Моноефірні сполуки та фармацевтична композиція для лікування вірусних інфекцій
Номер патенту: 41361
Опубліковано: 17.09.2001
Автори: Стокке Хелль Тургеір, ДАЛЕН Аре, МЮХРЕН Фінн, Бьорретсен Бернт







Формула / Реферат
1. Моноэфирное соединение формулы I:
Nu - О – Fa,
где О является кислородом, Nu является аналогом нуклеозида, Fa является ацильной группой мононенасыщенной С18 или С20 ω-9 жирной кислоты, причем эта жирная кислота этерифицирует гидроксильную группу в 5'- положении сахарной части нуклеозидного аналога или концевую гидроксильную группу на нециклической группе нуклеозидного аналога, и причем Nu представляется формулой II:
В – S,
где S является моносахаридным производным, выбираемым из:
1-β-D-рибофуранозы, 1-β-D -арабинофуранозы, 2-дезокси-1-β-D -рибофуранозы, 2, З – дидезокси -1-β-D -рибофуранозы, 4-гидрокси-метил-2-циклопентен-1-ила или 2, 3-дигидроксипропила; и В является гетероциклической кольцевой системой, выбираемой из следующих формул:
где R1 = NH2, Y = С=СН;
или
где R2 = Н, ОН, рз = Н, ОН, NH2;
или
где R4 = NH2, R5 = Н; R6 = СН3, F, I;
при условии, что когда S является 1-β-D - арабинофуранозой, В не может быть:
2. Соединение по п.1, где B-S является рибавирином.
3. Соединение по п.1 или п.2, где Fа является олеиновой кислотой, элаидиновой кислотой или эйкозеновой кислотой, цис- или транс-
4. Соединение по п.1, где B-S является рибавирином, a Fa является олеиновой кислотой.
5. Соединение по п.1, где B-S является рибавирином, а Fa является элаидиновой кислотой.
6. Соединение по п.1, где B-S является рибавирином, а Fa является эйкозеновой кислотой.
7. Фармацевтическая композиция для лечения вирусных инфекций, отличающаяся тем, что в качестве активного ингредиента содержит эффективное количество соединения по любому из пунктов 1-6, а также фармацевтически приемлемый носитель или наполнитель.
8. Соединение формулы I по п.1 для получения фармацевтической композиции для лечения вирусной инфекции.
Текст
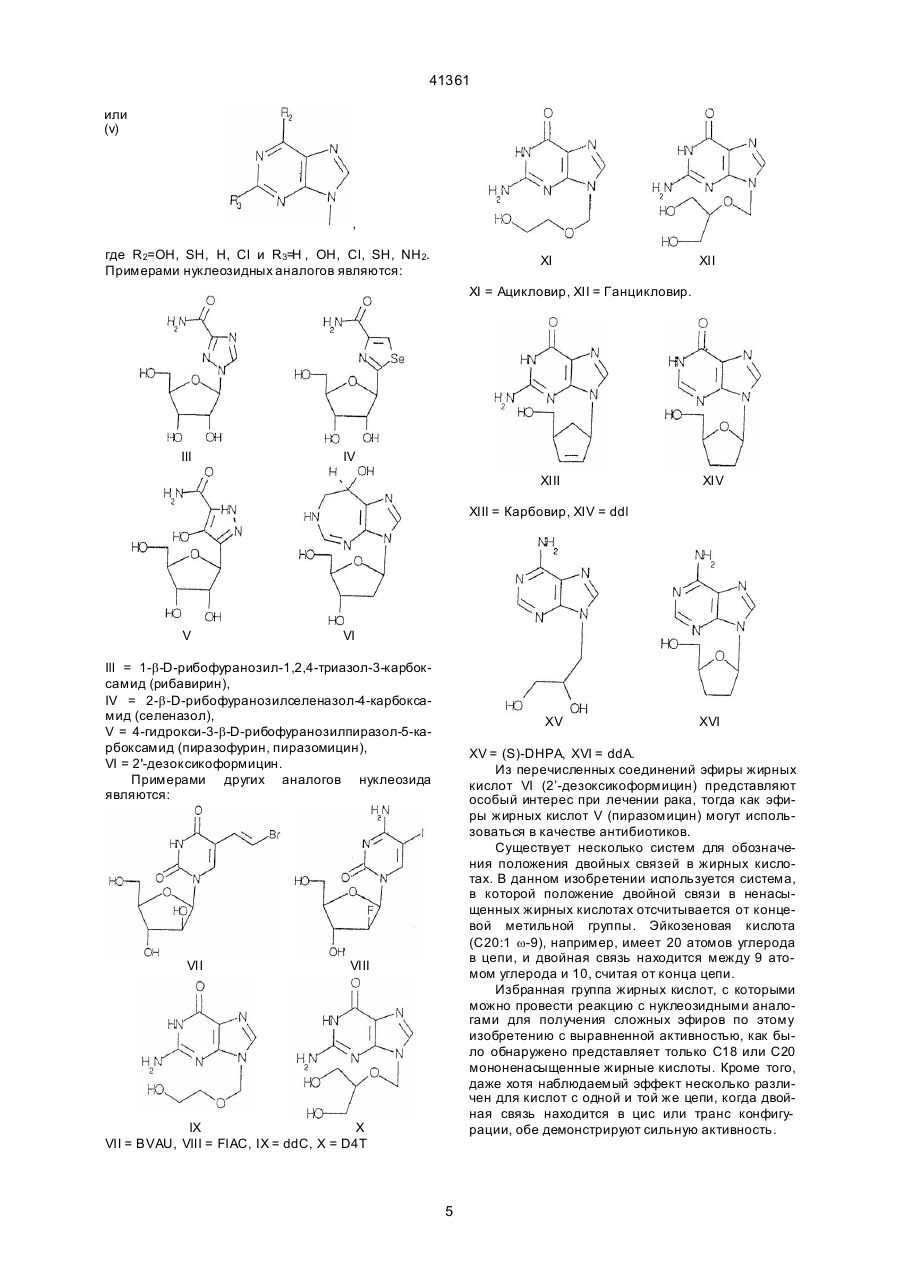
1. Моноэфирное соединение формулы I: 41361 ное количество соединения по любому из пунктов 8. Соединение формулы I по п. 1 для получения 1-6, а также фармацевтически приемлемый носифармацевтической композиции для лечения витель или наполнитель. русной инфекции. ___________________________________ Данное изобретение относится к группе новых соединений общей формулы I: Ме ханизмы, лежащие в основе некоторых из наиболее важных вирусных заболеваний, различны. При поражении ВИЧ инфекцией заражаются и разрушаются Т-хелперные клетки больного. Это приводит к состоянию иммунодефицита, что делает больного очень восприимчивым даже к инфекциям, с которыми в норме иммунная система справлялась без каких-либо опасных для больного эффектов. Вирус гепатита В инфицируе т клетки печени, и состояние больного может ухудшиться, когда имунная система старается освободить организм от эти х инфицированных клеток. Если инфекция не преодолевается иммунной системой на ранней стадии, результатом будет хронический гепатит. Больной, таким образом, будет ин фицированным в течение всей его жизни. У части больных хронический гепатит перейдет в цирроз или рак печени. При инфекциях, вызванных вирусом Herpes simplex, вирус проникает сначала в эпидермальные клетки. Вирус Herpes simplex доходит до нервного центра, где он пребывает латентным до того, как вызовет вспышку заболевания через какие-то интервалы времени. Хотя в большинстве случаев нет угрозы жизни, герпесная инфекция болезненна, и больной будет инфекционным каждый раз, когда появляется вспышка заболевания. У вируса папилломы, особенно, в генитальном тракте женщин, вирусный геном локализуется в ядрах эпителиальных клеток, но не интегрируется в клеточные хромосомы. Это является персистентным состоянием, а для некоторых, вызывающи х опухоли, штаммов интеграция, наконец, происходит, приводя к злокачественному перерождению. Вирусный геном в этом случае обладает определенным инициирующим действием в процессе, приводящем к раку. Если иммунной системе удается освободить организм от вируса на ранней стадии, это приводит к пожизненному иммунитету. С др угой стороны, если вирус слишком агрессивен и избежит действия иммунного аппарата, иммунитет не создается, и результатом является состояние постоянной инфекции. В результате существования различных механизмов стратегия терапии должна быть различной при этих состояниях. Непременной целью при лечении ВИЧ/СПИД должно быть освобождение организма больного от инфицирующего вируса. На данном этапе до этого, по-видимому, далеко. Однако многого можно достичь путем улучшения общего состояния больного. Снижение уровня инфицированности вирусом (вирусной нагрузки) должно увеличивать продолжительность бессимптомного периода и снижать инфицированность, которая крайне важна в отношении эпидемиологической ситуации. Все используемые в настоящее время противовирусные средства обладают токсическими побочными действиями, что делает сей Nu-О-Fa, где Nu является аналогом нуклеозида и представлен формулой II: B-S, в которой В представляет собой произвольно замещенную гетероциклическую кольцевую систему, S является моносахаридным производным, О представляет собой кислород, a Fa является ацильной группой мононенасыщенной C18 или С20 жирной кислоты. Изобретение также касается противовирусных фармацевтических и ветеринарних композиций, содержащих соединение с формулой I, одно или в комбинации с фармацевтически приемлемым носителем. Дополнительная часть этого изобретения представляет собой способ лечения больного человека или животного, страдающего вирусной инфекцией и снижения уровня инфицированности путем введения соединения формулы I. Подобным образом некоторые соединения формулы I могут использоваться в качестве антибиотика или при лечении paковых заболевании. Предпосылки создания изобретения. Большое число тяжелых заболеваний, таких как СПИД, гепатит В, герпес и гинекологический рак, как отдаленный результат папилломных выростов, вызываются вирусной инфекцией. Вирусы - это очень мелкие инфицирующие агенты, которые неспособны к независимой репликации и, таким образом, в отношении репликации зависят от клетки-хозяина. Генетический материал вируса представляет собой или РНК