Хімічні сполуки
Номер патенту: 110338
Опубліковано: 25.12.2015
Автори: Аквіно Крістофер Джозеф, Ву Юлінь, Кован Дейвід Джон, Коллінз Джон Лорен




































































Формула / Реферат
1. Сполука формули:

або її сіль, або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, якою є:
 .
.
3. Сполука за п. 2, де вказана сполука є кристалічною.
4. Сполука або сіль, як визначено в будь-якому з пп. 1-3, для застосування в лікуванні розладу обміну речовин у людини.
5. Фармацевтична композиція, що містить сполуку або сіль за будь-яким з попередніх пп. 1-4.
6. Спосіб лікування або попередження розладів обміну речовин, за яким вводять сполуку або сіль за будь-яким з попередніх пунктів.
7. Спосіб за п. 6, де зазначеним розладом обміну речовин є цукровий діабет (типу І та типу II) або ожиріння.
8. Застосування фармацевтичної композиції за п. 5 для лікування розладів обміну речовин у людини.
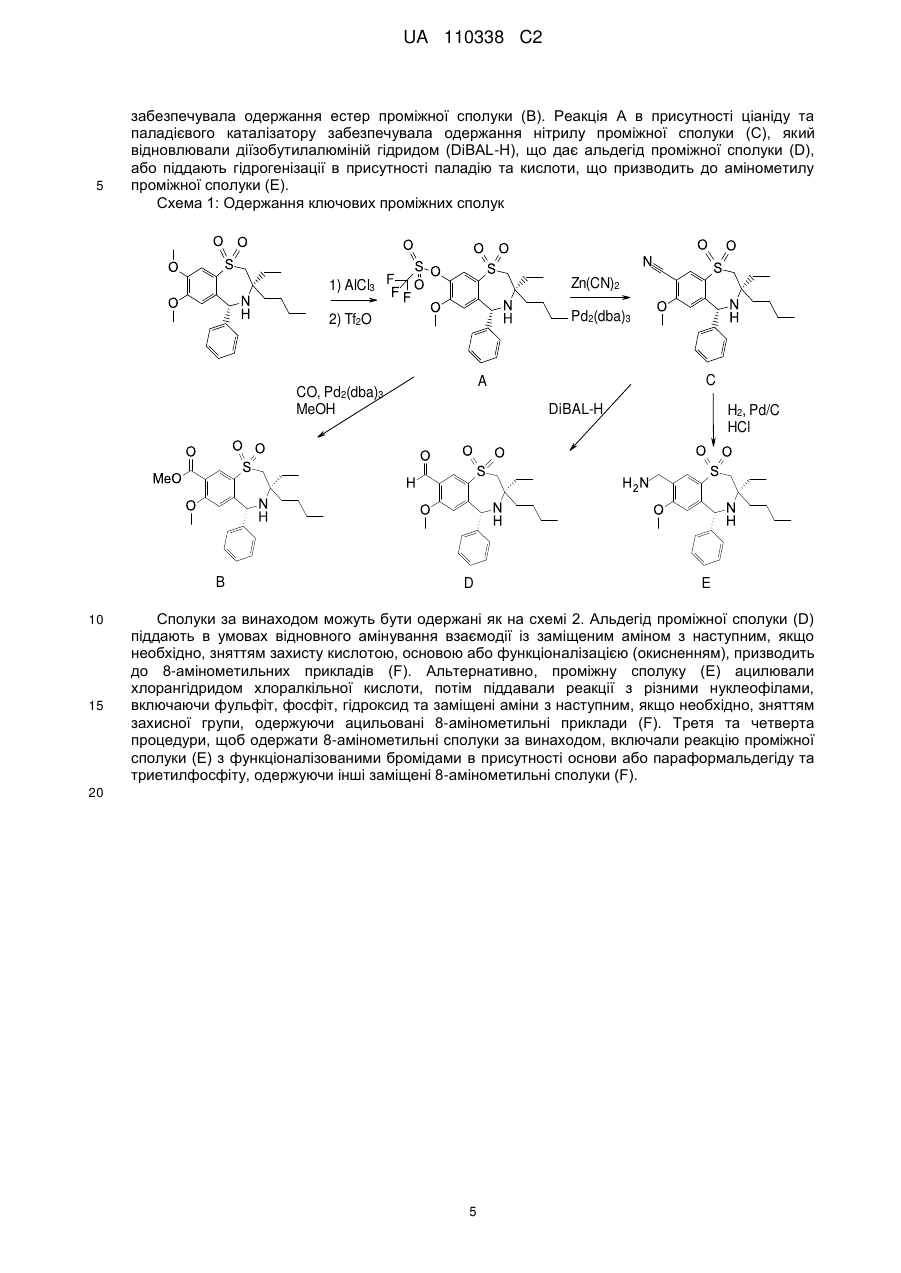
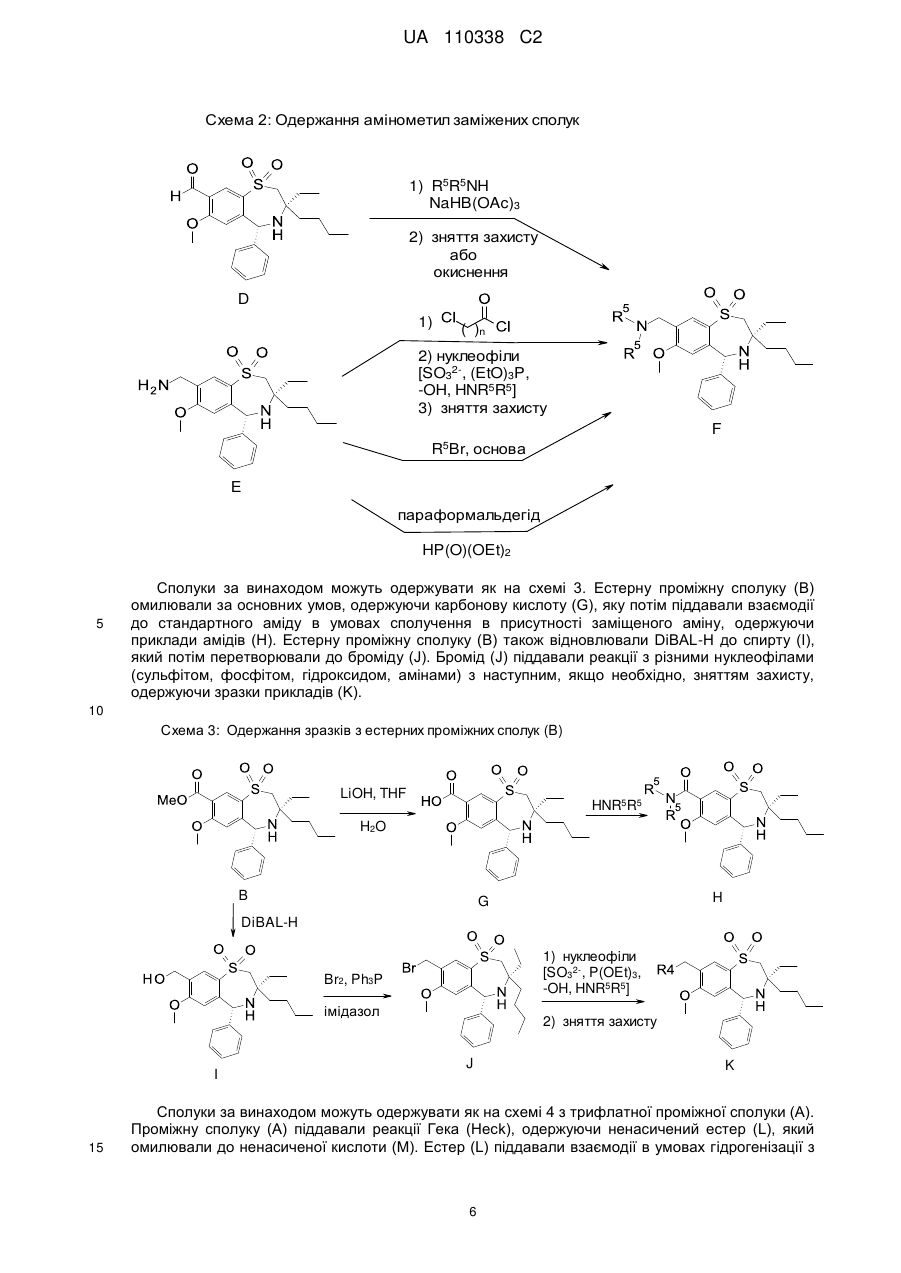
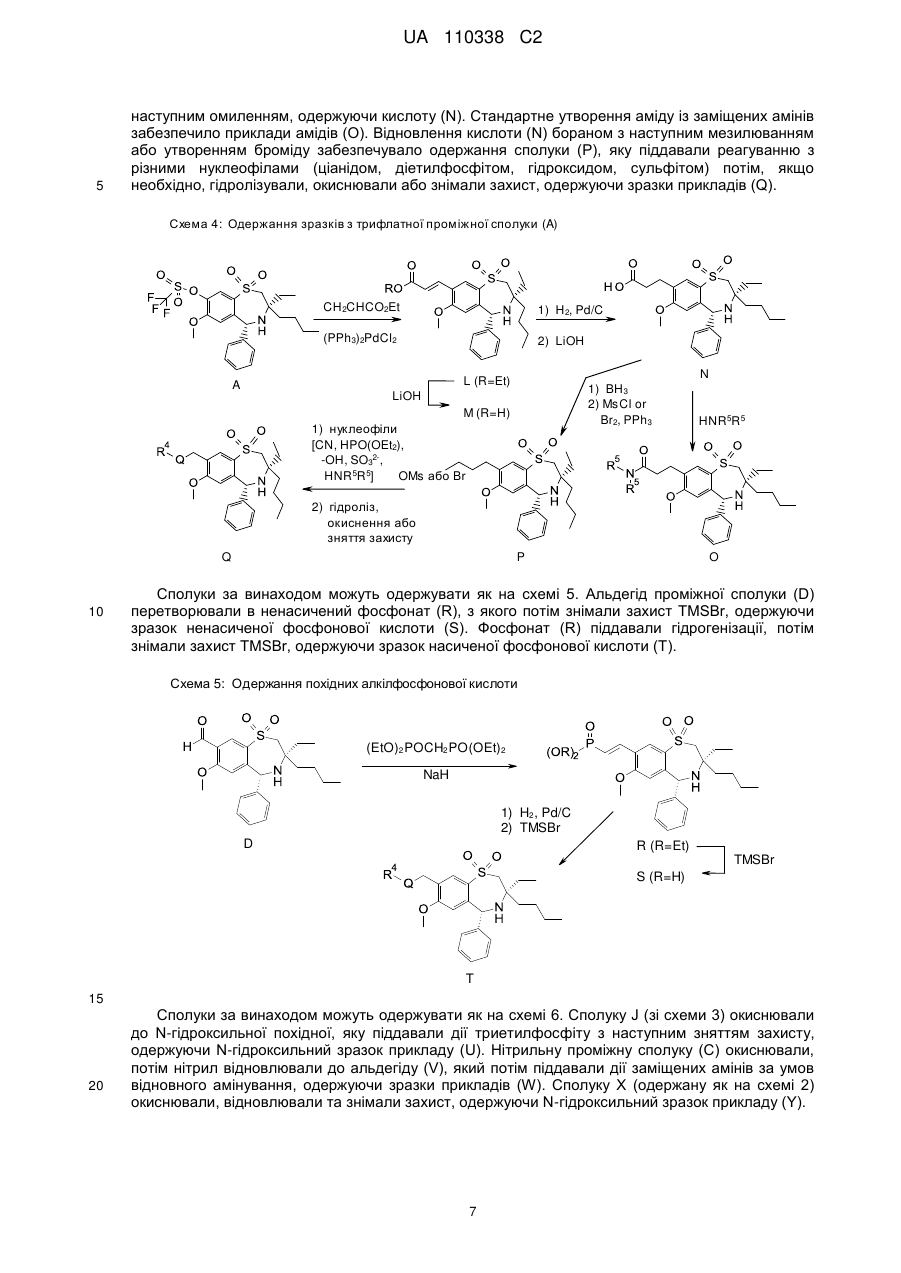
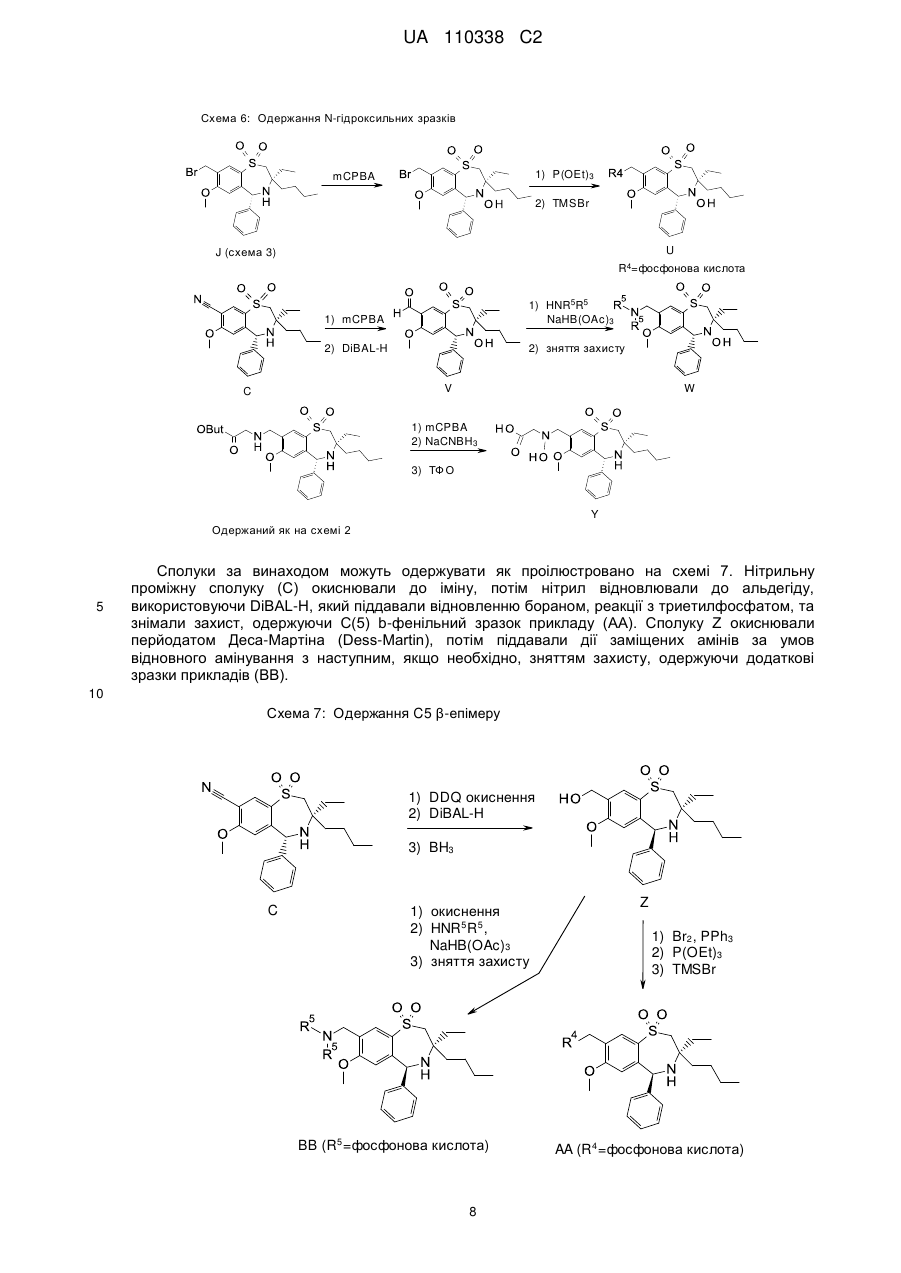
Текст
Реферат: Винахід стосується сполуки формули І та способів лікування розладів. O R 4 Q O S X R 3 R R 1 3 N R 2 I UA 110338 C2 (12) UA 110338 C2 UA 110338 C2 5 10 15 20 25 30 35 40 45 50 55 60 Галузь винаходу Представлений винахід стосується сполук, які є корисними для лікування та попередження розладів обміну речовин, включаючи цукровий діабет (тип I та тип II), ожиріння та пов'язаних з ним розладів, а також способів одержання сполук, фармацевтичних композицій, що містять ці сполуки, та терапевтичних застосувань таких сполук. Передумови створення винаходу Більше 200 мільйонів людей в усьому світі хворі на діабет. Всесвітня організація охорони здоров'я вважає, що 1,1 мільйона людей померло від діабету в 2005 році та прогнозує, що в усьому світі кількість випадків смерті від діабету подвоїться в період між 2005 і 2030 роками. Нові хімічні сполуки, які ефективно лікують діабет, могли б врятувати мільйони людських життів. Діабет відноситься до розладів обміну речовин, що виникають в результаті нездатності організму ефективно регулювати рівень глюкози. Приблизно 90 % всіх випадків діабету є результатом цукрового діабету 2 типу, тоді як решта 10 % є результатом цукрового діабету 1 типу, гестаційного діабету та латентного аутоімунного діабету зрілого віку (LADA). Всі форми цукрового діабету в результаті призводять до підвищеного рівня глюкози в крові і, якщо їх постійно не лікувати, то можуть збільшити ризик розвитку макросудинних (хвороба серця, інсульт, інші форми серцево-судинних захворювань) та мікросудинних [ниркова недостатность (нефропатія), сліпота від діабетичної ретинопатії, ушкодження нервів (діабетична нейропатія)] ускладнень. Цукровий діабет 1 типу, також відомий як ювенільний або інсулінозалежний цукровий діабет (IDDM), може виникнути в будь-якому віці, але найчастіше діагностується у дітей, підлітків або молодих дорослих. Цукровий діабет 1 типу викликається аутоімунним руйнуванням інсулінпродукуючих бета-клітин, що в результаті призводить до нездатності виробляти достатню кількість інсуліну. Інсулін контролює рівень глюкози в крові, сприяючи транспорту глюкози з крові в клітини для використання енергії. Недостатнє виробництво інсуліну буде призводити до зниження рівню глюкози в клітини і в результаті до накопичення глюкози в кров'яному потоці. Відсутність доступної глюкози в клітинах, в кінцевому результаті призведе до виникнення симптомів цукрового діабету 1 типу: поліурії (часті позиви до сечовипускання), полідипсії (спрага), постійного голоду, втрати ваги, зміни зору та втоми. Протягом 5-10 років з діагнозом цукровий діабет 1 типу інсулінпродукуючі бета-клітини пацієнта підшлункової залози повністю руйнуються та організм вже більше не може виробляти інсулін. В результаті, пацієнтам з цукровим діабетом 1 типу необхідно щоденно вводити інсулін протягом всього залишку свого життя. Цукровий діабет 2 типу, також відомий як неінсулінзалежний цукровий діабет (NIDDM) або діабет зрілого віку, виникає, коли підшлункова залоза виробляє недостатньо інсуліну та/або тканини стають стійкими до нормальних або високих рівнів інсуліну (резистентність до інсуліну), що в результаті призводить до надмірно високого рівню глюкози в крові. Безліч чинників може призвести до резистентності до інсуліну, в тому числі хронічно підвищений рівень глюкози в крові, генетика, ожиріння, відсутність фізичної активності, а також збільшення віку. На відміну від діабету 1 типу, симптоми цукрового діабету 2 типу є більш актуальними, та, як наслідок, хвороба може бути недіагностованою до кількох років після початку захворювання, з піком поширеності серед дорослих близько 45 років. На жаль, захворюваність на цукровий діабет 2 типу серед дітей збільшується. Першочерговою метою лікування цукрового діабету 2 типу є досягнення та підтримка контролю глікемії, щоб зменшити ризик мікросудинних (діабетична нейропатія, ретинопатія або нефропатія) та макросудинних (хвороба серця, інсульт, інші форми серцево-судинних захворювань) ускладнень. Нинішні керівні принципи для лікування цукрового діабету 2 типу від Американської Діабетичної Асоціації (ADA) та Європейської асоціації з вивчення діабету (EASD) [Diabetes Care, 2008, 31 (12), 1] - план змін способу життя, включаючи втрату ваги та збільшення фізичної активності як основний терапевтичний підхід для контролю діабету 2 типу. Однак, оскільки один цей підхід зазнає невдачі в більшості пацієнтів протягом першого року, провідні лікарі призначають препарати з плином часу. ADA й EASD рекомендують метформін, агент, який зменшує виробництво глюкози в печінці, як препарат рівню 1а, однак, у значного числа пацієнтів, що приймають метформін може виникнути шлунково-кишкові побічні ефекти та, в окремих випадках, потенційно смертельний молочнокислий ацидоз. Рекомендації щодо рівня 1b классу ліків включають сульфонілсечовини, які стимулюють секрецію інсуліну підшлункової залози за допомогою модуляції активності калієвого каналу, і екзогенний інсулін. У той час як препарати швидко й ефективно знижують рівень глюкози в крові, потрібно 1-4 ін'єкції інсуліну в день, та обидва агенти можуть призвести до небажаного збільшення ваги та потенційно смертельної гіпоглікемії. Рівень 2а рекомендацій включає новіші засоби, такі як тіазолідиндіони 1 UA 110338 C2 5 10 15 20 25 30 35 40 45 50 55 60 (TZD піоглітазон та розиглітазон), які підвищують чутливість до інсуліну м'язів, печінки та жирової тканини, а також GLP-1 аналоги, які підвищують постпрандіальну глюкозопосередковану секрецію інсуліну бета-клітинами підшлункової залози. У той час як TZD показали надійний та міцний контроль рівня глюкози в крові, побічні ефекти включають збільшення ваги, набряки, переломи кісток у жінок, загострення серцевої недостатності та потенційне збільшення ризику випадків серцево-судинної ішемії. GLP-1 аналоги також ефективно контролюють рівень глюкози в крові, проте, цей клас препаратів вимагає ін'єкційного введення та багато пацієнтів скаржаться на нудоту. Найостанніше