Амідні проліки гемцитабіну, їх композиції та застосування







Формула / Реферат
1. Сполука формули
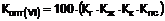 .
.
2. Сполука, яка є змішаним кристалом 1-(2,2-дифтор-2-деокси-b-D-рибофуранозил)-4-(2-пропіл-1-оксопентил)амінопіримідин-2-ону та n-толуолсульфонової кислоти.
3. Сполука за п. 2, яка є змішаним кристалом 1-(2,2-дифтор-2-деокси-b-D-рибофуранозил)-4-(2-пропіл-1-оксопентил)амінопіримідин-2-ону та гідрату n-толуолсульфонової кислоти (2:1:1).
4. Фармацевтична композиція, яка містить сполуку за будь-яким з пп. 1-3 та фармацевтично прийнятний наповнювач.
5. Фармацевтична композиція за п. 4, яка має кишковорозчинне покриття.
6. Спосіб лікування чутливих новоутворень у ссавців, який включає введення в організм ссавця, що потребує такого лікування, терапевтично ефективної кількості сполуки за будь-яким з пп. 1-3.
7. Спосіб за п. 6, де чутливе новоутворення вибране з групи, яку складають лімфома Т-лімфоцитів, саркома м'яких тканин, рак підшлункової залози, рак молочної залози, злоякісний лімфоматоз, незлоякісний лімфоматоз, недрібноклітинний рак легенів, рак яєчників та рак сечового міхура.
8. Застосування сполуки за будь-яким з пп. 1-3 для виготовлення лікарського засобу для лікування чутливих новоутворень.
9. Сполука, вибрана з-поміж сполук за будь-яким з п. 1, п. 2 або п. 3, для застосування як лікарський засіб.
10. Застосування сполуки, вибраної з-поміж сполук за будь-яким з п. 1, п. 2 або п. 3, для виготовлення лікарського засобу для лікування лімфоми Т-лімфоцитів, саркоми м'яких тканин, раку підшлункової залози, раку молочної залози, злоякісного лімфоматозу, незлоякісного лімфоматозу, недрібноклітинного раку легенів, раку яєчників та раку сечового міхура.
Текст
1. Сполука формули 2 86119 1 3 №6,555,518]. На даний час Gemzar® застосовують шляхом внутрішньовенної інфузії у дозі приблизно від 1000мг/м2 до 1250мг/м2 на протязі 30хв один раз на тиждень протягом до 7 тижнів, після чого лікування припиняють на тиждень для відпочинку пацієнта. Застосування гемцитабіну пероральним шляхом може бути обмежене його низькою біодоступністю при пероральному введенні, яка є наслідком метаболізму при першому проходженні. [Дивись Шиплі та ін. - L.A. Shipley et. al., "Метаболізм та розподіл гемцитабіну, онколітичного аналога деоксицитидину, в організмі мишей, пацюків та собак", Drug Metabolism & Disposition. 20(6): 849-855, 1992]. Крім того, при пероральному застосуванні гемцитабіну виникають ускладнення, оскільки він викликає негативні ефекти, що вимагають обмеження дози; вони полягають в ураженнях кишкового тракту, що характеризуються помірною або навіть значною втратою слизового епітелію (атрофічною ентеропатією) по всій довжині кишкового тракту у мишей, які одержували шляхом згодовування одну дозу [167мг/кг, 333мг/кг або 500мг/кг] гемцитабіну [Гортон та ін. - N.D. Horton et.al., «Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs", American Association for Cancer Research, Poster Presentation, Orlando, FL, March 27-31, 2004]. Порівнянні дози при внутрішньовенному введені не викликали смерті або токсичності для шлунково-кишкового тракту у вищезгаданому дослідженні на мишах. Способи виготовлення проліків та композицій для пролонгованого вивільнення гемцитабіну добре відомі в галузі. Приклади таких проліків та композицій з пролонгованим вивільненням можна знайти у заявках WO 04/0412303 "Gemcitabine Prodrugs, Pharmaceutical Compositions and Uses Thereof" на ім'я Геллопа та ін. [Gallop et al.]; WO 98/32762 "Gemcitabine Derivatives" на ім'я Мірена та ін. [Myhren et al.]; WO 02/09768 "Therapeutic polyesters and polyamides" на ім'я Уріх (Uhrich); WO 02/76476 "Terminally-branched polymeric linkers and polymeric conjugates as prodrug" на ім'я Грінуодда та ін. [Greenwald et al.]; WO 02/65988 "Terminallybranched polymeric linkers and polymeric conjugates as prodrug" на ім'я Чоу та ін. [Choe et al.]. Амідні похідні гемцитабіну описано у галузевій літературі як корисні проміжні продукти синтезу гемцитабіну [дивись, наприклад, Бріттон та ін. Britton et al., патент США №5,420,266, та Гріндлі та ін. - Grindley et al., патент США №5,464,826], а також корисні як групи проліків для застосування гемцитабіну [дивись, наприклад, Геллоп та ін. Gallop et al., заявка з WO 04/041203]. Продовжує існувати потреба у проліках гемцитабіну, які уможливлювали б пероральне застосування, проходили б через кишковий тракт без істотного розкладу та забезпечували б постачання гемцитабіну в уражений захворюванням орган при прийнятному рівні безпечності та ефективності. Автори цього винаходу несподівано