Заміщені похідні імідазопіридину, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі (варіанти), спосіб пригнічення секреції шлункової кислоти або лікування шлунково-кишкових запалювальних за
Номер патенту: 64741
Опубліковано: 15.03.2004
Автори: Нордберг Петер, Дальстром Мікаель, Амін Косрат, Старке Інгемар







Формула / Реферат
1. Заміщені похідні імідазопіридину формули І
або їх фармацевтично прийнятна сіль, де
R1 - СН3 або СН2ОН,
R2 - нижчий алкіл,
R3 - нижчий алкіл,
R4 - Н або галоген,
R5 - Н, галоген або нижчий алкіл,
Х - NH або О.
2. Сполука за п. 1, яка відрізняється тим, що у ній
R2 - С1-С4алкіл,
R3 - С1-С4алкіл,
R5 - Н, галоген або С1-С4алкіл, а
R1, R4 та Х визначено у п. 1.
3. Сполука за п. 1 або п. 2, яка відрізняється тим, що у ній
R2 - СН3 або СН2СН3,
R3 - СН3 або СН2СН3,
R4 - Н, Br, Cl або F,
R5 - Н, СН3, Br, Cl або F, а
R1 та Х визначені у п. 1.
4. Сполука за п. 3, яка відрізняється тим, що у ній
R5 - Н, СН3 або F, а
R1, R2, R3, R4 та Х визначені у п. 3.
5. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2,6-диметилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
6. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2,6-диметилбензиламіно)-3-гідроксиметил-2-метилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
7. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,3-диметил-8-(2,6-диметил-4-фторбензиламіно)імідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
8. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,6-диметил-8-(2,6-диметилбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
9. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,6-диметил-8-(2,6-диметил-4-фторбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
10. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2,6-диметил-4-фторбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
11. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,3-диметил-8-(2,6-диметил-4-хлорбензиламіно)імідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
12. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,6-диметил-8-(2-етил-6-метилбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
13. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2,6-діетилбензиламіно)-2,6-диметил-3-гідроксиметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
14. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2-етил-6-метилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
15. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2,6-диметил-4-фторбензилокси)-3-гідроксиметил-2-метилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
16. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,6-диметил-8-(2,6-диметилбензилокси)-3-гідроксиметилімідазо[1,2-а]піридин або його фармацевтично прийнятну сіль.
17. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 2,6-диметил-8-(2-етил-4-фтор-6-метилбензиламіно)-3-гідроксиметилімідазо-[1,2-а]піридин або його фармацевтично прийнятну сіль.
18. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що являє собою 8-(2-етил-4-фтор-6-метилбензиламіно)-2,3,6-триметилімідазо-[1,2-а]піридин або його фармацевтично прийнятну сіль.
19. Сполука за будь-яким з пп. 1-18, яка відрізняється тим, що є гідрохлоридною сіллю.
20. Сполука за будь-яким з пп. 1-19 або її фармацевтично прийнятна сіль, яка відрізняється тим, що призначена для використання у терапії.
21. Сполука за будь-яким з пп.1-19, яка відрізняється тим, що її використовують для виготовлення медикаменту, призначеного для пригнічення секреції шлункової кислоти.
22. Сполука за будь-яким з пп.1-19, яка відрізняється тим, що її використовують для виготовлення медикаменту, призначеного для лікування або профілактики інфікування Helicobacter pylori слизової оболонки шлунку людини і пристосованого для уведення у сполученні з щонайменше одним антимікробним агентом.
23. Спосіб отримання сполуки за будь-яким з пп. 1-19, який передбачає операцію
уведення сполуки загальної формули ІІ
,
де Х1 - NН2 або ОН, а R1 та R5 визначені, як для формули І,
у реакцію з сполуками загальної формули ІІІ
,
де R2, R3, R4 визначені, як для формули І, а Y - група, здатна відщеплюватись,
у нейтральному розчиннику у присутності основи або без неї.
24. Спосіб отримання сполуки за будь-яким з пп. 1-19, який передбачає операцію
уведення сполуки загальної формули IV
,
де R1, R5 визначені, як для формули І,
у реакцію з сполуками загальної формули V
,
де R2, R3, R4 визначені, як для формули І, бажано у присутності кислоти Льюїса, у інертному розчиннику, що дає сполуку формули VI
,
де R1, R2, R3, R4, R5 визначені, як для формули І, з подальшим відновленням цієї сполуки до сполуки загальної формули І, у якій Х - NH, за стандартних умов у інертному розчиннику.
25. Спосіб отримання сполуки за будь-яким з пп. 1-19, який передбачає операцію
уведення сполуки загальної формули VII
,
де Х1 - NН2 або ОН, а R5 визначено, як для формули І,
у реакцію з сполуками загальної формули ІІІ
,
де R2, R3, R4 визначені для формули І, а Y - група, здатна відщеплюватись, що дає сполуку формули VIII
,
де R2, R3, R4, R5 та Х визначені, як для формули І, з подальшим відновленням сполук загальної формули VIII, до сполук загальної формули І, у яких R1 - СН2ОН, у нейтральному розчиннику за стандартних умов.
26. Спосіб отримання сполуки за будь-яким з пп. 1-19, який передбачає операцію
уведення сполуки загальної формули ІХ
,
де R5 визначено, як для формули І, у реакцію з сполуками формули V
,
де R2, R3, R4 визначені, як для формули І, у присутності кислоти Льюїса, у інертному розчиннику, що дає сполуки загальної формули Х
,
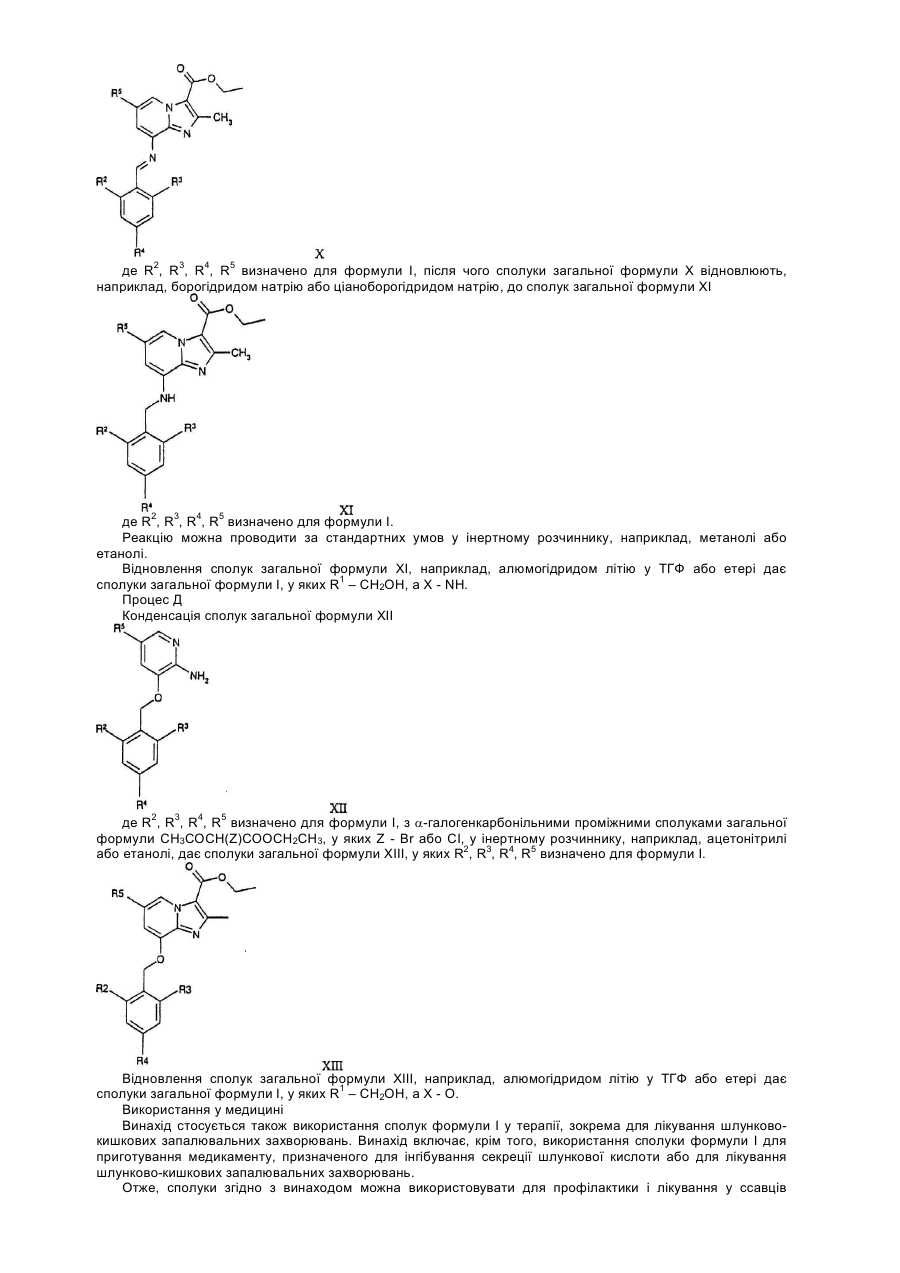
де R2, R3, R4, R5 визначені, як для формули І, з подальшим відновленням сполук формули Х у інертному розчиннику за стандартних умов до сполук загальної формули ХІ
,
де R2, R3, R4, R5 визначені, як для формули І, з подальшим відновленням сполук загальної формули ХI у інертному розчиннику за стандартних умов до сполуки загальної формули І, у якій R1 - СН2ОН, а Х – NH.
27. Спосіб отримання сполуки за будь-яким з пп. 1-19, який передбачає операцію
уведення сполуки загальної формули ХІІ
,
де R2, R3, R4, R5 визначені, як для формули І, у реакцію з сполуками загальної формули СН3СОСН(Z)СООСН2СН3, у яких Z - Br або Cl, у інертному розчиннику, що дає сполуки загальної формули ХІІІ
,
де R2, R3, R4, R5 визначені, як для формули І, з подальшим відновленням сполук загальної формули ХIІІ у інертному розчиннику за стандартних умов до сполук загальної формули І, у яких R1 - СН2ОН, а Х - О.
28. Фармацевтична композиція, яка містить сполуку за будь-яким з пп. 1-19 і фармацевтично прийнятний носій.
29. Спосіб придушення секреції шлункової кислоти або лікування шлунково-кишкових запалювальних захворювань у ссавця, включаючи людину, який передбачає уведення ефективної кількості сполуки за будь-яким з пп. 1-19.
30. Спосіб лікування або профілактики станів, пов’язаних з інфікуванням Helicobacter pylori слизової оболонки шлунку людини, який передбачає уведення ссавцю, включаючи людину, що потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-19 у сполученні з щонайменше одним антимікробним агентом.
31. Фармацевтична композиція, призначена для придушення секреції шлункової кислоти або лікування шлунково-кишкових запалювальних захворювань, у якій активним інгредієнтом є сполука за будь-яким з пп. 1-19.
32. Фармацевтична композиція, призначена для лікування або профілактики станів, пов’язаних з інфікуванням Helicobacter pylori слизової оболонки шлунку людини, у якій активним інгредієнтом є сполука за будь-яким з пп. 1-19 у сполученні з щонайменше одним антимікробним агентом.
