Похідні 1,2,3,4,-тетрагідрохіноксаліндіону та фармацевтична композиція на їх основі
Номер патенту: 44283
Опубліковано: 15.02.2002
Автори: Сісікура Дзун-Іті, Окада Масаміті, Цукамото Сін-іті, Сасамата Масао, Інамі Хіросі, Фудзі Міцуо, Сакамото Суіті








Формула / Реферат
1. Производное 1,2,3,4-тетрагидрохиноксалиндиона следующей формулы (1):
где символы в вышеуказанной формуле имеют, соответственно, следующие значения:
Х - атом азота или группа формулы СН;
R - имидазолильная группа или ди-(низший алкил)аминогруппа ;
R1 - (1) атом галогена, нитро-группа, циано-группа, карбоксильная группа, амино-группа, моно-. или ди-(низший алкил)амино-группа, низшая алканоильная группа, низшая алкилтио-группа, низшая алкилсульфинильная группа, низшая алкилсульфонильная группа или карбамоильная группа;
(2) низшая алкильная группа или низшая алкоксильная группа, которая может быть замещенной атомом (или атомами) галогена, карбоксильной группой (или группами), или арильной группой (или группами);
(3) фенилокси-группа, которая может быть замещена низшей алкоксикарбонильной группой или карбоксильной
группой;
R2 - гидроксильная группа, низшая алкоксильная группа, амино-группа, или моно- или ди-(низший алкил)амино-группа;
А - низшая алкиленовая группа, которая может быть замещенной, или группа формулы: -О-В- и
В - низшая алкиленовая группа;
при условии, что исключается случай, когда R представляет собой имидазолильную группу, R1 представляет собой циано-группу, А представляет собой этиленовую группу, a R2 является гидроксильной группой;
или таутомер вышеописанного производного, его соль, гидрат или сольват.
2. Соединение или соль по п. 1, где R представляет собой имидазолильную группу, а
R1 представляет собой:
(1) атом галогена, нитро-группу, циано-группу, карбоксильную группу, моно- или ди(низший алкил)амино-группу, низшую алкилсульфинильную группу, низшую алкилсульфонильную группу, или карбамоильную группу;
(2) низшую алкильную группу или низшую алкоксильную группу, которая может быть замещена карбоксильной группой или арильной группой;
(3) фенилокси-группу, которая может быть замещена низшей алкоксикарбонильной группой.
3. Соединение или его соль по п. 2, где R представляет собой 1-имидазолильную группу, Х представляет собой группу формулы СН; R1 представляет собой атом галогена, нитро-группу, трифторметильную группу, циано-группу, или бензилокси-группу.
4. Соединение по п. 1, выбранное из группы, включающей:
2-[2,3-Диоксо-7-(1Н-имидазол-1-ил)-6-нитро-1,2,3,4-тетраги.дрохиноксалин-1-ил] уксусной кислоты или ее соль,
2-[2,3-Диоксо-7-(1Н-имидазол-1-ил)-6-трифторметил-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусную кислоту или ее соль, и
2-[6-Бензилокси-2,З-диоксо-7-(1Н-имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусную кислоту или ее соль.
5. Фармацевтическая композиция, отличающаяся тем, что содержит в качестве активного ингредиента эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п. 5, отличающаяся тем, что является антагонистом глутаматного рецептора.
7. Фармацевтическая композиция по п. 5 или 6, отличающаяся тем, что является антагонистом NMDA-глицинового рецептора и/или антагонистом АМРА-рецептора.
8. Фармацевтическая композиция по п. 5 или 6, отличающаяся тем, что является ингибитором нейротоксичности, индуцируемой каиновой кислотой.
9. Фармацевтическая композиция по п. 5 или 6, которая является противоишемическим средством.
10. Фармацевтическая композиция по п. 5 или 6, которая является психотропным средством.
Текст
1 Производное 1,2,3,4тетрагидрохиноксалиндиона следующей формулы О) А-СОЙ 2 (і) где символы в вышеуказанной формуле имеют, соответственно, следующие значения X - атом азота или группа формулы СН, R - имидазолильная группа или ди-(низший алкил)аминогруппа , R1 - (1) атом галогена, нитро-группа, циано-группа, карбоксильная группа, амино-группа, моно- или ди-(низший алкил)амино-группа, низшая алканоильная группа, низшая алкилтио-группа, низшая алкилсульфинильная группа, низшая алкилсульфонильная группа или карбамоильная группа, (2) низшая алкильная группа или низшая алкоксильная группа, которая может быть замещенной атомом (или атомами) галогена, карбоксильной группой (или группами), или арильной группой (или группами), (3) фенилокси-группа, которая может быть замещена низшей алкоксикарбонильной группой или карбоксильной группой, R - гидроксильная группа, низшая алкоксильная группа, амино-группа, или моно- или ди-(низший алкил)амино-группа, А - низшая алкиленовая группа, которая может быть замещенной, или группа формулы -О-В- и В - низшая алкиленовая группа, при условии, что исключается случай, когда R представляет собой имидазолильную группу, R1 представляет собой циано-группу, А представляет собой этиленовую группу, a R2 является гидроксильной группой, илитаутомер вышеописанного производного, его соль, гидрат или сольват 2 Соединение или соль по п 1, где R представляет собой имидазолильную группу, а R1 представляет собой (1) атом галогена, нитро-группу, циано-группу, карбоксильную группу, моно- или ди(низший алкил)амино-группу, низшую алкилсульфинильную группу, низшую алкилсульфонильную группу, или карбамоильную группу, (2) низшую алкильную группу или низшую алкоксильную группу, которая может быть замещена карбоксильной группой или арильной группой, (3) фенилокси-группу, которая может быть замещена низшей алкоксикарбонильной группой 3 Соединение или его соль по п 2, где R представляет собой 1 -имидазолильную группу, X представляет собой группу формулы СН, R1 представляет собой атом галогена, нитро-группу, трифторметильную группу, циано-группу, или бензилокси-группу 4 Соединение по п 1, выбранное из группы, включающей 2-[2,3-Диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетраги дрохиноксалин-1-ил] уксусной кислоты или ее соль, 2-[2,3-Диоксо-7-(1 Н-имидазол-1 -ил)-6трифторметил-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусную кислоту или ее соль, и о го 00 44283 2-[6-Бензилокси-2,3-диоксо-7-(1 Н-имидазол-1 -ил)1,2,3,4-тетрагидрохиноксалин-1-ил] уксусную кислоту или ее соль 5 Фармацевтическая композиция, отличающаяся тем, что содержит в качестве активного ингредиента эффективное количество соединения по п 1 или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель 6 Фармацевтическая композиция по п 5, отличающаяся тем, что является антагонистом глутаматного рецептора 7 Фармацевтическая композиция по п 5 или 6, отличающаяся тем, что является антагонистом Настоящее изобретение относится к хиноксалиндионовому производному или его соли, которое обладает антагонистическим действием по отношению к глутаматному рецептору, высокой степенью сродства к АМРА-рецепторам, не являющимся NMDA-рецепторами, сильным ингибирующим действием, направленным против нейротоксичности, вызываемой каиновой кислотой, ингибирующим аудиогенный приступ действием, и высокой степенью растворимости Настоящее изобретение также относится к агенту, ингибирующему нейротоксичность, вызываемую действием каиновой кислоты, и содержащему в качестве эффективного ингредиента хиноксалиндионовое производное или его соль Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей хиноксалиндионовое производное или его соль и фармацевтически приемлемый носитель Предпосылки создания изобретения Известно, что аминокислоты, такие, как Lглутаминовая кислота и L-аспарагиновая кислота являются нейромедиаторами (нейротрансметерами) центральной нервной системы Иначе говоря, внеклеточная аккумуляция этих возбуждающих аминокислот и их непрерывное и чрезмерное стимулирующее действие на нервные ткани приводит к таким заболеваниям, как хорея Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, сенильная деменция, нейродегенерация, или недостаточность психической и двигательной функций, наблюдаемая после явлений церебральной ишемии, гипоксии или гипогликемии В соответствии с этим был сделан вывод, что, регулятор аномальной активности указанных возбуждающих аминокислот может быть использован для терапевтического лечения дегенерации нервной ткани или психических болезней Действие возбуждающих аминокислот опосредуется глутаматными рецепторами, которые представляют собой специфические постсинаптические или пресинаптические рецепторы В настоящее время, на основании электрофизиологических и нейрохимических исследований, указанные рецепторы можно разделить на три группы 1) NMDA (N-метил-О-аспартат) — рецептор, 2) не - NMDA -рецептор 4 NMDA-глицинового рецептора и/или антагонистом АМРА-рецептора 8 Фармацевтическая композиция по п 5 или 6, отличающаяся тем, что является ингибитором нейротоксичности, индуцируемой каиновой кислотой 9 Фармацевтическая композиция по п 5 или 6, которая является противоишемическим средством 10 Фармацевтическая композиция по п 5 или 6, которая является психотропным средством a) АМРА-рецептор (рецептор [2-амино-3-(3гидрокси-5-метил-4-изоксазолил) пропионовой кислоты]), b) рецептор каината 3) глутаматный рецептор метаболического действия Соединение настоящего изобретения является антагонистом глутаматного рецептора и обладает ингибирующей активностью, направленной против нейротоксического действия каиновой кислоты, а поэтому оно может быть использовано в качестве противоишемического или психотропного средства L-глутаминовая кислота или L-аспарагиновая кислота активируют вышеописанные глутаматные рецепторы, и опосредуют передачу возбуждающих эффектов Воздействие на нейроны избыточным количеством NMDA, AMPA или каиновой кислоты приводит к их гибели Сообщалось, что 2амино-5-фосфоновалериановая кислота или 2амино-7-фосфоногептановая кислота, которые являются избирательными антагонистами NMDAрецептора, обладают эффективным действием в экспериментальной модели животных с невропатией, эпилепсией или церебральной ишемией (J Pharmacology and Experimental Therapeutics, 250, 100 (1989), J Pharmacology and Experimental Therapeutics, 240, 737 (1987), или Science, 226, 850 (1984)) Сообщалось, что NMDA-рецепторные функции аллостерически регулируются глициновым рецептором (Eur J Pharmacol , 126, 303 (1986)), и в то же время сообщалось, что НА-966, который является антагонистом глицинового рецептора, обладает эффективным действием в экспериментальной модели животных с церебральной ишемией (Annual meeting of Society for Neuroscience, 1989) Кроме того, указывалось, что NBQX (6-нитро7-сульфамо-илбензо [f] хиноксалин), который является избирательным антагонистом АМРАрецептора, также обладает эффективным действием в экспериментальной модели животных с церебральной ишемией (Science, 247,571 (1990)) С другой стороны, было показано, что все не — NMDA -рецепторы, подвергнутые клонированию, обладают сродством к каиновой кислоте, и было сделано предположение, что среди этих ре 44283 цепторов, рецептор, имеющий низкую аффинность к каиновой кислоте (АМРА/каинатный рецептор), является ответственным за гибель нейронов во время ишемии, например, инфаркта головного мозга (Р С May & Р М Robison, J Neurochem , 60, 1171-1174 (19931) Этот АМРА/каинатный рецептор имеет высокую степень сродства к АМРА, однако, сайты связывания АМРА и каиновой кислоты пока неизвестны При этом, сообщалось, что АМРА и каиновая кислота обнаруживают различные электрофизиологические ответы на АМРА/каинатный рецептор Указывалось, что в тесте на нейротоксичность, проводимом с использованием системы культуры нервных клеток, сама каиновая кислота вызывает значительную гибель нервных клеток, тогда, как действие лишь одной АМРА является довольно слабым (Р С May & Р М Robison, J Neurochem , 60, 1171-1174 (1993)) В соответствии с этим, можно предположить, что гибель нейронов, вызываемая чрезмерным возбуждающим действием глутаминовой кислоты при ишемическом состоянии, будет интенсивно подавляться соединением, обладающим ингибирующим действием, направленным против токсичности, индуцируемой каиновой кислотой в системе культуры нервных клеток Имеется несколько работ, в которых описаны дикетохиноксалиновые производные, обладающие антагонистическим действием по отношению к NMDA-глициновому рецептору и/или АМРАрецептору (опубликованная нерассмотренная японская патентная заявка (Kokai) № 63-83074, опубликованная нерассмотренная японская патентная заявка (Kokai) № 63-258466, опубликованная нерассмотренная японская патентная заявка (Kokai) № 1 -153680, опубликованная нерассмотренная японская патентная заявка (Kokai) № 2-48578, опубликованная нерассмотренная японская патентная заявка (Kokai) № 2-221263, опубликованная нерассмотренная японская патентная заявка (Kokai) — 2-221264, публикация международного патента W092/07847, и публикация международного патента W0 93/08173) Описание изобретения Как будет более подробно описано ниже, соединение настоящего изобретения обладает антагонистическим действием по отношению к глутаматному рецептору на основе хиноксалина, высокой степенью аффинности к АМРАрецептору, не являющемуся NMDA-рецептором, интенсивным ингибирующим действием, направленным против нейротоксичности, обусловленной действием каиновой кислоты, супрессорной активностью, подавляющей рефлекторный слуховой эпилептический припадок, а также высокой степенью растворимости Кроме того, авторами настоящего изобретения были проведены исследования, относящиеся к дикетохиноксалиновым производным В результате этих исследований было установлено, что соединение, имеющее группу — A-COR2 в 1- или 4-положении дикетохиноксалиновой структуры, обладает высокоэффективным фармакологическим действием (действием, ингибирующим нейротоксичность каиновой кислоты, противосудорожным действием, предотвращающим рефлекторные слуховые эпилептические припадки, или т п) г а также высокой степенью растворимости, а поэтому указанное соединение обладает ценными свойствами, на основании которых было разработано настоящее изобретение Таким образом, настоящее изобретение относится к 1,2, 3,4-тетрагидрохиноксалиндионовому производному или 1,2,3,4-тетрагидропиридо[2,3-Ь] пиразиновому производному следующей формулы A-COR2 .0) R1 где символы, присутствующие в этой формуле, имеют, соответственно, следующие значения X — атом азота или группа формулы СН, R — имидазолильная группа или ди (низший алкил) аминогруппа R — (1) атом галогена, нитро-группа, цианогруппа, карбоксильная группа, амино-группа, моно- или ди (низший алкил) амино-группа, низшая алканоильная группа, низшая алкилтио-группа, низшая алкилсульфонильная группа, низшая алкилсульфонильная группа или карбамоильная группа, (2) низшая алкильная группа или низшая алкоксильная группа, которая может быть