відкрили нову амідну похідну гемцитабіну, яка проходить через ентероцити практично без пошкодження; гідролізується до гемцитабіну без значного нагро 86119 4 мадження деоксидифторуридину (dFdU), який є переважним метаболітом гемцитабіну в печінці; має при пероральному введенні нижчу токсичність, ніж гемцитабін; та зберігає відповідні профілі ефективності та безпечності при пероральному застосуванні. Цей винахід пропонує сполуку формули І Відповідно до іншого аспекту, цей винахід пропонує нові фармацевтичні композиції, які включають сполуку формули І та один або кілька фармацевтично прийнятних наповнювачів. Цей винахід пропонує також застосування сполуки формули І для лікування чутливих новоутворень у ссавців, які потребують такого лікування. Цей винахід пропонує застосування сполуки формули І для лікування чутливих вірусних інфекцій у ссавців, які потребують такого лікування. Цей винахід пропонує також застосування сполуки формули І для виготовлення лікарських засобів для лікування чутливих новоутворень або вірусних інфекцій. Крім того, цей винахід пропонує також спосіб одержання сполуки формули І. Детальний опис винаходу Терміни, які вживаються у цьому описі, мають вказані нижче значення. Термін "ссавець" вживається для позначення будь-якої з різноманітних теплокровних хребетних живих істот класу Mammalia, найбільш переважно людей, які характеризуються наявністю волосяного покриву на шкірі та наявністю у самок грудних залоз, які продукують молоко для вигодовування дитинчат. Термін "фармацевтично прийнятний наповнювач" означає фармацевтично прийнятний носій, розчинник або домішку, призначені для покращення характеристик фармацевтичної композиції. Такі наповнювачі мають бути сумісними з іншими інгредієнтами композиції та нешкідливими для істоти, в організм якої вводиться композиція, і добре відомі фахівцям у галузі; дивись, наприклад, монографію Ремінгтона [Remingtons Pharmaceutical Sciences, 19th Edition, Mack Publishing Company, 1995].Термін "змішаний кристал" означає фізичну асоціацію двох або кількох молекул, стабільність якої забезпечується нековалентною взаємодією. Один або кілька компонентів цього молекулярного комплексу забезпечують стабільну систему у кристалічних гратах. У деяких випадках домішкові молекули включаються у кристалічні грати у формі дегідратів або сольватів; дивись, наприклад, публікацію Альмарассона та ін. "Crystal Engineering of the Composition of Pharmaceutical Phases. Do Pharmaceutical Co-crystals Represent a New Path to Improved Medicines?" [Almarasson O., et. al., The 5 Royal Society of Chemistry, 1889-1896, 2004]. Змішані кристали, яким віддається перевага, включають n-толуолсульфонову кислоту та бензолфульфонову кислоту. Термін "фармацевтично прийнятний змішаний кристал" означає змішаний кристал, сумісний з іншими інгредієнтами композиції та нешкідливий для істоти, в організм якої вводиться композиція. Термін "чутливе новоутворення" означає аномальний ріст тканини в організмі ссавців, який піддається лікуванню шляхом перорального застосування сполуки формули І. Оскільки ці проліки гідролізуються до гемцитабіну, то очікується, що застосування цих проліків буде характеризуватися широким спектром активності стосовно різноманітних типів як твердих, так і нетвердих пухлин. До чутливих новоутворень переважно належать лімфома Т-лімфоцитів, саркома м'яких тканин, рак підшлункової залози, рак грудної залози, злоякісний лімфоматоз, незлоякісний лімфоматоз, недрібноклітинний рак легенів, рак яєчників та рак сечового міхура. Сполуки за цим винаходом корисні при лікуванні вірусних інфекцій, зокрема, вірусного гепатиту С (HCV). Термін "терапевтично ефективна кількість" означає кількість сполуки або композиції, яка спричиняє бажану біологічну або медичну реакцію тканини, системи або організму ссавця, якої домагається дослідник, медик або клініцист. Гемцитабін містить три функціональні групи, які піддаються дериватизації, а саме 3'- та 5'гідроксильні групи та N4-аміногрупу. Сполуку формули І можна одержати, застосовуючи відповідні групи захисту для блокування 3'- та 5'гідроксильних груп з подальшим ацилюванням N4аміногрупи. Типові групи захисту, добре відомі та загальноприйняті у галузі, описані в монографії Гріні та Вутса "Групи захисту в органічному синтезі" [Protecting Groups in Organic Synthesis. 