Похідні піридопіраноазепінів, спосіб їх одержання, лікарський засіб та фармацевтична композиція на їх основі
Номер патенту: 61164
Опубліковано: 17.11.2003
Автори: Галлі Фредерік, Жегам Самір, Локхед Алістер, Самсон Аксель







Формула / Реферат
1. Похідні піридопіраноазепінів, у формі чистого геометричного та оптичного ізомерів або їх суміші, загальної формули (І)
,
в якій
R1 - атом гідрогену, (С1-С4)алкіл, феніл(С1-С4)алкіл, фенілгідрокси(С1-С4)алкіл, фураніл(С1-С4)алкіл, або фуранілгідрокси(С1-С4)алкіл, R2 - атом гідрогену чи галогену, або трифлуорметил, ціаногрупа, гідроксил, нітрогрупа, ацетил, (С1-С6)алкіл або (С1-С6)алкоксил або група загальної формули NR4R5, в якій R4 - атом гідрогену або (С1-С4)алкіл, або (С1-С4)алканоїл, a R5 - атом гідрогену або (С1-С4)алкіл, або інакше R4 та R5 утворюють з атомом нітрогену, до якого вони приєднані, (С4-С7)-цикл, або феніл чи нафтил, що як варіант, заміщені атомом галогену або трифлуорметилом, трифлуорметоксилом, ціаногрупою, гідроксилом, нітрогрупою, ацетилом, (С1-С6)алкілом, (С1-С6)алкоксилом або метилендіоксилом, що приєднані у 2 та 3 позиціях фенільного кільця, а R3 - атом гідрогену чи галогену або (С1-С4)алкіл, у формі основи або кислотно-адитивної солі.
2. Спосіб одержання сполуки за п.1, який відрізняється тим, що сполуку загальної формули (IV)
,
в якій R2 та R3 визначено за п. 1, дегідратують у кислому середовищі з отриманням сполуки загальної формули (Іа)
,
в якій, за бажанням, замісники R2 та R3 модифікують і/або уводять замісник R1, який визначено за п. 1.
3. Лікарський засіб, який відрізняється тим, що складається зі сполуки за п. 1.
4. Фармацевтична композиція, яка відрізняється тим, що містить сполуку за п. 1 у поєднанні з ексципієнтом.
Текст
1 ПОХІДНІ піридопіраноазепінів, у формі чисто го геометричного та оптичного ізомерів або їх суміші, загальної формули (І) Ь в якій R2 та R3 визначено за п 1, депдратують у кислому середовищі з отриманням сполуки загальної формули (Іа) О В ЯКІЙ Ri - атом гідрогену, (Сі-С4)алкіл, феніл ( d С^алкіл, феніл гід рокси(Сі-С4)ал кіл, фураніл(Сг С4)алкіл, або фуранілпдрокси(Сі-С4)алкіл, R2 атом гідрогену чи галогену, або трифлуорметил, ціаногрупа, гідроксил, нітрогрупа, ацетил, ( d Сє)алкіл або (С-і-Сб)алкоксил або група загальної формули NR4R5, в якій R4 - атом гідрогену або ( d С4)алкіл, або (Сі-С4)алканоіл, a Rs - атом гідрогену або (С-і-С4)алкіл, або інакше R4 та Rs утворюють з атомом нітрогену, до якого вони приєднані, (С4С7)-цикл, або феніл чи нафтил, що як варіант, за Представлений винахід стосується сполук загальної формули (І) (І) в якій в якій, за бажанням, замісники R2 та R3 модифікують і/або уводять замісник Ri, який визначено за п1 3 Лікарський засіб, який відрізняється тим, що складається зі сполуки за п 1 4 Фармацевтична композиція, яка відрізняється тим, що містить сполуку за п 1 у поєднанні з ексципієнтом R I - атом гідрогену, (С-і-С4)алкіл, феніл(dС4)алкіл, феніл гід рокси(Сі-С4)ал кіл, фураніл(Сг С4)алкіл, або фураніл-пдрокси(Сі-С4)алкіл, R2 атом гідрогену чи галогену, або трифлуорметил, ціаногрупа, гідроксил, нітрогрупа, ацетил, ( d Сє)алкіл або (С-і-Сб)алкоксил або група загальної формули NR4R5, в якій R4 - атом гідрогену, або ( d С4)алкіл, або (Сі-С4)алканоіл, a Rs - атом гідрогену (О (О 61164 або (Сі-С4)алкіл, або інакше R4 та Rs утворюють з атомом нітрогену, до якого приєднані, (С4-С7)цикл, або феніл чи нафтил, що як варіант, заміщені атомом галогену або трифлуорметилом, трифлуорметоксилом, ціаногрупою, гідроксилом, нітрогрупою, ацетилом, (Сі-Сє)алкілом, (dСє)алкоксилом або метилендюксилом, що приєднані у 2 та 3 позиції фенільного кільця, a R3 - атом гідрогену або галогену або (Сі-С4)алкіл Сполуки загальної формули (І) можуть бути основами або кислотно-адитивними солями Крім того, атоми у позиціях 5а та 10а є асиметричними, тому сполуки можуть бути чистими геометричними та оптичними ізомерами або їх сумішами Згідно З винаходом, сполуки загальної формули (І) можливо виготовляти способом, що ілюстровано нижченаведеною схемою (П) 2-метилпіридин-З-ол загальної формули (II), в якій R2 та R3 визначено вище, реагує з алкіллітієм, тоді отриманий таким чином штермедіат конденсують з 1-азадицикло[2,2,2]октан-3-оном формули (III), при низькій температурі у такому апротонному