замещенной атомом (или атомами) галогена, карбоксильной группой (или группами) или арильнои группой (или арильными группами), (3) фенилокси-группа, которая может быть замещена низшей алкоксикарбонильнои группой или карбоксильной группой, R2 — гидроксильная группа, низшая алкоксильная группа, амино-группа или моно- или ди (низший алкил) амино-группа, А — низшая алкиленовая группа, которая может быть замещенной, или группа формулы — О — В—, и В — низшая алкиленовая группа, при условии, что исключается случай, когда £ представляет собой имидазолильную группу, R представляет собой циано-группу, А представляет собой этиленовую группу, a R является гидроксильной группой, либо к таутомеру вышеописанного производного, его соли, гидрату или сольвату Настоящее изобретение также относится к антагонисту глутаматного рецептора, содержащему в качестве эффективного ингредиента 1,2,3,4тетрагидрохиноксалин-дионовое производное или его фармацевтически приемлемую соль Более конкретно, настоящее изобретение относится к соединению, которое обладает антагонистическим действием по отношению к NMDA-глициновому рецептору и/или по отношению к АМРАрецептору, или активностью, ингибирующей каинат-индуцированную нейротоксичность, и которое 44283 8 может быть использовано в качестве противодиметилбу-тилтио 3,3-д иметилбутилтио 1ишемического или психотропного средства Кроме этилбутилтио 2-этилбутил-тио-, 1,1,2того, настоящее изобретение относится к фарматриметилпропилтио 1,2,2-триметилпропилтио- 1цевтической композиции, содержащей 1,2,3,4этил-1-метилпропилтио 1-этил-2-метилпропилтиотетра-гидрохиноксалиндионовое производное или группы, и т п Предпочтительной является алкилего соль и фармацевтически приемлемый носитио-группа, имеющая 1-3 атома углерода тель Иллюстративными примерами "низшей алкилНиже подробно описано соединение, предсульфонильной группы" являются метилсульфоставленное вышеуказанной общей формулой (1) нильная, этилсульфонильная, пропилсульфонильная, изопропилсульфонильная, бутилсульфоВ вышеприведенном определении общей нильная, изобутилсульфонильная, втор-бутилсуформулы (1), термин "низший", если это не оговольфонильная, трет-бутилсульфонильная, пентил рено особо, относится к линейной или разветв(амил) сульфонильная, изопентил-сульфонильленной углеродной цепи, содержащей 1-6 атомов ная, неопентилсульфонильная, трет-пентил сульуглерода фонильная, 1-метилбутилсульфонильная, 2-меИллюстративными примерами "низшей алтилбутилсульфонильная, 1,2-диметилпкильной группы" являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, третропилсульфонильная, гексилсульфонильная, изобутил, пентил (амил), изопентил, неопентил, третгексилсульфонильная, 1-метил пентилсульфонипентил, 1 -метилбутил, 2-метилбутил, 1,2льная, 2-метилпентилсульфонильная, 3-метилпедиметилпропил, гексил, изогексил, 1-метилпентил, нтилсульфонильная, 1,1-диметилбутилсульфони2-метил пентил, 3-метил-пентил, 1,1льная, 1,2-диметилбутилсульфонильная, 2,2д и метил бутил сул ьфон ильная, 1,3-диметилбутилдиметилбутил, 1,2-д и метилбутил, 2,2сульфонильная, 2,3-д и мети л бути л сул ьфон ильная, диметилбутил, 1,3-д и метилбутил, 2,33,3-д и метилбутилсул ьфон ильная, 1-этилбутилсудиметилбутил, 3,3-д и метилбутил, 1-этилбутил, 2льфонильная, 2-эти л бути л сул ьфон ильная, 1,1,2этилбутил, 1,1,2-три метил пропил, 1,2,2-тритриметилпропилсульфонильная, 1,2,2-триметилпМеТИЛПрОПИЛ, 1-ЭТИЛ-1-МеТИЛПрОПИЛ, 1-ЭТИЛ-2ропилсуль фонильная, 1-этил-1-метилпропилсуметилпропил и т п При этом, предпочтительной льфонильная, 1-этил-2-метилпропилсульфонильявляется алкильная группа, имеющая 1-3 атомов ная группы и т п предпочтительной является углерода алкилсульфонильная группа, имеющая 1-3 атома Используемый в настоящем описании термин углерода "моно- или ди (низший алкил) амино-группа" означает амино-группу, замещенную одной или двумя низшими алкильными группами, проиллюстрированными выше Иллюстративными примерами моно (низший алкил) амино-группы являются метиламино-, этиламино-, пропиламино-, изопропиламино-, бутиламино-, изобутиламино-, вторбутиламино-, трет-бутиламино-, пентил (амил) амино-, изопентиламино-, неопентиламино- и трет-пентиламино-группы? а примерами ди (низший алкил) амино-группы являются диметиламино-, этилметиламино-, диэтиламино-, дипропиламино-, диизопропиламино, дибутиламино- и диизобутиламино-группы Из них предпочтительными являются амино-, метиламино-, этиламино-, диметиламино- и диэтиламино-группы Иллюстративными примерами "низшей алканоильной группы" являются формильная, ацетильная, пропионильная, изопропионильная, бутирильная, изобутирильная, валерильная, изовалерильная, пивалоильная, гексаноильная группы и т п Используемый в настоящем описании термин "низшая алкилтио-группа означает вышеописанную тиоловую группу, в которой атом водорода замещен низшей алкильной группой Иллюстративными примерами низшей алкилтио-группы являются метилтио-, этилтио-, пропилтио-, изопропилтио-, бутилтио-, изобутилтио-, вторбутилтио-, третбутилтио-, пентил (амил) тио-, изопентилтио-, неопентилтио-, третпентилтио-, 1-метилбутилтио-, 2-метилбутилтио-, 1,2-диметилпропилтио-, гексилтио-, изогексилтио-, 1-метил пентилтио-, 2метилпентилтио-, 3-метил пентилтио 1,1диметилбутилтио1,2-диметилбутилтио 2,2диметилбутилтио 1,3-диметилбутилтио2,3 Иллюстративными примерами "низшей алкилсульфонильной группы" являются метилсульфонильная, этилсульфонильная, про пилсульфонильная, изопропилсульфонильная, бутилсульфонильная, изобутилсульфонильная, вторбутилсульфонильная, трет-бутилсульфонильная, пентил (амил) сульфонильная, изопентил-сульфонильная, неопентилсульфонильная, трет-пентилсульфонильная, 1-метилбутилсульфонильная, 2-метилбутилсульфонильная 1,2-диметилпропилсульфонильная, гексилсульфонильная, изо-гексил сул ьфон ильная, 1-метил пентилсульфонильная, 2-метил-пентилсульфонильная, 3-метил пентилсульфонильная, 1,1-диме-тилбутилсульфонильная, 1,2-диметилбутилсульфонильная, 2,2-диметилбутилсульфонильная, 1,3-диметилбутилсульфонильная, 2,3-д и мети л бути л сул ьфон ильная, 3,3-диметилбутилсульфонильная, 1этилбутилсул ьфон ильная, 2-этилбутилсульфонильная, 1,1,2-триметилпропилсульфонильная, 1,2,2-триметилпропилсульфонильная, 1-этил-1метилпропилсул ьфон ильная, 1-этил-2-ме-тилпропилсульфонильная группы и т п Предпочтительной является алкилсульфонильная группа, имеющая 1-3 атома углерода Примерами "низшей алкоксильной группы" являются метокси-, этокси-, пропокси-, изопропокси-, бутокси-, изобутокси-, втор-бутокси-, трет-бутокси, пентилокси (амилокси) —, изопентилокси-, третпентилокси-, неопентилокси-, 2-метил бутокси-, 1,2-диметилпропокси-, 1-этилпропокси-, гексилокси-группы и т п Из них предпочтительными являются метокси-, этокси-, пропокси- и изопропокси-группы Проиллюстрированные выше низшая алкиль 44283 ная группа или низшая алкоксильная группа могут быть замещенными в любом положении, по крайней мере, одним заместителем, выбранным из атома галогена, карбоксильной группы и арильной группы Иллюстративными примерами таких замещенных низших алкильных групп или низших алкоксильных групп являются три-галоген (низший алкил), карбокси (низший алкил), карбокси (низший алкоксил), арил (низший алкоксил), и т п При этом, предпочтительными являются тригалогенметильная, карбоксиметокси — и бензилоксигруппы Арильная группа, которая является заместителем, может быть сама замещенной атомом галогена или карбоксильной группой В качестве примера может служить карбоксибензилоксигруппа Используемый в настоящем описании термин "атом галогена" означает атом фтора, хлора, брома или йода Используемый в настоящем описании термин "арильная группа" означает, например, карбоциклическую арильную группу В качестве примеров арильной группы могут служить фенильная, нафтильная, антрильная или фенантрильная группы и тп Примерами "низшей алкиленовой группы" в А или В являются линейные или разветвленные алкиленовые группы, имеющие от 1 до 6 атомов углерода Иллюстративными примерами таких групп являются метиленовая, этиленовая, триметиленовая, метил метиленовая, д и мети л метиленовая, 1-метилэтиленовая, 2-мети л этиленовая, тетраметиленовая, 1-метилтриметиленовая, 2метилтриметиленовая, 3-метилтриметиленовая, 1-этилэтиленовая, 2-этилэтиленовая, 2,2-диметилэтиленовая, 1,1-диметил-этиленовая, этилметил метиленовая, пентаметиленовая, 1-метил-тетраметиленовая, 2-метилтетраметиленовая, 3метилтетраметиленовая, 4-метилтетраметиленовая, 1,1-диметилтриметиленовая, 2,2-диметилтриметиленовая, 3,3-диметилтриметиленовая, 1,3диметилтриметиленовая, 2,3-диметилтриметиленовая, 1,2-диметилтриметиленовая, 1,1,2-триметилэтиленовая, диэтилметиленовая, гексаметиленовая, 1 -метил пентаметиленовая, 1,1диметилтетраметиленовая, 2,2-диметилтетраметиленовая, 1,3-диметилтетраметиленовая, 1,4диметилтетраметиленовая группы и т п Примером заместителя для "низшей алкиленовой группы, которая может быть замещенной" является фенильная группа, которая может быть замещена нитро-группой В качестве иллюстративных примеров могут служить фенильная, 4нитрофенильная, 3-нитрофенильная, 2-нитрофенильная группы и т п , при этом, предпочтительной является 4-нитрофенильная группа Из групп, представленных R, предпочтительной является имидазолильная группа Предпочтительная группа, представленная R1, может быть определена следующим образом (1) атом галогена, нитро-группа, циано-группа, моно- или ди (низший алкил) амино-группа, низ 10 ший алкилсульфонильная группа, низший алкилсульфонильная группа или карбамоильная группа, (2) низшая алкильная или низшая алкоксильная группа, которая может быть замещена карбоксильной группой или арильной группой, и (3) фенилокси-группа, которая может быть замещена низшей алкоксикарбонильнои группой Более предпочтительным является соединение, где R представляет собой 1-имидазолильную группу, X представляет собой группу формулы СН, a R1 представляет собой атом галогена, нитро-группу, трифторметильную группу, цианогруппу, или бензилокси-группу Особенно предпочтительными соединениями являются 2 — [2,3-диоксо-7-(1 Н-имидазол-1 -ил) — 6нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусная кислота или ее соль, 2 — [2,3-диоксо-7-(1 Н-имидазол-1 -ил) — 6трифторметил-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусная кислота или его соль, и 2 — [6-бензилокси-2,3-диоксо-7-(1Н-имидазол1-ил) — 1,2,3,4 тетрагидрохиноксалин-1-ил] уксусная кислота или ее соль Соединение (1) настоящего изобретения имеет таутомер на основе дикетохиноксалиновой структуры Кроме того, это соединение может существовать в виде оптического изомера (оптически активного соединения, диастереомера, или т п ) в зависимости от вида группы Эти изомеры в чистой форме или смеси изомеров также входят в объем настоящего изобретения Соединение (1) настоящего изобретения образует соль с кислотой или основанием Примерами соли, образованной кислотой, являются соли присоединения кислоты, образованные неорганическими кислотами, например, минеральными кислотами, такими, как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота или фосфорная кислота, и органическими кислотами, такими, как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота или глутаминовая кислота Примерами соли, образованной основанием, являются соли, образованные неорганическими основаниями, такими, как натрий, калий, магний, кальций или алюминий, органическими основаниями, такими, как метиламин, этиламин или этаноламин, и основными аминокислотами, такими, как лизин, аргинин, или орнитин, или аммониевые соли Кроме того, соединение (1) настоящего изобретения может образовывать гидрат, сольват со спиртом, и обладает способностью к полиморфизму (Методы получения) Соединение настоящего изобретения может быть получено способом, проиллюстрированным нижеследующей реакционной схемой 44283 12 тонитрил, ацетон или тетрагидрофуран (ТГФ) Для ускорения реакции может быть добавлено основание, такое, как гидроксид натрия или гидроксид калия, либо соль меди Соединение (1) настоящего изобретения может быть также получено посредством реакции деблокирования соединения (IV) или реакции удаления R2 в соответствии со стандартной методикой Реакция деблокирования или удаления R2 может быть осуществлена методом, описанным в вышеуказанной книге, изданной Т W Greene и др , либо другим известным методом Так, например, может быть проведен гидролиз В качестве примера может служить кислотный гидролиз, проводимый в присутствии соляной кислоты или т п , и щелочное омыление, проводимое в присутствии основания, такого, как гидроксид натрия Метод 2 Соединения (1), (IX) или (X) настоящего изоблокирования бретения могут быть получены следующим спосоИЛИ уцажэнвд К?, есда бом это необходимо (1) Нитроанилиновое или нитроаминопиридиновое соединение (V) восстанавливают, в результате чего получают диаминовое соединение (VI) (2) Диаминовое соединение (VI) подвергают 1 2 где X, A, R и R являются такими, как они быреакции с оксалатгалогенидом (VII), в результате ли определены выше чего получают амидное соединение (VIII) и его Y представляет собой атом галогена, смесь Y' представляет собой атом галогена, кроме (3) Амидное соединение (VIII) и его смесь податома фтора, вергают реакции замыкания кольца, в результате 1 R представляет собой атом водорода и групчего получают Хиноксалиндионовое соединение пу, определенную выше для К, или пиридопиразиндионовое соединение (IX) или R представляет собой низшую алкильную (X) группу, (4) Хиноксалиндионовое соединение или пи2 Z , Z представляют собой атом водорода и ридопиразиндионовое соединение (IX) подвергают 3 группу формулы R COCO-, при условии, что оба известной реакции замещения в ароматическом Z1 и Z2 не являются атомами водорода одновреядре, в результате чего получают замещенное менно Хиноксалиндионовое соединение или пиридопиВ вышеуказанной формуле, примерами атома разиндионовое соединение (X) галогена являются атомы фтора, хлора, брома и (5) Реакцию деблокирования или удаления R2 йода осуществляют, как описано в Методе 1 Если в соединении присутствует гидроксильНиже описаны характерные примеры осуще2 ная группа, представленная R , то может быть ствления вышеуказанных стадий (1) — (4) использовано соединение, в котором карбоксильПримером реакции восстановления в стадии ная группа защищена защитной группой (1) может служить каталитическое восстановлеПримерами соединения с защищенной карние, осуществляемое стандартным способом с боксильной группой и его эквивалента являются использованием, например, скелетного никелевосложные эфиры, амиды и ортоэфиры, а также го