Текст
Винахід стосується нових сполук і їх терапевтично прийнятних солей, які придушують екзогенно або ендогенно стимульовану секрецію шлункової кислоти і, таким чином, можуть бути використані для профілактики і лікування шлунково-кишкових запалювальних захворювань. Винахід стосується також використання сполук згідно з винаходом у терапії, способів виготовлення таких нових сполук, терапевтичних композицій, у яких активним інгредієнтом є щонайменше одна сполука згідно з винаходом або її терапевтично прийнятна сіль, і використання активних сполук для виготовлення медикаментів для медичного використання, як зазначено вище. Відомо, що імідазо[1,2-а]піридини використовуються для лікування пептичних виразкових захворювань (див , наприклад, ЕР-В-003094, US 4 450 164, ЕР-В-0204285 та US 4 725 601, а також публікації J.J. Kaminski et al., The Journal of Medical Chemistry, vol. 28, 876-892, 1985; vol. 30, 2031-2046, 1987; vol. 30, 20472051, 1987; vol. 32, 1686-1700, 1989; vol. 34, 533-541, 1991). Похідну імідазопіридину, заміщену у позиції 8 2,4,6-(СН3)зС6Н2СН2О, описано у ЕР-В-0033094 і як "Сполуку No.49" у Kaminski et al., J. Med. Chem., vol. 28, 876-892, 1985. Проте, згідно з подальшими публікаціями, ця сполука не виявила сприятливих властивостей при випробуваннях на придушення секреції шлункової кислоти. Огляд фармакології щодо транспортування шлункової кислоти (Н+,К+-АТРаза) - див Sachs et al. (1995), Аnnu. Rev. Pharmacol. Toxicol., 35, 277-305). Було виявлено, що сполуки формули І, які є заміщеними похідними імідазопіридину і у яких фенільне угруповання заміщено нижчим алкілом у позиціях 2 та 6, особливо ефективно інгібують шлунково-кишкову Н+,К+-АТРазу і є, таким чином, інгібіторами секреції шлункової кислоти. або її фармацевтично прийнятної солі, де R1 – СН3 або СН2ОН, R2 - нижчий алкіл, R3 - нижчий алкіл, R4 - Н або галоген, R5 - Н, галоген або нижчий алкіл, X - NH або О. Тут і далі термін "нижчий алкіл" означає лінійну або розгалужену групу з 1-6, бажано 1-4 атомами карбону. Прикладами нижчого алкілу є метил, етил, n-пропіл, ізопропіл, n-бутил, ізобутил, sec-бутил, t-бутил і лінійно- або розгалужено-ланцюгові пентил і гексил. Бажаними нижчими алкілами є метил, етил, n-пропіл, ізопропіл, n-бутил, ізобутил, sec-бутил і t-бутил. Галоген - це фтор, хлор, бром і іод. Винахід включає чисті енантіомери, рацемічні суміші і нерівні суміші двох енантіомерів, усі можливі діастереомерні форми (чисті енантіомери, рацемічні суміші і нерівні суміші двох енантіомерів), а також похідні сполуки формули І, які мають біологічні властивості сполук формули І. Залежно від типу реакції кінцеві продукти формули І можуть бути у нейтральній або сольовій формі. Винахід включає вільну основу і солі цих кінцевих продуктів. Солі приєднання кислот нових сполук можуть бути у відомі способи перетворені у вільну основу основними агентами, наприклад, лугами або через іонообмін. Для приготування солей приєднання кислот бажано використовувати такі кислоти, які утворюють терапевтично прийнятні солі. Прикладами можуть бути гідрогалогенні кислоти, наприклад, гідрохлоридна, сульфурова кислота, фосфорна кислота, азотна кислота, аліфатичні, аліциклічні, ароматичні або гетероциклічні карбонові або сульфонові кислоти, наприклад, мурашина, оцтова, пропіонова, сукцинова, гліколева, молочна, яблучна, винна, лимонна, аскорбінова, малеїнова, гідроксималеїнова, піровиноградна, р-гідроксибензойна, ембонова, метансульфонова, етансульфонова, гідроксіетансульфонова, галогенбензолсульфонова, толуолсульфонова або нафталінсульфонова кислота. Згідно з винаходом, бажаними сполуками є сполуки формули І, у яких R2 – СН3 або СН2СН3, яких R3 СН3 або СН2СН3, R4 - Н, Вr, СІ або F, R5 - Н, СН3, Вr, СІ або F, бажано Н, СН3 або F. Особливо бажаними сполуками згідно з винаходом є: 8-(2,6-диметилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин, 8-(2,6-диметилбензиламіно)-3-гідроксиметил-2-метилімідазо[1,2-а]піридин, 2,3-диметил-8-(2,6-диметил-4-фторбензиламіно)імідазо[1,2-а]піридин, 2,6-диметил-8-(2,6-диметилбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин, 2,6-диметил-8-(2,6-диметил-4-фторбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин, 8-(2,6-диметил-4-фторбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин, 2,3-диметил-8-(2,6-диметил-4-хлорбензиламіно)імідазо[1,2-а]піридин, 2,6-диметил-8-(2-етил-6-метилбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин, 8-(2,6-діетилбензиламіно)-2,6-диметил-3-гідроксиметилімідазо[1,2-а]піридин, 8-(2-етил-6-метилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин, 8-(2,6-диметил-4-фторбензилокси)-3-гідроксиметил-2-метилімідазо[1,2-а]піридин, 2,6-диметил-8-(2,6-диметилбензилокси)-3-гідроксиметилімідазо[1,2-а]піридин, 2,6-диметил-8-(2-етил-4-фтор-6-метилбензиламіно)-3-гідроксиметилімідазо-[1,2-а]піридин, 8-(2-етил-4-фтор-6-метилбензиламіно)-2,3,6-триметилімідазо-[1,2-а]піридин. Приготування Для виготовлення сполук формули І винахід передбачає процеси А, Б, В, Г, Д. Процес А. Процес А включає такі операції. Сполуки загальної формули II де X1 - NH2 або ОН, a R1, R5 визначено для формули І, уводять у реакцію з сполуками загальної формули III де R2, R3, R4 визначено для формули І, a Y - група, здатна відщеплюватись, наприклад галогенова, тозилоксилова або мезилоксилова, з утворенням сполуки формули І. Реакцію зручно проводити у нейтральному розчиннику, наприклад, ацетоні, ацетонітрилі, диметоксіетані, метанолі, етанолі або ДМФ у присутності основи або без неї. Основою може бути, наприклад, гідроксид лужного металу, зокрема, гідроксид натрію або калію, карбонат лужного металу, зокрема, карбонат натрію або калію, або органічний амін, зокрема, тріетиламін. Процес Б Процес Б приготування сполук загальної формули І, у яких X - NH, включає такі операції. Сполуки загальної формули IV де R1, R5 визначено для формули І, уводять у реакцію з сполуками загальної формули V де R2, R3, R4 визначено для формули І, у присутності кислоти Льюїса, наприклад, хлориду цинку, і одержують сполуку формули VI де R1, R2, R3, R4, R5 визначено для формули І, після чого сполуки загальної формули VI відновлюють, наприклад, борогідридом натрію або ціаноборогідридом натрію, до сполуки загальної формули І, у якій X NH. Реакцію можна проводити за стандартних умов у інертному розчиннику, наприклад, метанолі або етанолі. Процес В Процес В, призначений для приготування сполук загальної формули І, у яких R1 - СН2ОН, передбачає такі операції. Сполуки загальної формули VII III де X1 - NH2 або ОН, a R5 визначено для формули І, уводять у реакцію з сполуками загальної формули де R2, R3, R4 визначено для формули І, a Y - група, здатна відщеплюватись, наприклад галогенова, тозилоксилова або мезилоксилова, і одержують сполуку формули VIII де R2, R3, R4, R5 тa X визначено для формули І. Цю реакцію зручно проводити у нейтральному розчиннику, наприклад, ацетоні, ацетонітрилі, диметоксіетані, метанолі, етанолі або ДМФ у присутності основи або без неї. Основою може бути, наприклад, гідроксид лужного металу, зокрема, гідроксид натрію або калію, карбонат лужного металу, зокрема, карбонат натрію або калію, або органічний амін, зокрема, тріетиламін. Відновлення сполук загальної формули VIII, наприклад, алюмогідридом літію у ТГФ або етері дає сполуки загальної формули І, у яких R1 – СН2ОН. Процес Г Процес Г, призначений для приготування сполук загальної формули І, у яких R1 –СН2ОН, а X - NH, передбачає такі операції. Сполуки загальної формули IX де R5 визначено для формули І, уводять у реакцію з сполуками загальної формули V де R2, R3, R4 визначено для формули І, у присутності кислоти Льюїса, наприклад, хлориду цинку до сполук формули X де R2, R3, R4, R5 визначено для формули І, після чого сполуки загальної формули X відновлюють, наприклад, борогідридом натрію або ціаноборогідридом натрію, до сполук загальної формули XI де R2, R3, R4, R5 визначено для формули І. Реакцію можна проводити за стандартних умов у інертному розчиннику, наприклад, метанолі або етанолі. Відновлення сполук загальної формули XI, наприклад, алюмогідридом літію у ТГФ або етері дає сполуки загальної формули І, у яких R1 – СН2ОН, а X - NH. Процес Д Конденсація сполук загальної формули XII де R2, R3, R4, R5 визначено для формули І, з a-галогенкарбонільними проміжними сполуками загальної формули СН3СОСН(Z)СООСН2СН3, у яких Z - Вr або СІ, у інертному розчиннику, наприклад, ацетонітрилі або етанолі, дає сполуки загальної формули XIII, у яких R2, R3, R4, R5 визначено для формули І. Відновлення сполук загальної формули XIII, наприклад, алюмогідридом літію у ТГФ або етері дає сполуки загальної формули І, у яких R1 – СН2ОН, а X - О. Використання у медицині Винахід стосується також використання сполук формули І у терапії, зокрема для лікування шлунковокишкових запалювальних захворювань. Винахід включає, крім того, використання сполуки формули І для приготування медикаменту, призначеного для інгібування секреції шлункової кислоти або для лікування шлунково-кишкових запалювальних захворювань. Отже, сполуки згідно з винаходом можна використовувати для профілактики і лікування у ссавців шлунково-кишкових запалювальних захворювань і шлункових захворювань, пов'язаних з кислотністю, наприклад, гастриту, виразки шлунку або дванадцятипалої кишки, рефлюкс-езофагіту і синдрому Цолінгера-Елісона. Ці сполуки можуть також використовуватись для лікування інших шлунково-кишкових захворювань, коли бажано інгібувати секрецію шлункової кислоти, наприклад, у пацієнтів з гастриномою або з гострою шлунковою кровотечею. Ці сполуки можуть бути корисними для пацієнтів, що проходять інтенсивне лікування, і для відвернення до- або післяопераційного всмоктування кислоти або стресової виразки. Типова денна доза активного компонента може варіюватись у широких межах залежно від різних факторів, зокрема, від індивідуальної потреби пацієнта, способу уведення і захворювання. Взагалі дози активного компонента для орального і парентерального уведення становлять від 5 до 1000мг. Фармацевтичні