или ДНК. При инфицировании организма вирус связывается с определенной клеткой-хозяином. После связывания вирус проникает через цитоплазматическую мембрану, и вир усный геном освобождается из вирусной частицы. Вирусный геном, обычно, транспортируется в клеточное ядро, где реплицируются новые вирусные геномы. В цитоплазме синтезируетоя новый вирусный белок, и новые частицы формируются или вблизи цитоплазматической или вблизи ядерной мембраны. Некоторые вирусы имеют геномный материал, который прямо (ДНК вируса) или косвенно (обратная транскрипция РНК, ретровирусы) включается в геномы клеток-хозяев. Внеклеточные вирусы нейтрализуются циркулирующими антителами, а клеточный иммунный аппарат может атаковать и удалять инфицированные клетки. Вирусы внутри инфицированных клеток ускользают от иммунного надзора, если вирусные антигены не представлены на поверхности клеток. Иммунная атака на инфицированные органы участвует в течение заболевания с помощью механизма, обычно называемого индуцированной вирусом иммунопатологией. 2 41361 час достаточно действенное лечение невозможным. Предполагается, что по всему миру существует от 250 до 300 миллионов носителей гепатита В. Известно, что у большого числа из них разовьются гепатомы или печеночная недостаточность в результате инфекций. Обещающие результаты при лечении состояния носительства в последние годы были получены путем индукции иммунного ответа интерфероном. Терапия, снижающая вирусную нагрузку, важна при этом режиме и, так как эффективное лечение острого гепатита В должно снижать число больных, у которых развивается состояние носительства. Недавно идентифицированный вирус гепатита С вызывает очень большое число случаев гепатита, из которых у большого числа развивается носительство. Предварительные исследования, по-видимому, показывают, что состояние носительства может быть прекращено с помощью таких же терапевтических режимов, что и для гепатита В. Herpes simplex 1 и 2 часто инфицирует людей, вызывая состояние носительства с рецидивами локальных инфекций. Генерализованные инфекции, включая энцефалит, редки, но являются катастрофой для больного. Существует большое индивидуальное разнообразие в частоте местных инфекций для тех больных, у которых поражения присутствуют на гениталиях или на лице, что создает серьезную проблему для здоровья, физически, психически и социально. Ни при одном из терапевтических режимов, разработанных до сих пор, не излечиваются латентные инфекции клеток центральной нервной системы. Цель терапии состоит, таким образом, в том, чтобы свести к минимуму клинические проявления рецидивов, как в отношении симптомов, так и продолжительности. Распространение инфекций, вызванных вирусом генитальной папилломы, очень сильно увеличилось в течение 1980-х годов. В настоящее время установлено, что некоторые генотипы являются онкогенными, то есть они инициируют изменения в клетке, которые после латентного периода приводят к paку. Вирус папилломы генитального тракта дает длительно присутствующие инфекции. Факторы, вызывающие злокачественную трансформацию поражений, еще недостаточно понятны, но предполагается, что большое значение имеет имунная система. Полагают, что поражения, которые демонстрируют прогрессирующее развитие в течение месяцев и лет, и являются теми, которые дают развитие рака. Генитальные папилломы, называемые кондиломами в, настоящее время лечатся физическими средствами, такими как хирургическое удаление, некротизирующие средства, удаление жидким азотом и тому подобное. Остроконечные кондиломы сначала являются доброкачественными опухолями с измененным набором ферментов, воздействующи х, между прочим, на метаболизм нуклеозидных аналогов. Нуклеозидные пролекарства воздействуют на эписомную пролеферацию вируса папилломы, индуцируя тем самым регрессию кондилом. Профилактическая вакцинация была успешной для острых инфекций, таких как полиомиелит, корь, эпидемический паротит и т. д, но никакой эффективной вакцинации не было разработано для многих др уги х тяжелых вирусны х инфекций. Даже хотя были предприняты интенсивные усилия по получению эффективных противовирусных химиотерапевтических препаратов в течение последних десятилетий, не было предложено удовлетворительного медикаментозного лечения для большинства вирусных заболеваний в настоящее время. Эти усилия были особенно велики из-за появления ВИЧ и родственных вирусных инфекций, которые распространяются по миру с тревожащей скоростью, однако эффекты, полученные с помощью средств, таких как азидотимидин (AЗT) и ацикловир (АЦВ), при СПИД и герпесе, можно охарактеризовать как частично успешные. Эти наиболее обещающие противовирусные средства являются производными природно встречающи хся нуклеозидов, которые были модифицированы или в основании или в сахаридной части. Они, однако, не обладали тем терапевтическим потенциалом, на который надеялись, так как они вызывают серьезные побочные эффекты у некоторых больных или демонстрируют слабый эффект или его отсутствие у др уги х. Кроме того, лечение этими средствами чрезвычайно дорого. По этим причинам только больные, страдающие очень тяжелыми вирусными инфекциями, такими как СПИД, получают такое лечение. Пациентов, страдающих менее серьезными, но также очень болезненными, вирусными инфекциями, часто оставляют без лечения, чтобы дать инфекции пройти своим путем. Нелеченный больной несет большую инфекционную нагрузку и создает риск заражения для своего окружения. Если он лечится противовирусным средством, то цель состоит в том, чтобы снизить инфекционную нагрузку так, чтобы дать возможность иммунной системе организма побороть инфекцию. Дополнительная цель состоит в том, чтобы снизить контагиозность и, таким образом, число новых больных и носителей. Таким образом, потребность в соединениях, обладающих лучшим терапевтическим индексом, очевидна. Потребность особенно велика при хронических и рецидивирующих вирусных инфекциях с опасной острой фазой или длительными болезненными эффектами на здоровье или самочувствие, таких как СПИД, гепатит В и С, инфекций герпесной группы и инфекций, вызванных вирусом папилломы. Аналогично, существует также потребность в противовирусных средствах, используемых при лечении животных, страдающи х вирусными заболеванияии. Чтобы улучшить эффект, были разработаны производные нуклеозидов, которые или модифицированы в азотном основании или в сахаридной части. В частности, были разработаны эфиры жирных кислот нуклеозидных аналогов, чтобы улучшить липофильность и достичь лучшего проникновения через мембрану. Так, из патента США 3984396 (Witkowski et. al.) известны эфиры рибавирина с ароматическими и насыщенными жирными кислотами, имеющими 1-18 атомов углерода. 