доповнення до списку препаратів рівню 2 є DPP-4 інгібітори, які, як і GLP-1 аналоги, підвищують глюкозопосередковану секрецію інсуліну з бета-клітин. На жаль, DPP-4 інгібітори лише в незначній мірі контролюють рівень глюкози в крові, а також довгострокову безпеку DPP-4 інгібіторів ще належить твердо встановити. Інші, менш визначені ліки для діабету 2 типу включають інгібітори α-глюкозидази, меглітиніди та аналоги аміліну. Очевидно, що нові препарати з покращеною ефективністю, довговічністю та профілем побічних ефектів, необхідні для пацієнтів з цукровим діабетом 2 типу. GLP-1 і GIP є пептидами, відомими як інкретіни, які виділяються L і К-клітинами, відповідно, із шлунково-кишкового тракту в кров після прийому поживних речовин. Дана важлива фізіологічна реакція служить основним сигнальним механізмом між концентрацією поживних речовин (глюкози/жирів) в шлунково-кишковому тракті та інших периферійних органах. Після секреції, обидва пептиди, що циркулюють, ініціюють сигнали в бета-клітинах підшлункової залози для підвищення глюкозостимульованої секреції інсуліну, яка, у свою чергу, контролює концентрацію глюкози в крові (дивись огляди: Diabetic Medicine 2007, 24(3), 223; Molecular and Cellular Endocrinology 2009, 297(1-2), 127; Experimental and Clinical Endocrinology & Diabetes 2001, 109(Suppl. 2), S288). Зв'язок між інкретіновими гормонами GLP-1 і GIP та цукровим діабетом 2 типу був широко вивчений. Більшість досліджень показують, що цукровий діабет 2 типу є пов'язаним з набутим дефектом GLP-1 секреції, а також активністю GIP (дивись Diabetes 2007, 56(8), 1951 та Current Diabetes Reports 2006, 6(3), 194). Застосування екзогенного GLP-1 для лікування пацієнтів з цукровим діабетом 2 типу суворо обмежена у зв'язку з його швидким розкладанням протеазою DPP-4. Багаторазово модифіковані пептиди були розроблені як GLP-1 міметики, які є стійкими до DPP-4 та демонструють більш тривалий період напіврозпаду, ніж ендогенний GLP-1. Як було показано, агенти з даним профілем є досить ефективними для лікування цукрового діабету 2 типу та включають ексенатід та ліраглутид, однак, дані агенти потребують ін'єкційного ведення. Пероральні агенти, які інгібують DPP-4, такі, як сітагліптін, вілдагліптін та саксагліптін, піднімають рівень інтактного GLP-1 та стримано контролюють рівні циркулюючої глюкози (дивись Pharmacology & Therapeutics 2010, 125(2), 328; Diabetes Care 2007, 30(6), 1335; Expert Opinion on Emerging Drugs 2008, 13(4), 593). Нові пероральні лікарські засоби, які підвищують секрецію GLP-1, були б бажаними для лікування цукрового діабету 2 типу. Показано, що жовчні кислоти підвищують секрецію пептидів з шлунково-кишкового тракту. Жовчні кислоти виділяються з жовчного міхура в тонкий кишечник після кожного прийому їжі для полегшення перетравлювання поживних речовин, зокрема жирів, ліпідів та жиророзчинних вітамінів. Жовчні кислоти також функціонують як гормони, які регулюють гомеостаз холестерину, енергію та гомеостаз глюкози через ядерні рецептори (FXR, PXR, CAR, VDR) та рецептор зв'язування G-протеїну TGR5 (дивись огляди: Nature Drug Discovery 2008, 7, 672; Diabetes, Obesity and Metabolism 2008, 10, 1004). TGR5 є членом родопсиноподібної підродини GPCR (клас А), що експресується в кишечнику, жовчному міхурі, жировій тканині, печінці та вибраних ділянках центральної нервової системи. TGR5 активується за допомогою багатьох жовчних кислот, зокрема літохолевої та дезоксихолевої кислоти, як найбільш потужних активаторів (Journal of Medicinal Chemistry 2008, 51(6), 1831). Обидві, як дезоксихолева, так і літохолева кислоти збільшують секрецію GLP-1 ентероендорцинової STC-1 клітинної лінії, зокрема, через TGR5 (Biochemical and Biophysical Research Communications 2005, 329, 386). Показано, що синтетичний агоніст TGR5 INT-777 збільшує кишкову секрецію GLP-1 in vivo у мишей (Cell Metabolism 2009, 10, 167). Крім того, показано, що солі жовчних кислот сприяють секреції GLP-1 з L клітин кишки в судинно перфузованій моделі товстої кишки щурів (Journal of Endocrinology 1995, 145(3), 521), а також GLP-1, пептид YY (PYY) і нейротензин в судинно перфузованій моделі клубової кишки щурів (Endocrinology 1998, 139(9), 3780). У людей, інфузія дезоксихолату в сигмовидну кишку викликає швидке та помітне збільшення в плазмі відповідних концентрацій PYY й ентероглюкагону (Gut 1993, 34 (9), 1219). Агенти, які збільшують концентрацію жовчних кислот або солей жовчних кислот в клубовій та товстій кишках, збільшують секрецію пептидів кишечника, включаючи, але не обмежуючись цим, GLP-1 та PYY. 2 UA 110338 C2 5 10 15 20 25 30 Жовчні кислоти синтезуються з холестерину в печінці, потім піддаються сполученню карбонової кислоти з амінною функціональною групою таурину та гліцину. Кон'юговані жовчні кислоти виділяються в жовчний міхур, де відбувається їх накопичення до споживання їжі. Після їжі, жовчний міхур скорочується та очищає свій вміст в дванадцятипалу кишку, де кон'юговані жовчні кислоти сприяють всмоктуванню холестерину, жирів та жиророзчинних вітамінів в проксимальних відділах тонкого кишечника (дивись огляди: Frontiers in Bioscience 2009, 14, 2584; Clinical Pharmacokinetics 2002, 41(10), 751; Journal of Pediatric Gastroenterology and Nutrition 2001, 32, 407). Кон'юговані жовчні кислоти продовжують текти через тонку кишку до дистального відділу клубової кишки, де 90 % реабсорбується в ентероцитах за допомогою апікального натрійзалежного транспортера жовчної кислоти (ASBT, також відомий як iBAT). Решта 10 % декон'югується в жовчні кислоти кишковими бактеріями в термінальному відділі клубової кишки та товстої кишки, з яких 5 %, потім пасивно реабсорбується в товстій кишці та решта 5 % їх виділяється з калом. Жовчні кислоти, що поглинаються ASBT в клубовій кишці, потім транспортуються в ворітну вену для рециркуляції в печінці. Даний дуже регульований процесс має назву ентерогепатична рециркуляція, і має важливе значення для загального підтримання в організмі загального набору жовчних кислот, тому кількість жовчних кислот, які синтезуються в печінці еквівалентна сумі жовчних кислот, які виділяються з фекаліями. Фармакологічні порушення реабсорбції жовчних кислот інгібітором ASBT призводить до підвищення концентрації жовчних кислот у товстій кишці та калі, і, як фізіологічний наслідок збільшується перетворення печінкового холестерину в жовчні кислоти, щоб компенсувати фекальні втрати жовчних кислот. Багато фармацевтичних компаній дотримуються цього механізму як стратегії для зниження рівня холестерину в сироватці крові у пацієнтів з дисліпідемією/ гіперхолестеринемією (дивись огляд: Current Medicinal Chemistry 2006, 13, 997). Важливо відзначити, що опосередковане ASBT інгібітором збільшення в товстій кишці концентрації жовчної кислоти/солі також збільшить кишкові GLP-1, PYY, GLP-2, та інші пептидні гормони секреції кишечника. Таким чином, інгібітори ASBT можуть бути корисні для лікування цукрового діабету 2 типу, діабету 1 типу, дисліпідемії, ожиріння, синдрому короткої кишки, хронічного ідіопатичного запору, синдрому подразненого кишечника (IBS), хвороби Крона і