застосовуються для лікування бактеріальних, грибкових та протозойних інфекцій у ссавців шляхом введення ссавцю терапевтично ефективної кількості сполук. Передбачається, що сполуками цього винаходу можна лікувати повний ряд патогенних мікроорганізмів, включаючи без обмежень ті організми, які є стійкими до антибіотиків. Спроможність сполук за Формулою І активувати численні компоненти імунної системи минає механізми резистентності, які, як звичайно визначається, зменшують сприйнятливість до антибіотиків, та через це лікування інфекцій сполуками за Формулою І у ссавців, спричинених такими резистентними мікроорганізмами, є особливо корисними у цьому винаході. В іншому аспекті винаходу сполуки за Формулою І застосовуються для лікування пухлин у ссавців шляхом введення ссавцю терапевтично ефективної кількості сполук. Як передбачається, пухлини та ракові хвороби, які можна лікувати, включають ракові хвороби, спричинені вірусом, та ефект може включати інгібування трансформації інфікованих вірусом клітин у неопластичний стан, інгібування розповсюдження вірусів від трансформованих клітин до інших здорових клітин та/або припинення росту трансформованих 79764 12 вірусом клітин. Очікується, що сполуки за Формулою І будуть корисними проти широкого спектру пухлин, включаючи, проте не обмежуючись ними, карциноми, саркоми та лейкози. Такий клас включає рак грудей, рак товстої кишки, рак сечового міхура, рак легенів, рак простати, рак шлунку та рак підшлункової залози та лімфобластичний та мієлоїдний лейкоз. В іншому аспекті винаходу спосіб лікування ссавця включає введення терапевтично та/або профілактично ефективної кількості фармацевтичного засобу, що містить сполуку винаходу. У цьому аспекті ефект може мати відношення до модуляції певної частини імунної системи ссавця, особливо до модуляції активності цитокінів Th1 та Th2, включаючи, проте не обмежуючись лише родиною інтерлейкінів, наприклад, від IL-1 до IL12, та інших цитокінів, таких як фактор некрозу пухлини α та інтерферон, включаючи інтерферональфа, інтерферон-тета та інтерферон гама, та їх подальших ефекторів. Там, де виникає модуляція цитокінів Тh1 та Th2, передбачається, що модуляція може включати стимуляцію як Th1 так і Th2, супресію як Th1, так і Th2, стимуляцію Th1 або Th2 та супресію іншого, або бімодальну модуляцію, при якій один вплив на рівні Th1/Th2 (такий як загальна супресія) виникає при високій концентрації, у той час коли інший вплив (такий як стимуляція Тh1 або Th2 та супресія іншого) виникає при низькій концентрації. В іншому аспекті винаходу фармацевтичні композиції, що містять сполуку за Формулою І, вводяться у терапевтично ефективній дозі ссавцю, який отримує протиінфекційні ліки, які не включено у Формулу І. В переважному аспекті цього винаходу фармацевтичні композиції, що містять сполуку за Формулою І, вводяться у терапевтично ефективній дозі разом з протиінфекційними ліками, які діють безпосередньо на інфікувальний агент, з метою інгібувати ріст або знешкодити інфікувальний агент. У переважному аспекті винаходу фармацевтична композиція, що включає терапевтично ефективну кількість сполуки згідно з Формулою І, пропонує поліпшену пероральну придатність та введення як імуномодулятора. В іншому переважному аспекті винаходу фармацевтична композиція, що включає терапевтично ефективну кількість сполуки за Формулою І, пропонує приховування активної структури, коли агент проходить крізь лімфоїдну тканину, що вистилає шлунок, тим самим мінімізуючи активацію цієї тканини, наслідком чого стає поліпшена пероральна переносність. Фіг.1 являє собою графічне зображення рівнів у плазмі ізаторибіну та інтерферону-альфа у мишей. Там, де наступні терміни застосовуються у цьому описі винаходу, вони застосовуються так, як визначено нижче: Терміни "що включає" та "включаючи" застосовуються тут у їхньому відкритому, не обмежувальному смислі. Термін "нуклеозид" позначає сполуку, що складається з будь-якої пентозної або модифіко 13 ваної пентозної складової, приєднаної до специфічного положення гетероциклу або до природного положення пурину (9-положення), або піримідину (1-положення), або до еквівалентної позиції в аналогу. Термін "пурин" позначає азотні біциклічні гетероцикли. Термін "піримідин" позначає азотні моноциклічні гетероцикли. Термін "D-нуклеозиди" позначає нуклеозидні сполуки, що мають D-рибозну цукрову складову (наприклад, аденозин). Термін "L-нуклеозиди" позначає нуклеозидні сполуки, що мають L-рибозну цукрову складову. Термін "алкіл", як застосовується тут, позначає алкільну грyпy з прямим або розгалуженим ланцюгом, яка має від 1 до 12 атомів вуглецю. Приклади алкільних груп включають метил (Me, який можна також структурно описати як "/"), етил (Et), n-пропіл, ізопропіл, бутил, ізобутил, сек.бутил, трет-бутил (tBu), пентил, ізопентил, третпентил, гексил, ізогексил тощо. Термін "алкокси" позначає -О-алкіл. Ілюстративні приклади включають метокси, етокси, пропокси тощо. Термін "галоген" представляє хлор, фтор, бром або йод. Термін "гало" представляє хлоро, фторо, бромо або йодо. Термін "циклоалкіл" позначає насичений або частково насичений, моноциклічний або сконденсований, або спірополіциклічний вуглецевий цикл, що має від 3 до 12 атомів у кільці. Ілюстративні приклади циклоалкільних гр уп включають наступні складові: Термін "гетероциклоалкіл" позначає моноциклічну, або сконденсовану, або спірополіциклічну кільцеву структуру, яка є насиченою або частково насиченою, та яка мають у кільці від 3 до 12 атомів на кільце, які вибрані з атомів С та гетероатомів Ν, Ο та S. Ілюстративні приклади гетероциклоалкільних груп включають 79764 14 Термін "арил" (Аг) позначає моноциклічний, або сконденсований, або спірополіциклічний ароматичний вуглецевий цикл (кільцеву стр уктуру, що має атоми у кільці, які усі являють собою вуглець), який має у кільці від 3 до 12 атомів на кільце. Ілюстративні приклади арильних груп включають наступні складові: Термін "заміщений" позначає, що визначена група або складова несе один або більше замісників. Термін "незаміщений" позначає, що визначена група не несе жодного замісника. Заміщений алкіл, циклоалкіл або гетероциклоалкіл є заміщеним одним або більше замісниками, включаючи галоген (F, СІ, Вг або І), нижчий (С1-6) алкіл, -ОН, -NO2, -CN, -СО2Н, -О-нижчий алкіл, -арил, -арил-нижчий алкіл, -СО2СН3, CONH2, -OCH 2CONH2, -NH2, -SO2NH2, галоалкіл(наприклад, -CF3, -CH2CF3), -О-галоалкіл (наприклад, -OCF3, -OCHF2) і тому подібні. Термін "імуномодулятор" позначає природний або синтетичний продукт, спроможній модифікувати нормальну або порушену імунну систему шля хом стимуляції або супресії. Термін "запобігання" позначає спроможність сполуки або композиції винаходу запобігати хворобі, визначеній тут, у пацієнтів, яким поставили діагноз цієї хвороби або які мають ризик розвитку такої хвороби. Термін також охоплює запобігання наступному розвитку хвороби у пацієнтів, які вже страждають від цієї хвороби або які вже мають симптоми такої хвороби. Термін "лікування" позначає: I) запобігання виникненню хвороби, розладу або стану у тварини, яка може бути схильною до хвороби, розладу та/або стану, проте у неї іще не діагностували наявність цієї хвороби; II) інгібування хвороби, розладу або стану, тобто припинення її розвитку, та III) ослаблення хвороби, розладу або стану, тобто спричинення регресіїї хвороби, розладу та/або стану. Терміни "a" та "b" вказують на специфічну стереохімічну конфігурацію замісника на асиметричному атомі вуглецю у хімічній структурі, як проілюстровано. Сполуки, описані тут, усі знаходяться у конфігурації D-фуранозилу. Сполуки винаходу можуть демонструвати явище та утомерії. Незважаючи на те, що Формула І не може виразно описати усі можливі таутомеричні форми, слід розуміти, що Формула І призначена для того, щоб представити будь-яку таутомеричну форму зображеної сполуки, та та 15 кож не слід обмежуватися лише специфічною формою сполуки, зображеної на структурній формулі. Наприклад, стосовно Формули І, зрозуміло, що незважаючи на те, чи зображено або не зображено замісників у їхній енольній формі або їхній кетоформі, вони представляють одну сполуку (як показано на прикладі нижче). Деякі сполуки винаходу можуть існувати як єдині стереоізомери (тобто, суттєво вільні від інших стереоізомерів), рацемати та/або суміші енантіомерів та/або діастереомерів. Ми наполягаємо, що усі такі єдині стереоізомери, рацемати та їхні суміші входять до обсягу цього винаходу. Переважно, щоб сполуки винаходу, які є оптично активними, застосовувалися в оптично чистій формі. Як взагалі зрозуміло для фахівців у галузі, оптично чиста сполука, що має один хіральний центр (тобто, один асиметричний атом вуглецю) є такою, що складається суттєво з одного з дво х можливих енантіомерів (тобто, є енантіомерно чистою), та оптично чиста сполука, що має більш ніж один хіральний центр, є такою, що вона є як діастереомерно чистою, так і енантіомерно чистою. Переважно, сполуки цього винаходу застосовуються у формі, яка є принаймні на 90% оптично чистою, тобто формою, що містить принаймні 90% єдиного ізомеру (80% надлишку енантіомеру ("е.