Похідні заміщеного 2-метилбензімідазолу, спосіб їх отримання, фармацевтична композиція та спосіб лікування шлунково-кишкових захворювань
Номер патенту: 64714
Опубліковано: 15.03.2004
Автори: Старке Інгемар, Амін Косрат, Дальстром Мікаель, Нордберг Петер







Формула / Реферат
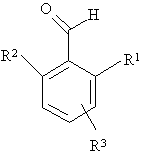
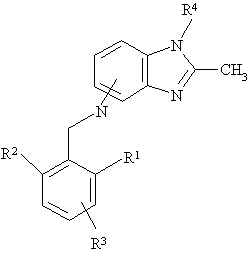
1. Сполука формули І
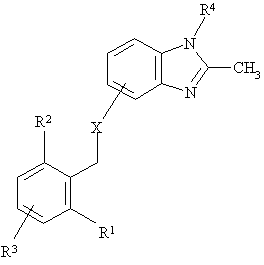 , (І)
, (І)
або її фармацевтично прийнятна сіль, в якій
R1 - нижчий алкіл,
R2 - нижчий алкіл,
R3, який знаходиться в положенні фенільного кільця 3, 4 або 5, -
(а) Н,
(б) галоген чи
(в) нижчий алкіл,
R4-
(а) Н чи
(б) нижчий алкіл,
X, що приєднано до гетероциклу в положеннях 4 чи 7, -
(а) NH чи
(б) О.
2. Сполука за п. 1, яка відрізняється тим, що Х приєднано до гетероциклу в положенні 4.
3. Сполука за п. 2, яка відрізняється тим, що R1 – СН3 чи СН2СН3, R2 – СН3 чи СН2СН3, R3- Н, 4-F чи 4-CI, a R4- H чи -СН3.
4. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметилбензиламіно)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
5. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметилбензилокси)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
6. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметил-4-флуорбензиламіно)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
7. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметил-4-флуорбензилокси)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
8. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметилбензиламіно)-1,2-диметилбензімідазол або його фармацевтично прийнятна сіль.
9. Сполука за п. 3, яка відрізняється тим, що вона -4-(2-етил-6-метилбензиламіно)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
10. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диетилбензиламіно)-2-метилбензімідазол або його фармацевтично прийнятна сіль.
11. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметил-4-флуорбензиламіно)-1,2-диметилбензімідазол чи його фармацевтично прийнятна сіль.
12. Сполука за п. 3, яка відрізняється тим, що вона -4-(2,6-диметил-4-флуорбензилокси)-1,2-диметилбензімідазол або його фармацевтично прийнятна сіль.
13. Сполука за будь-яким з пп. 1-12, яка відрізняється тим, що вона є гідрохлоридом або метансульфонатом.
14. Сполука за будь-яким з пп. 1-13, яка відрізняється тим, що її призначено для використання в терапії.
15. Сполука за будь-яким з пп. 1-13, яка відрізняється тим, що її використовують для виробництва медикаментів для інгібування секреції кислоти в шлунку.
16. Сполука за будь-яким з пп. 1-13, яка відрізняється тим, що її використовують для виробництва медикаментів для лікування запальних кишково-шлункових захворювань.
17. Сполука за будь-яким з пп. 1-13, яка відрізняється тим, що її використовують для виробництва медикаментів для лікування чи попередження станів, що включають інфікування Helicobacter Pilori слизової шлунку людини, причому вказана сіль пристосована для вживання в комбінації з щонайменше одним антимікробним засобом.
18. Спосіб виготовлення сполуки за будь-яким з пп.1-13, що включає реакцію Сполуки загальної формули II
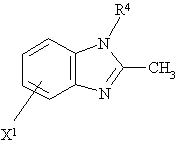 , (II)
, (II)
в якій Х1 - NH2 чи ОН, що приєднані до гетероциклу в позиції 4 чи 7, a R4 визначено для формули І, зі сполукою загальної формули ІІІ
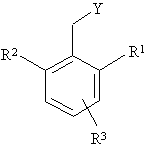 , (III)
, (III)
в якій R1, R2 та R3 визначено для формули І, a Y - здатна до відщеплення група, в інертному розчиннику разом з основою чи без неї з утворенням сполуки формули І.
19. Спосіб виготовлення сполуки за будь-яким з пп. 1-13, що включає
а) реакцію сполуки загальної формули IV
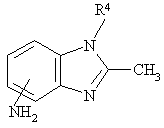 , (IV)
, (IV)
в якій групу NH2 приєднано до гетероциклу в положення 4 чи 7, а R4 визначено для формули І, зі сполукою загальної формули V
 , (V)
, (V)
в якій R1, R2, R3 визначено для формули І, у присутності кислоти Льюіса з утворенням сполуки формули VI
 , (VI)
, (VI)
в якій імінний нітроген приєднано до гетероциклу в положеннях 4 чи 7, а R4 визначено для формули І,
(б) відновлення сполуки формули VI в інертному розчиннику в стандартних умовах до сполуки загальної формули І.
20. Спосіб виготовлення сполуки за будь-яким з пп. 1, 2, 3 чи 8, в якій R4 - нижчий алкіл, що включає
алкілування сполуки формули І, в якій R4 - Н, в інертному розчиннику у присутності основи чи без неї сполукою загальної формули VII
R4X2 , (VII)
в якій R4 визначено для формули І, a X2 - здатна до відщеплення група, до сполуки формули І, в якій R4 - нижчий алкіл.
21. Фармацевтична композиція, що містить сполуку за будь-яким з пп. 1-13 та на додаток фармацевтично прийнятний носій.
22. Спосіб інгібування секреції кислоти в шлунку, що включає вживання ссавцем, включаючи людину, що потребує такого інгібування, ефективної кількості сполуки за будь-яким з пп. 1-13.
23. Спосіб лікування запальних кишково-шлункових захворювань, що включає вживання ссавцем, включаючи людину, що потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-13.