артриту. Деякі 1,4-тіазепіни є розкритими, наприклад, в WO 94/18183 та WO 96/05188. Вважається, що дані сполуки будуть корисними як інгібітори зворотного захоплення жовчних кислот клубової кишки (ASBT). Короткий опис винаходу Коротко, в першому аспекті представленого винаходу розкрито сполуку, формули І 35 O O R 4 Q S X R 3 R R 1 N R I 1 40 45 50 3 2 , де R є H, Cl, Br, N(CH3)2 або метокси; 2 R є H або OH; 3 кожен R незалежно є C1-6алкілом; X є CH2, C(O) або CH=CH; Q є C0-6алкілом; 4 5 5 5 5 5 5 R є OH, SO3H, CO2H, PO3H2, CONR R , NR R ; або NHC(O)CH2NR R ; 5 кожен R незалежно є H, OH, C1-6алкілом, C0-6алкілCO2H, C0-6алкілSO3H, C0-6алкілPO3H2, 7 C(O)C0-6алкілCO2H, C(O)C0-6алкілSO3H, C(O)C0-6алкілPO3H2, або CH(R )C0-6алкілCO2H; та 7 R є C0-6алкілCO2H, C0-6алкілOH, C0-6алкілSO3H або C0-6алкілPO3H2. В іншому аспекті представлений винахід розкриває фармацевтично прийнятні солі сполуки формули I. В іншому аспекті представлений винахід розкриває фармацевтичні композиції, що містять сполуку формули І або її фармацевтично прийнятну сіль. 3 UA 110338 C2 5 10 15 20 25 30 35 40 45 50 55 60 В іншому аспекті представлений винахід розкриває спосіб лікування або попередження розладів обміну речовин, включаючи цукровий діабет (типу I та типу II), ожиріння та пов'язані з ним розлади у людей, що включає введення сполуки формули І або її фармацевтично прийнятної солі. Детальний опис винаходу 1 Переважно, R є метокси. 2 Переважно, R є H. 3 3 Переважно, кожен R незалежно є C2-4 алкілом. Найбільш переважно, кожен R незалежно є етилом або н-бутилом. 3 Коли R групи є різними, карбон, до якого вони є приєднаними, є хіральним. В таких хіральних сполуках переважною стереохімічною конфігурацією при такому атомі карбону є R. Атом карбону, до якого є приєднаним незаміщене фенільне кільце, є хіральним. Переважною стереохімічною конфігурацією при такому атомі карбону є R. Переважно, X є CH2 або C(O). Найбільш переважно, X є CH2. Переважно, Q є C0-2алкілом. Найбільш переважно, Q є C0алкілом (тобто відсутній). 4 5 5 4 5 5 Переважно, R є PO3H2 або NR R . Найбільш переважно, R є NR R . 5 Переважно, один R є H та інший є OH, метилом, C1-4алкілCO2H, C0-2алкілSO3H, C17 2алкілPO3H2, C(O)CO2H, C(O)C1-2алкілSO3H, C(O)C1-2алкілPO3H2, або CH(R ) C0-1алкілCO2H. 5 7 Найбільш переважно, один R є H та інший є CH2CO2H, CH2SO3H, CH2CH2SO3H або CH(R )C01алкілCO2H. 7 7 Переважно, R є C0-1алкілCO2H або C0-2алкілSO3H. Найбільш переважно, R є C0-1алкілCO2H. Як використано в дагомк документі, "алкіл" може бути лінійним алкільним ланцюгом або розгалуженим алкільним ланцюгом, якщо, очевидно, не зазначено інше. Для огляду відповідних фармацевтично прийнятних солей дивись, наприклад, Berge et al, J. Pharm, Sci., 66, 1-19, 1977. У втіленні кислотні адитивні солі вибирають з гідрохлориду, гідроброміду, гідройодиду, сульфату, бісульфату, нітрату, фосфату, гідрогенфосфату, ацетату, бензоату, сукцинату, сахарату, фумарату, малеату, лактату, цитрату, тартрату, глюконату, камсилату, метансульфонату, етансульфонату, бензолсульфонату, п-толуїлсульфонату або памоату. У втіленні основні адитивні солі включають солі металу (такого як натрію, калію, алюмінію, кальцію, магнію та цинку) та солі амонію (такі як солі ізопропіламіну, діетиламіну, діетаноламіну). Інші солі (такі як трифлуорацетати та оксалати) можуть застосовувати у виробництві сполук формули (I) та їх фармацевтично прийнятних солей, та є включеними в межі винаходу. Кислотні та основні адитивні солі можуть бути одержані кваліфікованим хіміком шляхом обробки сполуки формули (I) відповідною кислотою або основою в прийнятному розчиннику, з наступною кристалізацією та фільтруванням. Спосіб лікування або попередження розладів обміну речовин може включати введення сполуки або солі за даним винаходом самостійно як моно-терапія. Сполуки та солі за даним винаходом, крім того, можуть застосовувати в комбінації з іншими терапевтичними агентами. Прийнятні агенти для застосування в комбінації включають, наприклад, підсилювачи чутливості до інсуліну, інгібітори абсорбції глюкози, бігуаніди, підсилювачи секреції інсуліну або метформін. Приклади Сполуки за даним винаходом можуть бути одержані, використовуючи різноманітні способи, включаючи добре відомі стандартні синтетичні способи. Ілюстративні загальні синтетичні способи представлені нижче, та далі конкретні сполуки за винаходом одержують в дослідних прикладах. В усіх схемах, описаних нижче, захисні групи для чутливих або реакційно здатних груп застосовують, де необхідно у відповідності із загальними принципами синтетичної хімії. Захисні групи використовують відповідно до стандартних способів органічного синтезу (T.W. Green and P.G.M. Wuts, (1991) Protecting Groups in Organic Synthesis, John Wiley & Sons, включений шляхом посилання стосовно до захисних груп). Данні групи видаляють на прийнятній стадії синтезу сполуки, використовуючи способи, що є без зусиль очевидним для кваліфікованого фахівця в даній галузі з рівня техніки. Вибір способів, а також умов реакції та послідовність їх виконання буде ухгодженим з одержанням сполук за представленим винаходом. Сполуки за винаходом можуть бути легко одержані відповідно до схем 1-9 кваліфікованим фахівцем в даній галузі. Як проілюстровано на схемі 1, декілька ключових проміжних сполук можуть одержувати з (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін 1,1діоксид (Brieaddy, L.E. WO9605188, 1996). Деметилювання опосередковане кислотами Льюїса з наступою реакцією з ангідридом трифлатної кислоти забезпечувало одержання трифлатної проміжної сполуки (A). Реакція A з монооксидом карбону та паладієвим каталізатором в MeOH 4 UA 110338 C2 5 забезпечувала одержання естер проміжної сполуки (B). Реакція A в присутності ціаніду та паладієвого каталізатору забезпечувала одержання нітрилу проміжної сполуки (C), який відновлювали діїзобутилалюміній гідридом (DiBAL-H), що дає альдегід проміжної сполуки (D), або піддають гідрогенізації в присутності паладію та кислоти, що призводить до амінометилу проміжної сполуки (E). Схема 1: Одержання ключових проміжних сполук 1) AlCl3 Zn(CN)2 2) Tf2O Pd2(dba)3 B 10 15 C A CO, Pd2(dba)3 MeOH DiBAL-H D H2, Pd/C HCl E Сполуки за винаходом можуть бути одержані як на схемі 2. Альдегід проміжної сполуки (D) піддають в умовах відновного амінування взаємодії із заміщеним аміном з наступним, якщо необхідно, зняттям захисту кислотою, основою або функціоналізацією (окисненням), призводить до 8-амінометильних прикладів (F). Альтернативно, проміжну сполуку (E) ацилювали хлорангідридом хлоралкільної кислоти, потім піддавали реакції з різними нуклеофілами, включаючи фульфіт, фосфіт, гідроксид та заміщені аміни з наступним, якщо необхідно, зняттям захисної групи, одержуючи ацильовані 8-амінометильні приклади (F). Третя та четверта процедури, щоб одержати 8-амінометильні сполуки за винаходом, включали реакцію проміжної сполуки (E) з функціоналізованими бромідами в присутності основи або параформальдегіду та триетилфосфіту, одержуючи інші заміщені 8-амінометильні сполуки (F). 