3 41361 В настоящее время неожиданно было обнаружено, что избранная группа эфиров жирных кислот противовирусных н уклеозидных аналогов, где жирная кислота является мононенасыщеной C18 или С20 кислотой, дает намного улучшенный эффект. Хотя известно, что как нуклеозиды и нуклеозидные аналоги, сами по себе, так и также некоторые ненасыщенные жирные кислоты; сами по себе, проявляют противовирусное действие, величина эффектов, достижимых с помощью соединений данного изобретения, показывает, что это не аддитивное, а, скорее, синергидное действие, которое специфично для соединений с формулой I. Механизм, стоящий за этими эффектами, в настоящее время неизвестен. Вероятно, нельзя считать, что они возникают только благодаря действию на уровне мембраны или благодаря эффекту доставки к мишени действия. Кроме того, также ясно, как будет видно из биологических примеров, включенных сюда, что эффективность достигается с помощью этих соединений в системах, где более низкие эффекты могут достигаться с помощью исходных нуклеозидных соединений. Соединения этого изобретения могут быть представлены общей формулой I: 1-b-D-рибофуранозы, 1-b-D-арабинофуранозы, 2-дезокси-1-b-D-рибофуранозы 2,3-дидезокси-1b-D-рибофуранозы, 2,3-дидегидро-2,3-дидезокси1-b-D-рибофуранозы, 2-дезокси-2-фтор-1-b-D-арабинофуразы, 2,3-дидезокси-3-азидо-1-b-D-рибофуранозы или 4-гидроксиметил-2-циклопентен-1-ила, или выбирается из группы из 2-гидроксиэтоксиметила, 4-гидрокси-3-(гидроксиметил)-бутила, 2-гидрокси-1-(гидроксиметил)-этоксиметила, 2,3-дигидроксипропокси- или 2,3-дигидроксипропила; и В является гетероциклической кольцевой системой, выбираемой из (І) X R N 1 N N R R1 1 N A A N , где X=0, , Н, R 1=Н2, СН3 , CН3О и А=Н, , , СН3, О, или (ii) Nu–О–Fa, где О является кислородом, Nu является аналогом нуклеозида и представлен формулой II: B-S, в которой В представляет собой произвольно замещенную гетероциклическую кольцевую систему, а S является моносахаридным производным и Fa является ацильной группой мононенасыщенной С18 или С20 жирной кислоты. Природные нуклеозиды, названные так благодаря их присутствию в РНК и ДНК, являются молекулами, содержащими гетероциклическое основание, такое как цитозин, урацил, тимидин, аденин или гyaнин, связанное с рибозой или 2-дезоксирибозным елементом. В нуклеозидных аналогах или основание или рибозный элемент модифицированы. Например, рибозная единица может быть заменена другим сахаридным элементом или нециклической цепью. Модифицированное основание может относиться или к соединениям, выбираемым из групп пиримидинов или пуринов, замещенных иначе, чем упомянутые здесь природные основания, или произвольно замещенным гетеро-(моно или поли)циклическим кольцевым системам, которые не являются пиримидином или пурином. Жирная кислота зтерифицируется с гидроксильной группой сахаридной части нуклеозидного аналога или с гидроксильной группой на нециклической группе нуклеозидного аналога. Нуклеозидные аналоги, которые могут быть выбраны в качестве Nu в cоединениях с формулой I, могут быть, предпочтительно, представлены формулой II: В-S, X X , где X=NH, S, О и Y=Н, ОН, F, Cl, Br, I, NH 2, CH2CN, CºN, или (iii) или (iv) , (II) где R4=H, NH2, NHOH, NНСОСН3, NHCH3, NHNH2 и R5=H, F, Cl, Br, І, СН3, CF3, и R6=СН3, F, I, CH=CHBr, CH 2OH, CH 2NH2, CºCH, где S является или моносахаридным производным, выбираемым из: 4 41361 или (v) , где R2=ОН, SH, Н, Сl и R3=Н , ОН, Cl, SH, NH2. Примерами нуклеозидных аналогов являются: XI XII XI = Ацикловир, XII = Ганцикловир. III IV XIII XIV XIII = Карбовир, XIV = ddl V VI Ill = 1-b-D-рибофуранозил-1,2,4-триазол-3-карбокcамид (рибaвирин), ІV = 2-b-D-рибофуранозилселеназол-4-карбоксамид (селеназол), V = 4-гидрокси-3-b-D-рибофуранозилпиразол-5-карбоксамид (пиразофурин, пиразомицин), VI = 2'-дезоксикоформицин. Примерами других аналогов нуклеозида являются: VII XV XVI XV = (S)-DHPA, XVI = ddA. Из перечисленных соединений эфиры жирных кислот VІ (2’-дезоксикоформицин) представляют особый интерес при лечении рака, тогда как эфиры жирных кислот V (пиразомицин) могут использоваться в качестве антибиотиков. Существует несколько систем для обозначения положения двойных связей в жирных кислотах. В данном изобретении используется система, в которой положение двойной связи в ненасыщенных жирных кислотах отсчитывается от концевой метильной группы. Эйкозеновая кислота (С20:1 w-9), например, имеет 20 атомов углерода в цепи, и двойная связь находится между 9 атомом углерода и 10, считая от конца цепи. Избранная группа жирных кислот, с которыми можно провести реакцию с нуклеозидными аналогами для получения сложных эфиров по этому изобретению с выравненной активностью, как было обнаружено представляет только С18 или С20 мононенасыщенные жирные кислоты. Кроме того, даже хотя наблюдаемый эффект несколько различен для кислот с одной и той же цепи, когда двойная связь находится в цис или транс конфигурации, обе демонстрируют сильную активность. VIII IX X VII = BVAU, VIII = FIAC, IX = ddC, X = D4T 5 41361 Те С18 или С20 w-9 жирные кислоты, которые, связанные с нуклеозидными аналогами, дают удивительно повышенный эффект, являются следующими: олеиновая кислота (С18:1, w-9, цис), элаидиновая кислота (С18, w-9, транс), эйкозеновая кислота (С20:1, w-9, цис) и (С20:1, w9, транс). Предпочтительные представители соединений по этому изобретению перечислены ниже: сложный эфир рибавирина и олеиновой кислоты, эфир рибавирина и элаидиновой кислоты, эфир рибавирина и цис-эйкозеновой кислоты, эфир рибавирина и транс-эйкозеновой кислоты, эфир селеназола и олеиновой кислоты, эфир селеназола и элаидиновой кислоты, эфир селеназола и цисэйкозеновой кислоты, эфир селеназола и трансэйкозеновой, эфир пиразомицина и олеиновой кислоты, эфир