Сполуки, що інгібують дипептидилпептидазу-iv і фармацевтична композиція, яка містить їх як активний агент
Номер патенту: 103181
Опубліковано: 25.09.2013
Автори: Йім Хієон Дзоо, Лі Кіу Воонг, Парк Ван Су, Лі Сунг-Хак, Лі Хеє Бонг, Лі Чанг-Сеок, Лі Чжеік, Кім Кіоунг-Хеє, Мінь Чангхі








Формула / Реферат
1. Сполука, представлена формулою 1:
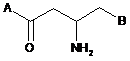 , (1)
, (1)
де
А вибирають із замісника формули 2:
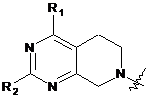 , (2)
, (2)
де
R1 являє собою водень або CF3, і
R2 вибирають із групи, що складається з водню, заміщеного або незаміщеного С1-С10алкілу, заміщеного або незаміщеного С3-С10циклоалкілу, заміщеного або незаміщеного С3-С10гетероциклоалкілу, заміщеного або незаміщеного С4-С8арилу й заміщеного або незаміщеного С3-С7гетероарилу; і
замісника формули 3:
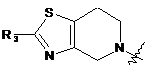 , (3)
, (3)
де R3 вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу; і
В вибирають із групи, що складається із замісника формули 4:
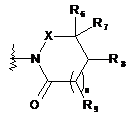 , (4)
, (4)
де
X являє собою -CR4R5- або -СО-, де R4 і R5, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R4 і R5 являє собою гідрокси,
R6, R7, R8 і R9, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу, і
n дорівнює 0 або 1;
замісника формули 5:
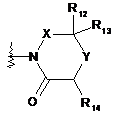 , (5)
, (5)
де
X являє собою -CR10R11- або -СО-, де R10 і R11, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R10 і R11 являє собою гідрокси,
R12, R13 і R14, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу, і
Y являє собою кисень або сірку;
замісника формули 6:
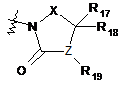 , (6)
, (6)
де
X являє собою -CR15R16- або -СО-, де R15 і R16, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R15 і R16 являє собою гідрокси,
Z являє собою -СН- або кисень, за умови, що, коли Z являє собою кисень, R19 відсутній, і
R17, R18 і R19, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу; і
замісника формули 7:
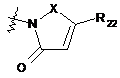 , (7)
, (7)
де
X являє собою -CR20R21- або -CO, де R20 і R21, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R20 і R21 являє собою гідрокси, і
R22 являє собою заміщений або незаміщений С1-С4алкіл;
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, в якій, коли алкіл, циклоалкіл, гетероциклоалкіл, арил або гетероарил є заміщеним, замісник являє собою С1-С10алкіл або галоген.
3. Сполука за п. 2, в якій галоген являє собою фтор.
4. Сполука за п. 1, в якій А у формулі 1 являє собою замісник, представлений формулою 2, де R1 являє собою водень або CF3 і R2 вибирають із групи, що складається з водню й С1-С5алкілу, C3-C6циклоалкілу, С3-С10гетероциклоалкілу, С4-С8арилу й С3-С7гетероарилу, кожний з яких необов'язково заміщений галогеном.
5. Сполука за п. 4, в якій, коли R2 являє собою незаміщений або галогензаміщений С3-С10гетероциклоалкіл або незаміщений або галогензаміщений С3-С7гетероарил, вказаний гетероциклоалкіл або гетероарил являє собою будь-яку групу, вибрану із групи, що складається з фурану, тіофену, піролу, піролідину, імідазолу, піразолу, піразоліну, оксазолу, оксазоліну, ізоксазолу, ізоксазоліну, тіазолу, тіазоліну, ізотіазолу, ізотіазолідину, тіадіазолу, тіадіазоліну, тетрагідрофурану, тетрагідротіофену, імідазолідину, піразолідину, оксазолідину, ізоксазолідину, тіазолідину, ізотіазолідину, тіадіазолідину, сульфолану, пірану, дигідропірану, тетрагідропірану, піридину, піридинону, піридазину, піразину, піримідину, піперидину, піперазину, морфоліну, піридазинону, тетразолу, триазолу, триазолідину й азепіну.
6. Сполука за п. 4, в якій R2 вибирають із групи, що складається із трифторметилу, пропілу, бутилу, трет-бутилу, циклобутилу, піридину, фурану, метоксіетилу, тіофену й 4-фторфенілу.
7. Сполука за п. 1, в якій В у формулі 1 являє собою замісник, представлений формулою 4, де X являє собою -(СН-ОН)- або -CO-, R6 і R7, кожний, незалежно вибирають із групи, що складається з водню, фтору й незаміщеного С1-С4алкілу, і R8 і R9, кожний, незалежно являють собою водень.
8. Сполука за п. 1, в якій сполука являє собою сполуку формули 1а
 , (1a)
, (1a)
де А і В є такими, як визначено для формули 1.
9. Сполука за п. 1, в якій сполука являє собою будь-яку з наступних сполук:
1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтop-(6R)-гiдpoкcипіпepидин-2-oн,
1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-(6S)-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-пропіл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-(2-циклобутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он,
1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіпeридин-2-он,
1-{(S)-2-аміно-4-[2-(4-фторфеніл)-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-3,3-дифторпіперидин-2,6-діон,
1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5-фтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5-фтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-пропіл-5-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-5-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-он,
1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]-3-метилпіперидин-2,6-діон і
1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-3-метилпіролідин-2,6-діон.
10. Фармацевтична композиція для інгібування дипептидилпептидази-IV (DPP-IV), яка містить сполуку формули 1 за п. 1 або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій, розріджувач і наповнювач або будь-яке їх поєднання.
11. Фармацевтична композиція за п. 10, де композиція призначена для лікування або запобігання захворюванню, пов'язаному з DPP-IV.
12. Фармацевтична композиція за п. 11, в якій захворювання, пов'язане з DPP-IV, являє собою діабет або ожиріння.
Текст
Реферат: У даному винаході описуються сполуки формули (1), як зазначено в описі, що мають чудову інгібіторну активність проти дипептидилпептидази-IV (DPP-IV), способи їх одержання й фармацевтичні композиції, що містять їх як активний агент. A B O NH2 (1) UA 103181 C2 (12) UA 103181 C2 UA 103181 C2 40 Галузь техніки, до якої належить винахід Даний винахід належить до нових сполук, які мають чудову інгібіторну активність проти дипептиділпептидази-IV (DPP-IV), до способів їх одержання і до фармацевтичних композицій, які містять їх як активний агент. Рівень техніки Діабет надає значні негативні серйозні впливи на здоров'я людей і супроводжується різними ускладненнями. Є два головні типи діабету: цукровий діабет типу I, який відрізняється зниженою здатністю до вироблення інсуліну або її відсутністю через руйнування клітин підшлункової залози, і цукровий діабет типу II, який відрізняється дефіцитом інсуліну і резистентністю до інсуліну з інших причин. Захворюваність цукровим діабетом типу II становить 90 % або більше від загальної кількості пацієнтів з діабетом. Репрезентативні приклади діабетичних ускладнень включають гіперліпідемію, гіпертонію, ретинопатію і ниркову недостатність (Paul Zimmer, et al, Nature, 2001, 414, 782). Сульфонілсечовини (стимуляція секреції інсуліну в підшлунковій залозі), бігуаніди (інгібування продукції глюкози в печінці), інгібітори α-глюкозидази (пригнічення поглинання глюкози в кишечнику), і тому подібне, використовують як антидіабетичні агенти. Останнім часом, агоністи проліфератор-активованих рецепторів гамма пероксисом (PPARγ) (триазолідиндіони, підвищення чутливості до інсуліну) привертають велику увагу як перспективні антидіабетичні терапевтичні засоби. Однак ці антидіабетичні лікарські засоби мають такі негативні побічні впливи, як гіпоглікемія, збільшення маси тіла, і тому подібне (David Е. Moller, Nature, 2001, 414, 821). У даний час, є гостра необхідність в розробці терапевтичних засобів проти діабету із зменшеними побічними впливами, зокрема, не викликаючих гіпоглікемії і збільшення маси тіла. Тим часом, недавно виявлено, що миші, нокаутні за дипептиділпептидазою-IV (DPP-IV), підтримують активність глюкагон-подібного білка 1 (GLP-1) і високі рівні інсуліну, що призводить до зменшення рівнів глюкози в крові, якщо говорити про терапевтичну корисність DPP-IV як антидіабетичного агента (Marguet D. et al, Natl. Acad. Sci. USA, (2000) 97, 6874-6879). GLP-1 залучений до диференціації і росту β-клітин підшлункової залози in vivo і грає важливу роль в продукції і секреції інсуліну. GLP-1 дезактивується під дією DPP-IV, і інгібітори DPP-IV як повідомляється, збільшують секрецію інсуліну за допомогою інгібування дезактивації GLP-1. Інгібітори DPP-IV також розробляються як лікарські засоби проти ожиріння, оскільки GLP-1 вносить внесок у відчуття насичення і наповненості у щурів і сповільнює кишкове переварювання їжі, призводячи до втрати маси. Крім того, багато дослідників і наукових установ також продемонстрували, що інгібітори DPP-IV контролюють рівні глюкози і ліпідів в крові в експериментах на тваринних моделях (Pospislik J. A., et al, Diabetes, (2002) 51, 943-950). У цьому відношенні, інгібітори DPP-IV можуть розглядатися як потенційно корисні агенти для лікування діабетичних захворювань. До теперішнього часу, велика кількість досліджень відносно виявлення і розробки поліпшених інгібіторів DPP-IV зосереджені на речовинах, в яких ціаногрупа вводиться в піролідинове кільце. Наприклад, WO 00/34241 описує інгібітори DPP-IV, представлені наступною формулою 45 , де R являє собою заміщений адамантил, і n знаходиться в межах від 0 до 3. Інші інгібітори можна знайти в WO 04/064778, WO 03/004498, WO 03/082817, і тому подібне. Описані в WO 04/064778 DPP-IV являють собою інгібітори DPP-IV, представлені наступною формулою 5 10 15 20 25 30 35 , 1 UA 103181 C2 5 де Ar являє собою незаміщений або заміщений феніл; R 15, R16 і R17, кожний, незалежно являють собою водень або алкіл; і U, V і W, кожний, незалежно являють собою азот, кисень, або заміщений азот або вуглець. У доповнення до цього, WO 03/004498 пропонує інгібітори DPP-IV, представлені наступною формулою , де Ar являє собою незаміщений або заміщений феніл; R 18 являє собою водень або алкіл і Т являє собою азот або заміщений вуглець. Крім того, WO 03/082817 описує інгібітори DPP-IV, представлені наступною формулою 10 15 20 25 30 35 40 , де Ar являє собою незаміщений або заміщений феніл; R 19, R20 і R21, кожний, незалежно являють собою водень або алкіл і Q являє собою азот або заміщений вуглець. У публікації WO 06/104356, що належить до інгібіторів DPP-IV, яка належить автору даної заявки і озаглавленій "Dipeptidyl peptidase-IV inhibiting compounds", автори даного винаходу демонструютьширокі і вичерпні впливи сполук, представлених формулою I', нижче. Сполуки формули I' являють собою циклічні сполуки, що мають кільця, з'єднані за допомогою амідного зв'язку, які подібні тим сполукам інгібіторів DPP-IV, які наведені в розглянутих вище публікаціях патентів; однак молекулярні структури, заміщені фенільною групою, яка представлена як Ar або Z, у вказаних вище документах, повністю відрізняються від структур, заміщених насиченим або ненасиченим, 5-членним або 6-членним циклічним залишком, як в даному винаході. Крім того, наскільки відомо авторам, в даній галузі ще немає опису інгібіторів DPP-IV відповідно до даного винаходу, що мають молекулярну структуру, де лактамне кільце заміщене в фенільному положенні. Крім того, автори даного винаходу виявили, що сполуки формули I', описані в попередньому патенті авторів даного винаходу, можуть демонструвати значно поліпшену інгібіторну активність і селективність по відношенню до DPP-IV за допомогою введення амінів, що мають високі інгібіторні впливи на DPP-IV, в замісник А і введення різних замісників (таких як гідрокси або карбонільна група) в замісник В. Зокрема, приймаючи до уваги останнє повідомлення, яке демонструє, що низька селективність відомих сполук по відношенню до інших дипептиділпептидаз може призводити до різноманітних негативних впливів (Lankas, G. R. et al., Potential Importance of Selectivity over Dipeptidyl Peptidases 8 and 9, Diabetes, 2005, 548, 2988-1994), даний винахід забезпечує вкрай значні результати з точки зору чудової фармацевтичної ефективності і високої селективності сполук за даним винаходом по відношенню до DPP-IV. Крім того, сполуки інгібітори DPP-IV за даним винаходом, які мають вказану вище хімічну структуру, і способи їх одержання не описані в літературі. Суть винаходу Внаслідок різноманітних широких і інтенсивних досліджень і експериментів для розв'язання проблем, як описано вище, і для виявлення сполук, що мають чудову інгібіторну активність і селективність по відношенню до DPP-IV, автори даного винаходу виявили, як буде проілюстровано нижче, що сполуки, які мають необов'язково заміщену структуру лактамного кільця, є ефективними як інгібітори DPP-IV. Даний винахід завершений на основі цих даних. 