20 5 UA 110338 C2 Схема 2: Одержання амінометил заміжених сполук 1) R5R5NH NaHB(OAc)3 2) зняття захисту або окиснення D 1) ( )n 2) нуклеофіли [SO32-, (EtO)3P, -OH, HNR5R5] 3) зняття захисту F R5Br, основа E параформальдегід HP(O)(OEt)2 5 Сполуки за винаходом можуть одержувати як на схемі 3. Естерну проміжну сполуку (B) омилювали за основних умов, одержуючи карбонову кислоту (G), яку потім піддавали взаємодії до стандартного аміду в умовах сполучення в присутності заміщеного аміну, одержуючи приклади амідів (H). Естерну проміжну сполуку (B) також відновлювали DiBAL-H до спирту (I), який потім перетворювали до броміду (J). Бромід (J) піддавали реакції з різними нуклеофілами (сульфітом, фосфітом, гідроксидом, амінами) з наступним, якщо необхідно, зняттям захисту, одержуючи зразки прикладів (K). 10 Схема 3: Одержання зразків з естерних проміжних сполук (B) LiOH, THF HNR5R5 H2O B H G DiBAL-H 1) нуклеофіли [SO32-, P(OEt)3, -OH, HNR5R5] Br2, Ph3P імідазол I 15 2) зняття захисту J K Сполуки за винаходом можуть одержувати як на схемі 4 з трифлатної проміжної сполуки (A). Проміжну сполуку (A) піддавали реакції Гека (Heck), одержуючи ненасичений естер (L), який омилювали до ненасиченої кислоти (M). Естер (L) піддавали взаємодії в умовах гідрогенізації з 6 UA 110338 C2 5 наступним омиленням, одержуючи кислоту (N). Стандартне утворення аміду із заміщених амінів забезпечило приклади амідів (O). Відновлення кислоти (N) бораном з наступним мезилюванням або утворенням броміду забезпечувало одержання сполуки (P), яку піддавали реагуванню з різними нуклеофілами (ціанідом, діетилфосфітом, гідроксидом, сульфітом) потім, якщо необхідно, гідролізували, окиснювали або знімали захист, одержуючи зразки прикладів (Q). Схема 4: Одержання зразків з трифлатної проміжної сполуки (A) CH2CHCO2Et 1) H 2, Pd/C (PPh 3)2PdCl 2 2) LiOH N L (R=Et) A 1) BH 3 2) MsCl or Br2, PPh 3 LiOH M (R=H) 1) нуклеофіли [CN, HPO(OEt2), -OH, SO32-, HNR 5R5] OMs або Br HNR5R5 2) гідроліз, окиснення або зняття захисту Q 10 P O Сполуки за винаходом можуть одержувати як на схемі 5. Альдегід проміжної сполуки (D) перетворювали в ненасичений фосфонат (R), з якого потім знімали захист TMSBr, одержуючи зразок ненасиченої фосфонової кислоти (S). Фосфонат (R) піддавали гідрогенізації, потім знімали захист TMSBr, одержуючи зразок насиченої фосфонової кислоти (T). Схема 5: Одержання похідних алкілфосфонової кислоти (EtO)2POCH2 PO(OEt)2 NaH 1) H2, Pd/C 2) TMSBr D R (R=Et) TMSBr S (R=H) T 15 20 Сполуки за винаходом можуть одержувати як на схемі 6. Сполуку J (зі схеми 3) окиснювали до N-гідроксильної похідної, яку піддавали дії триетилфосфіту з наступним зняттям захисту, одержуючи N-гідроксильний зразок прикладу (U). Нітрильну проміжну сполуку (C) окиснювали, потім нітрил відновлювали до альдегіду (V), який потім піддавали дії заміщених амінів за умов відновного амінування, одержуючи зразки прикладів (W). Сполуку X (одержану як на схемі 2) окиснювали, відновлювали та знімали захист, одержуючи N-гідроксильний зразок прикладу (Y). 7 UA 110338 C2 Схема 6: Одержання N-гідроксильних зразків 1) P(OEt)3 mCPBA 2) TMSBr U J (схема 3) R4=фосфонова кислота 1) mCPBA 1) HNR5R5 NaHB(OAc)3 2) DiBAL-H 2) зняття захисту V C W 1) mCPBA 2) NaCNBH3 3) ТФО Y Одержаний як на схемі 2 5 Сполуки за винаходом можуть одержувати як проілюстровано на схемі 7. Нітрильну проміжну сполуку (C) окиснювали до іміну, потім нітрил відновлювали до альдегіду, використовуючи DiBAL-H, який піддавали відновленню бораном, реакції з триетилфосфатом, та знімали захист, одержуючи C(5) b-фенільний зразок прикладу (AA). Сполуку Z окиснювали перйодатом Деса-Мартіна (Dess-Martin), потім піддавали дії заміщених амінів за умов відновного амінування з наступним, якщо необхідно, зняттям захисту, одержуючи додаткові зразки прикладів (BB). 10 Схема 7: Одержання C5 β-епімеру 1) DDQ окиснення 2) DiBAL-H 3) BH3 C 1) окиснення 2) HNR5 R5, NaHB(OAc)3 3) зняття захисту BB (R5=фосфонова кислота) 8 Z 1) Br2, PPh3 2) P(OEt)3 3) TMSBr AA (R4 =фосфонова кислота) UA 110338 C2 5 10 15 Сполуки за винаходом можуть одержувати як проілюстровано на схемі 8. Перетворення норлейцину до етилового естеру з наступним утворенням іміну з бензальдегідом та алкілуванням з йодбутаном забезпечувало одержання проміжної сполуки (CC). Гідроліз іміну з наступним відновленням LAH та утворенням сульфату забезпечувало одержання аміносульфатної проміжної сполуки (DD). Ацилювання 2-бром-1,4-біс(метилокси)бензолу бензоїлхлоридом з наступним селективним деметилюванням забезпечувало одержання фенолу проміжної сполуки. Ацилювання з наступним перегрупуванням Ньюмана-Кворта (Newman-Kwart) забезпечувало одержання проміжної сполуки (EE). В одній ємності зняття захисту з (EE) та алкілування аміносульфатом (DD) призводить до утворення продукту (FF). Міжмолекулярна циклізація до іміну з наступним карбонілюванням опосередкованим паладієм забезпечувало одержання естерної проміжної сполуки, яку відновлювали LAH, одержуючи проміжну сполуку (GG). Відновлення іміну з наступним окисненням сульфіду забезпечувало одержання проміжної сполуки (HH). Перетворення (HH) до зразків прикладів за винаходом (II та JJ) могло досягатися відповідно до хімічних процесів, зображених на схемах 2 та 3, відповідно. Схема 8: Одержання C(3)-дибутильних зразків 1) EtOH, SOCl2 2) бензальдегід 1) вод. HCl 2) LAH 3) NaH, йодбутан 3) ClSO2 OH CC 1) бензоїлхлорид TfOH DD 1) ClC(S)NEt2 2) D, Ph2O 2) BCl3 EE 1) BH3 1) H , толуол, D 2) CO, MeOH Pd2(dba)3 2) H2 O2, ТФО KOH EtOH 3) LAH + GG FF HH 1) окиснення 2) HNR5 R5 NaHB(OAc)3 3) зняття захисту 1) Br2, PPh3 2) P(OEt)3 3) TMSBr JJ 20 25 Сполуки за винаходом можуть одержувати як проілюстровано на схемі 9. Деметилювання (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепіну 1,1діоксиду (Brieaddy, L.E. WO9605188, 1996) з наступним утворенням три флату та амінуванням опосередкованим паладієм забезпечувало одержання похідної (KK). Деметилювання, утворення три флату та ціанування опосередковане паладієм призводило до одержання нітрилу (LL). Відновлення нітрилу DiBAL-H до альдегіду (MM) з наступною взаємодією із заміщеними амінами за умов відновного амінування та, якщо необхідно, зняттям захисту забезпечувало одержання зразків прикладів (NN). 9 UA 110338 C2 Схема 9: Одержання 7-N,N-диметильних зразків 1) AlCl3 2) Tf2O 1) AlCl3 2) Tf2O 2) HNMe2 Pd2(dba)3 3) Zn(CN)2 Pd2(dba)3 KK 1) HNR5R5 NaHB(OAc)3 DIBAL 2) зняття захисту . 