е.") або надлишку діастереомеру ("d.e.")), більш переважно принаймні 95% (90% надлишку енантіомеру або надлишку діастереомеру), навіть більш переважно принаймні 97,5% (95% надлишку енантіомеру або надлишку діастереомеру) та найбільш переважно принаймні 99% (98% надлишку енантіомеру або надлишку діастереомеру). Крім того, Формула І призначена охоплювати сольватовані, а також несольватовані форми ідентифікованих структур. Наприклад, Формула І включає сполуки вказаної структури як у гідратній, так і у негідратній формах. Інші приклади сольватів включають структури у комбінації з ізопропанолом, етанолом, метанолом, ДМСО (DMSO), етилацетатом, оцтовою кислотою або етаноламіном. Окрім сполук за Формулою І винахід включає фармацевтично прийнятні проліки, фармацевтично активні метаболіти та фармацевтично прийнятні солі таких сполук та метаболітів. "Фармацевтично прийнятні проліки" являють собою сполуку, яка може перетворюватися за певними фізіологічними умовами або через сольволіз у визначену сполуку або у фармацевтично прийнятну сіль такої сполуки до того, як вона буде демонструвати свої фармакологічні властивості. Звичайно проліки мають такий склад, щоб 79764 16 досягти поліпшеної хімічної стійкості, поліпшеної прийнятності та сприятливості, поліпшеної біологічної придатності, подовження тривалості дії, поліпшеної селективності щодо органів, удосконаленої технології виготовлення ліків (наприклад, підвищеної розчинності у воді) та/або зниження побічних ефектів (наприклад, токсичності). Проліки можна легко одержати зі сполук за Формулою І, застосовуючи способи, відомі у галузі, такі як описані у [Burger's Medicinal Chemistry and Drug Chemistry, 1, 172-178, 949-982 (1995). Дивись також Bertolini et al., J. Med. Chem., 40, 2011-2016 (1997); Shan, et al., J. Pharm. Sci., 86 (7), 765-767; Bagshawe, DrugDev. Res., 34, 220-230 (1995); Bodor, Advances in Drug Res., 13, 224-331 (1984); Bundgaard, Design ofProdrugs (Elsevier Press 1985); Larsen, Design and Application ofProdrugs, Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991); Dear et al., J. Chromatogr. B, 748,281-293 (2000); Spraul et al., J. Pharmaceutical & Biomedical Analysis, 10,601-605 (1992); та Prox et al., Xenobiol, 3,103-112(1992)]. Вираз "Фармацевтично активний метаболіт" призначений позначати фармакологічно активний продукт визначеної сполуки або її солі, який виробляється як наслідок метаболізму у тілі. Після потрапляння у тіло більшість ліків являють собою субстрати для хімічних реакцій, які можуть змінювати їхні фізіологічні властивості та біологічні ефекти. Ці метаболічні конверсії, які звичайно впливають на полярність сполук за Формулою І, змінюють спосіб, за яким ліки розповсюджуються у тілі та виводяться з нього. Однак, у деяких випадках метаболізм ліків є необхідним для терапевтичного ефекту. Наприклад, протиракові ліки класу анти-метаболітів повинні перетворюватися на їхні активні форми, після того, як вони потраплять у ракову клітину. Оскільки більшість ліків зазнають метаболічної трансформації певного типу, біохімічні реакції, які відіграють роль у метаболізмі ліків, можуть бути численними та різноманітними. Головне місце метаболізму ліків - це печінка, проте інші тканини можуть також брати участь. Характерною ознакою багатьох цих трансформацій є те, що продукти метаболізму або "метаболіти" є більш полярними, ніж первинні ліки, хоча і полярні ліки іноді утворюють менш полярний продукт. Речовини з високими коефіцієнтами розподілення ліпід/вода, які легко проходять крізь мембрани, також легко дифундують назад з сечі ниркового канальця крізь клітини епітелію ниркового канальця у плазму. Отже, такі речовини звичайно мають низький нирковий кліренс та тривалу стійкість у тілі. Якщо ліки перетворюються у ході метаболізму у більш полярну сполуку, сполуку з більш низьким коефіцієнтом розподілення, їхня реабсорбція з канальця значно знизиться. Крім того, специфічні секреторні механізми для аніонів та катіонів у проксимальних ниркових канальцях та у паренхіматозних печінкових клітинах діють на високополярні речовини. Як специфічний приклад, фенацетин (ацетофенетидин) та ацетанілід обидва являють собою 17 м'які болезаспокійливі та жарознижувальні агенти, проте вони трансформуються у тілі у більш полярний та більш ефективний метаболіт, ргідроксиацетанілід (ацетамінофен), який широко застосовується сьогодні. Коли дозу ацетаніліду дають пацієнтові, наступні метаболіти досягають максимуму та розкладаються у плазмі послідовно. Протягом першої години ацетанілід являє собою головний компонент плазми. Протягом другої години, коли рівні ацетаніліду падають, концентрація ацетамінофену метаболіту досягає максимуму. Зрештою, через декілька годин головним компонентом плазми стає наступний метаболіт, який є інертним та може виводитися з тіла. Отже, концентрація одного або більше метаболітів у плазмі, а також самих ліків, може бути важливою з фармакологічної точки зору. "Фармацевтично прийнятна сіль" призначена позначати сіль, що зберігає біологічну ефективність вільних кислот та лугів визначеної сполуки та яка не є небажаною з біологічної точки зору або з будь-якої іншої точки зору. Сполука винаходу може мати достатньо кислотну, достатньо лужну або обидві функціональні грyпи та внаслідок цього може реагувати з будь-якими з ряду неорганічних або органічних лугів та неорганічних або органічних кислот, утворюючи фармацевтично прийнятну сіль. Приклади фармацевтично прийнятних солей включають ті солі, які одержуються шляхом реакції сполук цього винаходу з мінеральною або органічною кислотою або неорганічним лугом, такі як сульфати, піросульфати, бісульфати, сульфіти, бісульфіти, фосфати, моногідрофосфати, дигідрофосфати, метафосфати, пірофосфати, хлориди, броміди, йодиди, ацетати, пропіонати, деканоати, каприлати, акрилати, форміати, ізобутирати, капроати, гептаноати, пропіолати, оксалати, малонати, сукцинати, суберати, себакати, фумарати, малеати, бутен-1,4діоати, гексин-1,6-діоати, бензоати, хлорбензоати, метилбензоати, динітробензоати, гідроксибензоати, метоксибензоати, фталати, сульфонати, ксиленсульфонати, фенілацетати, фенілпропіонати, фенілбутирати, ци трати, лактати, gгідроксибутирати, гліколати, тартрати, метансульфонати, пропансульфонати, нафтален-1сульфонати, нафтален-2-сульфонати та манделати. Якщо сполука винаходу є лугом, бажану фармацевтично прийнятну сіль можна одержати за будь-яким придатним способом, доступним у галузі, наприклад, шляхом обробки вільного лугу неорганічною кислотою, такою як хлористоводнева кислота, бромистоводнева кислота, сірчана кислота, азотна кислота, фосфорна кислота тощо, або органічною кислотою, такою як оцтова кислота, малеїнова кислота, бурштинова кислота, мигдальна кислота, фумарова кислота, малонова кислота, піровиноградна кислота, щавлева кислота, гліколева кислота, саліцилова кислота, піранозидилова кислота, така як глюкуронова кислота або галактуронова кислота, альфаоксокислота, така як лимонна кислота або винна кислота, амінокислота, така як аспарагінова кислота або глютамінова кислота, ароматична кис 79764 18 лота, така як бензойна кислота або корична кислота, сульфонова кислота, така як ртолуолсульфокислота або етансульфонова кислота тощо. Якщо сполука винаходу є кислотою, бажану фармацевтично прийнятну сіль можна одержати за будь-яким придатним способом, наприклад, шляхом обробки вільної кислоти неорганічним або органічним лугом, таким як амін (первинний, вторинний або третинний), гідроксид лужного металу або гідроксид лужноземельного металу тощо. Ілюстративні приклади придатних солей включають органічні солі, що походять з амінокислот, таких як гліцин та аргінін, аміаку, первинних, вторинних та третинних амінів, циклічних амінів, таких як піперидин, морфолін та піперазин, та неорганічні солі, що походять з натрію, кальцію, калію, магнію, марганцю, заліза, міді, цинку, алюмінію та літію. У випадку, коли агенти являють собою тверді речовини, фахівці у галузі зрозуміють, що сполуки та солі винаходу можуть існувати у різних кристалічних або поліморфних формах, усі з яких, як передбачається, знаходяться в об'ємі цього винаходу та визначених формул. Наступний аспект цього винаходу спрямований на фармацевтичну композицію, що включає фармацевтично прийнятний носій або розріджувач та терапевтично ефективну кількість сполуки за Формулою І, фармацевтично прийнятної сілі, гідрату, естеру, сольвату, проліків, метаболіту або стереоізомеру. Сполуки за Формулою І є корисними у виробництві фармацевтичних складів, що включають їх ефективну кількість у поєднанні з або як домішка до наповнювачів або носіїв, придатних як для ентерального, так і для парентерального застосування. По суті, склади цього винаходу, придатні для перорального введення, можуть бути у формі окремих