3rd edition, Theodora Greene, Peter Wuts (WileyInterscience) 1999]. Ацилювання Ж-аміногрупи можна виконати шляхом проведення реакції з хлорангідридом або ангідридом карбонової кислоти або з карбоновою кислотою у присутності агента сполучення, наприклад, Ν,Νдициклогексилкарбодііміду (DCC), N-eтил-N'-(3диметиламінопропіл)-карбодііміду (EDC), 1,1карбоніл-діімідазолу (CDI) або інших аналогічних реагентів, добре відомих фахівцям у галузі органічної хімії. За альтернативним способом, сполуку формули І можна одержати без застосування груп захисту. У такому випадку утворюються суміші моно-, ди- та триадуктів, і бажаний продукт можна виділити з такої суміші. Подані нижче приклади ілюструють синтез сполук за цим винаходом. Усі вихідні матеріали та реагенти добре відомі та прийняті в галузі та є наявними на ринку або можуть бути без утруднень одержані способами, описаними у цій заявці. Спосіб одержання гемцитабіну (2',2'-дифтор-2'деоксицитидину) розкрито, наприклад, у патенті №4,808,614. Приклад 1 86119 6 1-(2,2-дифтор-2-деокси-b-D-рибофуранозил)4-(2-пропіл-1-оксопентил)-амінопіримідин-2-он Розчиняли 2',2'-дифтор-2'-деоксицитидин (10,0г, 38,0ммоль) у безводному піридині (100мл) та охолоджували до 0°С при перемішуванні в атмосфері азоту. Додавали краплями хлортриметилсилан (24,0мл, 190,0ммоль), підтримуючи внутрішню температуру суміші нижче 5°С. Продовжували перемішування при 0°С протягом 2год. В окремій колбі розчиняли 2-пропілпентанову кислоту (6,0г, 41,8ммоль) у безводному ацетонітрилі (100мл). Додавали невеличкими порціями 1,1карбонілдіімідазол (6,8г, 41,8ммоль) протягом 30хв та перемішували протягом 2год. Додавали цей ацетонітрильний розчин краплями до піридинового розчину при 0°С та давали реакційній суміші нагрітися до температури навколишнього середовища. Нагрівали реакційну суміш при 45°С протягом ночі, потім охолоджували до 30-35°С, додавали 100мл абсолютного етанолу та нагрівали при 45°С протягом 30хв. Додавали 50мл води, та нагрівали при 45°С протягом 5год, після чого охолоджували до температури навколишнього середовища та концентрували у вакуумі. Неочищений залишок розподіляли між етилацетатом та водою. Підкислювали до рН~2 фосфорною кислотою, та відділяли органічний шар. Водний шар екстрагували додатковою кількістю етилацетату. Об'єднували органічні розчини, промивали насиченим розчином бікарбонату натрію та насиченим розчином хлориду натрію, сушили над сульфатом магнію та концентрували у вакуумі. Очищали хроматографією на силікагелі (120г) при елююванні градієнтом від 30% до 60% етилацетату у дихлорметані. Виділяли бажаний продукт у вигляді білої крихкої піни (11,2 г, вихід 77%). MS (ES): m/z 390,3=[M+H]+. MS (ES): m/z 388,3=[M-H]+. 1 Я ЯМР (400МГц, DMSO-d6) δ 0,83 (t, 6H), 1,151,36 (m, 6H), 1,46-1,55 (m, 2H), 2,60 (dddd, 1H, J=14,4Гц, 9,6Гц, 5,6Гц, 5,6Гц), (ddd, 1H, J=12,6Гц, 6,2Гц, 3,6Гц), 3,77-3,81 (m, 1H), 3,87 (dt, 1H, J=8,4, 3,0 Гц), 4,12-4,22 (m, 1H), 5,27 (t, 1H, J=5,6 Гц), 6,15 (t, 1H, J=7,4Гц), 6,29 (d, 1H, J=6,4Гц), 7,31 (d, 1H, J=7,2Гц), 8,23 (d, 1H, J=8,0Гц), 11,03 (s, 1H). Приклад 2 Змішаний кристал гідрату 1-(2,2-дифтор-2деокси-b-D-рибофуранозил)-4-(2-пропіл-1оксопентил)амінопіримідин-2-ону з nтолуолсульфоновою кислотою (2:1:1) Розчиняли 0,709г (1,82ммоль) сполуки за Прикладом 1 у 9мл метанолу. В окремій колбі готували вихідний 0,25Μ водний розчин nтолуолсульфонової кислоти. Додавали краплями 3,6мл (0,9ммоль) цього водного розчину. Додавали приблизно 5 мл води, і перемішували суміш при кімнатній температурі (~30хв) до випадання осаду. 7 Осаджену тверду речовину відділяли фільтруванням під вакуумом та сушили на повітрі. Аналітичний аналіз змішаного кристалу: Готували розчини n-толуолсульфонової кислоти, гемцитабіну та сполуки за Прикладом 1 відомих концентрацій. Аналізували пробу змішаних кристалів n-толуолсульфонової кислоти для визначення компонентного складу. Для співвідношення N-(2-пропіл-пентаноїл)]2',2'-дифтор-2'-деокси-цитидин/nтолуолсульфонова кислота/вода 2:1:1 визначали відсоток n-толуолсульфонової кислоти: теоретично 17,8% n-толуолсульфонової кислоти; знайдено 19,1% n-толуолсульфонової кислоти. РХВЕ: Колонка: Waters Atlantis dC18, 3,0мкм, внутрішній діаметр 4,6мм, довжина 150мм. Температура колонки 50°С. УФ-детектор, довжина хвилі UV248 нм. 1) Рухома фаза A. 5/95 ацетонітрил/вода + 0,1% тетрафтороцтової кислоти (TFA) B. 50/50 ацетонітрил/вода + 0,1% TFA 2) Градієнт Час Розчинник (хв) %А %B 0 100 0 5 0 100 8 0 100 8,01 100 0 11 кінець циклу Приклад 3 Змішаний кристал гідрату 1-(2,2-дифтор-2деокси-b-D-рибофуранозил)-4-(2-пропіл-1оксопентил)амінопіримідин-2-ону з бензолсульфоновою кислотою (1:1) До 5мл етилацетату додавали 550мг сполуки за Прикладом 1. Нагрівали суміш приблизно до 55°С. Додавали 1 молярний еквівалент бензолсульфонової кислоти у формі вихідного розчину. Додавали додатково приблизно 10мл етилацетату, в разі потреби при обробленні ультразвуком для подрібнення осаду. Суспензії давали охолодитися до кімнатної температури, та відділяли тверду фазу фільтруванням під вакуумом. Одержаний продукт сушили на повітрі. Т.