розчиннику, як тетрапдрофуран Отримують сполуку загальної формули (IV), в яку можливо, за бажанням, уводити або модифікувати и замісниками R2 та R3 згідно з будь-яким способом, що відомий фахівцям Сполуку загальної формули (IV) далі депдратують, що супроводжується перегрупуванням, у кислотному середовищі, наприклад, метансульфоновій кислоті або сульфатній кислоті при підвищеній температурі Отримують сполуку загальної формули (Іа), яку можливо модифікувати замісниками R2 та R3 та/або уводити замісник г' згідно з будь-яким способом, що відомий фахівцям ВИХІДНІ сполуки формул (II) та (III) комерційне доступні (R2=R3-H) або їх можна виготовити згідно з відомими способами Нижченаведені приклади ілюструють виготовлення деяких сполук винаходу Мікроаналіз на елементи, спектри ІЧ та ЯМР, а також, у деяких випадках, рентгенодифракційні спектри підтверджують структури отриманої сполуки Числа у дужках у титлах прикладів відповідають числам у першій колонці нижченаведеної таблиці У назвах сполук, риска "-" є частиною назви, та риска "_" слугує тільки для переносу при закінченні строки, її є треба відкидати у відсутності переносу, та не можна заміняти нормальною рискою чи пробілом Приклад 1 (Сполука №1) П дрохлорид (транс)-5а,6,7,9,10,11-гексапд ро8,10а-метанпіридо[2',3' 5,6]пірано[2,3-сі]азепіну (2 1) 1 1 3-[(3-пдроксипіридин-2-іл)метил]-Іазадицикло[2,2,2]октан-3-ол 52,9г (484 ммоль) 2-метил-З-пдроксипіридину у розчині 1300мл тетрапдрофурану уводять у тригорлу колбу на 2000мл під аргоном Розчин охолоджують до -56°С та краплями додають 750мл (975ммоль) 1,ЗМ розчину 1-метилпропіллітію у циклогексані протягом 3 годин, підтримуючи температуру нижче -50°С При закінченні додавання, температурі дають підвищитися до -4°С протягом 45 хвилин та суміш далі знов охолоджують до 58°С до додавання 60,6г (484 ммоль) 1 азадицикло[2,2,2]октан-3-он у розчині 250мл тетрапдрофурану краплями протягом 40 хвилин Температурі дають підвищитися до зовнішньої та перемішування продовжують протягом 20 годин Реакційну суміш охолоджують до 4°С та пдролізують додаванням 110мл водного розчину 36% пдрохлоридної кислоти 400мл води додають, двом фазам дають відстоятися та органічну фазу екстрагують водою Водні фази поєднують, суміш охолоджують до 4°С та додають концентрований водний розчин гідроксиду натрію до рН 8,4 Отриманий осад фільтрують та сушать у вакуумі при 80°С Так отримали 62,5г продукту Темп плавл 270-272°С 12 Пдрохлорид (транс)-5а,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3d]a3enm (2 1) 2,34г (10 ммоль) 3-[(3-пдроксипіридин-2іл)метил]-1-азадицикло[2,2,2]окган-3-олу у розчині 10мл метансульфонової кислоти уводять у колбу на 50мл та гріють при 180°С протягом 48 годин Реакційну суміш охолоджують та виливають на лід її роблять лужною додаванням концентрованого водного розчину гідроксиду натрію та екстрагують хлороформом Органічну фазу сушать MgSO4 та концентрують під зниженим тиском Залишок очищають хроматографією на колонці з силікагелем, елююючи 90/10/1 сумішшю хлороформу, метанолу та аміаку Продукт отримують у формі основи, яку перетворюють у сіль додаванням розчину пдрохлоридної кислоти у етанолі Виділяють 1,55Г пдрохлориду Темп плавл >300°С Приклад 2 (Сполука №2) Пдрохлорид (5aS, 10aR)-5a,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сІ]азепіну (2 1) 2,1 (3-R,5R}-(-)-0,0'-flH6eH3Oin-LTapTpaT(5aS,10aR)-5a,6,7,9,10,11-reKcariflpo-8,10aметанпіридо[2',3' 5,6]пірано[2,3-сі]азепіну (1,2) 15,335г (0,0709 моль) (транс)-5а,6,7,9,10,11гексапдро-8,10а-метанліридо[2',3' 5,6]пірано[2,3 61164 сі]азепіну у 50мл етилацетату уводять у колбу на нолу та аміаку Отримують 1,2г продукту Темп 500мл Розчин 50,83г (0,142 моль) (3R,5R)-(-)-0,0'плавл 157-159°С дибензоіл-І_-винноі кислоти у 50мл етилацетату Приклад 4 (Сполука №28) додають, розчинник випарюють під зниженим тисГідробромід(транс)-(-)-2-бром-5а,6,7,9,10,11ком та залишок розчиняють у 885мл 7/3 суміші гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3води та етанолу при КИП'ЯТІННІ ПІД зворотним холосі]азепіну (1 1) дильником Після охолодження, отримані кристали 41 (3-R,5R)-(-)-0,0'-flH6eH3Oin-Lзбирають фільтруванням та перекристалізовують TapTpaT(5aS,10aR]-2-6poM-5a,6,7,9,10,11у 50мл гарячого пропан-2-олу гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сі]азепіну (1 2) Після охолодження отримують 13,7г кристалів Темп плавл 145-148°С, a D --104,3° (с=0,5, 0,3г (1 ммоль) (транс)-2-бром-5а,6,7,9,10,11МеОН) гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сі]азепіну у розчині Юмл етилацетату уводять у 22 Гідрохлорид (5aS,10aR)-5a,6,7,9,10,11колбу на 50мл, 0,358г (1 ммоль) 0,0'-(-)-дибензоілгексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3L-винноі кислоти у розчині Змл етилацетату додаd]a3enmy (2 1) ють, розчинник випарюють під зниженим тиском та Обробка попередньої сполуки водним розчизалишок перекристалізовують у 5мл гарячого проном карбонату калію з наступною екстракцією з пан-2-олу Після охолодження отримані кристали дихлорметану дали 3,1г (0,0143 моль) сполуки у збирають фільтруванням та сушать у вакуумі формі основи Темп плавл 69-71 °С Отримують 0,12г кристалів a D 2 0 =75,4°(c=1, МеОН) Темп плавл 200°С a D --106° (с=0,5, МеОН) Цю основу розчиняють у Юмл етанолу у колбі 42 Гідробромід (транс)-(-)-2-бромна 50мл, додають бмл (О.ОЗОмоль) розчину 6М 53,6,7,9,10,11-гексапдро-8,10апдрохлоридної кислоти у пропан-2-олі, суміш конметанпіридо[2',3' 5,6]пірано[2,3-сі]азепіну (1 1) центрують до суха під зниженим тиском, залишок переносять знов у 40мл пропан-2-олу, суміш гріПеретворення у основу проводять обробкою ють до кипіння та додають 5мл етанолу Після попередньої сполуки водним розчином гідроксиду охолодження отримані кристали збирають фільтнатрію, з наступною екстракцією з дихлорметаном руванням та сушать під зниженим тиском 0,3г (1 ммоль) основи розчиняють у ЗОмл пропан-2олу у колбу на 100мл, 0,36мл (2 ммоль) розчину Отримують 3,4г білих кристалів 33% пдробромідної кислоти у оцтовій кислоті доТемп плавл 330°С, a D --85,3° (с=1, МеОН) дають Після охолодження до 4°С, отримані крисПриклад 3 (Сполука №4) тали збирають фільтруванням та сушать у вакуумі (транс)-2-бром-5а,6,7,9,10,11-гексапдро-8,10аОтримують 0,25г білих кристалів метанпіридо[2',3' 5,6]пірано[2,3-сі]азепін Темп плавл 350-352°С, a D =-76,3° (с=0,5, 3 1 3-[(6-бром-3-пдроксипіридин-2-іл)метил]-1 МеОН) азадицикло[2,2,2]октан-3-ол Приклад 5 (Сполука №24) 52,23г (0,223 моль) 3-[(3-пдроксипіридин-2Гідрохлорид (транс)-(-)-2-хлор-5а,6,7,9,10,11іл)метил]-1 -азадицикло[2,2,2 ]октан-3-олу суспенгексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3дують у 500мл води при температурі довкілля і сі]азепіну (2 1) уводять у колбу на ЮООмл Додають 26,7г (0,669 моль) гідроксиду натрію у розчині 350мл води та 0,2г (0,68 ммоль) (транс)-(-)-2-бром26,5г (0,223моль) броміду калію та суміш перемі5з,6,7,9,10,11-гексзпдро-8,10зшують, доки не відбувається розчинення перед метанпіридо[2',3' 5,6]nipaHo[2,3-d]a3enmy розчинядодаванням краплями 11,5мл (0,223моль) брому ють у 4мл концентрованого водного розчину пдропротягом 2 годин хлоридної кислоти та гріють при 180°С у закритих трубах протягом 48 годин Водну фазу випарюють Суміш перемішують протягом 18 годин при та залишок перекристалізовують у пропан-2-олі температурі довкілля, далі реакційну суміш нейтОтримують 0,075г кристалів Темп плавл 339ралізують додаванням 23мл оцтової кислоти, охо344°С, aD20=-81° (c=0,5, МеОН) лоджують на льодяній бані та отриманий осад фільтрують Маточники концентрують та отриманий Приклад 6 (Сполука №27) осад розтирають у пропан-2-олі, фільтрують та Гідробромід (транс)-2-ціано-5а,6,7,9,10,11промивають Отримують 27,9г продукту Темп гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3плавл 215-221 °С сі]азепіну (1 1) 3,2 (транс)-2-бром-5а,6,7,9,10,11-гексапд ро0,45г (1,52 ммоль) (транс)-2-бром8,10а-метанпіридо[2',3' 5,6]пірано[2,3-сі]азепін 5а,6,7,9,10,11-гексапдро-8,10а-метанпіридо [2і,3' 5,6]mpaHo[2,3-d]a3eniHy розчиняють у 8мл 6,1г 3-[(6-бром-3-пдроксипіридин-2-іл)метил]піридину у колбі на 50мл, додають 0,205г (2,29 1-азадицикло[2,2,2]октан-3-олу та 50мл концентммоль) ціаніду купрумута суміш гріють до кипіння рованої сульфатної кислоти уводять у колбу на протягом ЗО годин Додають 75мл дихлорметану ЮОмл Суміш гріють при 130°С протягом 72 годин, та органічну фазу промивають 45мл насиченого потім охолоджують до температури довкілля та водного розчину хлориду амонію, далі 75мл води виливають на лід Водну фазу роблять лужною Після сушки та концентрації органічної фази під додаванням концентрованого