композиції Винахід включає також фармацевтичні композиції, що містять як активний інгредієнт щонайменше одну сполуку згідно з винаходом, або її фармацевтично прийнятну сіль. У цих композиціях сполуки згідно з винаходом можуть використовуватись разом з іншими активними інгредієнтами, наприклад, антибіотиками, зокрема з амоксиліном. При клінічному застосуванні фармацевтичні композиції з сполуками згідно з винаходом можуть бути призначені для орального, ректального, парентерального або іншого способу уведення. Фармацевтична композиція містить сполуку згідно з винаходом у сполученні з одним або більше фармацевтично прийнятними інгредієнтами. Носієм може бути твердий, напівтвердий або рідкий розріджувач або капсула. Такі фармацевтичні препарати також включено у винахід. Звичайно активні сполуки складають 0,1-95% (за масою), бажано 0,1-20% (за масою) композиції для парентерального уведення і, бажано, 0,1-50% (за масою) композиції для орального уведення. Фармацевтичні композиції з сполуками згідно з винаходом можуть виготовлятись у формі дозованих одиниць для орального уведення, які являють собою суміш обраної сполуки з твердими інгредієнтами у вигляді порошку, наприклад, лактозою, сахарозою, сорбітолом, манітолом, крохмалем, амілопектином, похідними целюлози, желатином, або іншим придатним для цього інгредієнтом, а також з дезінтегруючими і змащуючими агентами, наприклад, стеаратом магнію, стеаратом кальцію, стеарилфумаратом і поліетиленкгліколевими вісками. Суміш гранулюють або пресують у таблетки. М'які желатинові капсули можуть містити суміш активної сполуки (або сполук) згідно з винаходом і рослинної олії, жиру або іншого носія, придатного для застосування з такими капсулами. Жорсткі желатинові капсули можуть містити гранули активної сполуки або активну сполуку у сполученні з твердими інгредієнтами у вигляді порошку, наприклад, лактозою, сахарозою, сорбітолом, манітолом, картопляним або кукурудзяним крохмалем, амілопектином, похідними целюлози або желатином. Дозовані одиниці для ректального уведення можна виготовляти у формі (і) супозиторіїв, що містять активний агент у суміші з нейтральною жировою основою, (іі) желатинових ректальних капсул, що містять активний агент у суміші з рослинною олією, парафіновою олією або іншим носієм, придатним для застосування з такими капсулами, (ііі) заздалегідь виготовлених мікроклізм, або (iv) сухих композицій для мікроклізм, які перед уведенням перетворюють у належний розчин. Рідкі композиції для орального уведення готують у вигляді сиропів або суспензій, наприклад, розчинів або суспензій з вмістом активного інгредієнта від 0,1 до 10% (за масою) і рештою, якою можуть бути цукор або цукрові спирти і суміш етанолу, води, гліцеролу, пропіленгліколю і поліетиленгліколю. За бажанням у такі рідкі композиції можна додати забарвлюючі агенти, смакові добавки, сахарин і карбоксиметилцелюлозу або інший загущувач. Рідкі композиції для орального уведення можна готувати у вигляді сухого порошку, який перед уведенням має бути розчинений у придатному розчиннику. Розчини для парентерального уведення готують розчиненням сполуки згідно з винаходом у фармацевтично прийнятному розчиннику з бажаною концентрацією від 0,1 до 10% (за масою). Ці розчини можуть також містити стабілізуючі і/або буферні інгредієнти і виготовлятись у формі одиночних доз - ампул або склянок. Розчини для парентерального уведення також можна виготовляти у вигляді сухого порошку, який перед уведенням має бути розчинений у придатному розчиннику. Сполуки згідно з винаходом можна використовувати разом з іншими активними інгредієнтами у композиціях, призначених для лікування або профілактики станів, викликаних інфікуванням Helicobacter pylori слизової оболонки шлунку людини. Такими інгредієнтами можуть бути антимікробні агенти, зокрема: b-лактамові антибіотики, наприклад, амоксицилін, ампіцилін, цефалотин, цефаклор або сефіксим, макроліди, наприклад, еритроміцин або кларитроміцин, тетрацикліни, наприклад, тетрациклін або доксициклін, аміноглікозиди, наприклад, гентаміцин, канаміцин або амікацин, хінолони, наприклад, норфлоксацин, ципрофлоксин або еноксацин, інші препарати, наприклад, метронідазол, нітрофурантоїн або хлорамфенікол, або препарати, що містять вісмутові солі, наприклад, субцитрат, субсаліцилат, субкарбонат, субнітрат або субгалат вісмута. Приклади 1. Приготування сполук згідно з винаходом Приклад 1.1. Гідрохлорид 8-(2,6-диметилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридину. Суміш 0,9г (5,1ммоль) 8-(2,6-диметилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридину, 0,84г (6,2ммоль) хлориду цинку (II) і 0,83г (6,2ммоль) 2,6-диметилбензальдегіду у 50мл метанолу з перемішуванням обробляють 0,39г (6,2ммоль) ціаноборгідриду натрію і витримують під зворотним холодильником 3год. Метанол випаровують під зниженим тиском і залишок розчиняють у метиленхлориді і 2М гідроксиду натрію (40мл). Органічний шар відокремлюють, висушують над сульфатом натрію і випаровують під зниженим тиском. Залишок двічі очищують хроматографією на колонці силікагелю з елюентами а) етилацетат/метиленхлорид (1:2) і б) метанол/метиленхлорид (1:20). Маслянистий продукт розчиняють у діетиловому етері, обробляють сумішшю діетилового етеру і НСІ, осаджену сіль відфільтровують і одержують 0,6г (36%) бажаної речовини. 1 Н- ЯМР (300MHz, CDCl3): d 2,33 (s, 3Н), 2,38 (s, 3Н), 2,45 (s, 6Н), 2,50 (s, 3Н), 4,40 (d, 2Н), 6,40 (bs, 1H), 7,95-7,15 (m, 4H). Приклад 1.2. 2,3-диметил-8-(2,6-диметилбензиламіно)-6-фторімідазо[1,2-а]піридин. Суміш 0,16г (0,89ммоль) 8-аміно-2,3-диметил-6-фторімідазо[1,2-а]піридину, 0,14г (1,04ммоль) хлориду цинку (II) і 0,14г (1,04ммоль) 2,6-диметилбензальдегіду у 50мл метанолу з перемішуванням обробляють 0,065г (1,04ммоль) ціаноборгідриду і витримують під зворотним холодильником протягом 7год. До охолодженої реакційної суміші 0,5М NaOH (20мл), осаджену тверду речовину відфільтровують і очищують хроматографією на колонці силікагелю з елюентом метанол/метиленхлорид (1:10). Кристалізація з петролейного етеру дає 0,1г (38%) бажаной сполуки. 1 Н-ЯМР (300MHz, CDCI3): d 2,30 (s, ЗН), 2,34 (s, ЗН), 2,40 (s, 6H) 4,35 (d, 2H), 4,95 (bs, Ш), 6,15 (dd, IH), 7,0-7,20 (m, 4H). Приклад 1.3. 2,3-диметил-8-(2,6-діетилбензиламіно)імідазо[1,2-а]піридин. 0,33г (2,0ммоль) 8-аміно-2,3-диметилімідазо[1,2-а]піридину і 0,36г (2,2ммоль) 2,6-диметилбензальдегіду розчиняють у 7мл метанолу. Малими порціями додають 0,30г (2,2ммоль) ZnCl2 і потім 0,14г (2,2ммоль) NaBH3CN. Суміш витримують під зворотним холодильником у аргоні протягом 3год., охолоджують і вливають у водний розчин 1М NaOH (10мл). Утворену жовту суспензію екстрагують ДХМ (3х25мл) і об'єднані органічні розчини промивають розсолом, висушують над Na2SO4 і видаляють. Маслянистий осад (0,4г) очищують флеш-хроматографією (ДХМ-ЕЮАс 0%-20% ЕtOАс) і одержують 0,34г маслянистого продукта, який обробляють гексаном (2мл) і одержують 0,14г (23%) білуватих кристалів. 1 Н-ЯМР (300MHz, CDCI3): d 7,2-7,3 (2Н, m), 7,1 (2Н, d), 6,7 (1H, t), 6,2 (1H, d), 4,8 (1H, b), 4,4 (2Н, d), 2,7 (4Н, q), 2,3 (6Н, tдва сингл.), 1,2 (6Н, t). Приклад 1.4. 8-(2,6-диметилбензилокси)-3-гідроксиметил-2-метилімідазо[1,2-а]піридин. Суміш 0,89г (5,0ммоль) 8-гідрокси-3-гідроксиметил-2-метилімідазо[1,2-а]піридин, 1,5г карбонату натрію, 0,4г іодиду натрію, 0,7г (4,5ммоль) 2,6-диметилбензилхлориду і 60мл ацетону перемішують протягом ночі. Додають ще 1г карбонату натрію і реакційну суміш витримують під зворотним холодильником протягом 2год., після чого фільтрують і розчинник видаляють у вакуумі. Залишок суспендують у СН2СІ2/МеОН (100:5) і фільтрують. Випаровування розчинника у вакуумі дає залишок, який очищують флеш-хроматографією з елюентом СН2СІ2-МеОН (100:4). Фракції збирають, рекристалізують з CH2CI2/CH3CN і одержують 0,37г бажаної сполуки. 1 Н ЯМР (300MHz, CDCI3): d 7,87 (d, J=7,6Hz, 1H), 7,15-7,08 (m, 1H), 7,0 (d, J=7,6Hz, 2H), 6,73 (t, J=7,6Hz, 1H), 6,63 (d, J=7,6Hz, 1H), 5,23 (s, 2H), 4,83 (s, 2H), 2,4 (s, 6H), 2,28 (s, 3H). Приклад 1.5. 2,3-диметил-8-(2,6-диметилбензиламіно)імідазо[1,2-а]піридин. Суміш 0,7г (4,34ммоль) 8-аміно-2,3-диметилімідазо[1,2-а]піридину, 2,0г карбонату натрію, 0,3г іодиду натрію, 0,671г (4,34ммоль) 2,6-диметилбензилхлориду і 30мл ацетону перемішують протягом ночі. Реакційну суміш фільтрують і розчинник видаляють у вакуумі. Залишок розчиняють у метиленхлориді і промивають водним NaНСО3. Органічний шар відділяють і випаровують розчинник. Сирий продукт очищують флеш-хроматографією з елюентом СН2СІ2/МеОН (100:4) і одержують 0,7г бажаної сполуки. 1 Н ЯМР (300MHz, CDCI3): d 7,25 (d, J=7,7Hz, 1H), 7,14-7,09 (m, 1H), 7,03 (d, J=7,7Hz, 2H), 6,73 (t, J=7,7Hz, 1H), 6,21 (d, J=7,7Hz, 1H), 4,79 (br "t", 1H), 4,34 (d, J=4,5Hz, 2H), 2,38 (s, 6H), 2,34 (s, 6H). Приклад 1.6. 2,3-диметил-8-(2,6-диметилбензилокси)імідазо[1,2-а]піридин. Суміш 1,2г (7,41ммоль) 8-гідрокси-2,3-диметилімідазо[1,2-а]піридину, 1,145г (7,41ммоль) 2,6диметилбензилхлориду, 2,0г карбонату натрію, 0,3г іодиду натрію і 50мл ацетону витримують під зворотним холодильником протягом 3год. Додають метиленхлорид, реакційну суміш фільтрують і розчинник видаляють у вакуумі. Залишок розчиняють у СН2СІ2, промивають водним NaНСО3, висушують над Nа2СО3 і випаровують. Залишок хроматографують на силікагелі з елюентом СН2СІ2/МеОН (100:5) і одержують 0,7г бажаної сполуки (з етилацетату-етеру). 1 Н ЯМР (300МНz, СDСI3): d 7,56 (d, J=6,6Нz, 1Н), 7,1 (t, J=6,6Hz, 1H), 6,94-6,85 (m, 3Н), 6,73 (d, J=6,6Hz, 1Н), 2,31 (s, 3Н), 2,26 (s, 3Н), 2,24 (s, 6H). Приклад 1.7. 2.3-диметил-8-(2-етил-6-метилбензиламіно)імідазо[1,2-а]піpидин. 0,3г (1,86ммоль) 8-аміно-2,3-диметилімідазо[1,2-а]піридину і 0,31г (1,84ммоль) 2-етил-6метилбензилхлориду розчиняють у 5мл диметоксіетану. Додають 0,3г (2,8ммоль) карбонату натрію і 0,2г (1,2ммоль) іодиду натрію і суміш витримують під зворотним холодильником протягом 4год. Розчинник випаровують і залишок хроматографують на колонці силікагелю з елюентом метиленхлорид/етилацетат (60:40), одержуючи 230мг (42%) бажаної сполуки. 