Глюкопіранозилзаміщені похідні бензонітрилу
Номер патенту: 94087
Опубліковано: 11.04.2011
Автори: Зауер Ахім, Хіммельсбах Франк, Айкельманн Петер, Томас Лео, Екхардт Маттіас








Формула / Реферат
1. Глюкопіранозилзаміщена похідна бензонітрилу формули І
 , І
, І
у якій
R3 означає метоксигрупу, етоксигрупу, ізопропоксигрупу, циклобутоксигрупу, циклопентоксигрупу, циклогексоксигрупу, (S)-тетрагідрофуран-3-ілоксигрупу, (R)-тетрагідрофуран-3-ілоксигрупу, тетрагідропіран-4-ілоксигрупу або 1-ацетилпіперидин-4-ілоксигрупу,
або її похідна, в якій одна або більша кількість гідроксигруп β-D-глюкопіранозильної групи ацилована групами, вибраними з (С1-С18-алкіл)-карбонілу, (С1-С18-алкіл)-оксикарбонілу, фенілкарбонілу і феніл-(С1-С3-алкіл)-карбонілу;
включаючи їх таутомери, стереоізомери і їх суміші; і їх фізіологічно прийнятні солі.
2. Глюкопіранозилзаміщена похідна бензонітрилу за п. 1, яка відрізняється тим, що її вибирають з групи, що включає:
(1) 4-(β-D)-глюкопіраноз-1-іл)-2-[4-((S)-тетрагідрофуран-3-ілокси)-бензил]-бензонітрил,
(2) 4-(β-D)-глюкопіраноз-1-іл)-2-[4-((R)-тетрагідрофуран-3-ілокси)-бензил]-бензонітрил,
(3) 2-(4-циклопентилоксибензил)-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(4) 2-(4-циклобутоксибензил)-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(5) 2-(4-циклогексоксибензил)-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(6) 2-[4-(тетрагідропіран-4-ілокси)-бензил]-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(7) 2-[4-(1-ацетилпіперидин-4-ілокси)-бензил]-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(8) 2-(4-метоксибензил)-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(9) 2-(4-етоксибензил)-4-(β-D-глюкопіраноз-1-іл)-бензонітрил,
(10) 4-(β-D-глюкопіраноз-1-іл)-2-(4-ізопропоксибензил)-бензонітрил.
3. Глюкопіранозилзаміщена похідна бензонітрилу за п. 1 або 2 у формі її фізіологічно прийнятних солей з неорганічними або органічними кислотами.
Текст
1. Глюкопіранозилзаміщена похідна бензонітрилу формули І C2 2 UA 1 Дійсний винахід відноситься до глюкопіранозилзаміщених похідних бензонітрилу загальної формули І 3 у якій група R3 визначена нижче в дійсному винаході, включаючи їх таутомери, стереоізомери, їх суміші і їх солі. Дійсний винахід також відноситься до фармацевтичних композицій, що містять сполуку формули І, пропоновану в дійсному винаході, а також до застосування сполуки, пропонованої в дійсному винаході, для приготування фармацевтичної композиції, призначеної для лікування або попередження метаболічних порушень. Крім того, дійсний винахід відноситься до способів приготування фармацевтичної композиції, а також сполуки, пропонованої в дійсному винаході. У літературі сполуки, які надають інгібуючу дію на натрійзалежний співпереносник глюкози SGLT2, запропоновані для лікування захворювань, переважно - діабету. Глюкопіранозилзаміщені ароматичні групи і їх одержання і їх можлива активність як інгібіторів SGLT2 описані в заявці WO 2005/092877 і в цитованих в ній публікаціях. Завданням дійсного винаходу є одержання нових глюкопіранозилзаміщених похідних бензонітрилу, переважно таких, які активні по відношенню до натрійзалежного співпереносника глюкози SGLT, переважно - SGLT2. Іншим завданням дійсного винаходу є одержання глюкопіранозилзаміщених похідних бензолу, які надають значнішу інгібуючу дію на натрійзалежний співпереносник глюкози SGLT2 in vitro і/або in vivo, ніж відомі структурно схожі сполуки, і/або володіють кращими фармакологічними або фармакокінетичними характеристиками. Іншим завданням дійсного винаходу є одержання нових фармацевтичних композицій, які застосовні для попередження і/або лікування метаболічних порушень, переважно - діабету. Інші завдання дійсного винаходу стануть зрозумілі фахівцеві з приведених вище і нижче даних. Першим об'єктом дійсного винаходу є глюкопіранозилзаміщені похідні бензонітрилу формули І у якій R3 означає водень, фтор, хлор, бром, йод, метил, етил, пропіл, ізопропіл, бутил, втор-бутил, ізобутил, трет-бутил, 3-метилбут-1-іл, циклопропіл, циклобутил, циклопентил, циклогексил, дифторометил, трифторометил, пентафтороетил, 2гідроксіетил, гідроксиметил, 3-гідроксипропіл, 2гідрокси-2-метилпроп-1-іл, 3-гідрокси-3-метилбут1-іл, 1-гідрокси-1-метилетил, 2,2,2-трифторо-1гідрокси-1-метилетил, 2,2,2-трифторо-1-гідрокси-1трифторометилетил, 2-метоксіетил, 2-етоксіетил, гідроксигрупу, метилоксигрупу, етилоксигрупу, ізопропілоксигрупу, дифторометилоксигрупу, трифторометилоксигрупу, циклобутилоксигрупу, циклопентилоксигрупу, циклогексилоксигрупу, (S)тетрагідрофуран-3-ілоксигрупу, (R)тетрагідрофуран-3-ілоксигрупу, тетрагідропіран-4ілоксигрупу, 1-ацетилпіперидин-4-ілоксигрупу, 2метилоксіетилоксигрупу, метилсульфаніл, метил 94087 4 сульфініл, метилсульфоніл, етилсульфініл, етилсульфоніл, триметилсиліл або ціаногрупу, або його похідна, в якій одна або більша кількість гідроксигруп β-D-глюкопіранозильної групи ацилують групами, вибраними з групи, що включає (С1-C18-алкіл)-карбоніл, (С1-C18-алкіл)оксикарбоніл, фенілкарбоніл і феніл-(С1-С3-алкіл)карбоніл; включаючи їх таутомери, стереоізомери і їх суміші; і їх фізіологічно прийнятні солі. Сполуки, пропоновані в дійсному винаході, і їх фізіологічно прийнятні солі володіють цінними фармакологічними характеристиками, зокрема, надають інгібуючу дію на натрійзалежний співпереносник глюкози SGLT, переважно - SGLT2. Крім того, сполуки, пропоновані в дійсному винаході, можуть надавати інгібуючу дію на натрійзалежний співпереносник глюкози SGLT1. В порівнянні з можливою інгібуючою дією на SGLT1 сполуки, пропоновані в дійсному винаході, переважно селективно інгібують SGLT2. Дійсний винахід також відноситься до фізіологічно прийнятних солей сполук, пропонованих в дійсному винаході, з неорганічними або органічними кислотами. Дійсний винахід також відноситься до фармацевтичних композицій, що містять щонайменше одну сполуку, пропоновану в дійсному винаході, або фізіологічно прийнятну сіль, пропоновану в дійсному винаході, необов'язково спільно з одним або більшою кількістю інертних носіїв і/або розчинників. Дійсний винахід також відноситься до застосування щонайменше однієї сполуки, пропонованої в дійсному винаході, або її фізіологічно прийнятної солі для приготування фармацевтичної композиції, яка застосовна для лікування або попередження захворювань або патологічних станів, на які можна впливати шляхом інгібування натрійзалежного співпереносника глюкози SGLT, переважно SGLT2. Дійсний винахід також відноситься до застосування щонайменше однієї сполуки, пропонованої в дійсному винаході, або її фізіологічно прийнятної солі для приготування фармацевтичної композиції, яка застосовна для лікування одного або більшої кількості метаболічних порушень. Іншим об'єктом дійсного винаходу є застосування щонайменше однієї сполуки, пропонованої в дійсному винаході, або однієї з її фізіологічно прийнятних солей для приготування фармацевтичної композиції, призначеної для попередження дегенерації бета-клітин панкреатичних острівців і/або для поліпшення і/або відновлення функціональної активності бета-клітин панкреатичних острівців. Іншим об'єктом дійсного винаходу є застосування щонайменше однієї сполуки, пропонованої в дійсному винаході, або однієї з її фізіологічно прийнятних солей для приготування фармацевтичної композиції, призначеної для попередження, уповільнення, затримання або лікування захворювань або патологічних станів, обумовлених аномальним накопиченням жиру в печінці, у пацієнта, що потребує його. 5 Дійсний винахід також відноситься до застосування щонайменше однієї сполуки, пропонованої в дійсному винаході, або її фізіологічно прийнятної солі для приготування фармацевтичної композиції, призначеної для інгібування натрійзалежного співпереносника глюкози SGLT, переважно -SGLT2. Дійсний винахід також відноситься до способу приготування фармацевтичної композиції, пропонованої в дійсному винаході, що характеризується тим, що сполуку, пропоновану в дійсному винаході, або одну з її фізіологічно прийнятних солей включають в один або більшу кількість інертних носіїв і/або розчинників за допомогою нехімічної методики. Дійсний винахід також відноситься до способу одержання сполук загальної формули І, пропонованих в дійсному винаході, що характеризується тим, що а) для одержання сполук загальної формули І, які визначені вище і нижче в дійсному винаході, сполука загальної формули II у якій R' означає Н, С1-С4-алкіл, (С1-C18-алкіл)карбоніл, (С1-C18-алкіл)-оксикарбоніл, арилкарбоніл і арил-(С1-С3-алкіл)-карбоніл, де алкільні або арильні групи можуть бути моно- або полізаміщені галогеном; R8a, R8b, R8c, R8d незалежно один від одного означають водень або алільну групу, бензильну групу, (С1-С4-алкіл)-карбоніл, (С1-С4-алкіл)оксикарбоніл, арилкарбоніл, арил-(С1-С3-алкіл)карбоніл і арил-(С1-С3-алкіл)-оксикарбоніл або групу RaRbRcSi, або кетальну або ацетальну групу, переважно - алкіліденову або арилалкіліденову кетальну або ацетальну групу, де в кожному випадку дві сусідні групи R8a, R8b, R8c, R8d можуть утворити циклічну кетальну або ацетальну групу або 1,2-ді-(С1-С3-алкокси)-1,2-ді-(С1-С3-алкіл)етиленовий місток, де вказаний вище етиленовий місток разом з двома атомами кисню і двома зв'язаними з ними атомами вуглецю піранозного кільця утворюють заміщене діоксанове кільце, переважно - 2,3-диметил-2,3-ді-(С1-С3-алкокси)-1,4діоксанове кільце, і де алкільні, алільні, арильні і/або бензильні групи можуть бути моно- або полізаміщені галогеном або С1-С3-алкоксигрупою, і де бензильні групи також можуть бути заміщені ді-(С1С3-алкіл)-аміногрупою; і Ra, Rb, Rc незалежно один від одного означають С1-С4-алкіл, арил або арил- С1-С3-алкіл, де арильні або алкільні групи можуть бути моно- або полі заміщені галогеном; де арильні групи, вказані у визначенні відмічених вище груп, означають фенільні або нафтильні групи, переважно - фенільні групи; і в якій група R3 визначена вище і нижче в дійсному винаході; вводять в реакцію з відновним реагентом у присутності кислоти Льюїса або Бренстеда, і будь 94087 6 які захисні групи, що містяться, відщеплюють одночасно або послідовно; або b) для одержання сполук загальної формули І, сполука загальної формули III у якій R8a, R8b, R8c, R8d і R3 визначені вище і нижче в дійсному винаході, за умови, що щонайменше один замісник вибраний з групи, що включає R8a, R8b, R8c, R8d не означає водень; захисні групи R8a, R8b, R8c, R8d, що не означають водень, відщеплюють; і при необхідності одержана таким чином сполука загальної формули І шляхом ацилування перетворюють на відповідний ацил загальної формули І, і/або при необхідності відщеплюють будь-яку захисну групу, що використовується в описаних вище реакціях, і/або при необхідності одержану таким чином сполуку загальної формули І розділяють на її стереоізомери і/або при необхідності одержану таким чином сполуку загальної формули І перетворюють на її солі, для застосування у фармацевтиці переважно - в її фізіологічно прийнятні солі. Іншим об'єктом дійсного винаходу є нові проміжні продукти, описані на схемах реакцій і в експериментальній частині нижче в дійсному винаході. Об'єктами дійсного винаходу, зокрема, сполуки, фармацевтичні композиції і їх застосування, є глюкопіранозилзаміщені похідні бензонітрилу загальної формули І, визначені вище і нижче в дійсному винаході, або їх похідні, включаючи їх таутомери, стереоізомери і їх суміші, і їх фізіологічно прийнятні солі. Переважно, якщо всі гідроксигрупи β-Dглюкопіранозильної групи є незаміщеними або лише гідроксигрупа атома O-6 β-Dглюкопіранозильної групи заміщена, як це визначено. Переважні замісники вибрані з групи, що включає (С1-С8-алкіл)-карбоніл, (С1-С8-алкіл)оксикарбоніл і фенілкарбоніл. Ще переважніші замісники вибрані з групи, що включає ацетил, метоксикарбоніл і етоксикарбоніл, переважніше ацетил і етоксикарбоніл. Позначення в структурних формулах, що використовуються вище і нижче в дійсному винаході, в яких зв'язок замісника циклічної групи, такий як, наприклад, фенільне кільце, направлений до центру циклічної групи, якщо не вказане інше, означає, що замісник може бути приєднаний в будь-якому вільному положенні циклічної групи, що містить атом Н. Сполуки, пропоновані в дійсному винаході, можна отримати за методиками синтезу, які в цілому відомі. Сполуки переважно отримують за приведеними нижче методиками, пропонованими в дійсному винаході, які детальніше описані нижче в дійсному винаході. 