24. Спосіб лікування чи попередження станів, що включають інфікування Helicobacter Pіlоrі слизової шлунку людини, що включає вживання ссавцем, включаючи людину, що потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-13, причому вказану сіль вживають в комбінації з щонайменше одним антимікробним засобом.
25. Фармацевтична композиція для використання при інгібуванні секреції кислоти в шлунку, в якій активним інгредієнтом є сполука за будь-яким з пп. 1-13.
26. Фармацевтична композиція для використання при лікуванні запальних кишково-шлункових захворювань, в якій активним інгредієнтом є сполука за будь-яким з пп.1-13.
27. Фармацевтична композиція для використання при лікуванні чи попередженні станів, що включають інфікування Helicobacter Pіlоrі слизової шлунку людини, в якій активним інгредієнтом є сполука за будь-яким з пп. 1-13 у комбінації з щонайменше одним антимікробним засобом.
Текст
Згідно з винаходом запропоновані нові сполуки та їх фармацевтично прийнятні солі, що інгібують екзочи ендогенно стимульовану секрецію шлункової кислоти, які тому можна використовувати для попередження та лікування запальних кишково-шлункових захворювань. Згідно з іншим аспектом запропоновано для використовувати сполуки згідно з винаходом в терапії, а також спосіб виготовлення таких сполук, фармацевтичні композиції, що містять як активний інгредієнт щонайменше одну сполуку згідно з винаходом та використання активних сполук у виробництві медикаментів для вищеозначеного медичного застосування. Бензімідазольні похідні нижченаведеної формули, що активні як антивиразкові засоби, розкрито в ЕР-В0266326 та US 510686; де R1 - арильна група формули в якій кожний з R7, R8 та R9 незалежно один від одного - Н, С1-6алкіл чи галоген, R2 - H, R3 - Η чи С1-6алкіл, n - від 0 до 6, R4, R5 та R6 - Η чи С1-6алкіл, А - алкілен, в якому до 6 атомів карбону, що може бути розірваним таким гетероатомом, як О чи N. Для огляду фармакології насосу шлункової кислоти (Н+К+-АТФази), див. Sachs et al. (1995) Annu.Rev.Pharmacol.Toxicol. 35:277-305. Несподівано було виявлено, що сполуки формули І, які є заміщеними бензімімдазольними похідними, в яких феніл заміщено в 2- та 6-положеннях нижчими алкілами, особливо ефективні як інгібітори кишковошлункової Н+К+-АТФази, а тому як інгібітори секреції кислоти в шлунку. Згідно з одним аспектом винаходу запропоновано сполуки загальної формули І або їх фармацевтично прийнятні солі, в яких R1 та R2 незалежно один від одного - нижчий алкіл, R3 у позиції фенільного кільця 3, 4 або 5 - (а) Н, (б) галоген чи (в) нижчий алкіл, R4 - (а) Н чи (б) нижчий алкіл, X, що приєднано до гетероциклу в положеннях 4 чи 7 - (a) NH чи (б) О. Використаний термін "нижчий алкіл" означає лінійний чи розгалужений алкіл з числом атомів карбону від 1 до 6. Прикладами таких нижчих алкілів є метил, етил, н-пропіл, iзо-пропіл, н-бутил, iзо-бутил в-бутил. т-бутил, а також лінійні чи розгалужені пентил та гексил. Термін "галоген" означає флуор, хлор, бром чи йод. Чисті енантіомери, рацемічні та інші суміші двох енантіомерів входять до рамок винаходу. Повинно бути зрозуміло, що можливі діастереомерні форми (чисті енантіомери, рацемічні та інші суміші двох енантіомерів) входять до рамок винаходу. Включено до винаходу також похідні сполук формули І, що мають біологічну функцію сполуки формули І. В залежності від способу виготовлення кінцевий продукт формули І отримують як нейтральний або у формі солі. Як вільна основа, так і сіль цього кінцевого продукту входять до рамок винаходу. Солі приєднання кислот до сполук згідно з винаходом відомими способами можна перетворити у вільні основи лугами або іонообміном. Отримані вільні основи можуть також утворювати солі з органічними та неорганічними кислотами. Кращими кислотами при отриманні терапевтично прийнятних солей приєднання кислот є така гідрогалогенідна кислота, як гідрохлоридна, а також сульфатна, фосфатна, нітратна, такі аліфатичні, аліциклічні, ароматичні, гетероциклічні або сульфонові кислоти, як мурашкова, оцтова, пропіонова, янтарна, гліколева, молочна, яблучна, винна, лимонна, аскорбінова, малеїнова, гідроксималеїнова, пірувинна, пгідроксибензойна, ембонова, метансульфонова, етансульфонова, гідроксіетансульфонова, галогенбензолсульфонова, толуолсульфонова або нафталінсульфонова. Кращими сполуками згідно з винаходом є ті, у формулі І яких R1 - СН3 чи СН2СН3, R2 - СН3 чи СН2СН3, R3 - Н, 4-F чи 4-СІ, R4 - H чи -СН3, а X, що приєднано до гетероциклу в положенні 4 - NH чи О. Найкращі сполуки згідно з винаходом включають: 4-(2,6-диметилбензиламіно)-2-метилбензімідазол, 4-(2,6-диметилбензилокси)-2-метилбензімідазол, 4-(2,6-диметил-4-флуорбензиламіно)-2-метилбензімідазол, 4-(2,6-диметил-4-флуорбензилокси)-2-метилбензімідазол, 4-(2,6-диметилбензиламіно)-1,2-диметилбензімідазол, 4-(2-етил-6-метилбензиламіно)-2-метилбензімідазол, 4-(2,6-диетилбензиламіно)-2-метилбензімідазол, 4-(2,6-диметил-4-флуорбензиламіно)-1,2-диметилбензімідазол, 4-(2,6-диметил-4-флуорбензилокси)-1,2-диметилбензімідазол, Згідно