пл. 171°С. Сполука формули І та її сольвати придатні для перорального застосування та за нормальних умов застосовуються перорально, таким чином, пероральному застосуванню віддається перевага. Фармацевтичні композиції виготовляють способами, добре відомими у фармацевтичній галузі. Носієм або наповнювачем може бути твердий, напівтвердий або рідкий матеріал, який може відігравати роль носія або середовища для активного інгредієнта. Придатні носії або наповнювачі добре відомі в галузі. Фармацевтична композиція може бути пристосована для перорального, інгаляційного, парентерального або місцевого використання та може вводитися в організм пацієнта у формі таблеток, капсул, аерозолів, інгаляційних препаратів, супозиторіїв, розчинів, суспензій тощо. Сполу 86119 8 ки за цим винаходом можна вводити в організм пероральним шляхом, наприклад, спільно з інертним розріджувачем або у капсулах, або у пресованій формі (таблетках). Для цілей терапевтичного застосування згадані сполуки можуть бути введені в наповнювачі та застосовуватися у формах таблеток, пастилок, капсул, еліксирів, суспензій, сиропів, вафель, жувальних гумок тощо. Відповідно до варіанта, якому віддається перевага, ці препарати містять як активний інгредієнт щонайменше 1% сполуки за цим винаходом, але вміст згаданої сполуки може варіювати залежно від конкретної форми та може становити від 1% до приблизно 90% маси одиниці лікарської форми. Кількість сполуки, присутньої в композиції, має бути такою, щоб забезпечити відповідне дозування. Композиції та препарати за цим винаходом, яким слід віддавати перевагу, можна визначити способами, добре відомими фахівцям. Таблетки, пілюлі, капсули, пастилки тощо можуть також містити одну або кілька перелічених нижче допоміжних речовин: в'яжучих, наприклад, повідону, гідроксипропілцелюлози, мікрокристалічної целюлози або желатину; наповнювачів або розріджувачів, наприклад, крохмалю, лактози, мікрокристалічної целюлози або дикальційфосфату; розпушувальних речовин, наприклад, кроскармелози, кросповідону, крохмальгліколяту натрію, кукурудзяного крохмалю тощо; змащувальних агентів, наприклад, стеарату магнію, стеаринової кислоти, тальку або гідрогенізованих рослинних олій; ковзних агентів, наприклад, колоїдного діоксиду кремнію; змочувачів, наприклад, лаурилсульфату натрію та полісорбату-80; та підсолоджувачів, наприклад, сахарози, аспартаму або сахарину, або смакоароматичних домішок, наприклад, м'яти, метилсаліцилату або апельсинового ароматизатора. Якщо формою дозованої одиниці є капсула, то вона може містити, на додаток до матеріалів вищезгаданих типів, рідкий носій, наприклад, поліетиленгліколь або олію. Інші форми дозованих одиниць можуть містити різноманітні інші матеріали, які модифікують фізичну форму дозованої одиниці, наприклад, покриття. Перевага віддається дозованим формам з кишковорозчинним покриттям. Таким чином, таблетки або пілюлі можуть бути покриті оболонками з цукру, гідроксипропілметилцелюлози, поліметакрилатів або інших покривних речовин. Сиропи можуть містити, окрім активних сполук, сахарозу як підсолоджувач та певні консерванти, барвники та пігменти, а також ароматизатори. Матеріали, що застосовуються при виготовленні цих різноманітних композицій, мають бути фармацевтично чистими та нетоксичними у застосовуваних кількостях. Сполуки формули І, як правило, ефективні у широкому діапазоні дозування. Наприклад, добові дози (у формі однієї дози або кількох часткових доз) за нормальних умов лежать у межах від приблизно 15мг/добу до приблизно 200мг/добу, більша перевага віддається дозам приблизно 85мг/добу. У деяких випадках більш ніж адекватними можуть бути рівні дозування, нижчі від нижньої межі вищезазначеного діапазону, тоді як в 9 інших випадках без будь-яких шкідливих побічних ефектів можуть застосовуватися ще більші дози, і, отже, вищезазначений діапазон дозування не призначений для будь-якого обмеження обсягу цього винаходу. Мається на увазі, що реальну застосовувану кількість сполуки визначає лікар з урахуванням релевантних обставин, у тому числі стану, що підлягає лікуванню, обраного шляху введення, конкретної застосовуваної сполуки чи сполук, віку, маси тіла та індивідуальної реакції пацієнта та тяжкості симптомів захворювання у пацієнта. Випробування хімічної стабільності при різних значеннях рН Хімічну стабільність при різних значеннях рН випробовували із застосуванням напівавтоматичної