2 UA 103181 C2 5 10 15 20 25 30 З цієї причини метою даного винаходу є створення сполук інгібіторів DPP-IV, що мають структуру лактамного кільця, яке є необов'язково заміщеним, зокрема, гідрокси або карбонілом. Іншою метою даного винаходу є створення способів одержання цих сполук. Крім того, метою даного винаходу є створення фармацевтичних композицій, що містять ці сполуки, і способів лікування або запобігання захворюванням, пов'язаним з DPP-IV, що включають введення ефективної кількості цих сполук суб'єкту, потребуючому цього. Відповідно до аспекту даного винаходу, вказані вище і інші цілі можуть досягатися за допомогою створення сполук інгібітора дипептиділпептидази-IV (DPP-IV), представленого формулою 1: , де А вибирають із замісника формули 2: , де R1 являє собою водень або CF3, і R2 вибирають з групи, що складається з водню, заміщеного або незаміщеного C 1-C10 алкілу, заміщеного або незаміщеного C3-C10 циклоалкілу, заміщеного або незаміщеного C 4-C8 арилу і заміщеного або незаміщеного C3-C7 гетероарилу; і замісника формули 3: , де R3 вибирають з групи, що складається з водню, галогену і заміщеного або незаміщеного C1-C4 алкілу; і В вибирають з групи, що складається із замісника формули 4: , де X являє собою -CR4R5- або -CO-, де R4 і R5, кожний, незалежно являє собою водень або гідрокси, за умови, що щонайменше один з R4 і R5 являє собою гідрокси, R6, R7, R8 і R9, кожний, незалежно вибирають з групи, що складається з водню, галогену і заміщеного або незаміщеного C1-C4 алкілу, і n дорівнює 0 або 1; замісника формули 5: , 3 UA 103181 C2 5 10 15 20 25 30 35 40 45 де X являє собою -CR10R11- або -CO-, де R10 і R11, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R10 і R11 являє собою гідрокси, R12, R13 і R14, кожний, незалежно вибирають з групи, що складається з водню, галогену і заміщеного або незаміщеного C1-C4 алкілу, і Y являє собою кисень або сірку; замісника формули 6: , де X представляє собою -CR15R16- або -CO-, де R15 і R16, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R15 і R16 являє собою гідрокси, R17, R18 і R19, кожний, незалежно вибирають з групи, що складається з водню, галогену і заміщеного або незаміщеного C1-C4 алкілу, і Z являє собою -CH- або кисень, за умови, що коли Z являє собою кисень, R19 відсутній; і замісника формули 7: , де X являє собою -CR20R21- або -CO-, де R20 і R21, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R20 і R21 являє собою гідрокси, і R22 являє собою заміщений або незаміщений C1-C4 алкіл. Відповідно до різних експериментів, здійснених авторами даного винаходу, підтверджується, що сполуки формули 1 демонструють значне поліпшення інгібіторної активності і селективності по відношенню до DPP-IV за рахунок введення аміногруп, які мають чудові інгібіторні впливи по відношенню до DPP-IV, в замісник А і введення певної групи замісника (такої як гідрокси або карбонільна група) як замісник В в існуючу кільцеву структуру. Крім того, сполуки за даним винаходом можуть утворювати адитивні солі з фармацевтично прийнятними кислотами. Термін "фармацевтично прийнятна сіль" означає форму сполуки, яка не спричиняє значного подразнення в організмі, в який вона вводиться, і не погіршує біологічної активності і властивостей сполуки. Приклади фармацевтичної солі можуть включати кислотно-адитивні солі сполуки з кислотами, здатними до утворення нетоксичної кислотно-адитивної солі, що містить фармацевтично прийнятні аніони, наприклад, з неорганічними кислотами, такими як хлористоводнева кислота, сірчана кислота, азотна кислота, фосфорна кислота, бромистоводнева кислота і йодистоводнева кислота; з органічними карбоновими кислотами, такими як винна кислота, мурашина кислота, лимонна кислота, оцтова кислота, трихлороцтова кислота, трифтороцтова кислота, глюконова кислота, бензойна кислота, молочна кислота, фумарова кислота, малеїнова кислота і саліцилова кислота; або з сульфоновими кислотами, такими як метансульфонова кислота, етансульфонова кислота, бензолсульфонова кислота і птолуолсульфонова кислота. Зокрема, приклади фармацевтично прийнятних солей карбонових кислот включають солі з лужними металами або лужноземельними металами, такими як літій, натрій, калій, кальцій і магній, солі з амінокислотами, такими як лізин, аргінін і гуанідин, і солі з органічними основами, такими як дициклогексиламін, N-метил-D-глюкамін, трис(гідроксиметил)метиламін, діетаноламін, холін і триетиламін. Сполука формули 1 відповідно до даного винаходу може перетворюватися на її сіль, за допомогою звичайних способів, відомих в даній галузі. Якщо не вказано іншого, термін "сполука формули 1" означає сполуки формули 1 самі по собі, а також їх фармацевтично прийнятні солі. Як використовується в цьому документі, термін "алкіл" означає радикал, який не має ненасичених груп і складається з атомів вуглецю і водню. Алкільний радикал може бути 4 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 55 60 лінійним або розгалуженим. Приклади алкілу можуть включати, але не обмежуючись цим, метил, етил, пропіл, ізопропіл, гексил, трет-бутил і втор-бутил. Нижчий алкіл являє собою C1-C10 алкіл (наприклад, алкіл, що має 1-10 атомів вуглецю в лінійному або розгалуженому алкільному основному ланцюгу). Алкіл може бути необов'язково заміщеним. Коли він є заміщеним, алкіл може бути заміщений чотирма або менше замісниками в будь-якій точці приєднання (при будь-якому атомі вуглецю). Коли алкіл є заміщеним іншим алкілом, він, як передбачається, також згадується як "розгалужений алкіл". Як використовується в цьому документі, термін "циклоалкіл" належить до алкільних груп, які містять 3-15 атомів вуглецю, переважно, 3-8 атомів вуглецю, без утворення або зв'язаних подвійних зв'язків, що чергуються між атомами вуглецю. Циклоалкіл може містити 1-4 кільця. Приклади циклоалкілу можуть включати циклопропіл, циклобутил, циклопентил, циклогексил і адамантил. Ілюстративні замісники циклоалкілу можуть включати галоген, C1-C10 алкіл, C1-C10 алкокси, C1-C10 алкіл гідрокси, аміно, нітро, ціано, тіол і C1-C10 алкілтіо. Гетероциклоалкіл являє собою насичені або насичені, 7-11-членні біциклічні гетероциклічні кільцеві системи або стабільні неароматичні 3-8-членні моноциклічні гетероциклічні кільцеві системи, або може являти собою конденсовані, спіро або місточкові кільцеві системи, де кільцеві атоми вуглецю замінені гетероатомами, такими як азот (N), сірка (S) або кисень (О). Кожне гетероциклічне кільце складається з одного або декількох атомів вуглецю і 1-4 гетероатомів. Гетероциклоалкіл може приєднуватися на будь-якому ендоциклічному атомі вуглецю, коли це призводить до утворення стабільної структури. Переважні приклади гетероциклічних груп можуть включати, але не обмежуючись цим, фуран, тіофен, пірол, піролін, піролідин, оксазол, тіазол, імідазол, імідазолін, імідазолідин, піразол, піразолін, піразолідин, ізотіазол, триазол, тіадіазол, піран, піридин, піперидин, морфолін, тіоморфолін, піридазин, піримідин, піразин, піперазин і триазин. Приклади гетероциклічних кілець можуть включати, але не обмежуючись цим, 3-7-членні моноциклічні гетероциклічні кільця, такі як піперидинільні, піранільні, піперазинільні, морфолінільні, тіоморфолінільні і тетрагідрофуранільні, і більш переважно, 3-7-членні моноциклічні гетероциклічні кільця. Термін "арил" належить до ароматичної групи, яка має щонайменше одне кільце, що має зв'язану пі (π) електронну систему і включає карбоциклічну арильну (наприклад, фенільну) і гетероциклічну арильну (наприклад, піридинову) групи. Цей термін, як передбачається, включає моноциклічні або маючі конденсовані кільця поліциклічні (тобто групи з кільцями, які мають загальні сусідні пари атомів вуглецю) групи. Арильна група може необов'язково містити 1-4 гетероатомів (наприклад, азот (N), сірку (S) або кисень (О)) в карбоциклічному або ароматичному кільці, і така арильна система, що містить гетероатом, також згадується як "гетероарил". Приклади арильних або гетероарильних груп можуть включати, але не обмежуючись цим, феніл, нафтил, піридил, піримідил, піроліл, ізотіазоліл, триазоліл, тетразоліл, піразоліл, оксазоліл, ізоксазоліл, піразиніл, піридазиніл, триазиніл, хіназолініл, тіазоліл, бензотіофеніл, фураніл, імідазоліл і тіофеніл. У сполуці формули 1 відповідно до даного винаходу, коли алкіл, циклоалкіл, гетероциклоалкіл, арил або гетероарил є заміщеним, замісник може являти собою C 1-C10 алкіл або галоген, особливо переважно, фтор-заміщений C1-C10 алкіл. В одному з переважних варіантів здійснення даного винаходу, А в формулі 1 являє собою замісник, представлений формулою 2, де R1 являє собою водень або CF3, і R2 вибирають з групи, що складається з водню і C1-C5 алкілу, C3-C6 циклоалкілу, C3-C10 гетероциклоалкілу, C4C8 арилу і C3-C7 гетероарилу, кожний з яких може бути необов'язково заміщений галогеном. Коли R2 являє собою незаміщений або галоген-заміщений C3-C10 гетероциклоалкіл або незаміщений або галоген-заміщений C3-C7 гетероарил, гетероциклоалкіл або гетероарил можуть являти собою будь-яку групу, вибрану з групи, що складається з фурану, тіофену, піролу, піролідину, імідазолу, піразолу, піразоліну, оксазолу, оксазоліну, ізоксазолу, ізоксазоліну, тіазолу, тіазоліну, ізотіазолу, ізотіазолідину, тіадіазолу, тіадіазоліну, тетрагідрофурану, тетрагідротіофену, імідазолідину, піразолідину, оксазолідину, ізоксазолідину, тіазолідину, ізотіазолідину, тіадіазолідину, сульфолану, пірану, дигідропірану, тетрагідропірану, піридину, піридинону, піридазину, піразину, піримідину, піперидину, піперазину, морфоліну, піридазинону, тетразолу, триазолу, триазолідину і азепіну. У більш переважному варіанті здійснення даного винаходу, R 2 може вибиратися з групи, що складається з трифторметилу, пропілу, бутилу, трет-бутилу, циклобутилу, піридину, фурану, метоксіетилу, тіофену і 4-фторфенілу. Крім того, В в формулі 1 являє собою замісник, представлений формулою 4, де X являє 5 UA 103181 C2 собою -(CH-OH)- або -CO-, R6 і R7, кожний, незалежно вибирають з групи, що складається з водню, фтору і незаміщеного C1-C4 алкілу, і R8 і R9, кожний, незалежно являє собою водень. Сполуки за даним винаходом охоплюють їх ізомери. Особливо переважними є сполуки, де NH2-заміщений атом вуглецю утворює стереогенний центр, як показано в структурі формули 1a 5 10 15 20 25 30 35 40 45 50 55 , де А і В є такими, як визначено для формули 1. Особливо переважно, необмежувальні приклади сполук формули 1 відповідно до даного винаходу можуть включати наступні сполуки: 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5- дифтор-(6R)-гідроксипіперидин-2-он, 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5-дифтор-(6S)-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-циклобутил-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)бутил]-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)бутил]-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он, 1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-{(S)-2-аміно-4-[2-(4-фторфеніл)-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-3,3- дифторпіперидин-2,6-діон, 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-5-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)бутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-5-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин7-іл)бутил]-3-метилпіперидин-2,6-діон і 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-3-метилпіролідин-2,6-діон. Сполуки за даним винаходом або їх фармацевтично прийнятні солі можуть знаходитися в формі гідратів або сольватів. Крім того, даний винахід передбачає спосіб одержання сполук формули 1. Ілюстративний спосіб одержання сполук формули 1 включає взаємодію сполуки формули 8 зі сполукою формули 9 і видалення амінозахисної групи P 1 6 UA 103181 C2 5 10 15 20 25 30 35 , AH2F1 (9). У формулах 8 і 9, А і В є такими, як визначено вище; P1 являє собою аміно-захисну групу; і F1 відсутній або являє собою хлористоводневу