водного розчину зниженим тиском, отримують 0,22г очікуваного гідроксиду натрію до рН 10 та екстрагують хлоропродукту Його розчиняють у пропан-2-олі та обформом Органічні фази сушать над MgSO4 та робляють одним еквівалентом пдробромідної кисконцентрують під зниженим тиском Залишок очилоти у 33% розчині у оцтовій кислоті Після охолощають хроматографією на колонці з силікагелем, дження, відділення кристалів фільтруванням та елююючи 90/10/4 сумішшю дихлорметану, мета 61164 сушки у вакуумі отримують 0,21г продукту Темп плавл 329-332°С Приклад 7 (Сполука №10) Гідробромід (транс)-2-(4-метилфеніл)5а,6,7,9,10,11-гексапдро-8,10аметанпіридо[2',3' 5,6]пірано[2,3-сі]азепіну (2 1) 0,3г (1 ммоль) (транс)-2-бром-5а,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сі]азепіну у бмл толуолу, 0,193г (1,4 ммоль) 4метилфенілборонової кислоти, 0,072г (0,06 ммоль) тетракіс(трифеніл)фосфшпаладію, 1мл (2 ммоль) карбонату натрію у 2М водному розчині та 0,05мл етанолу уводять у реактор на Юмл, та реакційну суміш гріють до кипіння протягом 72 годин Після відстоювання, органічну фазу переносять на силікагель та елююють 97/3/0,3 сумішшю дихлорметану, метанолу та аміаку Отримують 0,31г продукту, який перетворюють у сіль двома еквівалентами гідробромідної кислоти у розчині в оцтовій кислоті Темп плавл 355°С Приклад 8 (Сполука №5) Гідрохлорид (транс)-11-метил-5а,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сІ]азепіну (2 1) (Транс)-5а,6,7,9,10,11-гексапдро-8,10аметанпіридо[2',3' 5,6]пірано[2,3-сі]азепін у 20мл безводного тетрапдрофурану уводять у тригорлу колбу на ЮОмл, реакційну суміш охолоджують до 78°С, додають краплями 1,2мл (Зммоль) 2,5М буТИЛЛІТІЮ у гексані, та перемішування продовжують при -78°С протягом ЗО хвилин Додають 0,19мл (Зммоль) иодметанута суміші дають повільно нагрітися до температури довкілля перед додаванням ЮОмл води та екстрагуванням дихлорметаном Органічну фазу сушать MgSO4, випарюють під зниженим тиском та залишок очищають хроматографією на колонці з силікагелем, елююючи 90/10/1 сумішшю дихлорметану, метанолу та аміаку Отриманий продукт обробляють двома еквівалентами пдрохлоридної кислоти у розчині пропан-2-олу та виділяють фільтруванням 0,15г кристалів Темп плавл >330°С Приклад 9 (Сполука №9) Гідробромід (транс)-а-фуран-З-іл5а,6,7,9,10,11-гексзпдро-10зН-8,10зметанпіридо[2',3' 5,6]пірано[2,3-сі]азепін-11метанолу (2 1) 0,43г (2ммоль) (транс)-5а,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сі]азепіну обробляють фуран-3-карбоксальдепдом в описаних у прикладі 8 умовах Після перетворення у сіль 2 еквівалентами гідробромідної кислоти у оцтовій кислоті, отримують 0,3г сполуки Темп плавл 69-73°С з розкладанням Приклад 10 (Сполука №26) Гідробромід (транс)-2-4-дибром-5а,6,7,9,10,11гексапдро-8,10а-метанпіридо[2',3' 5,6]пірано[2,3сі]азепіну (1 1) 10 1 3-[(4,6-дибром-3-пдроксипіридин-2 8 іл)метил]-1-азадицикло[2,2,2]октан-3-ол Розчин 24г (0,426моль) гідроксиду калію у 600мл води уводять у колбу на 2000мл, додають 50,0г (0,213моль) 3-[(3-пдроксипіридин-2-іл)-метил]-1азадицикло[2,2,2 ]октан-3-олу, а потім краплями протягом 40 хвилин додають розчин 10,93мл (0,213моль) брому та 152,4г (1,280моль) броміду калію у 600мл води, та суміш перемішують при температурі довкілля протягом 16 годин рН суміші доводять до 7,5 додаванням оцтової кислоти і перемішують протягом 1 години, фільтрують, отриману тверду речовину сушать, переносять у ЮООмл етанолу, та отриману суспензію гріють протягом 2 годин Після охолодження, осад збирають Отримують 21,24г твердої речовини Темп плавл 260-265°С 10 2 Гідробромід (транс)-2-4-дибром5а,6,7,9,10,11-гексзпдро-8,10зметанпіридо[2',3' 5,6]пірано[2,3-сі]азепіну (1 1) 10г (25ммоль) 3-[(4,6-дибром-3пдроксипіридин-2-іл)метил]-1азадицикло[2,2,2]октан-3-олу уводять у колбу на 500мл, додають 150мл концентрованої сульфатної кислоти та 3,6г (25ммоль) пентоксиду фосфору і суміш гріють при 150°С протягом 48 годин, охолоджують, виливають на 300г льоду, доводять рН до 10 додаванням аміаку та суміш екстрагують хлороформом Органічну фазу сушать сульфатом натрію та фільтрують, розчинник випарюють під зниженим тиском і залишок очищають хроматографією на колонці з силікагелем, елююючи 98/2/0,2 сумішшю хлороформу, метанолу та аміаку Після перетворення отриманої твердої речовини