РХВЕ. Виготовляли проби сполуки формули І у концентрації 100мкг/мл у п'яти буферних розчинах, які відповідали діапазону рН у шлунковокишковому тракті (рН1-рН8). Проби вміщували в автоматичний пристрій введення проб для РХВЕ, в якому підтримували температуру 40°С. Проби повторно вводили у колонку для РХВЕ через годинні інтервали на протязі періоду до 24год, використовуючи колонку для РХВЕ, яка забезпечувала відділення сполуки формули І від гемцитабіну. Для визначення стабільності контролювали зміни площі піка сполуки формули І у часі за допомогою УФ детектора та зіставляли одержані значення з початковою площею піка. Через 4год у діапазоні рН1-8 розкладалося з утворенням гемцитабіну менше ніж 25% сполуки за Прикладом 1. Фармакокінетичні випробування Фармакокінетика в організмі мишей Фармакокінетичні профілі гемцитабіну та сполуки формули І визначали з використанням самців мишей лінії CD-1 після перорального введення у дозах, які відповідали приблизно 10мг/кг гемцитабіну. Піддослідних тварин (n=3 для кожного значення часу, дози та сполуки) умертвляли через 0,08год, 0,25год, 0,5год, 1год, 2год та 6год після введення дози та відбирали системні проби крові в оброблені EDTA пробірки, які містили тетрагідроуридин (кінцева концентрація у крові 0,5мМ) для запобігання подальшого метаболізму гемцитабіну. У момент часу 0,08год умертвляли додатково 3 тварини для збирання крові на вході печінки. Плазму відділяли центрифугуванням та зберігали до аналізу в замороженому вигляді. Концентрації гемцитабіну та проліків у плазмі визначали комбінованим методом рідинної хроматографії та масспектрометрії (LC/MS/MS). Фармакокінетичні параметри обчислювали, застосовуючи програмне забезпечення WinNonlin [Pharsight Corp., Mountain View, CA]. Фармакокінетичні параметри кожного виду проліків зіставляли з параметрами, визначеними для випадку перорального введення гідрохлориду гемцитабіну із застосуванням аналогічної схеми експерименту. Сполука за Прикладом 1, введена перорально мишам лінії CD-1, екстенсивно гідролізувалася in vivo з вивільненням гемцитабіну. Кількість гемцитабіну у плазмі мишей лінії CD-1 при введенні сполуки за Прикладом 1 збільшувалася у порівнянні з випадком безпосереднього перорального введен 86119 10 ня гідрохлориду гемцитабіну. Засвоєння неушкоджених проліків підтверджується відносно високими концентраціями сполуки за Прикладом 1 у плазмі крові, взятої на вході печінки через 0,08год після перорального введення. Фармакокінетичне випробування на мавпах Визначали фармакокінетичні профілі гемцитабіну та сполуки формули І в організмі мавп виду Cynomolgus після перорального та внутрішньовенного введення за перехресною схемою експерименту. Сполуки вводили у дозах, які відповідали приблизно 10мг/кг гемцитабіну. Відбирали проби крові в оброблені EDTA пробірки, які містили тетрагідроуридин (кінцева концентрація у крові 0,5мМ) у визначені моменти часу на протязі інтервалу до 48год. У періоди перорального введення тварин попередньо обробляли ранітидином (внутрішньовенно, 5мг/кг). Плазму відділяли центрифугуванням та зберігали до аналізу в замороженому вигляді. Концентрації гемцитабіну та сполуки формули І у плазмі визначали комбінованим методом LC/MS/MS. Фармакокінетичні параметри обчислювали, застосовуючи програмне забезпечення WinNonlin [Pharsight Corp., Mountain View, CA]. Сполука за Прикладом 1, введена внутрішньовенно мавпам виду Cynomolgus, екстенсивно гідролізувалася in vivo з вивільненням гемцитабіну. Кількість гемцитабіну в організмі мавп виду Cynomolgus при пероральному введенні Сполуки за Прикладом 1 збільшувалася приблизно у 5 разів у порівнянні з випадком безпосереднього перорального введення гідрохлориду гемцитабіну. Випробування гідролізу Проба на гомогенаті тонкої кишки Для визначення стійкості сполуки формули І до ферментного гідролізу у кишечнику виготовляли неочищені гомогенати клітин епітелію тонкої кишки з відрізків верхнього відділу тонкої кишки мишей лінії CD-I, собак-гончаків, мавп виду Cynomolgus та людини. Гомогенати з організмів мишей та собак виготовляли зі свіжовідібраних тканин, а гомогенати з організмів мавпи та людини - з попередньо заморожених тканин. Клітини обережно зіскрібали з відрізків кишок, нагромаждували та гомогенізували у 50мМ ацетатному буфері, застосовуючи прилад Polytron (PT-10-85). Концентрації протеїнів визначали стандартним спектрофотометричним способом. Виготовлені гомогенати зберігали до використання при -70°С. Швидкість гідролізу сполуки формули І у гомогенатах тонкої кишки (SIH) визначали шляхом інкубування сполуки формули І (100мкМ) з SIH (загальна концентрація