1 Н-ЯМР (300MHz, CDCl3): d 1,22 (t, ЗН), 2,35 (s, 6Н), 2,39 (s, 3Н), 2,70 (q, 2H), 4,35 (d,2H), 4,81 (t, 1H) 6,21 (d, 1H), 6,73 (t, 1H), 7,01-7,10 (m,2H), 7,13-7,19 (m, 1H), 7,24 (d, 1H). Приклад 1.8. 6-бром-2,3-диметил-8-(2,6-диметилбензиламіно)імідазо[1,2-а]піридин. Суміш 1,2г (5,0ммоль) 8-аміно-6-бромо-2,3-диметилімідазо[1,2-а]піридину, 0,772г (5,0ммоль) 2,6диметилбензилхлориду, 0,8г карбонату натрію, 0,2г іодиду натрію і 45мл ацетону перемішують протягом ночі, після чого додають ще 0,285г 2,6-диметилбензилхлориду і реакційну суміш витримують під зворотним холодильником протягом 5год. Додають ацетон, реакційну суміш фільтрують і розчинник видаляють у вакуумі. Залишок розчиняють у СН2СІ2, промивають водним NaHСО3, висушують над Nа2СО3 і випаровують. Сирий продукт розчиняють у етилацетаті і додають петролейний етер. Фільтрування і випаровування розчинника дають залишок, з якого рекристалізацією з етилацетату одержують 1,45г бажаної сполуки. 1 H ЯMP (300MHz, CDCI3): d 7,37 (d,J=1,5Hz, 1Н), 7,15-7,09 (m, 1H), 7,04 (d, J=7,5Hz, 2H), 6,28 (d, J=1,5Hz, 1H), 4,88 ("t", 1H), 4,33 (d, J=4,13Hz, 2H), 2,38 (s, 6H), 2,3 (s, 3H), 2,29 (s, 3H). Приклад 1.9. 8-(2,6-диметилбензиламіно)-3-гідроксиметил-2-метилімідазо[1,2-a]піридин Розчин 40мл (136ммоль) вітриду у 25мл толуолу краплями додають до очищеного азотом розчину 8,0г (23,7ммоль) 3-карбоетокси-8-(диметилбензиламіно)-2-метилімідазо[1,2-а]піридину у 100мл толуолу. Видаляють льодяну ванну і реакційну суміш перемішують при кімнатній температурі протягом 105хвил., після чого охолоджують до 0°С і гасять водою (36мл). Суміш фільтрують, органічний шар промивають водним NаНСО3, висушують над Nа2СО3 і концентрують. Додають 20мл ацетонітрилу і фільтруванням відбирають кристалічний продукт, який двічі промивають ацетонітрилом і після висушування у вакуумі одержують 5,6г бажаної сполуки. 1 Н ЯMP (300MHz, CDCl3): d 7,58 (d, J=7,1Hz, 1Н), 7,15-7,1 (m, 1Н), 7,05 (d, J=7,1Hz, 2H), 6,74 (t, J=7,1Hz, 1H), 6,28 (d, J=7,1Hz, 1H), 4,84 (br t, J=4,5Hz, 1H), 4,8 (s, 2H), 4,35 (d, J=4,5Hz, 2H), 2,4 (s, 6H), 2,2 (s, 3H). Приклад 1.10. 6-хлор-2,3-диметил-8-(2,6-диметилбензиламіно)імідазо[1,2-а]піридин. Суміш 0,894г (4,57ммоль) 8-аміно-6-хлор-2,3-диметилімідазо[1,2-а]піридину, 0,77г (5,7ммоль) 2,6диметилбензальдегіду, 1,08г (7,92ммоль) ZnCl2, 0,36г (5,7ммоль) NaB(CN)H3 і 35мл МеОН витримують під зворотним холодильником протягом 3,5год. Додають 2,6-диметилбензальдегід (0,25г у 4мл МеОН), 0,55г ZnCI2 і 0,35г NaB(CN)H3 і реакційну суміш витримують під зворотним холодильником протягом ще 4год. Додають 150мл 1М NaOH і 50мл води, екстрагують СН2СІ2, висушують над Na2SO4 і випаровуванням розчинника одержують твердий залишок. Сирий продукт розчиняють у етилацетаті і додають етер. Фільтрування і випаровування розчинника дають залишок, з якого рекристалізацією з етилацетату одержують 0,52г бажаної сполуки. 1 Н ЯMP (300MHz, CDCl3): d 7,28 (d, J=1,7Hz, 1Н), 7,15-7,1 (m, 1Н), 7,04 (d, J=12Hz, 2Н), 6,2 (d, J=1,7Hz, 1H), 4,89 (br "t", 1H), 4,33 (d, J=4Hz, 2H), 2,37 (s, 6H), 2,33 (s, 3H), 2,32 (s, 3H). Приклад 1.11. 2,3-диметил-8-(2,6-диметил-4-фторбензиламіно)імідазо[1,2-а]-піридин. 0,5г (3,1ммоль) 8-аміно-2,3-диметилімідазо[1,2-а]піридину розчиняють у 6мл ацетонітрилу. До розчину додають 0,67г (3,1ммоль) 2,6-диметил-4-фторбензилброміду і 0,47г (3,4ммоль) карбонату калію і суміш витримують під зворотним холодильником протягом 16год. Додають 12мл метиленхлориду і 20мл розчину хлориду натрію, органічний шар відділяють, сушать над сульфатом натрію і випаровують під зниженим тиском. Сирий продукт очищують хроматографією з елюентом етилацетат/петролейний етер (1:1) і одержують 400мг бажаної сполуки у вигляді твердої речовини. 1 Н-ЯМР (300MHz, CDCl3): d 2,3 (s, 6Н), 2,3 (s, 6Н), 4,2 (d, 2H), 4,65 (b, 1H), 6,15 (d, 1Н), 6,65-6,75 (m, 3H), 7,2 (d, 1H). Приклад 1.12. 2,6-диметил-8-(2,6-диметилбензиламіно)-3-гідроксиметилімідазо[1,2-а]піридин. Розчин 0,4г (1,1ммоль) 3-карбоетокси-2,6-диметил-8-(2,6-диметилбензиламіно)імідазо[1,2-а]піридину у 10мл толуолу охолоджують льодяною водою і через 30хвил. додають 2,1г (6,6ммоль) 65%-го Red-AL у толуолі. Краплями додають 10мл розчину солі Рошеля (тетрагідрат тартрату натрію-калію, 35г/250мл води), додають 10мл толуолу, органічний шар відділяють, сушать над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на силікагелі з елюентом ДХМ/етанол (9:1) і одержують 0,21г (62%) бажаної сполуки. 1 Н-ЯМР (300MHz, CDCl3): d 1,65 (s, 1Н ), 2,30 (d, 6H ), 2,38 (s, 6Н), 4,37 (d, 2H), 4,75 (s, 1H), 4,85 (s, 2H), 6,15 (s, 1H ), 7,0-7,15 (m, 3H), 7,40 (s, 1H). Приклад 1.13. 2,6-диметил-8-(2,6-диметил-4-фторбензиламіно)-3-гідрокси-метилімідазо[1,2-а]-піридин Розчин 0,4г (1,1ммоль) 3-карбоетокси-2,6-диметил-8-(2,6-диметил-4-фторбензиламіно)імідазо[1,2а]піридину у 10мл толуолу охолоджують льодяною водою і через 30хвил. додають 2,1г (6,6ммоль) 65%-го Red-AL. Розчин перемішують при кімнатній температурі протягом 2год., після чого краплями додають 10мл розчину солі Рошеля (тетрагідрат тартрату натрію-калію, 35г/250мл води). Додають 10мл толуолу, органічний шар відділяють, сушать над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на силікагелі з елюентом ДХМ/етанол (95:5) і одержують 0,3г (83%) бажаної сполуки. 1 Н ЯМР (300MHz, CDCI3): d 2,26 (s, 3Н), 2,33 (s, 3Н), 2,37 (s, 6H), 4,28 (d, 2H), 4,70 (s, 1H), 4,82 (s, 2H), 6,14 (s, 1H), 6,75 (d, 2H), 7,42 (s, 1H). Приклад 1.14. 8-(2,6-диметил-4-фторбензиламіно)-2,3,6-триметилімідазо[1,2-а]-піридин. Суміш 0,5г (2,85ммоль) 8-аміно-2,3,6-триметилімідазо[1,2-а]піридину, 0,7г (3,4ммоль) 2,6-диметил-4фторбензильроміду, 0,6г (4,6ммоль) карбонату натрію, 0,1г іодиду натрію і 15мл ацетонітрилу з перемішуванням витримують під зворотним холодильником протягом ночі. Розчинник видаляють під зниженим тиском і залишок розчиняють у ДХМ і промивають водою. Органічний шар сушать над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом гексан/етилацетат (2:1). Маслянистий продукт розчиняють у діетиловому етері і осаджену сіль відфільтровують, одержуючи 0,55г (56%) бажаної сполуки. 1 Н ЯМР (300MHz, CDCI3): d 2,22 (s, 3Н), 2,30 (d, 12Н), 4,23 (d, 2Н), 4,68 (s, 1Н), 6,05 (s, 1Н), 6,70 (d,2H), 7,00 (s, 1H). Приклад 1.15. 2,3-диметил-8-(2,6-диметил-4-хлорбензиламіно)імідазо[1,2-а]-піридин. Суміш 4-хлор-2,6-диметилбензилброміду, 2-хлор-4,6-диметилбензилброміду (1,1г, 4,68ммоль) і 4,65ммоль 8-аміно-2,3-диметилімідазо[1,2-а]піридину розчиняють у 15мл диметоксіетану. Додають 1,0г (9,4ммоль) карбонату натрію і 0,5г (3,0ммоль) іодиду калію і суміш витримують під зворотним холодильником протягом 4год. Розчинник випаровують і залишок очищують хроматографією на силікагелі з елюентом метиленхлорид/етилацетат (70:30) і одержують 70мг бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,35 (s, 6H), 4,29 (d, 2H), 4,74 (f, 1H), 6,19 (d, 1H), 6,72 (t, 1H), 7,04 (s, 2H), 7,25 (d, 1H). Приклад 1.16. 2,6-диметил-8-(2-етил-6-метилбензиламіно)-3-гідроксиметил-імідазо[1,2-а]піридин. 1,0г (2,8ммоль) 3-карбоетокси-2,6-диметил-8-(2-етил-6-метилбензил-аміно)імідазо[1,2-а]піридину додають до 25мл ТГФ і перемішують при 5°С. Протягом 1,5год. порціями додають алюмогідрид літію (0,5г, 13ммоль) таким чином, щоб температура залишалась нижче 10°С. Сухі речовини видаляють фільтруванням і ретельно промивають ТГФ і метиленхлоридом. Фільтрат і промивний розчин об'єднують, сушать і розчинник випаровують під зниженим тиском. Залишок розчиняють у метиленхлориді і промивають водою, органічний шар відділяють, сушать над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на силікагелі з елюентом етилацетат/метиленхлорид (1:1) і після кристалізації з суміші петролейний етер/діетиловий етер (1:1) одержують 0,37г (41%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,25 (t, 3H), 2,25 (s, 3H), 2,35 (s, 3H), 2,40 (s, 3H), 2,70 (q, 1H), 4,35 (d, 2H), 4,75 (bs, 1H), 4,80 (s, 2H), 6,15 (s, 1H), 7,05-7,25 (m, 3H), 7,40 (s, 1H). Приклад 1.17. 8-(2,6-діетилбензиламіно)-2,6-диметил-3-гідроксиметилімідазо-[1,2-а]піридин. Розчин 1,75г (4,6ммоль) 3-карбоетокси-2,6-диметил-8-(2,6-діетилбензиламіно)імідазо[1,2-а]піридину у 30мл ТГФ обробляють алюмогідридом літію (0,7г, 18,5ммоль) при кімнатній температурі протягом 3,5год. Через 4год. реакція завершується, і її обережно гасять, додаючи краплями воду (0,7мл), водний гідроксид натрію (0,7мл, 15%) і знову воду (2мл). Суміш екстрагують хлороформом і органічний шар концентрують, залишок рекристалізують з етанолу, білий кристалічний продукт фільтрують, промивають діетиловим етером і після висушування у вакуумі одержують 1,5г (96%) бажаної сполуки. 1 Н-ЯМР (500МГц, CDCL3): d 1,23 (t, 6H), 1,99 (s, 1H), 2,25 (s, 3H), 2,33 (s, 3H), 2,73 (q, 4H), 4,34 (d, 2H), 4,80 (s, 3H), 6,13 (s, 1H), 7,09 (d, 2H), 7,22 (t, 1H) 7,40 (s, 1H). Приклад 1.18. 8-(2,6-диметилбензиламіно)-2,3,6-триметилімідазо[1,2-а]-піридин. Суміш 0,5г (2,8ммоль) 8-аміно-2,3,6-триметилімідазо[1,2-а]піридину, 0,7г (4,3ммоль) 2,6диметилбензальдегіду і 0,44г (3,0ммоль) хлориду цинку (II) і у 50мл метанолу з перемішуванням обробляють 0,19г (3,0ммоль) ціаноборгідриду і витримують під зворотним холодильником протягом 20год. Метанол випаровують під зниженим тиском і залишок розчиняють у дихлорметилені і воді. Органічний шар відділяють, висушують над сульфатом натрію і випаровують під зниженим тиском. Залишок хроматографують на силікагелі з елюентом дихлорметилен/етилацетат (1:1) і одержують 0,42г бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,25 (t, 6H), 2,28 (s, 3H), 2,30 (s, 3H), 2,33 (s, 3H), 2,71 (q, 4H), 4,36 (d, 2H), 4,84 (s, 1H), 6,10 (s, 1H), 7,04-7,23 (m, 4H). Приклад 1.19. 