кислоту, сірчану кислоту або трифтороцтову кислоту. [Схема реакції 1] На Схемі реакції 1, А, В і P1 є такими, як визначено вище; і G1H являє собою хлористоводневу кислоту, сірчану кислоту або трифтороцтову кислоту. Реагенти і умови реакції a: EDCI, HOBT, Et3N або AH2G1H b: G1H Конкретно, сполука 1 може бути одержана таким чином. Спочатку, бажану аміногрупу вводять в сполуку формули 8 за допомогою реакції конденсації з використанням EDC і HOBT, при цьому утворюється сполука 10 у вигляді аміду. Потім аміно-захисну групу P1 видаляють з одержанням сполуки 1, з використанням сильної кислоти, такої як TFA або HCl, коли амінозахисна група P1 являє собою Boc, або використовують H2/Pd/С або TMSI, коли аміно-захисна група P1 являє собою Cbz, або використовують Et 2NH, коли аміно-захисна група P1 являє собою Fmoc. На стадії реакції a), амін А може бути одержаний за допомогою використання способів, описаних в WO 04/064778, WO 03/004498, WO 03/082817, WO 06/104356 і тому подібне, або, в іншому випадку, може являти собою комерційно доступний амін. Тим часом, сполука формули 8 може бути одержана, як правило, за допомогою Схеми реакції 2 нижче. [Схема реакції 2] На Схемі реакції 2, В і P1 є такими, як визначено вище; і R являє собою бензил, метил, етил або ізопропіл. Реагенти і умови реакції a: ClCO2i-Bu, NMM, ТГФ; NaBH4 або H2O b: BH, DIAD, PPh3 або ТГФ с: колонковий хроматограф з NaBH4, MeOH або SiO2 d: Pd/C, H2 (бензиловий ефір), LiOH-H2O або MeOH-H2O (метиловий, етиловий або ізопропіловий ефір) Конкретно, карбонову кислоту формули 11 перетворюють в безводний ефір, а потім складноефірну сполуку відновлюють в метанолі з використанням боргідриду натрію (NaBH 4), 7 UA 103181 C2 5 10 15 20 25 щоб отримати при цьому сполуку 12 у вигляді первинного спирту. Продукт конденсації одержаного первинного спирту і бажаної імідної сполуки, тобто сполуки 13, може бути одержаний з використанням DIAD і PPh3. Потім аміно-захисну групу P1 видаляють з одержанням сполуки 8, з використанням сильної кислоти, такої як TFA або HCl, коли аміно-захисна група P1 являє собою Boc, або використовують H2/Pd/C або TMSI, коли аміно-захисна група P1 являє собою Cbz, або використовують Et2NH, коли аміно-захисна група P1 являє собою Fmoc. Тим часом, замісник В в Формулі 8 може бути одержаний, як правило, згідно зі схемою реакції 3-1 або схемою реакції 3-2 нижче. [Схема реакції 3-1] На схемі реакції 3-1, Ra і Rb, кожний, незалежно являє собою водень або галоген. Реагенти і умови реакції a: етилакрилат, порошок Cu, TMEDA, ТГФ b: NH3 в MeOH с: NaOEt, EtOH; HCl Конкретно, сполуку складного діефіру формули 14 одержують за допомогою реакції приєднання Міхаеля сполуки формули 14 з етилакрилатом, і сполуки формули 16, що містить амідну групу, одержують за допомогою взаємодії одержаного складного діефіру з аміаком. Як можна побачити, реакція відбувається за карбонільною групою, суміжною з галогеном, між двома ефірами, через тяжіння електрона одного або декілька атомів галогену, що міститься у взаємодіючій сполуці. Нарешті, здійснюють цикліацію в етанолі в присутності етоксиду натрію, з одержанням солі, що містить натрій, приєднаний до імідного атома азоту. Обробка солі безводною сильною кислотою дає бажану імідну сполуку формули 17. Одержаний нерозчинний твердий NaCl видаляють за допомогою простого фільтрування. [Схема реакції 3-2] 8 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 55 60 На схемі реакції 3-2, n дорівнює 0 або 1; R' являє собою метил або водень; R" являє собою гідрокси або аміно; і Rc являє собою C1-C4 алкіл. Реагенти і умови реакції a: Ac2O b: NH4OH, ТГФ с: Ac2O Якщо R' являє собою метил, одержану сполуку потім гідролізують за допомогою взаємодії з сильною основою, такою як гідроксид літію або гідроксид калію, перед використанням. Додавання оцтового ангідриду як розчинника і реагенту до сполуки дикислоти формули 18 призводить до утворення дуже активної хімічно проміжної сполуки в формі циклічного ангідриду. Сполуку формули 20 одержують за допомогою розмикання кільця проміжної сполуки за допомогою аміаку. Завдяки відсутності замісника, що є сильним акцептором електронів, на відміну від схеми реакції 3-1, аміак може взаємодіяти з атомами вуглецю обох кетонів і з цієї причини одержують два типи амінокислотних сполук. Ці амінокислотні сполуки перешкоджають протіканню подальшої реакції і тому можуть безпосередньо використовуватися без додаткового виділення. Нарешті, бажану імідну сполуку формули 21 одержують за допомогою циклізації сполуки 20 в формі амінокислоти за допомогою додавання оцтового ангідриду, як показано на стадії (a). Якщо не стверджується іншого, вихідні сполуки, які використовуються в даному винаході, відомі в даній галузі або можуть синтезуватися з відомих сполук за допомогою способів, відомих самі по собі або схожих з ними способів. Сполуки формули 1 можуть виділятися і очищуватися від продуктів реакції за допомогою звичайних способів, таких як перекристалізація, іонний електрофорез, колонкова хроматографія на силікагелі або хроматографія на іонообмінній смолі і тому подібне. Як описано вище, сполуки відповідно до даного винаходу, вихідні речовини для їх одержання і проміжні сполуки можуть синтезуватися за допомогою різних способів, таким чином, ці способи повинні інтерпретуватися як включені в межі даного винаходу з точки зору одержання сполук формули 1. Крім того, даний винахід передбачає фармацевтичну композицію для інгібування DPP-IV, що містить сполуку формули 1 або її фармацевтично прийнятну сіль. Фармацевтична композиція може містити сполуку за даним винаходом і інші хімічні компоненти, такі як розріджувачі, носії, і тому подібне. З цієї причини, фармацевтична композиція може містити фармацевтично прийнятний носій, розріджувач або наповнювач або будь-яке їх поєднання, якщо це необхідне. Фармацевтична композиція полегшує введення сполуки в організм суб'єкта in vivo. Різні методики введення сполук відомі в даній галузі і включають, але не обмежуючись цим, пероральне введення, введення за допомогою ін'єкції, аерозольне, парентеральне і місцеве введення. Термін "носій" означає хімічну сполуку, яка полегшує введення сполуки в клітини або тканини. Наприклад, диметилсульфоксид (ДМСО) являє собою носій, що зазвичай використовується, оскільки він полегшує поглинання багатьох органічних сполук в клітинах або тканинах організму. Термін "розріджувач" визначає хімічну сполуку, розбавлену у воді, яка буде розчиняти композицію, що представляє інтерес, а також стабілізувати біологічно активну форму композиції. Солі, розчинені в буферних розчинах, використовують як розріджувачі в даній галузі. Один з буферних розчинів, що зазвичай використовуються, являє собою фосфатний сольовий буферний розчин (PBS), оскільки він повторює сольові умови тілесних рідин людини. Оскільки буферні солі можуть контролювати pН розчину при низьких концентраціях, буферний розріджувач рідко модифікує біологічну активність сполуки. Термін "фізіологічно прийнятний" визначає носій або розріджувач, який не є шкідливим для біологічної активності і фізичних властивостей сполуки. Сполука за даним винаходом може приготовлятися в різних фармацевтичних дозованих формах відповідно до передбачуваного використання. При одержанні фармацевтичних композицій відповідно до даного винаходу, активний агент, більш конкретно, сполука формули 1 або її фармацевтично прийнятна сіль, може змішуватися з одним або декількома фармацевтично прийнятними носіями, які можуть вибиратися залежно від дозованої форми, яка повинна приготовлятися. Наприклад, фармацевтична композиція відповідно до даного винаходу може приготовлятися у вигляді дозованих форм, придатних для ін'єкції або перорального введення. Сполуки за даним винаходом можуть бути приготовані звичайним способом, з використанням відомих фармацевтично прийнятних носіїв і наповнювачів, і бути представлені в 9 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 55 60 стандартних дозованих формах або в багатодозових контейнерах. Препарати можуть приймати такі форми як розчини, суспензії або емульсії в масляних або водних зв'язувальних речовинах, і можуть містити звичайні диспергувальні, суспендувальні або стабілізуючі агенти. Альтернативно, активний агент може знаходитися в порошкоподібній формі для розведення за допомогою стерильної, апірогенної води, перед використанням. Сполуки за даним винаходом можуть також приготовлятися у вигляді супозиторіїв, що містять звичайні основи для супозиторіїв, такі як олія какао або інші гліцериди. Тверді дозовані форми для перорального введення включають капсули, таблетки, пілюлі, порошки і гранули. Переважні дозовані форми являють собою капсули і таблетки. Є переважним, щоб таблетки і пілюлі мали покриття. Тверді дозовані форми для перорального введення можуть бути одержані за допомогою змішування сполуки за даним винаходом як активний агент з одним або декількома інертними розріджувачами, такими як сахароза, лактоза, крохмаль, і тому подібне, і носіями, такими як мастильні речовини (наприклад, стеарат магнію), розпушувачами, зв'язувальними речовинами і тому подібне. Якщо це необхідно, сполуки за даним винаходом і композиції, що містять їх, можуть вводитися в поєднанні з іншими фармацевтичними агентами, наприклад, з іншими антидіабетичними агентами. Коли препарат представлений у вигляді стандартної дозованої форми, сполука формули 1 як активний агент переважно може міститися в стандартній дозі приблизно від 0,1 до 1500 мг. Дозована кількість сполуки формули 1 буде залежати від маси і віку суб'єкта, від природи і тяжкості захворюванні і від думки приписуючого лікаря. Для введення дорослому, необхідна дозована кількість буде знаходитися в діапазоні від 1 до 500 мг на день, залежно від частоти і сильнодії дози. Для внутрішньом'язового або внутрішньовенного введення дорослим вистачить загальної дозованої кількості приблизно від 5 до 300 мг на день. Для деяких пацієнтів щоденна доза буде вищою, ніж ця. Даний винахід передбачає способи лікування або запобігання захворюванням, що включає аномальну активність DPP-IV, за допомогою використання ефективних кількостей сполук формули 1. Репрезентативні приклади захворювань, пов'язаних з DPP-IV, включають, але ні в якому разі не обмежуючись цим, діабет, ожиріння і тому подібне, як описано вище. Серед іншого, даний винахід є переважним для лікування і запобігання цукровому діабету типу II. Термін "лікування" означає припинення або уповільнення розвитку захворювань, коли сполуки формули 1 або композиції, що містять їх, вводяться суб'єктам, що демонструють симптоми захворювань. Термін "запобігання" означає припинення або уповільнення симптомів захворювань, коли сполуки формули 1 або композиції, що містять їх, вводяться суб'єктам, що не демонструє симптомів захворювань, але що має високий ризик розвитку симптомів захворювань. Докладний опис переважних варіантів здійснення Тепер, даний винахід буде описуватися більш детально з посиланнями на Приклади, що ідуть далі. Ці приклади наводяться тільки для ілюстрації даного винаходу і не повинні розглядатися як такі, що обмежують межі і суть даного винаходу. Приклад одержання 1: Синтез гідрохлориду 2,4-біс-трифторметил-5,6,7,8тетрагідропіридо[3,4-d]піримідину (1) Синтез трет-бутилового ефіру 3-гідроксипіперидин-1-карбонової кислоти 1,0 еквівалент дибутилдикарбонату (159 г) в 200 мл метиленхлориду повільно додають до суміші 3-гідроксипіперидина гідрохлориду (100 г, 0,73 моль) і 1,1 еквіваленти триетиламіну (111 мл) в метиленхлориді (800 мл) при кімнатній температурі. Потім одержану суміш додатково перемішують протягом 2 годин. Після завершення реакції, реакційний розчин промивають водним розчином 1,0 н. хлористоводневої кислоти (1,0 л) і органічний шар концентрують, з одержанням 138 г (вихід: 94 %) вказаної в заголовку сполуки у вигляді білого твердого продукту. 