7 Похідні глюкози формули II, пропоновані в дійсному винаході, можна синтезувати з Dглюконолактону або його похідної шляхом приєд 94087 8 нання необхідного бензилбензолу у формі металоорганічної сполуки (схема 1). Схема 1: Приєднання металоорганічної сполуки до глюконолактону Реакцію, представлену на схемі 1, переважно проводять з застосуванням як вихідної речовини галогенованого бензилбензолу загальної формули IV, в якій Hal означає хлор, бром або йод. R1 на схемі 1 означає ціаногрупу або групу, яку потім можна перетворити на ціаногрупу, таку як хлор, бром, карбоксигрупу, етерифіковану карбоксигрупу, карбоксамідну групу або її похідну, боронову або силільну групу, захищену або замасковану альдегідну групу, таку як, наприклад, ацетальну або тіазольну, або захищену або замасковану аміногрупу, таку як, наприклад, нітрогрупу. Реагент Гриньяра або літійвмісний реагент на основі бензилбензолу (V) можна отримати з відповідного хлорованого, бромованого або йодованого бензилбензолу IV за допомогою так званої реакції обміну галоген-метал або шляхом впровадження металу по зв'язку вуглець-галоген. Обмін галогенметал для синтезу відповідної літієвої сполуки V можна провести, наприклад, з літійорганічною сполукою, такою як, наприклад, н-, втор- або третбутиллітій. Аналогічну магнієву сполуку також можна отримати за допомогою обміну галоген-метал з підходящим реагентом Гриньяра, таким як, наприклад, ізопропіл- або втор-бутилмагнійбромід або -хлорид, або діізопропіл- або ди-вторбутилмагній без присутності або у присутності додаткової солі, такої як, наприклад, хлорид літію, який може прискорювати металювання; конкретну переметалюючу магнійорганічну сполуку також можна отримати in situ з підходящих попередників (див., наприклад, Angew. Chem. 2004, 116, 33963399 і Angew. Chem. 2006, 118, 165-169 і цитовані в них посилання). Крім того, також можна використовувати комплекси магнійорганічних сполук, одержані, наприклад, об'єднанням бутилмагнійхлориду або -броміду або ізопропілмагнійхлориду або броміду і бутиллітію (див., наприклад, Angew. Chem. 2000, 112, 2594-2596 і Tetrahedron Lett. 2001, 42, 4841-4844 і цитовані в них посилання). Реакції обміну галоген-метал переважно проводять при температурі від 40 до -100°С, особливо переважно - від 10 до -80°С, в інертному розчиннику або в їх сумішах, таких як, наприклад, діети ловий ефір, діоксан, тетрагідрофуран, толуол, гексан, диметилсульфоксид, дихлорометан або їх суміші. Одержані таким чином магнієві або літієві похідні сполук необов'язково можна переметалювати солями металів, такими як, наприклад, трихлорид церію, хлорид або бромід цинку, хлорид або бромід індію, з утворенням альтернативних металоорганічних сполук (V), придатних для приєднання. Альтернативно, металоорганічну сполуку V також можна отримати шляхом впровадження металу по зв'язку вуглець-галоген гетероароматичної сполуки IV. Елементарними металами, підходящими для цього перетворення, є літій і магній. Впровадження можна провести в розчинниках, таких як, наприклад, діетиловий ефір, діоксан, тетрагідрофуран, толуол, гексан, диметилсульфоксид і їх суміші, при температурі в діапазоні від -80 до 100°С, переважно - від -70 до 40°С. У разі, коли до активації металу самовільна реакція не протікає, може бути потрібна активація, така як, наприклад, обробка 1,2-дибромоетаном, йодом, триметилсилілхлоридом, оцтовою кислотою, хлористоводневою кислотою і/або обробка ультразвуком. Приєднання металоорганічної сполуки V до глюконолактону або його похідних (VI) переважно проводять при температурі від 40 до -100°С, особливо переважно - від 0 до -80°С, у інертному розчиннику або в їх сумішах і отримують сполуку формули II. Всі вказані вище реакції можна провести на повітрі, хоча переважне проведення в атмосфері інертного газу, такого як аргон і азот. Металювання і/або реакцію сполучення також можна провести в мікрореакторах і/або мікрозмішувачах, в яких забезпечуються високі швидкості обміну; наприклад, аналогічно способам, описаним в WO 2004/076470. Розчинниками, підходящими для приєднання метальованої фенільної групи V до відповідним чином захищеного глюконолактону VI, є, наприклад, діетиловий ефір, диметоксіетан, бензол, толуол, метиленхлорид, гексан, тетрагідрофуран, діоксан, N-метилпіролідон і їх суміші. Реакції приєднання можна провести без додавання яких-небудь допоміжних речовин або в разі повільний взаємодіючих компонентів реакції спо 9 94087 лучення - у присутності промотору, такого як, наприклад BF3OEt2 або Me3SiCl (див. М. Schlosser, Organometallics in Synthesis, John Wiley & Sons, Chichester/New York/Brisbane/Toronto/Singapore, 8 1994). Переважними значеннями замісників R на схемі 1 є бензил, заміщений бензил, аліл, триалкілсиліл, особливо переважно - триметилсиліл, триізопропілсиліл, аліл, 4-метоксибензил і бензил. Якщо два сусідніх замісники R8 зв'язані один з одним, то переважно, якщо ці два замісники є частиною бензиліденацеталю, 4метоксибензиліденацеталю, ізопропілкеталю або утворюють діоксан з 2,3-диметоксибутиленом, який по положеннях 2 і 3 бутану зв'язаний з сусідніми атомами кисню піранози. Група R' переважно означає водень, С1-С4-алкіл, С1-С4-алкілкарбоніл або С1-С4-алкоксикарбоніл, особливо переважно водень, метил або етил. Групу R' вводять після приєднання металоорганічної сполуки V або її похідної до глюконолактону VI. Якщо R' означає водень або С1-С4-алкіл, то розчин реакційної суміші обробляють спиртом, таким як, наприклад, метанол або етанол, або водою у присутності кислоти, такої як, наприклад, оцтова кислота, метансульфонова кислота, толуолсульфонова кислота, сірчана кислота, трифторооцтова кислота або хлористоводнева кислота. R' також можна приєднати після одержання воденьвмісної сполуки II за реакцією аномерної гідроксигрупи з підходящим електрофільним реагентом, таким як, наприклад, метилйодид, диметилсульфат, етилйодид, діетилсульфат, ацетилхлорид або оцтовий ангідрид, у присутності основи, такої як, наприклад, триетиламін, етилдіізопропіламін, карбонат натрію 10 або калію, або цезію, гідроксид натрію, або калію, або цезію. Гідроксигрупу також можна депротонувати до додавання електрофільного реагенту, наприклад, за допомогою гідриду натрію. Під час 8 введення R' захисні групи R можуть відщепитися, якщо вони нестабільні за умов проведення реакції, що використовуються, що приводить до відповідної протонованої сполуки, тобто до сполуки II, в якій R8 означає Н. Синтез галогенароматичної сполуки формули IV можна провести за стандартними методиками перетворень в органічній хімії або щонайменше за методиками, приведеними в спеціальній літературі з органічного синтезу (див., наприклад, J. March, Advanced Organic Reactions, Reactions, Mechanisms, and Structure, 4th Edition, John Wiley & Sons, Chichester/New York/Brisbane/Toronto/Singapore, 1992 і цитовану в ній літературу). Крім того, застосування перехідних металів і металоорганічних сполук для синтезу ароматичних сполук детально описане в різних монографіях (див., наприклад, L. Brandsma, S.F. Vasilevsky, H.D. Verkruijsse, Application of Transition Metal Catalysts in Organic Synthesis, SpringerVerlag, Berlin/Heidelberg, 1998; M. Schlosser, Organometallics in Synthesis, John Wiley & Sons, Chichester/New York/Brisbane/Toronto/Singapore, 1994; P.J. Stang, F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 1997 і цитовані в них посилання). Стратегії синтезу, описані нижче, демонструють це як приклад. Крім того, за допомогою таких же синтетичних підходів агліконовий фрагмент також можна об'єднати з вже наявним піранозним фрагментом. Схема 2: Синтез діарилкетонного фрагменту На схемі 2 представлено одержання попередника, який можна використовувати для синтезу галогенароматичної сполуки формули IV з використанням як вихідної речовини бензоїлхлориду і другої ароматичної групи за допомогою ацилування за Фріделем-Крафтсом і його варіантів. R1 на схемі 2 означає ціаногрупу або групу, яку потім можна перетворити на ціаногрупу, таку як хлор, бром, карбоксигрупу, етерифіковану карбоксигрупу, карбоксамідну групу або її похідну, захищену або замасковану альдегідну групу, таку як, наприклад, тіоацетальну або тіазольну, або захищену або замасковану аміногрупу, таку як, наприклад, нітрогрупу. Цю класичну реакцію проводять з бі льшою кількістю субстратів і зазвичай проводять у присутності каталізатора, який використовують в каталітичних або стехіометричних кількостях, такого як, наприклад, АlСl3, FeCl3, йод, залізо, ZnCl2, сірчана кислота або трифторометансульфонова кислота. Замість бензоїлхлориду також можна використовувати відповідну карбонову кислоту, ангідрид, складний ефір або бензонітрил. Реакції переважно проводять в хлорованих вуглеводнях, таких як, наприклад, дихлорометан і 1,2дихлороетан, при температурі від -30 до 120°С, переважно - від 30 до 100°С. Проте також можливі реакції у відсутності розчинника і реакції в мікрохвильовій печі. 11 94087 12 Схема 3: Відновлення діарилкетонів і діарилметанолів в діарилметани На схемі 3 замісник R означає С1-С3-алкіл або арил і R1 означає ціаногрупу або групу, яку потім можна перетворити на ціаногрупу, таку як хлор, бром, карбоксигрупу, етерифіковану карбоксигрупу, карбоксамідну групу або її похідну, боронову або силільну групу, захищену або замасковану альдегідну групу, таку як, наприклад, ацетальну або тіазольну, або захищену або замасковану аміногрупу, таку як, наприклад, нітрогрупу. З використанням як вихідної речовини діарилкетону або діарилметанолу за одну або дві стадії реакції можна отримати діарилметан. Діарилкетон можна відновити в діарилметан в дві стадії через відповідний діарилметанол або в одну стадію. У двостадійному варіанті кетон відновлюють відновним реагентом, таким як, наприклад, гідрид металу, такий як, наприклад, NaBH4, LiAlH4 або iВu2АlН, і отримують спирт. Одержаний спирт відновним реагентом, таким як, наприклад, Et3SiH, NaBH4 або Ph2SiClH, можна перетворити на шуканий дифенілметан у присутності кислоти Льюїса, такої як, наприклад, BF3-OEt2, ІnСl3 або АlСl3 або кислоти Бренстеда, такої як, наприклад, хлористоводнева кислота, сірчана кислота, трифторооцтова кислота або оцтова кислота. Одностадійну реакцію з використанням як вихідної речовини кетону для одержання дифенілметану можна провести, наприклад, з використанням силану, такого як, наприклад, Et3SiH, борогідриду, такого як, наприклад, NaBH4 або гідриду алюмінію, такого як, LiAlH4, у присутності кислоти Льюїса або Бренстеда, такої як, наприклад, BF3-OEt2, трис(пентафторофеніл)боран, трифторооцтова кислота, хлористоводнева кислота, хлорид алюмінію або ІnСl3. Реакції переважно проводять в розчинниках, таких як, наприклад, галогеновані вуглеводні, такі як, дихлорометан, толуол, ацетонітрил або їх суміші при температурі від -30 до 150°С, переважно - від 20 до 100°С. Відновлення воднем у присутності каталізатора на основі перехідного металу, такого як, наприклад, Pd на деревному вугіллі є іншою можливою методикою синтезу. Також можливе відновлення за Вольфом-Кішнером і його варіантах. Спочатку кетон піддають перетворенню за допомогою гідразину або його похідної, такої як, наприклад, 1,2біс(трет-бутилдиметилсиліл)гідразин, в гідразон, який в сильнолужному середовищі і при нагріванні розкладається з утворенням дифенілметану і азоту. Реакцію можна провести в одну стадію або, після виділення гідразону або його похідної, в дві окремі стадії. Підходящі основи включають, наприклад, КОН, NaOH і KOtBu в розчинниках, таких як, наприклад, етиленгліколь, толуол, ДМСО (диметилсульфоксид), 2-(2-бутоксіетокси)-етанол або трет-бутанол; також можливі реакції у відсутності розчинника. Реакції можна провести при температурі від 20 до 250°С, переважно - від 80 до 200°С. Альтернативою відновленню в лужному середовищі за Вольфом-Кішнером є відновлення за Клеменсеном, яке протікає в кислому середовищі і яке також можна використовувати в дійсному винаході. Гідроксигрупу діарилметанолу також можна спочатку перетворити на групу, що легше відщеплюється, таку як, наприклад, хлорид, бромід, йодид, ацетат, карбонат, фосфат, або сульфат; подальша стадія відновлення з утворенням діарилметану детально описана в літературі органічної хімії. 13 94087 14 Схема 4: Синтез діарилметанового фрагменту і можливих його попередників На схемі 4 R1 означає ціаногрупу або групу, яку потім можна перетворити на ціаногрупу, таку як хлор, бром, карбоксигрупу, етерифіковану карбоксигрупу, карбоксамідну групу або її похідну, боронову або силільну групу, захищену або замасковану альдегідну групу, таку як, наприклад, ацетальну або тіазольну, або захищену або замасковану аміногрупу, таку як, наприклад, нітрогрупу. Термін "Alk" означає С1-С4-алкіл і кожен замісник R незалежно один від одного вибраний з групи, що включає Н, С1-С3-алкіл і С1-С3-алкоксигрупу. На схемі 4 представлений синтез діарилметанів і можливих його попередників з використанням як вихідної речовини метальованої фенільної групи. Літій- і магнійзаміщені ароматичні сполуки можна синтезувати з хлорованих, бромованих або йодованих ароматичних сполук за допомогою реакції обміну галоген-метал, наприклад, з бутиллітієм, ізопропілмагнійгалогенідом або діізопропілмагнієм, або шляхом впровадження елементарного металу по зв'язку галоген-вуглець. Відповідну борозамі щену сполуку, таку як, наприклад, боронову кислоту, ефір боронової кислоти або діалкіларилборан можна отримати з цих метальованих фенільних груп за реакцією з електрофільним реагентом, що містить бор, таким як, наприклад, ефір боронової кислоти або його похідна. Крім того, борильовану ароматичну сполуку також можна отримати з відповідного галогенованого або псевдогалогенованого попередника і сполуки, що містить два атоми бору, або борану за реакцією, що каталізується перехідним металом, наприклад, паладієм (див., наприклад, Tetrahedron Lett. 2003, p.4895-4898 і цитовані в них посилання). Літій- і магнійзаміщені феніли сполуки приєднують до бензальдегідів (стадія 3) і бензойних кислот або їх похідних (стадія 4), таких як ефіри бензойної кислоти, бензаміди, такі як, наприклад, типу Вайнреба, бензонітрил або бензоїлхлориди. Цю реакцію можна проводити без додаткового каталізу перехідним металом і переметалювання іншим металом, таким як, наприклад, церій, індій або цинк; інколи переважне 15 94087 застосування одного з останніх варіантів. Арилборонові кислоти можна приєднати до бензальдегідів за допомогою родієвого каталізатора і отримати відповідний діарилметанол (див., наприклад, Adv. Synth. Catal. 