пиразомицина и элаидиновой кислоты, эфир пиразомицина и цис-эйкозеновой кислоты, эфир пиразомицина и транс-эйкозеновой кислоты, эфир 2’-дезокcикоформицина и олеиновой колоты, эфир 2'-дезоксикоформицина и элаидиновой кислоты, эфир 2'-дезоксикоформицина и цис-эйкозеновой кислоты, эфир 2'-дезоксикоформицина и транс-эйкозеновой кислоты, эфир d4T и цис-эйкозеновой кислоты, эфир d4Т и трансэйкозеновой кислоты, эфир ddС и олеиновой кислоты, эфир ddС и элаидиновой кислоты, эфир ddС и цис-эйкозеновой кислоты, эфир ddС и транс-эйкозеновой кислоты, эфир ddI и олеиновой кислоты, эфир ddI и элаидиновой кислоты, эфир ddI и цис-эйкозеновой кислоты, эфир ddI и транс-эйкозеновой кислоты, эфир карбовира и олеиновой кислоты, эфир карбовира и элаидиновой кислоты, эфир карбовира и цис-эйкозеновой кислоты, эфир карбовира и транс-эйкозеновой кислоты, эфир (S)-DНРА и олеиновой кислоты, эфир (S)-DHPA и элаидиновой кислоты, эфир (S)DНРА и цис-эйкозеновой кислоты, (S)-DНРА и транс-эйкозеновой кислоты, эфир BV AR AU и олеиновой кислоты, эфир BV AR AU и элаидиновой кислоты, эфир BV ARAU и цис-эйкозеновой кислоты, эфир BV AR AU и транс-эйкозеновой кислоты, эфир BVDU и олеиновой кислоты, эфир BVDU и элаидиновой кислоты, эфир BVDU и цисэйкозеновой кислоты, эфир BVDU и транс-эйкозеновой кислоты, эфир FIAC и олеиновой кислоты, эфир FIAC и элаидиновой кислоты, эфир FIAC и цис-эйкозеновой кислоты, эфир FIAC и транс-эйкозеновой кислоты, эфир EICAR и олеиновой кислоты, эфир EICAR и элаидиновой кислоты, эфир EICAR и цис-эйкозеновой кислоты, эфир EICAR и транс-эйкозеновой кислоты, эфир 2’F’AR A-рибавирина и олеиновой кислоты, эфир 2’F’AR A-рибавирина и элаидиновой кислоты, эфир 2’-F’ARA-рибавирина и цис-эйкозеновой кислоты, эфир 2’-F’ARA-рибавирина и транс-эйкозеновой кислоты. Их формулы будут ясны из фигуры 4. Соединение по этому изобретению проявляют противовирусное действие, и данное изобретение, таким образом, включает фармацевтические или ветеринарные композиции, содержащие, по крайней мере, одно вещество с формулой I, отдельно или в сочетании с фармацевтически приемлемым носителем или наполнителем. В оставшейся части текста и в формуле изобретения фармацевтиче ская композиция будет использоваться для обозначения композиций, применяемых при лечении как больных людей, так и животных. Кромо того, по-видимому, нуклеозидные аналоги с некоторыми из мопоненасыщенных жирных кислот будут особенно подходящими для лечения некоторых вирусных инфекций. Так, очевидно, что эфиры жирных кислот, по этому изобретению, рибавирина особенно подходят для лечения герпесных инфекций. Также очевидно, что соединения этого изобретения или композиции, их содержащие, также применимы при лечении заболеваний у людей, вызываемых аденовирусами, вирусами группы А и В, респираторным синцитиальным вирусом (РСВ), цитомегаловирусом (ЦМВ), вирусами папилломы, бунья-вирусами, аренавирусами и ВИЧ. Как упоминалось, выработка необходимого иммунного ответа для преодоления вирусной инфекции, такой как гепатит, может индуцироваться в некоторых случаях путем совместного введения с интерфероном. Кроме тoгo, очевидно, что сложные эфиры жирных кислот, по этому изобретению, или содержащие их композиции также применимы для лечения млекопитающих (не людей), птиц, например, цыплят и индюшек, и холоднокровных животных, например, рыб, страдающих инфекциями, вызываемыми: бычьими герпесвирусами 1, 2, 3, 4, лошадиными герпесвирусами 1, 2, 3, свинными герпесвирусами 1, 2, фазаньими герпесвирусами 1, 2 (болезнь Марека) и IPN (инфекционный некроз поджелудочной железы) вирус. В зависимости oт того, какую вирусную инфекцию нужно лечить и на какой стадии находится инфекционное заболевание или является ли больной человеком или животным, может применяться системное или местное введение веществ. Для местного применения могут быть приготовлены лекарственные формы веществ, известные в данной области технологии, для применения на коже или слизистой в любой подходящей форме. Для местного применения соединения формулы І могут приготавливаться в виде мази, крема, геля, настойки, аэрозоля, лосьона или тому подобного, содержащего соединения формулы I в смеси с инертными, твердыми и жидкими, носителями, которые обычны в препаратах для местного применения. Особенно подходит использование лекарственной формы, которая защищает активный ингредиент от окисления и деградации. Фармацевтичексие препараты, содержащие соединения формулы I, могут также вводиться системно или перорально, или парентерально. При энтеральном введении соединения формулы I могут быть приготовлены в лекарственной форме, например, такой как мягкие или твердые желатиновые капсулы, таблетки, гранулы, крупинки или порошки, драже, сиропы, суспензии или растворы. Для парентерального введения препараты соединений формулы I пригодны в виде инъекционных или инфузионных растворов, суспензий или эмульсий. Препараты могут содержать инертные или фаркодинамически активные добавки. Таблетки 6 41361 или гранулы, например, могут содержать ряд связывающих ве ществ, наполняющих материалов, вещества-носители или разбавители. Жидкие препараты могут быть представлены, например, в форме стерильного раствора. Капсулы могут содержать вещество-наполнитель или загущающее ве щество в дополнение к активному ингредиенту. Кроме того, обычно используются улучшающие вкус и запах добавки, а также вещества, применяемые в качестве консервирующи х, стабилизирующих, поддерживающих влажность и эмульгирующи х средств, соли для изменения осмотического давления, буферы и другие добавки могут также присутствовать. Дозы, в которых препараты по этому изобретению применяются, будут меняться в соответствии со способом применения и путем введения, а также в соответствии с потребностями больного. В основном, суточная дозировка для системной терапии средних взрослых больных или животных будет около 0,1-100 мг/кг веса тела/сутки, предпочтительно, 1-20 мг/кг/сутки. Для местного применения подходящая мазь может содержать от 0,1 до 10% по весу от терапевтической рецептуры, предпочтительно, 0,5-5% по весу. Если желательно, фармацевтический препарат соединения формулы I может содержать антиоксидант, например, токоферол, -метил-токоферамин, бутилированный