1 H ЯМР (500 МГц, СDCl3) δ 1,35-1,55 (11H, м), 1,75 (1Н, м), 1,88 (1Н, м), 3,08 (2H, м), 3,53 (1H, м), 3,73 (2H, ушир. д, J=5,6 Гц). (2) Синтез трет-бутилового ефіру 3-оксопіперидин-1-карбонової кислоти трет-Бутиловий ефір 3-гідроксипіперидин-1-карбонової кислоти (4,0 г, 20 ммоль), синтезований в розділі (1), розчиняють в змішаному розчині (2:1:2, 100 мл) толуолу, води і етилового ефіру, і 1,0 % моль TEMPO (31 мг), і до нього послідовно додають NaBr (2,3 г). Суміш NaHCO3 (4,7 г) і NaOCl (5 %, 36 мл) повільно додають до суміші за умов охолоджування (0-4 °C). Після завершення реакції, органічний шар сушать над безводним сульфатом магнію і фільтрують. Відфільтрований органічний шар концентрують при зниженому тиску з одержанням 3,8 г (вихід: 95 %) вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,47 (с, 9H), 1,98 (м, 2H), 2,47 (т, J=6,7 Гц, 2H), 3,59 (т, J=6,1 Гц, 10 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 55 60 2H), 4,00 (ушир.с, 2H). (3) Синтез трет-бутилового ефіру 3-оксо-4-(2,2,2-трихлорацетил)піперидин-1-карбонової кислоти трет-Бутиловий ефір 3-оксопіперидин-1-карбонової кислоти (7,7 г, 38,4 ммоль), синтезований в розділі (2), розчиняють в безводному ТГФ (60 мл) і одержаний розчин додають при перемішуванні до 1,0 M LHMDS (46 мл) за умов охолоджування (-10 °С - -5 °C), з подальшим перемішуванням протягом 30 хв. До суміші додають по краплях етилтрифторацетат (6,6 г) за таких самих умов охолоджування. Реакційну суміш повільно нагрівають до кімнатної температури і реакцію зупиняють за допомогою додавання насиченого кислотного водного розчину (100 мл). Реакційний розчин екстрагують два рази етилацетатом (100 мл), сушать над безводним сульфатом магнію і фільтрують. Екстракт відганяють при зниженому тиску, з одержанням 9,5 г (вихід: 80 %) вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,48 (с, 9H), 2,58 (ушир.т, 2H), 3,57 (т, J=5,8 Гц, 2H), 4,23 (ушир.с, 2H). (4) Синтез трет-бутилового ефіру 2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7(8H)-карбонової кислоти трет-Бутиловий ефір 3-оксо-4-(2,2,2-трихлорацетил)піперидин-1-карбонової кислоти (2,96 г, 10 ммоль), синтезований в розділі (3), розчиняють в піридині (10 мл) і повільно додають при перемішуванні до трифторацетамідину (2,0 г) при 80 °C. Після завершення реакції, піридин видаляють за допомогою відгонки при зниженому тиску. Одержаний концентрат розчиняють в метиленхлориді і промивають водним розчином 1,0 н. хлористоводневої кислоти. Органічний шар відганяють при зниженому тиску, з одержанням 3,24 г вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,50 (с, 9H), 3,12 (ушир.т, 2H), 3,78 (т, J=5,8 Гц, 2H), 4,85 (с, 2H). (5) Синтез гідрохлориду 2,4-біс-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину трет-Бутиловий ефір 2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7(8H)карбонової кислоти (7,5 г, 20 ммоль), синтезований в розділі (4), розчиняють в 100 мл етилацетату, який насичують газоподібною хлористоводневою кислотою, і суміш перемішують при кімнатній температурі протягом 1 години. Розчинник і газоподібну хлористоводневу кислоту, що залишилася, відганяють при зниженому тиску, і одержаний коричневий твердий продукт суспендують, використовуючи трет-бутилметиловий ефір, протягом 1 години. Залишок фільтрують, з одержанням 5,1 г вказаної в заголовку сполуки. 1 H ЯМР (400 МГц, CD3OD) δ 3,41 (т, J=6,4 Гц, 2H), 3,68 (т, J=6,4 Гц, 2H), 4,66 (с, 2H). Приклад одержання 2: Синтез гідрохлоридних похідних 2,4-біс-трифторметил-5,6,7,8тетрагідропіридо[3,4-d]піримідину Аналогічно процедурі прикладу одержання 1, синтезують наступні похідні. (1) гідрохлорид 2-пропіл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (2) гідрохлорид 2-трет-бутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (3) гідрохлорид 2-циклобутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (4) гідрохлорид 2-піридин-4-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (5) гідрохлорид 2-фуран-2-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (6) гідрохлорид 2-(2-метоксіетил)-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (7) гідрохлорид 2-тіофен-3-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (8) гідрохлорид 2-(4-фторфеніл)-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину Приклад одержання 3: Синтез 3,3-дифторпіперидин-2,6-діону (1) Синтез діетилового ефіру 2,2-дифторпентандіонової кислоти Комерційно доступний етилбромдифторацетат (16 мл, 120 ммоль) і етилакрилат (11 мл, 100 ммоль) розчиняють в тетрагідрофурані (100 мл), і до нього послідовно додають мідний порошок (15,2 г, 240 ммоль) і TMEDA (16,7 мл, 110 ммоль) при кімнатній температурі. Після перемішування протягом приблизно 8 годин, реакцію зупиняють за допомогою додавання насиченого водного розчину 1,0 н. хлористоводневої кислоти і екстрагують два рази толуолом. Реакційний розчин відганяютьпри зниженому тиску, і концентровану вказану в заголовку сполуку безпосередньо використовують на подальшій стадії без додаткового очищення. 1 H ЯМР (500 МГц, СDCl3) δ 1,25 (т, J=7,2 Гц, 3H), 1,35 (т, J=7,2 Гц, 3H), 2, 41 (м, 2H), 2,52 (м, 2H), 4,15 (кв, J=7,2 Гц, 2H), 4,32 (кв, J=7,2 Гц, 2H). (2) Синтез етилового ефіру 4-карбамоїл-4,4-дифтормасляної кислоти Діетиловий ефір 2,2-дифторпентандіонової кислоти (21 г), синтезований в розділі (1), розчиняють в етанолі (50 мл) і до нього повільно додають 2,0 M метанольний розчин аміаку. Суміш перемішують при кімнатній температурі протягом 30 хв і відганяють при зниженому тиску, з одержанням 18,2 г вказаної в заголовку сполуки у вигляді в'язкого масла, яке використовують на подальшій стадії без додаткового очищення. 11 UA 103181 C2 1 5 10 15 20 25 30 35 40 45 50 55 60 H ЯМР (500 МГц, СDCl3) δ 1,25 (т, J=7,2 Гц, 3H), 2, 43 (м, 2H), 2,54 (м, 2H), 4,14 (кв, J=7,2 Гц, 2H), 5,92 (ушир. с, 1H), 6,30 (ушир. с, 1H). (3) Синтез 3,3-дифторпіперидин-2,6-діону Етиловий ефір 4-карбамоїл-4,4-дифтормасляної кислоти (18 г), синтезований в розділі (2), розчиняють в етанолі (100 мл) і до нього повільно додають етанольний розчин 1,0 н. етоксиду натрію (105 мл). Після перемішування протягом 3 годин при кімнатній температурі, pН суміші доводять до 3-4 за допомогою додавання 3,0 н. хлористоводневої кислоти в діоксані, і одержаний NaCl видаляють за допомогою фільтрування. Дуже в'язке масло суспендують за допомогою діетилового ефіру (100 мл). Одержаний білий твердий продукт фільтрують і сушать, з одержанням 11,4 г вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, ДМСO-d6) δ 2,56 (м, 2H), 2,71 (т, J=6,8 Гц, 2H), 11,60 (ушир. с, 1H). Приклад одержання 4: Синтез (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2-гідрокси-6оксопіперидин-1-іл)масляної кислоти (1) Синтез ізопропілового ефіру (3S)-трет-бутоксикарбоніламіно-4-гідроксимасляної кислоти Комерційно доступний 4-ізопропіловий ефір (2S)-трет-бутоксикарбоніламінобурштинової кислоти (2,8 г, 10,3 ммоль) розчиняють в тетрагідрофурані (10 мл) і до нього додають Nметилморфолін (1,13 мл). Суміш охолоджують до -10 °C. Ізобутілхлорформіат (1,40 мл) додають до суміші, яку потім перемішують протягом 10 хв з утворенням активного ефіру. Після видалення одержаної амонієвої солі хлористоводневої кислоти за допомогою фільтрування, до фільтрату додають по краплях NaBH4 (0,59 г) у воді і реакцію зупиняють за допомогою додавання насиченого розчину NH4Cl. Одержану реакційну суспензію екстрагують два рази етилацетатом, і органічний шар сушать над безводним сульфатом магнію і фільтрують. Фільтрат відганяють при зниженому тиску, з одержанням 2,51 г (вихід: 95 %) вказаної в заголовку сполуки у вигляді світло-жовтого масла. 1 H ЯМР (400 МГц, СDCl3) δ 1,22 (д, J=6,8, 6H), 1,44 (с, 9H), 2,54-2,62 (м, 2H), 3,65 (д, J=4,0, 2H), 3,99 (ушир.с 1H), 5,02 (м, 1H), 5,36 (ушир.с, 1H). (2) Синтез ізопропілового ефіру (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6діоксопіперидин-1-іл)масляної кислоти Ізопропіловий ефір (3S)-трет-бутоксикарбоніламіно-4-гідроксимасляної кислоти (5,3 г, 20 ммоль), синтезований в розділі (1), і 3,3-дифторпіперидин-2,6-діон (3,6 г, 26 ммоль), синтезований в прикладі одержання 3, розчиняють в метиленхлориді (20 мл) і охолоджують до температури від 0 до 5 °C. Послідовно додають трифенілфосфін (6,3 г) і 2,2 M DEAD (11 мл) в толуолі і температуру реакції повільно підіймають до температури навколишнього середовища. Після перемішування протягом 4 годин, реакційну суміш безпосередньо піддають колонковій хроматографії без конкретної стадії зупинки, отримуючи, таким чином, 5,5 г (вихід: 70 %) вказаної в заголовку сполуки. 1 H ЯМР (400 МГц, СDCl3) δ 1,22 (м, 6H), 1,35 (с, 9H), 2,33-2,56 (м, 4H), 2,87 (м, 2H), 3,70 (ушир, д, J=11,8, 1H), 4,07-4,19 (м, 3H), 5,04 (м, 1H), 5,11 (ушир, д, J=9,8, 1H). (3) Синтез ізопропілового ефіру (3S)-трет-бутоксикарбоніл-4-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти Ізопропіловий ефір (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6-діоксопіперидин-1іл)масляної кислоти (5,5 г, 14 ммоль), синтезований в розділі (2), розчиняють в змішаному розчиннику тетрагідрофурану і метанолу (2:1, 60 мл) і охолоджують до температури від 0 до 5 °C. Додають 1,0 еквівалент NaBH4 (530 мг) і суміш перемішують протягом 30 хв, підтримуючи при цьому температуру охолоджування. Реакцію зупиняють за допомогою додавання насиченого водного розчину NH4Cl, і реакційну суміш очищають за допомогою колонкової хроматографії, з одержанням 4,0 г вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,22 (м, 6H), 1,41 (ушир. с, 9H), 2,15 (м, 1H), 2,45-2,70 (м, 5H), 3,05 (м, 0,4 Н), 3,42 (м, 0,6 Н), 3,80 (м, 0,6 Н), 4,10-4,35 (м, 2,4 Н), 4,88-5,10 (м, 2H), 5,35-5,60 (м, 1,6 Н), 6,15 (ушир. с, 0,4 Н). (4) Синтез (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2-гідроксі-6-оксопіперидин-1іл)масляної кислоти Ізопропіловий ефір (3S)-трет-бутоксикарбоніл-4-(3,3-дифтор-2-гідрокси-6-оксопіперидин-1іл)масляної кислоти (4,0 г, 10 ммоль), синтезований в розділі (3), розчиняють в змішаному розчиннику тетрагідрофурану, H2O і метанолу (1:1:1, 30 мл) і до нього додають літій гідроксид гідрат (630 мг) при кімнатній температурі. Після 2 годин, реакційний розчин розбавляють 100 мл дистильованої води, і домішки відмивають метиленхлоридом (50 мл). Основний водний розчин доводять до pН 3,0-4,0 за допомогою додавання сильного кислотного водного розчину, і одержану білу суспензію екстрагують два рази метиленхлоридом. Органічний екстракт відганяють при зниженому тиску, з одержанням 1,9 г концентрованої вказаної в заголовку 12 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 сполуки, яке використовують на подальшій стадії без додаткового очищення. 1 H ЯМР (500 МГц, ДМСO-d6) δ 1,31 (ушир. с, 9H), 2,05 (м, 1H), 2,20-2,50 (м, 5H), 2,85 (м, 0,6H), 3,05 (м, 0,4H), 3,60 (м, 2H), 3,98 (м, 1), 4,80 (м, 1H), 6,50 (ушир. с, 0,4H), 6,75 (ушир. с, 0,6H), 7,02 (м, 1H), 12,05 (ушир. с, 1H). Приклад одержання 5: Синтез трет-бутилового ефіру [3(S)-(2,4-біс-трифторметил-5,8дигідро-6H-піридо[3,4-d]-піримідин-7-іл)-1-(3,3-дифтор-2-гідроксі-6-оксопіридин-1-ілметил)-3оксопропіл]карбамінової кислоти (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (2,6 г, 4 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2,4-бістрифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (1,4 г), синтезований в прикладі одержання 1, розчиняють в метиленхлориді (50 мл) і охолоджують до температури від 0 до 4 °C. До них послідовно додають HOBT (650 мг), діізопропілетиламін (2,2 мл) і EDCI (1,20 г), і температуру реакції повільно підвищують до кімнатної температури. Після перемішування протягом 6 годин, реакційний розчин промивають водним розчином 1,0 н. хлористоводневої кислоти, і органічний шар відганяють при зниженому тиску, з одержанням 2,6 г вказаної у заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,31 (м, 9H), 2,14 (м, 1H), 2,30-2,85 (м, 3H), 2,90-3,30 (м, 3H), 3,45 (м, 0,5H), 3,60-4,30 (м, 3H), 4,75-5,05 (м, 2H), 5,95-6,50 (м, 1H). 