одиниць, таких як капсули, крохмальні капсули, таблетки, пастилки, при цьому кожна містить попередньо визначену кількість активного інгредієнта, у формі порошку або гранул, у формі розчину або суспензії у водній рідині або неводній рідині, або у формі емульсії масла у воді або емульсії води у маслі. Активний інгредієнт може також бути у формі болюсу, електуарію (кашки) або пасти. Композиція може бути звичайно приготована у виді стандартної дозованої форми, такої як таблетка, капсула, водна суспензія або розчин. Такі склади звичайно включають тверді, напівтверді або рідкі носії. Приклади носіїв включають лактозу, декстрозу, цукрозу, сорбіт, маніт, крохмалі, аравійську камедь, фосфат кальцію, мінеральне масло, масло какао, олія какаового дерева, кокосове масло, альгінати, трагакант, желатин, сироп, метилцелюлозу, сорбітанполікосіетилен-монолаурат, метил-гідроксибензоат, пропіл-гідрокси-бензоат, тальк, стеарат магнію тощо. Особливо переважні варіанти технології виготовлення лікарських засобів включають таблетки та желатинові капсули, які включають активний інгредієнт разом з (а) розріджувачами, таким 19 як лактоза, декстроза, цукроза, маніт, сорбіт, целюлоза, висушений крохмаль зернових та гліцин, та/або мастильними речовинами, такими як діоксид кремнію, тальк, стеаринова кислота, її магнієва або кальцієва сіль та поліетиленгліколь. Таблетки можуть також містити зв'язувальні речовини, такі як алюмосилікат магнію, крохмальна паста, желатин, трагакант, метилцелюлоза, натрієва карбозиметилцелюлоза та полівінілпіролідон, носії, такі як лактоза та крохмаль зернових, дезінтегратори, такі як крохмалі, агар, альгінова кислота або її натрієва сіль, та спінювальні суміші, та/або абсорбенти, барвники, ароматизатори та підсолоджувані. Композиції винаходу можуть бути стерилізованими та/або містити ад'юванти, такі як консерванти, стабілізатори, речовини, що сприяють набуханню, або емульгатори, стимулятори розчину, солі для регулювання осмотичного тиску та/або буфери. Крім того, композиція може також містити інші терапевтично цінні речовини. Водні суспензії можуть містити емульгатори та суспендувальні речовини, поєднані з активним інгредієнтом. Усі пероральні форми дози можуть, крім того, містити підсолоджувачі, та/або ароматизатори, та/або барвники. Ці композиції готують за звичайними способами змішування, гранулювання або нанесення покриття, відповідно, та вони містять приблизно від 0,1 до 75% активного початку, переважно приблизно від 1 до 50% активного початку. Таблетку можна виготовляти шляхом стискання або пресування активного початку необов'язково разом з одним або більше додатковими інгредієнтами. Стиснені таблетки можна приготувати шляхом стискання у придатному пристрої активного початку у вільній плинній формі, такій як порошок або гранули, необов'язково змішаного із зв'язувальним агентом, мастильним агентом, інертним розріджувачем, поверхнево-активним агентом або диспергатором. Пресовані таблетки можна виготовити шляхом пресування у придатному пристрої суміші активного початку у вигляді порошку та придатного носія, зволоженого інертним рідким розріджувачем. При парентеральному введенні композиція буде звичайно у стандартній дозованій стерильній формі для ін'єкцій (водний ізотонічний розчин, суспензія або емульсія) з фармацевтично прийнятним носієм. Такі носії переважно є нетоксичними, прийнятними для парентерального введення та містять нетерапевтичні розріджувачі або розчинники. Приклади таких носіїв включають воду, водні розчини, такі як фізіологічний розчин (ізотонічний розчин хлориду натрію), розчин Рингера, розчин декстрози та розчин Хенкса, та неводні носії, такі як 1,3-бутандіол, нелеткі олії (наприклад, кукурудзяну олію, бавовняну олію, арахісову олію, кунжутн у олію та синтетичний моно- або ди-гліцерид), етилолеат та ізопропілмеристат. Олеагінові суспензії можна приготувати за способами, відомими у галузі, застосовуючи придатні диспергатори або зволожувачі та суспендувальні агенти. Серед прийнятних розчинників або суспендувальних середовищ є стерильні нелеткі олії. З цією метою можна застосовувати будь-яку 79764 20 змішану нелетку олію. Жирні кислоти, такі як олеїнова кислота та її гліцеридні похідні, включаючи оливкову олію та рицинову олію, особливо у їхні х поліоксиетилованих формах, є також корисними у приготуванні ліків, які можна вводити шляхом ін'єкцій. Такі масляні розчини або суспензії можуть також містити довголанцюгові спиртові розріджувачі або диспергатори. Стерильний фізіологічний розчин являє собою переважний носій, та сполуки часто є суттєво водорозчинними, щоб їх можна було виготовити як розчини для усіх передбачених випадків. Носій може містити невелику кількість домішок, таких як речовини, які підвищують розчинність, ізотонічність та хімічну стійкість, наприклад, антиоксиданти, буфери та консерванти. При ректальному введенні композиція буде звичайно виготовлятися у стандартній дозованій формі, такій як супозиторії або крохмальні капсули. Ці композиції можна приготувати шля хом змішування сполуки з придатними неподразнювальними наповнювачами, які є твердими при кімнатній температурі, але стають рідкими при ректальній температурі, так що вони розтануть у прямій кишці, виділяючи при цьому сполуку. Звичайні наповнювачі включають кокосову олію, бджолиний віск та поліетиленгліколі або інші жирні емульсії або суспензії. Склади, придатні для нозального або трансбукального введення (такі як саморухомі порошкорозподілювальні склади) можуть включати приблизно від 0,1% до приблизно 5% (мас.%) активного початку або, наприклад, приблизно 1% (мас.%) активного початку. Крім того, деякі склади можна сформувати у під'язикову пастилку або таблетку. Крім того, сполуки можна вводити місцево, особливо коли стани, які слід лікувати, о хоплюють ділянки або органи, які є легко доступними для місцевого застосування, включаючи розлади очей, шкіри або нижнього кишкового тракту. Для місцевого застосування до ока або офтальмологічного застосування сполуки можуть входити до складу мікронізованих суспензій в ізотонічному стерильному фізіологічному розчині з відрегульованим рН або, переважно, до складу розчину в ізотонічному стерильному фізіологічному розчині з відрегульованим рН з або без консерванту, такого як бензилалконійхлорид. Альтернативно, сполуки можуть входити до складу мазей, наприклад вазелінових. Для місцевого застосування до шкіри, сполуки можуть входити до складу придатних мазей, що містять сполуки, суспендовані або розчинені, наприклад, у сумішах з одним або більше з наступних інгредієнтів: мінеральне масло, рідкий вазелін, білий вазелін, пропіленгліколь, поліоксиетилен сполука, поліоксипропіленова сполука, емульгувальний віск та вода. Альтернативно, сполуки можуть входити до складу придатних лосьйонів або кремів, що містять активну сполуку, суспендовану або розчинену у, наприклад, сумішах з одним або більше з наступних інгредієнтів: мінеральне масло, сорбітан-моностеарат, полісорбат 60, цетилестерний віск, цетеариловий 21 спирт, 2-октилдодеканол, бензиловий спирт та вода. Місцеве застосування у нижньому кишковому тракті можна здійснювати у складах ректальних супозиторіїв (дивись вище) або у складах, придатних для клізми. Склади можна звичайно представити у стандартній дозованій формі та можна приготувати за будь-якими способами, відомими у галузі фармації. Усі способи включають етап поєднання активного початку з носієм, який складається з одного або більше додаткових інгредієнтів. Взагалі, склади готують шля хом однорідного та ретельного приведення активного початку у сполучання з рідким носієм або тонко подрібненим твердим носієм або з обома, а потім, якщо необхідно, формування продукту у бажаний склад. Фармацевтична композиція цього винаходу застосовується у кількості, що є терапевтично ефективною, та кількості, що застосовуються, можуть залежати від бажаного профілю виділення, концентрації фармацевтичної композиції, необхідної для сенсибілізації ефекту, та тривалості терміну, протягом якого фармацевтична композиція повинна виділятися для лікування. Сполуки за Формулою І цього винаходу переважно вводяться у вигляді капсули або таблетки, що містить єдину або розділену дозу сполуки, або у вигляді стерильного розчину, суспензії або емульсії для парентерального введення у єдиній або розділеній дозі. Сполуки винаходу застосовуються у композиції у кількостях, що є терапевтично ефективними. Незважаючи на те, що ефективна кількість сполук за Формулою І буде залежати від певної сполуки, що застосовується, кількості цих сполук, що змінюються від приблизно 1% до приблизно 65%, легко залучалися до системи доставки рідкого або твердого носія. Для медичного застосування кількість, яка необхідна для того, щоб сполука за Формулою І досягла терапевтичного ефекту, буде різнитися залежно від певної сполуки, яка вводиться, шляху введення, ссавця, який зазнає лікування, та певного розладу відносно хвороби, з якою мають справу. Придатна