2001, p.343-350 і цитовані в них посилання). Крім того, арилборонові кислоти, їх ефіри, діалкілборани або арилтрифтороборати можна ввести в реакцію сполучення з бензоїлхлоридами при каталізі перехідним металом, таким як, наприклад, паладій, його комплекс або сіль, і отримати діарилкетони. Метальовані фенільні групи можна ввести в реакцію з бензильними електрофільними реагентами, такими як, бензилхлориди, -броміди або -йодиди, і отримати діарилметани. Літієві або магнієві похідні фенільних сполук ефективно вступають в реакцію, але не завжди обов'язково у при 16 сутності перехідного металу, такого як, наприклад, мідь, залізо або паладій (див., наприклад, Org. Lett. 2001, 3, 2871-2874 і цитовані в ній посилання). Переметалювання із заміною літію або магнію, наприклад, на бор, олово, кремній або цинк, дає, наприклад, відповідні ароматичні боронові кислоти, станани, силани або сполуки цинку відповідно, які можна ввести в реакцію з бензильними електрофільними реагентами, наприклад, бензилгалогенідами, -карбонатами, -фосфатами, сульфонатами або ефірами карбонових кислот. Реакцію проводять у присутності перехідного металу, наприклад, паладію, нікелю, родію, міді або заліза (див., наприклад, Tetrahedron Lett. 2004, p.8225-8228 і Org. Lett. 2005, p.4875-4878 і цитовані в них посилання). Схема 5: Введення ціаногрупи На схемі 5 представлені можливі шляхи приєднання ціаногрупи до центральної фенільної групи на різних стадіях синтезу шуканої молекули. Ціаногрупу можна ввести за допомогою каталізованої перехідним металом реакції сполучення відповідного джерела ціаногрупи, такого як, наприклад, ціанід натрію, калію, цинку або міді, з галогенованою або псевдогалогенованою фенільною групою. Підходящий каталізатор можна приготувати з перехідних металів, таких як, наприклад, паладій, родій, нікель, залізо або мідь, які можна використовувати в елементній формі, такій як, наприклад, паладію на вугіллі, у вигляді солей, таких як, наприклад, хлорид, бромід або ацетат паладію, або комплексів, наприклад, з фосфінами, такими як, наприклад, трифенілфосфін, три-третбутилфосфін або dppf, або з алкенами, такими як, наприклад, дибензиліденацетон. Активний каталі затор можна утворити in situ або до додавання до реакційної суміші. Може бути корисним використання добавок, таких як, наприклад, цинк в елементному вигляді або у вигляді солі (див. Tetrahedron Lett. 2005, 46, 1849-1853 і Tetrahedron Lett. 2005, 46, 1815-1818 і цитовані в них посилання). Реакції відповідних сполук цинку, магнію або літію, які можна отримати з хлорованих, бромованих або йодованих сполук за реакцією обміну галоген-метал або шляхом впровадження відповідного металу в зв'язок з галогеном, з електрофільним реагентом, що містить ціаногрупу, таким як, наприклад, птолілсульфонілціанід, бромоціан або 2піридилціанат, є іншим ефективним шляхом введення ціаногрупи (див., наприклад, Synth. Commun. 1996, 3709-3714 і цитовані в ній посилання). Схема 6: Введення ціаногрупи за допомогою похідної альдегіду або карбонової кислоти 17 94087 Альтернативною методикою введення ціаногрупи є синтез з використанням як вихідної речовини альдегіду або карбоксаміду (схема 6). Альдегідну групу можна ввести саму по собі, в захищеному або замаскованому вигляді. Поширеними захисними групами альдегідної групи є ацетальні, але також можна використовувати інші захисні групи (див. Т. W. Greene, P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, 1999). Підходящими засобами маскування альдегідної групи є, наприклад, олефіни і тіазоли. Альдегідну групу можна перетворити на ціаногрупу, наприклад, з використанням гідроксиламіну в комбінації, наприклад, з мурашиною кислотою, концентрованою хлористоводневою кислотою, поліфосфорною кислотою або сумішшю піридин-толуол. Проміжний оксим, що утворюється за таких умов проведення реакції, можна виділити до дегідратації з одержанням кінцевого продукту. Також можна використовувати 18 альтернативні гідроксиламіни, такі як, наприклад, біс-трифтороацетилгідроксиламін і NH2OSO3 і без використання додаткових реагентів отримати нітрил. Іншими реагентами, що використовуються, є, наприклад, NH4PO4H2 і нітропропан в оцтовій кислоті, триметилсилілазид і 8,8-диметилсіркодіімід. Підходящими попередниками нітрилу можуть бути і карбоксаміди. Перетворення можна провести дегідратуючими реагентами, такими як, наприклад, ангідрид трифторооцтової кислоти, пентаоксид фосфору, РОСl3, комбінація ССl4-фосфін, комбінація Сl3СОСl-амін, реагент Бурджеса, реагент Вісмайера, SOCl2 або хлороангідрид ціанурової кислоти. З використанням як вихідної речовини відповідного моноалкілованого карбоксаміду, карбонової кислоти, її ефіру або хлороангідриду карбонової кислоти, одержання нітрилу також можливе в однореакторному режимі без виділення якогонебудь проміжного продукту. Схема 7: Введення ціаногрупи за допомогою анілінового попередника Загальноприйнятою методикою введення ціаногрупи є так звана реакція Зандмайера з ціанідом міді і відповідною діазонієвою сполукою, яку можна отримати діазотуванням відповідної похідної аніліну. Синтез діазонієвих сполук і подальше видалення діазогрупи детально описані в літературі з органічної хімії. Схема 8: Альтернативний синтез діарилметанового фрагменту Альтернативний шлях формування діарилметанового фрагменту представлений на схемі 8. У ньому використовується ортофторозаміщений бензонітрил, який або є у продажу, або його можна отримати за вказаними вище методиками. Орто-фторозаміщений бензонітрил в лужному середовищі вводять в реакцію з алкілфенілацетатом, що містить замісник R3 (див., наприклад, J. Org. Chem. 55, 1990, 4817-4821; J. Heterocycl. Chem, 32, 1995, 1461-1466), а потім розщеплюють складний ефір і проводять декарбоксилування (див., наприклад, J. Heterocycl. Chem, 32, 1995, 1461-1466; Org. Prep. Proced. Int. 37, 2005, 550-555) або пряме деалкоксикарбонілування (див., наприклад, J. Med. Chem. 46, 2003, 52495257; Angew. Chem. Int. Ed. 47, 2004, 6493-6496). Для одержання сполук загальної формули І в способі а), пропонованому в дійсному винаході, сполука загальної формули II 19 у якій R' і R3 є такими, як визначено вище в дійсному винаході, і R8a, R8b, R8c, R8d є такими, як визначено вище в дійсному винаході, і незалежно один від одного означають, наприклад, ацетил, півалоїл, бензоїл, трет-бутоксикарбоніл, бензилоксикарбоніл, аліл, триалкілсиліл, бензил або заміщений бензил, або в кожному випадку дві сусідні групи R8a, R8b, R8c, R8d утворюють бензиліденацетальну, ізопропіліденкетальну або 2,3диметоксибутиленову групу, яка по положеннях 2 і 3 бутиленової групи зв'язана з атомами кисню піранозного кільця і утворює з ним заміщений діоксан, яку можна отримати, як описано вище в дійсному винаході, вводять в реакцію з відновним реагентом у присутності кислоти Льюїса або Бренстеда. Відновні реагенти, підходящі для цієї реакції, включають, наприклад, силани, такі як триетил-, трипропіл-, триізопропіл- або дифенілсилан, борогідрид натрію, ціаноборогідрид натрію, борогідрид цинку, борани, алюмогідрид літію, діізопропілалюмінійгідрид і йодид самарію. Відновлення проводять за відсутності або у присутності підходящої кислоти Бренстеда, такої як, наприклад, хлористоводнева кислота, толуолсульфонова кислота, трифторооцтова кислота або оцтова кислота, або кислоти Льюїса, такої як, наприклад, ефірат трифториду бору, триметилсилілтрифлат, тетрахлорид титану, тетрахлорид олова, трифлат скандію або йодид цинку. Залежно від відновного реагенту і кислоти реакцію можна провести в розчиннику, такому як, наприклад, метиленхлорид, хлороформ, ацетонітрил, толуол, гексан, діетиловий ефір, тетрагідрофуран, діоксан, етанол, вода або їх суміші при температурі від -60 до 120°С. Особливо підходяща комбінація реагентів включає, наприклад, триетилсилан і ефірат трифториду бору, яку зазвичай використовують в ацетонітрилі або дихлорометані при температурі від -60 до 60°С. Крім того, для описаних перетворень можна використовувати водень у присутності каталізатора на основі перехідного металу, такого як, наприклад, паладій на деревному вугіллі або нікель Ренея, в розчинниках, таких як, тетрагідрофуран, етилацетат, метанол, етанол, вода або оцтова кислота. Альтернативно, для одержання сполук загальної формули І способом b), пропонованим в дійсному винаході, відщеплюють захисні групи від сполуки загальної формули III 94087 20 у якій R3 є таким, як визначено вище в дійсному винаході, і R8a-R8d означають одну із захисних груп, визначених вище в дійсному винаході, таку як, наприклад, ацильна, арилметильна, алільна, ацетальна, кетальна або силільна група, і яку можна отримати наприклад, відновленням із сполуки формули II, як описано вище в дійсному винаході. Слід розуміти, що при проведенні вказаних вище синтезів можна замінювати одну або декілька з груп R8a-R8d. Будь-яку ацильну, що використовується, захисну групу відщеплюють, наприклад, гідролітично у водному розчиннику, наприклад, у воді, суміші ізопропанол/вода, оцтова кислота/вода, тетрагідрофуран/вода або діоксан/вода, у присутності кислоти, такої як трифторооцтова кислота, хлористоводнева кислота або сірчана кислота, або у присутності основи лужного металу, такої як гідроксид літію, гідроксид натрію або калію, або апротонно, наприклад, у присутності йодотриметилсилану при температурі від 0 до 120°С, переважно при температурі від 10 до 100°С. Трифтороацетильну групу переважно відщеплюють шляхом обробки кислотою, такою як хлористоводнева кислота, необов'язково у присутності розчинника, такого як оцтова кислота, при температурі від 50 до 120°С або шляхом обробки розчином гідроксиду натрію необов'язково у присутності розчинника, такого як тетрагідрофуран або метанол, при температурі від 0 до 50°С. Будь-яку ацетальну, що використовується, або кетальну захисну групу відщеплюють, наприклад, гідролітично у водному розчиннику, наприклад, у воді, суміші ізопропанол/вода, оцтова кислота/вода, тетрагідрофуран/вода або діоксан/вода, у присутності кислоти, такої як трифторооцтова кислота, хлористоводнева кислота або сірчана кислота, або апротонно, наприклад, у присутності йодотриметилсилану, при температурі від 0 до 120°С, переважно - при температурі від 10 до 100°С. Триметилсилільну групу відщеплюють, наприклад, у воді, у водній суміші розчинників або в нижчому спирті, такому як метанол або етанол, у присутності основи, такої як гідроксид літію, гідроксид натрію, карбонат калію або метоксид натрію. У водні або спиртні розчинники корисно включати кислоти, такі як, наприклад, хлористоводнева кислота, трифторооцтова кислота або оцтова кислота. Для відщеплення в органічних розчинниках, таких як наприклад, діетиловий ефір, тетрагідрофуран або дихлорометан, також корисно використовувати фториди, такі як, наприклад, тетрабутиламонійфторид. Бензильну, метоксибензильну і бензилоксикарбонільну групу переважно відщеплюють гідрогенолітично, наприклад, воднем у присутності каталізатора, такого як паладій/деревне вугілля, в підходящому розчиннику, такому як метанол, етанол, етилацетат або льодяна оцтова кислота, необов'язково з додаванням кислоти, такої як хлористоводнева кислота, при температурі від 0 до 100°С, але переважно - при температурі навколишнього середовища, рівного від 20 до 60°С, і при тиску водню, рівному від 1 до 7 бар, але переважно - від 3 до 5бар. Проте 2,4-диметоксибензильну 21 групу переважно відщеплюють в трифторооцтовій кислоті у присутності анізолу. Трет-бутильну або трет-бутилоксикарбонільну групу переважно відщеплюють шляхом обробки кислотою, такою як трифторооцтова кислота або хлористоводнева кислота або шляхом обробки йодотриметилсиланом, необов'язково з використанням розчинника, такого як метиленхлорид, діоксан, метанол або діетиловий ефір. При проведенні реакцій, описаних вище в дійсному винаході, будь-які активні