у сіль одним еквівалентом гідробромідної кислоти у оцтовій кислоті, отримують 3,53г пдроброміду Темп плавл 320°С з розкладанням Нижченаведена таблиця ілюструє ХІМІЧНІ структури та фізичні властивості деяких сполук винаходу У колонках позначками "Ri" та "R2", "СєНб", "СбН4'' та "СбНЗб" позначають, ВІДПОВІДНО, незаміщені, монозаміщені або дизаміщені фенільні групи Замісники та їх позиції показано "С4Н3О" означає фуран-3-іл "2-СюН7" означає нафталш-2-іл Колонками "5а,10а" позначено конфігурацію хіральних центрів 5а та 10а, а "+/-" означає рацемат У колонці "сіль", "-" означає сполуку у стані основи, "НСІ" означає гідрохлорид, "НВг" означає гідробромід, "dbl_" означає дибензоіл-І_-тартрат та "dbD" означає дибензоіл-О-тартрат Показано молярні співвідношення кислота основа У колонці "Темп плавл (°С)", "(d)" означає темп плавл з розкладанням (І) 61164 № 1 Ri H R2 н R3 н 5а, 10а (+/-) 2 H н н S.R 3 4 5 6 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 н н Вг н н н н н н н н н н н н н н н н н н н R.S (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) (+/-) СНз сн2сн, н н н н н н н н н н н н н н н н н н н C6H4-4-OCF3 C6H4-4-CF3 C6H4-3-NO2 С6Н4-3-СОСНз С6Нз-3,4-(ОСН2О) C6H3-3,5-(CF3)2 C6H4-4-F СбН4-4-СбН5 2-С10Н7 Br Br СІ СІ СІ Br CN Br + + Вг н н Сполуки винаходу піддано дослідженням, які продемонстрували їх терапевтичні властивості Отже, вони були досліджені стосовно їх спорідненості до нікотинових рецепторів, що містять субелемент очРг способами, що описано Anderson та Агпепс, Eur J, Pharmacol (1994) 253 261, та Hall et al, Brain Res (1993) 600 127 Самців щурів Sprague Dawley масою 150-200г обезголовлюють та увесь мозок швидко видаляють, гомогенізують у 15 об'ємах 0.32М розчину сахарози при 4°С, а потім центрифугують при ЮООд протягом 10 хвилин Пелету відкидають, а супернатант центрифугують при 20,000 g протягом 20 хвилин при 4°С Пелету збирають та гомогенізують за допомогоюмлина Pohtron™ у 15 об'ємах бідистиляту при 4°С, а потім центрифугують при 8000д протягом 20 хвилин Пелету відкидають і супернатант та "покрив кольору шкіри буйвола" центрифугують при 40,000д протягом 20 хвилин, пелету збирають, ресуспендують у 15мл бідистиляту при 4°С та центрифугують ще раз при більше 40,000д перед її зберіганням при -80°С На добу експерименту, тканину повільно розтоплюють та суспендують у 3 об'ємах буферу 150мкл цієї суспензії мембран інкубують при 4°С протягом 120 хвилин у присутності 10Омкл 1 нМ [3Н]цитизину у кінцевому об'ємі буферу 500мкл, у присутності або відсутності сполуки, що підлягає тестуванню Реакцію зупиняють фільтруванням на фільтрах Whatman GF/B™, попередньо оброблених поліетиленіміном, фільтри промивають два рази 5мл буферу при 4°С, та вимірюють рідинною сцинтиграфією радіоактивність, що залишилася на фільтрі Неспецифічне приєднання визначають у присутності ЮмкМ (-)-нікотину, неспецифічне приєднання представляє 75-85% 10 СІЛЬ н а 21 dbL1 1 н а 21 н а 21 НВг21 — НВг21 НВг21 НВг21 НВг21 dbL1 1 dbD1 1 н а 21 НСІ 21 НВг1 1 НВг1 1 НВг1 1 Мр (°С) >300 145-148 69-71 330 69-71 157-159 >330 170 (d) 120-121 120 203 (d) 260 310 102-103 302 350 182 200 214-217 280-281 339-344 82-85 320 (d) 329-332 350-352 CtD С) -104 3с=0 5, МеОН -75 4с=1, МеОН -85 3с=1, МеОН +75 с=1, МеОН -106с=0 7, МеОН +108с=0 4, МеОН -81 с=0 5, МеОН +94 1 с=0 5, СНСІз -76 3 с=0 5, МеОН загального приєднання, зібраного на фільтрі Для кожної концентрації досліджених сполук визначають процент інгібування специфічного приєднання [ Н]цитизину, а далі розраховують IKso, концентрацію сполуки, що інгібує 50% специфічного приєднання Величини IKso найактивніших сполук винаходу знаходяться між 0,08 та 1 мкМ Сполуки винаходу також досліджено на їх спорідненість стосовно нікотинових рецепторів що містять субелемент а7, згідно зі способами, що описано Marks and Collins, J Pharmacol Exp Ther (1982) 22 554 та Marks et al, Моль Pharmacol (1986) 30 427 Самців щурів Sprague Dawley масою 150-200г обезголовлюють та увесь мозок швидко видаляють, гомогенізують у 15 об'ємах 0.32М розчину сахарози при 4°С, а потім центрифугували при ЮООд протягом 10 хвилин Пелету відкидають, а супернатант центрифугують при 8000д протягом 20 хвилин при 4°С Пелету збирають та гомогенізують за допомогоюмлина Pohtron™ у 15 об'ємах бідистиляту при 