8-(2-етил-6-метилбензиламіно)-2,3,6-триметилімідазо[1,2-а]піридин. Суміш 0,5г (2,8ммоль) 8-аміно-2,3,6-триметилімідазо[1,2-а]піридину, 0,45г (3ммоль) 2-етил-6метилбензальдегіду і 0,4г (3ммоль) хлориду цинку (II) у 50мл метанолу з перемішуванням обробляють 0,19г (3,0ммоль) ціаноборгідриду і витримують під зворотним холодильником протягом 20год. Метанол випаровують під зниженим тиском і залишок розчиняють у дихлорметилені і воді. Органічний шар відділяють, висушують над сульфатом натрію і випаровують під зниженим тиском. Залишок хроматографують на силікагелі з елюентом дихлорметилен/метанол (10:1) і одержують 0,28г (33%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,22 (t, 3H), 2,32 (s, 6H), 2,34 (s, 3H), 2,38 (s, 3H), 2,72 (q, 2H), 4,33 (d, 2H), 4,77 (s, 1H), 6,08 (s, 1H), 7,03-7,19 (m, 4H). Приклад 1.20. 8-(2,6-диметил-4-фторбензилокси)-3-гідроксиметил-2-метил-імідазо[1,2-а]піридин. Алюмогідрид літію (0,31г, 8,4ммоль) додають до 30мл ТГФ і протягом 30хвил. краплями додають 1,5г (4,2ммоль) 3-карбоетокси-8-(2,6-диметил-4-фторбензилокси)-2-метилімідазо[1,2-а]піридину у 30мл ТГФ, після чого додають водний гідроксид натрію (0,31мл, 15%) і воду (0,93мл). Тверді компоненти видаляють фільтруванням і ретельно промивають сумішшю метанол/метиленхлорид (1:1). Фільтрат і промивний розчин об'єднують і під зниженим тиском видаляють розчинники. Залишок хроматографують на силікагелі з елюентом метиленхлорид/метанол (9:1) і після обробки залишку ацетонітрилом одержують 0,9г (69%) бажаной сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,25 (s, 3H), 2,35 (s, 6H), 4,85 (d, 2H), 5,1 (t, 1H), 5,2 (s, 2H), 6,8-7,05 (m, 4H), 7,95 (d, 1H). Приклад 1.21. 6-бром-8-(2,6-диметил-4-фторбензиламіно)-3-гідроксиметил-2-метилімідазо[1,2а]піридин. 70мг LiBH4 протягом 4год. краплями додають розчину 100мг (0,23ммоль) 6-бром-3-карбоетокси-8-(2,6диметил-4-фторбензилокси)-2-метилімідазо[1,2-а]-піридину у 30мл ТГФ, який витримують під зворотним холодильником, після чого реакційну суміш гасять розведеною НСІ і додають метиленхлорид. Органічний шар відділяють, висушують і випаровують у вакуумі. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/етилацетат (100:10) і одержують 0,40г (44%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 7,72 (s, 1H), 6,75 (d, 2H), 6,35 (s, 1H), 4,9 (t, 1H), 4,8 (s, 2H), 4,3 (d, 2H), 2,35 (s, 6H), 2,25 (s, 3H). Приклад 1.22. 2,6-диметил-8-(2,6-диметилбензилокси)-3-гідроксиметилімідазо-[1,2-а]піридин. 3мл (10,2ммоль) вітриду у 3мл толуолу краплями додають до очищеного азотом розчину 0,68г (1,93ммоль) 3-карбоетокси-2,6-диметил-8-(диметилбензилокси)імідазо[1,2-а]піридину у 15мл толуолу. Видаляють льодяну ванну і реакційну суміш перемішують при кімнатній температурі протягом 2год. 15хвил., після чого охолоджують до 0°С і гасять водою (6мл). Додають метиленхлорид/метанол і суміш фільтрують. Видаляють у вакуумі розчинник і залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/метанол (100:5) і одержують 0,35г (58%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 7,65 (s, 1H), 7,10 (t, 1H), 7,0 (d, 2H), 6,50 (s, 1H), 5,2 (s, 2H), 4,8 (s, 2H), 2,4 (s, 6H), 2,35 (s, 3H), 2,25 (s, 3H). Приклад 1.23. 8-(2,6-диметил-4-фторбензиламіно)-3-гідроксиметил-2-метил-імідазо[1,2-а]піридин. Розчин 0,6г (1,7ммоль) 3-карбоетокси-8-(2,6-диметил-4-фторбензил-аміно)-2-метилімідазо[1,2а]піридину у 30мл толуолу охолоджують льодяною водою і протягом 30хвил. додають 2,1г (6,6ммоль) 65%го Red-AL у толуолі. Розчин перемішують протягом 1год. при кімнатній температурі і потім краплями додають 25мл розчину солі Рошеля (тетрагідрат тартрату натрію-калію, 35г/250мл води). Органічний шар відділяють, водний шар промивають метиленхлоридом. Об'єднані органічні розчини, сушать над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на силікагелі з елюентом ДХМ/етанол (95:5) і одержують 0,42г (79%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,15 (s, 3H), 2,35 (s, 6H), 4,30 (d, 2H), 4,75 (s, 2H), 4,85 (t, 1H), 6,25 (d, 1H), 6,70-6,80 (m, 3H), 7,55 (d, 1H). Приклад 1.24. 2,6-диметил-8-(2-етил-4-фтор-6-метилбензиламіно)-3-гідрокси-метилімідазо[1,2а]піридин. До 0,08г (2,1ммоль) LiAlН4 у 15мл ТГФ додають розчин 0,4г (1,0ммоль) 3-карбоетокси-2,6-диметил-8-(2етил-4-фтор-6-метилбензиламіно)імідазо[1,2-а]-піридину у 15мл ТГФ. Після перемішування суміші при кімнатній температурі протягом 4год. краплями додають 0,1мл води, потім 0,1мл 15%-го гідроксиду натрію і ще 0,3мл води. Сухі речовини видаляють фільтруванням і ретельно промивають ТГФ. Фільтрат і промивний розчин об'єднують, сушать і розчинник видаляють під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/метанол (9:1) і кристалізацією з ацетонітрилу одержують 0,32г (89%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,2 (t, 3H), 2,2 (s, 3H), 2,35 (s, 3H), 2,4 (s, 3H), 2,75 (q, 2H), 4,3 (d, 2H), 4,75 (bs, 3H), 6,15 (s, 1H), 6,75-6,85 (m, 2H), 7,45 (s, 1H). Приклад 1.25. 8-(2-етил-4-фтор-6-метилбензиламіно)-2,3,6-триметилімідазо-[1,2-а]піридин. 0,38г (2,16ммоль) 8-аміно-2,3,6-триметилімідазо[1,2-а]піридину і 0,50г (2,16ммоль) 2-етил-4-фтор-6метилбензилброміду розчиняють у 10мл диметоксіетану. Додають 0,4г (3,8ммоль) карбонату натрію і 0,2г (1,2ммоль) іодиду калію і суміш витримують під зворотним холодильником протягом 6год. Розчинник випаровують і залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/етилацетат (60:40), одержуючи 203мг (29%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,21 (t, 3H), 2,32 (s, 6H), 2,33 (s, 3H), 2,37 (s, 3H), 2,71 (q, 2H), 4,28 (d, 2H), 4,68 (t, 1H), 6,06 (s, 1H), 6,73-6,80 (m, 2H), 7,05 (s, 1H). Приклад 1.26. 2,3-диметил-8-(2,6-диметил-4-фторбензилокси)імідазо-[1,2-а]-піридин. 1,7г (10ммоль) 2,3-диметил-8-гідроксіімідазо[1,2-а]піридину, 2,3г (10ммоль) 2,6-диметил-4фторбензилброміду, 2,6г (28ммоль) карбонату натрію і 0,5г (0,3ммоль) іодиду натрію додають до 75мл ацетону і суміш витримують під зворотним холодильником протягом 6год. Додають метиленхлорид, суміш фільтрують і розчинник випаровують під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/етилацетат (1:2), одержуючи 0,85г (28%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,36 (s, 3H), 2,38 (s, 9H), 5,15 (s, 2H), 6,57 (d, 1H), 6,68-6,75 (m, 3H), 7,46 (d, 1H). Приклад 1.27. 2,6-диметил-8-(2-етил-6-метилбензилокси)імідазо-[1,2-а]-піридин. 0,8г (5ммоль) 2,3-диметил-8-гідроксіімідазо[1,2-а]піридину, 0,25г (1,7ммоль) 2-етил-6метилбензилхлориду, 1,2г (11ммоль) карбонату натрію і 0,25г (1,7ммоль) іодиду натрію додають до 40мл ацетону і суміш витримують під зворотним холодильником протягом 5год. Випаровують ацетон і залишок розчиняють у метиленхлориді і промивають водою. Органічний розчин сушать і випаровують під зниженим тиском. Залишок двічі очищують хроматографією на колонці силікагелю з елюентомами а) метиленхлорид/етилацетат (1:2) і метиленхлорид/етилацетат (2:1) одержуючи 0,02г (1,4%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,2 (t, 3H), 2,36 (s, 3H), 2,38 (s, 3H), 2,40 (s, 3H), 2,74 (q, 2H), 5,21 (s, 2H), 6,59 (d, 1H), 6,7 (t, 1H), 7,04 (m, 2H), 7,17 (d, 1H) 7,45(d,1H). Приклад 1.28. 8-(2-етил-6-метилбензилокси)-3-гідроксиметил-2-метилімідазо-[1,2-а]-піридин. До 0,08г (2,1ммоль) LiAIH4 у 25мл ТГФ додають розчин 1,0г (2,8ммоль) 3-карбоетокси-8-(2-етил-6метилбензилокси)-2-метилімідазо[1,2-а]-піридину у 25мл ТГФ. Після перемішування суміші при кімнатній температурі протягом 2год. краплями додають 0,2мл води, потім 0,2мл 15%-го гідроксиду натрію і ще 0,6мл води. Сухі речовини видаляють фільтруванням і розчинник видаляють під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/метанол (9:1) і кристалізацією з діетилового етеру одержують 0,52г (60%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,2 (t, 3H), 2,25 (s, 3H), 2,4 (s, 3H), 2,75 (q, 2H), 4,75 (s, 2H), 5,2 (s, 2H), 6,656,75 (m, 2H), 7,0-7,2 (m, 3H), 7,85 (d, 1H). ТАБЛИЦЯ Сполуки прикладів 1.1 - 1.28 Приклад 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 R1 СН3 СН3 H CH2OH СН3 СН3 СН3 СН3 R2 СН3 СН3 СН2СН3 СН3 СН3 СН3 СН3 СН3 R3 СН3 СН3 СН2СН3 СН3 СН3 СН3 СН2СН3 СН3 R4 H H H H H H H H R5 СН3 F H H H H H Вr X NH NH NH O NH O NH NH 1.9 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 1.22 1.23 1.24 1.25 1.26 1.27 1.28 СН2ОН СН3 СН3 СН2ОН СН2ОН СН3 СН3 СН2ОН СН2ОН СН3 СН3 СН2ОН СН2ОН СН2ОН СН2ОН СН2ОН СН3 СН3 СН3 СН2ОН СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН2СН3 СН2СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН3 СН2СН3 СН2СН3 СН2СН3 СН2СН3 СН3 СН3 СН3 СН3 СН2СН3 СН2СН3 СН3 СН2СН3 СН2СН3 H H F H F F СІ H H H H F F H F F F F H H H СІ H СН3 СН3 СН3 H СН3 СН3 СН3 СН3 H Вr СН3 H СН3 СН3 H H H NH NH NH NH NH NH NH NH NH NH NH O NH O NH NH NH O O O 1. Приготування проміжних сполук Приклад 2.1. 2,6-диметил-4-фторбензилбромід. Суміш 5г (0,04ммоль) 3,5-диметилфторбензолу, 15г параформальдегіду, 70мл гідробромідної кислоти (30% у оцтовій кислоті) і 25мл оцтової кислоти перемішують при кімнатній температурі протягом 4,5год. До суміші додають воду і петролейний етер і органічний шар відділяють, сушать над безводним сульфатом натрію і обережно піддають випарюванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з петролейним етером як елюентом і одержують 3,7г (43%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,5 (s, 6H), 4,55 (s, 2H), 6,75 (d, 2H). Приклад 2.2. 2-етил-6-метилбензилхлорид. 1,0г (6,67ммоль) 2-етил-6-метилбензилового спирту розчиняють у 10мл метиленхлориду. Додають 1,0г (8,5мл) тіонилхлориду і суміш перемішують при кімнатній температурі протягом ночі. Реакційну суміш піддають випарюванню, залишок розчиняють у метиленхлориді і фільтрують через 5г силікагелю. Випаровування фільтрату дає 1,0г (89%) бажаної сполуки (масло). 1 Н-ЯМР (300МГц, CDCL3): d 1,29 (t, 3H), 2,46 (s, 3H), 2,76 (q, 2H), 4,71 (s, 2H), 7,0-7,2 (m, 3H). Приклад 2.3. 8-аміно-2,3,6-триметилімідазо[1,2-а]піридин. До розчину 2,0г (16ммоль) 2,3-діаміно-5-метилпіридину у 100мл етанолу додають 2,4г (16ммоль) 3бром-2-бутанону. Реакційну суміш витримують під зворотним холодильником протягом 2год. Під зниженим тиском випаровують етанол і залишок обробляють метиленхлоридом і розчином бікарбонату. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Маслянистий залишок очищують хроматографією на колонці силікагелю з елюентом метанол/метиленхлорид (1:20) і одержують 1,05г (37%) бажаної сполуки. 