групи, що містяться, такі як етинільну групу, гідроксигрупу, аміногрупу, алкіламіногрупу або іміногрупу, на час проведення реакції можна захистити звичайними захисними групами, які повторно відщеплюють після проведення реакції. Наприклад, захисною групою етинільної групи може бути триметилсилільна або триізопропільна група. Як захисну групу також можна використовувати 2-гідроксіізопроп-2-ільну групу. Наприклад, захисною групою гідроксигрупи може бути триметилсилільна, ацетильна, тритильна, бензильна або тетрагідропіранільна група. Захисними групами для аміногрупи, алкіламіногрупи або іміногрупи можуть бути, наприклад, формільна, ацетильна, трифтороацетильна, етоксикарбонільна, трет-бутоксикарбонільна, бензилоксикарбонільна, бензильна, метоксибензильна або 2,4-диметоксибензильна група. Крім того, одержані сполуки загальної формули І можна розділити на їх енантіомери і/або діастереоізомери, як зазначено вище в дійсному винаході. Так, наприклад, цис/транс-суміші можна розділити на цис- і транс-ізомери, і сполуки, що містять щонайменше один оптично активний атом вуглецю, можна розділити на їх енантіомери. Так, наприклад, цис/транс-суміші можна розділити за допомогою хроматографії на цис- і трансізомери, одержані сполуки загальної формули І, які виявляються рацематами, можна розділити згідно відомих методик (див. Allinger N. L. and Eliel E. L. in "Topics in Stereochemistry", Vol.6, Wiley Interscience, 1971) на їх оптичні антиподи і сполуки загальної формули І, що містять щонайменше 2 оптично активних атома вуглецю, можна розділити на їх діастереоізомери з використанням відмінностей їх фізико-хімічних характеристик згідно відомих методик, наприклад, за допомогою хроматографії і/або фракційної кристалізації, і, якщо ці сполуки виходять в рацемічній формі, то потім їх можна розділити на енантіомери, як вказано вище. Енантіомери переважно розділяють на колонках з хіральними фазами або шляхом перекристалізації з оптично активного розчинника або за реакцією з оптично активною речовиною, яка утворює солі або похідні, такі як, наприклад, складні ефіри або аміди з рацемічною сполукою, переважно - її складні ефіри і активовані похідні або спирти, і розділення одержаної таким чином суміші діастереоізомерів солей або похідних, наприклад, на підставі відмінності їх розчинностей, хоча вільні антиподи можна виділити з чистих діастереоізомерів солей або похідних шляхом обробки підходящими реагентами. Зазвичай оптично активними кислотами, що використовуються, є, наприклад, D 94087 22 і L-форми винної кислоти або дибензоїлвинної кислоти, ді-о-толуолвинної кислоти, яблучної кислоти, мигдалевої кислоти, камфорсульфонової кислоти, глутамінової кислоти, аспарагінової кислоти або хінінової кислоти. Оптично активним спиртом може бути, наприклад, (+) або (-)-ментол і оптично активною ацильною групою в амідах може бути, наприклад, (+)або (-)ментилоксикарбонільна група. Крім того, сполуки формули І можна перетворити на їх солі, для використання у фармацевтиці, переважно - у фізіологічно прийнятні солі з неорганічними або органічними кислотами. Кислоти, які можна використовувати для цієї мети, включають, наприклад, хлористоводневу кислоту, бромистоводневу кислоту, сірчану кислоту, метансульфонову кислоту, фосфорну кислоту, фумарову кислоту, янтарну кислоту, молочну кислоту, лимонну кислоту, винну кислоту і малеїнову кислоту. Крім того, одержані сполуки можна перетворити на суміші, наприклад, суміші складу 1:1 або 1:2 з амінокислотами, переважно - з альфаамінокислотами, такими як пролін або фенілаланін, які можуть володіти особливо сприятливими характеристиками, такими як високий ступінь кристалічності. Сполуки, пропоновані в дійсному винаході, також переважно отримувати за методиками, описаними в наведених нижче прикладах, які для цієї мети можна комбінувати з методиками, відомими фахівцеві в даній галузі техніки з літератури, наприклад, з методиками, описаними в WO 98/31697, WO 01/27128, WO 02/083066, WO 03/099836 і WO 2004/063209. Дійсний винахід також відноситься до нових проміжних сполук, описаних на схемах реакцій вище в дійсному винаході і описаним в експериментальному розділі нижче в дійсному винаході. Зокрема, наступні проміжні сполуки є додатковим об'єктом дійсного винаходу: 23 де R8a-R8d визначені вище в дійсному винаході і переважно означають Η або ацетил; R' визначений вище в дійсному винаході і переважно означає Н, метил або етил; Аlk означає С1-С4-алкіл, переважно - метил або етил; R1 визначений вище в дійсному винаході і переважно означає Вr або CN, найпереважніше - CN; R3 визначений вище в дійсному винаході, наприклад, циклопропіл або циклобутил, і переважно вибраний з групи, що включає хлор, бром, метил, етил, н-пропіл, ізопропіл, циклопропіл, циклобутил, циклопентил, гідроксигрупу, ціаногрупу (S)тетрагідрофуран-3-ілоксигрупу, (R)тетрагідрофуран-3-ілоксигрупу, С3-С7циклоалкілоксигрупу, С1-С3-алкілоксигрупу, гідроксигрупу; LG означає групу, що відщеплюється, таку як Br, I, -O-(SO2)-CF3, переважно - -O-(SO2)-CF3; 94087 24 U означає СІ, Вr, І, -О-СО-С1-С4-алкіл, -ОС(=О)-О-С1-С4-алкіл або -ОРО(О-С1-С4-алкіл)2; переважно - Вr. Як вже відмічено, сполуки загальної формули І, пропоновані в дійсному винаході, і їх фізіологічно прийнятні солі володіють цінними фармакологічними характеристиками, зокрема, інгібуючою дією на натрійзалежний співпереносник глюкози SGLT, переважно - SGLT2. Біологічні характеристики цих нових сполук можна досліджувати так, як описано нижче: Здатність пропонованих у винаході сполук інгібувати активність SGLT2 можна продемонструвати експериментально з використанням клітин лінії СНО-K1 (АТСС №CCL 61) або в іншому варіанті клітин лінії НЕК293 (АТСС №CRL-1573), стабільно трансфікований експресуючим вектором pZeoSV (Invitrogen, реєстраційний номер L36849 в базі даних EMBL), який містить кДНК для кодуючої послідовності натрійзалежного співпереносника глюкози типу 2 людини (реєстраційний номер у банку генів NM_003041) (CHO-hSGLT2 або HEKhSGLT2). Клітини цих ліній забезпечують натрійзалежне перенесення всередину клітин міченого 14 С α-метилглюкопіранозиду (14С-АМГ, фірма Amersham). Дослідження SGLT-2 проводять таким чином: Клітини CHO-hSGLT2 культивують в середовищі Хема F12 (Bio Whittaker), доповненому 10% фетальної телячої сироватки і зеоцином в концентрації 250мкг/мл (Invitrogen), а клітини HEK293hSGLT2 культивують в середовищі МДСІ (модифіковане за способом Дульбекко середовище Ігли), доповненому 10% фетальної телячої сироватки і зеоцином в концентрації 250мкг/мл (Invitrogen). Клітини відділяють від колб для культивування шляхом двократного промивання забуференим фосфатом фізіологічним розчином (ЗФР) і подальшої обробки сумішшю трипсин/ЕДТК (етилендіамінотетраоцтова кислота). Після додавання культурального середовища клітини відділяють центрифугуванням, потім повторно суспендують в культуральному середовищі і підраховують в цитометрі з технологією CASY. Потім клітини висівають в білий, покритий полі-D-лізином 96-лунковий планшет по 40000 клітин на лунку і інкубують протягом ночі при 37°С і 5% СО2. Потім клітини двічі промивають 300мкл буферу для аналізу (збалансований сольовий розчин Хенкса, 137мМ NaCl, 5,4мМ KСl, 2,8мМ СаСl2, 1,2мМ MgSO4 і 10мМ HEPES (N-2-гідроксіетилпіперазин-N'-2етансульфонова кислота) (рН7,4), 50мкг/мл гентаміцину). Потім в кожну лунку додають по 250мкл буфера для аналізу і по 5мкл досліджуваної сполуки і планшети інкубують протягом подальших 15хв. в термостаті. Як негативний контроль використовують 5мкл 10% ДМСО. Реакцію ініціюють додаванням в кожну лунку по 5мкл 14С-АМГ (0,05мкКи). Після 2год. інкубації при 37°С і 5% СО2 клітини повторно промивають за допомогою 300мкл ЗФР (20°С), після чого ліолізують шляхом додавання 25мкл 0,1н. NaOH (протягом 5хв. при 37°С). Потім в кожну лунку додають по 200мкл сцинтилятору MicroScint20 (фірма Packard) і інкубують протягом ще 20хв. при 37°С. Після завер 25 шення цієї інкубації в детекторі Topcount (Packard) за допомогою програми вимірювання сцинтиляції, обумовленою 14С, вимірюють радіоактивність поглиненого 14С-АМГ. Для визначення селективності по відношенню до SGLT1 людини проводять аналогічне дослідження, в якому в клітинах СНО-K1 або НЕК293 експресується кДНК для hSGLT1 (реєстраційний номер у банку генів NM000343) замість кДНК для hSGLT2. Сполуки, пропоновані в дійсному винаході, можуть, наприклад, володіти значеннями ЕС50, рівними менше 1000нМ, переважно - менше 200нМ, найпереважніше - менше 50нМ. З врахуванням їх здатності інгібувати активність SGLT сполуки, пропоновані в дійсному винаході, і їх відповідні фармацевтично прийнятні солі придатні для лікування і/або попереджувального лікування всіх тих патологічних станів або захворювань, на які можна впливати шляхом інгібування активності SGLT, переважно - активності SGLT2. Тому сполуки, пропоновані в дійсному винаході, є особливо підходящими для попередження або лікування захворювань, переважно - метаболічних порушень, або патологічних станів, таких як цукровий діабет типу 1 і типу 2, ускладнення при діабеті (такі як, наприклад, ретинопатія, нефропатія або невропатія, діабетична стопа, виразки, макроангіопатії), метаболічний ацидоз або кетоз, реактивна гіпоглікемія, гіперінсулінемія, порушення обміну глюкози, резистентність до інсуліну, метаболічний синдром, дисліпідимії різного генезу, атеросклероз і родинні захворювання, ожиріння, підвищений артеріальний тиск, хронічна серцева недостатність, набряк і гіперурикемія. Ці сполуки також придатні для попередження дегенерації бетаклітин, такої як, наприклад, апоптоз або некроз бета-клітин панкреатичних острівців. Ці сполуки також придатні для поліпшення або відновлення функціональної активності клітин підшлункової залози, і для збільшення кількості і розміру бетаклітин панкреатичних острівців. Сполуки, пропоновані в дійсному винаході, також можна використовувати як діуретики або гіпотензивні засоби і вони придатні для попередження і лікування гострої ниркової недостатності. Шляхом введення сполуки, пропонованої в дійсному винаході, можна зменшити або подавити аномальне накопичення жиру в печінці. Тому іншим об'єктом дійсного винаходу є спосіб попередження, уповільнення, затримання або лікування захворювань або патологічних станів, обумовлених аномальним накопиченням жиру в печінці, у пацієнта, що потребує його, що характеризується тим, що вводять сполуку або фармацевтичну композицію, пропоновану в дійсному винаході. Захворювання або патологічні стани, обумовлені аномальним накопиченням жиру в печінці, переважно вибрані з групи, що включає загальну жирову інфільтрацію печінки, неалкогольну жирову інфільтрацію печінки (НАЖП), неалкогольний стеатогепатит (НАСГ), викликану гіпераліментацією жирову інфільтрацію печінки, діабетичну жирову інфільтрацію печінки, викликану алкоголем жирову інфільтрацію печінки або токсичну жирову інфільтрацію печінки. 94087 26 Зокрема, сполуки, пропоновані в дійсному винаході, включаючи їх фізіологічно прийнятні солі, застосовні для попередження або лікування діабету, переважно - цукрового діабету типу 1 і типу 2 і/або ускладнень при діабеті. Крім того, сполуки, пропоновані в дійсному винаході, є особливо підходящими для попередження або лікування надлишкової маси, ожиріння (включаючи ожиріння класу І, класу II і/або класу III), вісцелярного ожиріння і/або абдомінального ожиріння. Дози, необхідні для забезпечення відповідної активності з метою лікування або попередження, зазвичай залежать від сполуки, яку необхідно вводити, від пацієнта, характеру і важкості захворювання або патологічного стану і методики і частоти введення і визначаються лікарем, що лікує. Доцільна доза може складати від 1 до 100 мг, переважно - від 1 до 30мг при внутрішньовенному введенні і від 1 до 1000мг, переважно - від 1 до 100мг при пероральному введенні і в кожному випадку засіб вводять від 1 до 4 разів на добу. Для цієї сполуки, пропоновані в дійсному винаході, необов'язково можна приготувати спільно з іншими активними речовинами, спільно з одним або більшою кількістю звичайних інертних наповнювачів і/або розчинників, наприклад, з кукурудзяним крохмалем, лактозою, глюкозою, мікрокристалічною целюлозою, стеаратом магнію, полівінілпіролідоном, лимонною кислотою, винною кислотою, водою, сумішшю вода/етанол, вода/гліцерин, вода/сорбіт, вода/поліетиленгліколь, пропіленгліколем, цетилстеариловим спиртом, карбоксиметилцелюлозою або жировмісними речовинами, такими як затверджений жир, або їх прийнятними сумішами, з одержанням звичайних галенових препаратів, таких як таблетки без покриття або з покриттям, капсули, порошки, суспензії або супозиторії. Сполуки, пропоновані в дійсному винаході, також можна використовувати спільно з іншими активними речовинами, переважно - для лікування і/або попередження вказаних вище захворювань і патологічних станів. Інші активні речовини, які придатні для таких