4°С, а потім центрифугують при 8000д протягом 20 хвилин Пелету відкидають, а супернатант та "покрив кольору шкіри буйвола" центрифугують при 40,000д протягом 20 хвилин Пелету збирають, ресуспендують у 15 об'ємах бідистиляту при 4°С та центрифугують один раз при більше 40,000д перед її зберіганням при -80°С На добу експерименту, тканину повільно розтоплюють та суспендують у 5 об'ємах буферу 150мкл цієї суспензії мембран попередньо інкубують при 37°С протягом ЗО хвилин, у темряві, у присутності або відсутності сполуки, що підлягає тестуванню Мембрани далі інкубують протягом 60 хвилин при 37°С, у темряві, у присутності 50мкл 1нМ [3Н]абунгаротоксину у кінцевому об'ємі 250мкл буферу 61164 11 20мМ Гепес з 0,05% поліетиленімшу Реакцію зупиняють фільтруванням на фільтрах Whatman GF/B™, попередньо оброблених, поліетиленіміном Фільтри промивають два рази 5мл буферу при 4°С і вимірюють рідинною сцинтиграфією радіоактивність, що залишилася на фільтрі Неспецифічне приєднання визначають у присутності а-бунгаротоксину при кінцевій концентрації 1мкМ, неспецифічне приєднання представляє приблизно 60% загального приєднання, зібраного на фільтрі Для кожної концентрації досліджуваних сполук, визначають процент інгібування специфічного приєднання [ Н]а-бунгаротоксину, а далі розраховують IKso, концентрацію сполуки, яка інгібує 50% специфічного приєднання Величини IKso сполук винаходу знаходяться між 1 та 20мкМ Сполуки винаходу аналогічно досліджували стосовно їх спорідненості до периферійних нікотинових рецепторів ганглюзного типу згідно зі способом, що описано Houghthng et al, Мої Pharmacol (1995) 48 280-287 Здатність сполук до заміщення [3Н]-епібатидину з мембран надниркової залози від корів показує їх спорідненість до цього рецептору Надниркові залози від корів, що зберігали при -80°С, розтоплюють та гомогенізують за допомогоюмлина Pohtron™ у 20 об'ємах 50мМ буферу Трис НСІ при рН 7,4, при 4°С, далі їх центрифугують при 35,000д протягом 10 хвилин Супернатант відкидають, а пелету ресуспендують у ЗО об'ємах 50мМ буферу Трис НСІ при 4°С та повторно гомогенізують перед повторним центрифугуванням при 35,000д протягом 10 хвилин Кінцеву пелету переносять у 10 об'ємів буферу Трис НСІ при 4°С ЮОмкл мембран або Юмг свіжої тканини шкубують при 24°С протягом З годин у присутності 50мкл 0,66 нМ [3Н]епібатидину у кінцевому об'єм 250мкл буферу, у присутності або відсутності сполуки, що підлягає тестуванню Реакцію зупиняють розведенням зразків 50мкМ Tns НСІ буферуб при рН 7,4 і при 4°С, а тоді фільтруванням на фільтрах Whatman GF/B™, попередньо оброблених поліетиленіміном Фільтри промивають два рази 5мл буферу та вимірюють рідинною сцинтиграфією радіоактивність, що залишилася на фільтрі Неспецифічне приєднання визначають у присутності (-)нікотину при 2мМ кінцевій концентрації, неспецифічне приєднання представляє 30-40% загального приєднання, зібраного на фільтрі Для кожної концентрації досліджених сполук визначають процент інгібування специфічного приєднання [3Н]-епібатидину, а далі розраховують IKso, концентрацію сполуки, яка інгібує 50% специфічного приєднання Величини IKso найактивніших сполук винаходу лежать між 9 та 20мкМ Результати попередніх тестів показують, що деякі сполуки винаходу є селективними лігандами для субелементів (Х4Р2 нікотинових рецепторів Сполуки винаходу наприкінці досліджували m vivo, що продемонструвало їх терапевтичні властивості Отже, наприклад, їх досліджували у моделі гарячої пластини, згідно зі способом Eddy та 12 Leimbach, J Pharmacol Exp Ther (1953)107 385393, з метою дослідження та КІЛЬКІСНОГО аналізу можливої обезболювальної дії Мишей масою 2030г піддавали тепловій стимуляції контактом лап з пластиною, що підтримують при ПОСТІЙНІЙ температурі 57,5°С на термостатованій водяній бані Час реакції на біль, яку виявляють за вилизуванням лап або стрибками, вимірюють Отже, після періоду попередньої обробки, яку проводять підшкірним або пероральним шляхом (кожна група має ВІСІМ тварин з однаковою попередньою обробкою), мишей поміщають кожну окремо на планшет та вимірюють час реакції на біль Тварин видаляють з планшету негайно після виявлення болю Максимальний час експозиції до стимулу складає 30с Величину часу реакції зі стандартними похибками ( s e m ) представлено для кожної групи