1 H ЯМР (500 МГц, СDCl3) δ 1,31 (м, 9H), 2,14 (м, 1H), 2,30-2,85 (м, 3H), 2,90-3,30 (м, 3H), 3,45 (м, 0,5H), 3,60-4,30 (м, 3H), 4,75-5,05 (м, 2H), 5,95-6,50 (м, 1H). Приклад одержання 6: Синтез трет-бутилового ефіру [(S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-(2-пропіл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти (3S)-трет-Бутоксикарбоніламіно-4-(3,3-дифтор-2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (20 мг, 0,06 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-пропіл-4трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (22 мг), синтезований в прикладі одержання 2-(1), розчиняють в диметилформальдегіді (2 мл) і охолоджують до температури від 0 до 4 °C. До них послідовно додають HOBT (650 мг), діізопропілетиламін (2,2 мл) і EDCI (1,20 г) і температуру реакції повільно підвищують до кімнатної температури. Після перемішування реакційного розчину протягом 10 годин, додають водний розчин хлориду амонію, і органічний шар екстрагують етилацетатом. Залишок очищують за допомогою препаративної ТШХ, з одержанням 7,4 мг (виход: 22 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, СDCl3) δ 0,98-1,02 (м, 3H), 1,26-1,30 (м, 2H), 1,40-1,46 (м, 9H), 1,75-1,86 (м, 3H), 2,17 (м, 1H), 2,55-2,69 (м, 3H), 2,88-3,10 (м, 5H), 3,49-4,31 (м, 4H), 4,70-4,96 (м, 3H). MS (m/e) 602 (M+Na). Приклад 1: Синтез гідрохлориду 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Hпіридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-(6R)-гідроксипіперидин-2-ону трет-Бутиловий ефир [3(S)-(2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)-1-(3,3-дифтор-2-гідроксі-6-оксопіридин-1-ілметил)-3-оксопропіл]карбамінової кислоти (2,0 г), синтезований в прикладі одержання 5, представляє собою діастереомерну суміш, яку потім розділюють на два ізомери за допомогою колонкової хроматографії на силікагелі. З розділених ізомерів, ізомер, що має відносно високу полярність, розчиняють в етилацетаті (10 мл), обробляють комерційно доступним 4,0 н. хлористим воднем в діоксані і перемішують протягом 30 хв. Реакційний розчин відганяють при зниженому тиску, і в'язке масло суспендують за допомогою трет-бутилметилового ефіру (10 мл). Одержану суспензію фільтрують і сушать, з одержанням 500 мг вказаної у заголовку сполуки. 1 H ЯМР (500 МГц, ДМСO-d6) δ 2,16 (м, 1H), 2,35-2,55 (м, 2H), 2,78-2,88 (м, 2H), 2,95-3,18 (м, 2H), 3,40 (м, 1H), 3,70 (м, 1H), 3,82 (м, 2H), 4,90 (м, 2H), 7,30 (ушир. с, 1H), 7,95 (м, 3H). Приклад 2: Синтез гідрохлориду 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Hпіридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-(6S)-гідроксипіперидин-2-ону 13 UA 103181 C2 5 10 15 20 25 30 35 Аналогічно прикладу 1, діастереомерну суміш, синтезовану в прикладі одержання 5, піддають колонковій хроматографії для відділення ізомеру з відносно низькою полярністю, який потім розчиняють в етилацетаті (10 мл), з подальшою обробкою розчином комерційно доступного 4,0 н. хлористого водню в діоксані, і перемішують протягом 30 хв. Реакційний розчин відганяють при зниженому тиску, і одержаному концентраті суспендують за допомогою третбутилметилового ефіру (10 мл). Одержану суспензію фільтрують і сушать, з одержанням 250 мг вказаної в заголовку сполуки. 1 H ЯМР (500 МГц, ДМСO-d6) δ 2,13 (м, 1H), 2,35-2,55 (м, 2H), 2,75-2,90 (м, 2H), 2,98 (ушир. с, 1H), 3,12 (ушир. с, 1H), 3,50-3,74 (м, 2H), 3,80 (м, 3H), 3,82 (м, 2H), 4,90 (м, 2H), 7,25 (ушир. с, 1H), 7,95 (м, 3H). Приклад 3: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Сполуку, синтезовану в прикладі одержання 6, обробляють розчином комерційно доступного 4,0 н. хлористого водню в діоксані і перемішують протягом 1 години. Реакційний розчин відганяють при зниженому тиску і очищають за допомогою препаративної ТШХ, з одержанням 4,7 мг (вихід: 77 %) вказаної в заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 0,87-0,91 (м, 3H), 1,72-1,80 (м, 3H), 2,45-2,64 (м, 6H), 2,81-3,06 (м, 4H), 3,25-3,80 (м, 5H), 4,19-4,78 (м, 3H); MS (m/е) 480 (M+1). Приклад одержання 7: Синтез трет-бутилового ефіру [(S)-3-(2-трет-бутил-4-трифторметил5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-1-(3,3-дифтор-2-гідроксі-6-оксопіперидин-1-ілметил)3-оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (26 мг, 0,07 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-трет-бутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (30 мг), синтезований в прикладі одержання 2-(2), конденсують, з одержанням 15,5 мг (вихід: 36 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,38-1,42 (м, 18H), 2,16-2,24 (м, 2H), 2,58-2,72 (м, 5H), 2,993,12 (м, 3H), 3,81-3,89 (м, 3H), 4,30 (м, 1H), 4,74-4,97 (м, 3H); MS (m/e) 616 (M+Na). Приклад 4: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро6H-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 10,5 мг (вихід: 76 %) вказаної у заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 7. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,30 (д, 9H), 2,07 (ушир., 1H), 2,42-2,56 (м, 4H), 2,61-2,98 (м, 3H), 3,21-3,30 (м, 2H), 3,70-3,80 (м, 3H), 4,69-4,79 (м, 3H); MS (m/e) 494 (M+1). Приклад одержання 8: Синтез трет-бутилового ефіру [(S)-3-(2-цикло-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)-1-(3,3-дифтор-2-гідроксі-6-оксопіперидин-1-ілметил)-3 14 UA 103181 C2 5 10 15 20 25 30 35 оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (26 мг, 0,07 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-циклобутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (30 мг), синтезований в прикладі одержання 2-(3), конденсують, з одержанням 14,6 мг (вихід: 34 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,39 (м, 9H), 1,96-2,24 (м, 4H), 2,38-2,74 (м, 9H), 2,99-3,12 (м, 3H), 3,82-3,89 (м, 4H), 4,30 (м, 1H), 4,78-4,97 (м, 3H); MS (m/e) 614 (M+Na). Приклад 5: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-циклобутил-4-трифторметил-5,8-дигідро6H-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, кількісно одержують 13 мг вказаної у заголовку сполуки з використанням сполуки прикладу одержання 8. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,87-2,02 (м, 3H), 2,26-2,52 (м, 9H), 2,89-2,99 (м, 2H), 3,21-3,25 (м, 2H), 3,70-3,80 (м, 4H), 3,99-4,80 (м, 3H); MS (m/e) 492 (M+1). Приклад одержання 9: Синтез трет-бутилового ефіру [(S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-3-(2-піридин-4-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідрокси-6-оксопіперидин-1-іл)масляну кислоту (24 мг, 0,07 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-піридин-4-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (30 мг), синтезований в прикладі одержання 2-(4), конденсують, з одержанням 13,5 мг (вихід: 32 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,37 (м, 9H), 2,16-2,26 (м, 2H), 2,59-2,88 (м, 5H), 3,01-3,22 (м, 3H), 3,37-3,94 (м, 3H), 4,30 (м, 1H), 4,92-5,01 (м, 3H), 8,28 (м, 2H), 8,75 (м, 1H); MS (m/e) 637 (M+Na). Приклад 6: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Сполуку, синтезовану в прикладі одержання 9, обробляють комерційно доступним 4,0 н. хлористим воднем в діоксані і перемішують протягом 1 години. Оскільки вказана в заголовку сполука представляє собою діастереомерну сполуку, реакційний розчин відганяють при зниженому тиску і розділюють на два ізомери за допомогою препаративної ТШХ. З розділених ізомерів, одержують 2 мг вказаної у заголовку сполуки з відносно низькою полярністю. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17 (м, 1H), 2,58-2,68 (м, 4H), 2,77-2,82 (м, 1H), 3,11-3,21 (м, 2H), 3,34-3,70 (м, 2H), 3,91-4,00 (м, 3H), 4,77-5,06 (м, 3H), 8,44 (д, 2H, J=6,0 МГц), 8,76 (д, 2H, J=4,8 МГц); MS (m/e) 515 (M+1). Приклад 7: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-ону 15 UA 103181 C2 5 10 15 20 25 30 35 Сполуку, синтезовану в прикладі одержання 9, обробляють комерційно доступним 4,0 н. хлористим воднем у діоксані й перемішують протягом 1 години. Оскільки вказана в заголовку сполука являє собою діастереомерну сполуку, реакційний розчин відганяють при зниженому тиску й розділяють на два ізомери за допомогою препаративної ТШХ. З розділених ізомерів одержують 1,8 мг вказаної в заголовку сполуки з відносно високою полярністю. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17 (м, 1H), 2,53-2,65 (м, 4H), 2,74-2,80 (м, 1H), 3,06-3,20 (м, 2H), 3,34-3,66 (м, 2H), 3,82-4,00 (м, 3H), 4,77-5,00 (м, 3H), 8,43 (д, 2H, J=6,0 Мгц), 8,76 (д, 2H, J=4,4 Мгц); MS (m/e) 515 (M+1). Приклад одержання 10: Синтез трет-бутилового ефіру [(S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)-3-оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (25 мг, 0,07 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-фуран-2-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (30 мг), синтезований у прикладі одержання 2-(5), конденсують, з одержанням 18,1 мг (вихід: 43 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,28 (м, 9H), 2,13 (м, 1H), 2,39-2,47 (м, 3H), 2,61-2,76 (м, 2H), 2,89-3,03 (м, 3H), 3,56-3,80 (м, 3H), 4,31 (м, 1H), 4,70-4,86 (м, 3H), 6,55-6,56 (м, 1H), 7,28 (д, 1H, J=4,0 Мгц), 7,52-7,57 (м, 1H); MS (m/e) 626 (M+Na). Приклад 8: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро6H-піридо[3,4-d]піримідин-7-іл)бутил]-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 6, два ізомери виділяють зі сполуки, синтезованої в прикладі одержання 10. З виділених ізомерів одержують 4,7 мг вказаної у заголовку сполуки, що має відносно низьку полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17 (м, 1H), 2,45-2,81 (м, 5H), 3,04-3,17 (м, 2H), 3,33-3,72 (м, 2H), 3,87-3,96 (м, 3H), 4,90-5,00 (м, 3H), 6,68 (с, 1H), 7,41 (д, 1H, J=3,2 Мгц), 7,79 (с, 1H); MS (m/e) 504 (M+1). Приклад 9: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро6H-піридо[3,4-d]піримідин-7-іл)бутил]-4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 6, два ізомери виділяють зі сполуки, синтезованої в прикладі одержання 10. З виділених ізомерів одержують 4,8 мг вказаної у заголовку сполуки, що має відносно високу полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17 (м, 1H), 2,51-2,76 (м, 5H), 3,03-3,11 (м, 2H), 3,34-3,70 (м, 2H), 3,84-3,93 (м, 3H), 4,82-4,90 (м, 3H), 6,68 (с, 1H), 7,41 (д, 1H, J=3,6 Мгц), 7,79 (с, 1H); MS (m/e) 504 (M+1). 16 UA 103181 C2 5 10 15 20 25 30 35 Приклад одержання 11: Синтез трет-бутилового ефіру {(S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-[2-(2-метоксіетил)-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл]-3-оксопропіл}карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (61 мг, 0,17 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-(2-метоксіетил)-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (65 мг), синтезований у прикладі одержання 2-(6), конденсують, з одержанням 38,2 мг (вихід: 37 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,31-1,44 (м, 9H), 2,17-2,24 (м, 2H), 2,58-3,74 (м, 4H), 2,88-3,37 (м, 10H), 3,85-3,98 (м, 4H), 4,29 (м, 1H), 4,75-4,96 (м, 3H); MS (m/e) 618 (M+Na). Приклад 10: Синтез гідрохлориду 1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 6, виділяють два ізомери зі сполуки, синтезованої в прикладі одержання 11. З виділених ізомерів одержують 6,2 мг вказаної у заголовку сполуки, що має відносно низьку полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,18 (м, 1H), 2,62-2,78 (м, 4H), 3,02-3,42 (м, 10H), 3,68-4,06 (м, 5H), 4,80-4,90 (м, 3H); MS (m/e) 496 (M+1). Приклад 11: Синтез гідрохлориду 1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 6, два ізомери виділяють зі сполуки, синтезованої в прикладі одержання 11. З виділених ізомерів одержують 9,1 мг вказаної у заголовку сполуки, що має відносно високу полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,21 (м, 1H), 2,60-2,64 (м, 3H), 2,87-3,37 (м, 11H), 3,61-4,02 (м, 5H), 4,79-4,99 (м, 3H); MS (m/e) 496 (M+1). Приклад одержання 12: Синтез трет-бутилового ефіру {(S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-3-(2-тіофен)-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (39 мг, 0,11 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-тіофен-3-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (50 мг), синтезований у прикладі одержання 2-(7), конденсують, з одержанням 26 мг (вихід: 38 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,40 (с, 9H), 2,16-2,25 (м, 2H), 2,59-2,75 (м, 5H), 3,01-3,13 (м, 3H), 3,83-3,93 (м, 3H), 4,31 (м, 1H), 4,88-4,98 (м, 3H), 7,51-7,53 (м, 1H), 7,89 (д, 1H, J=4,4 Мгц), 8,40 (с, 1H); MS (m/e) 642 (M+Na). Приклад 12: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-ону 17 UA 103181 C2 5 10 15 20 25 30 35 Аналогічно процедурі прикладу 6, два ізомери виділяють зі сполуки, синтезованої в прикладі одержання 12. З виділених ізомерів одержують 6,2 мг вказаної у заголовку сполуки, що має відносно низьку полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,18 (м, 1H), 2,58-2,67 (м, 4H), 2,77-2,81 (м, 1H), 3,04-3,12 (м, 2H), 3,34-3,42 (м, 2H), 3,81-3,96 (м, 3H), 4,90-4,97 (м, 3H), 7,52-7,55 (м, 1H), 7,89 (д, 1H, J=5,2 Мгц), 8,40-8,41 (м, 1H); MS (m/e) 520 (M+1). Приклад 13: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 6, два ізомери виділяють зі сполуки, синтезованої в прикладі одержання 12. З виділених ізомерів одержують 5,8 мг вказаної у заголовку сполуки, що має відносно високу полярність. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17 (м, 1H), 2,55-2,65 (м, 4H), 2,80-2,85 (м, 1H), 3,04-3,12 (м, 2H), 3,46-3,51 (м, 1H), 3,66-3,74 (м, 1H), 3,86-3,95 (м, 3H), 4,85-4,93 (м, 3H), 7,52-7,55 (м, 1H), 7,89 (д, 1H, J=5,2 Мгц), 8,40-8,41 (м, 1H); MS (m/e) 520 (M+1). Приклад одержання 13: Синтез трет-бутилового ефіру {[S)-1-(3,3-дифтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-[2-(4-фторфеніл)-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл]-3-оксопропіл}карбамінової кислоти Аналогічно процедурі прикладу одержання 5, (3S)-трет-бутоксикарбоніламіно-4-(3,3-дифтор2-гідроксі-6-оксопіперидин-1-іл)масляну кислоту (30 мг, 0,09 ммоль), синтезовану в прикладі одержання 4, і гідрохлорид 2-(4-фторфеніл)-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4d]піримідину (25 мг), синтезований у прикладі одержання 2-(8), конденсують, з одержанням 10,5 мг (вихід: 20 %) вказаної у заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,41-1,44 (м, 9H), 2,57-2,61 (м, 6H), 3,12-3,14 (м, 3H), 3,91-4,00 (м, 4H), 4,86-4,96 (м, 4H), 7,16-7,18 (м, 2H), 8,46-8,49 (м, 2H); MS (m/e) 654 (M+Na). Приклад 14: Синтез гідрохлориду 1-{(S)-2-аміно-4-[2-(4-фторфеніл)-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл]-4-оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 8,8 мг (вихід: 93 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 13. 1 H ЯМР (500 МГц, МeOH-d4) 82,55-2,64 (м, 6H), 3,00-3,38 (м, 5H), 3,86-3,93 (м, 2H), 4,84-4,92 (м, 3H), 7,21-7,24 (м, 2H), 8,48-8,51 (м, 2H); MS (m/e) 532 (M+1). Приклад одержання 14: Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6діоксопіперидин-1-іл)масляної кислоти (1) Синтез бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4-гідроксимасляної кислоти 4-Бензиловий ефір (S)-2-трет-бутоксикарбоніламіно-бурштинової кислоти (545 мг, 1,69 ммоль) розчиняють у тетрагідрофурані (ТГФ), до них додають N-метилморфолін (NMM, 0,19 мл, 18 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 1,77 ммоль) і ізобутилхлорформіат (0,23 мл, 1,77 ммоль) при 0 °C, з наступним перемішуванням при цій температурі протягом 30 хв. Через 30 хв, отриману білу тверду сіль видаляють за допомогою фільтрування через целіт, і розчин боргідриду натрію (96 мг, 2,53 ммоль) у дистильованій воді (2 мл) додають до отриманого прозорого фільтрату без додаткового концентрування при кімнатній температурі. Після перемішування протягом 1 години при кімнатній температурі, ТГФ відганяють при зниженому тиску. Реакційну суміш розбавляють етилацетатом і реакцію зупиняють за допомогою додавання 1 н. HСl. Органічний шар екстрагують етилацетатом і очищають за допомогою колонкової хроматографії (етилацетат:гексан=1:1), з одержанням 468 мг (вихід: 90 %) вказаної у заголовку сполуки. 1 H ЯМР (500 МГц, СDCl3) δ 1,42 (с, 9H), 2,67 (д, 2H, J=5,5 Гц), 3,67-3,68 (м, 2H), 3,99-4,01 (м, 1H), 5,12 (с, 2H), 5,29 (ушир., 1H), 7,33-7,36 (м, 5H); MS (m/e) 332 (M+Na). (2) Синтез бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6діоксопіперидин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(2), 1,86 г (вихід: 86 %), одержують зазначене в заголовку сполука з використанням бензилового ефіру (S)-4огідроксимасляної кислоти (1,5 г, 4,9 ммоль), синтезованої в розділі (1) і 3,3-дифторпіперидин-2,6-діону (880 мг, 5,9 ммоль), синтезованого в прикладі одержання 3. 1 H ЯМР (500 МГц, СDCl3) δ 1,37 (с, 9H), 2,43 (м, 2H), 2,62-2,67 (м, 2H), 2,88-2,89 (м, 2H), 3,71 (д, 1H, J=11 Гц), 4,2 (м, 2H), 5,17 (с, 2H), 5,19 (д, 1H), 7,36-7,37 (м, 5H). (3) Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6-діоксопіперидин-1-іл)масляної кислоти Бензиловий ефір (S)-3-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6-діоксопіперидин-1іл)масляної кислоти (310 мг, 0,70 ммоль), синтезований у розділі (2), розчиняють у метанолі й до нього додають 10 % Pd/C (31 мг, 0,1 екв.). Суміш перемішують протягом 1 години при 1 атм. газоподібного водню. Після завершення реакції, твердий матеріал видаляють за допомогою фільтрування через целіт, з одержанням 260 мг сирої вказаної у заголовку сполуки. Приклад одержання 15: Синтез трет-бутилового ефіру [(S)-3-(2,4-біс-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)-1-(3,3-дифтор-2,6-діоксопіперидин-1-ілметил)-3оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 5, одержують 6,6 мг (вихід: 13 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3,3-дифтор-2,6діоксопіперидин-1-іл)масляної кислоти (30 мг, 0,09 ммоль), синтезованої в прикладі одержання 14, і гідрохлориду 2,4-біс-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (23 мг, 0,009 ммоль), синтезованого в прикладі одержання 1. 1 H ЯМР (500 МГц, СDCl3) δ 1,37 (с, 9H), 2,42 (м, 2H), 2,76-2,95 (м, 4H), 3,14-3,28 (м, 2H), 3,714,22 (м, 5H), 4,85-5,00 (м, 2H), 5,52-5,55 (м, 1H); MS (m/e) 626 (M+1). Приклад 15: Синтез 1-[(S)-2-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)-4-оксобутил]-3,3-дифторпіперидин-2,6-діону Аналогічно процедурі прикладу 3, одержують 6,4 мг (вихід: 97 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 15. 1 H ЯМР (500 МГц, МeOH-d4) δ 2,51-2,54 (м, 2H), 2,85-2,91 (м, 4H), 3,12-3,21 (м, 3H), 4,10-4,20 (м, 4H), 4,95 (м, 2H); MS (m/e) 504 (M+1). Приклад одержання 16: Синтез 3-фторпіперидин-2,6-діону (1) Синтез діетилового ефіру 2-фторпентандіонової кислоти Аналогічно процедурі прикладу одержання 3-(1), одержують 1,86 г (вихід: 30 %) вказаної в заголовку сполуки з використанням комерційно доступного етилового ефіру бромфтороцтової кислоти (7,1 мл, 60 ммоль) і етилакрилату (3,3 мл, 30 ммоль). 1 H ЯМР (400 МГц, СDCl3) δ 1,25-1,37 (м, 6H), 2,17-2,37 (м, 2H), 2,51-2,57 (м, 2H), 4,14-4,33 (м, 4H), 4,93-5,08 (м, 1H). (2) Синтез етилового ефіру 4-карбамоїл-4-фтормасляної кислоти Аналогічно процедурі прикладу одержання 3-(2), одержують 2,2 г сирої вказаної в заголовку сполуки з використанням діетилового ефіру 2-фторпентандіонової кислоти (2,4 г, 11,7 ммоль), 19 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 синтезованого в розділі (1). 1 H ЯМР (400 МГц, СDCl3) δ 1,21-1,33 (м, 3H), 2,13-2,38 (м, 2H), 2,47-2,52 (м, 2H), 4,13-4,18 (м, 2H), 4,89-5,08 (м, 1H), 5,90-6,32 (ушир., 1H). (3) Синтез 3-фторпіперидин-2,6-діону Аналогічно процедурі прикладу одержання 3-(3), одержують 720 мг (вихід: 44 %) вказаної в заголовку сполуки з використанням етилового ефіру 4-фтормасляної кислоти (2,2 г, 12,5 ммоль), синтезованого в розділі (2). 1 H ЯМР (400 МГц, МeOH-d4) δ 2,18-2,41 (м, 2H), 2,71-2,75 (м, 2H), 5,11-5,27 (м, 1H); MS (m/e) 132 (M+1). Приклад одержання 17: Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (1) Синтез ізопропілового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2,6діоксопіперидин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(2), одержують 139 мг сирої вказаної в заголовку сполуки з використанням 3-фторпіперидин-2,6-діону (47 мг, 0,36 ммоль), синтезованого в прикладі одержання 16, і ізопропілового ефіру (3S)-трет-бутоксикарбоніламіно4-гідроксимасляної кислоти (78,4 мг, 0,3 ммоль), синтезованої в прикладі одержання 4-(1). 1 H ЯМР (400 МГц, СDCl3) δ 1,24-1,39 (м, 6H), 1,44 (с, 9H), 2,24-2,35 (м, 2H), 2,48-2,71 (м, 2H), 2,85-2,91 (м, 2H), 3,73 (д, 1H, J=4,8 Гц), 4,09-4,20 (м, 2H), 4,99-5,31 (м, 3H); MS (m/e) 397 (M+Na). (2) Синтез ізопропілового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(3), одержують 109 мг (вихід: 78 %) вказаної в заголовку сполуки з використанням ізопропілового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3фтор-2,6-діоксопіперидин-1-іл)масляної кислоти (139 мг, 0,37 ммоль), синтезованого в розділі (1). MS (m/e) 399 (M+Na). (3) Синтез (S)-3-трет-бутоксикарбоніламіно-4-f3-фтор-2-гідроксі-6-оксопіперидин-1іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(4), одержують 54 мг (вихід: 60 %) вказаної в заголовку сполуки з використанням ізопропілового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3фтор-2-гідроксі-6-оксопіперидин-1-іл)масляної кислоти (102 мг, 0,27 ммоль), синтезованого в розділі (2). MS (m/e) 357 (M+Na). Приклад одержання 18: Синтез трет-бутилового ефіру [(S)-3-(2-трет-бутил-4-трифторметил5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-1-(3-фтор-2-гідроксі-6-оксопіперидин-1-ілметил)-3оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, одержують 42 мг (вихід: 65 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (38 мг, 0,11 ммоль), синтезованої в прикладі одержання 17, і гідрохлориду 2-трет-бутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (49 мг, 0,16 ммоль), синтезованого в прикладі одержання 2-(2). 1 H ЯМР (400 МГц, МeOH-d4) δ 1,41 (д, 18H), 2,17 (м, 2H), 2,45 (м, 2H), 2,71 (м, 2H), 3,00 (м, 1H), 3,11 (м, 2H), 3,92 (м, 3H), 4,11 (м, 1H), 4,88-4,91 (м, 3H) 5,43-5,55 (м, 1H); MS (m/e) 598 (M+Na). Приклад 16: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5-фтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 6,0 мг (вихід: 18 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 18. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,42 (с, 9H), 2,00-2,50 (м, 4H), 2,81-3,11 (м, 5H), 3,50-3,93 (м, 4H), 4,84-5,10 (м, 3H) 5,48-5,62 (м, 1H); MS (m/e) 476 (M+1). Приклад одержання 19: Синтез трет-бутилового ефіру [(S)-1-(3-фтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4 20 UA 103181 C2 5 10 15 20 25 30 35 40 d]піримідин-7-іл)-3-оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, одержують 49 мг (вихід: 79 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (35 мг, 0,11 ммоль), синтезованої в прикладі одержання 17, і гідрохлориду 2-фуран-2-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (45 мг, 0,15 ммоль), синтезованого в прикладі одержання 2-(5). MS (m/e) 608 (M+Na). Приклад 17: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-5-фтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 15 мг (вихід: 35 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 19. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,17-3,14 (м, 9H), 3,70-3,89 (м, 4H), 4,86-4,87 (м, 3H) 5,43-5,55 (м, 1H), 6,68 (м, 1H), 7,41 (м, 1H), 7,79 (м, 1H); MS (m/e) 486 (M+1). Приклад одержання 20: Синтез трет-бутилового ефіру [(S)-1-(3-фтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-3-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, одержують 13 мг (вихід: 15 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (48 мг, 0,14 ммоль), синтезованої в прикладі одержання 17, і гідрохлориду 2-тіофен-3-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (65 мг, 0,20 ммоль), синтезованого в прикладі одержання 2-(7). MS (m/e) 624 (M+Na). Приклад 18: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 1,4 мг (вихід: 12 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 20. 1 H ЯМР (400 МГц, МeOH-d4) δ 2,2-2,3 (м, 4H), 2,50 (м, 1H), 3,00-3,15 (м, 4H), 3,74 (м, 1H), 3,88 (м, 3H), 4,88 (м, 3H), 5,51 (м, 1H), 7,53 (м, 1H), 7,88 (м, 1H), 8,39 (м, 1H); MS (m/e) 502 (M+1). Приклад одержання 21: Синтез трет-бутилового ефіру [(S)-1-(3-фтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-3-(2-пропіл-5-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, одержують 15 мг (вихід: 18 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (47 мг, 0,14 ммоль), синтезованої в прикладі одержання 17, і гідрохлориду 2-пропіл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (56 мг, 0,20 ммоль), синтезованого в прикладі одержання 2-(1). MS (m/e) 584 (M+Na). Приклад 19: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-5-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-ону 21 UA 103181 C2 5 10 15 20 25 30 35 40 45 Аналогічно процедурі прикладу 3, одержують 2,3 мг (вихід: 18 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 21. 1 H ЯМР (400 МГц, МeOH-d4) δ 0,95-0,99 (м, 3H), 1,82-1,84 (м, 2H), 2,18 (м, 2H), 2,44-2,56 (м, 3H), 2,74 (м, 1H), 2,90-3,07 (м, 5H), 3,76-3,89 (м, 4H), 4,84 (м, 3H), 5,54 (м, 1H); MS (m/e) 462 (M+1). Приклад одержання 22: Синтез трет-бутилового ефіру [(S)-1-(3-фтор-2-гідроксі-6оксопіперидин-1-ілметил)-3-оксо-3-(2-піридин-4-іл-5-трифторметил-5,8-дигідро-6H-піридо[3,4d]піримідин-7-іл)пропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, одержують 55 мг (вихід: 65 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-фтор-2-гідроксі-6оксопіперидин-1-іл)масляної кислоти (33 мг, 0,10 ммоль), синтезованої в прикладі одержання 17 і гідрохлориду 2-піридин-4-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (44 мг, 0,14 ммоль), синтезованого в прикладі одержання 2-(4). MS (m/e) 619 (M+Na). Приклад 20: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-5-трифторметил5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-5-фтор-6-гідроксипіперидин-2-ону Аналогічно процедурі прикладу 3, одержують 12 мг (вихід: 24 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 22. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,30-3,21 (м, 8H), 3,37-4,20 (м, 5H), 5,00 (м, 3H), 8,43 (с, 2H), 8,76 (с, 2H); MS (m/e) 519 (M+Na). Приклад одержання 23: Синтез 3-метилпіперидин-2,6-діону Оцтовий ангідрид додають до комерційно доступної 2-метилдіової кислоті (1 г, 6,8 ммоль) при кімнатній температурі, і суміш перемішують при 60 °C протягом 8 годин при нагріванні зі зворотним холодильником. Після підтвердження завершення реакції за допомогою ТШХ оцтовий ангідрид, що залишився, видаляють при зниженому тиску. Концентровану сполуку розчиняють у тетрагідрофурані й до нього повільно додають водний розчин аміаку (1,7 мл, 14,6 ммоль) при 0 °C, з наступним перемішуванням протягом 8 годин при кімнатній температурі. Після завершення реакції водний розчин аміаку, що залишився, видаляють при зниженому тиску й додають оцтовий ангідрид, з наступним нагріванням зі зворотним холодильником при 60 °C протягом 8 годин. Оцтовий ангідрид, що залишився, видаляють при зниженому тиску. Концентрат очищають за допомогою колонкової хроматографії (етилацетат:гексан=1:1), з одержанням 682 мг (вихід: 78 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,24-1,28 (м, 3H), 1,71-1,77 (1Н), 2,04-2,08 (м, 1H), 2,57-2,65 (м, 3H); MS (m/e) 128 (M+1). Приклад одержання 24: Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,6діоксопіперидин-1-іл)масляної кислоти (1) Синтез бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,6діоксопіперидин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(2), одержують 129 мг (вихід: 88 %) вказаної в заголовку сполуки з використанням 3-метилпіперидин-2,6-діону (53 мг, 0,42 ммоль), синтезованого в прикладі одержання 23, і бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4гідроксимасляної кислоти (107 мг, 0,35 ммоль), синтезованого в прикладі одержання 14-(1). 1 H ЯМР (400 МГц, СDCl3) δ 1,30-1,35 (м, 3H), 1,47 (с, 9H), 1,78-2,01 (м, 2H), 2,53-2,85 (м, 5H), 3,72 (м, 1H), 4,13-4,36 (м, 2H), 5,12-5,23 (м, 3H), 7,30-7,39 (м, 5H), MS (m/e) 441 (M+Na). 22 UA 103181 C2 5 10 15 20 25 30 35 40 45 50 (2) Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,6-діоксопіперидин-1-іл)масляної кислоти Бензиловий ефір (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,6-діоксопіперидин-1іл)масляної кислоти (218 мг, 0,52 ммоль), синтезований у розділі (1), розчиняють у метанолі й до нього додають 10 % Pd/C (22 мг, 0,1 екв.), з наступним перемішуванням протягом 1 години при тиску 1 атм. газоподібного водню. Після завершення реакції, Pd на куті видаляють за допомогою фільтрування через целіт. Залишок очищають за допомогою препаративної ТШХ, з одержанням 59 мг (вихід: 35 %) вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,28 (м, 3H), 1,42 (с, 9H), 1,80-1,99 (м, 2H), 2,38-2,87 (м, 5H), 3,72 (м, 1H), 4,22-4,44 (м, 2H); MS (m/e) 351 (M+Na). Приклад одержання 25: Синтез трет-бутилового ефіру [(S)-1-(3-метил-2,6-діоксопіперидин-1ілметил)-3-оксо-3-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6H-піридо[3,4-d]піримідин-7іл)пропіл]карбаминової кислоти Аналогічно процедурі прикладу одержання 6, одержують 47 мг (вихід: 88 %) вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,6діоксопіперидин-1-іл)масляної кислоти (29 мг, 0,09 ммоль), синтезованої в прикладі одержання 24, і гідрохлориду 2-тіофен-3-іл-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (40 мг, 0,12 ммоль), синтезованого в прикладі одержання 2-(7). 1 H ЯМР (400 МГц, МeOH-d4) δ 1,24-1,28 (м, 3H), 1,38 (с, 9H), 1,68-2,04 (м, 2H), 2,69-2,94 (м, 5H), 3,02-3,17 (м, 2H), 3,91 (м, 2H), 3,72 (м, 1H), 4,11-4,20 (м, 3H), 4,80-4,90 (м, 2H), 7,53 (м, 1H), 7,91 (м, 1H), 8,41 (м, 1H); MS (m/e) 618 (M+Na). Приклад 21: Синтез гідрохлориду 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)бутил]-3-метилпіперидин-2,6-діону Аналогічно процедурі прикладу 3, одержують 23 мг (вихід: 65 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 25. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,23-1,31 (м, 3H), 1,72-2,17 (м, 2H), 2,64-2,72 (м, 5H), 3,04-3,13 (м, 2H), 3,55-3,68 (м, 2H), 3,87-3,93 (м, 2H), 4,17 (м, 1H), 4,90 (м, 2H), 7,53 (м, 1H), 7,90 (м, 1H), 8,41 (м, 1H); MS (m/e) 496 (M+1). Приклад одержання 26: Синтез 3-метилпіролідин-2,5-діону (1) Синтез 2-метилбурштинової кислоти Комерційно доступний диметилметилсукцинат (1,57 г, 9,8 ммоль) розчиняють у змішаному розчиннику з тетрагідрофурану й води, 3:1, і додають гідроксид літію (4 г, 98 ммоль), і перемішують протягом 2 днів при 40C при нагріванні зі зворотним холодильником. Після підтвердження завершення реакції за допомогою ТШХ, залишок відганяють при зниженому тиску, з одержанням вказаної у заголовку сполуки. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,21-1,23 (д, 3H, J=7,2 Гц), 2,43-2,48 (м, 1H), 2,64-2,70 (м, 1H), 2,80-2,89 (м, 1H). (2) Синтез 3-метилпіролідин-2,5-діону Аналогічно процедурі прикладу одержання 23, одержують 250 мг вказаної в заголовку сполуки з використанням 2-метилбурштинової кислоти, синтезованої в розділі (1). 1 H ЯМР (400 МГц, МeOH-d4) δ 1,19-1,29 (м, 3H), 2,32-2,40 (м, 1H), 2,71-2,96 (м, 2H). Приклад одержання 27: Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,5діоксопіролідин-1-іл)масляної кислоти (1) Синтез бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,5діоксопіролідин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 4-(2), одержують 230 мг сирої вказаної в заголовку сполуки з використанням 3-метилпіролідин-2,5-діону (61 мг, 0,54 ммоль), синтезованого в прикладі одержання 26, і бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4гідроксимасляної кислоти (140 мг, 0,45 ммоль), синтезованого в прикладі одержання 14-(1). 1 H ЯМР (400 МГц, СDCl3) δ 1,31-1,39 (м, 3H), 1,43 (с, 9H), 2,29-2,39 (м, 1H), 2,62-2,92 (м, 4H), 3,70 (м, 1H), 4,18 (м, 2H), 5,13-5,14 (м, 2H), 5,24 (ушир., 1H), 7,35-7,36 (м, 5H); MS (m/e) 427 (M+Na). 23 UA 103181 C2 5 10 15 20 25 30 35 40 (2) Синтез (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,5-діоксопіролідин-1-іл)масляної кислоти Аналогічно процедурі прикладу одержання 23-(2), одержують 62 мг вказаної в заголовку сполуки з використанням бензилового ефіру (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,5діоксопіролідин-1-іл)масляної кислоти (218 мг, 0,52 ммоль), синтезованого в розділі (1). MS (m/e) 337 (M+Na) Приклад одержання 28: Синтез трет-бутилового ефіру [(S)-3-(2-трет-бутил-4-трифторметил5,8-дигідро-6H-піридо[3,4-d]піримідин-7-іл)-1-(3-метил-2,5-діоксопіролідин-1-ілметил)-3оксопропіл]карбамінової кислоти Аналогічно процедурі прикладу одержання 6, кількісно одержують 74 мг вказаної в заголовку сполуки з використанням (S)-3-трет-бутоксикарбоніламіно-4-(3-метил-2,5-діоксопіролідин-1іл)масляної кислоти (39 мг, 0,13 ммоль), синтезованої в прикладі одержання 27, і гідрохлориду 2-трет-бутил-4-трифторметил-5,6,7,8-тетрагідропіридо[3,4-d]піримідину (52 мг, 0,18 ммоль), синтезованого в прикладі одержання 2-(2). 1 H ЯМР (400 МГц, МeOH-d4) δ 1,24-1,28 (м, 3H), 1,38-1,42 (м, 18H), 2,27-2,92 (м, 5H), 3,003,12 (м, 2H), 3,63-4,59 (м, 5H), 4,82-4,86 (м, 2H); MS (m/e) 578 (M+Na). Приклад 22: Синтез гідрохлориду 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8дигідро-6H-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]-3-метилпіролідин-2,6-діону Аналогічно процедурі прикладу 3, одержують 32 мг (вихід: 61 %) вказаної в заголовку сполуки з використанням сполуки, синтезованої в прикладі одержання 28. 1 H ЯМР (400 МГц, МeOH-d4) δ 1,26-1,29 (м, 3H), 1,42 (с, 9H), 2,33-2,38 (м, 1H), 2,57-2,67 (м, 2H), 2,90-2,92 (м, 2H), 3,00-3,10 (м, 2H), 3,55-3,37 (м, 3H), 3,84-3,90 (м, 2H), 4,82-4,86 (м, 2H); MS (m/e) 456 (M+1). Експериментальний приклад 1: Аналіз інгібіторної активності відносно DPP-IV Дипептиділпептидазу-IV (DPP-IV), відому як серинпротеаза, одержують за допомогою модифікації відомого способу (Tanaka T. et al, Proc. Natl. Acad. Sci. USA, (1994) 91, 3082-3086), який включає стадії клонування, очищення з використанням Baculovirus і активування. DPP-IV використовують для дослідження фармацевтичної ефективності кандидатів в інгібітори в такий спосіб. Клоновану DPP-IV експресують в Baculovirus, який очищають за допомогою нікелевої колонки, а потім піддають діалізу. Інгібітори, синтезовані в прикладах, досліджують для визначення їх інгібіторної активності відносно DPP-IV з використанням флуоресцентного субстрату, AC-GLY-PRO-AFC. Ферментативні реакції здійснюють при різних концентраціях інгібіторів, використовуючи 100 мкM AC-GLY-PRO-AFC при 25 °C у буферному розчині, що містить 50 мМ HEPES (pН 7,4), при цьому концентрація DPP-IV становить 7,1 нм. Значення IC50 для інгібіторів визначають за допомогою вимірювання величини флуоресценції, що випускається таким чином, на флуоресцентному спектрометрі після здійснення ферментативної реакції протягом 1 години, а потім обчислюють концентрацію інгібіторів, що демонструє 50 % інгібування ферментативної реакції в цілому. Спектри флуоресцентного спектрометра MAX Geminixs (Molecular Device Co.) використовують як флуоресцентний спектрометр, і довжини хвиль порушення й випускання встановлюють на 400 нм і 505 нм, відповідно. Отримані результати наводяться в таблиці 1, нижче. 24 UA 103181 C2 Таблиця 1 Приклад 1 Приклад 3 Приклад 5 Приклад 7 Приклад 9 Приклад 11 Приклад 13 Приклад 15 Приклад 17 Приклад 19 Приклад 21 5 10 15 20 25 30 35 IC50 (нM) 36 20 17 63 93 160 195 2500 81 409 >2500 Приклад 2 Приклад 4 Приклад 6 Приклад 8 Приклад 10 Приклад 12 Приклад 14 Приклад 16 Приклад 18 Приклад 20 Приклад 22 IC50 (нM) 5 39 10 6 14 10 57 197 159 206 >2500 Експериментальний приклад 2: Аналіз інгібіторної активності відносно DPP-H, VIII і IX Для порівняння селективності деяких сполук із прикладів 1-22 відносно ферменту DPP і відомих інгібіторів DPP-IV, інгібіторну активність відносно ферментів у цих досліджуваних сполук вимірюють у такий спосіб. - Вимірювання інгібіторної активності відносно DPP-II: Одержують і очищають DPP-II. Використовуючи Gly-pro-afc у якості субстрату, інгібітори DPP-IV, як наведено в таблиці 2 нижче, аналізують на інгібіторну активність відносно DPP-II. Ферментативні реакції здійснюють для різних концентрацій інгібіторів DPP-IV, використовуючи 0,018 мкг DPP-II і 100 мкM Gly-pro-afc у буферному розчині, що містить 50 мМ оцтової кислоти (pН 4,5), при кімнатній температурі. Значення IC50 інгібіторів визначають за допомогою відстеження швидкості реакції з використанням флуоресцентного спектрометра протягом 60 хв і аналізуючи результати з використанням Tablecurve 2D (v 5.01). Інгібітори DPP-IV аналізують із використанням флуоресцентного спектрометра при довжинах хвиль порушення й випускання 405 нм і 505 нм, відповідно. - Вимірювання інгібіторної активності відносно DPP-VIII: Одержують і очищають DPP-VIII. Використовуючи Gly-pro-afc як субстрат, інгібітори DPP-IV, як наведено в таблиці 2 нижче, аналізують на інгібіторну активність відносно DPP-VIII. Ферментативні реакції здійснюють для різних концентрацій інгібіторів DPP-IV, використовуючи 0,0049 мкг DPP-VIII і 100 мкM Gly-pro-afc у буферному розчині, що містить 50 мМ HEPES (pН 7,5), при кімнатній температурі. Значення IC50 для інгібіторів визначають за допомогою відстеження швидкості реакції з використанням флуоресцентного спектрометра протягом 60 хв і аналізуючи результати з використанням Tablecurve 2D (v 5.01). Інгібітори DPP-IV аналізують із використанням флуоресцентного спектрометра при довжинах хвиль порушення й випускання 405 нм і 505 нм, відповідно. - Вимірювання інгібіторної активності й відносно DPP-IX: Одержують і очищають DPP-IX. Використовуючи Gly-pro-afc як субстрат, інгібітори DPP-IV, як наведено в таблиці 2 нижче, аналізують на інгібіторну активність стосовно DPP-IX. Ферментативні реакції здійснюють для різних концентрацій інгібіторів DPP-IV, використовуючи 200 нг DPP-IX і 100 мкM Gly-pro-afc у буферному розчині, що містить 50 мМ HEPES (pН 7,5), при кімнатній температурі. Значення IC 50 для інгібіторів визначають за допомогою відстеження ходу швидкості реакції в часі, з використанням флуоресцентного спектрометра й аналізуючи результати з використанням Tablecurve 2D (v 5.01). Інгібітори DPP-IV аналізують із використанням флуоресцентного спектрометра при довжинах хвиль порушення й випускання 405 нм і 505 нм, відповідно. Отримані результати аналізу наводяться в таблиці 2, нижче. 