комбінацій, включають, наприклад, такі, які підсилюють терапевтичну дію антагоніста SGLT, пропонованого в дійсному винаході, при його введенні в разі одного з вказаних вище показань і/або які дозволяють понизити дозування антагоніста SGLT пропонованого в дійсному винаході. Терапевтичні засоби, які придатні для таких комбінацій, включають, наприклад, антидіабетичні засоби, такі як метформін, сульфонілсечовини (наприклад, глібенкламід, толбутамід, глімепірид), натеглінід, репаглінід, тіазолідиндіони (наприклад, росиглітазон, піоглітазон), агоністи і антагоністи рецептора PPAR-гамма (наприклад, GI 262570), модулятори рецептора PPAR-гамма/альфа (наприклад, KRP 297), інгібітори альфа-глюкозидази (наприклад, акарбоза, воглібоза), інгібітори DPP-IV (наприклад, LAF237, МK-431), альфа2-антагоністи, інсулін і аналоги інсуліну, GLP-1 і аналоги GLP-1 (наприклад, ексендин-4) або амілін. Перелік також включає інгібітори протеїнтирозинфосфатази 1, речовини, що впливають на дерегуляцію продуку 27 вання глюкози в печінці, такі як, наприклад, інгібітори глюкозо-6-фосфатази, фруктозо-1,6бісфосфатази, глікогенфосфорилази, антагоністи глюкагонового рецептора і інгібітори фосфоенолпіруваткарбоксикінази, глікогенсинтазикінази або піруватдегідрогенази, засоби, що знижують вміст ліпідів в крові, такі як, наприклад, інгібітори ГМГКоА-редуктази (наприклад, симвастатин, аторвастатин), фібрати (наприклад, безафібрат, фенофібрат), нікотинову кислоту і її похідні, агоністи PPARальфа, агоністи PPAR-дельта, інгібітори АСАТ (наприклад, авасиміб) або інгібітори всмоктування холестерину, такі як, наприклад, езетиміб, речовини, що зв'язують жовчні кислоти, такі як, наприклад, холестирамін, інгібітори клубового транспорту жовчних кислот, сполуки, що підвищують рівень ліпопротеїнів високої щільності в крові, такі як інгібітори СЕТР або регулятори АВС1, або активні речовини для лікування ожиріння, такі як сибутрамін, тетрагідроліпстатин, дексфенфлурамін, аксокін, антагоністи канабіноїдного рецептора 1, антагоністи рецептора МСН-1, агоністи рецептора МС4, антагоністи NPY5 або NPY2 або β3-агоністи, такі як SB-418790 або AD-9677, а також агоністи рецептора 5НТ2с. Крім того, є підходящими комбінації з лікарськими засобами, що впливають на підвищений артеріальний тиск, хронічну серцеву недостатність або атеросклероз, такими як, наприклад, антагоністи ангіотензину II (А-ІІ) або інгібітори АСЕ, інгібітори ЕСЕ, діуретики β-блокатори, Са-антагоністи, гіпотензивні засоби, що володіють центральною дією, антагоністи альфа-2-адренорецептора, інгібітори нейтральної ендопептидази, інгібітори агрегації тромбоцитів та інші або їх комбінації. Прикладами антагоністів рецептора ангіотензину II є кандесартан, цилексетил, калій-лосартан, епросартанмезилат, валсартан, телмісартан, ірбесартан, ЕХР-3174, L-158809, ЕХР-3312, олмесартан, медоксоміл, тазосартан, КТ-3-671, GA-0113, RU64276, EMD-90423, BR-9701 тощо. Антагоністи рецептора ангіотензину II переважно використовуються для лікування або попередження підвищеного артеріального тиску і ускладнень при діабеті, часто у поєднанні з діуретиком, таким як гідрохлоротіазид. Для лікування або попередження подагри застосовні комбінації з інгібіторами синтезу сечової кислоти або засобами, що сприяють виведенню з організму сечової кислоти. Для лікування або профілактики ускладнень при діабеті можна використовувати комбінації з антагоністами ГАМК-рецептора, блокаторами Naканалів, топіраматом, інгібіторами протеїнкінази С, інгібіторами кінцевих продуктів глікозирування або інгібіторами альдозоредуктази. Дозування компонентів комбінацій, вказаних вище, переважно складає від 1/5 від мінімальної дози, що зазвичай рекомендується, до 1/1 від дози, що зазвичай рекомендується. Тому, іншим об'єктом дійсного винаходу є використання сполуки, пропонованої в дійсному винаході, або фізіологічно прийнятної солі такої сполуки в комбінації щонайменше з однією з активних речовин, описаних вище як компоненти комбінації, 94087 28 для приготування фармацевтичної композиції, яка застосовна для лікування або попередження захворювань або патологічних станів, на які можна впливати шляхом інгібування натрійзалежного співпереносника глюкози SGLT. Ними переважно є метаболічні захворювання, переважно - одне з перерахованих вище захворювань або патологічних станів, найпереважніше - діабет або ускладнення при діабеті. Застосування сполуки, пропонованої в дійсному винаході, або її фізіологічно прийнятної солі, в комбінації з іншою активною речовиною можна проводити одночасно або по черзі через певні проміжки часу, але переважно - через невеликі проміжки часу. Якщо їх вводять одночасно, то ці дві активні речовини вводять пацієнтові спільно; якщо їх вводять по черзі через певні проміжки часу, то ці дві активні речовини вводять пацієнтові через проміжки часу, менші або рівні 12год., але переважно - менші або рівні 6год. Тому іншим об'єктом дійсного винаходу є фармацевтична композиція, яка включає сполуку, пропоновану в дійсному винаході, або фізіологічно прийнятну сіль такої сполуки і щонайменше одну з активних речовин, описаних вище як компоненти комбінації, необов'язково спільно з одним або більшою кількістю інертних носіїв і/або розчинників. Так, наприклад, фармацевтична композиція, пропонована в дійсному винаході, включає комбінацію сполуки, пропонованої в дійсному винаході, або фізіологічно прийнятної солі такої сполуки і щонайменше одного антагоніста рецептора ангіотензину II необов'язково спільно з одним або більшою кількістю інертних носіїв і/або розчинників. Сполука, пропонована в дійсному винаході, або її фізіологічно прийнятна сіль, і комбінована з нею додаткова активна речовина можуть обоє міститися в одномупрепараті, наприклад, таблетці або капсулі, або окремо входити в два однакових або різних препарати, наприклад, у вигляді так званого набору компонентів. У тексті вище і нижче атоми Η гідроксигруп вказані в структурних формулах не у всіх випадках. Наведені нижче приклади призначені для ілюстрації дійсного винаходу без накладення обмежень. Терміни "кімнатна температура" і "температура навколишнього середовища" використовуються, як взаємозамінні, і означають температуру, рівну приблизно 20°С. Одержання вихідних сполук: Приклад І 4-Бромо-3-гідроксиметил-1-йодобензол Оксалілхлорид (13,0мл) додають до охолодженого льодом розчину 2-бромо-5-йодобензойної кислоти в СН2Сl2 (200мл). Додають ДМФ (диметилформамід) (0,2мл) і розчин перемішують при кімнатній температурі протягом 6год. Потім розчин концентрують при зниженому тиску і залишок розчиняють в ТГФ (тетрагідрофуран) (100мл). Одержаний розчин охолоджують в бані з льодом і пор 29 ціями додають LiВН4 (3,4г). Баню, що охолоджує, видаляють і суміш перемішують при кімнатній температурі протягом 1год. Реакційну суміш розбавляють з допомогою ТГФ і обробляють 0,1Μ хлористоводневою кислотою. Потім органічний шар відділяють і водний шар екстрагують етилацетатом. Об'єднані органічні шари сушать (Na2SO4) і розчинник випаровують при зниженому тиску і отримують неочищений продукт. Вихід: 47,0г (99% від теоретичного виходу) Приклад II 4-Бромо-3-хлорометил-1-йодобензол Тіонілхлорид (13мл) додають до суспензії 4бромо-3-гідроксиметил-1-йодобензолу (47,0г) в дихлорометані (100мл), що містить ДМФ (0,1мл). Суміш перемішують при температурі навколишнього середовища протягом 3год. Потім розчинник і надлишок реагенту видаляють при зниженому тиску. Залишок розтирають з метанолом і сушать. Вихід: 41,0г (82% від теоретичного виходу) Приклад III 4-Бромо-1-йодо-3-феноксиметилбензол Фенол (13г), розчинений в 4Μ розчині КОН (60мл), додають до 4-бромо-3-хлорометил-1йодобензолу (41,0г), розчиненого в ацетоні (50мл). Додають Nal (0,5г) і одержану суміш перемішують при 50°С протягом ночі. Потім додають воду і одержану суміш екстрагують етилацетатом. Об'єднані екстракти сушать і розчинник випаровують при зниженому тиску. Залишок очищають за допомогою хроматографії на силікагелі (циклогексан/етилацетат 19:1). Вихід: 38,0г (79% від теоретичного виходу) Приклад IV (5-Бромо-2-хлорофенілУ(4-метоксифеніл)метанон 38,3мл Оксалілхлориду і 0,8мл диметилформаміду додають до суміші 100г 5-бромо-2хлоробензойної кислоти в 500мл дихлорометану. Реакційну суміш перемішують протягом 14год., потім фільтрують і відділяють від всіх летких компонентів в роторному випаровувачі. Залишок розчиняють в 150мл дихлорометану, одержаний розчин охолоджують до -5°С, і додають 46,5г анізолу. Потім порціями додають 51,5г трихлориду алюмінію, так щоб температура не перевищувала 5°С. Розчин перемішують протягом 1год. при температурі від 1 до 5°С і потім виливають на подрібнений 94087 30 лід. Органічну фазу відділяють, і водну фазу екстрагують дихлорометаном. Об'єднані органічні фази промивають 1Μ хлористоводневою кислотою, двічі 1Μ розчином гідроксиду натрію і розсолом. Потім органічну фазу сушать над сульфатом натрію, розчинник видаляють і залишок перекристалізовують з етанолу. Вихід: 86,3г (64% від теоретичного виходу) Мас-спектр (ІЕР+ (іонізація електророзпиленням)): m/z=325/327/329 (Вr+Сl) [М+Н]+ Приклад V 1-Бромо-4-хлоро-3-(4-метоксибензил)-бензол Розчин 86,2г (5-бромо-2-хлорофеніл)-(4метоксифеніл)-метанону і 101,5мл триетилсилану в 75мл дихлорометану і 150мл ацетонітрилу охолоджують до 10°С. Потім при перемішуванні додають 50,8мл ефірату трифториду бору, так щоб температура не перевищувала 20°С. Розчин перемішують протягом 14год. при температурі навколишнього середовища, потім додають ще 9мл триетилсилану і 4,4мл ефірату трифториду бору. Розчин перемішують протягом ще 3год. при 4550°С і потім охолоджують до температури навколишнього середовища. Додають розчин 28г гідроксиду калію в 70мл води і одержану суміш перемішують протягом 2год. Органічну фазу відділяють і водну фазу екстрагують ще три рази діізопропіловим ефіром. Об'єднані органічні фази двічі промивають 2Μ розчином гідроксиду калію і один раз розсолом і потім сушать над сульфатом натрію. Після випаровування розчинника залишок промивають етанолом і сушать при 60°С. Вихід: 50,0г (61% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=310/312/314 (Br+Cl) [M+H]+ Приклад VI 4-(5-Бромо-2-хлоробензил)-фенол Розчин 14,8г 1-бромо-4-хлоро-3-(4метоксибензил)-бензолу в 150мл дихлорометану охолоджують в бані з льодом. Додають 50мл 1Μ розчину триброміду бору в дихлорометані і одержаний розчин перемішують протягом 2год. при температурі навколишнього середовища. Потім розчин повторно охолоджують в бані з льодом і по краплях додають насичений водний розчин карбонату калію. При температурі навколишнього середовища значення рН суміші доводять до 1 водним розчином 1Μ хлористоводневої кислоти, органічну фазу відділяють і водну фазу три рази екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник повністю видаляють. Вихід: 13,9г (98% від теоретичного виходу) 31 H] Мас-спектр (ІЕР-): m/z=295/297/299 (Br+Cl) [MПриклад VII [4-(5-Бромо-2-хлоробензил)-фенокси1-третбутилдиметилсилан Розчин 13,9г 4-(5-бромо-2-хлоробензил)фенолу в 140мл дихлорометану охолоджують в бані з льодом. Потім додають 7,54г третбутилдиметилсилілхлориду в 20мл дихлорометану, а після цього 9,8мл триетиламіну і 0,5г 4диметиламінопіридину. Одержаний розчин перемішують протягом 16год. при температурі навколишнього середовища і потім розбавляють за допомогою 100мл дихлорометану. Органічну фазу двічі промивають 1Μ водним розчином хлористоводневої кислоти і один раз водним розчином гідрокарбонату натрію і потім сушать над сульфатом натрію. Після видалення розчинника залишок фільтрують через силікагель (циклогексан/етилацетат 100:1). Вихід: 16,8г (87% від теоретичного виходу) Мас-спектр (IE (іонізація електронним ударом)): m/z=410/412/414 (Br+Cl) [М]+ Приклад VIII 2,3,4,6-Тетракіс-о-(триметилсиліл)-Dглюкопіранон Розчин 20г D-глюконо-1,5-лактону і 98,5мл Nметилморфоліну в 200мл тетрагідрофурану охолоджують до -5°С. Потім по краплях додають 85мл триметилсилілхлориду, так щоб температура не перевищувала 5°С. Потім розчин перемішують протягом 1год. при температурі навколишнього середовища, 5год. при 35°С і повторно протягом 14год. при температурі навколишнього середовища. Після додавання 300мл толуолу розчин охолоджують в бані з льодом і додають 500мл води, так щоб температура не перевищувала 10°С. Органічну фазу відділяють і промивають водним розчином дигідрофосфату натрію, водою і розсолом. Розчинник видаляють і залишок піддають азеотропній сушці толуолом. Вихід: 52,5г (чистота приблизно 90%) Мас-спектр (ІЕР+): m/z=467 [М+Н]+ Приклад IX 1-Бромо-4-(1-метокси-D-глюкопіраноз-1-іл)-2(феноксиметил)-бензол 94087 32 2Μ Розчин iPrMgCl в ТГФ (11мл) додають до сухого LiCl (0,47г), суспендованому в ТГФ (11мл). Суміш перемішують при кімнатній температурі до повного розчинення LiCl. Цей розчин в атмосфері аргону по краплях додають до розчину 4-бромо-1йодо-3-феноксиметилбензолу (8,0г) в тетрагідрофурані (40мл), охолодженому до -60°С. Розчин нагрівають до -40°С