катализатора Ренея или катализатора "паллапроизводные, описанные в "Protective Groups in дий-на-угле", и восстановление металлом с исOrganic Synthesis, 2nd ed , edited byT W Greene & пользованием измельченного в порошок железа P G M Wuts, John Willey & Sons, INC , Chapter 5 или цинковой пыли (1990)" Примером реакции амидирования в стадии (2) Метод 1 является реакция между диаминовым соединениСоединения (I) или (IV) настоящего изобретеем (VI) и оксалатгалогенидом (VII) в количестве, ния могут быть получены посредством реакции соответствующем данной реакции, которую прогалогенного соединения (П) и имидазола (III), взяводят в растворителе, таком, как хлороформ или тых в соответствующих данной реакции количестТГФ, при температуре от -10 С до 60 С, а предпочвах, при этом указанная реакция может быть протительно от О С до комнатной температуры ведена в растворителе или в отсутствие Для ускорения реакции предпочтительно дорастворителя при размешивании, и при темперабавить щелочь такую, кактриэтиламин туре от -10 С до 150 С, а предпочтительно от 20 С Реакцию замыкания кольца в стадии (3) осудо120°С ществляют, например, путем нагревания амидного Эту реакцию обычно осуществляют при насоединение (VIII) и его смеси в вышеуказанном гревании в растворителе, таком, как диметилфоррастворителе, либо путем нагревания в присутстмамид (ДМФ), диметилсульфоксид (ДМСО), аце11 13 44283 вий кислотного катализатора, такого, как соляная кислота Примерами реакции замещения в ароматическом ядре, в стадии (4), где R1 в соединении (IX) является водородом, могут служить метод воздействия азотной кислотой или ее солью на хиноксалиндионовое соединение или пиридопиразиндионовое соединение (IX) в серной кислоте, в смеси уксусного ангидрида и уксусной кислоты или в смеси серной кислоты, уксусного ангидрида и уксусной кислоты, и метод осуществления реакции между хиноксалиндионовым соединением или пиридопиразиндионовым соединением и тетрафторборатом нитрония в органическом растворителе, таком, как сульфолан или ацетонитрил при температуре 0°С или при нагревании Метод 3 Соединения (1), (IX) или (X) настоящего изобретения могут быть получены следующим способом (a) Нитроанилидное соединение восстанавливают как описано в (2) Метода 2, в результате чего получают аминоанилидное соединение (XII) (b) Аминоанилидное соединение (XII) подвергают реакции замыкания кольца, как описано в (3) Метода 2, в результате чего получают хиноксалиндионовое соединение (IX) или (X) Последующий процесс получения соединения (1) настоящего изобретения осуществляют в соответствии со вторым способом получения Соединение, имеющее в качестве R1 нитрогруппу, может быть получено, например, посредством реакции нитрования, которой подвергают хиноксалиндионовое соединение (IX), где R1 является атомом водорода, и которую осуществляют, как описано в (4) Метода 2 Метод 4 Соединение, в котором R1 является аминогруппой, может быть получено посредством реакции восстановления соединения, в котором R1 является нитро-группой, как описано в (2) Метода 2 Альтернативно, это соединение может быть получено посредством реакции деблокирования с использованием защищенной амино-группы, представленной R1, как описано в Методе 2 или 3 в соответствии с известными способами Соединение, где R1 является моно- или ди (низший алкил) — амино-группой, может быть получено посредством реакции соединения, в котором R1 является амино-группой, с галоген (низший алкил) — содержащим соединением, как описано в Методе 1 Альтернативно, это соединение может быть получено посредством реакции альдегидного соединения (такого, как формалин) с аминовым соединением в растворителе или в отсутствие растворителя, предпочтительно в присутствии кислоты, в условиях аналогичных условиям осуществления стадии (1) Метода 2 Метод 5 Соединение, в котором R2 является аминогруппой или моно- или ди (низший алкил) аминогруппой, может быть получено путем реакции амидирования карбокси-соединения (1), осуществляемой известным способом Альтернативно, это соединение может быть получено посредством реакции эфир-амид ного обмена между эфирным соединением (IV), (IX) 14 или (X) и соответствующим амином или аммиаком Реакцию амидирования карбокси-соединения (1) осуществляют, например, путем превращения этого соединения в хлор-ангидрид с использованием, например, тионилхлорида, с последующим добавлением соответствующего количества амина или аммиака Реакцию эфир-амидного обмена осуществляют, например, посредством реакции сложноэфирного соединения с концентрированным водным аммиаком при температуре от -10°С до комнатной температуры Метод 6 Соединение (II), в котором А является алкиленокси-группой, может быть получено посредством реакции соответствующего гидроксихоноксалин2,3-диона с соответствующим алкилирующим агентом в присутствии основания Эта реакция может быть осуществлена, в основном, в растворителе, таком, как ДМФ, ДМСО, ТГФ, ацетонитрил или ацетон В качестве основания предпочтительно использовать органическое основание (например, карбонат калия или гидрид натрия) Метод 7 Соединение (1), в котором R1 является карбамоильной группой, может быть получено известным способом путем обработки соотвествующего производного (1), имеющего в качестве R цианогруппу, в кислотных или основных условиях Так, например, это соединение может быть получено посредством реакции циано-производного с кислотой, такой, как соляная кислота, серная кислота или муравьиная кислота, либо посредством реакции в основных условиях, например, в водном растворе перекиси водорода или гидроксида натрия Полученные таким образом соединения настоящего изобретения выделяют и очищают в виде свободного соединения или его соли Выделение и очистку соединений настоящего изобретения осуществляют традиционными химическими методами, такими, как экстракция, концентрирование, выпаривание, кристаллизация, фильтрация, перекристаллизация, различные способы хроматографии и т п Соединение настоящего изобретения обладает высокой степенью аффинности к АМРАрецептору, сильной активностью, подавляющей нейротоксичность, индуцированную каиновой кислотой, и противосудорожным действием, направленным на подавление рефлекторных слуховых эпилептических припадков у мыши DBA /2 Поэтому соединение настоящего изобретения, обладающее ценными фармацевтическими свойствами, может быть использовано, в частности, как психотропное средство для предупреждения и лечения хореи Гентингтона, болезни Паркинсона, эпилепсии, болезни Альцгеймера или сенильной деменции, а также как противоишемическое средство для предупреждения и лечения состояний, связанных с гибелью клеток, вызываемой церебральной ишемией, гипоксией, временной остановкой сердца, дегенерацией нервной ткани, наблюдаемой после гипогликемии или эпилептического припадка, и недостаточностью психической и дви 15 44283 гательной функции Метод испытаний Определение активности, ингибирующей связывание с [3Н] АМРА и подавляющей нейротоксическое действие каиновой кислоты и рефлекторные слуховые эпилептические припадки, осуществляли следующим образом 1) Измерение активности, ингибирующей связывание [3Н]-АМРА При охлаждении ледяной водой, 0,5 мл (всего) реакционной жидкой смеси, содержащей около 45 иМ [ Н]-АМРА (2-амино-3-(3-гидрокси-5-метил-4изоксазолил) пропионовая кислота), около 300 мг образца мозговой оболочки крысы, и испытуемое соединение, подвергали реакции в течение 45 минут Количество [3Н]-АМРА, связанное с АМРАрецептором, измеряли методом фильтрации За количество специфического связывания принимали часть (от общего связанного количества), замещенную 10 мкМ хискваловой кислотой Оценку испытуемого соединения проводили путем определения степени ингибирования специфического связывания В результате такого определения было, например, установлено, что величина к' для соединения Примера 1 составляет 0,73 мкМ, для соединения Примера 9 она составляет 0,093 мкМ, а для соединения Примера 19 эта величина составляет 0,070 мкМ, что свидетельствует о том, что эти соединения являются сильными ингибиторами 2) Измерение активности, ингибирующей нейротоксичность каиновой кислоты Ингибирующее действие соединения настоящего изобретения, направленное на подавление нейротоксичности каиновой кислоты, исследовали с использованием системы первичной культуры нейронов гиппокампа плода крысы (1) Условия инкубирования Из головного мозга 18-20-дневного плода крысы вырезали гиппокамп и обрабатывали папаином и ДНКазой 1 для диспергирования клеток Полученные таким образом клетки помещали на среду MEM, содержащую 10%-ную сыворотку, а затем инокулировали на 48-луночный планшет, предварительно обработанный поли-1-лизином в концентрации 4 х 10 кл/см 2 Через 24 часа сыворотку заменяли бессывороточной средой Замену среды проводили два раза в неделю Клетки культивировали в течение, по крайней мере, 6 дней, после чего проводили следующий тест (2) Ингибирование нейротоксичности, индуцируемой каиновой кислотой Нейротоксичность выражали в виде активности лактатде-гидрогеназы, высвобождаемой в культуральную среду вследствие гибели клеток В качестве контроля использовали нейроны, обработанные в течение 24 часов бессывороточной средой, содержащей 30 мкМ каиновой кислоты Нейроны подвергали воздействию каждого из испытуемых соединений вместе с 300 мкМ каиновой кислотой в течение 24 часов, после чего оценивали ингибирующее действие каждого из соединений, направленное на предотвращение гибели нейронов, вызываемой каиновой кислотой В результате этих испытаний было установлено, например, что IC50 для соединения Приме 16 ра 1 составляет 0,8 мкМ, 1Cso для соединения Примера 9 составляет 0,96 мкМ, a 1Cso для соединения Примера 19 составляет 0,48 мкМ, что свидетельствует о том, что эти соединения обладают сильным ингибирующим действием (3) Измерение ингибирующего действия, направленного на подавление рефлекторных слуховых эпилептических припадков у мышей DBA /2 Десять 21-28-дневных мышей-самцов подвергали слуховой стимуляции звуком в 12 кГц и 120 дБ в звуковой испытательной камере в течение 1 минуты или до тех пор, пока у мышей не возникнет тонической эпилептический припадок Испытуемое соединение суспендировали в 0,5%-ном растворе метил-целлюлозы или растворяли в физиологическим растворе, и полученную суспензию или раствор вводили мышам внутрибрюшинно за 15 минут до начала стимуляции звуком Эффективность лекарственного средства оценивали при появлении и в отсутствие припадков, и определяли минимальную эффективную дозу (MED) Результаты этих оценок показали, что соединение Примера 1, соединение Примера 9 и соединение Примера 19 подавляли слуховой эпилептический припадок у мышей при концентрациях 3 мг/кг, 10 мг/кг и 1 мг/кг, соответственно (4) Измерение растворимости Получение буфера К 0,1 М водному раствору первичного кислого фосфата калия добавляли 0,1 М водный раствор первичного кислого фосфата натрия, при этом получали буферы, которые имели рН 5, 6, 7 и 8, соответственно Измерение растворимости Примерно 5-миллиграммовые порции соединения настоящего изобретения точно взвешивали в четырех стеклянных лабораторных пробирках и добавляли 0,1 мл фосфатных буферов, имеющих рН 5, 6, 7 и 8, соответственно, после чего пробирки интенсивно встряхивали Растворимость определяли как величину, полученную из нижеследующего уравнения Растворимость (мг/мл) соединения настоящего изобретения = Масса соединения настоящего изобретения (мг), определенная путем взвешивания Объем (мл) фосфатного буфера, использованного для растворения соединения настоящего изобретения Результаты этих измерений представлены в Таблице 1 Соединение Примера 9 имело растворимость 4 100мкг/мл при рН 6 Таким образом, соединение настоящего изобретения имеет высокую растворимость даже при нейтральных или близких к нейтральным значениях рН В соответствии с этим, соединение настоящего изобретения может быть с успехом включено в качестве активного ингредиента в препараты для перорального введения, такие, как таблетки и капсулы, или в препараты для парентерального введения, например, для инъекций Кроме того, соединение настоящего изобретения обладает высокой, а поэтому прекрасной растворимостью в крови даже при клиническом введении, и не подвергается легкому осаждению в 17 44283 18 органах, что придает этому соединению особую мерами разбавителя для безводного раствора или безводной суспензии могут служить пропиленглиценность коль, полиэтиленгликоль, растительное масло, такое, как оливковое масло, спирт, такой, как этаТаблиця нол и "Полисорбат 80" (торговая марка) Кроме того, указанная композиция может также содеррН5 рН6 рН7 рН8 жать такие добавки, как агент для придания расПри2489(мк 2621 (мк 3641 (мк 5130мкг твору изотоничности, антисептическое средство, мер1 г/мл) г/мл) г/мл) /мл) смачивающий агент, эмульгирующий агент, диспергирующее средство, стабилизатор (например, Фармацевтический препарат, содержащий одлактоза), и солюбилизирующий агент или солюбино или несколько соединений настоящего изобрелизирующая добавка Эти композиции могут быть тения или его соли в качестве эффективного инстерилизованы, например, путем фильтрации чегредиента, получали с использованием носителя рез бактериальный фильтр, путем введения инили наполнителя и других добавок, которые обычсектицида, или путем облучения Препарат для но используются для изготовления лекарственноинъекций может быть также изготовлен в виде го средства стерильной твердой композиции, которая может Носители или наполнители, используемые быть затем, непосредственно перед ее использодля изготовления фармацевтических препаратов, ванием, растворена в стерильной воде или в стемогут быть твердыми или жидкими, например, рильном растворе, пригодном для инъекций такими, как лактоза, стеарат магния, крахмал, Вышеописанные композиции могут быть вветальк, желатин, агар, пектин, аравийская камедь, дены любым способом, например, перорально, в оливковое масло, кунжутное масло, какао-масло, форме таблеток, драже, капсул, гранул, порошков этиленгликоль и другие стандартные носители или растворов, парентерально, в виде инъекций, или наполнители таких, как внутривенные или внутримышечные В соответствии с настоящим изобретением, в инъекции, в виде суппозиториев, или чрезкожно качестве твердой композиции для перорального Вводимая доза должна быть определена отдельвведения могут быть использованы таблетки, поно в каждом конкретном случае в зависимости от рошки, гранулы и т п В такой твердой композиции симптомов заболевания, возраста или пола пациможет присутствовать одно или