з винаходом запропоновано також способи А-В виготовлення сполук загальної формули І. Спосіб А виготовлення сполук загальної формули І включає операції а) сполуки загальної формули II в якій X - NH2 або ОН, що приєднані до гетероциклу в положення 4 чи 7, a R4 визначено для формули І, реагують зі сполуками загальної формули III в якій R1, R2 та R3 визначено для формули І, a Y - така здатна до відщеплення група, як галоген, тозилокси- чи мезилокси група, з утворенням сполуки формули І. Зручним для проведення такої реакції є такий інертний розчинник, як ацетон, ацетонітрил, диметоксиетан, метанол, етанол чи диметилформамід разом з основою чи без неї. Основою є, наприклад, гідроксид такого лужного металу, як натрій та калій, такий алкоголят натрію, як метоксид чи етоксид, гідрид такого лужного металу, як натрій та калій, карбонат такого лужного металу, як натрій та калій, або такий органічний амін, як тріетиламін. Спосіб Б виготовлення сполук загальної формули І включає операції сполуки загальної формули IV в якій ΝΗ2 приєднано до гетероциклу в положення 4 чи 7, a R4 визначено для формули І, реагують зі сполуками загальної формули V в якій R1, R2, R3 визначено для формули І, у присутності кислоти Льюіса, наприклад, хлориду цинку, з утворенням сполуки формули VI в якій імінний нітроген приєднано до гетероциклу в положення 4 чи 7, a R4 визначено для формули І, а далі сполуки загальної формули VI відновлюють, наприклад, борогідридом чи ціаноборогідридом натрію до сполуки загальної формули І. Реакції можна проводити в стандартних умовах у такому інертному розчиннику, як метанол чи етанол. Спосіб В виготовлення сполук загальної формули І, в яких R4 - нижчий алкіл, включає операції алкілування сполук загальної формули І, в яких R4 - Н, у такому інертному розчиннику, як ацетон, ацетонітрил разом з основою чи без неї сполукою загальної формули VII R4X2 VII в якій R4 визначено для формули І, а X2 - така здатна до відщеплення група, як галоген, тозилокси- чи мезилокси група. Основою є, наприклад, гідроксид такого лужного металу, як натрій та калій, такий алкоголят натрію, як метоксид чи етоксид, гідрид такого лужного металу, як натрій та калій, карбонат такого лужного металу, як натрій та калій, або такий органічний амін, як тріетиламін. Згідно з подальшим аспектом винаходу запропоновано застосування сполук формули І у терапії, зокрема, проти запальних кишково-шлункових захворювань. Згідно з винаходом запропоновано застосування сполук формули І у виробництві медикаментів для інгібування секреції кислоти в шлунку або лікування запальних кишково-шлункових захворювань. Сполуки згідно з винаходом можна тому використовувати для попередження та лікування таких запальних кишково-шлункових та пов'язаних зі шлунковою кислотою захворювань ссавців, включаючи людину, як гастрити, виразка шлунка, виразка дванадцятипалої кишки, шлунково-стравохідний рефлюкс та синдром Золінгера-Еллісона. Крім того, сполуки можна застосовувати для лікування інших кишковошлункових розладів, при яких бажана антисекреторна шлункова дія, наприклад, у пацієнтів з ульцерогенною аденомою підшлункової залози та пацієнтів з гострою кишково-шлунковою кровотечею. Їх також можна використати для пацієнтів при інтенсивному лікуванні, а також до та після операції для попередження аспірації кислоти та стресового утворення виразки. Згідно з подальшим аспектом винаходу запропоновано фармацевтичні композиції, що включають щонайменше одну сполуку згідно з винаходом, або її фармацевтично прийнятну сіль в якості активного інгредієнту. Сполуки згідно з винаходом також можна використати у композиціях разом з іншими активними інгредієнтами, наприклад, таким антибіотиком, як амоксицилін. Сполуки згідно з винаходом для клінічного застосування можна вводити у фармацевтичні композиції для перорального, ректального, парентерального та інших способів вживання. Фармацевтичні композиції включають сполуки згідно з винаходом у комбінації з одним чи більше фармацевтично прийнятними інгредієнтами. Носії можуть бути твердими, напіврідкими та рідкими розріджувачами або капсулами. Ці фармацевтичні препарати складають подальший об'єкт винаходу. Звичайна кількість активного компоненту складає 0,1-95% від маси препарату, краще - 0,1-20% від маси препарату при парентеральному застосуванні і 0,1-50% від маси препарату при пероральному застосуванні. В препараті фармацевтичної композиції, що включає сполуку згідно з винаходом у формі одиничної дози для перорального застосування, вибрану сполуку можна змішувати з такими твердими порошковими інгредієнтами, як лактоза, сахароза, сорбіт, маніт, крохмаль, амілопектин, похідні целюлози, желатин або інші придатні інгредієнти, а також з такими дезинтегруючими та змащуючими засобами, як стеарат магнію чи кальцію, стеарилфумарат натрію, а також поліетиленгліколеві воски. Потім суміші гранулюють або пресують у таблетки. М'які желатинові капсули можна виготовити такими, що вміщують суміш активної сполуки або сполук згідно з винаходом, рослинної олії, жиру, чи