5 10 15 20 25 LL MM NN Скорочення HATU 1,1,3,3-тетраметилуронію гексафлуорфосфат ДХМ дихлорметан ДХЕ 1,2-дихлоретан ДІПЕА діізопропілетиламін ДМЕ 1,2-диметоксіетан ДМФ N, N-диметилформамід ДМСО диметилсульфоксид Et2O діетиловий етер EtOAc етилацетат EtOH етанол год. година HOAc оцтова кислота MeCN ацетонітрил MeOH метанол MTBE метил трет-бутиловий етер NMP N-метилпіролідинон PhH бензол PhMe толуол ТГФ тетрагідрофуран I. Одержання проміжних сполук Проміжні сполуки 1a та 1b: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро1,4-бензотіазепін-8-ол-1,1-діоксид (1a) та (3R, 5R)-3-бутил-3-етил-8-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-7-ол 1,1-діоксид (1b) O O HO O S O N H HO O O S N H 30 35 Спосіб 1: Розчин (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-1,1-діоксиду (200 г, 479 ммоль) (одержаний як в Brieaddy, L.E. WO9605188, 1996) в ДХЕ (1л) насичували HCl (газ.), потім обробляли хлоридом алюмінію (200 г, 1,5 моль) в одній порції. Реакційну суміш струшували, одночасно повільно охолоджуючи до температури навколишнього середовища, далі струшували протягом 2,5 год. Реакційну суміш додавали до суміші крига-H2O з енергійним струшуванням. Двофазну суміш обробляли 1N HCl (~200 мл), 10 UA 110338 C2 5 10 15 20 25 30 35 40 45 50 55 60 потім після струшування протягом 30 звилин фази розділяли. Органічну фазу відокремлювали, двічі промивали розбавленою HCl (1,5 л H2O/~200 мл 1N HCl), сушили над MgSO4, фільтрували та концентрували насухо, одержуючи регіоізомерну суміш 7-OH та 8-OH продуктів (185 г, 458 1 ммоль, 96 % вихід) у вигляді білої піни. H ЯМР продемонструвало 47:53 суміш 7/8-фенолів, відповідно. Регіоізомери розділяли шляхом хіральної хроматографії [нерухома фаза (CSP)целюлоза три(3,5-дихлорфенілкарбамат) – полімер, імобілізований на силікагель (CHIRALPAK ® IC , 20 мікрон, 20 см x 25 см), ДХМ та ізопропанол (98/2 об./об.) як рухома фаза], одержуючи проміжні сполуки 1a та 1b: Проміжна сполука 1b (більш швидкий пік елюювання): (3R, 5R)-3-бутил-3-етил-8(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-7-ол-1,1-діоксид (86,3 г, біла тверда 1 речовина, чистота = 98,2 %): H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,74 (т, J=7,0 Гц, 3 H), 0,79 (т, J=7,4 Гц, 3 H), 0,94-1,28 (м, 4 H), 1,30-1,53 (м, 2 H), 1,64-1,81 (м, 1 H), 1,91-2,10 (м, 1 H), 2,45 (д, J=9,8 Гц, 1 H), 3,04 (д, J=14,8 Гц, 1 H), 3,49 (д, J=14,8 Гц, 1 H), 3,80 (с, 3 H), 5,85 (д, J=10,0 Гц, 1 H), 6,06 (с, 1 H), 7,19-7,52 (м, 6 H), 9,93 (с, 1 H) Проміжна сполука 1a (більш повільний пік елюювання): (3R, 5R)-3-бутил-3-етил-7(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-ол-1,1-діоксид (72,5 г) у вигляді білої 1 твердої речовини: H ЯМР (400 МГц, ДМСО-д6) м.ч. 0,75 (т, J=7,1 Гц, 3 H), 0,79 (т, J=7,5 Гц, 3 H), 0,95-1,28 (м, 4 H), 1,31-1,53 (м, 2 H), 1,64-1,76 (м, 1 H), 1,98-2,10 (м, 1 H), 2,43 (д, J=9,8 Гц, 1 H), 3,06 (д, J=14,8 Гц, 1 H), 3,42 (с, 3 H), 3,49 (д, J=14,8 Гц, 1 H), 5,85 (д, J=9,8 Гц, 1 H), 6,04 (с, 1 H), 7,24-7,35 (м, 1 H), 7,35-7,48 (м, 5 H), 9,72 (с, 1 H). Спосіб 2: Розчин (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-1,1-діоксиду (1250 г, 2,99 моль) в 1,2-дихлоретані (6 л), що струшували, насичували газоподібним HCl при температурі навколишнього середовища (внутрішня температура підвищувалась до 39 °C під час даної стадії). Барботування газоподібного HCl через розчин продовжували протягом 5 хвилин після досягнення 39 °C. Хлорид алюмінію (1250 г, 9,37 моль) додавали порціями понад 40 хвилин з деяким зовнішнім охолодженням під час утримування температури реакції між 38-42 °C. Реакційну суміш струшували протягом 2 годин та потім повільно виливали на 10 кг суміші крига/вода. Дві додаткові серії 1250 г та 500 г (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-1,1-діоксида обробляли хлоридом алюмінію та HCl подібним способом. Три порції крига/вода, якими гасили реакційну суміш (7,19 моль об'єднаний теоретичний), об'єднували та фази розділяли. Водну фазу екстрагували CH2Cl2 (3 л) та об'єднані органічні шари промивали водою (3 × 4 л) та концентрували до початку кристалізації продукту. Суміш розбавляли гептаном та витримували зі струшуванням. Осад збирали фільтруванням, сушили на повітрі протягом 2 днів та потім сушили в вакуумній шафі при 55 °C протягом 48 годин, одержуючи приблизно 9:8 суміш (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-ол 1,1-діоксиду та (3R, 5R)-3-бутил-3-етил-8-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-7-ол 1,1діоксида (2,8 кг). Очистка суміші відповідно до процедури в спосібі 1 забезпечувала одержання 1235 грам (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8ол-1,1-діоксиду у вигляді білої твердої речовини. Спосіб 3: Розчин (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-1,1-діоксиду (11,1 кг, 26,58 моль) в ДХЕ (69,7 кг) обробляли водою (0,48 кг, 26,67 моль), далі регулювали температуру до 35°-40 °C. Розчин обробляли оксалілхлоридом (3,4 кг, 26,78 моль) та струшували протягом тридцяти хвилин. Потім розчин послідовно обробляли трихлоридом алюмінію (3,6 кг, 26,9 моль; 3,6 кг, 26,9 моль; 1,8 кг, 13,4 моль; 1,8 кг, 13,4 моль, 0,9 кг, 6,8 моль) даючи тридцять хвилин між обробками. Реакційну суміш струшували при 35°40 °C доки реакція завершиться. Реакційну суміш гасили шляхом повільного додавання реакційного розчину до води (119 кг). Органічну фазу відокремлювали, потім двічі екстрагували водою (60 кг) та один раз насиченим розчином хлориду натрію (30 кг). Розчин суміші продуктів в дихлоретані концентрували до кінцевої маси 39 кг. Даний розчин застосовували як вихідний розчин для препаративної хроматографії, як описано в способі 1. Фракції з бажаним продуктом частково концентрували до об'єму близько 12 л. Розчини потім додатково концентрували шляхом атмосферної перегонки до температура всередині ємності 80 °C. При охолодженні відбувалась кристалізація. Одержану початкову рідку суспензію розбавляли водою (6-12 л) та струшували протягом двох годин. Продукт збирали фільтрацією, промивали водою (4 л) потім сушили при 60 °C в вакуумі до постійної маси. З вищезазначеного одержували 4,1 кг (10,22 моль) (3R, 5R)-3-бутил-3-етил-8-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-7-ол-1,1-діоксиду (проміжна сполука 1b) та 3,9 кг (9,72 моль) of (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-ол-1,1-діоксиду (проміжна сполука 1a). 11 UA 110338 C2 5 10 Повторне використання проміжної сполуки 1b до (3R, 5R)-3-бутил-3-етил-7,8-біс(метилокси)5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-1,1-діоксида: Розчин (3R, 5R)-3-бутил-3-етил-8(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-7-ол-1,1-діоксиду (4,1 кг 10,22 моль) в ацетоні (19,7 кг) обробляли карбонатом калію (3,84 кг) та температуру регулювали до 25°30 °C. Загружали метилйодид (5,84 кг, 41,14 моль) та реакційну суміш витримували при 25°30 °C до завершення реакції. Реакційну суміш концентрували до ~ 8 л загального об'єму шляхом перегонки, потім загружали дихлоретан (31 кг). Реакційну суміш двічі екстрагували водою (20,8 кг) та далі загружали дихлоретан (26 кг). Розчин продукту концентрували до ~23 л, щоб одержати розчин прийнятний для повторного зняття захисту до проміжної сполуки 1a та 1b як описано вище в способі 3. Проміжна сполука 2: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонат F FO S O F O O O S N H O 15 20 25 30 35 40 45 50 Спосіб 1: До розчину (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-ол-1,1-діоксиду (105 г, 26 ммоль) та піридину (27,3 мл, 34 ммоль) в ДХМ, охолодженого до 0ºC, в атмосфері азоту повільно додавали трифлуорметансульфоновий ангідрид (57,1 мл, 34 ммоль) протягом понад 30 хвилин, в той же час підтримуючи температуру в середині в межах 5-10ºC. При завершенні додавання реакційну суміш струшували доки ТШХ та РХМС не демонструвало повного перетворення до продукту. До суміші повільно додавали H2O (250 мл), та суміш струшували протягом 10 хвилин, після чого шари розділяли. Водний шар екстрагували додатковий раз ДХМ, та об'єднані органічні фракції промивали 10 % HCl, сольовим розчином, потім сушили (Na2SO4), фільтрували та концентрували до половини об'єму. Додавали гексани доки розчин не стане мутним та почне відбуватися кристалізація. Далі тверду речовину відфільтровували з розчину, одержуючи названі сполуки (134,6 г, 95 %) у вигляді білої 1 твердої речовини, яку використовували без подальшої очистки: H ЯМР (400 МГц, CDCl3) м.