несколько активента Однако, в основном, для взрослого индивиных веществ, смешанных, по крайней мере, с оддуума, эта доза, в случае перопального введения, ним инертным разбавителем, таким, как лактоза, составляет в пределах от 1 до 1000 мг/день, а маннит, глюкоза, гидроксипропилцеллюлоза, микпредпочтительно от 50 до 200 мг/день и может рокристаллическая целлюлоза, крахмал, поливибыть введена один раз в день или порциями ненил пиррол идон или метасиликат алюминиясколько раз в день, или, в случае внутривенного магния Помимо инертного разбавителя, эта комвведения, эта доза составляет в пределах от 1 мг позиция может содержать добавки, например, до 500 мг в день, и может быть введена один раз в замасливатель, такой, как стеарат магния, дезиндень или порциями несколько раз в день, либо, в тегратор, такой, как кальций-содержащий гликолят случае непрерывного внутривенного введения, целлюлозы, стабилизатор, такой, как лактоза, или эта доза может вводиться непрерывно в течение солюбилизирующий агент или солюбилизирующая 1-24 часов в день Как уже указывалось выше, добавка, такая, как глутаминовая кислота или асвводимая доза может варьироваться в зависимопарагиновая кислота Таблетки или драже могут сти от ряда факторов, а поэтому, необходимо отбыть покрыты оболочкой, изготовленной из гастметить, что в некоторых случаях может оказаться ро- или энтеросолюбильного вещества, такого, как достаточной более низкая доза, чем дозы, опресахароза, желатин, гидроксипропилцеллюлоза, деленные вышеуказанными интервалами доз или фталат гидроксипропилметилцеллюлозы В качестве примеров жидкой композиции для перорального введения могут служить фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или элексиры, которые содержат обычно используемый в этих целях инертный разбавитель, такой, как очищенная вода или этанол Помимо инертного разбавителя, жидкая композиция может также содержать адъювант, такой, как солюбилизирующий агент или солюбилизирующая добавка, смачивающий агент или суспендирующий агент, подслащивающий агент, отдушка, ароматизирующее средство, или антисептическое средство Примерами композиции для парентерального введения путем инъекции могут служить стерильные водные и безводные растворы, суспензии и эмульсии В качестве разбавителя для водного раствора или водной суспензии могут быть использованы дистиллированная вода, пригодная для инъекций, и физиологический раствор При Примеры Более подробно настоящее изобретение описано в нижеследующих Примерах, при этом следует иметь в виду, что эти Примеры не должны рассматриваться как некое ограничение настоящего изобретения Кроме того, в этих Примерах также описано получение главных исходных соединений Пример 1 1) К смеси, содержащей 13,96 г гидрохлорида этилового эфира глицина, 30 мл тетрагидрофурана, 10,11 г триэтиламина и 30 мл диметилформамида, добавляли 15,91 г 2,4-дифторнитробензола, и эту смесь нагревали с обратным холодильником в течение трех часов в потоке газа аргона Реакционную смесь охлаждали и разводили этилацетатом, после чего, осажденное нерастворившееся вещество удаляли путем фильтрации Фильтрат концентрировали при пониженном давлении Полученный остаток растворяли в 19 44283 20 етилацетате, последовательно промывали водой 4) Смесь, содержащую 928 мг этил-2-(2, 3и солевым раствором, а затем осушали безводдиоксо-7-фтор-6-нитро-1,2,3,4ным сульфатом натрия, и концентрировали при тетрагидрохиноксалин-1-ил)ацетата, 450 мг имипониженном давлении Полученный остаток передазола и 10 мл диметилформамида, перемешикристаллизовывали из этанола, в результате чего вали в течение шести часов при температуре получали 17,32 г (71,5%) этилового эфира N-(2120°С После охлаждения реакционную смеет вынитро-5-фторфенил) глицина МС (m/z) 242 (М+) ливали в ледяную воду Полученные кристаллы собирали путем фильтрации, промывали водой, и Спектр ядерного магнитного резонанса (ЯМРосушали при пониженном давлении спектр) (CDCI3, внутренний стандарт-ТМС) 5 1,33 (ЗН, т, J-6,5 Гц), 4,04 (2Н, д, и=4,9Гц), 4,31 Полученное соединение добавляли к 5 мл 1 н (2Н, кв , J=6,5I~4), 6,34 (1Н, дд, J=2,4, 11,0Гц), 6,44 водного раствора гидроксида натрия при комнат(1Н, м), 8,25 (1Н, дд, J=6,1 и 9,7Гц), 8,55 (1Н, с) ной температуре, а затем, смесь перемешивали в течение 15 минут для гидролиза сложного эфира 2) Смесь, содержащую 6,52 г этилового эфиРеакционную смесь доводили до рН 3,5 путем ра І\І-(2-нитро~5-фторфенил) глицина, 100 мл тетдобавления 1 н соляной кислоты Осажденные рагидрофурана, 50 мл метанола и 500 мг 10% таким образом кристаллы собирали путем фильтпалладия-на-угле, перемешивали в атмосфере рации, промывали водой, и осушали при пониженводорода, в результате чего восстанавливали ном давлении, в результате чего получали 473 мг нитрогруппу Полученную реакционную смесь (40%) 2-[2, 3-диоксо-7-(1Н-имидазол-1-ил) — 6фильтровали, и фильтрат концентрировали при нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] уксуспониженном давлении К полученному остатку ной кислоты 1 НС1 1,2 Н20 добавляли 200 мл хлороформа и 7,54 мл триэтиламина, и эту смесь растворяли К полученной Т пл 225°С (с разлож) смеси, в атмосфере аргона, при охлаждении Элементный аналил для C13H9N5O6, — НС1 льдом и при перемешивании, по капле добавляли 1,2 Н20 раствор, содержащий 30 мл хлороформа в 7,35 г Вычислено С 40,11, Н 3,21, N17,99, CI 9,11 этилхлорглиоксилата После завершения добав(%) Найдено С 40,01, Н 3,11, N17,86, CI 9,02 (%) ления, перемешивание продолжали в течение МС (m/z) 332 (М+ + 1) ЯМР-спектр (flMCO-d6, одного часа при комнатной температуре, а затем внутренний стандарт — ТМС) смесь разводили хлороформом Разбавленный 5 4,89 (2Н, с), 7,88 (1Н, с), 8,01 (1Н, с), 8,08 раствор последовательно промывали водой, на(1Н, с), 8,28 (1Н, с), 9,43 (1Н, с), 12,89 (1Н, с), 13,1сыщенным водным раствором бикарбоната на14,2 (1Н, ш трия, насыщенным водным раствором хлорида Пример 2 аммония и солевым раствором, после чего осу1) 14,58 г (80%) метил-3-(5-фтор-2шали безводным сульфатом натрия, и концентринитрофениламино) — пропионата получали споровали при пониженном давлении К полученному собом, описанным в Примере 1-1, за исключением остатку добавляли 150 мл этанола и 1 мл концентого, что использовали 10,5 г гидрохлорида метитрированной соляной кислоты, а затем смесь налового эфира [3-аланина, 20 мл диметилформагревали с обратным холодильником в течение мида, 12,0 г 2,4-дифторнитробензола, 7,58 г триодного часа После охлаждения полученные криэтиламина и 20 мл тетра-гидрофурана сталлы собирали путем фильтрации и осушали МС (m/z) 242 (М+) при пониженном давлении, в результате чего поЯМР-спектр (CDCI3, внутренний стандарт — лучали 5,80 г (80%) этил-2-(7-фтор-2,3-диоксоТМС) 1,2,3,4-тетрагидрохинаксолин-1-ил) ацетата 5 2,73 (2Н, т, Т =6,6 Гц), 3,61 (2Н, т, J=6,6 Гц), МС (m/z) 266 (М+) 3,74 (ЗН, с), 6,36-6,42 (1Н, м), 6,52 (1Н, дд, Т =2,9 и 11,7 Гц), 8,22 (1Н, дд, J=5,9, 9,3 Гц), 8,34 (1Н, с) ЯМР-спектр (ДМСО-de, внутренний стандарт — ТМС) 5 1,22 (ЗН, т, J=7,3 Гц), 4,17 (2Н, кв , J = 2) 5,84 г (55%) этил-3-(2,3-диоксо-7-фтор7,3Гц), 4,95 (2Н, с), 7,70 (1Н,м), 7,22 (1Н, дд, J=5,4, 1,2,3,4-тетрагидрохиноксалин-1-ил) пропионата 8,8Гц), 7,36 (1Н, дд, J-2,4 и 11,2Гц), 12,20 (1Н, с) получали способом, описанным в Примере 1-2, за исключением того, что использовали 6,19 г ме3) В 15 мл концентрированной серной кислоты тил-3-(5-фтор-2-нитрофениламино) пропионата при температуре ниже 0°С растворяли 1,20 г МС (m/z) 280 (М+) этил-2-(7-фтор-2,3-ди-оксо-1,2,3,4-тетрагидрохинаксолин-1-ил) ацетата К полученному раствору, ЯМР-спектр (ДМСО-de, внутренний стандарт при перемешивании, по капле добавляли 0,21 мл — ТМС) дымящей азотной кислоты (d=l,52), и эту смесь 5 1,27 (ЗН, т, J=6,8 Гц), 2,79 (2Н, т, и=7,6Гц), перемешивали в течение 30 минут при той же 4,18 (2Н, кв , и=6,8Гц), 4,45 (2Н т, J=7,6 Гц), 6,94температуре Реакционную смесь выливали в во6,99 (1Н, м), 7,07 (1Н, дд, J=2,4, 9,7Гц), 7,30 (1Н, ду со льдом Осажденные таким образом кридд, J=4,5, 9,7Гц), 11,23 (1Н, с) сталлы собирали путем фильтрации, промывали 3) 1,58 г (91%) этил-3-(2,3-диоксо-7-фтор-6водой, и осушали при пониженном давлении, в нитро-1,2, — 3,4-тетриагидрохиноксалин-1-ил) результате чего получали 1,35 г (96%) этил-2-(7пропионата получали способом, описанным в фтор-6-нитро-2,3-диоксо-1,2,3,4-тетрагидро-хинокПримере 1-3, за исключением того, что использосалин-1-ил)ацетата МС (т/2) 312 (М+ + 1) ЯМРвали 1,50 г этил-3-(2,3-диоксо-7-фтор-1,2,3,4спектр (ДМСО-de, внутренний стандарт — ТМС) тетрагидро-хиноксалин-1-ил) пропионата 5 1,24 (ЗН, т, и=7,ЗГц), 4,18 (2Н, кв, и=7,ЗГц), МС (m/z) 326 (М+ * 1) ЯМР-спектр (flMCO-d6, 5,02 (2Н, с) 7,74 (1Н, д, и=13,5Гц), 7,95 (1Н, д, внутренний стандарт — ТМС) и=7,ЗГц), 12,4 (1Н, с) 5 1,21 (ЗН, т, и=7,1Гц), 2,67 (2Н, т, и=7,ЗГц), 21 44283 22 Элементный анализ для C15H13N5O6 HC1 4,08 (2H, кв, J=7,iriO, 4,32 (2H, т, и=13,ЗГц), 7,78 0,1 Н2О(%) (1Н, д, J=13,8 Гц), 7,89 (1Н, д, и=7,ЗГц), 12,3 (1Н, Вычислено С 45,32, Н 3,60, N 17, 62, CI 8,92 с) Найдено С 45,20, Н 3,68, N17,57, CI 8,96 4) 704 мг (48%) 32,3-диоксо-7-(1Н-имидазол-1MC(m/z) 360 (М+ + 1) ил) — 6-нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] пропионовой кислоты х 1НС1 х ІНгО получали ЯМР-спектр (ДМСО-de, внутренний стандарт способом, описанным в Примере 1-4, за исключе— ТМС) нием того, что использовали 1,20 г этил-3-(75 1,86 (дт, J=6,8, 7,4 Гц), 2,38 (2Н, т, и=6,8Гц), фтор-6-нитро-2,3-диоксо-1,2,3,44,12 (2Н, т, и=7,4Гц), 7,92 (1Н, с), 8,02 (1Н, с), 8,07 тетрагидрохиноксалин-1-ил) пропионата, 502 мг (1Н, с), 8,25 (1Н, с), 9,56 (1Н, с), 12,70 (1Н, с), 11,8имидазола и 10 мл диметилформамида 12,6 (1Н, шире) Т пл 281 -282°С (с разлож) Пример 4 Элементный анализ для C14H11N5O6 HC1 Ь^О Целевое соединение Примера 4 получали способом, описанным в Примере 1 (%) 1) 7,59 г (47%) этилового эфира ІЧ-(4-фтор-2Вычислено С 42, 07, Н 3,53, N 17,52, С1 8,87 нитрофенил) глицина получали с использованием Найдено С 41,98, Н 3,78, N 17,63, С1 8,65 9,25 г гидрохлорида этилового эфира глицина, MC(m/z) 346 (М+ + 1) 10,55 г (66,3 ммоль) 2,5-дифтор-нитробензола, 35 ЯМР-спектр (ДМСО-de, внутренний стандарт мл тетрагидрофурана, 9,29 мл триэтиламина, и 5 — ТМС) мл ДМФ ЯМР-спектр (flMCO-d6, внутренний стан5 2,65 (2Н, т, J=7,8 Гц), 4,29 (2Н, т, и=7,8Гц), дарт — ТМС) 7,91 (1Н, с), 8,05 (1Н, с), 8,07 (1Н, с), 8,25 (1Н, с), 9,57 (1Н, с), 12,72 (1Н, с), 12,1-13,2 (1Н, шире) 5 1,32 (ЗН, т, и=7,4Гц), 4,08 (2Н, д, и=5,5Гц), 4,29 (2Н, кв, и=7,4Гц), 6,68 (1Н, дд, Т=4,3, 9,1 Гц), Пример 3 7,24-7,29 (1Н, м), 7,92 (1Н, дд, J=3,0, 9,1 Гц), 8,27 1) 9,72 г (61%) этил-4-(5-фтор-2(1Н, шир , с ) нитрофениламино) бутирата получали способом, описанным в Примере 1-1, за исключением того, 2) Смесь, содержащую 7,41 г (30,6 ммоль) что использовали 9,55 г гидрохлорида этил-уэтилового эфира N - (4-фтор-2-нитрофенил) глиаминобутирата, 20 мл тетрагидрофурана, 6,73 г цина, 120 мл тетрагидрофурана, и 0,5 г 10% палтриэтиламина, 10 мл диметилформамида, и 9,45 г ладия-на-угле, перемешивали при комнатной тем2,4-дифторнитробензола пературе при нормальном давлении в атмосфере водорода После завершения реакции, катализаМС (m/z) 270 (М+) ЯМР-спектр (CDC13), внуттор удаляли путем фильтрации К фильтрату доренний стандарт — ТМС) бавляли 150 мл тетрагидрофурана и 19,5 мл три5 1,28 (ЗН, т, и=7,0Гц), 2,02-2,08 (2Н, м), 2,47 этиламина К полученной смеси при охлаждении (2Н, кв, и=7,1Гц), 3,35 (2Н, т, и=7,0Гц), 4,17 (2Н, Т, льдом по капле добавляли смесь, содержащую 19 и=6,0Гц), 6,35-6,41 (1Н, м), 6,52 (1Н, дд, Г=2,4, 11,6 г этилхлоргли-оксилата и 20 мл тетра гидрофураГц), 8,21 (1Н, дд, J=5,1, 9,2Гц) на, а затем перемешивали Реакционную смесь 2) 5,93 г (85%) этил-4-(2,3-диоксо-7-фторнагревали до комнатной температуры и переме1,2,3,4-тет-рагидрохиноксалин-1-ил)бутирата пошивали в течение ночи Осажденные таким обралучали способом, описанным в Примере 1-2, за зом кристаллы удаляли путем фильтрации, и этот исключением того, что использовали 6,43 г этилфильтрат концентрировали при пониженном дав4-(5-фтор-2-нитрофениламино)бутирата МС (m/z) лении К остатку добавляли 150 мл этанола и 1,5 294 (М+) ЯМР-спектр (ДМСО-de, внутренний станмл концентрированной соляной кислоты, и эту дарт — ТМС) смесь нагревали с обратным холодильником в 5 1,28 (ЗН, т, J=7, 4 Гц), 2,04-2,10 (2Н, м), 2,52 течение 4 часов После охлаждения, осажденные (2Н, т, J=6,7 Гц), 4,18-4,26 (2Н, м), 6,93-6,98 (1Н, кристаллы собирали путем фильтрации, промым), 7,26-7,35 (2Н, м), 11,59 (1Н, с) вали этанолом, и осушали при пониженном дав3) 2,81 г (92%) этил-4- (2,3-диоксо-7-фтор-6лении, в результате чего получали 6,77 г (83%) нитро-1, 2,3,4-тетрагидрохиноксалин-1-ил) бутираэтил-2-(2,3-диоксо-6-фтор-1,2,3,4та получали способом, описанным в Примере 1-3, тетрагидрохиноксалин-1-ил)ацетата ЯМР-спектр за исключением того, что использовали 2,64 г (ДМСО-de, внутренний стандарт — ТМС) этил-4(2,3-диоксо-7-фтор-1,2,3,4-тетрагидрохиноксалин-1-ил) бутирата МС (m/z) 340 (М+ 5 1,22 (ЗН, т, 3 = 7,3Гц), 4,17 (2Н, kb, J= 7,3 + 1) Гц), 4,97 (2Н, с), 7,00 (1Н, дд, J= 3,0, 9,2Гц), 7,027,06 (1Н, м), 7,35 (1Н, дд, J= 4,9, 9,2 Гц), 12,25 (1Н, ЯМР-спектр (ДМСО-сіє, внутренний стандарт — ТМС) с) 5 1,29 (ЗН, т, J =7,ЗГц), 1,98-2,05 (2Н, м), 2,52 3) 2,74г (96%) этил-2-(2,3-диоксо-6-фтор-7(2Н, т, и=6,7Гц), 4,15-4,24 (4Н, м), 7,56 (1Н, д, нитро-1,2,3,4-тетрагидрохиноксалин-1-ил)ацетата J=12,9riO, 8,03 (1Н, д, и=6,7Гц) получали с использованием 2,45 г (9,21 миллимоль) этил-2-(2,3-диоксо-6-фтор-1,2, 3,44) 1,42г (80%) 4-[2,3-диоксо-7- (1Н-имидазолтетрагидрохиноксалин-1-ил)ацетата, 15 мл кон1-ил) -6-нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] центрированной серной кислоты, и 0,5 мл дымямасляной кислоты 1НС1 0,1 НгО получали спосощей азотной кислоты ЯМР-спектр (ДМСО-de, бом, описанным в Примере 1-4, за исключением внутренний стандарт — ТМС) того, что использовали 1,50 г этил-4-(7-фтор-6нитро-2, З-диоксо-1,2,3, 4-тетрагидрохиноксалин5 1,23 (ЗН, т, J= 6,7Гц), 4,19 (2Н, kb, J= 6,7Гц), 1-ил) бутирата, 632 мг имидазола, и 10 мл диме5,05 (1Н, с), 7,19 (1Н, д, J= 11,6Гц), 8 ДО (1Н, д, = тилформамида Т пл >300°С 6,7Гц), 12,68 (1Н, с) 23 44283 24 4) 2,24 г (96%) этил-2-[2,3-диоксо-6-(1Нли при пониженном давлении, в результате чего имидазол-1 -ил) -7-нитро-1,2,3, 4получали 90 мг (89%) 2-[2,3-диоксо-7-(1Нтетрагидрохиноксалин-1-ил] ацетата получали с имидазол-1 -ил) — 6-нитро-1,2,3, 4использованием 2, 02 г этил-2-(2,3-диоксо-6тетрагидрохиноксалин~1-ил] ацетамида 0,5 ІЧНз фтор-7-нитро-1,2,3,4-тетрагидрохиноксалин-1-ил) 