іншого придатного середовища для м'яких желатинових капсул. Тверді желатинові капсули можуть вміщувати гранули активної сполуки. Тверді желатинові капсули можуть вміщувати також активну сполуку в комбінації з такими твердими порошковими інгредієнтами, як лактоза, сахароза, сорбіт, маніт, картопляний чи кукурудзяний крохмаль, амілопектин, похідні целюлози або желатин. Одиничні дози для ректального застосування можна виготовити (і) у формі супозиторієв з вмістом змішаної з нейтральною жировою основою активної речовини, (іі) у формі желатинових ректальних капсул з вмістом змішаної з рослинною олією, вазеліновим маслом чи іншим придатним для желатинових ректальних капсул середовищем активної речовини, (ііі) у формі готової для вживання мікроклізми, або (іν) у формі сухої композиції для мікроклізми, яку безпосередньо перед вживанням треба розбавити придатним розчинником. Рідкі препарати для перорального застосування можна виготовити як сиропи або суспензії, наприклад розчини чи суспензії з вмістом активного компоненту 0,1-20% за масою та цукру чи цукрозамінюючих спиртів та суміші етанолу, води, гліцерину, пропіленгліколю та поліетиленгліколю. За бажанням, такі рідкі препарати можуть містити забарвлюючі, смакові засоби, сахарин та карбоксиметилцелюлозу чи інший загущуючий засіб. Рідкі препарати для перорального застосування можна також виготовити як сухі порошки, які безпосередньо перед вживанням треба розбавити придатним розчинником. Розчини для парентерального застосування можна виготовити як розчини сполуки згідно з винаходом у фармацевтично прийнятному розчиннику, краще у концентрації 0,1-10% за масою, які можуть також містити стабілізатори та/або буферуючі інгредієнти та бути розподіленими на одиничні дози у формі ампул чи склянок. Розчини для парентерального застосування можна виготовити також як сухі препарати, які безпосередньо перед вживанням треба розбавити придатним розчинником. Типова добова доза активної речовини варіює в широких межах і залежить від різних факторів, наприклад, індивідуальної потреби кожного пацієнта, шляху вживання, а також захворювання. Взагалі, парентеральні та пероральні дози знаходяться в межах 5-1000мг активної речовини на добу. Сполуки згідно з винаходом також можна використати у композиціях разом з іншими активними інгредієнтами, наприклад, для лікування чи профілактики станів, що включають інфікування Helicobacter Pilori слизової шлунку людини. Такими іншими активними інгредієнтами можуть бути антимікробні засоби, зокрема такі b-лактамні антибіотики, як амоксицилін, ампіцилін, цефалотин, цефаклор чи цефіксим, такі макроліди, як еритроміцин чи кларитроміцин такі тетрацикліни, як тетрациклін чи діоксициклін, такі аміноглікозиди, як гентаміцин, канаміцин чи амікацин, такі хінолони, як норфлокацин, ципрофлокацин чи еноксацин, такі інші, як метронідазол, нітрофурантоїн чи хлорамфенікол, або препарати, що містять такі солі бісмуту, як субцитрати, субсаліцилати, субкарбонати, субнітрати чи субгалати. Виготовлення сполук згідно з винаходом Приклад 1.1 Синтез 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу 6,0г (41ммоль) 4-аміно-2-метилбензімідазолу розчинили в 120мл ацетонітрилу і додали 6,3г (41ммоль) 2,6-диметилбензилхлориду, 16,0г (150ммоль) карбонату натрію та каталітичну кількість йодиду натрію, потім під зворотним холодильником кип'ятили протягом 3 годин. Фільтруванням видаляли карбонат натрію і промивали метиленхлоридом. Випарюванням розчинника у вакуумі отримали маслоподібний залишок, який флеш-хроматографували на силікагелі метиленхлоридом/метанолом (10:1) і кристалізували залишок обробкою етилацетатом, одержавши 3,3г (30%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,35 (s, 3Н), 2,50 (s, 3Н), 4,35 (s, 2H), 4,45 (bs, 1H), 6,50 (d, 1H), 6,75 (d, 1H), 6,95-7,15 (m, 4H). Приклад 1.2 Синтез 4-(2,6-диметилбензилокси)-2-метилбензімідазолу 0,59г (4ммоль) 4-гідрокси-2-метилбензімідазолу розчинили в 15мл ацетонітрилу і додали 0,52г (4ммоль) 2,6-диметилбензилхлориду та 0,16г (4ммоль) карбонату натрію (розчиненого в 1мл води), потім під зворотним холодильником кип'ятили протягом 2 годин. Випарили розчинник під зниженим тиском, залишок розчинили в метиленхлориді і промили 2М NaOH, відділили органічний шар, висушили його сульфатом натрію та випарили розчинник під зниженим тиском. Маслоподібний залишок флеш-хроматографували на силікагелі, елююючи метиленхлоридом з 5% метанолу, і кристалізували залишок з ацетонітрилу, одержавши 0,18г (18%) потрібної сполуки. 