Непараметричний варіативний аналіз (Kruskal-Walhs) проводять для кожної групи Тест Уїлкінсона (Wilcoxon) дозволяє порівняти кожну оброблену групу з контрольною ВІДМІННОСТІ вважають статистичне значущими при порозі 5% Час цієї реакції помітно зростав під дією аналгетиків головним чином з центральною дією Сполуки винаходу показують активність у цьому тесті при дозах між 3 та 30мг/кг штраперитонально або перорально Ці результати свідчать про можливість використання сполук при лікуванні або попередженні розладів, які пов'язані з дисфункцією нікотинових рецепторів, особливо на рівні центральної нервової системи або шлунково-кишкової системи На рівні центральної нервової системи, ці розлади включають погіршання пізнавальної здатності, точніше погіршання пам'яті, але також погіршання уваги, пов'язане з хворобою Альцгеймера, з патологічним старінням (Age Associated Memory Impairment, AAMI), з хворобою Паркінсона, з монголізмом (синдром Дауна), з алкогольним синдромом Корсакова, та з васкулярною деменцією (мульти-інфарктною деменцією, MID) Сполуки винаходу можливо могли б бути корисними при лікуванні рухових розладів, спостерігаємих при хворобі Паркінсона або інших неврологічних захворюваннях, як-то хорея Хантингтона, синдром Туретта, пізньої дискінезм та гіперкінези Сполуки згідно з винаходом можливо забезпечуватимуть цілюще або симптоматичне лікування судинних порушень мозку та випадків гіпоксії мозку, їх можна використовувати у випадку психіатричних патолопй шизофренії, депресії, тривожності, нападів паніки, нав'язливої поведінки Вони можуть попереджати симптоми відвикання від тютюну, алкоголю та різних викликаючих залежність речовин, як-то кокаїн, ЛСД, конопля та бензодіазепіни Нарешті, їх можна використовувати для лікування болю На рівні шлунково-кишкової системи, сполуки винаходу можна було б використовувати при лікуванні хвороби Крона, виразкового коліту, синдрому подразненого кишечника та ожиріння Для цієї дії, сполуки винаходу можна представляти будь-якою прийнятною для ентерального, парентерального або транедермального за 13 61164 14 стосування композицією, наприклад, таблетками, приклад, сиропами або ампулами, трансдермапокритими цукром таблетками, м'якими чи тверльними пластирами, тощо, у поєднанні з придатдими желатиновими капсулами, придатними для ними ексципієнтами, та у дозах, що дозволяють ІН'ЄКЦІЙ ЧИ ПИТТЯ суспензіями або розчинами, на Комп'ютерна верстка О Воробей застосування кожної доби 0,01-20мг/кг Підписне Тираж39 прим Міністерство освіти і науки України Державний департамент інтелектуальної власності, Львівська площа, 8, м Київ, МСП, 04655, Україна ДП "Український інститут промислової власності", вул Сім'ї Хохлових, 15, м Київ, 04119
ДивитисяДодаткова інформація
Назва патенту англійськоюPyridopyranoazepines derivatives, a method for preparation thereof, a medicine and a pharmaceutical composition based thereon
Автори англійськоюGalli Frederic
Назва патенту російськоюПроизводные пиридопираноазепинов, способ их получения, лекарственное средство и фармацевтическая композиция на их основе
Автори російськоюГалли Фредерик
МПК / Мітки
МПК: A61P 25/16, A61P 1/00, A61P 29/00, A61P 25/22, A61P 25/20, A61P 25/34, A61P 25/00, A61P 25/28, A61P 3/04, A61K 31/439, A61P 1/04, A61P 25/18, A61P 25/32, C07D 491/22, A61P 25/24, A61P 25/36, A61P 25/30
Мітки: похідні, композиція, засіб, піридопіраноазепінів, одержання, спосіб, основі, фармацевтична, лікарський
Код посилання
<a href="https://ua.patents.su/7-61164-pokhidni-piridopiranoazepiniv-sposib-kh-oderzhannya-likarskijj-zasib-ta-farmacevtichna-kompoziciya-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні піридопіраноазепінів, спосіб їх одержання, лікарський засіб та фармацевтична композиція на їх основі</a>
Похідні 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3н)-ону як ліганди рецептора 5-ht4, спосіб їх одержання, фармацевтична композиція та лікарський засіб на їх основі

Номер патенту: 57071
Опубліковано: 16.06.2003
Автори: Неделек Ален, Жегам Самір, Локхед Алістер, Галлі Фредерік, Галле Т'єррі, Самсон Аксель
МПК: C07D 451/04, A61P 1/00, A61P 13/00, A61P 43/00, A61K 31/46, A61P 25/00, A61P 9/00
Мітки: одержання, 5-ht4, рецептора, фармацевтична, композиція, основі, лікарський, ліганди, похідні, 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3н)-ону, засіб, спосіб