1 Н-ЯМР (300МГц, DMSO-d6): d 2,15 (s, 3H), 2,25 (s, 3H), 2,3 (s, 3H), 5,45 (bs, 2H), 6,05 (s, 1H), 7,20 (s, 1H). Приклад 2.4. 2-аміно-5-фтор-3-нітропіридин. До розчину 8,6г (77ммоль) 2-аміно-5-фторпіридину у 40мл концентрованої сульфатної кислоти при температурі 3°С краплями протягом 30хвил. додають 3,25мл (77ммоль) паруючої азотної кислоти. Реакційну суміш перемішують при кімнатній температурі протягом 1год. і при температурі 55°С протягом 1год. Суміш виливають у льод, нейтралізують 10М гідроксидом натрію і екстрагують метиленхлоридом. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Залишок двічі очищують хроматографією на колонці силікагелю з елюентами (і) метанол/метиленхлорид (1:20) і (іі) діетиловий етер/петролейний етер (1:1) і одержують 0,44г (3,6%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 6,65 (bs, 3H), 8,20 (dd, 1H), 8,35 (d, 2H). Приклад 2.5. 2,3-діаміно-5-фторпіридин. До розчину 0,42г (2,3ммоль) 2-аміно-5-фтор-3-нітропіридину і 1,6г (28ммоль) порошку заліза у 10мл етанолу додають 0,5мл (28ммоль) води і 27мкл (0,32ммоль) гідрохлоридної кислоти. Реакційну суміш витримують під зворотним холодильником протягом 1год., додають ще 0,2г (3,6ммоль) порошку заліза і суміш витримують під зворотним холодильником протягом 30хвил., після чого фільтрують через броунмілерит. Випарювання під зниженим тиском розчинника дає 0,3г (100%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 3,55 (bs, 2H), 4,1 (bs, 2H), 6,7 (dd, 1H), 7,5 (d, 1H). Приклад 2.6. 8-аміно-2,3-диметил-6-фторімідазо[1,2-а]піридин. Суміш 0,3г (2,4ммоль) 2,3-діаміно-5-фторпіридину і 0,36г (2,4ммоль) 3-бром-2-бутанону у 20мл етанолу витримують під зворотним холодильником протягом 10год. Залишок розчиняють у метиленхлориді і обробляють розчином бікарбонату. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом метанол/метиленхлорид (1:20) і одержують 0,16г (37%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 2,3 (s, 3H), 2,4 (s, 3H), 4,6 (bs, 2H), 6,2 (dd, 1H), 7,2 (dd, 1H). Приклад 2.7. 8-аміно-6-бром-2,3-диметилімідазо[1,2-а]піридин. Розчин 4,0г (21,29ммоль) 2,3-діаміно-5-бромпіридину і 3,7г (24,48ммоль) 3-бром-2-бутанону у 40мл етанолу витримують під зворотним холодильником протягом ночі. Після охолодження до кімнатної температури відфільтровують кристалічну речовину, яку промивають етанолом і етером. Кристали розчиняють у метиленхлориді і нейтралізують водним NаНСО3. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском, одержуючи 2,3г бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 7,39 (d, J=1,7Гц, 3H), 6,36d, J=1,7Гц, 3Н), 4,5 (bs, 2H), 2,35 (s, 3H), 2,3 (s, 3H). Приклад 2.8. 3-карбоетокси-8-(диметилбензиламіно)-2-метилімідазо[1,2-а]-піридин. Суміш 6,08г (27,74ммоль) 8-аміно-3-карбоетокси-2-метилімідазо[1,2-а]-піридину, 4,5г (29,13ммоль) 2,6диметилбензилхлориду, 4,32г (43,7ммопь} карбонату натрію, 0,7г іодиду натрію у 120мл ацетону перемішують протягом 30год., після чого відфільтровують кристалічну речовину, яку розчиняють у дихлометані і після фільтрування піддають випарюванню під зниженим тиском, одержуючи 7,0г бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 8,66 (d, J=11Гц, 1Н), 7,16-7,1 (m, 1H), 7,05 (d, J=11Гц, 2H), 6,87 (t, J=11Гц, 1H), 6,45 (d, J=11Гц, 1H), 4,86 ("t", 1Н), 4,4 (q, J=7Гц, 2Н), 4,35 (d, J=3,6Гц, 2Н), 2,65 (s, 3H), 2,35 (s, 6H), 1,4 (t, J=7Гц, 3Н). Приклад 2.9. 8-аміно-6-хлор-2,3-диметилімідазо[1,2-а]піридин. Суміш 5,26г (36,64ммоль) 2,3-діаміно-5-хлорпіридину і 6,2г (41,06ммоль) 3-бром-2-бутанону у 60мл етанолу витримують під зворотним холодильником протягом ночі. Після охолодження до кімнатної температури відфільтровують кристалічну речовину, яку промивають етанолом і етером. Кристали розчиняють у метиленхлориді і нейтралізують водним NaHСО3. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском, одержуючи 3,0г бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 7,29 (d, J=1,5Гц, 1H), 6,26 (d, J=1,5Гц, 1Н), 4,55 (bs, 2H), 2,4 (s, 3H), 2,3 (s, 3H). Приклад 2.10. 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]-піридин. Суміш 4,0г (32,5ммоль) 2,3-діаміно-5-метилпіридину і 5,9г (36,0ммоль) етилхлорацетоацетату у 75мл абсолютного етанолу з перемішуванням витримують під зворотним холодильником протягом ночі. Під зниженим тиском випаровують етанол, залишок розчиняють у 2М НСІ,промивають тричі діетиловим етером, доводять pH до 9 і тричі екстрагують ДХМ. Органічний шар сушать над безводним сульфатом натрію і піддають випарюванню. Залишок очищують хроматографією на колонці силікагелю з елюентом ДХМ/метанол (95:5) і одержують 2,0г (28%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,42 (t, 3H), 2,28 (s, 3H), 2,65 (s, 3H), 4,40 (q, 2H), 4,47 (s, 2H), 6,40 (s, 1H), 8,55 (s, 1H). Приклад 2.11. 3-карбоетокси-2,6-диметил-8-(2,6-диметилбензиламіно)імідазо-[1,2-а]піридин. Суміш 1,2г (5,1ммоль) 8-аміно-2,6-диметилімідазо[1,2-а]піридину, 0,84г (6,2ммоль) хлориду цинку (II) і 0,84г (6,2ммоль) 2,6-диметилбензальдегіду у 50мл метанолу з перемішуванням обробляють 0,39г (6,2ммоль) ціаноборгідриду натрію і витримують під зворотним холодильником 5год. Метанол випаровують під зниженим тиском і залишок розчиняють у ДХМ і 2М гідроксиду натрію (40мл). Органічний шар відокремлюють, висушують над сульфатом натрію і випаровують під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом петролейний етер (40-60/ізопропіловий етер (8:2) і одержують 0,8г (44%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 1,44 (t, 3H), 2,35 (d, 9H), 2,60 (s, 3H), 4,33 (d, 2H), 4,40 (q, 2H), 4,6 (s, 1H), 6,60 (s, 1H), 7,10 (d, 2H), 7,25 (m, 1H), 8,50 (s, 1H). Приклад 2.12. 3-карбоетокси-2,6-диметил-8-(2,6-диметилбензиламіно)імідазо-[1,2-а]піридин. Суміш 1,2г (5,1ммоль) 8-аміно-2,6-диметилімідазо[1,2-а]піридину, 0,84г (6,2ммоль) хлориду цинку (II) і 0,84г (6,2ммоль) 2,6-диметилбензальдегіду у 50мл метанолу з перемішуванням обробляють 0,39г (6,2ммоль) ціаноборгідриду натрію і витримують під зворотним холодильником 5год. Метанол випаровують під зниженим тиском і залишок розчиняють у ДХМ і 2М гідроксиду натрію (40мл). Органічний шар відокремлюють, висушують над сульфатом натрію і піддають випаріванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом петролейний етер (40-60/ізопропіловий етер (8:2) і одержують 0,8г (44%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 1,44 (t, 3H), 2,35 (d, 9H), 2,60 (s, 3H), 4,33 (d, 2H), 4,40 (q, 2H), 4,6 (s, 1H), 6,60 (s, 1H), 7,10 (d, 2H), 7,25 (m, 1H), 8,50 (s, 1H). Приклад 2.13. 3-карбоетокси-2,6-диметил-8-(2,6-диметил-4-фторбензиламіно)-імідазо[1,2-а]піридин. Суміш 1,1г (4,7ммоль) 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]-піридину, 1,2г (5,7ммоль) 2,6диметил-4-фторбензилброміду, 1,0г (7,5ммоль) карбонату калію, 0,1г іодиду натрію у 15мл ацетонітрилу з перемішуванням витримують під зворотним холодильником протягом ночі. Після випаровування під зниженим тиском розчинника залишок розчиняють у ДХМ і промивають водою. Органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом петролейний етер (40-60/ізопропіловий етер (7:3) і одержують 0,8г (47%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 1,42 (t, 3H), 2,36 (s, 9H), 2,62 (2, 3H), 4,45 (d, 2H), 4,48 (q, 2H), 4,54 (s, 1H), 6,30 (s, 1H), 6,75 (d, 2H), 8,55 (s, 1H). Приклад 2.14. 4-хлор-2,6-диметилбензилбромід. Суміш 1,42г (0,01моль) 4-хлор-3,5-диметилбензолу, 0,31г (0,01моль) параформальдегіду додають до 2мл гідробромідної кислоти (33%) у оцтовій кислоті. Суміш перемішують при 70°С протягом ночі, після чого вливають у 25мл води і екстрагують діетиловим етером. Органічний шар промивають водою, сушать над сульфатом натрію і піддають випарюванню, одержуючи 1,1г продукту у вигляді масла. Спектр ЯМР показує, що ця речовина є сумішшю бажаної речовини і 2-хлор-4,6-диметилбензилброміду і її використовують без подальшого очищення. 1 Н-ЯМР (300МГц, CDCL3): d 2,28 (s, 6H), 4,51 (s, 2H), 7,04 (s, 2H). Приклад 2.15. 3-карбоетокси-2,6-диметил-8-(2-етил-6-метилбензиламіно)-імідазо[1,2-а]піридин. Суміш 1,4г (6ммоль) 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]піридину, 1,0г (7,4ммоль) ZnCl2, 0,9г (6,5ммоль) 2-етил-6-метилбензальдегіду, 0,41 (6,5ммоль) NaB(CN)H3 і 30мл МеОН витримують під зворотним холодильником 5год. Додають ще 0,2г ZnCI2 і 0,1г NaB(CN)H3 і витримують під зворотним холодильником ще 2год. Додають 2мл тріетиламіну і суміш перемішують протягом 10хвил. при кімнатній температурі. Розчинник випаровують під зниженим тиском і залишок очищують хроматографією на колонці силікагелю з метиленхлоридом як елюентом і одержують 1,1г (50%) бажаної речовини. 1 Н-ЯМР (3001 МГц, CDCL3): d 1,25 (t, 3H), 1,45 (t, 3H), 2,30 (s, 6H), 2,6 (s, 3H), 2,75 (q, 2H), 4,35 (d, 2H), 4,45 (q, 2H), 4,85 (bs, 1H), 6,35 (s, 1H), 7,0-7,25 (m, 3H), 8,5 (s, 1H). Приклад 2.16. 3-карбоетокси-2,6-диметил-8-(2,6-діетилбензиламіно)імідазо-[1,2-а]піридин. Суміш 2,02г (8,6ммоль) 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]піридину, 1,48г (10,8ммоль) хлориду цинку (II) і 2,17г (13,4ммоль) 2,6-діетилбензальдегіду у 50мл метанолу з перемішуванням обробляють 0,65г (10,3ммоль) ціаноборгідриду натрію і витримують під зворотним холодильником протягом ночі. Суміш охолоджують і вливають у 1М гідроксиду натрію (40мл). Осад фільтрують, промивають водою і очищують хроматографією на колонці силікагелю з елюентом ДХМ/метанол (95:5) і одержують 2,1г (64%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 1,23 (t, 6H), 1,42 (t, 3H), 2,38 (s, 3H), 2,61 (s, 3H), 2,72 (q, 4H), 4,34 (d, 2H), 4,40 (q, 2H), 4,83 (t, 1H), 6,32 (s, 1H), 7,11 (d, 2H), 7,24 (t, 1H), 8,51 (s, 1H). Приклад 2.17. 3-карбоетокси-8-(2,6-диметил-4-фторбензилокси)-2-метил-імідазо[1,2-а]піридин. Суміш 1,5г (6,8ммоль) 3-карбоетокси-8-гідрокси-2-метилімідазо[1,2-а]піридину, 1,6г (7,5ммоль) 2,6диметил-4-фторбензилброміду, 0,1г іодиду натрію, 1,9г (13,6ммоль) карбонату калію і 50мл ацетонітрилу витримують під зворотним холодильником протягом ночі. Розчинник видаляють у вакуумі, залишок розчиняють у СН2СІ2, промивають водою, висушують над Nа2SO4 і випаровують. Залишок хроматографують на силікагелі з елюентом гептан/ізопропіловий етер (1:2) і одержують 2,0г (83%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 1,45 (t, 3H), 2,4 (s, 6H), 2,7 (s, 3H), 4,45 (q, 2H), 5,2 (s, 2H), 6,7-6,9 (m, 4H), 9,0 (d, 2H). Приклад 2.18. 