1 Н ЯМР (300МГц, ДМСО-d6) d 2,3 (s, 6Н), 2,40 (s, 3Н), 5,25 (s, 2H), 6,9 (d, 1H), 7,05-7,15 (m, 4H), 7,2 (t, 1H). Приклад 1.3 Синтез 4-(2,6-диметил-4-флуорбензиламіно)-2-метилбензімідазолу 0,6г (4,1ммоль) 4-аміно-2-метилбензімідазолу розчинили в 10мл ацетонітрилу і додали 0,89г (4,1ммоль) 2,6-диметил-4-флуорбензилхлориду та 0,68г (4,9ммоль) карбонату калію, потім гріли при 70-80°С протягом 3 годин, охолоджували до кімнатної температури та додавали 25мл метиленхлориду та 25мл води. Органічний шар відділили, висушили сульфатом натрію та випарили розчинник під зниженим тиском. Залишок двічі перекристалізували з етилацетату та очищали хроматографією на силікагелі, елююючи метиленхлоридом з 10% метанолу, одержавши 0,26г (22%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,3 (s, 6H), 2,45 (s, 3Н), 4,25 (s, 2H), 4,35 (bs, 1H), 6,45 (d, 1H), 6,65 (d, 2H), 6,7 (d, 1H), 7,1 (t, 1H). Приклад 1.4 Синтез 4-(2,6-диметил-4-флуорбензилокси)-2-метилбензімідазолу 0,57г (3,8ммоль) 4-гідрокси-2-метилбензімідазолу розчинили в 4мл ацетонітрилу і додали розчин 0,17г (43ммоль) гідроксиду натрію (розчиненого в 1мл води), потім 0,82г (3,8ммоль) 2,6-диметил-4флуорбензилхлориду в 6мл ацетонітрилу та під зворотним холодильником кип'ятили протягом 80 хвилин, та додали 25мл метиленхлориду та 25мл води. Органічний шар відділили, висушили сульфатом натрію та випарили розчинник під зниженим тиском. Залишок перекристалізували з етилацетату та двічі очищали хроматографією на колонці з силікагелем, елююючи метиленхлоридом/метанолом (10:1), одержавши 0,066г (6%) потрібної сполуки. 1 Н ЯМР (300МГц, ДМСО-d6) d 2,3 (s, 6Н), 2,5 (s, 3Н), 5,05 (s, 2H), 6,7 (d, 2H), 6,8 (d, 1H), (t, 1H), 7,25 (d, 1H). Приклад 1.5 Синтез 4-(2,6-диметилбензиламіно)-1,2-диметилбензімідазолу 0,2г (0,75ммоль) 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу розчинили в 10мл ацетонітрилу і додали 0,05мл (0,82ммоль) метилйодиду та 0,2г (1,4ммоль) карбонату калію, потім перемішували при кімнатній температурі протягом 3 годин. Тверду фазу відфільтрували та промили метиленхлоридом, випарили розчинник та очищали хроматографією на силікагелі, елююючи метиленхлоридом/метанолом (10:1), і перекристалізували з ацетонітрилу одержавши 0,018г (9%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,4 (s, 6H), 2,55 (s, 3Н), 3,65 (s, 3Н), 4,4 (d, 2H), 4,55 (bs, 1H), 6,55 (d, 1H), 6,7 (d, 1H), 7,05 (d, 2H), 7,1 (t, 1H), 7,2 (t, 1H). Приклад 1.6 Синтез 4-(2-етил-6-метилбензиламіно)-2-метилбензімідазолу 0,4г (2,7ммоль) 4-аміно-2-метилбензімідазолу та 0,46г (2,7ммоль) 2-етил-6-метилбензилхлориду розчинили в 10мл диметоксіетану і додали, 0,5г (4,7ммоль) карбонату натрію та 0,2г (4,7ммоль) йодиду натрію, потім під зворотним холодильником кип'ятили протягом 2 годин. Фільтруванням видаляли неорганічні солі і промивали диметоксіетаном. Випарюванням розчинника досуха отримали залишок, який флеш-хроматографували на колонці з силікагелем, елююючи метиленхлоридом/етилацетатом (50:50), одержавши 0,21г потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 1,1 (t, 3Н), 2,35 (s, 3Н), 2,45 (s, 3Н), 2,70 (q, 2H), 4,35 (s, 2H), 4,45 (bs, 1H), 6,55 (d, 1H), 6,75 (d, 1H), 6,95-7,20 (m, 4H). Приклад 1.7 Синтез 4-(2-етил-6-метилбензиламіно)-2-метилбензімідазолу 0,79г (5,4ммоль) 4-аміно-2-метилбензімідазолу та 1,1г (6,6ммоль) 2-6-діетилбензилхлориду розчинили в 30мл метанолу і додали, 0,9г (6,6ммоль) хлориду цинку, а потім маленькими порціями 0,42г (6,6ммоль) NaBН3CN, під зворотним холодильником кип'ятили протягом 3 годин, потім перемішували при кімнатній температурі протягом 16 годин і виливали в 50мл водного 1М розчину NaOH. Утворену жовту суспензію екстрагували ДХМ та органічний розчин промивали розсолом, сушили сульфатом натрію та випарюванням розчинника під зниженим тиском отримали маслоподібний залишок, який перекристалізували з суміші етилацетату і ацетонітрилу одержавши 0,65г (41%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 1,2 (t, 6H), 2,5 (s, 3Н), 2,27 (q, 4H), 4,35 (d, 2H), 4,55 (bs, 1H), 6,05 (d, 1H), 6,75 (d, 1H), 7,0-7,25 (m, 4H), 9,0 (bs, 1H). Приклад 1.8 Синтез 4-(2,6-диметил-4-флуорбензиламіно)-1,2-диметилбензімідазолу 0,1г (0,35ммоль) 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу розчинили в 3мл диметоксіетану і додали 0,025г (0,63ммоль) твердого гідроксиду натрію та 0,005мл (0,016ммоль) броміду тетраметиламонію, потім перемішували при кімнатній температурі протягом 15 хвилин, додали 0,06г (0,42ммоль) метилйодиду і перемішували при кімнатній температурі протягом 2,5 годин. Розчинник випарили під зниженим тиском та очищали хроматографією на колонці з силікагелем, елююючи метиленхлоридом/етилацетатом (50:50), одержавши 0,08г (76%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,38 (s, 3Н), 2,53 (s, 3Н), 3,67 (s, 3Н), 4,32 (d, 2H), 4,44 (bs, 1H), 6,51 (d, 1H), 6,68 (d, 1H), 6,75 (d, 2H), 7,18 (t, 1H). Приклад 1.9 Синтез 4-(2,6-диметил-4-флуорбензилокси)-1,2-диметилбензімідазолу 0,18г (0,63ммоль) 4-(2,6-диметилбензилокси)-2-метилбензімідазолу розчинили в 6мл диметоксіетану і додали 0,026г (0,63ммоль) твердого гідроксиду натрію та 0,47мл (0,76ммоль) метиліодиду і перемішували при кімнатній температурі протягом 3,5 годин. Розчинник випарили та очищали залишок хроматографією на силікагелі, елююючи метиленхлоридом/метанолом (10:1). Хроматографією на силікагелі одної з отриманих фракцій з етилацетатом одержали 0,04г (21%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,40 (s, 6Н), 2,58 (s, 3Н), 3,70 (s, 3Н), 5,24 (s, 2H), 6,75 (d, 2H), 6,82 (d, 1H), 6,93 (d, 1H), 7,19 (t, 1H). Приклад 1.10 Виготовлення гідрохлориду 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу 0,5г (1,9ммоль) 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу розчинили в 20мл етилацетату та 6мл метанолу, додали 0,16мл (1,9ммоль) 12М гідрохлоридної кислоти і перемішували при температурі приміщення 5 хвилин. Фільтруванням видаляли осад солі і сушили, одержавши 0,45г (79%) потрібної сполуки як білі кристали. 