1,3 Н20 Т пл 245°С (с разлож) ацетата, 1,33 г имидазола, и 15 мл ДМФ ЯМРMC(m/z) 331 (М+ + 1) спеїсгр (ДМСО-de, внутренний стандарт — ТМС) ЯМР-спектр (ДМСО-de, внутренний стандарт — ТМС) 5 1,25 (ЗН, т, J = 7,3Гц), 4,21 (2Н, kb, J = 7,3Гц), 5,09 (1Н, с), 7,11 (1Н, с), 7,26 (1Н, с), 7,45 5 4,77 (2Н, с), 6,3-6,9 (1Н, шир с ), 7,07 (1Н, с), (1Н, с), 7,93 (1Н, с), 8,21 (1Н, с), 12,3-13,0 (1Н, 7,22-7,33 (2Н, м), 7,36 (1Н, с), 7,64 (1Н, с), 7,80шир с) 7,87 (2Н, м) 5) 1,88 г (84%) 22,3-диоксо-6-(1 Н-имидазол-1 Пример 7 ил) — 7-нитро-1,2,3,4-тетрагидрохиноксалин-1Смесь, содержащую 465 мг этил-2-[6-аминоиллуксусной кислоты -НС1 НгО получали с ис2,3-диоксо-7-(1 Н-имидазол-1 -ил) — 1,2,3,4пользованием 2,09 г (5,80 миллимоль) этил-2тетрагидрохиноксалин-1-ил]-ацетата, 160 мг фор12,3-диоксо-6-(1 Н-имидазол-1 -ил) — 7-нитромалина, 30 мл воды, 3 мл 1н соляной кислоты и 1,2,3,4-тетрагидрохиноксалин-1-ил] ацетата и 10 100 мг 20% палладия-на-угле, перемешивали в мл 1 н водного раствора гидроксида натрия Т пл атмосфере водорода в течение 8 часов, а затем 217-218°С фильтровали Фильтрат концентрировали при пониженном давлении Полученный остаток добавЭлементный анализ для C13H9N5O6 HC1 ляли к 4 мл 1 н водного раствора гидроксида наН2О (%) трия при комнатной температуре, и эту смесь Вычислено С 40,48, Н 3,14, N 18,16, С1 9,19 перемешивали в течение одного часа РеакционНайдено С 40,17, Н 3,04, N 18,08, С1 9,20 ную смесь доводили до рН 6 путем добавления 1 Пример 5 н соляной кислоты Осажденные таким образом 1) Смесь, содержащую 1,30 г этил-2- [2, 3кристаллы собирали путем фильтрации, промыдиоксо-7-(1 Н-имидазол-1 -ил) — 6-нитро-1,2,3,4вали водой, и осушали при пониженном давлении, тетрагидрохиноксалин-1-ил]-ацетата, 80 мл тетраа затем очищали на колонке с НР20, в результате гидрофурана, 20 мл метанола, и 600 мг 10% палчего получали 33 мг (6%) 22,3-диоксо-7-(1Нладия-на-угле, перемешивали в атмосфере водоимидазол-1-ил) — 6-метиламино-1,2,3, 4-тетрагидрода в течение 36 часов, после фильтрования рохиноксалин-1-ил] уксусной кислоты 1НС1 реакционной смеси, фильтрат концентрировали 1,45Н20 Тпл >300°С при пониженном давлении, в результате чего получали 1,02 г (86%) этил-2-[6-амино-2,3-диоксо-7MC(m/z) 316 (М+ + 1) (1 Н-имидазол-1-ил) — 1, 2, 3, 4ЯМР-спектр (ДМСО-de, внутренний стандарт тетрагидрохиноксалин-1-ил)-ацетата — ТМС) ЯМР-спектр (ДМСО-de, внутренний стандарт 5 2,65 (ЗН, с), 4,74 (2Н, с), 5,58 (1Н, шире), — ТМС) 6,57 (1Н, с), 7,51 (1Н, с), 7,78 (1Н, с), 7,84 (1Н, с), 9,24 (1Н, с), 12,18 (1Н, с), 13,0-13,1 (1Н, шире) 5 1,14-1,22 (ЗН, м), 4,09-4,18 (2Н, м), 4,88 (2Н, с), 5,05 (2Н, с), 6,71 (1Н, с), 7,11 (1Н, с), 7,16 (1Н, Пример 8 с), 7,30 (1Н, с), 7,74 (1Н, с), 12,11 (1Н, с) 395 мг (44%) 2-[6-диметиламино-2, З-диоксо-7(1 Н-имидазол-1-ил) -1,2,3, 42) К 1,5 мл 1 н водного раствора гидроксида натрия добавляли 150 мг этил-2-[6-амино-2,3тетрагидрохиноксалин-1-ил] уксусной кислоты диоксо-7-(1 Н-имидазол-1 -ил) -1,2,3,40.7Н2О получали способом, описанным в Примере тетрагидрохиноксалин-1-ил] ацетата при комнат1, за исключением того, что использовали 0,93 г ной температуре, и эту смесь перемешивали в этил-2- [б-амино-2, З-диоксо-7- (1 Н-имидазол-1-ил) течение двух часов Реакционную смесь доводили -1,2,3, 4-тетрагидрохинокса-лин-1-ил_|ацетата, до рН 6 путем добавления 1 н соляной кислоты 0,92 г формалина, 50 мл воды, 5 мл 1 н соляной Осажденные таким образом кристаллы собирали кислоты, и 600 мг 10% палладия-на-угле путем фильтрации, промывали водой, и осушали Т пл >300°С при пониженном давлении, в результате чего поМС (m/z) 329 (М+) лучали 112 мг (74%) 2г [6-амино-2,3-диоксо-7-(1НЯМР-спектр (ДМСО-de, внутренний стандарт имидазол-1-ил) — 1,2,3,4-тетра-гидрохиноксалин— ТМС) 1-ил] уксусной кислоты -1,7 Н2 300°С 5 2,40 (6Н, с), 4,87 (2Н, с), 6,97 (1Н, с), 7,10 MC(m/z) 302 (М+ + 1) (1Н, с), 7,31 (1Н, с), 7,42 (1Н, с), 7,88 (1Н, с), 12,14 (1Н,с) Спектр ядерного магнитного резонанса (ДМСО-de, внутренний стандарт — ТМС) Пример 9 5 4,78 (2Н, с), 5,04 (2Н, шире), 6,71 (1Н, с), 1) К смеси, содержащей 4,84 г (29 миллимоль) 7,11 (2Н, с), 7,29 (1Н, с), 7,73 (1Н, с), 12,09 (1Н, с) гидрохлорида трет-бутилового эфира глицина, 20 мл тетрагидрофурана, 4,1 мл триэтиламина, и 3 Пример 6 мл ДМФ, при ' перемешивании, по капле добавляК 6 мл водного раствора аммиака добавляли ли смесь 6,57 г 2, 4-дифтор-5100 мг этил-2- [2,3-диоксо-7-(1 Н-имидазол-1-ил) — трифторметилнитробензо-ла и 5 мл тетрагидро6-нитро-1,2,3,4-тетра-гидрохиноксалин-1-ил] ацефурана После перемешивания в течение трех тата при температуре -5 С, и полученную смесь часов к реакционной смеси добавляли 50 мл перемешивали в течение 3 часов при температуре этилацетата, и эту смесь фильтровали Фильтрат 0°С, а затем концентрировали при пониженном концентрировали при пониженном давлении Кондавлении Концентрат промывали водой и осуша 25 44283 26 центрат экстрагировали путем добавления этилда натрия и насыщенного водного раствора биацетата и воды Органический слой последовакарбоната натрия Полученный раствор очищали с тельно промывали водой, 1% водным раствором использованием "НР-20" (продукт, поставляемый бикарбоната натрия, и солевым раствором, а заMitsubishi Chemical Corporation, элюирующий растем осушали безводным сульфатом натрия, и творитель водЫметанол) Полученный таким обконцентрировали при пониженном давлении Осразом очищенный продукт перекристаллизовывататок очищали с помощью хроматографии на коли из 1 н водного раствора соляной кислоты, в лонке с силикагелем (элюирующей растворитель результате чего получали 1,31 г (40%) 2 - [2, 3гексан/эти л ацетат = 1 0 1,5), в результате чего подиоксо-7-(1Н-гимидазол-1-ил)-6-трифторметиллучали 9,54 г (98%) трет-бутилового эфира N-(51,2,3,4-тетрагидрохиноксалин-1-ил] уксусной кифтор-2-нитро-4-трифторметилфенил) глицина слоты 1HCI 1 Н2О ЯМР-спектр (CDCI3 внутренний стандарт-ТМС) Тпл 226-227°С 5 1,54 (9Н, с), 3,98 (2Н, д, J=4,9 Гц), 6,46 (1Н, МС (m/z) 354 (М+) д, J= 12,7 Гц), 8,54 (1Н, д, J=7,8 Гц), 8,76 (1Н, ЯМР-спектр (ДМСО-de, внутренний стандартшир с ) ТМС) 2) Смесь, содержащую 9,36 г (27,7 милли5 4,86 (1Н, с), 7,89 (1Н, с), 7,91 (1Н, д, J= 1,5 моль) трет-бутилового эфира ІЧ-(5-фтор-2-нитро-4Гц), 8,04 (1Н, с), 8,13 (1Н, с), 9,54 (1Н, с), 12,89 трифторметилфенил) глицина, 7,54 г имидазола и (1Н, с), 12,5-14,0 (1Н, шире) 30 мл ДМФ, перемешивали в течение 2 часов в Целевые соединения Примеров 10-12 получамасляной бане при температуре 60 С После охли способом, описанным в Примере 9 лаждения реакционную смесь концентрировали Пример 10 при пониженном давлении Остаток выливали в 1) 4,01 г (86%) трет-бутилового эфира N-(4воду и осажденное таким образом соединение ацетил-5-фтор-2-нитрофенил) глицина получали с собирали путем фильтрации Полученное соедииспользованием 2,50 г гидрохлорида третнение последовательно промывали водой и этибутилового эфира глицина, 20 мл тетрагидрофуловым эфиром, а затем осушали при пониженном рана, 5 мл диметилформамида, 2,08 мл триэтидавлении, в результате чего получали 8,95 г (84%) ламина и 3,0 г 2,4-дифтор-5-нитроацетофенона трет-бутилового эфира 1\1-[5-(1Н-ими-дазол-1-ил)— ЯМР-спектр (CDCI3, внутренний стандарт-ТМС) 2-нитро-4-трифторметилфенил] глицина 5 1,53 (9Н, с), 2,58 (ЗН, д, J= 4,3Гц), 3,98 (2Н, д, J=4,9 Гц), 6,34 (1Н, д, J=12,9 Гц), 8,90 (1Н, д, ЯМР-спектр (CDCI3, внутренний стандарти=8,2Гц), 10,7 (1Н, шире) ТМС) 5 1,52 (9Н, с), 4,02 (2Н, д, J =4,9Гц), 6,67 (1Н, 2) 3,80 г (87%) трет-бутилового эфира N - [4с), 7,14 (1Н, с), 7,21 (1Н, с), 7,64 (1Н, с), 8,66 (1Н, ацетил-5-(1 Н-имидазол-1 -ил)-2-нитрофенил] глис), 8,76 (1Н, шире) цина получали с использованием 3,77 г (12,1 ммоль) трет-бутилового эфира ІЧ-(4-ацетил-53) Смесь, содержащую 3,08г (7,98 ммоль) фтор-2-нитрофенил) глицина, 3,28 г имидазола и трет-бутилового эфира N-[5-(1 Н-имидазол-1-ил)-215 мл диметилформамида ЯМР-спектр (CDCI3, нитро-4-трифторметил-фенил] глицина, 100 мл внутренний стандарт-ТМС) тетрагидрофурана, 50 мл метанола и 350 мг 10% палладия-на-угле, перемешивали в атмосфере 5 1,53 (9Н, с), 2,14 (ЗН, с), 4,02 (2Н, д, водорода при нормальном давлении и при коми=5,1Гц), 6,57 (1Н, с), 7,09 (1Н, с), 7,25 (1Н, с), 7,63 натной температуре После завершения реакции, (1Н, с), 8,74 (1Н, с), 8,76 (1Н, шир с ) палладий-на-угле удаляли путем фильтрации, а 3) В данной стадии использовали третзатем фильтрат концентрировали при пониженном бутиловый эфир 1Ч-[4-ацетил-5-(1 Н-имидазол-1 давлении К полученному остатку добавляли 150 ил)-2-нитрофенил] глицина (3,63 г), 120 мл тетрамл хлороформа и 2,46 мл триэтиламина К полугидрофурака и 360 мг 10% палладия-на-углет и ченной смеси при перемешивании и при охлаждепроводили реакцию восстановления После этого нии льдом по капле добавляли смесь 2,29 г (16,7 получали 1,32 г (37%) 2-[6-ацетил-2, З-диоксо-7ммоль) этилхлорглиоксилата и 20 мл хлороформа (1 Н-имидазол-1-тл)-1,2,3,4-тетрагидрохиноксалинПосле завершения добавления реакционную 1 -ил] уксусной кислоты-1 НгО с использованием смесь оставляли для нагревания до комнатной 7,1 мл триэтиламина и 6,89 г этилхлорглиоксилатемпературы, а затем перемешивали в течение та ночи После этого добавляли 200 мл хлороформа Тпл 225-226°С (с разлож) ЯМР-спектр Полученную смесь последовательно промывали (ДМСО-de, внутренний стандарт-ТМС) водой и солевым раствором, осушали безводным 5 2,40 (ЗН, с), 4,90 (2Н, с), 7,73 (1Н, с), 7,81 сульфатом натрия и концентрировали при пони(1Н, с), 7,84 (1Н, с), 7,99 (1Н, с), 9,16 (1Н, с), 12,70 женном давлении К полученному остатку добав(1Н, с), 12,9-14,3 (1Н, шир с ) ляли 100 мл этанола и 2 мл 1 н соляной кислоты, Пример 11 и эту смесь нагревали с обратным холодильником 1) 2,27г (28%) 2-(4-трет-бутоксикарбонилметив течение 10 часов После охлаждения реакционламино-2-фтор-5-нитрофенокси) ацетата полуную смесь концентрировали при пониженном давчали с использованием 5,60 г (21, 4 миллимоль) лении К остатку добавляли 15 мл трифторэтил-2-(2,4-дифтор-5-нитрофенокси) ацетата, 3,59 уксусной кислоты, и смесь перемешивали при г гидрохлорида трет-бутилового эфира глицина, комнатной температуре в течение 6 часов Полу40 мл тетрагидрофурана, 10 мл диметилформаченную реакционную смесь концентрировали при мида, и 3 мл триэтиламина ЯМР-спектр (GDCI3, пониженном давлении, а затем доводили до рН=7 внутренний стандарт-ТМС) путем добавления 1 н водного раствора гидрокси5 1,31 (ЗН, т, и=6,8Гц), 1,52 (9Н, с), 3,94 (2Н, д, 27 44283 28 J=4,5I~4), 4,28 (2H, кв, J=6,8I~4), 4,64 (2Н, с), 6,45 мина К полученной смеси при перемешивании и (1Н, д, J=12,7f4), 7,83 (1Н, д, и=8,8Гц), 8,42 (1Н, при охлаждении льдом по капле добавляли смесь с)-2) 1,88 г (78% этил-[4-трет11,96г этилхлорглиоксилата и 20 мл тетрагидрофурана Реакционную смесь нагревали до комбутоксикарбонилметиламино-2-(1 Н-имидазол-1 натной температуры, а затем перемешивали в ил)-5-нитрофенокси] ацетата получали с испольтечение двух часов Осажденные таким образом зованием 2,13г (5,73ммоль) 2-(4-треткристаллы удаляли путем фильтрации, и этот бутоксикарбонилметиламино-2-фтор-5фильтрат концентрировали при пониженном давнитрофенокси) ацетата, 1,56г имидазола, и 15мл лении К остатку добавляли 100 мл этанола, и ДМФ полученную смесь нагревали с обратным холоЯМР-спеїсгр (CDCI3, внутренний стандартдильником в течение трех часов После охлаждеТМС) ния осажденные кристаллы собирали путем 5 1,31 (ЗН, т, = 8,7Гц), 1,52 (9Н, с), 3,98 (2Н, д, фильтрации, последовательно промывали этано=5,4Гц), 4,27 (2Н, кв, =8,7Гц), 4,66 (2Н, с), 6,65 (1Н, лом и диэтиловым эфиром, а затем осушали при с), 7,20 (1Н, с), 7,39 (1Н, с), 7,39 (1Н, с), 7,83 (1Н,пониженном давлении К полученному соединес), 8,01 (1Н, с), 8,37 (1Н, шир с ) нию добавляли 30 мл трифторуксусной кислоты, и 3) В данной стадии использовали этил-2-[4эту смесь перемешивали в течение ночи при комтрет-бутоксикарбонил метил) амино-2-(1Ннатной температуре Реакционную смесь конценимидазод-1-ил)-5-нитрофенокси] ацетат (1,75 г трировали при пониженном давлении К получен4,71 ммоль), 100 мл тетрегидрофурана и 0,3 г 10% ному остатку добавляли 10 н водный раствор палладия-на-угле, и проводили реакцию восстагидроксида натрия и 1 н водный раствор гидроновления 1,03 г (53%) 2-[6-карбоксиметокси-2, 3ксида натрия для доведения рН до значения 9-10, диоксо-7-(1 Н-имидазол-1 -ил)-1,2,3,4а затем перемешивали в течение ночи Для довететрагидрохиноксалин-1-ил] уксусной кислоты дения рН до 2-3, к реакционной смеси добавляли получали с использованием 3,3 мл триэтиламина концентрированную соляную кислоту и 1 н водный и 3,22 г этилхлорглиоксилата раствор соляной кислоты Осажденные кристаллы Т пл >300°С (с разлож) собирали путем фильтрации Полученное соедиМС (m/z) 360 (М+) нение перекристаллизовывали из 1 н водного расЯМР-спектр (ДМСО-de, внутренний стандарттвора соляной кислоты, в результате чего получаТМС) ли 3,65г (63%) 2-[6-карбокси-2,3-диоксо-7-(1Н5 4,81 (2Н, с), 4,88 (2Н, с), 7,09 (1Н, с), 7,81 имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1(1Н, с), 7,89 (1Н, с), 8,02 (1Н, с), 9,45 (1Н, с), 12,40 ил] уксусной кислоты 0,15 НСІ 1,5 НгО (1Н, с), 12,5-14,2 (1Н, шир с ) Пример 12 Т пл >300°С МС (m/z) 330 (М+) 1) 6,41 г (87%) трет-бутилового эфира N-(4ЯМР-спектр (ДМСО-de, внутренний стандартэтокси-карбонил-5-фтор-2-нитрофенил) глицина ТМС) получали с использованием 3,63 г гидрохлорида 5 4,92 (2Н, с), 7,18 (1Н, с), 7,43 (1Н, с), 7,56 трет-бутилового эфира глицина, 25 мл тетрагид(1Н, с), 7,83 (1Н, с), 8,08 (1Н, с), 12,48 (1Н, с) рофурана, 3,03 г триэтиламина, 5 мл диметилПример 13 формамида и 5,0 г (21,6 ммоль) этил-2, 4-дифтор1) К смеси, содержащей 6,86г 2,4-дифтор-55-нитробекзоата нитро-бензонитрила, 22,67г триэтиламина, 40мл ДМФ, и 40мл тетрагидрофурана, при охлаждении ЯМР-спектр (CDCI3, внутренний стандартльдом добавляли 5,20г гидрохлорида этилового ТМС) эфира глицина, и полученную смесь перемешива5 1,39 (ЗН, т, Г=8,3 Гц), 1,53 (9Н, с), 3,98 (2Н, ли при той же температуре в течение 4 часов К д, J=4,9 Гц), 4,37 (2Н, кв, J =8,3 Гц), 6,36 (1Н, д, реакционной смеси добавляли воду, и полученный J=12,7 Гц), 8,74 (1Н, шире), 8,88 (1Н,д, J=8,7 Гц) продукт экстрагировали хлороформом Экстракт 2) 7,01 г (97%) трет-бутилового эфира N-[4последовательно промывали водой и солевым этоксикар-бонил-5-(1 Н-имидазол-1 -ил)-2раствором, осушали безводным сульфатом нанитрофенил] глицина получали с использованием трия, и концентрировали при пониженном давле6,35 г (18,5 ммоль) трет-бутилового эфира N-(4нии Полученный остаток очищали с помощью