Формула / Реферат:
1. Похідні 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3Н)-ону у формі чистого геометричного чи оптичного ізомеру або суміші ізомерів загальної формули (І), (I)де R1 - (С1-С4)алкіл або (С3-С7)циклоалкілметил,Х1 - атом гідрогену чи галогену або (С1-С4)алкоксил, або інакшеOR1 та Х1 разом утворюють групу формул:-ОСН2О-, -O(СН2)2-, -O(СН2)3-, -O(СН2)2О- або -O(СН2)3О-,Х2 - атом гідрогену чи...
Похідні бензоїлгуанідину, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 48214
Опубліковано: 15.08.2002
Автори: Айкмайєр Крістіан, Бюргер Еріх, Роос Отто
МПК: C07D 295/185, A61P 35/00, A61K 31/4025, A61P 9/10, A61K 31/496, C07D 295/192, A61K 31/40, C07D 307/68, A61P 9/06, C07D 307/52, A61K 31/495, A61P 9/00, C07D 295/12, C07D 207/34, C07D 333/38, C07D 333/40, A61K 31/00, C07D 295/155
Мітки: одержання, спосіб, основі, бензоїлгуанідину, композиція, похідні, фармацевтична
Формула / Реферат:
1. Похідні бензоїлгуанідину загальної формули (І): деR1 означає алкіл з 1-8 атомами вуглецю,А означає групу:В означає групу (-CH2-)a або (-СО-)b, деа означає ціле число від 0 до 8, переважно 1, 2, 3 або 4,b означає 0, 1 або 2, переважно 1,R2 означає незаміщений або заміщений алкіл з 1-8 атомами вуглецю, незаміщений феніл, NR3R4, де R3 і R4 кожен означає алкіл з...
Похідні 8-азабіцикло[3.2.1]октан-3-метанаміну, фармацевтична композиція та лікарський засіб
Номер патенту: 58476
Опубліковано: 15.08.2003
Автори: МАРАБУ Бенуа, Машнік Дєвід, Севрен Мірей, Мерлі Жан-П'єр, Руа Жоселін, Де Перетті Даніель, Жорж Паскаль
МПК: A61P 25/32, A61P 25/22, A61K 31/46, A61K 31/36, A61P 25/06, A61K 31/439, A61P 1/14, C07D 451/02, A61P 15/00, A61P 25/24, A61P 3/04, A61P 25/18, A61P 43/00
Мітки: лікарський, фармацевтична, 8-азабіцикло[3.2.1]октан-3-метанаміну, похідні, засіб, композиція
Формула / Реферат:
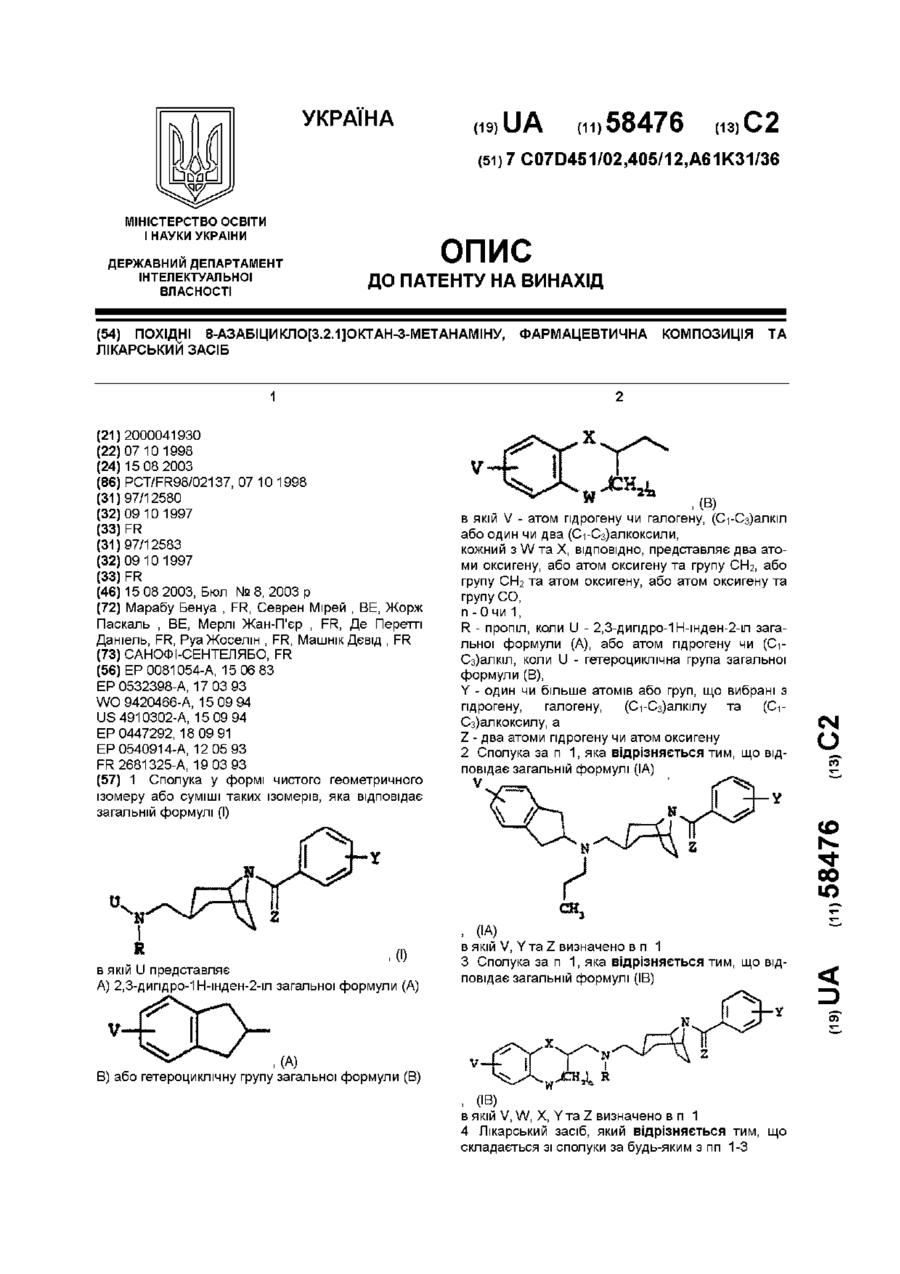
1. Сполука у формі чистого геометричного ізомеру або суміші таких ізомерів, яка відповідає загальній формулі (І), (I)в якій U представляєА) 2,3-дигідро-1Н-інден-2-іл загальної формули (А), (А)В) або гетероциклічну групу загальної формули (В), (В)в якій V - атом гідрогену чи галогену, (С1-С3)алкіл або один чи два (С1-С3)алкоксили,кожний з W та X, відповідно, представляє два атоми...
Похідні 3,3a,4,5-тетрагідро-1н-оксазоло[3,4-а]хінолін-1-ону, спосіб їх отримання, лікарський засіб та фармацевтична композиція

Номер патенту: 41916
Опубліковано: 15.10.2001
Автори: Пюш Фредерік, Жегам Самір, Кеніг Жан Жак, Зард Лідія, Бюрньє Філіпп
МПК: C12N 1/16, A61P 25/26, C07D 498/04, A61K 31/47, A61P 25/00, A61P 25/28, C12P 17/18, A61P 25/20, A61P 25/06, A61P 25/24, A61P 25/04
Мітки: засіб, лікарський, отримання, 3,3a,4,5-тетрагідро-1н-оксазоло[3,4-а]хінолін-1-ону, спосіб, композиція, похідні, фармацевтична
Формула / Реферат:
1. Производные 3,3а,4,5-тетрагидро-1Н-оксазоло[3,4-а]хинолин-1-она общей формулы (I),в которой n равно 0 или 1;R1 - атом водорода либо этенил, метил, этил, фенил, гидроксиметил или метоксиметил; а такжелибо R2 - метил, трифторметил или цианогруппа,R3 - атом водорода, гидроксил или бензилоксигруппа, аR4 и R5 - атомы водорода;либо R2 и R4 вместе образуют группу -(СН2)4-,R3 - гидроксил, а...
Похідні 2,1,3-бензотіадіазолу, спосіб їх отримання, фармацевтична композиція та лікарський засіб

Номер патенту: 51696
Опубліковано: 16.12.2002
Автори: Медерскі Вернер, Оссвальд Матіас, Дорш Дітер, Шмітгес Клаус, Крістадлер Маріа, Анзалі Сохейла, Вільм Клаудіа
МПК: A61P 9/10, C07D 285/14, A61K 31/433, A61P 11/06, A61P 9/12
Мітки: похідні, 2,1,3-бензотіадіазолу, фармацевтична, спосіб, засіб, лікарський, отримання, композиція
Формула / Реферат:
1. Производные 2,1,3-бензотиадиазола формулы I ,где R - группа формул , , ,X – SR1 – H, OH, OA;R2, R3, и R4 независимо друг от друга – незамещенный фенил или фенил, одно- или многократно замещенный Hal, OH, OA, NH2, NHA, NA2, или группа формулы,причем R2 может также обозначать А или циклоалкил;А - алкил с 1 - 6 С-атомами, в котором 1-7 атомов Н могут быть заменены на...
Попередній патент: Електричний кабель
Наступний патент: Фармацевтична композиція та спосіб її одержання
Випадковий патент: Спосіб очистки від монооксиду вуглецю альфа-олефінів і насичених вуглеводнів