ч. 0,76-0,96 (м, 6 H), 1,03-1,40 (м, 4 H), 1,40-1,67 (м, 4 H), 1,77-1,97 (м, 1 H), 2,10-2,28 (м, 1 H), 3,07 (д, J=14,8 Гц, 1 H), 3,48 (д, J=14,9 Гц, 1 H), 3,62 (c, 3 H), 6,09 (c, 1 H), 6,34 (c, 1 H), 7,32-7,50 (м, 5 + H), 7,96 (c, 1 H); ЕС-РХМС m/z 536 (M+H) . Спосіб 2: Піридин (441 мл, 5,45 моль) додавали до суспензії (3R, 5R)-3-бутил-3-етил-7(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-ол- 1,1-діоксиду (1099 г, 2,72 моль) в трет-бутилметиловому етері (16 л), що струшували, з охолодженням до 15 °C. Чистий (без домішок) ангідрид трифлатної кислоти (1076 г, 3,81 моль) по краплям додавали протягом понад 45 хвилин, в той же час підтримуючи температуру реакційної суміші в межах 14-16 °C. Наступне додавання, реакційну суміш струшували при температурі навколишнього середовища протягом 2 годин. Повільно додавали воду (6 л), та суміш інтенсивно струшували протягом 15 хвилин. Шари розділяли, та органічну фазу промивали 1:1 вода/сольовий розчин (5 л), сушили над MgSO4 та фільтрували. Фільтрат концентрували, щоб максимально видалити третбутилметиловий етер (видаляли близько 13-14 л). Одержану в результаті суспензію фільтрували, та відфільтрований осад промивали гептаном. Далі відфільтрований осад сушили в вакуумі протягом ночі при 40 °C, одержуючи (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонат у вигляді майже білої твердої речовини (1342 г). Другу порцію продукту одержували з маточного розчину (63 г). Об'єднаний вихід становив 1405 г. Спосіб 3: В реактор загружали 2,75 кг (6,8 моль) (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-ол-1,1-діоксиду, 19,9 кг 1,4-діоксану, 1,08 кг (13,6 моль) піридину при 25 °C та струшували при даній температурі до розчинення. Потім розчин охолоджували до близько 10 °C, куди далі додавали ангідрид трифлатної кислоти (2,8 кг, 9,9 моль) з такою швидкістю, щоб температура всередині становила приблизно 5-15 °C. Реакційну суміш струшували при даній температурі протягом близько 45 хвилин. Температуру сорочки потім підвищували до близько 25 °C, та реакційну суміш струшували при даній температурі до 12 UA 110338 C2 5 10 завершення реакції. Далі в реактор додавали толуол (16,3 кг) та 10 % сольовий розчин (8,3 л), та суміш перемішували, щонайменше, протягом 5 хвилин, після чого відстоювали протягом інших 5 хвилин. Водний шар видаляли. Додавали воду (8,3 кг), та суміш струшували протягом близько 5 хвилин, після чого відстоювали, щонайменше, протягом 10 хвилин. Водний шар видаляли, та промивання водою повторювали. Після наступного видалення водного шару, органічний шар зменшували до близько 11 л шляхом вакуумної перегонки. Додавали гептан (18,8 кг), та загальний об'єм знову зменшували шляхом вакуумної перегонки до близько 14 л. Добавляли інші 4 кг гептанів, та суміш повільно охолоджували до близько 5 °C протягом ночі. Продукт збирали фільтрацією та двічі промивали гептанами (3,6 кг та 4,0 кг). Корж віджимали досуха під N2 поверхневим шаром в вакуумі (~60 °C), одержуючи 3,47 кг (95 %) (3R, 5R)-3-бутил3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонату. Проміжна сполука 3: Метил (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-карбоксилат-1,1-діоксид 15 O O O S O N H O 20 25 30 До реакційної пробірки, що містила (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонат (0,103 г, 0,192 ммоль), Pd2(dba)3 (0,011 г, 0,012 ммоль), dppf (0,013 г, 0,024 ммоль) та триетиламін (0,040 мл, 0,288 ммоль) в ДМФ (1 мл) барботували газоподібний моно оксид карбону протягом періоду 15 хвилин. Додавали безводний MeOH (0,039 мл, 0,962 ммоль), та реакційну ємність запаювали в атмосфері моно оксиду карбону, потім нагрівали при 70ºC протягом ночі. Реакційну суміш охолоджували до кімнатної температури, та вміст розбавляли Et2O, потім виливали в H2O. Шари розділяли, водний шар екстрагували один додатковий раз Et 2O, та об'єднані органічні фракції промивали сольовим розчином, сушили (Na2SO4), фільтрували та концентрували. Сиру речовину хроматографували на силікагелі, використовуючи гексани/EtOAc, щоб одержати 1 названу сполуку (0,080 г, 91 %) у вигляді білої твердої речовини: H ЯМР (400 МГц, CDCl3) м.ч. 0,75-0,96 (м, 6 H), 0,99-1,36 (м, 4 H), 1,36-1,59 (м, 5 H), 1,86 (ддд, J=14,4, 12,0, 4,4 Гц, 1 H), 2,072,23 (м, 1 H), 3,02 (д, J=14,8 Гц, 1 H), 3,45 (д, J=14,9 Гц, 1 H), 3,57 (с, 3 H), 3,86 (с, 3 H), 6,08 (с, 1 + H), 6,27 (с, 1 H), 7,29-7,48 (м, 5 H), 8,52 (с, 1 H); ЕС-РХМС m/z 446 (M+H) . Проміжна сполука 4: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбонової кислоти 1,1-діоксид O O O S HO N H O 35 40 45 H2O (0,333 мл), MeOH (0,333 мл) та ТГФ (1 мл) додавали до метил (3R, 5R)-3-бутил-3-етил7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбоксилат-1,1-діоксиду (0,046 г, 0,103 ммоль) разом з LiOH (0,013 г, 0,310 ммоль), та суміш струшували протягом 2 годин при температурі навколишнього середовища, щоб гідролізувати до бензойної кислоти. Суміш концентрували до половини об'єму, потім додавали 6N HCl. Суміш екстрагували EtOAc (2X), промивали сольовим розчином, сушили (Na2SO4), фільтрували та концентрували в вакуумі, 1 одержуючи названу сполуку (0,039 г, 86 %): H ЯМР (400 МГц, CDCl3) м.ч. 0,67-0,99 (м, 6 H), 0,99-1,64 (м, 7 H), 1,87 (т, J=11,8 Гц, 1 H), 2,08-2,29 (м, 1 H), 3,09 (д, J=14,8 Гц, 1 H), 3,46 (д, J=14,9 Гц, 1 H), 3,70 (ш с, 3 H), 6,11 (ш с, 1 H), 6,33 (ш с, 1 H), 7,30-7,60 (м, 5 H), 8,73 (ш с, 1 H); + ЕС-РХМС m/z 432 (M+H) . 13 UA 110338 C2 Проміжна сполука 5: [(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл]метанол O O S HO N H O 5 10 15 До розчину метил (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбоксилат-1,1-діоксиду (0,275 г, 0,617 ммоль) в ДХМ (2 мл) при 0ºC в атмосфері азоту додавали 1M розчин DIBAL-H в толуолі (1,3 мл, 1,30 ммоль). Реакційну суміш нагрівали до температури навколишнього середовища та струшували протягом 1,5 год., потім додавали MeOH з наступною H2O. Реакційну суміш концентрували, далі знову розчиняли в EtOAc. Шари розділяли, водний шар екстрагували додатковий раз EtOAc, потім об'єднані органічні фракції промивали сольовим розчином, сушили (Na 2SO4), фільтрували та концентрували. Хроматографія на силікагелі, використовуючи гексани/EtOAc забезпечувала 1 одержання названої сполуки (0,238 г, 91 %): H ЯМР (400 МГц, CDCl3) м.