этоксикарбонил-5-фтор-2-нитрофенил) глицина, хроматографии на колонках с силикагелем (элю5,05 г имидазола, и 20 мл ДМФ ент гексан/эти л ацетат = 80 20), в результате чего ЯМР-спектр (CDCI3, внутренний стандартполучали 4,85г (49%) этилового эфира ІЧ-(4-цианоТМС) 5тфтор-2-нитрофенил)глицина ЯМР-спектр 5 1,18 (ЗН, т, Т=7,3 Гц), 1,52 (9Н, с), 4,01 (2Н, (CDCI3, внутренний стандарт-ТМС) д, J = 5,4Гц), 4,19 (2Н, кв, и=7,ЗГц), 6,58 (1Н, с), 7,07 (1Н, с) 7,19 (1Н, с), 7,60 (1Н, с), 8,75 (1Н, шир 5 1,22-1,48 (ЗН, м), 4,08 (2Н, д, J =5,3 Гц), с), 8,96 (1Н, с) 4,19-4,50 (2Н, м), 6,46 (1Н, д, J=11,3 Гц), 8,57 (1Н, д, 3=6,7 Гц), 8,80-9,05 (1Н, шир) 3) Смесь, содержащую 6,84 г (17,5 миллимоль) трет-бутилового эфира N-[42) Смесь, содержащую 2,50 г этилового эфира этоксикарбонил-5-(1 Н-имидазол-1 -ил)-2|\|-(4-циано-5-фтор-2-нитрофенил) глицина, 0,67 г нитрофенил] глицина, 150 мл тетрагидрофурана и имидазола, 7,40 г пиридина и 40 мл ДМСО пере0,5 г 10% палладия-на-угле, перемешивали при мешивали в течение четырех часов при темперанормальном давлении и при комнатной температуре 80°С После добавления воды реакционный туре в атмосфере водорода После завершения продукт экстрагировали хлороформом Экстракт реакции катализатор удаляли путем фильтрации последовательно промывали водой и солевым К этому фильтрату добавляли 12,3 мл триэтилараствором, а затем осушали безводным сульфа 29 44283 30 том натрия и концентрировали при пониженном тетрагидрофурана, и 2,42 г гидрохлорида этиловодавлении Полученный остаток очищали с помого эфира глицина ЯМР-спектр (CDCI3, внутренний щью колоночной хроматографии на силикагеле стандарт-ТМС) (элюент хлороформ метанол= 95 5), в результате 5 1,29-1,36 (ЗН, м), 2,20 (ЗН, с), 4,03 (2Н, д, J = чего получали 2,82 г (92%) этилового эфира N-[45,5 Гц), 4,24-4,34 (2Н, м), 6,32 (1Н, д, J=11,6 Гц), циано-5-(1 Н-имидазол-1 -ил)-2-нитрофенил] глици8,10 (1Н, д, J=7,9 Гц), 8,39 (1Н, шир) на ЯМР-спектр (ДМСО-de, внутренний стандарт2) 470 мг (39%) этилового эфира N-[5-(1HТМС) имидазол-1-ил)-4-метил-2-нитрофенил] глицина получали с использованием 1,00 г этилового эфи5 1,15-1,26 (ЗН, м), 4,10-4,21 (2Н, м), 4,45 (2Н, ра І\І-(5-фтор-4-метил-2-нитрофенил)-глицина, 266 д, J=5,5 Гц), 7,15-7,22 (2Н, м), 7,68-7,73 (1Н, м), мг имидазола и 10 мл пиридина 8,17 (1Н, с), 8,75 (1Н, с), 8,85-8,94 (1Н, м) 3) Смесь, содержащую 2,00 г этилового эфира ЯМР-спектр (CDCI3, внутренний стандарт1Ч-[4-циано-5-(1 Н-имидазол-1 -ил)-2-нитрофенил] ТМС) глицина, 40 мл тетрагидрофурана, 8 мл метанола 5 1,26-1,36 (ЗН, м), 2,15 (ЗН, с), 4,05 (2Н, д, и 200 мг 10% палладия-на-угле, перемешивали в J=5,5 Гц), 4,23-4,33 (2Н, м), 6,55 (1Н, с), 7,09 (1Н, атмосфере водорода для восстановления нитрос), 7,24 (1Н, с), 7,63 (1Н, с), 8,20 (1Н, с), 8,32 (1Н, группы После фильтрации реакционной смеси шир) фильтрат концентрировали при пониженном дав3) 221 мг (44%) этил-2-[2,3-диоксо-7-(1Нлении Затем к остатку добавляли 50мл хлороимидазол-1 -ил)-6-метил-1,2,3, 4форма и 4,43мл триэтиламина, в полученную тетрагидрохиноксалин-1-ил] ацетата получали с смесь, при охлаждении льдом, по капле вводили использованием 467 мг этилового эфира N-[5-(1H1,69 мл этилхлорглиоксилата После перемешиимидазол-1-ил)-4-метил-2-нитрофенил] глицина вания при комнатной температуре в течение 20 ЯМР-спектр (ДМСО-de, внутренний стандартчасов реакционную смесь разводили хлорофорТМС) мом Разведенную смесь промывали солевым 5 1,21 (ЗН, т, J=7,3 Гц), 2,17 (ЗН, с), 4,10-4,19 раствором, осушали безводным сульфатом на(2Н, м), 7,25 (1Н, с), 7,70 (1Н, с), 7,89 (1Н, с), 7,98 трия и концентрировали при пониженном давле(1Н, с), 12,45 (1Н, с) нии К полученному остатку добавляли 60 мл эта4) 180 мг (79%) 22,3-диоксо-7-(1 Н-имидазол-1нола и 0,5мл концентрированной соляной ил)-6-метил-1,2,3,4-тетрагидрохиноксалин-1-ил] кислоты, и эту смесь нагревали с обратным холоуксусной кислоты 1 HCL 0,4 НгО получали с исдильником в течение 3 часов После охлаждения пользованием 218 мг этил-22,3-диоксо-7-(1 Нобразовавшиеся кристаллы собирали путем имидазол-1 -ил)-6-метил-1,2,3, 4фильтрации и осушали при пониженном давлететрагидрохиноксалин-1-ил] ацетата нии, в результате чего получали 1,07 г (50%) этилТпл >300°С 2-и6-циано-2,3-диоксо-7-(1 Н-имидазол-1 -ил)MC(m/z) 301 (М+ + 1) 1,2,3,4-тетрагидрохиноксалин-1-ил] ацетата ЯМРЯМР-спектр (ДМСО-de, внутренний стандартспектр (ДМСО-de, внутренний стандарт-ТМС) ТМС) 5 1,15-1,25 (ЗН, м), 4,10-4,20 (2Н, м), 4,99 (2Н, 5 2,11 (ЗН, с), 4,83 (2Н, с), 7,25 (1Н, с), 7,72 с), 7,84 (1Н, с), 7,90 (1Н, с), 8,04 (1Н, с), 8,16 (1Н, (1Н, с), 7,90 (1Н, с), 7,99 (1Н, с), 9,34 (1Н, с), 12,46 с), 9,47 (1Н, с), 12,83 (1Н, с) (1Н, с), 13,1-13,3 (1Н, шир) 4) К 6 мл 1 н водного раствора гидроксида наПример 15 трия добавляли 672мг этил 2-[6-циано-2, 3-диоксо1) 0,59 г (20%) этилового эфира N-(4,57-(1 Н-имидазол-1-ил)-1,2,3,4-тетрагидрохиноксадифтор-2-нитрофенил) глицина получали с ислин-1-ил] ацетата при комнатной температуре, и пользованием 2,00 г 2,4,5-трифторнитробензола, эту смесь перемешивали один час для гидролиза 3,43 г триэтиламина, 25 мл ДМФ, 25 мл ТГФ, и сложного эфира Реакционную смесь доводили до 1,58 г гидрохлорида этилового эфира глицина рН 1 путем добавления 1 н соляной кислоты ЯМР-спектр (CDCI3, внутренний стандарт-ТМС) Осажденные таким образом кристаллы собирали 5 1,20-1,45 (ЗН, м), 4,03 (2Н, д, J=5,3 Гц), 4,15путем фильтрации, промывали водой, и осушали 4,47 (2Н, м), 6,48 (1Н, дд, J=12,2 и 6,6 Гц), 8,10 при пониженном давлении, в результате чего по(1Н, дд, Т=10,6и8,4 Гц), 8,3-8,6 (1Н, шир) лучали 417 мг (62%) 2-[6-циано-2,3-диоксо-7-(1Н2) 0,41 г (59%) этилового эфира ІЧ-[4-фтор-5имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1(1 Н-имидазол-1-ил)-2-нитрофенил] глицина полуил] уксусной кислоты 1,7 НгО чали с использованием 0,59 г этилового эфира N(4,5-дифтор-2-нитрофенил) глицина, 155 мг имиТ шт 283-285°С (с разлож) дазола, 1,80 г пиридина и 10 мл ДМСО ЯМРЯМР-спектр (ДМСО-de, внутренний стандартспектр (CDCI3, внутренний стандарт-ТМС) ТМС) 5 4,95 (2Н, с), 7,17 (1Н, с), 7,62 (2Н, с), 7,74 5 1,20-1,46 (ЗН, м), 4,03-4,48 (4Н, м), 6,66 (1Н, (1Н, с), 8,06 (1Н, с), 12,54 (1Н, с), 13,2-13,4 (1Н, д, J=6,1 Гц), 7,20-7,40 (2Н, м), 7,90 (1Н, шир), 8,10шир) 8,60 (2Н, м) Соединения Примеров 14-20 получали спосо3) 154 мг (35%) этил-2-[2,3-диоксо-6-фтор-7бом, описанным в Примере 13 (1 Н-имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин1-ил] ацетата получали с использованием 406 мг Пример 14 этилового эфира 1Ч-[4-фтор-5-(1 Н-имидазол-1 -ил)1) 2,70 г (61%) этилового эфира ІЧ-(5-фтор-42-нитрофенил] глицина ЯМР-спектр (ДМСО-de, метил-2-нитрофенил) глицина получали с испольвнутренний стандарт-ТМС) зованием 3,00 г 2, 4-дифтор-5-нитротолуола, 8,77 г триэтиламина, 40 мл диметил-формамида, 40 мл 5 1,16-1,29 (ЗН, м), 4,12-4,22 (2Н, м), 4,98 (2Н, 31 44283 32 с), 7,33 (1Н, д, J=10,8 Гц), 7,81-7,96 (2Н, м), 8,06 (1Н, д, J= 7,3Гц), 8,49 (1Н, шир с ) (1Н, с), 9,36 (1Н, с), 12,58 (1Н, с) 2) 3,50г (90%) этилового эфира ІЧ-[4-бром-5(1Н-имидазол-1-ил)-2-нитрофенил] глицина полу4) 90мг (61%) 22,3-диоксо-6-фтор-7-(1Нчали с использованием 3,36г (10,5 миллимоль) имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1этилового эфира І\І-(4-бром-5-фтор-2-нитрофенил) ил] уксусной кислоты 1 НгО получали с использоглицина, 2,86г имидазола и 20мл диметилванием 152мг этил-2-[2,3-диоксо-6-фтор-7-(1 Нформамида ЯМР-спектр (CDCI3 внутренний станимидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1дарт-ТМС) ил] ацетата Тпл >300°С 5 1,32 (ЗН, т, и=7,4Гц), 4,05 (2Н, дд, J=5,2, 14,9Гц), 4,29 (2Н, кв, и=7,4Гц), 6,64 (1Н, д, MC(m/z) 305 (М+ + 1) и=9,1Гц), 7,19 (1Н, с), 7,23 (1Н, с), 7,72 (1Н, с), 7,72 ЯМР-спектр (ДМСО-de, внутренний стандарт(1Н, с), 8,45 (1Н шир с ), 8,57 (1Н, с) ТМС) 5 4,93 (2Н, с), 7,13 (1Н, с), 7,21 (1Н, д, J=10,4 3) Смесь, содержащую 3,24г (8,78ммоль) этиГц), 7,55 (1Н, с), 7,65 (1Н, д, 7=6,7 Гц), 8,01 (1Н, с), лового эфира N-[4-6poM-5-(1 Н-имидазол-1 -ил)-212,36 (1Н, с), 13,0-13,2 (1Н, шир) нитрофенил] глицина, 70мл тетрагидрофурана, 50 мл метанола, и приблизительно 0,5г никелевого Пример 16 катализатора Ренея, перемешивали при комнат1) 0,66г (7%) этилового эфира N-(4-xnop-5ной температуре и нормальном давлении в атмофтор-2-нитрофенил) глицина получали с испольсфере водорода После завершения реакции казованием 6,32г 5-хлор-2,4-дифторнитробензола, тализатор удаляли путем фильтрации, и фильтрат 9,91г триэтиламина, 60мл ДМФ, и 60мл тетрагидконцентрировали при пониженном давлении К рофурана, и 4,56 г гидрохлорида этилового эфира остатку добавляли 100мл хлороформа и 3,1мл глицина ЯМР-спектр (CDCI3, внутренний стантриэтиламина К полученной смеси при охлаждедарт-ТМС) нии льдом и при перемешивании по капле добав5 1,16-1,47 (ЗН, м), 4,04 (2Н, д, J=5,3 Гц), 4,16ляли смесь 3,02г этилхлоргли-оксилата и 20мл 4,45 (2Н, м), 6,47 (1Н, д, J=1131 Гц), 8,25-8,67 (2Н, хлороформа Реакционную смесь нагревали до м) комнатной температуры, а затем перемешивали в 2) 0,44 г (57%) этилового эфира ІЧ-[4-хлор-5течение ночи Реакционную смесь разводили 150 (1Н-ими-дазол-1-ил)-2-нитрофенил] глицина полумиллилитрами хлороформа, последовательно чали с использованием 0,66 г этилового эфира Nпромывали водой, 5% водным раствором бикар(4-хлор-5-фтор-2-нитрофенил) глицина, 162 мг боната натрия и солевым раствором, а затем имидазола и 4 мл пиридина ЯМР-спектр (CDCI3, осушали безводным сульфатом натрия, и конценвнутренний стандарт-ТМС) трировали при пониженном давлении После до5 1,16-1,44 (ЗН, м), 4,07 (2Н, д, Т=5,2Гц), 4,16бавления 100 мл этанола и 1,5мл концентриро4,48 (2Н, м), 6,65 (1Н, с), 7,15-7,37 (2Н, м), 7,76 ванной соляной кислоты смесь нагревали с (1Н, с), 8,35-8,63 (2Н, м) обратным холодильником в течение б часов 3) 235 мг (50%) этил-2-[6-хлор-2,3-диоксо-7(1Н-имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалинПосле охлаждения реакционную смесь кон1-ил] ацетата получали с использованием 442 мг центрировали при пониженном давлении К этой этилового эфира 1\1-[4-хлор-5-(1Н-имидазол-1-ил)смеси добавляли 15мл 1 н водного раствора гид2-нитрофенил ] глицина ЯМР-спектр (ДМСО-de, роксида натрия, а затем перемешивали в течение внутренний стандарт-ТМС) двух часов Реакционную смесь доводили до рН = 2-3 путем добавления 1 н водного раствора соля5 1,17-1,24 (ЗН, м), 4,12-4,20 (2Н, м), 4,93 (2Н, ной кислоты, и осажденное таким образом соедис), 7,48 (1Н, с), 7,81 (1Н, с), 7,91-7,98 (2Н, м), 9,23 нение собирали путем фильтрации Полученное (1Н, с), 12,54 (1Н, с) соединение перекристаллизовывали из 1 н водно4) 163мг (79%) 2-[6-хлор-2,3-диоксо-7-(1Нго раствора соляной кислоты, в результате чего имидазол-1-ил)-1,2,3, 4-тетрагидрохиноксалин-1получали 2,02 г (55%) 2-[6-бром-2,3-диоксо-7-(1Нил] уксусной кислоты 0,7 НгО получали с испольимидазол-1-ил)-1, 2,3,4-тетрагидрохиноксалин-1зованием 225 мг этил-2-[6-хлор-2,3-диоксо-7-(1Нил] уксусной кислоты 1 НС1 0,8 НгО имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1ил] ацетата Т пл 283°С (с разлож) Тпл 295-298°С МС (m/z) 365, 367 (М*) MC(m/z) 321 (М+ + 1) ЯМР-спектр ДМСО-сіє, внутренний стандартТМС) ЯМР-спектр (ДМСО-de, внутренний стандартТМС) 5 4,84 (2Н, с), 7,75 (1Н, с), 7,93 (1Н, с), 8,02 (1Н, с), 8,03 (1Н, с), 9,48 (1Н, с), 12,75 (1Н, с) 12,65 4,90 (2Н, с), 7,15 (1Н, с), 7,37-7,47 (2Н, м), 14,0 (1Н, шир) 7,67 (1Н, с), 7,92 (1Н, с), 12,40 (1Н, с) Пример 17 Пример 18 1) 10,5г (60%) этилового эфира ІЧ-(4-бром-51) 0,52г (13%) этилового эфира ІЧ-(5-фтор-4фтор-2-нитрофенил)глицина получали с испольметокси-2-нитрофенил) глицина получали с исзованием 7,61 г (54,5 ммоль) гидрохлорида этилопользованием 2,78г 2,4-дифтор-5вого эфира глицина, 15 мл тетрагид-рофурана, нитрофенилметилового эфира, 4,45г триэтилами7,64мл триэтиламина, 10 мл ДМФ и 11,8г (49,6 на, 30мл ДМФ, 30мл ТГФ, и 2,05 г этилового эфимл моль) 5-бром-2,4-дифторнитробензола ЯМРра глицина ЯМР-спектр (CDCI3, внутренний станспектр (CDCI3, внутренний стандарт-ТМС) дарт-ТМС) 5 1,33 (ЗН, т, и=7,4Гц), 4,04 (2Н, д, J= 4,9Гц), 5 1,19-1,45 (ЗН, м), 3,80-4,46 (7Н, м), 6,45 (1Н, 4,30 (2Н, кв, J= 7,4Гц), 6,46 (1Н, д, Т= 10,9Гц), 8,44 д, J= 13,1Гц), 7,81 (1Н, д, J= 8,8Гц), 8,3-8,6 (1Н, 33 44283 34 шир) (2Н, м), 7,94-8,08 (1Н, м), 9,36-9,48 (1Н, м), 12,47 2) 200 мг (41%) этилового эфира N-[5-(1Hимидазол-1-ил)-4-метокси-2-нитрофенил] глицина 4) 86 мг (62%) 2-[6-бензилокси-2,3-диоксо-7получали с использованием 420 мг этилового (1 Н-имидазол-1 -ил)-1,2,3, 4эфира І\І-(5-фтор-4-метокси-2-нитро-фенил) глитетрагидрохиноксалин-1-ил] уксусной кислоты-0,5 цина, 420 мг имидазола, и 2,5 мл диметилформаHCL 1,4 Н2О получали с использованием 134 мг мида ЯМР-спеїсгр (CDCI3, внутренний стандартэтил-2-[6-бензилокси-2,3-диоксо-7-(1Н-имидазолТМС) 1-ил)-1,2,3,4-тетрагидрохиноксалин-1-ил] ацетата 5 1,27-1,37 (ЗН, м), 3,88 (ЗН, с), 4,09 (2Н, д, J= Т пл 275°С (с разлож) 5,5 Гц), 4,25-4,34 (2Н, м), 6,64 (1Н, с), 7,20 {1Н, с), МС (m/z) 393 (М+ + 1) 7,27 (1Н, с), 7,85-7,94 (2Н, м), 8,32-8,40 (1Н, м) ЯМР-спектр (ДМСО-de —, внутренний стан3) 46 мг (22%) этил-2-[2,3-диоксо-7-(1Ндарт-ТМС) имидазол-1-ил)-6-метокси-1,2,3,45 4,88 (2Н, с), 5,17 (2Н, с), 7,13 (1Н, с), 7,30тетрагидрохиноксалин-1-ил] ацетата получали с 7,48 (6Н, м), 7,64 (1Н, с), 7,74 (1Н, с), 8,66 (1Н, с), использованием 196 мг этилового эфира N-[5-(1H12,32 (1Н, с) имидазол-1-ил)-4-метокси-2-нитрофенил] глицина Пример 20 ЯМР-спеїсгр (ДМСО-de, внутренний стандарт1) 2,44г (99%) этилового эфира ІЧ-(5-фтор-4ТМС) метилсу-льфонил-2-нитрофенил) глицина получали с использованием 1,82г 2,4-дифтор-55 1,17-1,25 (ЗН, м), 3,85 (ЗН, с), 4,11-4,20 (2Н, нитрофенил метилсульфона, 0,78г триэтиламина, м), 4,94 (2Н, с), 7,09 (1Н, с), 7,75 (1Н, с), 7,79 (1Н, 4мл ДМФ, 8мл ТГФ и 1,07 г гидрохлорида этиловос), 7,92 (1Н, с), 9,24 (1Н, с), 12,37 (1Н, с) го эфира глицина ЯМР-спектр (CDCI3, внутренний 4) 33 мг (7,5%) 2-[2,3-диоксо-7-(1Н-имидазолстандарт-ТМС) 1-ил)-6-метокси-1,2,3,4-тетрагидрохиноксалин-1ил] уксусной кислоты 0,1 НСІ 1,5 НгО получали с 5 1,15-1,55 (ЗН, м), 3,19 (ЗН, с), 3,93-4,50 (4Н, использованием 44 мг этил-2-[2,3-диоксо-7-(1Нм), 6,49 (1Н, д, J =12,0Гц), 8,76-9,07 (2Н, м) имидазол-1-ил)-6-метокси-1,2,3,4-тетра2) 0,35г (67%) этилового эфира N-[5-(1Hгидрохиноксалин-1-ил] ацетата имидазол-1-ил)-4-метилсульфонил-2-нитрофенил] глицина получали с использованием 1,00 г этилоТ пл >300°С вого эфира ІЧ-(5-фтор-4-метилсульфонил-2MC(m/z) 317 (М+ + 1) нитрофенил) глицина, 0,85 г имидазола и 5 мл ЯМР-спектр (ДМСО-de, внутренний стандартдиметил-формамида ЯМР-спектр (ДМСО-de, ТМС) внутренний стандарт-ТМС) 