системна доза сполуки за Формулою І для ссавця, що страждає від, або імовірно страждає від будь-якого стану, описаного тут, звичайно становить діапазон від приблизно 0,1 до приблизно 100мг основи на кілограм ваги тіла. Зрозуміло, що звичайний лікар-фахівець або ветеринар-фахівець легко зможе визначити та призначити кількість сполуки, ефективну для бажаного профілактичного або терапевтичного лікування. При цьому лікар або ветеринар може застосовува ти внутрішньовенну кульку з наступною інфузією та з повторними введеннями, якщо це вважається необхідним. У способах цього винаходу сполуки можна вводити, наприклад, перорально, парентерально, шляхом інгаляції, місцево, через задній прохід, назально, трансбукально, під язик, вагінально, крізь шлунок або через імплантований резервуар у дозованих складах, які містять традиційні нетоксичні фармацевтично 79764 22 прийнятні носії, ад'юванти та наповнювачі. Парентеральне введення включає, проте не обмежується лише ними, наступні приклади: способи внутрішньовенної, підшкірної, внутрішньом'язової, інтраспинальної, внутрішньокісткової, інтраперитоніальної, вн утрішньооболонкової, внутрішньошлункової, внутрішньогрудної або внутрішньочерепної ін'єкції та інфузії, такої як за допомогою сабдуральної помпи. Переважними є інвазивні способи, зокрема пряме введення у зруйновану нервову тканину. Незважаючи на те, що сполуки за Формулою І можна вводити самостійно, бажано вводити їх як частину фармацевтичного складу. Для того, щоб бути терапевтично ефективними у мішенях центральної нервової системи, сполуки, що застосовуються у способах цього винаходу, повинні легко проникати крізь гематоенцефалічний бар'єр, коли їх водять периферійно. Сполуки, які не можуть проникати крізь гематоенцефалічний бар'єр, проте, можуть іще бути ефективними, коли їх вводять внутрішньошлунковим способом. Сполуки, що застосовуються у способах цього винаходу, можна вводити шляхом єдиної дози, численних розділених доз або шляхом тривалої інфузії. Оскільки сполуки є дрібними, легко дифундують та є відносно стійкими, вони добре відповідають тривалій інфузії. Помпа, зокрема, підшкірна або сабдуральна помпа, є переважною для тривалої інфузії. Для способів цього винаходу можна застосовувати будь-який ефективний режим введення, який регулює хронометраж та послідовність дози. Дози сполук переважно включають фармацевтичні стандартні дозовані форми, які включають ефективну кількість активної сполуки. Під ефективною кількістю розуміється кількість, що є достатньою для забезпечення підсиленої імунної реакції та/або отримання бажаних корисних ефектів завдяки введенню однієї або більше стандартної дозованої форми. Приклад щоденної одиничної дози для хазяїна-хребетного включає кількість від приблизно 0,001мг/кг до приблизно 50мг/кг. Звичайно, рівні дози від приблизно 0,1мг до приблизно 10000мг сполуки активного початку є корисними для лікування вищезазначених станів, при цьому переважні рівні становлять від приблизно 0,5мг до приблизно 2000мг. Специфічний рівень дози для будь-якого певного пацієнта буде варіюватися залежно від різноманітних факторів, включаючи активність специфічної сполуки, що застосовується, вік, вагу тіла, загальний стан здоров'я, стать та харчування пацієнта, час введення, швидкість виведення, будь-яку комбінацію сполуки з іншими ліками, суворість певної хвороби, яку лікують, та форму та шлях введення. Звичайно, результати експериментів in vitro стосовно визначення ефективності доз дають корисну рекомендацію щодо належних доз для введення пацієнтові. Досліди на тваринних моделях також можуть допомогти. Питання визначення належних рівнів доз є також добре відомими у науці. Сполуки та композиції можна вводити разом 23 з одним або більше терапевтичних агентів або (і) разом у єдиній структурі, або (іі) окремо в індивідуальних складах, розроблених для отримання оптимальних швидкостей виділення їхнього відповідного активного агента. Кожен склад може містити від приблизно 0,01% до приблизно 99,99% (мас.%), переважно від приблизно 3,5% до приблизно 60% (мас%), сполуки винаходу, а також один або більше фармацевтичних наповнювачів, таких як зволожувачі, емульгатори та буфери рН. Коли сполуки, що застосовуються у способах винаходу, вводяться у комбінації з одним або декількома іншими терапевтичними агентами, специфічні рівні дози для таких агентів будуть залежати від міркувань, таких, на які вказано вище для композицій та способів винаходу взагалі. Для способів цього винаходу будь-який режим введення, який регулює хронометраж та послідовність доставки сполуки, можна застосовувати та повторювати, якщо необхідно, щоб здійснити лікування. Такий режим може включати попереднє лікування та/або спільне введення з додатковими терапевтичними агентами. Агенти винаходу можна одержати, застосовуючи шля хи реакції та схеми синтезу, які описано нижче, застосовуючи загальні процедури, відомі у галузі, застосовуючи початкові матеріали, які є легко доступними. Синтез сполук, які не наведено як приклади, згідно з цим винаходом можна успішно виконати шляхом модифікацій, відомих фахівцям у галузі, наприклад, шляхом відповідного захисту гр уп, що уводяться, шляхом заміни на інші придатні реагенти, відомі у науці, або шляхом виконання звичайних модифікацій умов реакції. Альтернативно, зрозуміло, що інші реакції, відмінні від описаних тут та які є загально відомими у галузі, можна буде застосовувати для одержання інших сполук винаходу. Одержання сполук У схемах синтезу, описаних нижче, якщо не вказано інше, температури позначаються у грaдycax Цельсію, а усі частини та відсотки вказано за масою. Реагенти купували у комерційних постачальників, таких як Aldrich Chemical Company або Lancaster Synthesis Ltd., та їх застосовували без подальшого очищення, якщо не вказано інше. Тетрагідрофуран (THF) та Ν,Νдиметилфорамід (DMF) купували у Aldrich у пляшках Sure Seal та застосовували у тому вигляді, в якому їх отримали. Якщо інше не вказано, наступні розчинники та реагенти дистилювали в атмосфері сухого азоту. THF та ЕtO2 очищали дистиляцією від Na-бензофенонкетилу; СН2Сl2, діізопропіламін, піридин та Et3N очи щали дистиляцією від СаН2; MeON очи щали дистиляцією від, по-перше, P2O5, а потім від СаН2; МеОН очищали дистиляцією від Mg; PhMe, EtOAc та і-PrOAc очищали дистиляцією від СаН 2; TFAA очищали шляхом простої перегонки в атмосфері сухого аргону. Реакції, описані нижче, виконували взагалі в умовах позитивного тиску аргону при кімнатній температурі (якщо не зазначається інше) у безводних розчинах, а до реакційних колб пристосу 79764 24 вали гумові перегородки для введення субстратів та реагентів крізь шприць. Скляний посуд висушили у печі та/або висушили шля хом нагрівання. Реакції проаналізували шляхом тонкошарової хроматографії та їх припиняли, звертаючи увагу на витрату початкового матеріалу. Аналітичну тонкошарову хроматографію (ТШХ) виконували на силікагелевих пластинках з алюмінієвою основою 60 F254 (EM Science) розміром 0,2мм та за нею спостерігали за допомогою ультрафіолетового випромінювання (254нм) з наступним нагріванням з комерційною етаноловою фосфомолібденовою кислотою. Препаративну тонкошарову хроматографію (ТШХ) виконували на силікагелевих пластинках з алюмінієвою основою 60 F254 (EM Science) розміром 0,1мм та за нею спостерігали за допомогою ультрафіолетового випромінювання (254нм). Змішування звичайно виконували шляхом збільшення об'єму реакційної суміші удвічі розчинником, що застосовується у реакції, або екстракційним розчинником з наступним промиванням вказаними водними розчинами, застосовуючи 25% від об'єму, використаному при екстракції, якщо не вказано інше. Розчини продукту висушували над безводним Na2SO4 та/або Mg2SO 4 до фільтр ування та випаровування розчинників в умовах зниженого тиску на роторному випарнику та спостерігали як розчинники видалялися в умовах вакуум у. Колоночну хроматографію здійснювали з позитивним тиском, застосовуючи силікагель з пористістю у 230-400 меш або нейтральний оксид алюмінію з пористістю у 50200 меш. Гідрогеноліз виконували в умовах тиску, вказаного у прикладах, або при атмосферному тиску. Спектри 1Н-ЯМР реєстрували на апараті Varian Mercury-VX400 при 400МГц, а спектри 13СЯМР реєстрували при 75МГц. Спектри ЯМР отримали стосовно розчинів CDCl3 (надані у млн -1), застосовуючи хлороформ як контрольний стандарт (7,27млн -1 та 77,00млн -1), CD3OD (3,4 та 4,8млн-1 та 49,3млн -1), ДМСО-d6 або внутрішньо тетраметилсилан (0,00млн -1), коли він був доречним. Інші розчинники для ЯМР застосовувалися за необхідністю. Для позначення максимальної мультиплетності застосовуються наступні абревіатури: с (синглет), д (дублет), т (триплет), к (квартет), м (мультиплет), шир. (розширений), дд (дублет дублетів), дт (дублет триплетів). Константи взаємодії, якщо наводяться, визначаються у Герцах (Гц). Інфрачервоні (IR) спектри реєструвалися на спектрометрі FT-IR Spectrometer як чисті олії, як гранули КВг або як розчини CDCl3, та коли їх наведено, вони визначаються у хвильових числа х (см -1). Мас-спектри, які наведено, являють собою (+)-ES LC/MS (рідкісна хроматографія/масспектроскопія), яку проводив відділ аналітичної хімії фірми Anadys Pharmaceuticals, Inc. Елементний аналіз був проведений Atlantic Microlab, Inc., з м. Норкросс, штат Джорджія. Точки плавлення (Т. пл.) визначали на відкритому капілярному апараті, та вони не корегувалися. В наданих схемах синтезу та в описаних екс 25 79764 26 периментальних процедурах застосовано багато звичайних хімічних абревіатур, THF (тетрагідрофуран), DMF (Ν,Ν-диметилформамід), ЕtOАс (етилацетат), DMSO (ди-метилсульфоксид, ДМСО), DMAP (4-диметиламінопіридин), DBU (1,8-діазацикло[5.4.0.]