протеїнів 2,5-5мг/мл) в ацетатному буфері при рН 7,5 протягом часу до 6год. Концентрації гемцитабіну, вивільненого внаслідок гідролізу, визначали методом LC/MS після гасіння реакції ацетонітрилом. Швидкість гідролізу обчислювали для моменту часу 30хв при скринінгових експериментах та з нахилу лінійної частини кривих часової залежності ступеню гідролізу при дослідженні характеристик сполуки формули І. Сполука за Прикладом 1 у пробі на гомогенаті тонкої кишки виявила низьку швидкість гідролізу. Найповільніший гідроліз виявлено у гомогенатах мавп та людини, де після 6год інкубування у гем 11 цитабін перетворювалося менше ніж 3% загальної кількості сполуки. Проба на гідроліз у фракції S9 печінки Гідроліз сполуки формули І ферментами печінки визначали у випробуванні на гідроліз у печінці. Виготовляли гомогенати печінки мишей лінії CD-1, собак-гончаків, мавп виду Cynomolgus та людини. Печінкову тканину різали на дрібні шматки за допомогою ножиць, після чого гомогенізували у 50мМ ацетатному буфері протягом 1хв, застосовуючи прилад Polytron (РТ-10-85). З кожної проби виготовляли пост-мітохондріальні фракції (S9) шляхом ультрацентрифугування при 9000g при температурі 4°С протягом 10хв. Фракції S9 печінки мишей, собак та мавп виготовляли зі свіжовідібраних тканин, а фракції S9 людської печінки - з попередньо замороженої тканини. Після центрифугування збирали надосадову рідину та визначали концентрації протеїнів стандартним спектрофотометричним способом. Виготовлені фракції S9 зберігали до використання при -70°С. Швидкість гідролізу сполуки формули І у фракціях S9 печінки визначали шляхом інкубування сполуки формули І (10мкМ) з S9 (загальна концентрація протеїнів 2мг/мл) у сольовому розчині із фосфатним буфером при рН8,0 протягом часу до 6год. Концентрації гемцитабіну, вивільненого внаслідок гідролізу, визначали методом LC/MS після гасіння реакції ацетонітрилом. Швидкість гідролізу обчислювали для моменту часу 30хв при скринінгових експериментах та з нахилу лінійної частини кривих часової залежності ступеня гідролізу при дослідженні характеристик сполуки формули І. Сполука за Прикладом 1 гідролізувалася у фракціях S9 усіх вищезазначених організмів. Найшвидший гідроліз виявлено у гомогенатах мавп та людини, де після 6год інкубування у гемцитабін перетворювалося понад 35% загальної кількості сполуки. Токсикологічні випробування Чотиридобовий скринінг на мишах Для визначення токсичності, що спричиняє сполука формули І при щоденному пероральному введенні самкам мишей лінії CD-a на протязі 4 днів, профілі шлунково-кишкової токсичності сполуки формули І зіставляли з попередніми результатами дослідження шлунково-кишкової токсичності при пероральному введенні гемцитабіну у дозі 8мг/кг мишам на протязі 4 днів. Самкам мишей лінії CD-1 у віці 5-8 тижнів вводили шляхом згодовування дози сполуки формули І. Рівень дози було добрано так, що він відповідав за молярним еквівалентом 8мг/кг гемцитабіну. Застосовували об'єм дози 10мл/кг, і дози вводили один раз на день протягом 4 послідовних днів. Приблизно через 5-8год після четвертої дози виконували аутопсію. Оцінювали клінічні прояви, клінічну хімію, загальну патологію та обмежену гістопатологію (клубова кишка, порожня кишка та печінка). Після введення мишам сполуки за Прикладом 1 в еквівалентній дозі спостерігалося значне зниження тяжкості атрофічних змін або ентеропатії у порівнянні з гідрохлоридом гемцитабіну. 14-добове дослідження на мишах 86119 12 14-добове дослідження на мишах виконували з метою визначення наявності або відсутності негативного впливу сполуки формули І на організм мишей після 14 днів перорального введення (згодовування) та визначення концентрацій сполуки формули І та її метаболітів - гідрохлориду гемцитабіну та деоксидифторуридину - у плазмі після 1 дози або 14 доз. Самцям та самкам мишей лінії CD-1 у віці 9-12 тижнів вводили шляхом згодовування дози сполуки формули І. Діапазон доз вибирали з розрахунком на визначення максимальної переносної дози та токсичності, що обмежує дозу. Застосовували об'єм дози 10мл/кг, і дози вводили один раз на день. Оцінювали клінічні прояви, масу тіла, споживання корму, гематологію, клінічну хімію, концентрації сполуки формули І та її метаболітів - гідрохлориду гемцитабіну та деоксидифторуридину - у плазмі та патологію (у тому числі загальну патологію, масу органів та гістопатологію). Сполука за Прикладом 1 при введенні в молярно еквівалентних дозах спричиняє меншу ентеропатію, ніж гідрохлорид гемцитабіну, при приблизно подвоєному системному впливі у порівнянні з гідрохлоридом гемцитабіну. 