8-аміно-6-бром-3-карбоетокси-2-метилімідазо[1,2-а]-піридин. Суміш 2,5г (13,31ммоль) 2,3-діаміно-5-бромпіридину і 2,41г (14,64ммоль) етил-2-хлорацетоацетату у 35мл абсолютного етанолу витримують під зворотним холодильником протягом 14год. Під зниженим тиском випаровують етанол, залишок розчиняють у метиленхлориді і нейтралізують водним NaHCO3. Органічний шар відділяють, сушать і піддають випарюванню у вакумі. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/метанол (100:3,5) і одержують 1,55г (39%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 8,9 (s, 1H), 6,65 (s, 1H), 4,6 (bs, 2H), 4,4 (q, 2H), 2,65 (s, 3H), 1,4 (t, 3H). Приклад 2.19. 6-бром-3-карбоетокси-8-(2,6-диметил-4-фторбензиламіно)-2-метилімідазо[1,2-а]піридин. Суміш 2,06г (6,91ммоль) 8-аміно-6-бром-3-карбоетокси-2-метилімідазо[1,2-а]піридину, 1,05г (4,48ммоль) 2,6-диметил-4-фторбензилброміду, 0,45г іодиду натрію, 2,2г карбонату калію і 40мл ацетону витримують під зворотним холодильником протягом 22год. Реакційну суміш фільтрують, фільтрат промивають СН2СІ2, метиленхлоридний розчин промивають водою, висушують і піддають випарюванню у вакуумі. Залишок суспендують у етанолі/етері, фільтрують і одержують 1,15г (56%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 8,85 (s, 1H), 6,8 (d, 2H), 6,55 (s, 1H), 4,9 (t, 1H), 4,4 (q, 2H), 4,3 (d, 2H), 2,6 (s, 3H), 2,4 (s, 6H), 1,45 (t, 3H). Приклад 2.20. 3-(2,6-диметилбензилокси)-5-метил-2-нітропіридин. До 0,52г (8,02ммоль) 87%-го КОН і 0,15г q-іодиду у 6мл 95%-го етанолу додають розчин 1,2г (7,79ммоль) 3-гідрокси-5-метил-2-нітропіридину у 25мл етанолу. До утвореної суспензії калієвої солі краплями додають розчин 1,24г (8,02ммоль) 2,6-диметилбензилхлориду у 13мл етанолу. Реакційну суміш витримують під зворотним холодильником протягом 1год., додають ще 0,16г 87%-го КОН і 0,38г 2,6диметилбензилхлориду і витримують під зворотним холодильником протягом ще 70хвил. Суміш фільтрують і неорганічні солі промивають етанолом і метиленхлоридом. Органічний шар піддають випарюванню у вакуумі, залишок розчиняють у метиленхлориді, промивають водним NaHCO3, сушать і піддають випарюванню у вакуумі. Залишок суспендують у етері/ізопропанолі, фільтрують і одержують 1,72г (81%) бажаної речовини. 1 Н-ЯМР (300МГц, CDCL3): d 7,94 (s, 1H), 7,46 (s, 1H), 7,19 (t, 1H), 7,08 (d, 2H), 5,18 (s, 2H), 2,47 (s, 3H), 2,4 (s, 6H). Приклад 2.21. 2-аміно-3-(2,6-диметилбензилокси)-5-метилпіридин. Суміш 2,5г (13,31ммоль) 2,3-діаміно-5-бромпіридину і 2,41г (14,64ммоль) етил-2-хлорацетоацетату у 35мл абсолютного етанолу витримують під зворотним холодильником протягом 14год. Під зниженим тиском випаровують етанол, залишок розчиняють у метиленхлориді і нейтралізують водним NаНСО3. Органічний шар відділяють, сушать і піддають випарюванню у вакумі. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/метанол (100:3,5) і одержують 1,55г (39%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 8,9 (s, 1H), 6,65 (s, 1H), 4,6 (bs, 2H), 4,4 (q, 2H), 2,65 (s, 3H), 1,4 (t, 3H). Приклад 2.22. 3-карбоетокси-2,6-диметил-8-(2,6-диметилбензилокси)імідазо-[1,2-а]піридин. Суміш 1,0г (4,13ммоль) 2-аміно-3-(2,6-диметилбензилокси)-5-метилпіридину і 0,79г (4,55ммоль) етил-2хлорацетоацетату у 20мл абсолютного етанолу витримують під зворотним холодильником протягом 19год. Додають ще 0,25г етил-2-хлорацетоацетату і витримують під зворотним холодильником протягом ще 23год. У вакуумі випаровують розчинник, залишок розчиняють у метиленхлориді і промивають водним NаНСО3. Органічний шар сушать і піддають випарюванню у вакуумі. Залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/етилацетат (100:10) і одержують 0,68г (47%) бажаної сполуки. 1 Н-ЯМР (500МГц, CDCL3): d 8,8 (s, 1H), 7,15 (t, 1H), 7,04 (d, 2H), 6,71 (s, 1H), 5,22 (s, 2H), 4,41 (q, 2H), 2,67 (s, 3H), 2,41 (s, 6H), 2,39 (s, 3H), 1,42 (t, 3H). Приклад 2.23. 3-карбоетокси-8-(2,6-диметил-4-фторбензиламіно)-2-метил-імідазо[1,2-а]піридин. Суміш 1,0г (4,7ммоль) 8-аміно-3-карбоетокси-2-метилімідазо[1,2-а]піридину, 1,2г (5,7ммоль) 2,6диметил-4-фторбензилброміду, 0,1г іодиду натрію і 1,0г (7,5ммоль) карбонату калію у 15мл ацетонітрилу з перемішуванням витримують під зворотним холодильником протягом ночі. Після випаровування під зниженим тиском розчинника залишок розчиняють у ДХМ і промивають водою, органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом петролейний етер (40-60)/ізопропіловий етер (7:3) і одержують 1,2г (75%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,45 (t, 3H), 2,35 (s, 6H), 2,65 (s, 3H), 4,40 (d, 2H), 4,40 (q, 2H), 4,85 (t, 1H), 6,40 (d, 1H), 6,75 (d, 2H), 6,85 (t, 1H), 8,70 (d, 1H). Приклад 2.24. 2-етил-4-фтор-6-метилбензилбромід. Суміш 1,1г (0,008моль) 3-етил-1-фтор-5-метилбензолу, 1,5г (0,05моль) параформальдегіду, 4,1мл (0,017моль) гідробромідної кислоти (4,1М у оцтовій кислоті) і 2,5мл оцтової кислоти перемішують при кімнатній температурі протягом 40год. До суміші додають воду і петролейний етер (40-60), органічний шар відділяють, промивають водою, сушать над безводним сульфатом натрію і обережно під зниженим тиском піддають випарюванню, одержуючи 1,3г (72%) бажаного продукту у вигляді жовтого масла. 1 Н-ЯМР (300МГц, CDCL3): d 1,2 (t, 3H), 2,35 (s, 3H), 2,7 (q, 2H), 4,50 (s, 2H), 6,7-6,85 (m, 2H). Приклад 2.25. 3-карбоетокси-2,6-диметил-8-(2-етил-4-фтор-6-метилбензил-аміно)імідазо[1,2-а]піридин. Суміш 0,7г (3,0ммоль) 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]піридину, 0,8г (3,5ммоль) 2-етил4-фторбензилброміду, 0,1г іодиду натрію і 0,7г (4,8ммоль) карбонату калію у 15мл ацетонітрилу з перемішуванням витримують під зворотним холодильником протягом ночі. Після випаровування під зниженим тиском розчинника залишок розчиняють у ДХМ і промивають водою, органічний шар відділяють, сушать над сульфатом натрію і піддають випарюванню під зниженим тиском. Залишок очищують хроматографією на колонці силікагелю з елюентом петролейний етер (40-60)/ізопропіловий етер (7:3) і одержують 0,4г (35%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,25 (t, 3H), 1,45 (t, 3H), 2,4 (s, 6H), 2,65 (s, 3H), 2,75 (q, 2H), 4,3 (d, 2H), 4,4 (q, 2H), 4,75 (bs, 1H), 6,3 (s, 1H), 6,75-6,85 (m, 2H), 8,5 (s, 1H). Приклад 2.26. 3-карбоетокси-8-(2-етил-6-метилбензилокси)імідазо[1,2-а]-піридин. Суміш 0,92г (4,2ммоль) 8-аміно-3-карбоетокси-2,6-диметилімідазо[1,2-а]піридину, 0,7г (4,2ммоль) 2етил-6-метилбензилхлориду, 1,0г (9,4ммоль) карбонату натрію і каталітичної кількості іодиду калію у 40мл ацетонітрилу з перемішуванням витримують під зворотним холодильником протягом 4год. Після фільтрування і випаровування під зниженим тиском розчинника залишок очищують хроматографією на колонці силікагелю з елюентом метиленхлорид/етилацетат і одержують 1,0г (68%) бажаної сполуки. 1 Н-ЯМР (300МГц, CDCL3): d 1,2 (t, 3H), 1,4 (t, 3H), 2,4 (s, 3H), 2,65 (s, 3H), 2,75 (q, 2H), 4,4 (q, 2H), 5,25 (s, 2H), 6,85-6,9 (m, 2H), 7,05-7,25 (m, 3H), 8,95 (dd, 1H). Приклад 2.27. 3-етил-1-фтор-5-метилбензол. При температурі 0°С 40мл (64ммоль) метиллітію краплями додають до суспензії 6,42г (33,6ммоль) іодиду міді (І) у діетилетері (20мл). Після 30-хвилинного перемішування при 0°С прозорий безбарвний однорідний розчин купрату охолоджують до -78°С, після чого додають 5,15г (25,4ммоль) 1-фтор-5метилбензолу у 10мл діетилового етеру і повільно доводять температуру до -50°С. При цій температурі реакцію гасять буфером NH4CI/NH3 (50мл) і екстрагують діетиловим етером (3х50мл) і розсолом (100мл). Органічний шар сушать над MgSO4, фільтрують і після видалення розчинників одержують 3,3г (94%) бажаної сполуки. 1 Н-ЯМР (500МГц, CDCL3): d 1,22 (t, 3H), 2,32 (s, 3H), 2,60 (q, 2H), 6,69 (d, 2H), 6,78 (s, 1H). Біологічні тести 1. Експерименти in vitro Придушення секреції кислоти у ізольованих шлункових залозах кролика. Інгібуючу дію на секрецію кислоти у ізольованих шлункових залозах кролика виміряли згідно з Berglindh et al. (1976), Acta Phisiol. Scand., 97, 401-414. Визначення активності Н+,К+-АТРази Приготування везикул шлункової мембрани: везикули шлункової мембрани з Н+,К+-АТРазою були приготовлені з шлунку свині згідно з Saccomani et al. (1977), Biochim. Biophis. Acta, 465, 311-330. Проникні везикули: Фракцію мембрани розводили 1мМ PIPES/Tris, pH 7,4 до одержання 1%-ї конуентрації сахарози, гомогенізували і центрифугували при 100000g протягом 2год. Одержані гранули були суспендовані у воді і двічі ліофілізовані. Визначення активності Н+,К+-АТРази: Проникні везикули мембрани (2,5-5мкг) були інкубовані протягом 15хвил. при 37°С у 18мМ буфера PIPES/Tris, pH 7,4, з вмістом 2мМ МgСІ2, 10мМ КСІ і 2мМ АТП. Активність АТПази визначали за вивільненням неорганічного фосфату з АТР згідно з LeBel et al. (1978), Anal. Biochem., 85, 86-89. 