25 UA 103181 C2 Таблиця 2 Інгібітори Приклад 2 Приклад 5 Приклад 6 Приклад 8 Приклад 10 Приклад 12 a Приклад 83 b Vildagliptin (Norvatis) c Sitagliptin (Merck) Saxagliptin (BMS) DPP-II (мкM) >400 >161 >70 >80 >400 >50 >400 >10 >100 DPP-IV (нM) 5 17 10 6 14 10 18 62 18 DPP-VIII (мкM) >400 >400 >127 >400 >400 >400 15 >10 48 DPP-IX (мкM) >400 >400 >100 >130 >210 >110 124 1,3 >100 >400 13 0,17 0,061 a. Див. WO 06/104356, що належить авторам даного винаходу b. Дані опубліковано в IDF, 2006 c. Kim, D., et al, J. Med Chem, 2005, 48, 141-151 5 10 15 Як можна побачити з результатів таблиці 2, сполуки за даним винаходом демонструють чудову селективність відносно DPP-IV, у порівнянні з інгібіторами DPP-IV з WO 06/104356. Ці результати говорять, що потенційна токсичність сполук через побічні реакції може зводитися до мінімуму, як описувалося раніше. Промислова застосовність Як видно із усього наведеного вище, ясно, що нові сполуки відповідно до даного винаходу інгібують активність DPP-IV, призводячи, таким чином, до збільшення секреції інсуліну й, як наслідок, до зниження рівнів глюкози в крові. Відповідно, ці сполуки можуть використовуватися для лікування або запобігання захворювань, пов'язаних з DPP-IV, наприклад, діабету (зокрема, типу II), ожиріння й тому подібного. Хоча переважні варіанти здійснення даного винаходу описані для ілюстративних цілей, фахівці в даній галузі помітять, що різні модифікації, доповнення й заміни є можливими, без відхилення від меж і духу даного винаходу, як описано в прикладеній формулі винаходу. ФОРМУЛА ВИНАХОДУ 1. Сполука, представлена формулою 1: A 20 B NH2 O , (1) де А вибирають із замісника формули 2: R1 N R2 25 30 N N , (2) де R1 являє собою водень або CF3, і R2 вибирають із групи, що складається з водню, заміщеного або незаміщеного С1-С10алкілу, заміщеного або незаміщеного С3-С10циклоалкілу, заміщеного або незаміщеного С3С10гетероциклоалкілу, заміщеного або незаміщеного С4-С8арилу й заміщеного або незаміщеного С3-С7гетероарилу; і замісника формули 3: 26 UA 103181 C2 S R3 N N , (3) де R3 вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С 1С4алкілу; і В вибирають із групи, що складається із замісника формули 4: R6 R7 X N R8 n 5 10 O R9 , (4) де X являє собою -CR4R5- або -СО-, де R4 і R5, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R4 і R5 являє собою гідрокси, R6, R7, R8 і R9, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу, і n дорівнює 0 або 1; замісника формули 5: R 12 R 13 X N O 15 20 Y R 14 , (5) де X являє собою -CR10R11- або -СО-, де R10 і R11, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R10 і R11 являє собою гідрокси, R12, R13 і R14, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу, і Y являє собою кисень або сірку; замісника формули 6: R 17 X N R 18 Z O 25 R 19 , (6) де X являє собою -CR15R16- або -СО-, де R15 і R16, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R15 і R16 являє собою гідрокси, Z являє собою -СН- або кисень, за умови, що, коли Z являє собою кисень, R19 відсутній, і R17, R18 і R19, кожний, незалежно вибирають із групи, що складається з водню, галогену й заміщеного або незаміщеного С1-С4алкілу; і замісника формули 7: X N R 22 O 30 , (7) де X являє собою -CR20R21- або -CO, де R20 і R21, кожний, незалежно являють собою водень або гідрокси, за умови, що щонайменше один з R20 і R21 являє собою гідрокси, і R22 являє собою заміщений або незаміщений С1-С4алкіл; або її фармацевтично прийнятна сіль. 27 UA 103181 C2 5 10 15 20 2. Сполука за п. 1, в якій, коли алкіл, циклоалкіл, гетероциклоалкіл, арил або гетероарил є заміщеним, замісник являє собою С1-С10алкіл або галоген. 3. Сполука за п. 2, в якій галоген являє собою фтор. 4. Сполука за п. 1, в якій А у формулі 1 являє собою замісник, представлений формулою 2, де R1 являє собою водень або CF3 і R2 вибирають із групи, що складається з водню й С 1-С5алкілу, C3-C6циклоалкілу, С3-С10гетероциклоалкілу, С4-С8арилу й С3-С7гетероарилу, кожний з яких необов'язково заміщений галогеном. 5. Сполука за п. 4, в якій, коли R2 являє собою незаміщений або галогензаміщений С3С10гетероциклоалкіл або незаміщений або галогензаміщений С 3-С7гетероарил, вказаний гетероциклоалкіл або гетероарил являє собою будь-яку групу, вибрану із групи, що складається з фурану, тіофену, піролу, піролідину, імідазолу, піразолу, піразоліну, оксазолу, оксазоліну, ізоксазолу, ізоксазоліну, тіазолу, тіазоліну, ізотіазолу, ізотіазолідину, тіадіазолу, тіадіазоліну, тетрагідрофурану, тетрагідротіофену, імідазолідину, піразолідину, оксазолідину, ізоксазолідину, тіазолідину, ізотіазолідину, тіадіазолідину, сульфолану, пірану, дигідропірану, тетрагідропірану, піридину, піридинону, піридазину, піразину, піримідину, піперидину, піперазину, морфоліну, піридазинону, тетразолу, триазолу, триазолідину й азепіну. 6. Сполука за п. 4, в якій R2 вибирають із групи, що складається із трифторметилу, пропілу, бутилу, трет-бутилу, циклобутилу, піридину, фурану, метоксіетилу, тіофену й 4-фторфенілу. 7. Сполука за п. 1, в якій В у формулі 1 являє собою замісник, представлений формулою 4, де X являє собою -(СН-ОН)- або -CO-, R6 і R7, кожний, незалежно вибирають із групи, що складається з водню, фтору й незаміщеного С1-С4алкілу, і R8 і R9, кожний, незалежно являють собою водень. 8. Сполука за п. 1, в якій сполука являє собою сполуку формули 1а A B O 25 30 35 40 45 50 55 NH2 , (1a) де А і В є такими, як визначено для формули 1. 9. Сполука за п. 1, в якій сполука являє собою будь-яку з наступних сполук: 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]5,5-дифтop-(6R)-гiдpoкcипіпepидин-2-oн, 1-[(2S)-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]5,5-дифтор-(6S)-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-циклобутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-піридин-4-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7іл)бутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)бутил]4-оксобутил]-5,5-дифтор-6-гідроксипіперидин-2-он, 1-{(S)-2-аміно-4-[2-(2-метоксіетил)-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл]-4оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7іл)бутил]-5,5-дифтор-6-гідроксипіпeридин-2-он, 1-{(S)-2-аміно-4-[2-(4-фторфеніл)-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл]-4оксобутил}-5,5-дифтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2,4-біс-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4-оксобутил]3,3-дифторпіперидин-2,6-діон, 1-[(S)-2-аміно-4-(2-трет-бутил-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-(2-фуран-2-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7-іл)-4оксобутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-тіофен-3-іл-4-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7іл)бутил]-5-фтор-6-гідроксипіперидин-2-он, 1-[(S)-2-аміно-4-оксо-4-(2-пропіл-5-трифторметил-5,8-дигідро-6Н-піридо[3,4-d]піримідин-7іл)бутил]-5-фтор-6-гідроксипіперидин-2-он, 28
ДивитисяДодаткова інформація
Назва патенту англійськоюDipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as active agent
Автори англійськоюLee, Chang-Seok, Yim, Hyeon Joo, Kim, Kyoung-Hee, Lee, Jaeick, Lee, Sung-Hack, Lee, Kyu Woong, Lee, Hee Bong, Park, Wan Su, Min, Changhee
Автори російськоюЛи Чанг-Сеок, Йим Хиеон Дзоо, Ким Киоунг-Хее
МПК / Мітки
МПК: C07D 471/04
Мітки: містить, композиція, фармацевтична, інгібують, активний, сполуки, агент, яка, дипептидилпептидазу-iv
Код посилання
<a href="https://ua.patents.su/31-103181-spoluki-shho-ingibuyut-dipeptidilpeptidazu-iv-i-farmacevtichna-kompoziciya-yaka-mistit-kh-yak-aktivnijj-agent.html" target="_blank" rel="follow" title="База патентів України">Сполуки, що інгібують дипептидилпептидазу-iv і фармацевтична композиція, яка містить їх як активний агент</a>
Сполуки-інгібітори дипептидилпептидази-іv, фармацевтична композиція, яка містить вказані сполуки як активний інгредієнт

Номер патенту: 89396
Опубліковано: 25.01.2010
Автори: Кім Кіоунг-Хеє, Кім Сунгсуб, Квон Ох Хван, Кім Сунг Хо, Кох Дзонг Сунг, Йєом Зі-Хо, Бу Сєонг Чєол, Йєо Донг-Дзун, Кім Мін-Дзунг, Хур Гвонг-Чєунг, Коо Кі Донг, Хонг Санг Йонг, Хан Хеє Оон, Кім Гєун Тає, Лі Чанг-Сеок, Йім Хієон Дзоо, Кім Дзі Янг, Кім Хіє Дзін, Лім Донгчул
МПК: C07D 471/04
Мітки: сполуки, фармацевтична, дипептидилпептидази-iv, містить, інгредієнт, яка, вказані, активний, композиція, сполуки-інгібітори
Формула / Реферат:
1. Сполука формули (1) або її фармацевтично прийнятна сіль: , (1)дeА вибраний з групи, яка складається із замісників наступних формул з (2) по (7): , (2)де R1 являє собою водень або заміщений або незаміщений C1-C4алкіл; і X являє собою вуглець або азот;
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Чу-Моєр Маргарет Юхуа, Зембровскі Уільям Джеймс, Міларі Банавара Лакшман, Маррі Джеррі Ентоні
МПК: C07D 521/00, C07D 405/14, C07D 487/08, C07D 403/14, C07D 403/12, A61K 31/5377, C07D 451/02, C07D 403/04, C07D 491/10, C07D 471/04, C07D 409/12, C07D 471/08, C07D 401/14, C07D 451/06, C07D 401/12, C07D 498/04, C07D 239/42, A61P 43/00, A61K 31/517, C07D 491/04, C07D 417/14, A61P 3/10, C07D 401/04, A61K 31/506, C07D 409/14, A61P 9/10, C07D 413/12, C07D 405/12, C07D 513/10, A61K 31/53, C07D 487/04, C07D 491/20, C07D 417/12, C07D 491/048
Мітки: сорбітдегідрогенази, сполуки, інгібітори, композиція, проміжні, спосіб, проміжної, піримідини, фармацевтична, містить, одержання
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Сполуки піридопіримідинону, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 80578
Опубліковано: 10.10.2007
Автори: Кеньяр Даніель-Енрі, Ренар Пьєр, Пфейфер Брюно, Копп Маріна, Рауль Сільвен, Ланцелот Жан-Шарль
МПК: A61P 3/10, A61P 19/02, A61P 9/10, A61P 9/12, A61P 3/04, A61P 35/00, C07D 471/04
Мітки: яка, містить, одержання, композиція, фармацевтична, піридопіримідинону, сполуки, спосіб
Формула / Реферат:
1. Сполуки формули (І), (I)деR1 і R2, які можуть бути однаковими або відрізнятись, являють собою атом водню або алкільну групу або разом з атомом азоту, який несе їх, утворюють гетероцикл,R3 являє собою атом галогену, алкоксигрупу, необов'язково заміщену арильну групу або групу NR'1R'2, де R'1 і R'2, які можуть бути однаковими або відрізнятись,...
Заміщені сполуки бензо[e][1,4]оксазино[3,2-g]ізоіндолу, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 79163
Опубліковано: 25.05.2007
Автори: Кудер Жерар, Краус-Бертьє Лоранс, Пьєр Ален, Ікман Джон, Ренар Пьєр, Лепіфр Франк, Кеньяр Даніель-Енрі
МПК: A61P 35/00, C07D 497/00
Мітки: заміщені, фармацевтична, композиція, одержання, спосіб, містить, сполуки, яка, бензо[e][1,4]оксазино[3,2-g]ізоіндолу
Формула / Реферат:
1. Сполуки формули (І):,де:W1 являє собою, з атомами вуглецю, до яких він прикріплений, фенільну групу або піридильну групу,Ζ являє собою групу, вибрану з водню, галогену і груп лінійного або розгалуженого (С1-С6)алкілу, арилу, арил-(С1-С6)алкілу, в якому алкільна частина може бути лінійною або розгалуженою, арилокси, арил-(С1-С6)алкокси,...
Сполуки імідазопіридину, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 80579
Опубліковано: 10.10.2007
Автори: Рауль Сільвен, Копп Маріна, Кеньяр Даніель-Енрі, Ренар Пьєр, Ланцелот Жан-Шарль, Бізо-Еспіяр Жан-Гій, Пфейфер Брюно
МПК: A61P 9/10, C07D 471/04, A61P 3/10, A61P 3/06, A61P 3/04
Мітки: композиція, містить, фармацевтична, сполуки, одержання, яка, імідазопіридину, спосіб
Формула / Реферат:
1. Сполуки формули (І), (І)де:R1 являє собою атом водню, атом галогену або алкільну, полігалоалкільну, ціано, нітро, гідроксикарбонільну, алкоксикарбонільну, амінокарбонільну, алкіламінокарбонільну або діалкіламінокарбонільну групу,R2 являє собою атом водню, алкільну групу, арильну групу, що необов'язково заміщується, гетероарильну групу, що...
Попередній патент: Збалансована за віком система харчування для дітей
Наступний патент: Таблетки з плівковим покриттям, що містять як активний компонент дроспіренон, та спосіб їх одержання
Випадковий патент: Пружинний блок