ундек-7-ен), DCM (4(диціанометилен)-2-метил-6-(4-диметиламіностирил)-4H-піран), МСРВА(3хлорпероксибензойна кислота), EDC (і-(3диметиламінопропіл)-3-етилкарбодііміду гідро хлорид), HATU (О-(7-азабензотриазол-1-іл)1,1,3,3-тетраметилуронію гексафторфосфат), НОВТ (1-гідроксибензотриазолу гідрат), TFAA (трифтороцтовий ангідрид), руВОР (бензотриазол-і-ілокси)трипіролідинфосфонію гексафторфосфат), DIEA (діізопропілетиламін) тощо. Схема 1 зображує загальну процедуру одержання 5'-амінокислотних естерів 5-аміно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2,7-діону. У звичайній схемі синтезу, 2',3'-гідроксильні групи b-D-рибозної складової 5-аміно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2,7-діону, по-перше, захищають, переважно ацетонідом, як зображено у 2. Вільний 5'-гідроксил можна потім піддавати різним способам естерифікації з Nзахищеною амінокислотою, щоб утворити IIа. Азот амінокислотного естеру та 2',3'-гідроксили рибозної одиниці потім наражають на вплив різних умов депротекції, переважно одночасно, з наступним утворенням солі вільного аміну амінокислотного естеру, як зображено для II. Приклад 1: Дигідрохлорид 5-аміно-3-(5'-О-Lвалініл-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону (3) У гетерогенну суміш сполуки 1 (5,37г, 17,0ммоль, одержаної за способом, запропонованим у патенті США №5041426 (приклад 2), який у його повному обсязі включено сюди шляхом посилання) в ацетоні (40мл), яка містилася у скляній колбі Morton об'ємом 250мл, додавали послідовно 2,2-DMP (6,26мл, 50,9ммоль), ДМСО (6,6мл) та MeSO3H (220мкл, 3,39ммоль) при кімнатній температурі. Реакційну суміш сильно перемішували, доки вона не стала гомогенною та золотисто-жовтою, коли витратився діол. Аналіз шляхом тонкошарової хроматографії (SiO2, 10% МеОН-СНСl3) вказав, що реакція завершилася за 6 годин. Нерозчинені тверді речовини видалили шляхом гравітаційного фільтрування, застосовуючи гофрований фільтрувальний папір Whatman типу 1. Після цього фільтрат вилили у 10-кратний об'єм льодяної води (приблизно 400мл), внаслідок чого одразу відбулося осадження білої твердої речовини. Після нетривалого періоду змішування, NaHCO3 (285мг, 3,39ммоль), розчинений у воді (10мл), додавали, щоб нейтралізувати MeSO3H. Сильне перемішування у реакторі Morton тривало 15 хвилин, після чого суміш профільтрували крізь лійку зі скла з крупними отворами. Твердий матеріал промили льодяною водою (100мл), висушили на повітрі, потім сушили Етап 1: Одержання 5-аміно-3-(2',3'-Оізопропіліден-b-D-рибофуранозил)тіазоло[4,5а]піримідин-2,7-діону 27 далі в умовах високого вакууму при 65°С, внаслідок чого отримали 5,36г (88%) ацетоніду 2 у вигляді білої твердої речовини: Т. пл. 280-81°С; 1Н (DMSO-d6) δ 1,28 (с, 3Н), 1,47 (с, 3Н), 3,43-3,55 (м, 2Н), 3,95-3,99 (м, 1H), 4,77-4,80 (м, 1Н), 4,884,91 (м, 1H), 5,24-5,26 (м, 1H), 5,99 (с, 1H), 6,97 (шир. с, 2Н), 11,25 (с, 1H). Етап 2: Одержання 5-аміно-3-(2',3 '-Оізопропіліден-5'-N-трет-бутоксикарбоніл-Lвалініл)-b-D-рибофуранозил)тіазоло[4,5а]піримідин-2,7-діону (4) У розчин N-бутоксикарбоніл-(L)-валіну (671мг, 2,81ммоль) у тетрагідрофурані (9мл) при 0°С додали EDC (588мг, 3,07ммоль). Одержану гомогенну суміш перемішували протягом 45 хвилин при 0°С, і вона стала гетерогенною, та додали однією порцією ацетонід 2 з Етапу 1, описаного вище (1,00г, 2,81ммоль). Потім додали твердий DMAP (522мг, 4,27ммоль). Реакційну суміш залишили, доки вона не досягла кімнатної температури, та змішували протягом додаткових 5 годин, після чого концентрували при 25°С, застосовуючи роторний випарник, доки не отримали жовтий сироп. Залишок розчинили у ЕtOАс (50мл), розділили 1N НСl (10мл), потім нейтралізували кислоту насиченим водним NaHCO3 (10мл). Кислотну водну фазу потім екстрагували ЕtoАс (2×50мл), а потім розділили лужною водною фазою. Комбіновані органічні фази висушили над Na2SO4, профільтрували крізь невеликий шар SiO2 та концентрували, внаслідок чого отримали 1,480г (96%) складного ефіру 4 Вос-захищеної амінокислоти у вигляді піни: Т. пл. 158°С (розклад); 1Н (CDCl3) δ 0,86 (д, J=7,0, 3Н), 0,95 (д, J=7,0, 3Н), 1,35 (с, 3Н), 1,44 (с, 9Н), 1,56 (с, 3Н), 1,75 (шир. с, 1H), 2,08-2,19 (м, 1Н), 4,20-4,24 (м, 2Н), 4,30-4,37 (м, 1Н), 4,56 (дц, J=11,0, 5,9, 1H), 4,96 (дд, J=6,2, 3,7,1Н), 5,11 (шир. д, J= 8,8,1Н), 5,29 (шир. д, J= 6,6,1Н), 5,88 (шир. с, 2Н), 6,23 (с, 1Н). Етап 3: Одержання дигідрохлориду 5-аміно3'-(5'-O-L-валініл-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону (3) Потік газовидного НСl проходив крізь барботер з концентрованою H2SO4 та далі спрямовувався (крізь фритовану дисперсійну трубку) у 3горлову колбу Morton об'ємом 250мл, яка містила сухий ізопропілацетат (80мл) при 0°С, доки не отримали насичений розчин. До нього додали розчин естеру Вос-захищеної амінокислоти з вищезгаданого Етапу 2 (5,53г, 9,95ммоль) в ізопропілацетаті (30мл), внаслідок чого за 5 хвилин утворився осад у вигляді білої твердої речовини. Туди додали 10% (об'ємн.%) ІРА (11мл). Реакційну суміш нагріли до кімнатної температури, потім перемішували протягом 12 годин. Гетерогенну реакційну суміш розвели сухим толуолом (100мл). Внаслідок фільтрування із застосуванням лійки, виготовленої зі скла з порами середнього розміру, в атмосфері N2 отримали не зовсім білу аморфну тверду речовину. Після перетирання твердої речовини у сухому тетрагідрофурані здійснили фільтрування та сушіння у вакуумі при 65°С, внаслідок чого отримали 3,677г (81%) сполуки 3 із заголовку у вигляді білої твер 79764 28 дої речовини: Т. пл. 166-68°С (розклад); 1Я (DMSO-d6) δ 0,90 (д, J=7,0, 3Н), 0,94 (д, J=7,0, 3Н), 2,14-2,18 (м, 1Н), 3,83-3,85 (м, 1H), 3,96-4,00 (м, 1Н), 4,23-4,28 (м, 2Н), 4,42 (дц, J=11,7, 3,4,1Н), 4,75 (дд, J= 10,3, 5,5, 1H), 5,81 (д, J=4,4,1Н), 6,46 (шир. с, 3Н), 7,23 (шир. с, 2Н), 8,47 (с, 3Н), 11,5 (шир. с, 1H). Елементний аналіз для C15H21N5O7S×2НСl: Розраховано С, 36,89; Н, 4,75; СІ, 14,52; N, 14,34; S, 6,57; знайдено: С, 37,03: Н, 4,74; СІ, 14,26; N, 14,24; S, 6,42. Приклад 2: 3/2 Гідрохлорид 5-аміно-3-(5'-O-Lізолейцил-b-D-рибофуранозил)тіазоло[4,5-d] піримідин-2,7-діону (5) Етап 1: Одержання 5-аміно-3-(2',3'-Оізопропіліден-5'-N-трет-бутоксикарбоніл-Lізолещж)-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону (6) Способом, подібним до способу Етапу 2 Прикладу 1, одержали 5-аміно-3-(2',3'-Оізопропіліден-5'-N-трет-бутоксикарбоніл-Lізолейцил)-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діон 6 з виходом 93% з 5-аміно-3(2',3'-О-ізопропіліден-b-D-рибофуранозил)тіазоло[4,5-d]піримідин-2,7-діону 2 та N-третбутокси-L-ізолейцину 7 у вигляді не зовсім білої піни: 1Н ЯМР (400МГц, d6-DMSO) δ 11,29 (с, 1Н), 7,09 (д, J=8,0, 1Н), 7,02 (шир. с, 1H), 6,02 (с, 1H), 5,28 (д, J=6,2,1Н), 5,06 (шир. с, 1Н), 4,16-4,22 (м, 2Н), 3,85 (дд, J= 8,0,6,6,1Н), 1,68 (шир. с, 1Н), 1,47 (с, ЗН), 1,34 (с, 9Н), 1,29 (с, 3Н), 0,71-0,89 (м, 5Н). Етап 2: Одержання дигідрохлориду 5-аміно-3(5'-O-L-ізолейцил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2,7-діону (5) Способом, подібним до способу Етапу 3 Прикладу 1, одержали сполуку з заголовку у вигляді білої твердої речовини з вищезгаданого проміжного продукту, ви хід якої становив 80%: Т. пл. 173-174°С (розклад); 1НЯМР (400МГц, d6-DMSO) δ 11,41 (шир. с, 1H), 8,41 (шир. с, 3Н), 7,15 (шир. с, 2Н), 5,82 (д, J=4,8,1Н), 4,50-5,00 (м, 2Н), 4,40 (дд, J=11,7, 3,3, 1Н), 4,21-4,30 (м, 2Н), 3,91-4,0 (м, 2Н), 1,84-1,91 (м, 1H), 1,37-1,44 (м, 1H), 1,19-1,27 (м, 1Н), 0,80-0,87 (м, 6Н). Елементний аналіз для C16H23N5 O7S×3/2HCl: розраховано: С, 39,69; Η, 5,10; Ν, 14,47; СІ, 10,98; S, 6,62; знайдено: С, 39,05; Η, 5,13; Ν, 13,73; СІ, 11,08; S, 6,02. Приклад 3: Гідрохлорид 5-аміно-3-(5'-О-[a-Lтрет-бутилгліциніл]-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2,7-діону (8) 29 79764 Етап 1: Одержання 5-аміно-3-(2',3'-Оізопропіліден-5'-N-тpeт-бутоксикарбоніл-[a-Lтрет-бутилгліцил]-b-D-рибофуранозил)тіазоло[4,5-d]піримідин-2,7-діону (9) Способом, подібним до Етапу 2 Прикладу 1,5-аміно-3-(2',3'-О-ізопропіліден-5'-N-тpeтбутоксикарбоніл-[a-L-трет-бутилгліциніл]-b-Dрибофуранозил)-тіазоло[4,5-d]піримідин-2,7-діон (10) одержали з виходом 66% з 5-аміно-3-(2,3-Оізопропіліден-b-D-рибофуранозил)тіазоло[4,5d]піримідинон-2,7-діону 2 та