1 Н ЯМР (500МГц, MeOD) d 2,4 (s, 6H), 2,65 (s, 3Н), 2,75 (s, 3Н), 4,45 (s, 2H), 6,9 (d, 1H), 7,0 (d, 1H), 7,1 (d, 2H), 7,15 (t, 1H), 7,4 (t, 1H). Приклад 1.11 Виготовлення метансульфонату 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу 0,5г (1,9ммоль) 4-(2,6-диметилбензиламіно)-2-метилбензімідазолу розчинили в 20мл етилацетату та 7мл метанолу, додали 0,18г (1,9ммоль) метансульфонової кислоти і перемішували при температурі приміщення 5 хвилин. Фільтруванням видаляли осад солі і сушили, одержавши 0,58г (85%) потрібної сполуки як білі кристали. 1 Н ЯМР (500МГц, MeOD) d 2,4 (s, 6Н), 2,65 (s, 3Н), 2,75 (s, 3Н), 4,45 (s, 2H), 6,9 (d, 1H), 7,0 (d, 1H), 7,1 (d, 2H), 7,15 (t, 1H), 7,4 (t, 1H). Виготовлення інтермедіатів Приклад 2.1 Синтез 2,6-диметил-4-флуорбензилброміду Суміш 5,0г (40ммоль) 3,5-диметилфлуорбензолу, 15г параформальдегіду, 70мл 30% гідробромідної кислоти в оцтовій кислоті та 25мл оцтової кислоти перемішували при температурі приміщення 4,5 години, потім додали суміш води та петролейного етеру, органічний шар відокремлювали, сушили безводним сульфатом натрію та обережно випарювали під зниженим тиском. Залишок очищали хроматографією на силікагелі, елююючи петролейнимо етером, і одержали 3,7г (43%) потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 2,5 (s, 6H), 4,55 (s, 2H), 6,75 (d, 2H). Приклад 2.2 Синтез 2-етил-6-метилбензилхлориду 1,0г (6,67ммоль) 2-етил-6-метилбензилового спирту розчинили в 10мл метиленхлориду і додали 1,0г (6,5ммоль)тіонілхлориду, потім перемішували при температурі приміщення протягом ночі і випарювали. Залишок розчинили в метиленхлориді і профільтрували крізь 5г силікагелю, фільтрат випарювали і одержали 1,0г (89%) маслоподібної потрібної сполуки. 1 Н ЯМР (300МГц, CDCI3) d 1,29 (t, 3Н), 2,46 (s, 3Н), 2,76 (q, 2H), 4,71 (s, 2H), 7,0-7,2 (m, 3Н). Біологічні тести 3.1 Дослід in vitro Інгібування секреції кислоти ізольованою шлунковою залозою кроля. Інгібуючу дію на секрецію кислоти ізольованою шлунковою залозою кроля in vitro визначали як описано Berlindh et al. (1976) Acta Physiol. Scand. 97, 401-414. Сполуки з прикладів 1,1-1,7 виявили величини ІК50 менші за 1,6mΜ. Сполука (А) для порівняння (4-бензиламіно-2-метилбензімідазол) розкрита в п.1 ЕР-В-266326, в якій A - CH2NH, R2, R3, R4, R5 та R6 - H, n=1, а R1 - група формули II’, де R7, R8 та R9 - Н. Сполука (А) для порівняння виявила величину ІК50 12,9mΜ. Сполука (В) для порівняння (4-бензилокси-2-метилбензімідазол) розкрита в п.1 ЕР-В-266326, в якій А - СН2О, R2, R3, R4, R5 та R6-H, n=1, а R1 - група формули II', де R7, R8 та R9 - Н. Сполука (В) для порівняння виявила величину ІК50 7,8mΜ. З цього виходить, що сполуки згідно з винаходом виявляють значно кращий інгібуючий ефект на секрецію кислоти in vitro у порівнянні зі сполуками (А) та (В), що мають незаміщену фенільну групу. Тому можна зробити висновок, що посилення дії обумовлене заміщенням фенільної групи в сполуках згідно з винаходом нижчими алкілами в положеннях 2 та 6. 3.1 Дослід in wo Інгібуюча дія на секрецію кислоти у самиць пацюків. Використовували самиць пацюків Spracjue-Dawly, яких було опоряджено катетерізованими фістулами шлунку (порожнини) та верхньої частини дванадцятипалої кишки для збирання секрету шлунка і, відповідно, застосування тестуємих речовин. Відновлювальний період після хірургічного втручання перед тестуванням - 14 діб. Перед тестуванням тварини не отримували їжу, але не воду, протягом 20 годин. Шлунок кілька разів через катетер промивали водопровідною водою (37°С) та вводили підшкірно 6мл Рінгер-глюкози. Секрецію кислоти стимулювали інфузією протягом 2,5-4 годин (1,2мл/год., підшкірно) пентагастрину та карбахолу (20 та 110нмоль/год., відповідно), протягом цього часу секрет шлунку збирали 30-хвилинними фракціями. Тестуємі речовини чи середовище давали або за 60 хвилин до початку стимуляції (внутрішньовенно або інтрадуоденально в дозі 1мл/кг), або через 2 години після початку стимуляції (перорально в дозі 5мл/кг з закритим катетером шлунку). Час між дозою та стимулюванням міг збільшуватися, щоб визначити подовженість дії. Зразки шлункового соку титрували до рН 7,0 0,1Μ NaOH і розраховували вихід кислоти за концентрацією та об'ємом титруемого розчину. Подальші розрахунки базувалися на значеннях реакції групи з 4-6 пацюків. У випадку вживання протягом стимуляції вихід кислоти після вживання тестуємої сполуки або середовища виражали як фракційні реакції, приймаючи вихід кислоти за 30 хвилин до вживання тестуємої сполуки за 1,0. Процент інгібування розраховували за викликаними тестуємими речовинами чи середовищем фракційними реакціями. У випадку вживання до стимуляції процент інгібування розраховували безпосередньо за виходом кислоти, що визначали після вживання тестуємих сполук або середовища. Біопридатність для пацюків Використовували дорослих пацюків Sprague-Dawly, яких за 1-3 доби до цього було під анестезією опоряджено катетерами лівої сонної артерії. Пацюків, яких використовували для внутрішньовенних дослідів, також було під анестезією опоряджено катетерами шийної вени (Popovic (1960) J.Appl.Physiol. 