гидроксианизол, аскорбиновую кислоту или бутилированный гидрокситолуол. В изобретении, кроме того, раскрывается способ лечения вирусных инфекций, который включает введение, по крайней мере, одного соединения с формулой I больному человеку или животному при необходимости такого лечения. Кроме того, изобретение также включает способ лечения больного, при необходимости такого лечения, с помощью комбинации соединения с формулой I и интерферона. Биологическое действие Тканевая культура вирус IPN Препаратом вируса IРN (1000 бое) заражают и инкубируют при перемешивании в течение 1 часа при 200С монослой клеток CHSE-214. К клеткам добавляют небольшой объем среды для роста, содержащей противовирусное средство. Затем клетки культивируют (48 час) до появления положительных ЦПД в необработанном контроле. Затем клетки помещают в морозильник на ночь. После оттаивания и центрифугирования супернатант в 5 уровнях разведения добавляют к монослою свежеприготовленной клеточной культуры в 96-ячеечной плате. ЦПД регистрируется через 48 часов, и титр вируса рассчитывается как TCI50. На фигуре 1 показан ингибирующий эффект эфира элаидиновой кислоты и рибавирина при трех уровнях дозировки, и снижение титра вируса порядка 104 наблюдалось даже при низкой концентрации. А. Эксперименты Бляшечный метод: Тканевая культура вируса HSV 1/2 Препараты вирусов HSV 1 и НSV 2 (3-й пассаж клинического штамма) разводят до 250 и 100 бое/ячейку, соответственно, и затем ими зара жали клетки и инкубировали в течение 1 часа в культуре тканей различных клеточных линий. Инфицированные клетки затем культивируют в течение 48 часов с противовирусным средством. Культуры замораживают и оттаивают для выделения свободного вируса. Готовят разведения или 1/100, или 1/10000 и добавляют к свежим тканевым культурам. После инкубации в течение 1 часа добавляют карбоксиметилцеллюлозу (КМЦ) для предотвращения миграции вируса между клетками через среду. Распространение вируса путем клеточного контакта, однако, остается эффективным, приводя к образованию бляшек. Одна бляшка будет представлять одну вир усную частицу. Таким образом; подсчет бляшек дает точную оценку числа частиц инфицирующего вируса. На фигуре 2 показан подавляющий эффект рибавирина и рибавирин-5’-олеилового эфира на штамм HSV 2 (68495) в клеточной линии HLI. Этот штамм вируса относительно устойчив к самому рибавирину, что можно видеть по умеренному снижению титра вируса. Эта резистентность была даже более очевидна с другими линиями клеток, которые использовались при исследовании. Однако с клеточной линией, к которой относится фиг. 2, даже и так видно, что введение эфирной группы ненасыщенной жирной кислоты все же потенцирует активность с помощью факора 3. В. Эксперименты Инфекция, вызванная вирусом FLC у мышей Молодых (20-25 г) самок мышей NMRI инфицировали в/б препаратом ретровируса синдрома лейкемии Френда (Friend Leukemid Complex FLC). Лечение начиналось на 2-й день после заражения вирусом, и животные получали суточную дозу, равную 200 мкл 20 мкМ липосомной лекарственной формы испытуемых веществ в/б в течение 8 дней. Группы животных забивали через 13 и 20 дней после инфицирования. Регистрировали вес тела и селезенки животных. Инфекция, вызванная вирусом FLC, приводит за период в 7-10 суток к заметное увеличению веса селезенки, пo-видимому, из-за высокой концентрации лейкемических клеток. На фигуре 3 показано сравнение действия АЗТ, гла вного лекарства для лечения ретровирусных инфекций, ddС-элаидата и соответствующего насыщенного аналога, например, ddС-стеарата. Данные представлены в виде отношения вес тела/вес селезенки, причем большое число отражает высокий антиретровирусный эффект. Через 13 дней эффект АЗТ равен высоте значения для ddС-элаидата. Через 20 дней отношения для животных, леченных АЗТ, падают до уровня контролей, тогда как у животных, леченных ddС-элаидатом, остается высокое отношение. Зависимость от природы жирной кислоты явна, что можно видеть по низким (~ контрольным) значениям для производного стеариновой кислоты. Противовирусные сродства по этому изобретению готовили из исходного стандартного раствора мицелл в воде 1 мг/мл путем смешивания лецитина и активного ингредиента в соотношении 1:1 (вес/вес) в стерильной дистиллированной воде. 7 41361 Получение Вещества с формулой 1 можно, в основном, получить по следующему уравнению реакции: B - S - OH + FaX Основание - НХ концентрированный раствор добавляли порциями по 2 мл, примерно, через 8 час интервалы. После общего времени реакции в 60 часов растворители выпаривали под высоким вакуумом, и продукт очищали на колонке силикагеля с помощью 15% метанола в хлороформе в качестве системы растворителей. Гомогенные фракции выпаривали с получением 1,25 г (63%) вещества; указанного в заголовке, в виде белого твердого ве щества. 1 Н ЯМР (ДМСО-d6, 300 МГц) d: 9,8 (1H, S, H-5) 7,85 и 7,65 (1+1Н, S,NH2), 5,88 (1H, d, H-1), 5,65 (1H, d, OH-2’), 5,35 (1H, d, OH-3’), 5,32 (2Н, m, CH=CH), 4,35-4,25 (ЗН, m, Н-2’, Н-3’, Н-5’1), 4,154,0 (2Н, m, Н-4’, Н-5’2), 2,25 (2Н, t, CH2-COO), 1,95 (4H, m, CH 3-CH2), 13 С ЯМР (ДМСО-d6 , 75 МГц) d: 172,59 (COO), 160,15 (CONH2) 157,40 (C-3), 145,27 (C-5), 129,46 (CH=CH), 91,28 (C-1’), 81,51 (C-4’), 73,99 (C-2’), 7026 (C-3’), 63,55 (C-5’), 33,07, 31,13, 28,94, 28,68, 28,54, 28,43, 28,83, 28,24, 26,42, 24,17, 21,95 (CH2), 13,78 (CH3-CH2). Пример 2 1-(5’-O-|транс-9"-октадeценоил|-2,3-дидезоксиb-D-глицеропент-2-енофуранозил)тимин К раствору 1-(2,3-дидезокси-b-D-глицеро-пент2-енофуранозил)тимина (d4T) (0,83 г, 3,7´10-3 моля) и 10 мл безводного N,N-диметилформамида и 10 мл пиридина добавляли 2 мл концентрированного раствора транс-9-октадеценоилхлорида (1,39 г, 4,9´10-3 моля) в 4 мл дихлормотана, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре. Оставшийся концентрированный раствор добавляли порциями по 1 мл через, примерно, 8 час интервалы. После общего времени протекания реакции в 60 час растворители выпаривали под высоким вакуумом. Остаток растворяли в хлороформе и промывали водой. Высушенную (MgSO4) органическую фазу концентрировали, и сырой продукт очищали на колонке силикагеля с 7% метанола в хлороформе в качестве системы элюентов. Гомогенные фракции выпаривали с получением 1,26 г (70%) вещества, указанного в заголовке в виде белого твердого вещества. 