N-a-L-тpeтбутоксигліцину у вигляді не зовсім білої піни: 1H ЯМР (400МГц, d6-DMSO) δ 11,28 (шир. с, 1Н), 6,70-7,40 (м, 3Н), 6,02 (с, 1H), 5,30 (д, J= 6,2, 1H), 5,05 (шир. с, 1H), 4,17-4,24 (м, 3Н), 3,77 (д, J=8,4, 1H), 1,47 (с, 3Н), 1,33 (с, 9Н), 1,29 (с, 3Н), 0,85 (с, 9Н). Етап 2: Одержання 5-аміно-3-(5'-О-[a-L-третбутилгліцил]-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону (8) Способом, подібним до Етапу 3 Прикладу 1, одержали сполуку 8 з заголовку у вигляді білої твердої речовини з вищезазначеного проміжного продукту з виходом 80%: Т. пл. 202-203°С (розклад); 1НЯМР (400МГц, d6-DMSO) δ 11,35 (шир. с, 1Н), 8,31 (шир. с, 3Н), 7,08 (шир. с, 2Н), 5,83 (д, J=4,0,1Н), 5,45 (шир. с, 1H), 5,21 (шир. с, 1H), 4,77-4,82 (м, 1Н), 4,42 (дд, J=11,4,2,6, 1H), 4,234,28 (м,.1H), 3,96-4,04 (м, 1Н), 3,74 (с, 1Н), 0,97 (с, 9Н). Елементний аналіз для C16N23N5O7S×НСl: розраховано: С, 41,25; Η, 5,19; Ν, 15,03; СІ, 7,61; S, 6,88; знайдено: С, 40,41; Η, 5,41; Ν, 14,16; СІ, 7,01; S, 6,23. Приклад 4: Гідрохлорид 5-аміно-3-(5'-O-[a-LN-метилвалініл]-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону (11) 30 Етап 1: Одержання 5-аміно-3-(2',3'-Оізопропіліден-5'-N-тpeт-бутоксикарбоніл-[a-L-Nметилвалініл]-b-D-рибофуранозил)-тіазоло[4,5d]піримідин-2,7-діону (12) Способом, подібним до Етапу 2 Прикладу, 1, 5-аміно-3-(2',3'-О-ізопропіліден-5'-N-третбутоксикарбоніл-[a-L-N-метилвалініл]-b-Dрибофуранозил)-тіазоло[4,5-d]піримідин-2,7-діон (12) одержали з виходом 63% з 5-аміно-3-(2',3'-Оізопропіліден-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7-діону 2 та N-трет-бутокси-L-Nметилваліну 13 у вигляді не зовсім білої піни: 1Н ЯМР (400МГц, d6-DMSO) ротаметричний карбамат δ 11,28 (шир. с, 1Н), 7,00 (шир. с, 2Н), 6,02 (с, 1Н), 5,27 (д, J=6,6, 1Н), 5,04 (шир. с, 1Н), 4,144,28 (м, 3Н), 3,91 (д, J=9,5,1Н), 2,79 (шир. с, 3Н), 2,09 (шир. с, 1H), 1,46 (с, 3Н), 1,36 (с, 4,5Н), 1,32 (с, 4,5Н), 1,28 (с, 3Н), 0,78-0,89 (м, 6Н). Етап 2: Одержання гідрохлориду 5-аміно-3(5'-O-[a-L-N-метилвалініл]-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2,7-діону (11) Способом, подібним до Eтaпу 3 Прикладу 1, одержали сполуку 11 з заголовку у вигляді білої твердої речовини з невеликою кількістю домішок з вищезазначеного проміжного продукту з виходом 60%: Т. пл. >180°С (розклад); 1Н ЯМР (400МГц, d6-DMSO) δ 11,31 (шир. с, 1H), 9,05 (шир. с, 2Н), 7,05 (шир. с, 2Н), 5,83 (д, J=4,4,1Н), 5,46 (шир. с, 1H), 5,21 (шир. с, 1H), 4,76-4,82 (м, 1H), 4,42-4,48 (м, 1H), 4,28-4,38 (м, 1H), 4,22-4,28 (м, 1H), 3,94-4,04 (м, 2Н), 2,54 (шир. с, 3Н), 2,23 (шир. с, 1H), 0,98 (д, J=7,0, 3Н), 0,88 (д, J=7,0, 3Н). Елементний аналіз для C16H23N5O7S×НСl: розраховано: С, 41,25; Н, 5,02; N, 15,03; S, 6,88; СІ, 7,61; знайдено: С, 40,57; Н, 5,37; N, 13,57; S, 6,16; СІ, 7,29. Схема 2 Схема 2 показує загальну процедуру одержання 5-аміно-7-метокси-3-b-D-рибофуранозилтіазоло[4,5d]піримідин-2-онів та 5,7-діаміно-3-b-D-рибофуранозилтіазоло[4,5-d]піримідин-2-онів. Приклад 5: 5-Аміно-3-b-D-рибофуранозил-7метокси-тіазоло[4,5-d]піримідин-2-он (14) Безводну сполуку 1 (2,0г, 6,3ммоль) розчинили у сухому піридині в атмосфері аргону. Розчин охолодили до 0°С, після чого до суміші краплями до давали TFAA (13,3г, 63ммоль). Через 5 хвилин реакційну суміш розташували на масляній бані, яка мала температуру 60°С, на 1,5 години та здійснювали моніторинг шляхом тонкошарової хроматографії (SiO 2,20% МеОН-СНСІ3) з метою визна 31 79764 чення утворення катіону піридинію. 0,2 Rf початкового матеріалу перетворили на пляму базового матеріалу, що зазнав блакитної флюоресценції під впливом ультрафіолетового випромінювання з довжиною хвилі 254нм. Після перетворення на активований проміжний продукт щойно приготовлений розчин метоксиду натрію (1,8г Na, 78ммоль, 300мл метанолу) додали до реакційної суміші при 0°С. Реакційну суміш залишили нагріватися до кімнатної температури, та реакція тривала іще протягом 2 днів. Суміш потім загасили 1М NH 4CI (100мл) та екстрагували 25% ІРА-СНСl3 (5×100мл). Сирий матеріал профільтрували крізь пробку з силікагелю, а потім концентрували, внаслідок чого отримали 1,6г (75%) сполуки 14 з заголовку. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (SiO2 , вода, метанол, етилацетат, 5:10:85) у вигляді білої твердої речовини: Т. пл. >160°С (розклад); [М+Н]+ 330,9, [2М+Н]+ 661,1, [3 М+Н]+ 991,0; Rf=0,6 (20% МеОНСНСІ3); Т. пл. 200,4°С - 200,9°С; 1Н ЯМР (400МГц, d6-DMSO) δ 6,92 (с, 2Н), 5,86 (д, J=5,2, 1H), 5,28 (д, J=5,6, 1H), 4,96 (д, J=5,2, 1H), 4,78 (дц, J=10,8, 5,6,1H), 4,67 (т, J=6,0, 1H), 4,07-4,10 (м, 1H), 3,91 (c, 3H), 3,70-3,80 (м, 1H), 3,55-3,60 (м, 1H), 3,403,45 (м, 1H). Елементний аналіз для C11H14N4O6S: розраховано: С, 40,00; Η, 4,27; Ν, 16,96; S, 9,71; знайдено: С, 40,07; Η, 4,43; Ν, 16,71; S, 9,53. Приклад 6: 5,7-Діаміно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2-он (15) Безводну сполуку 1 (0,3г, 0,9ммоль) розчинили у сухому піридині в атмосфері аргону. Розчин охолодили до 0°С, після чого до суміші краплями додавали TFAA (1,2мл, 9,5ммоль). Через 5 хвилин реакційну суміш розташували на масляній бані, 32 яка мала температуру 60°С, на 1,5 години та здійснювали моніторинг шляхом тонкошарової хроматографії (20% МеОН-СНСІ3) з метою визначення утворення катіону піридинію. 0,2 Rf початкового матеріалу перетворили на пляму базового матеріалу, що зазнав блакитної флюоресценції під впливом ультрафіолетового випромінювання з довжиною хвилі 254нм. Після перетворення на активований проміжний продукт реакційну колбу розташували на льодяній бані. Після того, як температура урівноважилася, 30% водний NH3 (25мл) додавали краплями, доки не припинилося утворення тепла, а потім додали залишок. Протягом 5 хвилин утворювався продукт, на що вказувала аналітична тонкошарова хроматографія Rf 0,25 (SiO2, 20% МеОН-СНСl3). Колбу нагрівали до кімнатної температури протягом понад 30 хвилин, потім здійснили дегазацію водного розчину в умовах вакууму від роторного насоса та екстрагували 25% ІРА-СНСІ3 (5×100мл). Продукт зазнав флешхроматографії (SiO2, 10% МеОН-СНСl3), внаслідок чого отримали 55мг (17%) сполуки 15 з заголовку, яка мала невелику кількість домішок. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (SiO2, вода, метанол, етилацетат, 5:10:85) у вигляді білої твердої речовини: Т. пл. >155°С (розклад); [М+Н]+ 316,0; Rf=0,25 (SiO2,20% МеОН-СНСl3); 1Η Я МР (400МГц, d6DMSO) δ 6,76 (с, 2Н), 6,14 (с, 2Н), 5,85 (д, J=5,2, 1Н), 5,22 (д, J=4,8, 1H), 4,92 (д, J=2,8, 1Н), 4,704,83 (м, 2Н), 4,05-4,10 (м, 1H), 3,65-3,80 (м, 1Н), 3,52-3,62 (м, 1Н) 3,40-3,50 (м, 1Н). Елементний аналіз для C10H13N5O5S×1 /2Н2О: розраховано: С, 37,03; Η, 4,35; Ν, 21,59; S, 9,89; знайдено: С, 37,27; Η, 4,32; Ν, 20,43; S, 10,11. 33 79764 Приклад 7: 5-Аміно-7-метиламіно-3-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-он (18) Етап 1: Одержання 5-ацетиламіно-3-(2',3',5'три-О-ацетил-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7(6Н)-діону (16) Безводну сполуку 1 (8,0г, 39,5ммоль) розчинили у сухому піридині (65мл). DMAP (3,1г, 25,3ммоль) та ацетангідрид (19,1мл, 202,4ммоль) додавали послідовно. Реакції дозволили продовжуватися протягом 2 годин при кімнатній температурі, після чого її загасили насиченим NаНСО3 (100мл) та екстрагували DCM (3×200мл). Органічну фазу концентрували, а потім розтерли в порошок разом з простим ефіром. Внаслідок цього отримали 12,5г (103%) 5-ацетиламіно-3-(2,3,5-триО-ацетил-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2,7(6Н)-діону (16) у вигляді білої твердої речовини з невеликою кількістю домішок: Т. пл. 246,7-248,1°С; Rf=0,20 (SiO2, 50% ЕtOAс-СНСІ3); 1 Η ЯМР (400МГц, d6-DMSO) δ 12,23 (с, 1Н), 11,85 (с, 1Н), 5,97 (м, 2Н), 5,48 (τ, J=6,1Н), 4,35-4,40 (м, 1H), 4,25-4,31 (м, 1H), 4,08-4,18 (м, 1Н), 2,49 (с, 3Н), 2,07 (с, 3H), 2,01 (c, 3H), 2,00 (c,3H). Етап 2: Одержання 5-ацетиламто-7-(2,4,6триізопропіл-бензолсульфонілокси)-3-(2,3,5-три-Оацетил-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2-ону (17) Проміжний продукт з вищезазначеного Етапу 1 (500мг, 0,98ммоль) розчинили у DCM (15мл) при кімнатній температурі. У розчин додали DMAP (7,3 мг, 0,06 ммоль) та TEA (16мл, 11ммоль), а потім 2,4,6-триізопропілбензолсульфонілхлорид (454мг, 1,5ммоль). Через 1 годину, після завершення реакції, сиру суміш концентрували, а потім очищували шляхом фле ш-хроматографії (SiO2,10% EtOAcСНСl3), внаслідок чого отримали 690мг (92%) 5ацетиламіно-7-(2,4,6-триізопропілбензолсульфонілокси)-3-(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-ону 17 у вигляді спінювальної білої твердої речовини: 74,576,3°С; Rf=0,7 (SiO2, 20% EtOAc-СНСl3); 1Н (400МГц, d6-DMSO) δ 10,83 (с, 1H), 7,39 (с, 2H), 6,03 (д, J= 4,0, 1H), 5,91-5,96 (м, 1H), 5,69 (τ, J= 6,4, 1Η), 4,30-4,70 (м, 1H), 4,22-4,26 (м, 1H), 4,164,20 (м, 1H), 3,90-4,00 (м, 2Н), 2,97-3,01 (м, lH), 2,07 (с, 3Н), 2,06 (с, 3Н), 2,04 (с, 3Н), 1,88 (с, 3Н), 1,17-1,25 (м, 18Н). Етап 3: Одержання 5-ацетиламіно-7метиламіно-3-(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-ону (19) Проміжний продукт з вищезазначеного Етапу 2 (1,7г, 2,27ммоль) розчинили у діоксані (20мл) при кімнатній температурі. До нього додали 2,0 Μ розчин метиламіну (3,4мл, 6,8ммоль) у метанолі. За 2 години початковий матеріал витратився. Реакційну суміш концентрували, а потім очищували шляхом 34 флеш-хроматографії (SiO2, градієнтне елюювання, 20-80% EtOAc-СНСl3), внаслідок чого отримали 945мг (83%) чистої сполуки з заголовку у вигляді жовтої олії: [М+Н]+ 498,2, [2М+Н]+ 995,4; Rf 0,55 (10% СН3ОН-СНСl3); 1 ЯМР (400МГц, d6-D MSO) δ 10,13 (с, 1H), 7,70 (д, J=4,41, 1H), 5,95-6,02 (м, 2H), 5,69 (с, 1Н), 4,35-4,39 (м, 1H), 4,16-4,23 (м, 2Н), 2,90 (д, J=4,8, 3Н), 2,20 (с, 3Н), 2,07 (с, 3Н), 2,02 (с, ЗН), 2,00 (с, 3Н). Етап 4: Одержання 5-аміно-7-метиламіно-3-bD-рибофуранозил)тіазоло[4,5-d]пірамидин-2-ону (18) Проміжний продукт з вищезазначеного Етапу 3 (420мг, 0,85ммоль) розчинили у діоксані (4 мл) та до розчину додали 1Μ LiOH (8,5мл, 8,5ммоль). Оацетильні групи вилучили протягом 40 хвилин, внаслідок чого отримали проміжний продукт при Rf=15 (SiO2, 5% MeOH-EtOAc). За 2 години вилучили N-ацетил, на що вказували тонкошарова хроматографія Rf=0,20 (SiO2, 5% MeOH-EtOAc). Реакційну суміш нейтралізували стехіометричною кількістю оцтової кислоти, екстрагували 25% ІРАСНСІ3, а потім концентрували, внаслідок чого отримали 195мг (70%) сполуки 18. Аналітичний зразок сполуки 18 з заголовку отримали шляхом препаративної тонкошарової хроматографії (SiO2, вода-MeOH-EtOAc, 10:20:70) у вигляді білої твердої речовини: [М+Н]+ 330,0; Rf=0,20 (5% МеОНЕtOАс); Т. пл. >108°С; 1H ЯМР (400МГц, d6-DMSO) δ 7,06 (д, J=3,6, 1H), 6,24 (с, 2H), 5,85 (д, J=5,2, 1H), 5,22 (д, J=4,8, 1Н), 4,93 (д, J=5,2, 1Н), 4,704,80 (м, 2Н), 4,07 (д, J=4,8, 1H), 3,75 (д, J= 4,4, 1H), 3,5-3,6 (м, 1Н), 3,40-3,50 (м, 1Н), 2,82 (д, J=4,4, 3Н). Приклад 8: 5-Аміно-7-диметиламіно-3-b-Dрибофуранозилтіазоло [4,5-d]піримідин-2-он (20) Етап 1: Одержання 5-ацетиламіно-7диметиламіно-3-(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-ону Способом, подібним до Етапу 2 з Прикладу 7, отримали 5-ацетиламіно-7-диметиламіно-3(2',3',5'-три-O-ацетил-b-Dрибофуранозил)тіазоло[4,5-с/|піримідин-2-онз виходом 80% у вигляді жовтої олії: М+ 511,14; Rf=0,70 (SiO2,10% МеОН-СНСl3); 1Н ЯМР (400МГц, d6-DMSO) δ 10,15 (с, 1Н), 6,10-6,15 (м, 1H), 5,986,09 (м, 1Н), 5,566-5,70 (м, 1H), 4,35-4,40 (м, 1Н), 4,22-4,27 (м, 1Н), 4,14-4,08 (м, 1H), 3,18 (с, 6Н), 2,19 (с, 3Н), 2,08 (с, 3Н), 2,06 (с, ЗН), 1,99 (с, 3Н). Етап 2: Одержання 5-аміно-7-диметиламіно-bD-рибофуранозил)тіазоло[4,5-d]піримідин-2-ону (20) Способом, подібним до Етапу 3 з Прикладу 7, отримали сполуку 20 з виходом 82%. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (S1O2, вода-МеОН-ЕtOАс, 35 79764 10:20:70) у вигляді білої твердої речовини: [М+Н]+ 344,0; [2М+Н]+ 687,4; Т. пл. >112°С; Rf=0,20 (5% МеОН-ЕtOАс); 1H Я МР (400МГц, d6-D MSO) δ 6,27 (с, 2Н), 5,91 (д, J=4,8,1Н), 5,22 (д, J=6,0, 1H), 4,93 (д, J=5,2, 1H), 4,71-4,76 (м, 2Н), 4,07-4,09 (м, 1H), 3,7-3,8 (м, 1H), 3,5-3,6 (м, 1H), 3,5-3,6 (м, 1Н), 3,09 (с, 6Н). Елементний аналіз для C12H17N5O5S: розраховано: С, 41,98; Η, 4,99; Ν, 20,40; знайдено: С, 41,32; Η, 5,14; Ν, 18,59. Приклад 9: Моногідрохлоридна сіль 5-аміно-7циклопропіламіно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2-ону (21) Етап 1: Одержання 5-ацетиламіно-7циклопропіламіно-3-(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-ону Способом, подібним до Етапу 2 Прикладу 3, отримали 5-ацетиламіно-7-циклопропіламіно-3(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-он з виходом 80% у вигляді жовтої олії: Rf=0,45 (SiO2, 75% EtOAc-СНСl3); 1Η Я МР (400МГц, d6-D MSO) δ 10,11 (с, 1H), 7,87 (д, J=2,8,1Н), 5,98-6,01 (м, 1H), 5,70-5,76 (с, 1Н), 4,32-4,39 (м, 1H), 4,16-4,30 (м, 2H), 3,85 (с, 1H), 2,87 (с, 1Н), 2,25 (с, 3Н), 2,07 (с, 3Н), 2,06 (с, 3Н), 1,98 (с, 3Н), 0,73-0,76 (м, 2Н), 0,57-0,60 (м, 2Н). Етап 2: Одержання 5-аміно-7циклопропіламіно-3-b-Dрибофуранозилтіазоло[4,5-d]пірамідин-2-ону Способом, подібним до Етапу 3 Прикладу 7, отримали 5-аміно-7-циклопропіламіно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2-он з виходом 79%. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (S1O2, вода-МеОН-ЕtOАс, 10:20:70) у вигляді білої твердої речовини: Rf=0,20 (5% МеОН-ЕtOАс); Т. пл. >100°С; [М+Н]+ 356,0; 1H (400МГц, d6-D MSO) δ 7,24 (с, 1H), 6,28 (с, 2Н), 5,86 (д, J=5,6, 1H), 5,22 (д, J=6, 1H), 4,92 (д, J=5,2,1Н), 4,70-4,80 (м, 2Н), 4,05-4,10 (м, 1H), 3,7-3,8 (м, 1Н), 3,5-3,6 (м, 1H), 3,45-3,50 (м, 1Н), 2,8 (с, 1H), 0,68-0,70 (м, 2Н), 0,54-0,57 (м, 2Н). Етап 3: Одержання гідрохлоридної солі 5аміно-7-циклопропіламіно-3^-0рибофуранозилтіазоло[4,5-d]піримідин-2-ону (21) Сполуку із заголовку одержали шляхом додавання твердого матеріалу, приготовленого на вищезазначеному етапі 2, до сильно перемішуваного 4Μ розчину НСl у діоксані, внаслідок чого отримали сполуку із заголовку у вигляді білої твердої речовини Т. пл. >99°С; 1Н ЯМР (400МГц, d6-DMSO) δ 7,25 (д, 1H, J=2,8,1H), 6,23 (с, 2H), 5,87 (д, J=5,2, 1H), 5,21 (шир. с, 1Н), 4,98 (шир. с, 1H), 4,73-4,79 (м, 2H), 4,09 (т, J=5,6, 1H), 3,72-3,79 (м, 1H), 3,553,60 (м, 1Η), 3,45-3,37 (м, 1Н), 2,75-2,82 (м, 1H), 0,72-0,79 (м, 2Н), 0,55-0,63 (м, 2Н). Елементний аналіз для C13H17N5O5S×HCl: розраховано: С, 39,85; Η, 4,63; Ν, 17,87; СІ, 9,05; знайдено: С, 36 39,66; Η, 4,85; Ν, 16,57; СІ, 8,13. Приклад 10: 5-Аміно-7-циклопентиламіно-3-bD-рибофуранозилтіазоло[4,5-d]піримідин-2-он (22) Етап 1: Одержання 5-ацетиламіно-7піролідино-3-(2',3',5'-три-О-ацетип-b-Dрибофуранозип)тіазоло[4,5-d]піримідин-2-ону Способом, подібним до Етапу 2 Прикладу 7, отримали 5-ацетиламіно-7-піролідино-3-(2',3',5'три-О-ацетил-b-D-рибофуранозил)тіазоло[4,5d]піримідин-2-он з виходом 70%. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (S1O2, вода-МеОН-ЕtOАс, 10:20:70) у вигляді білої твердої речовини: Т.пл.>108°С (розклад); Rf=0,80 (10% вода та 20% метанол у етилацетаті); [М+Н]+ 384,0; 1Н ЯМР (400МГц, d6-DMSO) δ 7,00 (д, J=7,2, 1H), 6,17 (с, 2Н), 5,18 (д, J=5,2,1Н), 5,21 (д, J=5,6, 1H), 4,92 (д, J=5,6, 1H), 4,74-4,80 (м, 2Н), 4,30-4,35 (м, 1Н), 4,05-4,10 (м, 1Н), 3,70-3,80 (м,1H), 3,55-3,60 (м, 1H), 3,30-3,45 (м, 1H), 1,40-2,0 (м, 8Н). Етап 2: Одержання 5-аміно-7циклопентиламіно-3-b-Dрибофуранозилтіазоло[4,5-d]пipимідин-2-ону Способом, подібним до Етапу 3 Прикладу 7, отримали сполуку 22 із заголовку з виходом 70%. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (SiO2, водаМеОН-ЕtOАс, 10:20:70) у вигляді білої твердої речовини: Т. пл. >108°С (розклад); Rf=0,80 (10% вода та 20% метанол у етилацетаті); [М+Н]+ 384,0; 1Н ЯМР (400МГц, d6-DMSO) δ 7,00 (д, J=7,2, 1H), 6,17 (с, 2Н), 5,18 (д, J=5,2, 1H), 5,21 (д, J=5,6,1Н), 4,92 (д, J=5,6,1Н), 4,74-4,80 (м, 2Н), 4,30-4,35 (м, 1Н), 4,05-4,10 (м, 1H), 3,70-3,80 (м, 1H), 3,55-3,60 (м, 1Н), 3,30-3,45 (м, 1H), 1,40-2,0 (м, 8Н). Приклад 11: 5-аміно-7-піролідино-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2-он (23) Етап 1: Одержання 5-ацетиламіно-7піролідино-3-(2',3',5'-три-О-ацетил-b-Dрибофуранозил)тіазоло[4,5-d]піримідин-2-ону Способом, подібним до Етапу 2 Прикладу 7, отримали 5-ацетиламіно-7-піролідино-3-(2',3',5'три-O-ацетил-b-D-рибофуранозил)-тіазоло[4,5d]піримідин-2-он з виходом 79% у вигляді жовтої олії: [М+Н]+ 538,1; R f 0,80 (SiO2, вода-MeOH-EtOAc, 10:20:70); 1H ЯМР (400МГц, d6-D MSO) δ 10,04 (с, 1Η), 5,97-6,02 (м, 2H), 5,68 (с, 1H), 4,38 (дд, J=11,6, 3,6,1Н), 4,15-4,23 (м, 2Н), 3,58 (с, 4Н), 2,23 (с, 3Н), 2,08 (с, 3Н), 2,05 (с, 3Н), 1,98 (с, 3Н), 1,89 (с, 4Н). Етап 2: Одержання 5-аміно-7-піролідино-3-b-D 37 79764 рибофуранозилтіазоло[4,5-d]піримідин-2-ону Способом, подібним до Етапу 3 Прикладу 7, отримали сполуку 23 із заголовку з виходом 81%. Аналітичний зразок отримали шляхом препаративної тонкошарової хроматографії (SiO2, водаMeOH-EtOAc, 10:20:70) у вигляді білої твердої ре Приклад 12: Гідрохлорид 5-аміно-7циклопентиламіно-3-(5'-O-L-валініл-b-Dрибофуранозил)тіазоло [4,5-d] піримідин-2-ону (24) Шляхом сильного перемішування проміжний продукт В розчинили у розчині безводного хлориду водню в ізопропілацетаті при 0°С та залишили нагріватися до кімнатної температури. У гетерогенну суміш додали додатковий ізопропілацетат. Реакційна суміш перемішували протягом додаткових 12 годин. Додали толуол та продукт профільтрували та висушили в умовах вакуум у, внаслідок чого отримали бажану сіль 24 ди-НСl. Проміжні продукти одержують наступним чином: 5-аміно-7-циклопентиламіно-3-(2',3'-Оізопропіяіден-b-D-рибофуранозил)тіазоло[4,5d]тримідин-2-он (А) Сполуку А одержують згідно з процедурою Kini et al., шляхом перемішування суміші 5-аміно7-циклопентиламіно-3-b-Dрибофуранозилтіазоло[4,5-d]піримідин-2-ону 22 з ацетоном, ДМСО, метансульфоновою кислотою та надлишком диметоксипропану при 0°С, доки не витратиться початковий матеріал. Реакційну суміш додають у льодяну воду та нейтралізують 38 човини: Т.пл.