15, 727-728). Катетери було виведено на задню частину шиї. Зразки крові (0,1-0,4г) відбирали з сонної артерії періодично через інтервали до 5,5 годин після дачі дози, їх заморожували до аналізування тестуємої сполуки. Біопридатність визначали розрахунком показника між площею під кривою кров/концентрація плазми (AUC) після (і) інтрадуоденального (і.д.) або перорального (п.о.) та (іі) внутрішньовенного (в.в.) застосування від пацюків або собак, відповідно. Площу під кривою залежності концентрації від часу (AUC) визначали log/лінійним трапецеїдальним способом та екстраполювали на нескінченість поділом останньої визначеної концентрації в крові оцінкою величини константи на кінцевій фазі. Системну біопридатність (F%) після інтрадуоденального або перорального вживання розраховували як F(%)=(АUC(і.д. або п.о.)/(AUC(в.в.)·100 Інгібування секреції кислоти та біопридатність для притомних собак Використовували мисливських лабрадорів або собак Гаріера обох статей, яких було опоряджено дуоденальними фістулами для застосування тестуємих сполук або середовища та катетеризованими шлунковими фістулами або Гейденхайм-мішком для збирання шлункового секрету. Перед тестуванням тварини голодували приблизно 18 годин, але воду їм давали досхочу. Секрецію шлункової кислоти стимулювали до 6,5 годин інфузією дигідрохлориду гістаміну (12мл/год.) у дозі, що призводить до приблизно 80% від індивідуальної максимальної секреторної реакції, і збирали шлунковий сік послідовними 30-хвилинними фракціями. Тестуємі сполуки або середовище застосовували перорально, і.д. або в.в. через 1 або 1,5 години після початку інфузії гістаміну в об'ємі 0,5мл/кг маси тіла. У випадку перорального вживання тестуєму сполуку застосовували до секретуючого кислоту головного шлунку собак з Гейденхайм-мішком. Кислотність зразків шлункового соку визначали титруванням до рН 7,0 і розраховували вихід кислоти. Вихід кислоти за період збирання після застосування тестуємої сполуки чи середовища виражали як фракційні реакції, приймаючи вихід кислоти за 30 хвилин до вживання тестуємої сполуки за 1,0. Процент інгібування розраховували за викликаними тестуємими речовинами чи середовищем фракційними реакціями. Зразки крові для аналізу концентрації тестуємої сполуки в плазмі збирали через інтервали до 4 годин після введення дози. Плазму відділяли і заморожували протягом 30 хвилин після збирання, а потім аналізували. Системну біопридатність (F%) після перорального або і.д. застосування розраховували, як описано вище для пацюків.
ДивитисяДодаткова інформація
Назва патенту англійськоюDervatives of substituted 2-methyl benzimidazole, a method for preparing thereof, a pharmaceutical composition and a method for treatment of gastrointestinal diseases
Автори англійськоюNordberg Peter
Назва патенту російськоюПроизводные замещенного 2-метилбензимидазола, способ их получения, фармацевтическая композиция и способ лечения желудочно-кишечных заболеваний
Автори російськоюНордберг Петер
МПК / Мітки
МПК: A61K 31/4184, C07D 235/08, A61P 1/00
Мітки: захворювань, отримання, шлунково-кишкових, похідні, заміщеного, фармацевтична, 2-метилбензімідазолу, спосіб, лікування, композиція
Код посилання
<a href="https://ua.patents.su/7-64714-pokhidni-zamishhenogo-2-metilbenzimidazolu-sposib-kh-otrimannya-farmacevtichna-kompoziciya-ta-sposib-likuvannya-shlunkovo-kishkovikh-zakhvoryuvan.html" target="_blank" rel="follow" title="База патентів України">Похідні заміщеного 2-метилбензімідазолу, спосіб їх отримання, фармацевтична композиція та спосіб лікування шлунково-кишкових захворювань</a>
Заміщені похідні імідазопіридину, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі (варіанти), спосіб пригнічення секреції шлункової кислоти або лікування шлунково-кишкових запалювальних за

Номер патенту: 64741
Опубліковано: 15.03.2004
Автори: Амін Косрат, Дальстром Мікаель, Старке Інгемар, Нордберг Петер
МПК: C07D 471/04, A61K 31/437, C07D 235/00, C07D 221/00, A61P 1/04
Мітки: фармацевтична, шлунково-кишкових, запалювальних, похідні, шлункової, спосіб, варіанти, секреції, імідазопіридину, одержання, композиція, лікування, пригнічення, кислоти, основі, заміщені
Формула / Реферат:
1. Заміщені похідні імідазопіридину формули Іабо їх фармацевтично прийнятна сіль, деR1 - СН3 або СН2ОН,R2 - нижчий алкіл,R3 - нижчий алкіл,R4 - Н або галоген,R5 - Н, галоген або нижчий алкіл,Х - NH або О.2. Сполука за п. 1, яка відрізняється тим, що у нійR2 - С1-С4алкіл,R3 - С1-С4алкіл, R5 - Н, галоген або С1-С4алкіл, аR1, R4 та Х визначено у п....