1 Н ЯМР (ДМСО- d6 , 300 МГц) d: 11,35 (1Н, S, NН), 7,25 (1Н, S, Н-6), 6,81 (1Н, m, Н-1’), 6,38 (1Н, m, Н-3’), 6,0 (1Н, m, Н-2’), 5,35 (2Н, m, СН=СН ), 4,95 (1Н, m, Н-4’), 4,2 (2Н, m, Н-5’), 2,25 (2H, m, CH2-COO), 1,95 (4Н, m, CH 2-C=), 1,75 (ЗH, S, CH3), 1,45 (2Н, m, СH2-С-СОО), 1,25 (20Н, m, CH 2), 0,85 (3Н, t, CH3-CH2). Пример 3 5-О-(9’-транс-октадеценоил)-2,3-дидезоксицитидин К суспензии 2,3-дидезоксицитидина (ddC) (1,2 г, 5,7´10-3 моля) в 40 мл безводного N,N-диметилформамида, содержащего НСl (г) (6,3´ х10-3 моля) добавляли 1 мл концентрированного раствора транс-9-октадеценоилхлорида (1,7 г, 6,0´10-3 моля) в 6 мл дихлорметана, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре. Оставшийся концентрированный раствор добавляли порциями по 1 мл через, примерно, 3 час интервалы. После общего 60 часового времени протекания реакции растворители выпаривали под высоким вакуумом. B - S - O - Fa , где В, S, С и Fа дано определение выше, а Х может быть Сl, Вr, С-CO-R', где R' является Fа, СН3, -СН2CH3 или СF3, Х может также быть бензотриазольной частью бензотриазолового эфира жирной кислоты. Таким образом, реакция протекает путем ацилирования нуклеозидного аналога. Это выполняется с помощью использования подходящих реактивных производных жирных кислот, особенно, галогенангидридов кислот или ангидридов кислот. Реактивные производные жирных кислот могут быть предварительно получены или произведены in situ путем применения реагентов, таких как дициклогексилкарбодиимид (DСС) или О-(1Н-бензотриазол-1-ил) N,N,N’,N’,-тетраметилуронийтетрафторборат TBTU). Когда используется галогенангидрид кислоты, такой как хлорангидрид кислоты, к реакционной смеси добавляется третичноаминный катализатор, такой как триэтиламин, N,N-диметиланилин, пиридин или N,N-диметиламинопиридин, для связывания освобождаемой галогенводородной кислоты. Реакции, предпочтительно, проводятся в нереактивном растворителе, таком так N,N-диметилформамид или галогенированный углеводород; такой как дихлорметан. Если желательно, любой из вышеупомянуты х третичноаминных катализаторов может использоваться в качестве растворителя, причем нужно следить, чтобы он был в соответствующем избытке. Температура реакции может меняться в интервале от 0°С до 40°С, но предпочтительно ее поддерживать в интервале от 5° до 25°С. Через 24-60 часов реакция, по существу, завершится. Ход реакции можно прослеживать, применяя тонкослойную хроматографию (ТСХ) и соответствующие системы растворителей. Когда реакция завершается, что определяется TCX, продукт экстрагируется органическим растворителем и очищается с помощью хроматографии и/или перекристаллизации из подходящей системы растворителей. Если в нуклеозидном аналоге присутствуют более, чем одна гидроксильная группа или также аминогруппы, может быть получена смесь ацилированных соединений. Отдельные моно- и полиацилированные соединения могут быть выделены с помощью хроматографии, например. Изобретение иллюстрируется не лимитирующими примерами, которые следуют ниже. Пример 1 1-(5’-О|цис-9’’-октадеценоил|-b-D-рибофуранозил)-1,2,4-триазол-3-карбоксамид К раствору 1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамида (рибавирин) (0,95 г, 3,9´ x10-3 моль) в 10 мл безводного N,N-диметилформамид и 15 мл пиридина добавляли 2 мл концентрированного раствора цис-9-октадеценоилхлорида (1,34 г, 4,7´10-3 моль) в 6 мл дихлорметана, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре. Оставшийся 8 41361 Остаток растворяли в хлороформе и промывали водой. Высушенную (MgSO4) органическую фазу концентрировали, и остаток растворяли в гептане. Добавление петролейного эфира (40-60 мл) давало белое твердое вещество (0,7 г), которое собирали на холоде. Фильтрат очищали на колонке силикагеля, с которой элюировали метанолом (0-25%) в этилацетате. Гомогенные фракции выпаривали с получением еще 0,8 г ве щества, указанного в заголовке (1,5 г, 55%). 1 Н ЯМР (ДМСО- d6, 300 МГц) d: 7,63 (1Н, d, Н-6), 7,1 (2Н, d, NH2), 5,95 (1Н, m, Н-1’), 5,7 (1Н, d, Н-5), 5,35 (2Н, m, СН=СН), 4,25-4,10 (3Н, m, Н-4’ и H-5’), 2,3 (3H, m, H-2’ и CH2-COO), 1,45 (2Н, m, CH2-C-СОО), 1,25 (20Н, m, CH2), 0,85 (3Н, t, CH3-CH2). 13 С ЯМР (ДМСО-d6, 75 МГц): 172,67 (COO), 165,60 (CNH2 4) 155,04 (CО 2), 140,45 (CН 6), 130,00 (CH=CH), 93,62 (CН 5), 85,84 (C 1), 77,84 (C 4), 64,87 (C 5), 33,38, 31,96, 31,30, 29,02, 28,96, 28,86, 28,74, 28,52, 28,46, 28,36, 24,20, 22,11 (CH2), 31,76, 25,47 (CH 2’ и 3’), 13,90 (CH3). Пример 4 5-О-(9’-транс-октадеценоил)-5-иод-2’-дезоксиуридин К раствору 5-иод-2’-дезоксиуридина (1,0 г, 2,8´10-3 моля) в 10 мл безводного N,N-диэтилформамида и 15 мл пиридина добавляли 1 мл концентрированного раствора транс-9-октадеценоилхлорида (1,12 г, 3,95´10-3 моля) и 4 мл дихлорметана, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре. Остаток концентрированного раствора добавляли порциями по 1 мл через, примерно, 4 час интервалы. После общего 50-часового времени протекания реакции растворители выпаривали под высоким вакуумом, и продукт очищали на колонке силикагеля с использованием 10% метанола в хлороформе в качестве системы элюентов. Фракции, содержащие продукт, концентрировали, и остаток перекристаллизовывали из этанола с получением 1,43 г (82%) вещества, указанного в заголовке, в виде твердого белого вещества. 1 Н ЯМР (ДМСО- d6 , 300 МГц) d: 11,71 (1Н, S, NH), 7,95 (1Н, S, H-6), 6,08 (1Н, t, Н-1’), 5,4 (1Н, d, OH-3’), 5,35 (2Н, m, СН=СН ), 4,2 (3Н, m, Н-3’ и H-5’), 3,95 (1H, m, H-4’), 2,35 (2Н, t, CH2-СОО), 2,15 (2Н, m, H-2’), 1,95 (4H, m, CH2-C=), 1,50 (2Н, m, CH2-C-COO), 1,25 (20H, m, CH 2), 0,85 (3Н, t, CH2-C). 