7-добове дослідження на собаках 7-добове дослідження на собаках виконували з метою визначення профілю токсичності сполуки формули І при введенні собакам-гончакам протягом 7 днів та визначення концентрацій сполуки формули І та її метаболітів - гідрохлориду гемцитабіну та деоксидифторуридину - у плазмі після 1 дози або 7 доз. Самцям та самкам собак-гончаків у віці 6-48 місяців вводили перорально сполуку формули І за допомогою капсул. Діапазон доз вибирали з розрахунком на визначення максимальної переносної дози та токсичності, що обмежує дозу. Застосовували об'єм дози 1 мл/кг, і дози вводили один раз на день. Оцінювали клінічні прояви, масу тіла, споживання корму, температуру тіла, гематологію (у тому числі коагуляцію), клінічну хімію, дослідження сечі, концентрації сполуки формули І та її метаболітів - гідрохлориду гемцитабіну та деоксидифторуридину - у плазмі та патологію (у тому числі загальну патологію, масу органів та гістопатологію). Гемотоксичність та інші види токсичності, у тому числі gl-токсичність, відповідали характеристикам, описаним раніше для парентерального застосування гемцитабіну. Таким чином, жодний із цих проявів токсичності не є специфічним для перорального введення. Випробування in vivo Клітини пухлини товстої кишки НСТ-116 вирощували in vitro у стандартних умовах культивування тканини, збирали, промивали, та 5×106 клітин [суспензія 1:1 у середовищі Matrigel, Collaborative Biomedical Products, Inc.] вводили підшкірно у задню частину боку самок безшерстих мишей (одержаних від фірми Charles River, лінія CD-1 nu/nu, маса тіла 24-27г, опромінені дозою 450 рад в межах інтервалу 24год після імплантації). Перед початком лікування пухлинам давали вирости до 13 86119 об'єму приблизно 100мм3. Мишам згодовували (в об'ємі 10мл/кг) носій (контроль), сполуку формули І або гідрохлорид гемцитабіну у різних дозах у моменти часу, вказані в описі індивідуальних експериментів. Сполуки вводили або щоденно протягом 14 днів, або через день при загальній кількості 7 доз, або кожного третього дня при загальній кількості 4 дози. Для режиму щоденного дозування сполуку формули І розчиняли у 100мМ фосфатнонатрієвому буфера, рН6,0, а для режимів дозування через день та через два дні вводили до складу композиції, що містила 1% натрієвої карбоксиметилцелюлози, 0,5% лаурилсульфату натрію, 0,05% протиспінювача Antifoam 150 та 0,085% повідону. Гідрохлорид гемцитабіну для перорального введення розчиняли у фізіологічному сольовому розчині. Розміри пухлини вимірювали за допомогою Комп’ютерна верстка А. Рябко 14 кронциркуля, і об'єм пухлини (мм3) визначали за формулою l×w2×0,536, де l - більший, a w - менший із двох взаємно перпендикулярних розмірів. Усі дані (розміри пухлин та маса тіла тварин) визначали двічі на тиждень, починаючи з початку лікування, та аналізували за допомогою комп'ютеризованої системи вимірювання пухлин. Протипухлинна ефективність, що спостерігалася при застосуванні сполуки за Прикладом 1, була порівнянною з ефективністю, що досягалася при застосуванні еквівалентних доз гідрохлориду гемцитабіну. Однак лікування сполукою за Прикладом 1 спричиняло нижчу загальну токсичність у порівнянні з показниками для тварин, які одержували еквівалентну кількість гідрохлориду гемцитабіну. Підписне Тираж 28 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюAmide prodrug of gemcitabine, compositions and use thereof
Автори англійськоюBender David Michael, Remick David Michael
Назва патенту російськоюАмидные пролекарства гемцитабина, их композиции и применение
Автори російськоюБендер Девид Майкл, Ремик Дейвид Майкл
МПК / Мітки
МПК: C07H 19/073, A61P 35/00
Мітки: композиції, гемцитабіну, застосування, проліки, амідні
Код посилання
<a href="https://ua.patents.su/7-86119-amidni-proliki-gemcitabinu-kh-kompozici-ta-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">Амідні проліки гемцитабіну, їх композиції та застосування</a>
Похідні гемцитабіну, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 67736
Опубліковано: 15.07.2004
Автори: Мюрен Фінн, Саннволл Маріт Ліланн, Берретсен Бернт, ДАЛЕН Аре
МПК: C07H 19/06
Мітки: основі, одержання, спосіб, похідні, композиція, гемцитабіну, фармацевтична
Формула / Реферат:
1. Похідна гемцитабіну, яка має формулу (І):,де R1, R2 і R3 незалежно вибирають із водню і С18- і С20 - насичених і мононенасичених ацильних груп, за умови, що R1, R2 і R3 - усі не можуть бути воднем.2. Сполука згідно з пунктом 1, в якій тільки один із R1, R2 і R3 являє собою ацильну групу.3. Сполука згідно з пунктом 2, в якій моноацильний замісник знаходиться в 3'-О або 5'-O положенні цукрового фрагмента.4....