Похідні хіноліну, спосіб їх одержання, фармацевтична композиція та спосіб лікування захворювань, які виникають внаслідок патологічного запалення або аутоімунітету

Номер патенту: 64791
Опубліковано: 15.03.2004
Автори: Бьйорк Андерс, Фекс Томас, Хедлунд Гунар, Йонсон Стіг
МПК: C07D 215/22, C07D 215/56, A61K 31/47, C07D 215/18
Мітки: патологічного, захворювань, запалення, лікування, виникають, одержання, похідні, хіноліну, внаслідок, фармацевтична, аутоімунітету, спосіб, композиція
Формула / Реферат:
1. Похідні хіноліну загальної формули (І),(І)деR вибирають з групи: метил, етил, н-пропіл, ізопропіл, н-бутил та аліл;R' вибирають з групи: метил, метокси, фтор, хлор, бром, трифторметил та OCHxFy;де х = 0 - 2;у = 1 - 3 за умови, щох + у = 3;R" вибирають з групи: водень, фтор та хлор, за умови що...
Похідні 2-аміно-1,2,3,4-тетрагідронафталіну, які призначені для лікування серцево-судинних захворювань, спосіб їх отримання і фармацевтична композиція, що їх містить

Номер патенту: 26689
Опубліковано: 12.11.1999
Автори: САНТАНГЕЛО Франшеско, Семераро Клаудіо, МАРКІНІ Франшеско, БЕРТОЛІНІ Джорджо, МОНТАНАРІ Стефанія, КАЗАГРАНДЕ Чесаре
МПК: C07F 9/08, C07C 271/20, A61K 31/215, A61P 9/00, C07C 271/44, C07C 217/14, A61K 31/135, C07C 217/20, A61P 13/02, A61P 15/00, C07C 219/00, C07C 237/22, A61P 9/12, A61K 31/66, C07D 213/79, A61K 31/165, C07F 9/09, A61P 43/00, A61P 9/10, A61K 31/675
Мітки: фармацевтична, захворювань, спосіб, лікування, містить, отримання, композиція, серцево-судинних, призначені, похідні, 2-аміно-1,2,3,4-тетрагідронафталіну
Формула / Реферат:
1. Производные 2-амино-1,2,3,4-тетрагидронафталина формулы (I)в которой R1 и R2, которые одинаковы или различны, представляют атом водорода или группу OY';Y и Y', которые одинаковы или различны, представляют атом водорода или ацил, происходящий из возможно замещенной алифатической, ароматической или гетероароматической карбоновой кислоты, из возможно замещенной карбаминовой или угольной кислоты или из фосфорной кислоты...
Похідні 1-[2-(заміщений вініл)]-5н-2,3-бензодіазепіну, спосіб їх отримання (варіанти),проміжна сполука, фармацевтична композиція, спосіб її отримання та спосіб лікування захворювань центральної нервової системи

Номер патенту: 61873
Опубліковано: 15.12.2003
Автори: Дароці Казоне Клара, Хорват Каталін, Реітер Йожеф, Мате Дьордьнє, Дьєртьян Іштван, Білкєі-Горзо Андраш, Зольомі Габор, Байногель Юдіт, Хорват Едіт, Андраші Ференц, Бєрженьі Паль, Кьорьоші Єньо, Гацшальі Іштван, Шаламон Хашкане Цецилія, Хаморі Тамаш, Баконьі Анна, Моравчік Імре, Блашко Габор, Ваго Паль, Тіханьі Карой, ШІМІГ Дьюла, Сенкуті Естер, Ботка Петер, Едьєд Андраш
МПК: A61K 31/551, A61K 31/55, C07D 491/056, C07D 403/06, A61P 25/00, C07D 493/04, C07D 243/02, A61P 25/20, A61P 25/22, C07D 491/04, C07D 409/06, C07D 405/06
Мітки: композиція, 1-[2-(заміщений, отримання, нервової, спосіб, системі, вініл)]-5н-2,3-бензодіазепіну, похідні, сполука, варіанти),проміжна, лікування, захворювань, центральної, фармацевтична
Формула / Реферат:
1. Производные 1-[2-(замещенный винил)]-5Н-2,3-бензодиазепина общей формулы (I):, (I)гдеА и В вместе образуют группу формулы -C=N- или -CH-NH-,R1 представляет собой фенил, который содержит от 1 до 3 одинаковых или различных заместителей, выбранных из группы, включающей галоген, трифторметил или циано,R2 представляет собой водород,R3 и R4 вместе образуют 7,8- или 8,9-метилендиоксильную группу,и...
Похідні індолу, фармацевтична композиція та лікарський засіб для лікування захворювань або патологічного стану, опосередкованого мср-1

Номер патенту: 63968
Опубліковано: 16.02.2004
Автори: Баркер Ендрю Джон, Фолл Алан Веллінгтон, Кеттл Джейсон Грант
МПК: C07D 401/04, A61P 29/00, A61P 17/00, A61P 17/06, A61K 31/4433, A61P 9/10, A61P 9/00, A61K 31/4427, A61P 11/06, A61P 13/12, A61P 11/00, A61K 31/4439, A61P 37/06, C07D 413/12, A61K 31/00, A61P 1/00, C07D 409/04, C07D 209/42, A61K 31/404, A61P 43/00, A61K 31/403, A61K 31/443, C07D 405/06, A61P 19/02
Мітки: лікарський, композиція, лікування, опосередкованого, індолу, патологічного, фармацевтична, стану, засіб, мср-1, похідні, захворювань
Формула / Реферат:
1. Лікарський засіб для лікування захворювань або патологічного стану, опосередкованого МСР-1, який включає сполуку формули (І),де:R1 є незалежно вибраним з трифторметилу, С1-4алкілу, галоїду, гідрокси, С1-4алкокси, С1-4алканоїлу, С1-4алканоїлокси, карбокси, трифторметокси, аміно, ціано, С1-4алкіламіно, ді(С1-4алкіл)аміно, С1-4алканоїламіно, нітро, карбамоїлу, С1-4алкоксикарбонілу, тіолу, С1-4алкілсульфанілу,...