2. Експерименти in vivo Придушення секреції кислоти у самиць щура Були використані самиці щура лінії Sprague-Dawly. Їм були встановлені канюльовані фістули у шлунку і верхній частині дванадцятипалої кишки для збирання секрецій шлунку і уведення речовин, що випробувались. Випробування проводились через 14 днів періоду реконвалесценції після хірургічної операції. Перед випробуванням тваринам протягом 20год. не давали їсти, але давали пити. Шлунок багаторазово промивали через канюлю водопровідною водою (37°С) і підшкірно ввели Ringer-Glucose (6мл). Секрецію кислоти протягом 2,5-4год. стимулювали підшкірним уведенням пентагастрину (1,2мл/год.) і карбахолу (20 та 100нмоль/кг-год., відповідно). Протягом цього періоду збирали 30-хвилинні фракції секрецій шлунку. Препарати для випробування або носій уводили на 60-й хвилині (внутрішньовенно або у дванадцятипалу кишку, 1мл/кг) або на 2-й год. (оральне уведення (5мл/кг) з закриттям шлункової канюлі) після початку стимуляції. Інтервал між дозуванням і стимуляцією може бути збільшений для визначення тривалості дії. Зразки шлункового соку були титровані до pH 7,0 дією NaOH (0,1M) і вихід кислоти обчислювали як добуток об'єму титранта і концентрації. Подальші обчислення виконувались на основі групового середнього для 4-6 щурів. У випадку уведення під час стимуляції вихід кислоти під час періодів після уведення препаратів, що випробувались, був репрезентований як часткова реакція, що давало вихід кислоти 1,0 за 30-хвилинний період, що передував уведенню. Придушення у % обчислювали через часткові реакції, викликані препаратами і носієм. У випадку уведення перед стимуляцією придушення у % обчислювали безпосередньо за виходом кислоти, зареєстрованим після препарату, який випробували, і носія. Біозасвоюваність у щурів Були використані дорослі щури лінії Sprague-Dawly. За 1-3 дні до експериментів усі щури були підготовлені канюлюванням лівої каротидної артерії під анестезією. Щури для експериментів з внутрішньовенним уведенням також були канюльовані у яремну вену (Popovic (1960), J. Appl. Physiol., 15, 727-728) з виведенням у нижній частині шиї. Після уведення дози з інтервалами до 5,5год. з каротидної артерії були взяті зразки крові (0,1-0,4г), які були заморожені до закінчення аналізу сполуки, яку випробували. Біозасвоюваність оцінювали обчисленням відношення між площами під кривою залежності концентрацій крові та плазми після (і) внутрішньодуоденального або орального уведення і після (іі) внутрішньовенного (в.в.) уведення щуру або собаці, відповідно. Цю площу (AUC) визначали методом логарифмічно-лінійних трапецій з екстраполяцією на нескінченність шляхом ділення останньої визначеної концентрації у крові на константу швидкості видалення у останній фазі. Системну біозасвоюваність (F%) після після внутрішньодуоденального (в.д.) або орального (о.) уведення обчислюють за формулою F(%) = (АUС(в.д. або o.)/AUC(в.в.) x 100 Придушення секреції шлункової кислоти і біозасвоюваність у собаки у стані свідомості. Були використані собаки обох статей порід Лабрадор або Харієр. їм були поставлені дуоденальні фістули для уведення сполук, що випробувались, і носія і канюльовані шлункові фістули для збирання шлункової секреції у мішечки Хайденхайма. Перед випробуваннями собак протягом 18год. не годували, але давали воду. Секрецію шлункової кислоти протягом до 6,5год. стимулювали уведенням дигідрохлориду гістаміну (12мл/год.) у дозі, що викликала приблизно 80% індивідуальної максимальної секреторної реакції, а шлунковий сік збирали 30хвилинними послідовними фракціями. Сполуки, що випробувались, і носій уводили орально, внутрішньодуоденально або внутрішньовенно через 1 або 1,5год. після початку уведенння гістаміну у кількості 0,5мл/кг маси тіла. У випадку орального уведення сполуку уводили у виробляючий кислоту головний шлунок собаки з мішечком Хайденхайма. Кислотність зразків шлункового соку визначали титруванням до pH 7,0 і обчисленням виходу кислоти. Вихід кислоти за період збирання після уведення сполуки або носія репрезонтували як часткові реакції, що давало вихід кислоти 1,0 за 30-хвилинний період, що передував уведенню. Придушення у % обчислювали через часткові реакції, викликані препаратами і носієм. Зразки крові для визначення концентрації сполуки, яку випробували, відбирали з інтервалом до 4год. після дозування. Плазму відділяли і заморожували не пізніше, ніж через 30хвил. після збирання і пізніше аналізували. Системну біозасвоюваність (F%) після після внутрішньодуоденального або орального уведення обчислювали, як описано вище для щурів.
ДивитисяДодаткова інформація
Назва патенту англійськоюCompounds for inhibition of gastric acid secretion
Автори англійськоюNordberg Peter
Назва патенту російськоюСоединения для ингибирования секреции желудочной кислоты
Автори російськоюНордберг Петер
МПК / Мітки
МПК: C07D 471/04, C07D 221/00, A61P 1/04, C07D 235/00, A61K 31/437
Мітки: одержання, шлунково-кишкових, імідазопіридину, похідні, основі, лікування, пригнічення, варіанти, запалювальних, композиція, фармацевтична, шлункової, секреції, заміщені, спосіб, кислоти
Код посилання
<a href="https://ua.patents.su/18-64741-zamishheni-pokhidni-imidazopiridinu-sposib-kh-oderzhannya-varianti-farmacevtichna-kompoziciya-na-kh-osnovi-varianti-sposib-prignichennya-sekreci-shlunkovo-kisloti-abo-likuvannya-sh.html" target="_blank" rel="follow" title="База патентів України">Заміщені похідні імідазопіридину, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі (варіанти), спосіб пригнічення секреції шлункової кислоти або лікування шлунково-кишкових запалювальних за</a>
Оральна фармацевтична складова одинична дозована форма у вигляді таблетки, спосіб її одержання, упаковка у вигляді блістера та спосіб інгібування секреції шлункової кислоти і/або лікування шлунково-кишкових зап

Номер патенту: 41946
Опубліковано: 15.10.2001
Автори: БЕРГСТРАНД Понтус Йохн Арвід, ЛЕВГРЕН Курт Інгмар
МПК: A61P 1/04, A61K 31/444, A61K 9/26
Мітки: фармацевтична, вигляді, лікування, одержання, одинична, блістера, інгібування, шлунково-кишкових, секреції, складова, оральна, таблетки, зап, дозована, спосіб, шлункової, кислоти, форма, упаковка
Формула / Реферат:
1. Оральная фармацевтическая составная единичная дозированная форма в виде таблетки, включающая наполнители для таблеток и индивидуально покрытые слоями энтеросолюбильного покрытия единицы материала сердцевины, содержащие активное вещество в виде омепразола или одного из его индивидуальных энантиомеров или щелочной соли омепразола или щелочной соли одного из его индивидуальных энантиомеров, покрытые одним или более слоем(ями), по крайней...
Заміщені похідні 4-бісфеніл-4-гідроксимасляної кислоти як інгібітори матричної металопротеази, спосіб їх одержання, фармацевтична композиція та спосіб лікування на їх основі

Номер патенту: 57047
Опубліковано: 16.06.2003
Автори: Брубейкер Уільям Фредерік, Клуендер Гарольд С.Є., Зад'юра Лайза Марія, Бйорге Сьюзен М.
МПК: A61P 19/02, C07C 323/62, A61P 43/00, A61P 9/00, A61K 31/4035, C07C 235/84, C07D 209/48, A61P 13/00, A61P 29/00, C07C 323/56, C07C 59/00, C07C 323/61, A61P 19/08, C07C 235/42, A61P 1/02, C07C 231/00, C07C 51/347, A61P 27/02, A61P 7/02, A61P 35/04
Мітки: кислоти, лікування, спосіб, композиція, фармацевтична, 4-бісфеніл-4-гідроксимасляної, інгібітори, матричної, металопротеази, основі, одержання, заміщені, похідні
Формула / Реферат:
1. Сполука, яка має інгібуючу активність у відношенні до матричної металопротеази загальної формули,(I)деΤ є фармацевтично придатною замінною групою;х дорівнює 0, 1 або 2;m дорівнює 0 або цілому числу від 1 до 4;n дорівнює 0 або 1; іабоА і G обидва є СН2,абоА є хімічним зв'язком і G є...
N-заміщені азагетероциклічні карбонові кислоти та їх ефіри, спосіб їх одержання та фармацевтична композиція на їх основі (варіанти), спосіб її одержання та спосіб лікування нейрогенного запалення

Номер патенту: 47396
Опубліковано: 15.07.2002
Автори: Петерсен Ханс, Сонневальд Урсула, Йоргенсен Тіне Крогх, Андерсен Кнуд Ерік, Андерсен Хенрік Суне, Ольсен Уффе Банг, Грьонвальд Фредерік Крістіан
МПК: A61K 31/4433, C07D 211/78, A61K 31/445, A61P 11/00, C07D 211/60, A61K 31/00, C07D 403/06, A61P 17/00, C07D 413/06, A61P 43/00, A61P 29/00, A61P 1/00, A61K 31/535, C07D 409/06, A61K 31/4427, A61K 31/54, C07D 417/06, A61P 25/04, A61K 31/55, C07D 401/06
Мітки: n-заміщені, карбонові, лікування, ефіри, композиція, спосіб, основі, запалення, одержання, варіанти, кислоти, азагетероциклічні, фармацевтична, нейрогенного
Формула / Реферат:
1. N-замещенные азагетероциклические карбоновые кислоты и их эфиры формулы (I):, (I)в которой R1 и R2 независимо представляют собой атом водорода, атом галогена, трифторметил, С1 - С6 - алкильная или С1 - С6 - алкоксильная группа, Y - это группы > N-CH2-, > СН- CH2- или > С=СН-, в которых только подчеркнутый атом участвует в циклической системе, Х - это группы -О-, -S-, -CR7R8-, -СН2-СН2-, СН=СН-СН2-, -СН2-СН=СН-,...
Похідні камптотецину (варіанти), спосіб їх одержання (варіанти) та фармацевтична композиція на їх основі
Номер патенту: 49005
Опубліковано: 16.09.2002
Автори: Мерліні Лучо, Пенко Серджо, Цуніно Франко
МПК: A61P 35/00, A61K 31/4738, A61K 31/00, C07D 491/22, A61K 31/4745, A61K 31/47, A61P 43/00
Мітки: одержання, похідні, основі, композиція, фармацевтична, камптотецину, спосіб, варіанти
Формула / Реферат:
l. Похідні камптотецину формули (І) , (I)або їх фармацевтично сприйнятливі солі, в яких:R1 - -CN, -CH(CN)-R4, -CH=C(CN)-R4, -CH2-CH(CN)-R4, -C(=NOH)-NH2, -C(=NH)-NH2, -CH=C(NO2)-R4, -CH(CN)-R5, -CH(CH2NO2)-R5; 5-тетразоліл, 2-(4,5-дигідрооксазоліл), 1,2,4-оксадіазолін-3-іл-5-он;R2 - гідроген;R3 - гідроген, ОR6;R4 - гідроген, лінійний чи розгалужений С1-С6алкіл, -CN, COOR7;R5 - гідроген,...
Похідні дигідроксигексанової кислоти, фармацевтична композиція, що їх містить (варіанти), та спосіб лікування (варіанти)

Номер патенту: 57824
Опубліковано: 15.07.2003
Автори: Кес Джон Чарлз, Посс Крістофер Стенлі, Браун Метью Френк
МПК: A61K 31/498, A61P 43/00, C07C 231/00, A61P 37/06, C07D 215/54, A61P 37/08, A61K 31/47, A61P 9/10, C07D 241/52, A61P 31/06, A61P 31/08, A61P 29/00, A61P 31/12, C07D 409/12, C07D 241/44, A61P 31/18
Мітки: кислоти, композиція, похідні, спосіб, варіанти, фармацевтична, лікування, містить, дигідроксигексанової
Формула / Реферат:
1. Похідні дигідроксигексанової кислоти, які вибирають з групи, що містить:[4(R)-карбамоїл-1(S)-(3-хлорбензил)-2(S),7-дигідрокси-7-метилоктил]амід хіноксалін-2-карбонової кислоти;(1(S)-бензил-4(R)-карбамоїл-2(S),7-дигідрокси-7-метилоктил)амід 7,8-дифторхінолін-3-карбонової кислоти;(1(S)-бензил-4(R)-карбамоїл-2(S),7-дигідрокси-7-метилоктил)амід 6,7,8-трифторхінолін-3-карбонової...
Попередній патент: Композиція на основі диметилдисульфіду з замаскованим запахом
Наступний патент: Спосіб одержання розчину g-глобуліну, призначеного для внутрішньовенного введення, і продукт, що одержується у цей спосіб (варіанти)
Випадковий патент: Електролюмінесцентний екран колективного користування