ч. 0,76-0,93 (м, 6 H), 1,04-1,36 (м, 6 H), 1,36-1,59 (м, 6 H), 1,76-1,93 (м, 1 H), 2,05-2,24 (м, 2 H), 3,02 (д, J=14,8 Гц, 1 H),3,42 (д, J=14,8 Гц, 1 H), 3,54 (с, 3 H), 4,65 (квд, J=13,2, 6,3 Гц, 2 H), 6,07 (ш с, 1 H), 6,17 (с, 1 + H), 7,29-7,50 (м, 5 H), 8,03 (с, 2 H); ЕС-РХМС m/z 418 (M+H) . Проміжна сполука 6: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбонітрил-1,1-діоксид 20 O O NC S N H O 25 30 35 40 45 Спосіб 1: Суміш (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонату (0,535 г, 1,0 ммоль), Pd2(dba)3 (0,055 г, 0,06 ммоль), DPPF (0,066 г, 0,12 ммоль), цинкового порошку (0,004 г, 0,06 ммоль) та Zn(CN)2 (0,117 г, 0,99 ммоль) в ДМФ (10 мл) струшували при температурі навколишнього середовища протягом 15 хвилин під потоком азоту. Реакційну суміш нагрівали до 80ºC та струшували протягом 8 годин. Далі суміш охолоджували температури навколишнього середовища, видаляли ДМФ в вукуумі, додавали Et2O, та органічні фракції промивали 2N розчином NH4OH (водним), після чого сольовим розчином. Органічні фракції сушили (Na 2SO4), фільтрували та концентрували. Залишок розтирали з сумішшю гексани/EtOAc, та білу тверду 1 речовину збирали фільтрацією, одержуючи названу сполуку (0,386 г, 92 %): H ЯМР (400 МГц, CDCl3) м.ч. 0,73-0,97 (м, 6 H), 0,98-1,62 (м, 8 H), 1,77-1,92 (м, 1 H), 2,08-2,24 (м, 1 H), 3,01 (д, J=14,9 Гц, 1 H), 3,45 (д, J=14,9 Гц, 1H), 3,62 (с, 3 H), 6,08 (ш с, 1 H), 6,26 (с, 1 H), 7,32-7,49 (м, 5 + H), 8,28 (с, 1 H); ЕС-РХМС m/z 413 (M+H) . Спосіб 2: Розчин (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл трифлуорметансульфонату (1,346 кг, 2,51 моль) в ДМФ (2,5 л) загружали з Zn(CN)2 (440 г, 3,75 моль) та струшували, суміш дегазували шляхом пропускання N 2 через суміш протягом 60 хвилин. Додавали воду (25 мл), та дегазування продовжували протягом 30 хвилин. Додавали DPPF (14,5 г, 26,16 ммоль) та Pd 2(dba)3 (10,5 г, 11,47 ммоль) та реакційну суміш нагрівали до 80-85 °C, продовжуючи дегазування. Вважали, що реакція завершилася через 2 години при даній температурі. Після охолодження реакційної суміші до 60 °C, додавали толуол (500 мл) та суміш струшували протягом ночі з охолодженням до температури навколишнього середовища. Додавали толуол (8 л) та суміш промивали водою (4 × 2 л). Органічну фазу сушили над MgSO4 та фільтрували, та фільтрат струшували над PL 14 UA 110338 C2 5 10 15 20 25 BnSH смолою (460 г, щоб видалити домішки важкого металу) протягом 3 днів. Смолу видаляли фільтрацією, та фільтрат концентрували до кристалізації одержаного в результаті продукту. Додавали гептан (12 л) та одержані в результаті тверді речовини збирали фільтрацією та промивали гептаном. Білу тверду речовину далі сушили в вакуумі, одержуючи (3R, 5R)-3-бутил3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбонітрил-1,1-діоксид (1,010 кг). Спосіб 3: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-іл трифлуорметансульфонат (2160 г, 4,05 моль) та ціанід цинку (266 г, 2,23 моль) загружали в реактор з наступним додаванням води (1084 мл) та N-метилпіролідинону (10830 мл). Вміст знаходився під атмосферою азоту, потім тричі вакуумували. Реакційну суміш нагрівали до 100 °C таобробляли кашоподібним ацетатом паладію (4,8 г, 0,02 моль) та 1,1'біс(дифенілфосфіно)ферроценом (15,7 г, 0,03 моль) в NMP(50 мл). Реакційну суміш безпосередньо обробляли поліметилгідросілоксаном (PMHS, 22 г) в NMP (50 мл) та витримували при 100 °C протягом приблизно однієї години, коли ВЕРХ аналіз показав, що реакція завершилася. Реакційну суміш охолоджували приблизно до 35 °C та обробляли приготовленим розчином з води (8685 г), гідроксиду амонію (1958 г) та етанолу (17130 г) протягом приблизно понад десяти хвилин. Одержану в результаті суспензію нагрівали приблизно до 80 °C, щоб розчинити. Розчин охолоджували до 20 °C протягом приблизно понад 60 хвилин та утримували при 20 °C протягом приблизно 90 хвилин. Тверду речовину фільтрували, промивали приготовленим розчином 50 % етанол/вода (4330 мл), з наступним промиванням гептаном (4300 мл), та віджимали насухо. Тверду речовину сушили в вакуумній шафі при 50 °C, одержуючи (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро1,4-бензотіазепін-8-карбонітрил-1,1-діоксид (1482 г, 89 %). Проміжна сполука 7: (3R, 5R)-8-(бромметил)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-1,1-діоксид O O S Br N H O 30 35 40 Імідазол (0,153 г, 0,563 ммоль) розчиняли в ДХМ (2 мл), та розчин охолоджували до 0ºC. Додавали трифенілфосфін (0,295 г, 1,126 ммоль), після чого бром (0,058 мл, 1,126 ммоль). Розчин [(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-іл]метанолу (0,235 г, 0,563 ммоль) в ДХМ (1 мл) повільно додавали при 0ºC. Реакційну суміш струшували при 0ºC протягом 2 годин з наступним додаванням Na 2SO3 (вод.), та одержану в результаті суміш розділяли. Органічний шар сушили (Na 2SO4), фільтрували через шар силікагелю та концентрували, одержуючи густу олію, яка затвердівала при стоянні, 1 одержуючи названу сполуку (0,265 г, 84 %): H ЯМР (400 МГц, CDCl3) м.ч. 0,73-0,95 (м, 6 H), 1,02-1,36 (м, 5 H), 1,38-1,65 (м, 5 H), 1,76-1,92 (м, 1 H), 2,08-2,23 (м, 1 H), 3,02 (д, J=14,8 Гц, 1 H), 3,43 (д, J=14,8 Гц, 1 H), 3,57 (c, 3 H), 4,38-4,56 (м, 2 H), 6,06 (c, 1 H), 6,16 (c, 1 H), 7,28-7,45 (м, 5 + + H), 8,05 (c, 1 H); РХ-МС m/z 480 (M+H) , РХ-МС 482 (M+H+2) . Проміжна сполука 8: (3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-карбонітрил-1,1-діоксид O O S N N O OH 45 (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8карбонітрил-1,1-діоксид (6,00 г, 14,54 ммоль), розчинений в ДХМ (100 мл), обробляли m-CPBA (2,51 г, 11,20 ммоль) зі струшуванням при 22 °C протягом 3 годин. Додавали додаткову кількість 15 UA 110338 C2 5 10 m-CPBA (0,77 г, 3,44 ммоль), та суміш струшували додаткові 2 години. Реакційну суміш обробляли 10 % Na2SO3, потім енергійно струшували протягом 1 години. Органічний шар відокремлювали, сушили над MgSO4, фільтрували та концентрували насухо. Залишок очищали на 330 г силікагелю, елююючи від 10 до 60 % EtOAc/гексани, одержуючи назад вітрильну вихідну речовину (1,33 г, 3,23 ммоль, 22,2 % вихід) та бажаний (3R, 5R)-3-бутил-3-етил-4гідрокси-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбонітрил-1,1-діоксид (з 1 домішками) (1,99 г, 31,9 % вихід): H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,78 (т, J=7,1 Гц, 3 H), 0,85 (т, J=7,2 Гц, 3 H), 1,05-1,37 (м, 4 H), 1,38-1,51 (м, 1 H), 1,57-1,70 (м, 1 H), 1,78-1,89 (м, 1 H), 2,012,12 (м, 1 H), 3,40-3,53 (м, 2 H), 3,56 (с, 3 H), 6,28 (с, 1 H), 6,39 (с, 1 H), 7,34-7,58 (м, 5 H), 8,16 (с, 1 H), 8,26 (с, 1 H). Проміжна сполука 9: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбальдегід-1,1-діоксид O H O O S N H O 15 20 25 30 35 40 45 50 Спосіб 1: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбонітрил-1,1-діоксид (7,42 г, 17,99 ммоль), розчинений в ДХМ (150 мл) обробляли 1M DIBAL-H в толуолі (36,0 мл, 36,0 ммоль) при 0 °C зі струшуванням протягом 1 години, після чого РХМС показала повне перетворення. Реакційну суміш виливали на суміш крига/1N HCl та інтенсивно струшували протягом 1 години. Органічну фазу відокремлювали, сушили над MgSO4, фільтрували та концентрували, одержуючи жовту тверду речовину. Сирий продукт розчиняли в 80 мл гарячого EtOAc, до якого додавали 250 мл гексанів до помутніння. Суміші давали повільно охолонути до температури навколишнього середовища та потім охолоджували на крижаній бані. Одержаний в результаті