і потім додають 2,3,4,6тетракіс-O-(триметилсиліл)-D-глюкопіранон (10,7г, чистота 90%) в тетрагідрофурані (5мл). Одержаний розчин нагрівають до -5°С у бані, що охолоджує, і перемішують протягом ще 30хв. при цій температурі. Додають водний розчин NH4Cl і одержану суміш екстрагують етилацетатом. Об'єднані органічні екстракти сушать над сульфатом натрію і розчинник видаляють при зниженому тиску. Залишок розчиняють в метанолі (80мл) і обробляють метансульфоновою кислотою (0,6мл). Після перемішування розчину при 35-40°С протягом ночі розчин нейтралізують твердими NaHCO3 і метанол видаляють при зниженому тиску. Залишок розбавляють водним розчином NaHCO3 і одержану суміш екстрагують етилацетатом. Об'єднані екстракти сушать над сульфатом натрію і розчинник випаровують і отримують неочищений продукт, який вводять в реакцію відновлення без додаткового очищення. Вихід: 7,8г (93% від теоретичного виходу) Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі IX: (1) 1-Бромо-4-(1-метокси-D-глюкопіраноз-1-іл)2-фторобензол Приклад X 1-Бромо-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-2-(феноксиметил)-бензол Ефірат трифториду бору (4,9мл) додають до розчину 1-бромо-4-(1-метокси-D-глюкопіраноз-1іл)-2-(феноксиметил)-бензолу (8,7г) і триетилсилану (9,1мл) в дихлорометані (35мл) і ацетонітрилі (50мл) охолоджують до -20°С з такою швидкістю, щоб температура підтримувалася нижче -10°С. Одержаний розчин нагрівають при 0°С протягом 1,5год. і потім обробляють водним розчином гідрокарбонату натрію. Одержану суміш перемішують протягом 0,5год., органічний розчинник видаляють і залишок екстрагують етилацетатом. Об'єднані органічні шари сушать над сульфатом натрію і розчинник видаляють. Залишок розбавляють дихлорометаном (50мл) і піридином (9,4мл), до розчину послідовно додають оцтовий ангідрид (9,3мл) і 4-диметиламінопіридин (0,5г). Розчин перемішу 33 ють протягом 1,5год. при температурі навколишнього середовища і потім розбавляють дихлорометаном. Цей розчин двічі промивають 1Μ хлористоводневою кислотою і сушать над сульфатом натрію. Після видалення розчинника залишок перекристалізовують з етанолу і отримують продукт у вигляді безбарвної твердої речовини. Вихід: 6,78г (60% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=610/612 (Br) [M+NH4]+ Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі X: (1) 1-Бромо-4-(2,3,4,6-тетра-O-ацетил-Dглюкопіраноз-1-іл)-2-фторобензол Приклад XI 2-(Феноксиметил)-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрил Колбу, в яку поміщені 1-бромо-4-(2,3,4,6-тетраО-ацетил-D-глюкопіраноз-1-іл)-2-(феноксиметил)бензол (5,4г), ціанід цинку (1,0г), цинк (30мг), Рd2(дибензиліденацетон)3-СНСl3 (141мг) і тритрет-бутилфосфонійтетрафтороборат (111мг), продувають аргоном. Потім додають NMP (Nметилпіролідон) (12мл) і одержану суміш перемішують при кімнатній температурі протягом 18год. Після розбавлення етилацетатом суміш фільтрують і фільтрат промивають водним розчином гідрокарбонату натрію. Органічну фазу сушать (сульфат натрію) і розчинник видаляють. Залишок перекристалізовують з етанолу. Вихід: 4,10г (84% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=557 [M+NH4]+ Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі XI: (1) 2-Фторо-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрил Приклад XII 94087 34 2-Бромометил-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрил 33% Розчин бромистоводневої кислоти в оцтовій кислоті (15мл) додають до розчину 2фенілоксиметил-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрилу (0,71г) і оцтового ангідриду (0,12мл) в оцтовій кислоті (10мл). Одержаний розчин перемішують при 55°С протягом 6год. і потім охолоджують в бані з льодом. Реакційну суміш нейтралізують охолодженим водним розчином карбонату калію і одержану суміш екстрагують етилацетатом. Об'єднані органічні екстракти сушать над сульфатом натрію і розчинник видаляють при зниженому тиску. Залишок розбавляють сумішшю етилацетат/циклогексан (1:5) і осад відділяють фільтруванням і сушать при 50°С і отримують чистий продукт. Вихід: 0,52г (75% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=543/545 (Вr) [M+NH4]+ Приклад XIII 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-(4гідроксибензил)-бензол Розчин 4,0г [4-(5-бромо-2-хлоробензил)фенокси]-трет-бутилдиметилсилану в 42мл сухого діетилового ефіру охолоджують до -80°С у атмосфері аргону. До охолодженого розчину повільно додають 11,6мл охолодженого (приблизно -50°С) 1,7Μ розчину трет-бутиллітію в пентані і потім розчин перемішують протягом 30хв. при -80°С. Потім цей розчин шприцом, голка якого охолоджена твердим діоксидом вуглецю, по краплях додають до розчину 4,78г 2,3,4,6-тетракіс-О-(триметилсиліл)D-глюкопіранону в 38мл діетилового ефіру, охолодженого до -80°С. Одержаний розчин перемішують протягом 3год. при -78°С. Потім додають розчин 1,1мл метансульфонової кислоти в 35мл метанолу і одержаний розчин перемішують протягом ще 16год. при температурі навколишнього середовища. Потім розчин нейтралізують твердим гідрокарбонатом натрію, додають етилацетат і одержаний розчин концентрують при зниженому тиску. До розчину, що залишився, додають водний розчин гідрокарбонату натрію і чотири рази екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник випаровують. Залишок розчиняють в 30мл ацетонітрилу і 30мл дихлорометану і одержаний розчин охолоджують до -10°С. Після додавання 4,4мл триетилсилану по краплях додають 2,6мл ефірату трифториду бору, так щоб температура не 35 94087 перевищувала -5°С. Після завершення додавання розчин перемішують протягом ще 5год. при температурі від -5 до -10°С і потім реакцію зупиняють шляхом додавання водного розчину гідрокарбонату натрію. Органічну фазу відділяють і водну фазу чотири рази екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію, розчинник видаляють і залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол). Одержаний продукт є сумішшю β/α складу приблизно 6:1, яку можна розділити шляхом повного ацетилування гідроксигруп оцтовим ангідридом, піридином і 4-диметиламінопіридином в дихлорометані і перекристалізації одержаного ацетильованого продукту з етанолу. Одержаний таким чином чистий ацетильований β-продукт перетворюють на шукану сполуку шляхом видалення ацетильних груп в метанолі 4Μ розчином гідроксиду калію. Вихід: 1,6г (46% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=398/400 (СІ) [M+NH4]+ Приклад XIV 36 Мас-спектр (ІЕР+): m/z=451/453 (СІ) [М+Н]+ (3) 1-Хлоро-2-(4-циклобутилоксибензил)-4-(βD-глюкопіраноз-1-іл)-бензол Мас-спектр (ІЕР+): m/z=452/454 (СІ) [M+NH4]+ (4) 1-Хлоро-2-(4-циклогексилоксибензил)-4-(βD-глюкопіраноз-1-іл)-бензол Мас-спектр (ІЕР+): m/z=480/482 (СІ) [M+NH4]+ (5) 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-[4(тетрагідропіран-4-ілокси)-бензил]-бензол 1-Хлоро-2-(4-циклопентилоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензол 0,16мл Йодоциклопентану додають до суміші 0,25г 1-хлоро-4-(β-D-глюкопіраноз-1-іл)-2-(4гідроксибензил)-бензолу і 0,4г карбонату цезію в 2,5мл диметилформаміду. Суміш перемішують протягом 4год. при 45°С, потім додають ще 0,1г карбонату цезію і 0,05мл йодоциклопентану. Після додаткових 14год. перемішування при 45°С додають водний розчин хлориду натрію і одержану суміш екстрагують етилацетатом. Органічну фазу сушать над сульфатом натрію, розчинник видаляють і залишок очищають за допомогою силікагелю (дихлорометан/метанол 1:05:1). Вихід: 0,23г (78% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=466/468 (СІ) [M+NH4]+ Наступні сполуки отримують аналогічно одержанню в прикладі XIV: (1) 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-[4-((R)тетрагідрофуран-3-ілокси)-бензил]-бензол Реакцію проводять з тетрагідрофуран-3-іл-(S)толуол-4-сульфонатом як компонент реакції сполучення. Мас-спектр (ІЕР+): m/z=487/489 (СІ) [M+Na]+ (6) 2-[4-(1-Ацетилпіперидин-4-ілокси)-бензил]1-хлоро-4-(β-D-глюкопіраноз-1-іл)-бензол Реакцію проводять з 1-ацетил-4метилсульфонілоксипіперидином як електрофільний реагент. Мас-спектр (ІЕР+): m/z=506/508 (СІ) [М+Н]+ (7) 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-(4метоксибензил)-бензол Мас-спектр (ІЕР+): m/z=412/414 (СІ) [M+NH4]+ (8) 1-Хлоро-2-(4-етоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензол + + Мас-спектр (ІЕР ): m/z=451/453 (СІ) [М+Н] (2) 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-[4-((S)тетрагідрофуран-3-ілокси)-бензил]-бензол Реакцію проводять з тетрагідрофуран-3-іл-(R)толуол-4-сульфонатом як компонент реакції сполучення. Мас-спектр (ІЕР+): m/z=426/428 (СІ) [M+NH4]+ 37 (9) 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-(4ізопропоксибензил)-бензол Приклад XV 1-Хлоро-4-(β-D-глюкопіраноз-1-іл)-2-[4(трифторометилсульфоніл-окси)-бензил]-бензол 10мг 4-Диметиламінопіридину додають до розчину 0,38г 1-хлоро-4-(β-D-глюкопіраноз-1-іл)-2-(4гідроксибензил)-бензолу, 0,21мл триетиламіну і 0,39г N,N-біс-(трифторометансульфоніл)-аніліну в 10мл сухого дихлорометану. Розчин перемішують протягом 4год. при температурі навколишнього середовища і потім об'єднують з розсолом. Одержану суміш екстрагують етилацетатом, органічні екстракти сушать над сульфатом натрію і розчинник видаляють. Залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол 1:04:1). Вихід: 0,33г (64% від теоретичного значення) Мас-спектр (ІЕР+): m/z=530/532 (СІ) [M+NH4]+ Наступні сполуки отримують аналогічно одержанню в прикладі XV: (1) 1-Ціано-4-(β-D-глюкопіраноз-1-іл)-2-[4(трифторометилсульфоніл-окси)-бензил]-бензол Мас-спектр (ІЕР+): m/z=504 [М+Н]+ Приклад XVI 1-Хлоро-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-2-[4(трифторометилсульфонілокси)-бензил]-бензол До розчину 5,6г 1-хлоро-4-(β-D-глюкопіраноз-1іл)-2-[4-(трифторометилсульфонілокси)-бензил]бензолу в 75мл дихлорометану послідовно додають 7мл піридину, 7,8мл оцтового ангідриду і 0,12г 4-диметиламінопіридину. Розчин перемішують при температурі навколишнього середовища протягом 1год. Після додавання 50мл води одержану суміш перемішують протягом ще 5хв. Органічну фазу відділяють і промивають 1Μ водним розчином хлористоводневої кислоти і водним розчином гідрокарбонату натрію. Після сушки над сульфатом 94087 38 магнію і випаровування органічного розчинника отримують продукт у вигляді білої твердої речовини. Вихід: 7,0г (94% від теоретичного значення) + + Мас-спектр (ІЕР ): m/z=698/700 (СІ) [M+NH4] Наступні сполуки отримують аналогічно одержанню в прикладі XVI: (1) 1-Ціано-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-2-[4(трифторометилсульфонілокси)-бензил]-бензол Мас-спектр (ІЕР+): m/z=689 [M+NH4]+ Приклад XVII 1-Хлоро-2-(4-етинілбензил)-4-(β-Dглюкопіраноз-1-іл)-бензол 25мг Йодиду міді, 44мг біс-(трифенілфосфін)паладійдихлориду, 0,30мл триетиламіну і на закінчення 0,14мл триметилсилілацетилену в атмосфері аргону додають до розчину 0,32г 1-хлоро-4(β-D-глюкопіраноз-1-іл)-2-[4(трифторометилсульфонілокси)-бензил]-бензолу в 3мл диметилформаміду. Колбу щільно закривають і суміш перемішують протягом 8год. при 90°С. Потім додають ще 25мг біс-(трифенілфосфін)паладійдихлориду і 0,1мл триметилсилілацетилену і розчин перемішують протягом ще 10год. при 90°С. Потім додають водний розчин гідрокарбонату натрію, одержану суміш три рази екстрагують етилацетатом і об'єднані органічні фази сушать над сульфатом натрію. Після випаровування розчинника залишок розчиняють в 5мл метанолу і об'єднують з 0,12г карбонату калію. Суміш перемішують протягом 1год. при температурі навколишнього середовища і потім нейтралізують 1Μ хлористоводневою кислотою. Потім метанол випаровують, залишок об'єднують з розсолом і екстрагують етилацетатом. Зібрані органічні екстракти сушать над сульфатом натрію і розчинник видаляють. Залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол 1:05:1). Вихід: 0,095г (40% від теоретичного значення) Мас-спектр (ІЕР+): m/z=406/408 (СІ) [M+NH4]+ Приклад XVIII 39 1-Хлоро-2-(4-етилбензил)-4-(β-D-глюкопіраноз1-іл)-бензол 2,87г 1-хлоро-2-(4-етинілбензил)-4-(βD-глюкопіраноз-1-іл)-бензолу розчиняють в 10мл етилацетату і 5мл етанолу. Додають 0,3г 10% паладію на вугіллі і одержану суміш перемішують в атмосфері водню (1атм.) протягом ночі. Реакційну суміш фільтрують через целіт і фільтрат концентрують. Залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол 1:05:1). Вихід: 1,0г (34% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=410/412 (СІ) [M+NH4]+ Приклад XIX 1-Хлоро-2-[4-((S)-тетрагідрофуран-3-ілокси)бензил]-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-бензол До розчину 2,02г 1-хлоро-4-(β-D-глюкопіраноз1-іл)-2-[4-((S)-тетрагідрофуран-3-ілокси)-бензил]бензолу в 20мл дихлорометану послідовно додають 2,5мл піридину, 2,8мл оцтового ангідриду і 50мг 4-диметиламінопіридину. Розчин перемішують при температурі навколишнього середовища протягом 4год. Розчин розбавляють за допомогою 50мл дихлорометану, двічі промивають за допомогою 50мл 1Μ хлористоводневої кислоти і один раз розчином гідрокарбонату