>112,4°С (розклад); [М+Н]+ 370,3; 1Н ЯМР (400МГц, d6-OMSO) δ 6,22 (с, 2Н), 5,90 (д, J=4,8, 1H), 5,23 (д, J=5,2,1Н), 4,94 (д, J=4,4,1Н), 4,68-4,75 (м, 2Н), 4,08 (д, J=4,8,1Н), 3,71-3,76 (м,1H), 3,55 (шир.с, 5Н), 3,38-3,54 (м, 1H), 1,87 (с, 4Н). до рН 7 насиченим NaHCO3 та екстрагують ЕtOАс. Органічний шар концентрують та піддають колоночній xpoмaтoгрaфiї на діоксиді кремнію, внаслідок чого отримують 2',3'-захищений діоловий продукт. 5-аміно-7-циклопентиламіно-3-(5'-О-(N-(третбутоксикарбоніл)-і-валініл)-2',3'-О-ізопропіліден-bD-рибофуранозил)тіазоло[4,5-d]піримідин-2-он (В) У розчин 1,0 еквіваленту N-(третбутоксикарбоніл)-L-валіну у тетрагідрофурані при 0°С додають 1,1 еквівалента EDC. Після перемішування протягом 30 хвилин додають 1,0 еквівалент 5-аміно-7-циклопентил-3-(2',3'-Оізопропіліден-b-D-рибофуранозил)тіазоло-[4,5d]піримідин-2-ону (А) та 1,5 еквівалента DMAP. Реакційну суміш нагрівають до кімнатної температури та залишають перемішуватися протягом 5 годин, потім концентрують. Залишок розчиняють у ЕtOАс, розділяють 1N НСl та нейтралізують насиченим водним NаНСО3 (10мл). Водну фазу потім екстрагують ЕtOАс. Комбіновані органічні фази висушують над Na2SO4, фільтрують та випаровують в умовах вакуум у, внаслідок чого отримують проміжний продукт В, який очищують шляхом колоночної хроматографії на діоксиді кремнію. 39 79764 Біологічні випробування Спроможність сполук за Формулою І демонструвати переважні характеристики для перорального введення та індукувати імунні реакції, коли їх водять обраним способом, легко продемонстрували в експериментах з мишами та гончаками. Результати таких вимірювань для сполук за Формулою І можна порівняти з результатами подібних експериментів зі сполуками, описаними у літературі, на яку посилаються у цьому описі [наприклад, патенти США №5041426 та 4880784], для того, щоб побачити переваги сполук за Формулою І стосовно фармакокінетичних та фармакодинамічних властивостей. Концентрації інтерферону-альфа (Mu-IFN-a) у мишей Звичайна миша являє собою корисну систему для оцінювання ступеню, до якого винахід, описаний тут, забезпечує суттєве удосконалення при пероральному введенні сполуки 1 (ізаторибін). Не тільки можна було визначити концентрації ізаторибіну у плазмі, які є наслідком перорального введення згаданих проліків, але й також завдяки розширеному імунологічному дослідженню, що проводилося на мишах, були запропоновані реагенти, придатні для вимірювання рівнів інтерферону-альфа - цитокіну, який представляє інтерес, оскільки він відображує одну з бажаних біологічних активностей ізаторибіну. Ми застосовували мишачу систему у серії експериментів, які демонструють, що сполука 35'-валіновий естер сполуки 1 (вал-ізаторибін) викликає інтерферонну реакцію, суттєво поліпшену порівняно з реакцією внаслідок введення самого ізаторибіну. У таблиці 1 наведено результати аналізу для мишачого інтерферону-альфа у плазмі мишей, яким двічі вводили дозу ізаторибіну, змішаного з бікарбонатом натрію, на рівні 50мг/кг шляхом перорального введення. Очевидно, що ніякого інтерферону не можна було визначити навіть тоді, коли дозу повторювали з інтервалом у 4 години. Таблиця 1 Концентрація у плазмі (пг/мл) інтерферону альфа (Mu-IFN-a) у мишей піс ля дв ох пероральних доз ізаторибіну у 50мг/кг з інтерв алом 4 години Час, години 0,00 0,03 0,08 0,25 0,50 1,00 1,50 2,00 3,00 4,00 Індивідуальне значення BQL50 BQL25 BQL25 BQL50 BQL25 BQL25 BQL100 BQL25 BQL25 BQL25 Перша доза BQL125 BQL50 BQL250 BQL25 BQL25 BQL25 BQL25 BQL25 BQL25 BQL25 25 BQL BQL25 BQL25 BQL25 BQL75 BQL25 BQL25 BQL25 BQL25 BQL25 Середнє значення Середнє кв адратичне в ідхилення 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 40 4,03 4,08 4,25 4,50 5,00 5,50 6,00 7,00 8,00 BQL25 BQL25 BQL25 BQL50 BQL50 BQL37,5 BQL50 BQL50 BQL50 Друга доза BQL25 BQL25 BQL25 BQL25 25 BQL BQL25 BQL37,5 BQL50 50 BQL BQL50 BQL37,5 BQL37,5 BQL41,3 BQL37,5 BQL50 BQL50 BQL25 BQL50 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 BQLn - нижче підв ищеного порогу, який піддається в изначенню
ДивитисяДодаткова інформація
Назва патенту англійською3-b-d-ribofuranosylthiazolo[4,5-d]pyridimine nucleosides and uses thereof
Автори англійськоюRueden Erik J.
Назва патенту російською3-b- d-рибофуранозилтиазоло[4, 5-d]пиримидиновые нуклеозиды и их применение
Автори російськоюРуден Эрик Дж.
МПК / Мітки
МПК: A61K 31/7064, C07H 19/24, A61P 37/02
Мітки: застосування, 3-b-d-рибофуранозилтіазоло[4,5-d]піримідинові, нуклеозиди
Код посилання
<a href="https://ua.patents.su/23-79764-3-b-d-ribofuranoziltiazolo45-dpirimidinovi-nukleozidi-ta-kh-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">3-b-d-рибофуранозилтіазоло[4,5-d]піримідинові нуклеозиди та їх застосування</a>
Піридо[2,3-d]піримідинові та піримідо[4,5-d]піримідинові нуклеозиди, проліки та спосіб інгібування ракових клітин

Номер патенту: 72612
Опубліковано: 15.03.2005
Автори: Пєтшковский Збігнєв, Жирарде Жан-Лу, Ванг Гуанжі, Лау Джонсон, Гунік Есмір, Гонг Жі
МПК: A61P 35/00, A61K 31/70
Мітки: проліки, ракових, піримідо[4,5-d]піримідинові, інгібування, піридо[2,3-d]піримідинові, нуклеозиди, клітин, спосіб
Формула / Реферат:
1. Нуклеозидний аналог згідно з формулою (І) (І),деА - це О, S, CH2,R2, R2', R3 та R3' є незалежно вибраними з Н, F, OH, NH2, CN, N3, CONH2 та R, де R - це нижчий алкіл, нижчий алкеніл, нижчий алкініл або нижчий ацил, та він необов'язково містить принаймні один гетероатом та функціональну групу,або R2 та R2' разом, або R3 та R3' разом є вибраними з =СН2, =CHR", =CR"2, =NR", де R" - це...
Триазоло[4,5-d]піримідинові сполуки, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі та спосіб лікування порушень агрегації тромбоцитів

Номер патенту: 66801
Опубліковано: 15.06.2004
Автори: Спрінгторп Брайан, Інголл Ентоні, Гайл Сімон, Уілліс Пол
МПК: A61P 11/06, C07D 487/04, A61P 9/10, A61P 43/00, A61K 31/519, A61P 7/02, A61P 11/00
Мітки: триазоло[4,5-d]піримідинові, порушень, лікування, основі, спосіб, агрегації, варіанти, фармацевтична, тромбоцитів, композиція, одержання, сполуки
Формула / Реферат:
1. Триазоло[4,5-d]піримідинові сполуки загальної формули (I),деR1 являє собою C1-6 алкіл, C2-6 алкеніл, С2-6 алкініл, С3-8 циклоалкіл або фенільну групу, при цьому кожна група необов’язково заміщена одним чи кількома замісниками, вибраними з галогену, OR8, NR9R10, SR11 або C1-6 алкілу (який, в свою чергу, необов’язково заміщений одним чи кількома...
Застосування 4-н-1-бензопіран-4-онових похідних як інгібіторів проліферації клітин гладких м’язів

Номер патенту: 73110
Опубліковано: 15.06.2005
Автори: Паттерсон Уінстон Кемпбелл, Дюмон Дженніфєр А.
МПК: A61K 31/00
Мітки: м'язів, проліферації, 4-н-1-бензопіран-4-онових, гладких, інгібіторів, клітин, застосування, похідних
Формула / Реферат:
1. Застосування сполуки формули І (І),в якійR1 являє собою водень, алкіл, який має 1-6 атомів вуглецю, арил-С1-С4-алкіл; С1-С6-алкіл, який заміщений галогеном, гідрокси або карбокси; С3-С6-циклоалкіл, піридил, тієніл, С3-С6-циклоалкіл-С1-С4-алкіл, С2-С6-алкеніл, С2-С6-алкініл, феніл; феніл, моно- або полізаміщений галогеном, С1-С4-алкілом, С1-С4-алкокси, гідроксилом, карбоксилом, СОО-алкілом, CONH2, CONH-алкілом,...
Піроло(2,3-d)піримідинові сполуки як імуносупресори

Номер патенту: 74370
Опубліковано: 15.12.2005
Автори: Фленеґен Марк Едвард, Блюменкопф Тодд Ендрю, Манчгор Майкл Джон
МПК: A61P 17/00, A61P 35/00, A61P 5/00, C07D 487/04, A61K 31/5377, A61P 29/00, A61P 19/02, A61P 3/10, A61P 37/06, A61K 45/00, A61P 17/06, A61K 31/519, A61P 37/02, A61P 11/06, A61P 25/00, A61P 21/00, A61P 25/28, A61P 43/00, A61P 35/02, A61P 1/04
Мітки: сполуки, піроло(2,3-d)піримідинові, імуносупресори
Формула / Реферат:
1. Похідні піроло[2,3-d]піримідину формулиабо їх фармацевтично прийнятні солі;де R1 представляє групу формули,де у дорівнює 0;R4 є (С1-С6)алкіл;R5 є піперидинілом, який повинен бути заміщений від одної до п'яти групами, що складаються з (С1-С6)алкілу,...
Нуклеозиди з біциклічною цукровою складовою, олігонуклеотид та антивірусна фармацевтична композиція

Номер патенту: 61997
Опубліковано: 15.12.2003
Автор: Ванг Гуанжі
МПК: C07H 19/16, C07H 19/06, C07H 19/10, C07H 21/00, C07H 19/20, C07G 3/00, C07H 19/04
Мітки: фармацевтична, біциклічною, антивірусна, нуклеозиди, олігонуклеотид, композиція, цукровою, складовою
Формула / Реферат:
1. Нуклеозиди з біциклічною цукровою складовою, які мають загальну формулу,де Z являє собою метилен,R1 є незалежно вибраним з групи, що містить аденін, цитозин, гуанін, гіпоксантин, урацил, тимін;R2, R3 є незалежно вибраними з групи, що містить Н, ОН, DMTO, TBDMSO, ВnО, ТНРО, АсО, BzO, OP(NiPr2)O(CH2)2CN, ОРО3Н, дифосфат, трифосфат; R2 та R3 разом можуть бути PhCHO2, TIPDSO2 або DTBSO2.2. Олігонуклеотид, що...
Попередній патент: Пристрій для фільтрації молока
Наступний патент: Процес омолоджування багаторічної деревини кущів винограду
Випадковий патент: Спосіб виявлення вертебрально-базилярної недостатності у хворих з сенсоневральною приглухуватістю