5 3,81 (ЗН, с), 4,91 (2Н, с), 7,02 (1Н, с), 7,12 (1Н, с), 7,43-7,51 (2Н, м), 7,99 (1Н, с), 12,23 (1Н, с), 5 1,08-1,32 (ЗН, м), 2,86 (ЗН, с), 4,16 (2Н, kb, 13,0-13,3 (1Н, шир) J= 7,3Гц), 4,41 (2Н, д, и=6,0Гц), 7,09-7,23 (2Н, м), 7,43-7,51 (1Н, м), 7,83-7,90 (1Н, м), 8,72 (1Н, с), Пример 19 8,80-9,03 (1Н, м) 1) 454 мг (8%) этилового эфира N-(4бензилокси-5-фтор-2-нитрофенил) глицина полу3) 265 мг (72%) этил-212,3-диоксо-7-(1Нчали с использованием 4,41 г бензил-2,4-дифторимидазол-1-ил)-6-метилсульфонил-1,2,3,45-нитрофенилового эфира, 5,05 г триэтиламина, тетрагидрохиноксалин-1-ил]-ацетата получали с 30 мл ДМФ, 30 мл ТГФ, и 2,32 г гидрохлорида этииспользованием 345 мг этилового эфира N-[5-(1Hлового эфира глицина ЯМР-спектр (CDCI3, внутимидазол-1-ил)-4-метилсульфонил-2-нитрофенил] ренний стандарт-ТМС) глицина 5 1,32 (ЗН, т, J= 7,3Гц), 4,03 (2Н, д, и=5,5Гц), ЯМР-спектр (ДМСО-de, внутренний стандарт4,25-4,33 (2Н, м) 5,09 (2Н, с), 6,44 (1Н, д, J=12,2 ТМС) Гц), 7,30-7,48 (5Н, м), 7,99 (1Н, д, и=8,5Гц), 8,40 5 1,16-1,25 (ЗН, м), 3,18 (ЗН, с), 4,12-4,20 (2Н, (1Н, шир) м), 4,95 (2Н, с), 7,83 (1Н, с), 7,94-8,06 (ЗН, м), 9,37 (1Н, с), 12,75 (1Н, с) 2) 331мг (65%) этилового эфира N-[4бензилокси-5-(1 Н-имидазол-1 -ил)-2-нитрофенил] 4) 106мг (68%) 2-[2,3-диоксо-7-(1Н-имидазолглицина получали с использованием 448 мг эти1-ил)—6-метилсульфонил-1,2,3,4лового эфира І\І-(4-бензилокси-5-фтор-2тетрагидрохиноксалин-1-ил] уксусной кислоты 0,6 нитрофенил) глицина, 350 мг имидазола и 2 мл HCL 1,2 НгО получали с использованием 150 мг ДМФ ЯМР-сепктр (CDCI3, внутренний стандартэтил-2-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6ТМС) метилсульфонил-1,2,3,4-тетрагидрохиноксалин-1ил] ацетата 5 1,32 (ЗН, т, 7=7,3 Гц), 4,08 (2Н, Д, J=5,5 Гц), 4,25-4,34 (2Н, м), 5,09 (2Н, с), 6,63 (1Н, с), 7,20 (1Н, Т пл >300°С с), 7,25-7,40 (6Нг м), 7,92 (1Н, с), 8,00 (1Н, с), 8,31МС (m/z) 365 (М+ + 1) ЯМР-спектр (ДМСО-de, 8,37 (1Н, м) внутренний стандарт-ТМС) 3) 70 мг (66%) этил-2-[6-бензилокси-2,35 3,06 (ЗН, с), 4,88 (2Н, с), 7,59 (1Н, с), 7,82 диоксо-7-(1 Н-имидазол-1 -ил)-1,2,3,4(1Н, с), 7,88-7,98 (2Н, м), 8,84 (1Н, с), 12,68 (1Н, с), тетрагидрохиноксалин-1-ил] ацетата получали с 13,1-13,5 (1Н, шир) использованием 100 мг этилового эфира N-[4Пример 21 бензилокси-5-(1 Н-имидазол-1 -ил)-2-нитрофенил] Смесь, содержащую 1,63 г этил-2-[2,3-диоксоглицина ЯМР-спектр (ДМСО-de, внутренний стан7-фтор-6-нитро-1,2,3,4-тетрагидрохиноксалин-1дарт-ТМС) ил] ацетата, 4 мл 50% водного раствора диметиламина, и 10 мл диметилформамида, нагревали и 5 1,08-1,33 (ЗН, м), 4,16 (2Н, кв, и=7,ЗГц), 4,95 перемешивали в масляной бане (100С), в атмо(2Н, с), 5,19 (2Н, с), 7,15-7,52 (6Н, м), 7,75-7,92 35 44283 36 сфере аргона в течение 5 часов После охлаждедавлении, в результате чего получали 3,94г (60%) ния реакционную смесь концентрировали при поэтил-2-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-1,2,3,4ниженном давлении К полученному остатку дотетрагидрохиноксалин-1-ил] ацетата бавляли 15мл 1 н водного раствора гидроксида MC(m/z) 314 (М+) натрия, а затем перемешивали в течение 3 часов ЯМР-спектр (ДМСО-de, внутренний стандартпри температуре 30°С Реакционную смесь конТМС) центрировали при пониженном давлении до поло5 1,22 (ЗН, т, J =7,0Гц), 4,18 (2Н, кв, и=7,0Гц), вины первоначального объема, а затем добавля5,06 (2Н, с), 7,41 (1Н, д, 7=8,6Гц), 7,60 (1Н, дд, ли 3 н соляную кислоту для доведения рН этой J=2,5, 8,6Гц), 7,65 (1Н, с), 7,77 (1Н, д, J =25Гц), реакционной смеси до значения 5-6 Осажденные 8,13 (1Н, с), 9,24 (1Н, с), 12,47 (1Н, с) таким образом кристаллы собирали путем фильт2) При температуре, равной 0°С или ниже 0°С, рации, промывали 1 н соляной кислотой и осуша3,63 г этил-2-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)ли при пониженном давлении Полученные не1,2,3,4-тетрагид-рохиноксалин-1-ил] ацетата при очищенные кристаллы перекристаллизовывали из перемешивании растворяли в 15 мл дымящей 1 н соляной кислоты, в результате чего получали азотной кислоты Полученный раствор нагревали 742 мг (45%) 2-[7-диметиламино-6-нитро-1,2,3,4до комнатной температуры, перемешивали в тететрагидрохиноксалин-1-ил] уксусной кислоты 0,5 чение одного часа, и выливали в ледяную воду, Н2О после чего доводили до рН 2,0 путем добавления водного раствора гидроксида натрия Полученное Т пл >300°С нерастворившееся вещество удаляли путем MC(m/z) 309 (М+ + 1) фильтрования К этому фильтрату добавляли ЯМР-спектр (ДМСО-de, внутренний стандартводный раствор гидроксида натрия для доведения ТМС) рН полученной жидкости до значения, равного 5 2,82 (6Н, с), 4,97 (2Н, с), 6,83 (1Н, с), 7,45 около 6,5 Полученное нерастворившееся веще(1Н, с), 12,17 (1Н, шир с ) ство собирали путем фильтрации, промывали воПример 22 дой и осушали при пониженном давлении, в реСмесь, содержащую 2,94 г этил-2-[7-фтор-2,3зультате чего получали 1,07г (26%, чистота около диоксо-6-нитро-1,2,3,4-тетрагидрохиноксалик-1-ил] 95% (ВРЖХ-анализ)) этил-2-[2,3-диоксо-7-(1 Нацетата, 1,36г имидазола и 20мл диметилформаимидазол-1 -ил)-6-нитро-1,2,3,4мида, перемешивали при нагревании при темпететрагидрохиноксалин-1-ил] ацетата ратуре 120°С в течение 3 часов После охлаждения реакционную смесь выливали в ледяную воду Осажденные таким образом кристаллы собирали путем фильтрации, промывали водой и осушали при пониженном давлении, в результате чего получали 3,45г (99%) этил-2-[2,3-диоксо-7-(1Нимидазол-1 -ил)-6-нитро-1,2,3,4тетрагидрохиноксалин-1-ил)-ацетата 0,65 Н2О Т пл 159-160°С (с разлож) МС (m/z) 359 (М+) ЯМР-спектр (ДМСО-de, внутренний стандартТМС) 5 1,21 (ЗН, т, J = 7,1Гц), 4,16 (2Н, kb, J= 7,1 Гц), 5,03 (2Н, с), 7,09 (1Н, с), 7,39 (1Н, с), 7,75 (1Н, с), 7,87 (1Н, с), 8,00 (1Н, с), 12,60 (1Н, шир с ) Пример 23 1) Смесь, содержащую 5,85г (20 миллимоль) этил-2-[5-(1 Н-имидазол-1 -ил)-2-нитрофенил] ацетата, 250 мл тетрагидрофурана и 1,5 г 10% палладия-на-угле, перемешивали при комнатной температуре в атмосфере водорода После восстановления нитро-группы катализатор удаляли путем фильтрации К этому фильтрату добавляли 11,3мл триэтиламина К полученной смеси при охлаждении льдом и при перемешивании по капле добавляли смесь 11 г этилхлорглиоксилата и 30мл тетрагидрофурана После завершения добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи Нерастворившееся вещество удаляли путем фильтрации Фильтрат концентрировали при пониженном давлении К полученному остатку добавляли 150 мл этанола, и эту смесь нагревали с обратным холодильником в течение пяти часов в атмосфере аргона После охлаждения образовавшийся осадок собирали путем фильтрации, промывали этанолом, и осушали при пониженном 3) Смесь, содержащую 2,12г этил-2-[2,3диоксо-7-(1 Н-имидазол-1 -ил)—6-нитро-1,2,3,4тетрагидрохиноксалин-1-ил] ацетата и 13мл 1 н водного раствора гидроксида натрия, перемешивали при комнатной температуре в атмосфере аргона После завершения реакции реакционную смесь доводили до рН 3,0 путем добавления 0,5 мл концентрированной соляной кислоты и соответствующего количества 1 н соляной кислоты Осажденные таким образом кристаллы собирали путем фильтрации, промывали 1н соляной кислотой и осушали при пониженном давлении Полученное неочищенное соединение в виде кристаллов перекристаллизовывали из 1 н соляной кислоты, в результате чего получали 1,90г (84%) 2-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетрагидрохиноксалин-1-ил] уксусной кислоты 1НС1_-1Н2О Т пл 248-250°С (с разлож) МС (m/z) 331 (М+) ЯМР-спектр (ДМСО-de, внутренний стандартТМС) 5 4,90 (2Н, с), 7,89 (1Н, с), 8,03 (1Н, с), 8,09 (1Н, с) 8,33 (1Н, с), 9,48 (1Н, с), 12,96 (1Н, с) Пример 24 К 66 мл 1 н водного раствора гидроксида натрия при охлаждении льдом постепенно добавляли 8,5 г соединения Примера 23 для его растворения К полученному раствору при охлаждении льдом постепенно добавляли 44мл 1 н водного раствора соляной кислоты Осажденные кристаллы собирали путем фильтрации и осушали, в результате чего получали 6,8 г 2-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] уксусной кислоты в виде кристаллов 37 44283 38 Элементный анализ для C13H9N5O6 0,2 НоО реакционную смесь концентрировали при пониженном давлении до 1/3 первоначального объема К полученному остатку добавляли 100мл этиловоВычислено С 46,63, Н 2,83, М 20,92, го эфира Осажденное таким образом нераствоНайдено С 46,51, Н 2,91, N21,00 рившееся вещество собирали путем фильтрации ЯМР-спектр (ДМСО-de, внутренний стандарти осушали при пониженном давлении, в результаТМС) те чего получали 2,17г (19%) изопропил-52,35 4,95 (2Н, с), 7,10 (1Н, с), 7,40 (1Н, с), 7,73 диоксо-7-фтор-1,2,3,4-тетрагидрохиноксалин-1(1Н, с), 7,88 (1Н, с), 8,00 (1Н, с), 12,58 (1Н, с), 13,4 ил]-валерата (1Н, шир с ) Пример 25 МС (m/z) 322 (М+) ЯМР-спектр (flMCO-d6, внутренний стандарт-ТМС) 1) К смеси, содержащей 5,06г (32,9 миллимоль) 2,4-дифторнитробензола, 5,24г гидрохлори5 1,16 (6Н, д, J=6, ІГц), 1,60-1,62 (4Н, м), 2,32 да 5-аминовалериановой кислоты и 20мл тетра(2Н, т, и=6,7Гц), 4,01-4,08 (2Н, м), 4,87 (1Н, кв, гидрофурана добавляли 9,24мл три-этиламина, и J=6,H~LO, 7,01-7,05 (1Н, м), 7,18 (1Н, дд, J=5,5, эту смесь нагревали с обратным холодильником в 8,6Гц), 7,32 (1Н, дд, J=2,7,1iriO, 12,0 (1Н, с) течение 18 часов После охлаждения реакционную 4) В 10 мл концентрированной серной кислосмесь разводили этилацетатом, и нерастворивты, охлажденной в бане из льда/метанола, при шееся вещество удаляли путем фильтрации перемешивании растворяли 1,96г (6,07 миллиФильтрат концентрировали при пониженном давмоль) изопропил-5-[2,3-диоксо-7-фтор-1,2,3,4лении Полученный остаток перекристаллизовытетрагидрохиноксалин-1-ил] валерата К полученвали из этил ацетата/этил о во го эфира, в результаной смеси при температуре, равной-5 С или ниже, те чего получали 4,73г (56%) 5-(3-фтор-6по капле добавляли 300 мкл дымящей азотной нитрофенил) аминовалериановой кислоты кислоты, а затем смесь перемешивали в течение 30 минут Реакционную смесь выливали в ледяМС (m/z) 256 (М+) ную воду Осажденное таким образом нераствоЯМР-спектр (ДМСО-de, внутренний стандартрившееся вещество собирали путем фильтрации, ТМС) промывали водой, и осушали при пониженном 5 1,78-1,83 (4Н, м), 2,44-2,48 (2Н, м), 3,29 (2Н, давлении, в результате чего получали 2,06г (93%) кв, J=7,0 Гц), 3,34-3,39 (1Н, м), 6,45-6,49 (1Н, м), изопропил-5-[2,3-диоксо-7-фтор-6-нитро-1,2,3,48,16-8,23 (1Н, м) тетрагидрохиноксалин-1-ил] валерата МС (m/z) 2) Смесь, содержащую 2,19г (8,55ммоль) 5-(3367 (М+) ЯМР-спектр (ДМСО-de, внутренний станфтор-6-нитрофенил)аминовалериановой кислоты, дарт-ТМС) 100 мл 2-пропанола, и 5мл 4 н соляной кислоты в диоксане, нагревали с обратным холодильником в 5 1,16 (6Н, д, и=6,ЗГц), 1,61-1,63 (4Н, м), 2,30течение двух часов После охлаждения, реакци2,32 (2Н, м), 4,02-4,14 (2Н, м), 4,88 (1Н, kb, онную смесь концентрировали при пониженном и=6,ЗГц), 7,66 (1Н, д, J= 13,7Гц), 7,90 (1Н, д, J= давлении Остаток очищали с помощью колоноч7,3Гц), 12,2 (1Н, с) ной хроматоографии на силикагеле (проявляющий 5) Смесь, содержащую 1,88г (5,11 ммоль) изорастворитель гексан/эти л ацетат = 9 1-6 1), в репропил-5-[2,3-диоксо-7-фтор-6-нитро-1,2,3,4зультате чего получали 2,55г (стехиометрич , колтетрагидрохиноксалин-1-ил] валерата, 765мг имиво) изопропил-5-(3-фтор-6-нитрофенил) аминовадазола, и 15мл ДМФ, перемешивали при нагревалериата нии в масляной бане (70°С) в течение десяти часов в потоке аргона Реакционную смесь MC(m/z) 298 (М+) ЯМР-спектр (CDCI3, внутохлаждали до комнатной температуры и конценренний стандарт-ТМС) трировали при пониженном давлении до полови5 1,24 (6Н, д, и=6,4Гц), 1,76-2,82 (4Н, м), 2,36 ны от первоначального объема Полученный оста(2Н, т, и=6,8Гц), 3,28 (2Н, д, и=5,4Гц), 5,03 (1Н, кв, ток выливали в ледяную воду Осажденное и=6,4Гц), 6,33-6,39 (1Н, м), 6,47 (1Н, дд, J=2,5, нерастворившееся вещество собирали путем 11,7Гц 8,17-8,23 (1Н, м) фильтрации, промывали водой и осушали при 3) Смесь, содержащую 2,53 г (8,48 ммоль) пониженном давлении Приблизительно к 2,01 г изопропил-5-(3-фтор-6-нитрофенил) аминовалеполученного соединения в потоке газа аргона дората, 100 мл тетрагидрофурана, и 380 мг 10% бавляли 8 мл тетрагидрофурана и 20 мл 1 н водпалладия-на-угле, перемешивали при комнатной ного раствора гидроксида натрия, и эту смесь петемпературе и при нормальном давлении в атморемешивали в течение 5 часов Полученную сфере водорода в течение 5 часов После заверреакционную смесь доводили до рН = 5-6 путем шения реакции катализатор удаляли путем добавления водного раствора соляной кислоты фильтрации К фильтрату добавляли 5,94мл триПосле осаждения нерастворившегося вещества этиламина, и эту смесь перемешивали при охлажреакционную смесь нагревали до получения гомодении льдом в атмосфере аргона К полученному генного раствора, а затем фильтровали Фильтрат раствору, при перемешивании, по капле добавляконцентрировали при пониженном давлении Поли смесь, состоящую из 5,49г этилхлорглиоксилалученный остаток перекристаллизовывали из 10 та и 15мл тетрагидрофурана После перемешивамл воды, в результате чего получали 1,15г 5-[2,3ния в течение одного часа, нерастворившееся диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро-1,2,3,4вещество удаляли путем фильтрации, и этот тетрагидрохиноксалин-1-ил] валериановой кислофильтрат концентрировали при пониженном давты 1HCL 0,2Н 2 0(54%) Т пл 236-237°С лении К полученному остатку добавляли 150мл этанола, и смесь нагревали с обратным холоЭлементный анализ для C16H15N5O6 HC1 дильником в течение 18 часов После охлаждения 0,2Н2О (%) 39 44283 40 Вычислено С 46,49, Н 4,00, N 16,94, CI 8,56 соляной кислоты Осажденные таким образом кристаллы собирали путем фильтрации, промыНайдено С 46,34, Н 3,95, N 16,88, CI 8,69 вали водой и осушали при пониженном давлении, Пример 26 в результате чего получали 138 мг (81%) 2-[69,64г (86%) этил-6-(5-фтор-2карбамоил-2,3-диоксо-7-(1 Н-имидазол-1 -ил)нитрофениламино) гексаноата получали спосо1,2,3,4-тетрагидрохиноксалин-1-ил] уксусной кибом, описанным в Примере 1-1, за исключением слоты 1,4 Н20 того, что использовали 7,38г гидрохлорида этил-6аминогексаноата, 100мл тетрагидрофурана, Т п л >300°С 26,3мл триэтиламина, 35мл диметилформамида и Элементный анализ для C14H11N5O5 1,4 Н2О 6,00 г 2,4-дифторнитробензола МС (m/z) 298 (М+) Вычислено С 47,44, Н 3,92, N 19,76 ЯМР-спектр (CDCI3, внутренний стандартНайдено С 47,36, Н 3,82, N 19,88 ТМС) МС (m/z) 330 (М+ + 1) ЯМР-спектр (flMCO-d6, 5 1,10-1,97 (9Н, м), 2,34 (2Н, т, и=6,5Гц), 3,10внутренний стандарт-ТМС) 3,42 (2Н, м), 4,14 (2Н, кв, J =7,1 Гц), 6,22-6,62 (2Н, 5 4,94 (2Н, с), 7,04 (1Н, с), 7,26-7,34 (2Н, м), м), 8,00-8,35 (2Н, м) 7,45 (1Н, с), 7,51 (1Н, с), 7,72-7,80 (2Н, м), 12,39 2) 1,75г (32%) этил-6-(2,3-диоксо-7-фтор1,2,3,4-тетрагидрохиноксалин-1-ил) гексаноата Пример 28 получали способом, описанным в Примере 1-2, за 1) В 16мл концентрированной серной кислоты исключением того, что использовали 5,00г этил-6растворяли 3,29г (11,8ммоль) этил-4-(2,4(5-фтор-2-нитрофениламино) гексаноата МС дифторфенокси) бензоата при температуре, не (m/z) 323 (М+ + 1) превышающей 5 С К полученной смеси при перемешивании добавляли 520 микролитров дымящей ЯМР-спектр (ДМСО-de, внутренний стандартазотной кислоты при температуре-5С или ниже, а ТМС) затем эту смесь перемешивали в течение 30 ми5 0,95-1,83 (9Н, м), 2,10-2,45 (2Н, м), 3,86-4,28 нут при той же температуре Полученную реакци(4Н, м), 6,90-7,46 (ЗН, м), 11,95-12,15 (1Н, шир) онную смесь выливали в ледяную воду, а нерас3) 1,04г (91%) этил-6-(2,3-диоксо-7-фтор-6творившееся осажденное вещество собирали нитро-1,2,3,4-тетрагидрохиноксалин-1-ил) гексапутем фильтрации Полученное соединение расноата получали способом, описанным в Примере творяли в хлороформе Затем раствор последова1-(3), за исключением того, что использовали 1,00г тельно промывали водой и солевым раствором, этил-6-(2,3-диоксо-7-фтор-1,2,3,4-теосушали безводным сульфатом натрия, и концентрагидрохиноксалин-1-ил) гексаноата трировали при пониженном давлении ПолученMC(m/z) 368 (М+ + 1) ный остаток очищали посредством колоночной ЯМР-спектр (ДМСО-de, внутренний стандартхроматографии на силикагеле (проявляющий расТМС) творитель гексангэтилацетат = 8 1), в результате 5 0,90-1,82 (9Н, м), 2,02-2,50 (2Н, м), 3,76-4,33 чего получали 2,42г (63%) этил-4-(2,4-дифтор-5(4Н, м), 7,66 (1Н, д, J= 13,7Гц), 7,90 (1Н, д, J= нитрофенокси)-бензоата ЯМР-спектр (CDCI3, 7,4Гц), 12,13-12,40 (1Н, шир) внутренний стандарт-ТМС) 4) 318 мг (74%) 6-[2,3-диоксо-7-(1Н-имидазол1-ил)-6-нитро-1,2,3, 4-тетрагидрохиноксалин-1-ил] 5 1,40 (ЗН, т, J=7,3 Гц), 4,38 (2Н, кв, Г=7,ЗГц), гексановой кислоты О.