натрію. Після сушки над сульфатом натрію розчинник випаровують і отримують продукт. Вихід: 2,53г (91% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=642/644 (СІ) [M+Na]+ Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі XIX: (1) 1-Хлоро-2-[4-((R)-тетрагідрофуран-3ілокси)-бензил]-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-бензол (2) 1-Хлоро-2-(4-циклопентилоксибензил)-4(2,3,4,6-тетра-О-ацетил-β-D-глюкопіраноз-1-іл)бензол Мас-спектр (ІЕР+): m/z=634/636 (СІ) [M+NH4]+ (3) 1-Хлоро-2-(4-циклобутилоксибензил)-4(2,3,4,6-тетра-O-ацетил-β-D-глюкопіраноз-1-іл)бензол 94087 40 (4) 1-Хлоро-2-(4-циклогексилоксибензил)-4(2,3,4,6-тетра-О-ацетил-β-D-глюкопіраноз-1-іл)бензол (5) 1-Хлоро-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-2-[4-(тетрагідропіран-4-ілокси)бензил]-бензол (6) 2-[4-(1-Ацетилпіперидин-4-ілокси)-бензил]1-хлоро-4-(2,3,4,6-тетра-О-ацетил-β-Dглюкопіраноз-1-іл)-бензол (7) 1-Хлоро-2-(4-метоксибензил)-4-(2,3,4,6тетра-O-ацетил-β-D-глюкопіраноз-1-іл)-бензол Мас-спектр (ІЕР+): m/z=585/587 (СІ) [M+NH4]+ (8) 1-Хлоро-2-(4-етоксибензил)-4-(2,3,4,6тетра-O-ацетил-β-D-глюкопіраноз-1-іл)-бензол (9) 1-Хлоро-2-(4-ізопропоксибензил)-4-(2,3,4,6тетра-О-ацетил-β-D-глюкопіраноз-1-іл)-бензол 41 (10) 1-Хлоро-2-(4-етилбензил)-4-(2,3,4,6-тетраО-ацетил-β-D-глюкопіраноз-1-іл)-бензол 94087 42 1-Хлоро-2-(4-щанобензил)-4-(2,3,4,А6-тетра-Оацетил-β-D-глюкопіраноз-1-іл)-бензол Тетракіс(трифенілфосфін)паладій(0) (0,13г) в атмосфері Аr додають в колбу, що містить 1хлоро-4-(2,3,4,6-тетра-О-ацетил-β-D-глюкопіраноз1-іл)-2-[4-(трифторометилсульфонілокси)-бензил]бензол (0,80г) і ціанід цинку (0,14г). Суміш перемішують при 100°С протягом 3год. Після охолодження до кімнатної температури додають етилацетат і одержану суміш фільтрують, промивають водним розчином NaHCO3, сушать (сульфат натрію) і розчинник видаляють. Залишок перекристалізовують з етанолу. Вихід: 0,45г (69% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=580/582 (СІ) [M+Na]+ Приклад XXII (11) 2-(4-Ацетоксибензил)-1-хлоро-4-(2,3,4,6тетра-О-ацетил-β-D-глюкопіраноз-1-іл)-бензол Мас-спектр (ІЕР+): m/z=608/610 (СІ) [M+NH4]+ Приклад XX 1-Хлоро-2-(4-метилбензил)-4-(2,3,4,6-тетра-Оацетил-β-D-глюкопіраноз-1-іл)-бензол Діїзобутилалюмінійгидрід (54мкл, 1моль/л в толуолі) в атмосфері Аr додають до суміші 1,1'біс(дифенілфосфіно)фероцен-дихлоропаладію(ІІ) (22мг) з ТГФ (3мл) і охолоджують в бані з льодом. Суміш перемішують в бані з льодом протягом 0,5год. і потім послідовно додають 1-хлоро-4(2,3,4,6-тетра-O-ацетил-β-D-глюкопіраноз-1-іл)-2[4-(трифторометилсульфонілокси)-бензил]-бензол (0,60г) і Me2Zn (0,88мл, 1моль/л в толуолі). Баню з льодом видаляють і суміш кип'ятять із зворотним холодильником протягом 2,5год. Після охолодження до кімнатної температури додають 1Μ хлористоводневу кислоту і одержану суміш екстрагують етилацетатом. Зібрані екстракти сушать над сульфатом натрію і розчинник видаляють. Залишок очищають за допомогою хроматографії на силікагелі (циклогексан/етилацетат 1:02:1). Вихід: 0,25г (52% від теоретичного значення) Приклад XXI 4-Циклопропілфенілборонова кислота 2,5Μ н-Бутиллітій в гексані (14,5мл) по краплях додають до 1-бромо-4-циклопропілбензолу (5,92г) в ТГФ (14мл) і толуолу (50мл), охолодженим до 70°С. Одержаний розчин перемішують при -70°С протягом 30хв. потім додають триізопропілборат (8,5мл). Розчин нагрівають до -20°С і потім обробляють 4Μ водним розчином хлористоводневої кислоти (15,5мл). Реакційну суміш нагрівають до кімнатної температури і потім органічну фазу відділяють. Водну фазу екстрагують етилацетатом і об'єднані органічні фази сушать (сульфат натрію). Розчинник випаровують і залишок промивають сумішшю ефіру з циклогексаном і отримують продукт у вигляді безбарвної твердої речовини. Вихід: 2,92г (60% від теоретичного виходу) Мас-спектр (ІЕР-): m/z=207 (СІ) [М+НСОО]Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі XXII: (1) 4-Дифторометоксифенілборонова кислота Мас-спектр (ІЕР-): m/z=233 (СІ) [М+НСОО]На відміну від описаної вище методики сполуку отримують з 4-дифторометокси-1-йодобензолу з використанням iPrMgCl для утворення арилпохідної металу і взаємодії цього проміжного продукту з триметилборатом. (2) 4-Дифторометилфенілборонова кислота Мас-спектр (ІЕР+): m/z=172 (СІ) [М+Н]+ На відміну від описаної вище методики сполуку отримують з 4-дифторометил-1-йодобензолу (одержаного з 4-йодобензальдегиду з використанням діетиламінотрифториду сірки (ДАТС) в дихлорометані) з використанням iPrMgCl для утворення арилпохідної металу і взаємодії цього проміжного продукту з триметилборатом. 43 Приклад XXIII 1-Бромо-4-ціано-3-(4-метоксибензил)-бензол Суміш 25г етил-(4-метоксифеніл)-ацетату, 27,4г 1-бромо-4-ціано-3-фторобензолу і 20мл Nметилпіролідин-2-ону повільно додають до 31,4г трет-бутоксиду калію в 130мл N-метилпіролідин-2ону, підтримуючи температуру нижче 10°С. Після перемішування протягом 1год. при кімнатній температурі додають 100мл метанолу і 137мл 1Μ водного розчину гідроксиду натрію і суміш перемішують протягом ночі при кімнатній температурі. Метанольну фракцію випаровують, залишок підлужнюють 1Μ водним розчином гідроксиду натрію і екстрагують трет-бутилметиловим ефіром. Водну фазу підкисляють 4Μ хлористоводневою кислотою і кілька разів екстрагують етилацетатом. Об'єднані етилацетатні екстракти випаровують і залишок разом з 120мл Ν,Ν-диметилформаміду і 24,9г карбонату калію нагрівають при 100°С протягом 1год. Реакційну суміш розбавляють водним розчином бікарбонату натрію і кілька разів екстрагують етилацетатом. Об'єднані екстракти випаровують і залишок кристалізують з метанолу. Вихід: 13г (33% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=319/321 (Вr) [M+NH4]+ Приклад XXIV Етил-4-циклопропілфенілацетат Отримують з етил-4-бромофенілацетату за допомогою каталізованої перехідним металом реакції сполучення з циклопропілбороновою кислотою з використанням трициклогексилфосфонійтетрафтороборату, ацетату паладію, фосфату калію в толуолі і води за методикою, описаною в публікації Tetrahedron Lett. 2002, 43, 6987-6990. Мас-спектр (ІЕР+): m/z=205 [М+Н]+ Одержання кінцевих сполук: Приклад 1 94087 44 чинника залишок розчиняють в 5мл метанолу. Додають 4мл 4Μ водного розчину гідроксиду калію і розчин перемішують при температурі навколишнього середовища протягом 3год. Розчин нейтралізують 1Μ хлористоводневою кислотою і метанол випаровують. Залишок екстрагують етилацетатом, об'єднані екстракти сушать над сульфатом натрію і розчинник видаляють при зниженому тиску. Залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол 4:1). Вихід: 0,35г (49% від теоретичного значення) Мас-спектр (ІЕР+): m/z=442 [М+Н]+ Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі 1: (2) 4-(β-D-глюкопіраноз-1-іл)-2-[4-((R)тетрагідрофуран-3-ілокси)-бензил]-бензонітрил (3) 2-(4-Циклопентилоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил (4) 2-(4-Циклобутилоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил (5) 2-(4-Циклогексилоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил (6) 2-[4-(Тетрагідропіран-4-ілокси)-бензил]-4(β-D-глюкопіраноз-1-іл)-бензонітрил 4-(β-D-глюкопіраноз-1-іл)-2-[4-((S)тетрагідрофураніл-3-окси)-бензил]-бензонітрил Суміш 1,00г 1-хлоро-2-[4-((S)тетрагідрофураніл-3-окси)-бензил]-4-(2,3,4,6тетра-О-ацетил-β-D-глюкопіраноз-1-іл)-бензолу, 0,16 ціаніду натрію і 0,35г броміду нікелю з 2,5мл К-метил-2-піролідинону протягом 15хв. нагрівають в мікрохвильовій печі при 220°С. Після охолодження до кімнатної температури додають воду і одержану суміш екстрагують етилацетатом. Після сушки над сульфатом натрію і випаровування роз (7) 2-[4-(1-Ацетилпіперидин-4-ілокси)-бензил]4-(β-D-глюкопіраноз-1-іл)-бензонітрил 45 (8) 2-(4-Метоксибензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=403 [M+NH4]+ Цю сполуку також отримують з 1-бромо-4ціано-3-(4-метоксибензил)-бензолу (приклад XXIII) і 2,3,4,6-тетракіс-О-(триметилсиліл)-Dглюкопіранону аналогічно тому, як описано в прикладі XIII, за тим виключенням, що реакцію з третбутиллітієм проводять в тетрагідрофурані при 87°С і що реакцію з сумішшю метанол/метансульфонова кислота проводять при 55°С. (9) 2-(4-Етоксибензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=417 [M+NH4]+ (10) 4-(β-D-глюкопіраноз-1-іл)-2-(4ізопропоксибензил)-бензонітрил Мас-спектр (ІЕР+): m/z=431 [M+NH4]+ (11) 2-(4-Етилбензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=401 [M+NH4]+ (12) 4-(β-D-глюкопіраноз-1-іл)-2-(4гідроксибензил)-бензонітрил Сполуку отримують з 2-(4-ацетоксибензил)-1хлоро-4-(2,3,4,6-тетра-О-ацетил-β-D-глюкопіраноз1-іл)-бензолу Мас-спектр (ІЕР+): m/z=389 [M+NH4]+ Сполуку також отримують перацетилуванням 2-(4-метоксибензил)-4-(β-D-глюкопіраноз-1-іл)бензонітрилу (сполука 1(8)) з подальшим розщеп 94087 46 люванням простого ефіру трибромідом бору і дезацетилуванням. (13) 4-(β-D-глюкопіраноз-1-іл)-2-(4метилбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=387 [M+NH4]+ (14) 2-(4-Ціанобензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=398 [M+NH4]+ Приклад 15 4-(β-D-глюкопіраноз-1-іл)-2-(4метоксіетоксибензил)-бензонітрил 2-Бромоетилметиловий ефір (85мкл) додають до суміші 4-(β-D-глюкопіраноз-1-іл)-2-(4гідроксибензил)-бензонітрилу (0,30г) і карбонату цезію (0,39г) в 3мл диметилформаміду. Суміш перемішують при 80°С протягом 16год., потім додають воду і розсіл. Одержану суміш екстрагують етилацетатом, об'єднані екстракти сушать над сульфатом натрію і розчинник видаляють при зниженому тиску. Залишок очищають за допомогою хроматографії на силікагелі (дихлорометан/метанол 1:05:1). Вихід: 0,19г (49% від теоретичного виходу) Мас-спектр (ІЕР+): m/z=430 [М+Н]+ Приклад 16 4-(β-D-глюкопіраноз-1-іл)-2-(4трифторометоксибензил)-бензонітрил У заповнену Аr колбу поміщають 2бромометил-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрил (0,25г), 4трифторометокси-фенілборонову кислоту (0,20г), карбонат калію (0,26) і суміш складу 3:1 дегазованого ацетону і води (4мл). Суміш перемішують при кімнатній температурі протягом 5хв., потім її охолоджують в бані з льодом. Потім додають паладійдихлорид (5мг) і реакційну суміш перемішують протягом 16год. при температурі навколишнього середовища. Потім суміш розбавляють розсолом і екстрагують етилацетатом. Об'єднані екстракти сушать над сульфатом натрію і розчинник вида 47 ляють при зниженому тиску. Залишок розчиняють в метанолі (9мл) і обробляють 4Μ водним розчином гідроксиду калію (1мл). Одержаний розчин перемішують при температурі навколишнього середовища протягом 1год. і потім нейтралізують 1Μ хлористоводневою кислотою. Метанол випаровують і залишок розбавляють розсолом і екстрагують етилацетатом. Зібрані органічні екстракти сушать над сульфатом натрію, і розчинник видаляють. Залишок хроматографують на силікагелі (дихлорометан/метанол 1:08:1). Вихід: 0,145г (69% від теоретичного значення) Мас-спектр (ІЕР+): m/z=457 [M+NH4]+ Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі 16: (17) 4-(β-D-глюкопіраноз-1-іл)-2-(4трифторометилбензил)-бензонітрил 94087 48 (22) 4-(β-D-глюкопіраноз-1-іл)-2-[4-(3метилбут-1-іл)-бензил]-бензонітрил Мас-спектр (ІЕР+): m/z=443 [M+NH4]+ (23) 2-(4-Фторобензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=391 [M+NH4]+ (24) 2-(4-Хлоробензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=441 [M+NH4]+ (18) 4-(β-D-глюкопіраноз-1-іл)-2-(4ізопропілбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=415 [M+NH4]+ (19) 4-(β-D-глюкопіраноз-1-іл)-2-(4-третбутилбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=429 [M+NH4]+ (20) 4-(β-D-глюкопіраноз-1-іл)-2-(4триметилсилілбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=445 [M+NH4]+ (21) 4-(β-D-глюкопіраноз-1-іл)-2-(4метилсульфанілбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=419 [M+NH4]+ Мас-спектр (ІЕР+): m/z=407/409 (СІ) [M+NH4]+ (25) 2-(4-Дифторометоксибензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил Мас-спектр (ШР+): m/z=439 [M+NH4]+ (26) 2-(4-Дифторометилбензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил Мас-спектр (ІЕР+): m/z=423 [M+NH4]+ (27-1) 2-(4-Циклопропілбензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил Мас-спектр (ІЕР+): m/z=413 [M+NH4]+ Сполуку отримують відповідно до прикладу 16 і замість 4-трифторометоксифенілборонової кислоти використовують едукт (XXII) 4циклопропілфенілборонової кислоти. Сполуку (27) також отримують за допомогою каталізованої перехідним металом реакції сполучення 2-(4-бромобензил)-4-(2,3,4,6-тетра-О 49 ацетил-β-D-глюкопіраноз-1-іл)-бензонітрилу або 1ціано-4-(2,3,4,6-тетра-О-ацетил-β-D-глюкопіраноз1-іл)-2-[4-(трифторометилсульфонілокси)-бензил]бензолу (сполука XVI(l)) з циклопропілбороновою кислотою з використанням трициклогексилфосфонійтетрафтороборату, ацетату паладію, фосфату калію в толуолі і води за методикою, описаною в публікації Tetrahedron Lett. 2002, 43, 6987-6990, з подальшим дезацетилуванням продукту сполучення за допомогою КОН в метанолі. Сполуку (27) також