13 С ЯМР (ДМСО-d6 , 75 МГц) d: 172,16 (COO), 159,90 (C = O 4) 149,50 (C =О 2), 143,89 (C-6), 129,51 (CH=CH), 84,43 (C-4'), 83,65 (C-1’), 69,66 (C-3’), 69,21 (C-5), 63,06 (C-5’), 38,85 (C-2’), 33,01, 31,45, 30,79, 28,51, 28,34, 28,22, 28,01, 27,92, 27,85, 27,46, 23,93, 21, 60 (CH2), 13,43 (CH 3-C). Пример 5 5-О-(9-транс-октадеценоил)-2,З-дидезоксиинозин Раствор транс-9-октадоценоиновой кислоты (0,33 г, 1,17´10-3 моля), О-(1Н-бензотриазол-1ил) , , , - те траметилуронийтетрафторбората (ТВТU) (0,52 г, 1,6´10-3 моля) и диизопропилэтиламина (0,5 мл) в 6 мл безводного N,N-диметилформамида перемешивали в атмосфере азота при комнатной температуре. Добавляли 2,3-дидезоксиинозина (0,19 г, 0,8´10-3 моля), и реакци онную смесь перемешивали в течение 60 часов. Растворитель удаляли под высоким ваккумом, и остаток растворяли в хлороформе и промывали водой. Высушенную (MgSO4) органическую фазу концентрировали, и сырой продукт очищали на колонке силикагеля, с которой элюировали с помощью 15% метанола в хлороформе. Гомогенные фракции выпаривали с получением 0,25 г (62%) вещества , указанного в заголовке, в виде слегка коричневатого воскоподобного твердого вещества. 1 ЯМР (ДМСО-d6 , 300 МГц) d: 12,35 (1Н, S, Н), 8,22 (1Н, S, Н-2), 8,05 (1Н, S, Н-8), 6,25 (1Н, dd, Н-1’), 5,32 (2Н, m, СН=СH), 4,35-4,10 (3H, m, Н-4’ и Н-5’), 2,45 (2H, m, H-2’), 2,25 (2Н, m, СH2-СОО), 2,10 (2Н, m, H-3’), 1,95 (4H, m , CH 2-C=), 1,45 (2Н, m, CH2C-COO), 1,25 (20H, m, CH 2), 0,85 (ЗН, t, CH3-C). 13 C ЯМР (ДМСO-d6, 75 МГц) d: 172,65 (COO), 156,57 (C-6), 147,63 (C-3), 145,62 (C-2), 138,07 (C-8), 129,98 (C=C), 124,38 (C-5), 84,33 (С-1’), 78,54 (C-4’), 64,91 (C-5’), 33,25, 31,94, 31,36, 28,99, 28,93, 28,84, 28,71, 28,50, 28,39, 28,32, 24,34, 22,09 (CH2), 31,27, 26,00 (CH 2’ и 3’), 13,88 (CH3). Пример 6 (S)-9-(2-гидрокси-З-О-|9’-транс-октадеценоил|пропил)-аденин К раствору (S)-9-(2,3-дигидроксипропил)аденина (0,8 г, 3,8´10-3 моля), транс-9-октадеценоиновой кислоты (1,2 г, 4,25´10-3 моля) и N,N-диметиламинопиридина (50 мг) в 20 мл безводного N,N-диметилформамида добавляли дициклогексилкарбодиимид (0,83 г, 4,0´10-3 моля), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 40 часов. Растворитель выпаривали под высоким вакуумом и остаток очищали с помощью повторной хроматографии на колонке силикагеля с использованием 5-15% метанола в хлороформе в качестве системы элюентов. Один набор гомогенных фракций концентрировали с получением 0,7 г (38%) вещества, указанного в заголовке в виде твердого белого вещества. 1 ЯМР (ДМСО-d6 , 300 МГц) d: 8,12 (1Н, S , Н-8), 8,05 (1Н, S, Н-2), 7,20 (2H, brS, NH2), 5,48 (1H, d, ОН 2’), 5,35 (2Н, m, СН=СН), 4,3 (1Н, m, Н-1’), 4,15 (ЗН, m, Н-3’ и Н-1’), 3,95 (1Н, m, Н-2’), 2,25 (ЗН, t, СН2-СОО), 1,95 (4Н, m, СH2-С=), 1,45 (2Н, m, СН2-С-СОО), 1,25 (20Н, m, СH2), 0,85 (ЗН, t, CH3C). 13 С ЯМР (ДМСО-d6, 75 МГц) d: 172,69 (COO), 155,94 (C-6), 152,30 (C-2), 149,65 (C-4), 141,48 (C-8), 130,02 (CH=CH), 118,57 (C-5), 66,52 (C-2’), 65,44 (С-3’), 46,10 (C-1’), 33,48, 31,98, 31,31, 29,02, 28,87, 28,75, 28,60, 28,52, 23,40, 24,37, 22,13 (CH2), 13,93 (CH3). Из другого набора фракций получали 0,35 г (20%) изомерного продукта (з-9-(3-гидрокси-2-О|9’-транс-октдеценoил|-пропил)-аденин. 1 ЯМР (ДМСО-d6, 300 МГц) d: 8,12 (1H, S, Н-8), 8,05 (1H, S, H-2), 7,20 (2H, br S, NH2), 5,35 (2Н, m, СH=СH), 5,12 (1H, t, ОН 3’), 4,35 (1Н, m, Н-2’), 4,3 (1Н, m, Н-1’), 4,05 (1Н, m, Н-1’), 3,5 (2Н, m, Н-3’), 2,15 (2Н, t, CH2-COO), 1,95 (4Н, m, CH2-C=), 1,45 (2H, m, СH2-С-СОО), 1,25 (20Н, m, CH3), 0,85 (3Н, t, CH3-C). 9 41361 13 C ЯМР (ДМСО-d6, 75 МГц) d: 172,17 (COO), 155,96 (C-6), 152,41 (C-2), 149,78 (C-4), 141,11 (C-8), 130,02 (CH=CH), 118,52 (C-5), 72,26 (C-2), 60,16 (C-3), 43,21 (C-1 ), 33,49, 31,99, 31,32, 29,02, 28,88, 28,75, 28,53, 28,35, 24,21, 22,13 (CH2), 13,93 (CH3). Пример 7 5’-О-(транс-9"-октадеценоил)-5-этенил-1-b-Dрибофуранозилимидазол-4-карбоксамид К раствору 5-этенил-1-b-D-рибофуранозилимидазол-4-карбоксамида (EICAR) (0,10 г, 0,37´ x10-3 моля) в 5 мл безводного N,N-диметилформамида и 5 мл пиридина добавляли 1 мл концентрированного раствора транс-9-октадеценоилхлорида (0,13 г, 0,45´10-3 моля) в 3 мл дихлорметана, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре. Оставшийся концентрированный раствор добавляли порциями по 1 мл через, примерно, 3-часовые интервалы. После общего времени реакции, равного 50 часам, растворитель выпаривали под высоким вакуумом и продукт очищали на колонке силикагеля с 15% метанола в хлороформе в качестве системы элюентов. Гомогенные фракции выпаривали с получением 30 мг (15%) вещества, указанного в заголовке в виде твердого белого вещества. 1 ЯМР (ДМСО-d6, 300 МГц) d: 8,05 (1Н, S, Н-2), 7,35 и 7,25 (2Н, S+S, H2), 5,65 (1Н, d, Н-1’), 5,6 (1Н, br d, ОН-2’), 5,35 (3Н, m, СН=СН и ОН-3’), 4,91 (1H, S, CºCH), 4,3 (1H, m, H-2’), 4,2 (2Н, m, H-5’), 4,05 (2H, m, H-3’ и Н-4’), 2,30 (2Н, t, CH2COO), 1,95 (4H, m, CH 2-C=), 1,45 (2H, m, CH 2-C-СОО), 1,25 (20H, m, CH2), 0,85 (3Н, t, CH3). 13 C ЯМР (ДМСО-d6, 75 МГц) d: 172,76 (COO), 162,57 (CO H2), 139,80 (C-5), 135,82 (C-2), 130,08 (CH=CH), 115,51 (C-4), 91,39 (-Cº), 88,75 (С-1’), 81,78 (C-4’), 73,91 (C-2’), 71,46 (ºCH), 70,09 (C-3’), 63,57 (C-5’), 33,66, 31,95, 31,29, 29,00, 28,84, 28,72, 28,55, 28,50, 28,42, 28,36, 24,49, 22,12 (CH2), 13,97 (CH3). Следуя процедуре из примеров 1-7, могут быть получены дополнительные соединения формулы 1 из исходных материалов, указанных далее: Таблица B-S-OH+FaX ¾¾¾¾¾ B-S-OFa (1) (2) (3) Пример № 8 9 10 11 12 13 14 15 Реагент (1) Реагент (2) 2-b-D-рибофуранозилселеназол-4карбоксамид 4-гидрокси-3-b-Dрибофуранозилпиразол-5-карбоксамид Олеиновая кислота Олеиновая кислота Элаидиновая кислота Элаидиновая кислота цис-11-эйкозеноиновая кислота 2’-дезоксикоформицин Продукт (3) 2-(5’-О-олеил-b-D-рибофуранозол-4карбоксамид 4-гидрокси-3-(5’-О-олеил-b-D-рибофуранозил)-пиразол-5-карбоксамид 5’-О-элаидил-2’-дезоксикоформицин 5’-О-элаидил-2’-дезокси-2’-фтор-5иод-1-b-D-арабинофуранозилцитозин 5’-О-(цис-11-эйкозеноил)-(Е)-5-(2(Е)-5-(2-бромвинил)-2’-дезокси-уридин бромвинил)-2’-дезоксиуридин 1-b-D-арабинофуранозил-(Е)-5-(2Олеиновая кислота 5’-О-(олеоил)-1-b-D-арабинофурабромвинил)урацил нозил-(Е)-5-(2-бромвинил)урацил 9-(4-гидроксиметил-2-циклопентенил)9-(4-олеоилметил-2-циклопентенил)Олеиновая кислота гуанин гуанин транс-112’-дезокси-2’-фтор-5-О-(транс-11"2’-дезокси-2’-фтор-1-b-D-арабинофураэйкозеноиновая эйкозеноил)-1-b-D-арабинофуранозилнозил-1,2,4-триазол-3-карбоксиамид кислота 1,2,4-триазол-3-карбоксамид 2’-дезокcи-2’-фтор-5-иод-1-b-D-арабинофуранозилцитозин Замечено, что может быть получено больше продуктов по всем возможным перестановкам с помощью реакции соединений, названных в качестве реагента (1) и реагента (2), например, 5’-О олеил-2’-дезоксикоформицин, 5’-О-(цис-11"-эйкозеноил)-2’-дезоксиноформицин и 5’-О-(транс-11"эйкозеноил)-2’-дезоксиноформицин. 10 Фиг. 1 41361 11 Фиг. 2 41361 12 Фиг. 3 41361 13 41361 Фиг. 4 14 41361 Фиг. 4 (продолжение) 15 41361 Фиг. 4 (продолжение) 16 41361 Фиг. 4 (продолжение) 17 41361 Фиг. 4 (продолжение) 18 41361 Фиг. 4 (продолжение) 19 41361 Фиг. 4 (продолжение) 20 41361 Фиг. 4 (продолжение) 21 41361 Фиг. 4 (продолжение) 22 41361 Фиг. 4 (продолжение) 23 41361 Фиг. 4 (продолжение) 24 41361 Фиг. 4 (продолжение) 25 41361 Фиг. 4 (продолжение) 26 41361 Фиг. 4 (продолжение) 27 41361 __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 50 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 28