ЭЬЬО получали способом, 7.02 (2Н, д, и=9,2Гц), 7,21 (1Н, т (дд), и=9,8Гц), описанным в Примере 1-(4), за исключением того, 7,91 (1Н, т (дд), и=7,6Гц), 8,08 (2Н, д, и=9,2Гц) МС что использовали 392мг этил-6-(2,3-диоксо-7(тг/х) 323 (М+) фтор-6-нитро-1,2,3,4-тетрагидрохиноксалин-1-ил) 2) Смесь, содержащую 2,38 г (7,36 ммоль) гексаноата, 160мг имидазола и 2,5 мл ДМФ этил-4-(2,4-дифтор-5-нитрофенокси) бензоата, Т пл 120-123°С 1.03 г гидрохлорида этилового эфира глицина, 30 мл тетрагидрофурана, 10мл диметилформамида, Элементный анализ для C17H17N5O6 0,9 Н2О и 2,06 мл триэтиламина, нагревали с обратным (%) холодильником в течение 10 часов После охлажВычислено С 50,60 Н 4,70, N 17,35 дения реакционную смесь разводили этилацетаНайдено С 50,63, Н 4,38, N 17,32 том Нерастворившееся вещество удаляли путем MC(m/z) 388 (М+ + 1) фильтрации, а затем фильтрат концентрировали ЯМР-спектр (ДМСО-de, внутренний стандартпри пониженном давлении Полученный остаток ТМС) растворяли в этилацетате Полученный раствор 5 1,20-1,32 (2Н, м), 1,48-1,59 (2Н, м), 1,77-1,89 последовательно промывали водой и солевым (2Н, м), 2,19 (2Н, Т, J= 7,3 Гц), 4,34 (2Н, т, J= раствором, а затем осушали безводным сульфа7,3Гц), 7,09 (1Н, с), 7,43 (1Н, с), 7,90 (1Н, с), 8,11 том натрия, и концентрировали при пониженном (1Н, с), 8,51 (1Н, с), 8,66 (1Н, с), 11,97 (1Н, с) давлении Полученный остаток очищали с помоПример 27 щью колоночной хроматографии на силикагеле К смеси, содержащей 0,19 мл 30%-ного водно(проявляющий растворитель гекго раствора перекиси водорода и 1,2 мл 1 н водносан/дихлорметан/этилацетат = 8 2 1), в результате го раствора гидроксида натрия добавляли 150 мг чего получали 1,81 г этилового эфира N-4-[(42-[6-циано-2,3-диоксо-7-(1 Н-имидазол-1 -ил)этоксикарбонилфенокси)-5-фтор-2-нитрофенил] 1,2,3,4-тетрагидрохиноксалин-1-ил] уксусной киглицина (61%) слоты, и эту смесь перемешивали при комнатной температуре в течение 30 минут Реакционную смесь доводили до рН 1 путем добавления 1 н МС (m/z) 406 (М+) ЯМР-спектр (CDCI3, внутренний стандарт-ТМС) 41 44283 42 1) В 20мл диметилформамида растворяли 5 1,34 (ЗН, т, J =7,ЗГц), 1,38 (ЗН, т, J=7,0l~4), 500мг 7-фтор-1 -гидрокси-6-нитро-2,3 (1 Н,4Н)4.09 (2Н, д, J=2,9I~4), 4,32 (2Н, кв, и=7,ЗГц), 4,36 хиноксалиндиона К полученному раствору добав(2Н, kb, и=7,0Гц), 6,52 (2Н, д, и=12,2Гц), 6,95 (2Н, ляли 83 мг гидрида натрия, и смесь перемешивад, J =9,1 Гц), 8,03 (2Н, д, J=9, ІГц), 8,11 (1Н, д, ли в течение 10 минут К реакционной смеси дои=11,6Гц), 8,51 (1Н, шире) бавляли 218мл этил б ром ацетата, а затем 3) Смесь, содержащую 1,78 г (4,39ммоль) этипроводили реакцию в течение двух дней Полулового эфира 1\1-[4-(4-этоксикарбонилфенокси)-5ченную реакционную смесь выливали в насыщенфтор-2-нитрофенил]-глицина, 896мг имидазола и ный водный раствор хлорида аммония, а затем 20мл ДМФ, перемешивали при нагревании в мастри раза экстрагировали хлороформом Органичеляной бане (70°С) в течение 12 часов После охский слой концентрировали, после чего конценлаждения реакционную смесь концентрировали трат перекристаллизовывали из 2-пропанола, в при пониженном давлении Полученный остаток результате чего получали 481мг этил-2-[(2,3растворяли в этилацетате Затем раствор последиоксо-7-фтор-6-нитро-1,2,3,4довательно промывали водой и солевым раствотетрагидрохиноксалинил) окси] ацетата ром, осушали безводным сульфатом натрия, и концентрировали при пониженном давлении ПоMC(m/z) 328 (М+ + 1) лученный остаток очищали с помощью хроматоЯМР-спектр (ДМСО-de, внутренний стандартграфии на колонках с силикагелем (проявляющий ТМС) растворитель хлороформ + 0-1% метанол) и по5 1,23 (ЗН, т, и=7,2Гц), 4,19 (2Н, кв, и=7,2Гц), лучали 1,49г (75%) этилового эфира N-[4-(44,96 (2Н, с), 7,73 (1Н, д, и=12,4Гц), 7,91 (1Н, д, этоксикарбонилфенокси)-5-(1 Н-имидазол-1 -ил)-2нитрофенил] глицина 2) Этил-2-[(2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6МС (m/z) 454 (М+) нитро-1,2,3,4-тетрагидрохиноксалинил) окси] ацеЯМР-спектр (ДМСО-de, внутренний стандарттат получали способом, описанным в Примере 4ТМС) (4), за исключением того, что использовали этил2-[(2,3-диоксо-7-фтор-6-нитро-1,2,3,45 1,34 (ЗН, т, Т=7,ЗГц), 1,37 (ЗН, т и=7,2Гц), тетрагидрохиноксалинил) окси] ацетат 4,14 (2Н, д, J=4,9I~4), 4,31 (2Н, кв, J=7,3 Гц), 4,33 (2Н, kb, и=7,2Гц), 6,74 (1Н, с), 6,88 (2Н, д, и=9,2Гц), MC(m/z) 376 (М+ + 1) 7.10 (1Н, с), 7,22 (1Н, с), 7,83 (1Н, с), 7,95 (2Н, д, ЯМР-спектр (ДМСО-de, внутренний стандарт7=9,2Гц) 8,11 (1Н, с), 8,46-9,48 (1Н, м) ТМС) 4) Смесь, содержащую 1,46г (3,19ммоль) эти5 1,16 (ЗН, т, и=5,2Гц), 4,16 (2Н, кв, и=5,2Гц), лового эфира 1\1-[4-(4-этоксикарбонилфенокси)-54,97 (2Н, с), 7,05 (1Н, с), 7,11 (1Н, с), 7,45 (1Н, с), (1 Н-имидазол-1 -ил)-2-нитрофенил] глицина, 100 7,78 (1Н, с), 7,95 (1Н, с) мл тетрагидрофурана, и 410мг 10% палладия-на3) 2-[(2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6угле, перемешивали при комнатной температуре и нитро-1,2,3,4-тетрагидрохиноксалинил) окси] укпри нормальном давлении в атмосфере водорода сусной кислоты 2 НгО получали способом, опив течение 6 часов После завершения реакции санным в Примере 4-(5), за исключением того, что катализатор удаляли путем фильтрации К фильтиспользовали этил-2-[(2,3-диоксо-7-фтор-6-нитрорату добавляли 2,02мл триэтиламина, и эту смесь 1,2,3, 4-тетрагидрохиноксанолинил) окси] ацетат охлаждали льдом в потоке газа аргона К реакциТпл >300°С онной смеси при перемешивании по капле добавЯМР-спектр (ДМСО-de, внутренний стандартляли смесь 1,37мл этилхлорглиоксилата и 15мл ТМС) тетрагидрофурана Полученную реакционную 5 4,41 COOH \ го 10 н CH3SO/ Таблиця 6 Таблиця 4 Хишнеская структурная формула Зример 11 Химическая стсуктурная форі^ла -V ЯОСССНгО соон /СООЯ %Ао н .соон 22 п м 12 н HOOC .соон :00Н 25 O2N 13 ^•схюн 14 Я 25 Сі -•СООН н .соон IS Н н NC ґх н 0 11 44283 47 Таблиця 7 Зрймер Хишчэская структурная формуда соосаШ 28 H 5 C,OOC^O " g ° 0 СООН 29 /СООН ЗО OjN Н 1% С ••^^»- СООН Примеры композиций Ниже приводится пример получения композиции настоящего изобретения в качестве фармацевтического препарата Лиофилизованныи препарат в одном сосуде 31 U2N 48 (1Н-имидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин1-ил] уксусная кислота, 3) 5-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетрагидрохиноксалин-1-ил]-2,2-диметилпентановая кислота, 4) Этил-5-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6нитро-1,2,3,4-тетрагидрохиноксалин-1-ил] валерат, 5) 5-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетрагидрохиноксалин-1-ил] пентанамид, 6) 5-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетрагидрохиноксалин-1-ил]-2,4-диметилпентановая кислота, 7) 5-[2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6-нитро1,2,3,4-тетрагидрохиноксалин-1-ил]-4фенилпентановая кислота 8) 4-[(2,3-диоксо-7-(1 Н-имидазол-1 -ил)-6нитро-1,2,3,4-тетрагидрохиноксалинил)окси] масляная кислота н Таблица 8 32 Соединение Примера 1 Лимонная кислота D-маннитол /СООН 33 50 мг (0,5 %) 210 мг (2,1 %) 100 мг (1,0%) 10 мл N0 Каждое из нижеприведенных соединений может быть получено способом, в основном, аналогичным способам получения, проиллюстрированным в настоящем описании или в Примерах, либо эти соединения могут быть получены способами, имеющими небольшие модификации, очевидные для каждого специалиста 1) 2-[2,3-диоксо-7-(4-карбоксифенокси)-6-(1 Нимидазол-1-ил)-1,2,3,4-тетрагидрохиноксалин-1ил] уксусная кислота, 2) 2-[2,3-диоксо-7-(4-карбоксибензилокси)-6 В 800 мл воды последовательно добавляли для растворения 5 г соединения Примера 1, 21 г лимонной кислоты и 10 г D-маннитола Затем добавляли воду до получения полного количества 1000 мл После стерильной фильтрации, 10миллилитровую порцию раствора выливали в сосуд из темного стекла и лиофилизовали, в результате чего получали концентрированный препарат для инъекций, который необходимо растворить непосредственно перед использованием ДП «Український інститут промислової власності» (Укрпатент) вул Сім'ї Хохлових, 15, м Київ, 04119, Україна (044) 456 - 20 - 90
ДивитисяДодаткова інформація
Автори англійськоюTsukamoto Sin-iti, Fudzi Mitsuo
Автори російськоюЦукамото Син-ити, Фудзи Мицуо
МПК / Мітки
МПК: C07D 241/44, C07D 521/00
Мітки: похідні, основі, 1,2,3,4,-тетрагідрохіноксаліндіону, композиція, фармацевтична
Код посилання
<a href="https://ua.patents.su/24-44283-pokhidni-1234-tetragidrokhinoksalindionu-ta-farmacevtichna-kompoziciya-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні 1,2,3,4,-тетрагідрохіноксаліндіону та фармацевтична композиція на їх основі</a>
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
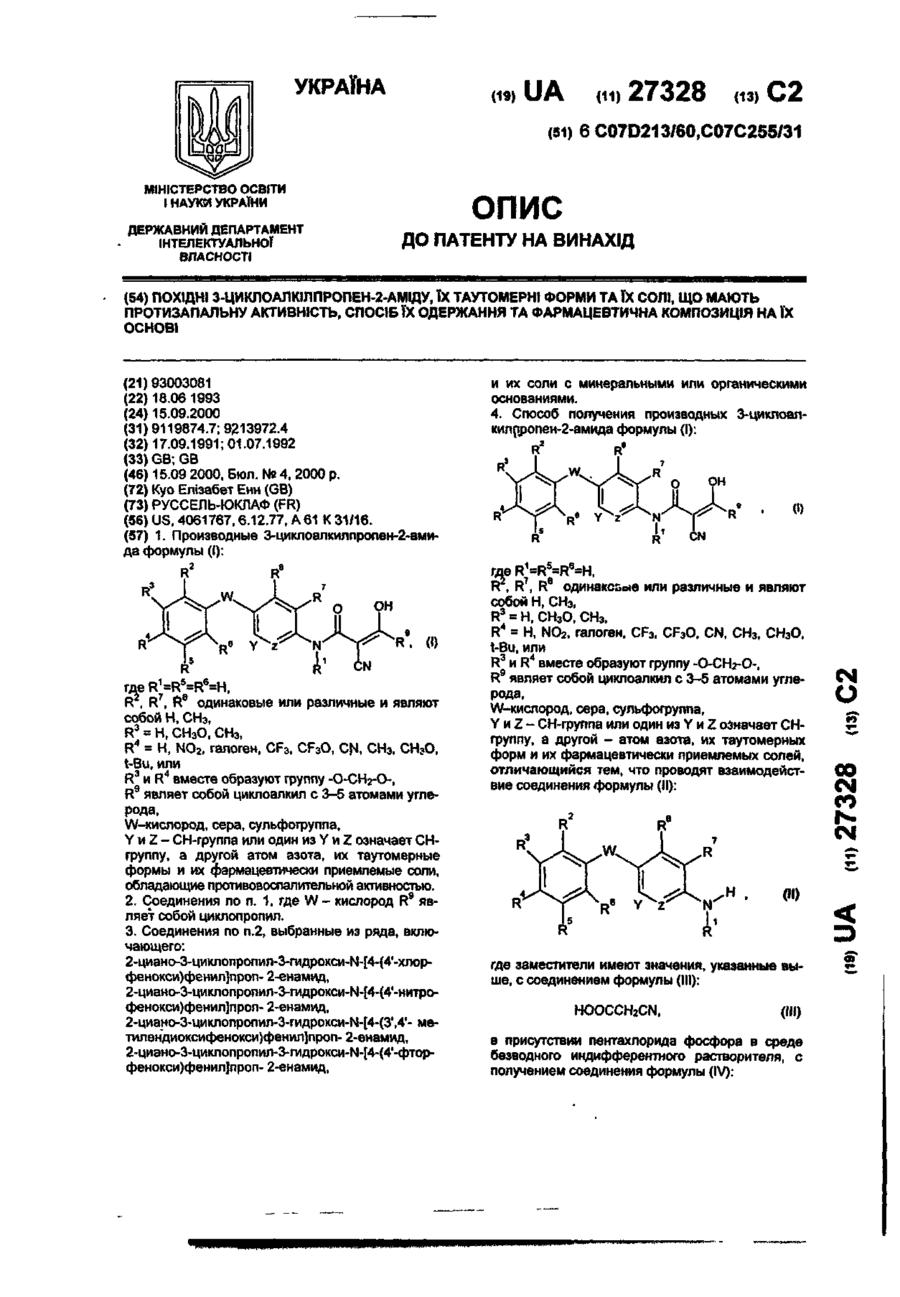
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: спосіб, фармацевтична, таутомерні, одержання, основі, 3-циклоалкілпропен-2-аміду, солі, похідні, протизапальну, мають, форми, активність, композиція
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...
Похідні оксазолідин-2-ону, спосіб їх одержання і фармацевтична композиція на їх основі.

Номер патенту: 43848
Опубліковано: 15.01.2002
Автори: Мельцер Гвідо, Ганте Йоахім, Бернотат-Данієловскі Сабіне, Раддатц Петер, Вурцігер Ханнс, Юращік Хорст
МПК: A61P 9/10, C07D 413/06, C07D 263/20, A61K 31/4166, A61P 9/08, C07D 233/32, A61P 19/10, A61P 7/02, A61P 13/02, C07D 413/10, C07D 413/14, A61K 31/445, A61P 15/00, A61K 31/495, A61P 29/00, A61K 31/496, C07D 277/14, A61K 31/415
Мітки: спосіб, похідні, композиція, одержання, оксазолідин-2-ону, фармацевтична, основі
Формула / Реферат:
1. Производные оксазолидин-2-она формулы I:где Х обозначает О,Y обозначаетилиR1 обозначаетилиR2 и R3 каждый, независимо друг от друга, обозначает Н, А или бензил, А обозначает алкил с 1-6 С-атомами,D обозначает амидиногруппу, аминометил, аминогидроксиминометил, 5-метил-1,2,4-оксадиазолидин-3-ил или гуанидинометил,r и s, независимо друг от друга, обозначают...
Похідні пенему, що є антибіотиками, і фармацевтична композиція на їх основі
Номер патенту: 27339
Опубліковано: 15.09.2000
Автори: Івата Хіромітсу, Накатсука Такасі, Танака Ріє
МПК: C07D 499/00, A61K 31/431, A61K 31/428
Мітки: похідні, основі, антибіотиками, композиція, пенему, фармацевтична
Текст:
...(8)-2-тетрагидрофурилкарбониnoKCHMeTnn(5R,6S)-6-[(R)-1- гидроксиэтил]-2-{(Р)-2-тетрагидрофурил]пенем-3-карбоксилат желтое масло 3466, 2975, 2676,1786, 1719,1323 1,34(ЗН,д,>5,8Гц), 1.52-2,16 (6Н,м),2,182,34 (1Н, м),2,38-2,52 (1Н. м),3,71 (1Н, gg f J=1,3r4, 6,6 Гц), 3.814,06 (4Н,м), 4,15-4,28 (1Н, м),4,45-4,56 (1Н, м)5,31 (1Н.Т, J=7,3 Гц), 5,50 (1Н, д, J=2 Гц), 5,88(2Н,дд,>5,9Гц, 14,5 Гц) 5 (5-МЄТИЛ-2-ОКСО-1.3-диоксоленил-4)...
Похідні гексагідроазепіну, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 27222
Опубліковано: 15.08.2000
Автори: Понселе Мартін, Меньян Жан-П'єр, Сантуссі Вінсен, Поль Раймон, Лявастр Серж
МПК: A61P 25/18, C07D 295/067, C07D 295/08, C07D 295/088, A61K 31/55, C07D 295/108, C07D 295/10, C07D 295/073, C07D 295/06, C07D 295/084
Мітки: композиція, основі, гексагідроазепіну, похідні, спосіб, фармацевтична, одержання
Формула / Реферат:
1 Производные гексагидроазепина общей формулы (I):где А - группа, выбранная из -СОСН2-, -СН(ОН)СН2-, -СН=СН-(цис), -С=С-,X - водород или атом хлора, Y - циклогексильная или фенильная группа, причем:если Y - циклогексильная группа, то X является атомом хлора,если Y - фенильная группа, то X является водородом и А представляет собой -СН==СН-(цис), -СΞС-, и их соли с фармацевтически приемлемыми...
Фторовані похідні 17b-заміщеного-4-aзa-5a-андростан-3-ону і фармацевтична композиція на їх основі

Номер патенту: 41291
Опубліковано: 17.09.2001
Автори: Енріко Ді Салле, Акілле Панцері, Марчелла Несі
МПК: A61P 5/28, C07J 73/00, C12N 9/99, A61P 35/00, A61K 31/58
Мітки: 17b-заміщеного-4-aзa-5a-андростан-3-ону, фторовані, композиція, основі, похідні, фармацевтична
Формула / Реферат:
1. Фторированные производные 17β-замещенного-4-аза-5α-андростан-3-она общей формулы (I):гдесимвол ------- независимо представляет одинарную или двойную связь;В- связь;R-водород, С1-С4 алкильная группа;R1-водород, С1-С6 алкильная группа, незамещенная или замещенная одним или более атомами фтора, или бензильная группа;r2 - водород, атом фтора, С1-С6 алкильная группа, незамещенная или...
Попередній патент: Магнітотерапевтичний апарат-“спектр-2″
Наступний патент: Лікувальний засіб для запобігання або лікування мігрені та інгібування нейронної менінгеальної транссудації, який містить агоніст 5-нт1f
Випадковий патент: Тензочутливий перетворювач