можна отримати за реакцією 2-фторо-4-(2,3,4,6-тетра-О-ацетил-Dглюкопіраноз-1-іл)-бензонітрилу (сполука ХІ(1)) і етил-4-циклопропілфенілацетату (сполука XXIV) в лужному середовищі з подальшим гідролізом складного ефіру і декарбоксилуванням. (27-2) 2-(4-Циклобутилбензил)-4-(β-Dглюкопіраноз-1-іл)-бензонітрил Сполуку отримують відповідно до прикладу (27-1), в якому, наприклад, замість 4циклопропілборонової кислоти використовують 4циклобутилборонову кислоту (яку можна отримати аналогічно тому, як описано в прикладі XXII). Мас-спектр (ІЕР+): m/z=427 [M+NH4]+ (28) 4-(β-D-глюкопіраноз-1-іл)-2-(4-проп-1ілбензил)-бензонітрил Мас-спектр (ІЕР+): m/z=415 [M+NH4]+ Приклад 29 4-(β-D-глюкопіраноз-1-іл)-2-(4йодобензилУбензонітрил 1Μ Розчин монохлориду йоду в дихлорометані (0,9мл) додають до 4-(β-D-глюкопіраноз-1-іл)-2-(4триметилсилілбензил)-бензонітрилу (0,26г), розчиненого в дихлорометані (5мл). Розчин перемішують при кімнатній температурі протягом 1год. і потім реакцію зупиняють шляхом додавання водного розчину Na2S2O3 і водного розчину NaHCO3. Органічну фазу відділяють і водну фазу екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник видаляють. Залишок хроматографують на силікагелі (дихлорометан/метанол 1:08:1). Вихід: 0,15г (88% від теоретичного значення) Мас-спектр (ІЕР+): m/z=499 [M+NH4]+ 94087 50 Вказані нижче сполуки можна отримати аналогічно тому, як описано в прикладі 29: (30) 2-(4-Бромобензил)-4-(β-D-глюкопіраноз-1іл)-бензонітрил Мас-спектр (ІЕР+): m/z=451/453 [M+NH4]+ Сполуку отримують за методикою, описаною в прикладі 29, з використанням брому в дихлорометані. Приклад 31 4-(β-D-глюкопіраноз-1-іл)-2-(4пентафтороетилбензил)-бензонітрил Колбу, в яку поміщені 4-(2,3,4,6-тетра-Оацетил-β-D-глюкопіраноз-1-іл)-2-(4-йодобензил)бензонітрил (0,16г), пентафтороетилтриметилсилан (0,14г), KF (43мг), CUI (0,16г), ДМФ (2мл), в атмосфері Аr нагрівають при 60°С протягом 24год. Потім додають водний розчин NaHCO3 і одержану суміш екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник видаляють. Залишок розчиняють в метанолі (8мл) і обробляють 4Μ розчином КОН (0,8мл). Розчин перемішують при кімнатній температурі протягом 1год. і потім розбавляють водним розчином NaHCO3. Після видалення метанолу при зниженому тиску залишок екстрагують етилацетатом, об'єднані органічні екстракти сушать і розчинник видаляють. Залишок хроматографують на силікагелі (дихлорометан/метанол 1:08:1). Вихід: 0,08г (69% від теоретичного значення) Мас-спектр (ІЕР+): m/z=491 [M+NH4]+ Приклад 32 4-(β-D-глюкопіраноз-1-іл)-2-(4-метилсульфінілбензил)-бензонітрил 35% Водний розчин пероксиду вуглецю (48мкл) додають до 4-(β-D-глюкопіраноз-1-іл)-2-(4метилсульфанілбензил)-бензонітрилу (83мг) в 1,1,1,3,3,3-гексафтороізопропанолі (2мл). Одержаний розчин перемішують при температурі навколишнього середовища протягом 1год. і потім реакцію зупиняють шляхом додавання водного розчину Na2S2O3 і водного розчину NaHCO3. Органічну фазу відділяють і водну фазу екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник видаляють. Залишок 51 хроматографують на силікагелі (дихлорометан/метанол 1:05:1). Вихід: 24мг (28% від теоретичного значення) Мас-спектр (ІЕР+): m/z=418 [М+Н]+ Приклад 33 4-(β-D-глюкопіраноз-1-іл)-2-(4метилсульфонілбензил)-бензонітрил 3-Хлоропероксибензойную кислоту (70%, 0,14г) додають до 4-(β-D-глюкопіраноз-1-іл)-2-(4метилсульфанілбензил)-бензонітрилу (100мг) в 94087 52 дихлорометані (2мл), охолодженому в бані з льодом. Баню, що охолоджує, видаляють і одержаний розчин перемішують при температурі навколишнього середовища протягом 1год. Після додавання водного розчину Na2S2O3 і водного розчину NaHCO3 органічну фазу відділяють і водну фазу екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і розчинник видаляють. Залишок хроматографують на силікагелі (дихлорометан/метанол 1:08:1). Вихід: 68мг (63% від теоретичного значення) Мас-спектр (ІЕР+): m/z=451 [M+NH4]+ Наступні сполуки також можна отримати аналогічно тому, як описано в наведених вище прикладах або за іншими методиками, відомими з літератури: 53 Нижче описані деякі приклади препаратів, в яких термін "активна речовина" означає одну або більшу кількість сполук, пропонованих в дійсному винаході, включаючи їх проліки або солі. У випадку однієї з комбінацій з однією або більшою кількістю додаткових активних речовин, описаних вище, термін "активна речовина" також включає додаткові активні речовини. Приклад А Таблетки, що містять 100мг активної речовини Склад: 1 таблетка містить: активна речовина 100,0мг лактоза 80,0мг кукурудзяний крохмаль 34,0мг полівінілпіролідон 4,0мг стеарат магнію 2,0мг 220,0мг. Методика приготування: Активну речовину, лактозу і крохмаль змішують і рівномірно зволожують водним розчином полівінілпіролідону. Після пропускання вологої композиції через сито (отвори розміром 2,0мм) і сушки в стелажній сушарці при 50°С її повторно пропускають через сито (отвори розміром 1,5мм) і додають змащуючу речовину. Готову суміш пресують і отримують таблетки. Маса таблетки: 220мг Діаметр: 10мм, біпланарна, з фасками з обох боків і насічкою з одного боку. Приклад Б Таблетки, що містять 150мг активної речовини Склад: 1 таблетка містить: активна речовина 150,0мг порошкоподібна лактоза 89,0мг кукурудзяний крохмаль 40,0мг колоїдний діоксид кремнію 10,0мг полівінілпіролідон 10,0мг стеарат магнію 1,0мг 300,0мг. Приготування: 94087 54 Активну речовину змішують з лактозою, кукурудзяним крохмалем і діоксидом кремнію і зволожують 20% водним розчином полівінілпіролідону і пропускають через сито з отворами розміром 1,5мм. Гранули, висушені при 45°С, повторно пропускають через те ж сито і змішують вказаною кількістю стеарата магнію. Таблетки пресують з суміші. Маса таблетки: 300мг Пуансон: 10мм, плоский Приклад В Капсули з твердого желатину, що містять 150мг активної речовини Склад: 1 капсула містить: активна речовина 150,0мг кукурудзяний крохмаль (висушений) приблизно 180,0мг лактоза (порошкоподібна) приблизно 87,0мг стеарат магнію 3,0мг приблизно 420,0мг. Приготування: Активну речовину змішують з інертними наповнювачами, пропускають через сито з отворами розміром 0,75мм і перемішують до однорідного стану в підходящому апараті. Готову суміш розфасовують в капсули з твердого желатину розміру 1. Вміст капсули: приблизно 320мг Оболонка капсули: капсула з твердого желатину розміру 1. Приклад Г Супозиторії, що містять 150мг активної речовини Склад: 1 Супозиторій містить: активна речовина 150,0мг поліетиленгліколь 1500 550,0мг поліетиленгліколь 6000 460,0мг поліоксіетиленсорбітанмоностеарат 840,0мг 2000,0мг. Приготування: Після плавлення маси супозиторію в ній рівномірно розподіляють активну речовину і розплав виливають в охолоджені форми. 55 94087 Приклад Γ Ампули, що містять 10мг активної речовини Склад: активна речовина 10,0мг 0,01н. хлористоводнева кислота скільки потрібний бідистильована вода до 2,0мл. Приготування: Активну речовину розчиняють у необхідній кількості 0,01н. НСI, розчин роблять ізотонічним шляхом додавання хлориду натрію, стерильно фільтрують і поміщають в ампули об'ємом 2мл Комп’ютерна верстка Т. Чепелева 56 Приклад Д Ампули, що містять 50мг активної речовини Склад: активна речовина 50,0мг 0,01н. хлористоводнева кислота скільки потрібний бідистильована вода до 10,0мл. Приготування: Активну речовину розчиняють у необхідній кількості 0,01н. НСI, розчин роблять ізотонічним шляхом додавання хлориду натрію, стерильно фільтрують і поміщають в ампули об'ємом 10мл. Підписне Тираж 23 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюGlucopyranosyl-substituted benzonitrile derivatives
Автори англійськоюEckhardt Matthias, Eickelmann Peter, Himmelsbach Frank, Sauer Achim, Thomas Leo
Назва патенту російськоюГлюкопиранозилзамещенные производные бензонитрила
Автори російськоюЭкхардт Маттиас, Айкельманн Петер, Химмельсбах Франк, Зауэр Ахим, Томас Лео
МПК / Мітки
МПК: C07D 309/10, A61K 31/351
Мітки: похідні, бензонітрилу, глюкопіранозилзаміщені
Код посилання
<a href="https://ua.patents.su/28-94087-glyukopiranozilzamishheni-pokhidni-benzonitrilu.html" target="_blank" rel="follow" title="База патентів України">Глюкопіранозилзаміщені похідні бензонітрилу</a>
Спосіб розділення 4-[4-(диметиламіно)-1-(4′-фторфеніл)-1-гідроксибутил]-3-(гідроксиметил)-бензонітрилу у вигляді рацемічної або нерацемічної суміші енантіомерів на окремі енантіомери

Номер патенту: 41919
Опубліковано: 10.06.2009
Автори: Хубер Флоріан Антон Мартин, Дансер Роберт Джеймс, Де Фавері Карла
МПК: A61K 31/00
Мітки: розділення, вигляді, нерацемічної, суміші, енантіомери, окремі, рацемічної, 4-[4-(диметиламіно)-1-(4'-фторфеніл)-1-гідроксибутил]-3-(гідроксиметил)-бензонітрилу, спосіб, енантіомерів
Формула / Реферат:
1. Спосіб розділення 4-[4-(диметиламіно)-1-(4'-фторфеніл)-1-гідроксибутил]-3-(гідроксиметил)-бензонітрилу у вигляді рацемічної або нерацемічної суміші енантіомерів на окремі енантіомери, який включає стадію фракційної кристалізації 4-[4-(диметиламіно)-1-(4'-фторфеніл)-1-гідроксибутил]-3-(гідроксиметил)-бензонітрилу у формі солі з (+)-(S,S)- або (-)-(R,R)-енантіомером О,О'-ди-n-толуоїл-винної кислоти, який відрізняється тим, що фракційну...
Гідрохлорид 4-[[4-[[4-(2-ціаноетеніл)-2,6-диметилфеніл]аміно]-2-піримідиніл]аміно]бензонітрилу

Номер патенту: 92467
Опубліковано: 10.11.2010
Автори: Стевенс Поль Теодоор Агнес, Копманс Алекс Герман, Вандекрюйс Роже Петрус Гереберн, Пеетерс Йозеф
МПК: A61K 31/505, A61P 31/18
Мітки: гідрохлорид, 4-[[4-[[4-(2-ціаноетеніл)-2,6-диметилфеніл]аміно]-2-піримідиніл]аміно]бензонітрилу
Формула / Реферат:
1. Тверда фармацевтична композиція, що містить фармацевтично прийнятний носій і як активний інгредієнт терапевтично ефективну кількість сполуки формули (І), (I)її N-оксиду або стереохімічно ізомерної форми.2. Фармацевтична композиція за п. 1, яка відрізняється тим, що сполука формули (І) являє собою сполуку формули (І-а)
Фумарат 4-[[4-[[4-(2-ціаноетеніл)-2,6-диметилфеніл]аміно]-2-піримідиніл]аміно]бензонітрилу
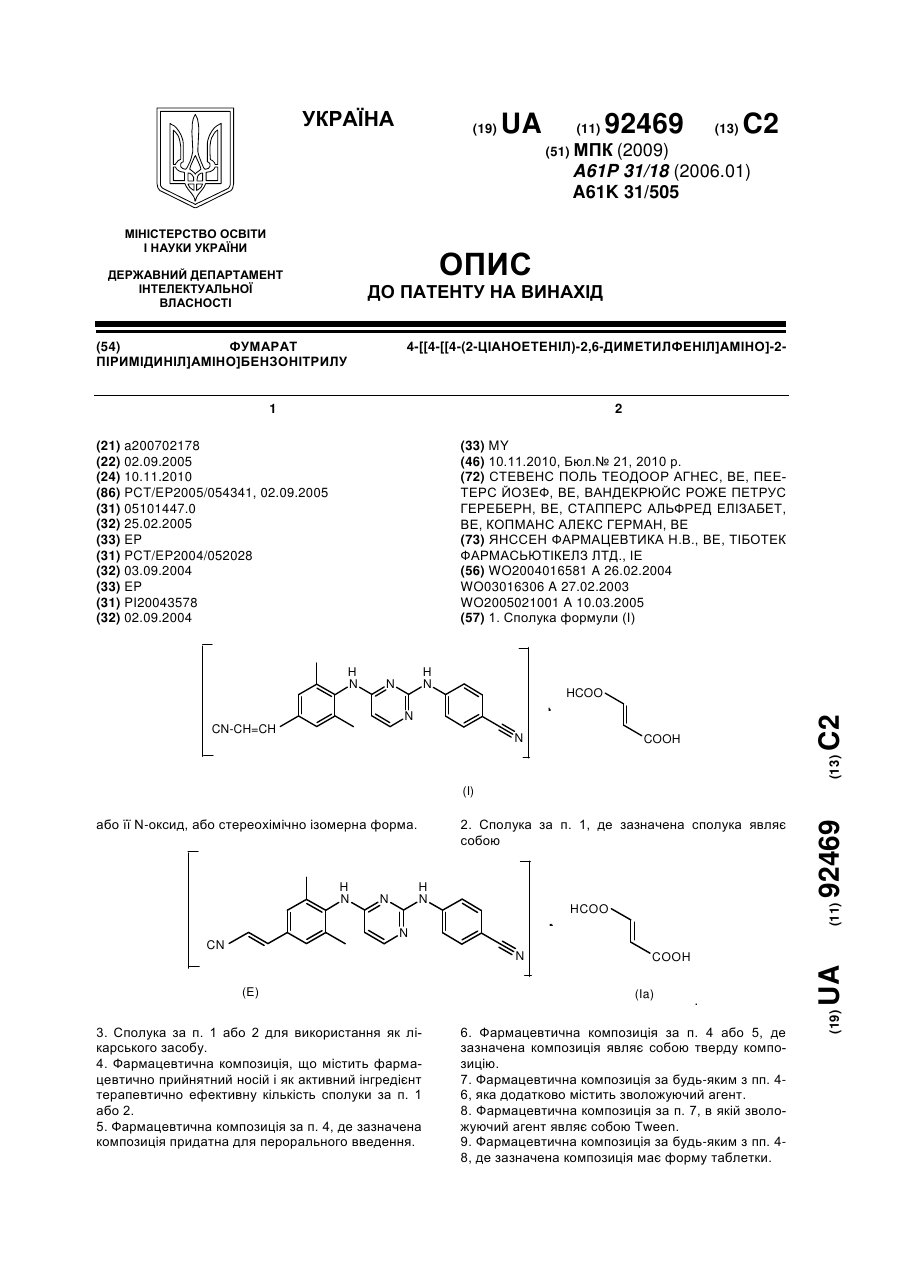
Номер патенту: 92469
Опубліковано: 10.11.2010
Автори: Стевенс Поль Теодоор Агнес, Стапперс Альфред Елізабет, Копманс Алекс Герман, Пеетерс Йозеф, Вандекрюйс Роже Петрус Гереберн
МПК: A61P 31/18, A61K 31/505
Мітки: фумарат, 4-[[4-[[4-(2-ціаноетеніл)-2,6-диметилфеніл]аміно]-2-піримідиніл]аміно]бензонітрилу
Формула / Реферат:
1. Сполука формули (І)або її N-оксид, або стереохімічно ізомерна форма. 2. Сполука за п. 1, де зазначена сполука являє собою. 3. Сполука за п. 1 або 2 для використання як лікарського засобу. 4. Фармацевтична...
Фармацевтичні композиції, що містять похідні 3-аміноазетидину, похідні і спосіб їхнього одержання

Номер патенту: 72319
Опубліковано: 15.02.2005
Автори: Боушард Херве, Філош Бруно, Грізоні Серж, Ашард Даніель, Хіттінгер Огюстін, Букерель Жан, Міерс Мішель
МПК: A61K 31/4025, A61P 11/06, A61P 1/08, A61P 23/00, A61P 11/00, C07D 205/00, A61P 1/14, A61K 31/4427, A61P 9/08, A61K 31/4709, A61P 37/00, C07D 401/12, A61P 1/00, A61P 25/16, A61K 31/4178, A61P 27/02, A61P 9/02, A61P 9/00, A61K 31/506, A61P 25/28, A61P 25/02, A61P 25/08, A61P 29/00, A61K 47/00, C07D 403/12, A61K 31/397, A61P 25/18, A61P 25/22, A61P 25/00, A61P 35/00, A61P 25/24, A61P 25/20, C07D 401/06, A61P 25/14, A61P 5/00, C07D 403/08, A61P 3/04, C07D 409/12
Мітки: спосіб, одержання, фармацевтичні, похідні, композиції, їхнього, 3-аміноазетидину, містять
Формула / Реферат:
1. Фармацевтична композиція, що містить як активний інгредієнт сполуку формули: , (І)у якійR1 означає радикал –NНСОR4 або -N(R5)-Y-R6,Υ означає CO або SO2,R2 і R3, однакові або різні, означають або ароматичний радикал, вибраний з фенілу, нафтилу і інденілу, що можуть бути незаміщеними або заміщеними одним або декількома замісниками:...
Похідні ди- або трифторметансульфоніланіліду, спосіб їх одержання і гербіциди, що містять вказані похідні як активні інгредієнти

Номер патенту: 56338
Опубліковано: 15.05.2003
Автори: Янасігава Кацутада, Йосімура Такумі, Накатані Масао, Тамару Масатосі, Оно Юкімаса, Дандзо Такесі
МПК: C07D 239/52, A01N 41/06, A01N 43/54, C07C 317/36
Мітки: гербіциди, вказані, активні, спосіб, містять, трифторметансульфоніланіліду, одержання, похідні, дії, інгредієнти
Формула / Реферат:
1. Похідне ди- або трифторметансульфоніланіліду, представлене наступною загальною формулою:,де R1 являє атом водню, алкільну групу або алкоксіалкільну групу; і R2 являє атом водню, коли R1 являє атом водню або алкільну групу, і являє атом водню або атом фтору, коли R1 являє алкоксіалкільну групу, або його сіль.2. Похідне ди- або трифторметансульфоніланіліду або його сіль за п. 1, в якому R1 являє атом водню.3....
Попередній патент: Протиінфекційний розчин, який містить сполуку піридо(3,2,1-ij)бензоксадіазинового типу
Наступний патент: Композиція з інгібувальною дією по відношенню до уреази та спосіб її одержання
Випадковий патент: Комбінований очисник вороху коренеплодів цикорію