Модифікований циклоспорин, який можна використовувати як проліки, і його застосування

Номер патенту: 75136
Опубліковано: 15.03.2006
Автори: Амель Арно, Муттер Манфред, Венгер Роланд, Юблер Франсіс
МПК: A61P 31/18, A61K 38/00, A61P 43/00, A61P 27/02, C07K 7/64
Мітки: застосування, модифікований, проліки, можна, використовувати, циклоспорин
Формула / Реферат:
1. Проліки, що являють собою циклічний ундекапептид, в якому пептидний ланцюг включає щонайменше один амінокислотний залишок загальної формули (І):, (І)в якій:- атом вуглецю Сa утворює один із зв'язків в кільці ундекапептиду;- кожний із замісників Y являє собою атом водню, або разом вони утворюють зв'язок;- замісники R1 і R3, незалежно...
Застосування бактерійної комбінованої композиції для отримання дієтичної або фармацевтичної композиції для профілактики або лікування гіпероксалурії

Номер патенту: 71621
Опубліковано: 15.12.2004
Автор: де Сімоне Клаудіо
МПК: A61K 31/661, A61K 38/22, C12N 1/20, A61K 9/14, A61K 35/02, A23L 1/03, A61K 35/74, A61P 3/00, A61P 13/12, A61P 13/02, A61K 38/43, A61K 9/02, A23L 1/30, A61K 31/4415, A61K 31/519, A61K 9/20, A61K 31/375, A01K 13/00, A61K 9/48, A61P 13/04, A61P 13/10, A61K 33/06, A61K 31/785, A61K 45/00
Мітки: комбінованої, композиції, профілактики, лікування, фармацевтично, гіпероксалурії, отримання, бактерійної, застосування, дієтичної
Формула / Реферат:
1. Застосування бактерійної комбінованої композиції, що включає штами бактерій(a) Streptococcus thermophilus в суміші з(b) щонайменше одним штамом бактерій, вибраним з групи, що складається з Lactobacillus brevis, Lactobacillus acidophilus, Lactobacillus plantarum, Bifidobacterium infantis, Bifidobacterium longum і Bifidobacterium breve або їх сумішей,для отримання дієтичної, фармацевтичної або ветеринарної композиції для...
Застосування (-) (3-тригалометилфенокси)(4-галофеніл) похідних оцтової кислоти для лікування інсулінорезистентності, діабету другого типу та фармацевтичні композиції

Номер патенту: 74147
Опубліковано: 15.11.2005
Автори: Ласкі Кенет Л., Лу Джіан
МПК: A61P 3/10, A61K 38/28, A61P 9/10, A61K 45/00, A61K 31/64, A61P 27/02, A61K 31/455, A61P 19/06, A61P 25/02, A61P 15/00, A61K 31/216, A61P 3/04, A61P 3/06, A61K 31/215, A61P 5/48, A61K 45/06, A61K 31/425, A61P 13/12, A61P 9/00
Мітки: кислоти, похідних, типу, композиції, фармацевтичні, діабету, оцтової, інсулінорезистентності, застосування, лікування, другого, 3-тригалометилфенокси)(4-галофеніл
Формула / Реферат:
1. Застосування (-)стереоізомера сполуки формули І , (І)де:R вибраний з групи, яка складається з гідрокси, нижчого аралкокси, динижчого алкіламінонижчого алкокси, нижчого алканамідо нижчого алкокси, бензамідонижчого алкокси, уреїдонижчого алкокси, N’нижчого алкілуреїдонижчого алкокси, карбамоїлнижчого алкокси, галофеноксизаміщеного нижчого...
Спосіб одержання гідрохлориду гемцитабіну

Номер патенту: 48152
Опубліковано: 15.08.2002
Автор: БЕРГЛУНД Ричард Алан
МПК: C07H 19/06
Мітки: одержання, гемцитабіну, спосіб, гідрохлориду
Формула / Реферат:
1. Спосіб одержання гідрохлориду гемцитабіну, який включає:a) відщеплення захисних груп від -1-(2'-деокси-2',2'-дифтор-3',5'-ди-О-бензоїл-D-рибофуранозил)-4-амінопіримідин-2-ону каталітичною кількістю алкіламіну у присутності метанолу або етанолу у середовищі, практично вільному від води;b)...
Попередній патент: Пристрій для дистанційної прокладки гнучкого трубопроводу
Наступний патент: Пристрій для фальцювання розгорток картонного паковання
Випадковий патент: Екстракт з рослини scabiosa arvensis з педикуліцидною дією