ДивитисяДодаткова інформація
Назва патенту англійськоюMonoether compounds and a pharmaceutical composition for the treatment of viral infections
Автори англійськоюBORRETZEN BERN, Dalen Are, MYHREN FINN, STOKKE KJELL TORGEIR
Назва патенту російськоюМоноэфирные соединения и фармацевтическая композиция для лечения вирусных инфекций
Автори російськоюБерретсен Бернт, Дален Аре, Мюхрен Финн, Стокке Хелль Тургеир
МПК / Мітки
МПК: C07H 19/16, A61K 31/7042, A61K 31/7076, A61K 31/70, C07H 19/04, A61P 31/12, C07H 19/056, A61K 31/7064, A61K 31/708, A61K 31/7072, A61K 31/7068, C07H 19/06, C07H 19/052, A61K 31/7052
Мітки: лікування, композиція, вірусних, моноефірні, фармацевтична, інфекцій, сполуки
Код посилання
<a href="https://ua.patents.su/28-41361-monoefirni-spoluki-ta-farmacevtichna-kompoziciya-dlya-likuvannya-virusnikh-infekcijj.html" target="_blank" rel="follow" title="База патентів України">Моноефірні сполуки та фармацевтична композиція для лікування вірусних інфекцій</a>
Фармацевтична композиція для лікування вірусних інфекцій, спосіб отримання композиції, спосіб лікування або профілактики віл-інфекції у людини

Номер патенту: 29429
Опубліковано: 15.11.2000
Автори: Фурман Філліп Аллен, Пейнтер Джордж Роберт
МПК: A61K 31/70, A61K 31/505
Мітки: профілактики, композиції, композиція, спосіб, фармацевтична, віл-інфекції, вірусних, інфекцій, людини, лікування, отримання
Текст:
...агентом /например.натрий гликоллат крахмала, сшитый по видон, сши тая натрий карбоксиметил целлюлоза/, поверхностно-активными или диспергирующим агентом. Формованные таблетки могут быть изготовлены при помощи формования смеси порошкообразного соединения, увлажненной инертным жидким разбавителем, при помоши соответствующего средства. Таблетки могут быть покрыты или на них может быть нанесено рифление,и им могут быть приданы...
Похідні індолу, проміжні сполуки, фармацевтична композиція та спосіб лікування

Номер патенту: 39866
Опубліковано: 16.07.2001
Автори: Новаковскі Йоланта Т., Мейкор Джон Е.
МПК: A61P 9/12, A61P 25/20, A61P 25/24, A61K 31/425, A61P 43/00, A61P 25/26, A61P 25/04, C07D 417/14, C07D 417/12, C07D 413/14, A61K 31/42, A61P 3/04, C07D 413/12
Мітки: похідні, композиція, індолу, лікування, спосіб, сполуки, фармацевтична, проміжні
Формула / Реферат:
1. Производные индола общей формулы I:, (I)где А - прямая связь или С1-С4-алкил, R1 - водород или С1-С6-алкил, W, X, Y, Z каждый, независимо, представляет кислород, серу, азот или углерод при условии, что, по меньшей мере, один из W, X, Y или Z является азотом, один из R2, R3, R4 и R5 представляет С1-С6-алкил, фенил, С1-С3-алкилфенил, а другие...
Спосіб лікування вірусних респіраторних інфекцій

Номер патенту: 19158
Опубліковано: 25.12.1997
Автори: Овчарєнко Алєксандр Валєнтіновіч, Жірнов Олєг Пєтровіч
МПК: A61P 11/00, A61K 38/57
Мітки: спосіб, респіраторних, вірусних, лікування, інфекцій
Формула / Реферат:
1. Способ лечения вирусных респираторнмх инфекций, включающий воздействие на организм ингибиторами протеиназ, отличающийся тем, что осуществляют непосредственное воздействие на инфекционный процесс через респираторный тракт аэрозолем ингибиторов протеиназ, полученным распылением сухого вещества ингибиторов или их водного раствора, содержащего активное вещество в концентрации не менее 300 калликреинингибирующих единиц на мл.2. Способ по...
Спосіб профілактики і лікування гострих респіраторних вірусних інфекцій у дітей

Номер патенту: 12354
Опубліковано: 02.12.1996
Автори: Осинська Лариса Феліксівна, Лук'янова Олена Михайлівна, Антіпкін Юрій Генадієвич, Чернишев Віктор Павлович, Омельченко Людмила Іванівна, Починок Тетяна Вікторівна, Шнідерманн Габріела, Хеуссер Пітер
МПК: A61K 45/00
Мітки: лікування, респіраторних, профілактики, інфекцій, гострих, дітей, вірусних, спосіб
Формула / Реферат:
Способ профилактики и лечения острых респираторных вирусных инфекций у детей путем использования иммуностимулирующих препаратов растительного происхождения, отличающийся тем, что для детей часто болеющих используют препарат искадор растительного происхождения, который вводят в область лопатки два раза в неделю с интервалом в два дня по схеме: первая неделя - 0,001 мг, вторая - 0,01 мг, третья - 0,1 мг, четвертая - 1 мг, пятая - 5 мг.
Похідні хінолон- і нафтиридонкарбонової кислоти у вигляді суміші ізомерів або окремих ізомерів, фармацевтична композиція, що має антибактеріальну активність та проміжні сполуки

Номер патенту: 35554
Опубліковано: 16.04.2001
Автори: ФІЛІПС Томас, ГРОХЕ Клаус, ХАЛЛЕР Інго, БРЕММ Клаус Дітер, КРЕБС Андреас, ШЕНКЕ Томас, ПЕТЕРСЕН Уве, Ендерманн Райнер, МЕТЦГЕР Карл-Георг
МПК: C07D 519/00, C07D 498/04, C07D 471/04
Мітки: похідні, сполуки, антибактеріальну, фармацевтична, активність, нафтиридонкарбонової, ізомерів, окремих, має, суміші, вигляді, кислоти, композиція, проміжні, хінолон
Формула / Реферат:
1. Производные хинолон- и нафтиридонкарбоновой кислоты общей формулы (I),(І)где:А = СН, CF, CCl, С-ОСН3, N,X1 = Н, Hal, NH2, СН3,R1 = С1-С3-алкил, циклопропил, фенил, который может быть от одного до трех раз замещен галогеном, илиА и R1 вместе означают С-О-СН2-СН(СН3)-,R2 = Н или С1-С3-алкил,В = остаток...
Попередній патент: Безголковий шприц і спосіб терапевтичного лікування із використанням безголкового шприца (варіанти)
Наступний патент: Біоцидна композиція, спосіб її одержання і спосіб запобігання грибковому або водоростевому ураженню матеріалів
Випадковий патент: Пристрій для хірургічного лікування аліментарно-конституціонального ожиріння