осад фільтрували, промивали холодним 20 % EtOAc/гексани, та сушили на повітрі, одержуючи (3R, 5R)-3-бутил-3-етил-7(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбальдегід-1,1-діоксид (4,00 г, 53,5 % вихід) у вигляді білої кристалічної твердої речовини. Маточний розчин концентрували насухо, та залишок очищали на 120 г силікагелю, елююючи 20 до 40 % EtOAc/гексани, 1 одержуючи додатковий продукт у вигляді білої твердої речовини: H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,74 (т, J=7,0 Гц, 3 H) 0,81 (т, J=7,4 Гц, 3 H) 0,96-1,29 (м, 4 H) 1,33-1,54 (м, 2 H) 1,68-1,81 (м, 1 H) 2,01-2,13 (м, 1 H) 2,80 (д, J=9,8 Гц, 1 H) 3,13 (д, J=15,0 Гц, 1 H) 3,58 (с, 3 H) 3,63 (д, J=15,0 Гц, 1 H) 5,98 (д, J=9,8 Гц, 1 H) 6,27 (с, 1 H) 7,32-7,40 (м, 1 H) 7,40-7,49 (м, 4 H) 8,23 (с, 1 H) 10,27 + (с, 1 H); РХ-МС (ES ) m/z 416,3 [M+H] Спосіб 2: 1,5 M розчин Dibal-H в толуолі (1,467 л, 2,20 моль) додавали протягом понад 50 хвилин до розчину (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбонітрил-1,1-діоксиду (673,3 г, 1,63 моль) в CH2Cl2 (3,3 л), що струшували, в той же час підтримуючи температуру реакції нижче 0 °C. Реакційну суміш струшували протягом 30 хвилин з охолодженням до -10 °C. Реакційну суміш гасили дуже повільним додаванням 1M HCl (8 л) протягом понад 1,25 год., в той же час підтримуючи температуру реакції нижче +15 °C (обережно: додавання перших 500 мл HCl є деже екзотермічним!). Додавали толуол (7 л), та суміш струшували протягом 3 годин. Водну фазу видаляли та органічний шар промивали 1M HCl (5 л). Об'єднані водні шари екстрагували CH 2Cl2 (2,5 л) та CH2Cl2 і CH2Cl2/толуол шари об'єднували та промивали 10 % Na2CO3 (5 л), сушили над MgSO4, фільтрували та частково концентрували до утворення густої кашиці. Кашицю розбавляли гептаном (2-3 об'єми) та витримували при струшуванні протягом 15 хвилин. Тверді речовини збирали фільтрацією та промивали гептаном. Відфільтрований корж сушили на повітрі протягом 3 годин та потім сушили в вакуумній шафі протягом ночі при 50 °C, одержуючи (3R, 5R)-3-бутил-3-етил-7(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбальдегід-1,1-діоксид у вигляді білої твердої речовини (533 г). Спосіб 3: (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбальдегід-1,1-діоксид (2402 г, 5,82 моль) та толуол (20714 г) додавали до реактора та струшували при 40 °C, утворюючи розчин. Потім розчин охолоджували приблизно до -35 °C та обробляли розчином DIBAL-H (1,5 M в толуол, 4472 г, 7,85 моль), в той же час підтримуючи температуру всередині нижче ніж -30 °C. Реакційну суміш струшували приблизно 16 UA 110338 C2 5 10 15 при -30 °C протягом однієї години до завершення, та потім гасили повільним додаванням 1N розчину HCl (приблизно 40 кг) до pH 1,0. Реакційну суміш нагрівали приблизно до 30 °C та струшували протягом однієї години. Струшування припиняли та нижній водний шар відокремлювали, а органічний двічі промивали 1N розчином HCl (кожен раз по 7200 мл), в той же час підтримуючи температуру приблизно на 40 °C. Водні шари об'єднували та знову промивали толуолом (4126 г). Об'єднані органічні шари промивали насиченим водним розчином хлориду натрію (9000 мл) та потім випаровували при зниженому тиску приблизно до 12 літрів. Далі розчинник випаровували при атмосферному тиску до кінцевого об'єму приблизно 7 літрів та обробляли гептанами (16420 г). Вміст охолоджували приблизно до 15 °C та струшували протягом приблизно двох годин. Тверду речовину розтирали вручну та вимивали з реактора гептанами (2 × 3260 г) та збирали фільтрацією. Зібрану тверду речовину промивали додатковою кількістю гептанів (3180 г), потім сушили при 50 °C, одержуючи (3R, 5R)-3-бутил-3етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбальдегід-1,1-діоксид (2128 г, 88 %) Проміжна сполука 10: диметил 3-({[(3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-1,1діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}аміно)пентандіоат O O O O S O N H O N O OH 20 25 30 (3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбальдегід-1,1-діоксид (298 мг, 0,691 ммоль) та диметил-3-амінопентандіоат (181 мг, 1,036 ммоль) (одержували як в Journal of the American Chemical Society 2005, 127, 247) загружали в ДХМ (4 мл) та струшували протягом 30 хвилин. Реакційну суміш обробляли NaHB(OAc)3 (293 мг, 1,381 ммоль) та струшували при 22 °C протягом 16 годин, після чого РХМС показала, що реакція завершилась. Суміш розбавляли ДХМ, двічі промивали H 2O, сушили над MgSO4, фільтрували та концентрували насухо. Залишок очищували на 40 г силікагелю, елююючи 20 до 100 % EtOAc/гексани, одержуючи диметил 3-({[(3R, 5R)-3-бутил-3-етил-4гідрокси-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8іл]метил}аміно)пентандіоат (304 мг, 0,515 ммоль, 74,5 % вихід) у вигляді олії янтарного кольору: + РХ-МС (ES ) m/z 591,3 [M+H]. Проміжна сполука 11: діетил {(E)-2-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]етеніл}фосфонат O O O S O P O N H O 35 40 Тетраетилметандіілбіс(фосфонат) (645 мг, 2,24 ммоль), розчинений в ТГФ (7 мл), обробляли NaH (83 мг, 2,075 ммоль, 60 % дисперсія в олії) зі струшуванням при 22 °C протягом 30 хвилин. Додавали (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбальдегід-1,1-діоксид (300 мг, 0,722 ммоль), та суміш струшували протягом 1 години при температурі навколишнього середовища, після чого РХМС показала завершення перетворення. Суміш гасили H2O, розділяли між EtOAc та сольовим розчином, та фази відокремлювали. Органічну фазу сушили над MgSO 4, фільтрували та концентрували до олії. Залишок очищували на 40 г силікагелю, елююючи 40 до 100 % EtOAc/гексани, одержуючи діетил {(E)-2-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4 17 UA 110338 C2 + бензотіазепін-8-іл]етеніл}фосфонат (342 мг, 86 % вихід) у вигляді безбарвної олії: РХ-МС (ES ) m/z 550,4 [M+H]. Проміжна сполука 12: диметил N-{[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}-L-цистинат 5 O O H2 N S 15 20 O O S N H N H O O O 10 S (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8карбальдегід-1,1-діоксид (231 мг, 0,556 ммоль) та диметил L-цистинат (95 мг, 0,278 ммоль) загружали в ДХМ (8 мл) та обробляли TEA (0,077 мл, 0,556 ммоль) при температурі навколишнього середовища протягом 1 години. Додавали NaHB(OAc) 3 (295 мг, 1,390 ммоль), та реакційну суміш струшували протягом 16 години. Суміш розподіляли між ДХМ та насиченим розчином NaHCO3, та фази розділяли. Водну фазу екстрагували ДХМ, та органічні фази об'єднували, сушили над MgSO4, фільтрували та концентрували до прозорої олії. Сиру речовину очищували на 40 г силікагелю, елююючи 20 до 100 % EtOAc/гексани, одержуючи диметил N-{[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,41 бензотіазепін-8-іл]метил}-L-цистинат (167 мг, 45,0 % вихід): H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,67-0,88 (м, 12 H) 0,96-1,29 (м, 8 H) 1,32-1,53 (м, 4 H) 1,66-1,80 (м, 2 H) 2,00-2,13 (м, 2 H) 2,54 (д, J=10,0 Гц, 2 H) 2,57-2,70 (м, 2 H) 2,93-3,13 (м, 6 H) 3,42 (с, 7 H) 3,46-3,78 (м, 13 H) 5,94 (д, J=9,6 + Гц, 2 H) 6,07 (с, 2 H) 7,26-7,36 (м, 2 H) 7,37-7,46 (м, 8 H) 7,87-8,01 (м, 2 H); РХ-МС (ES ) m/z 668.22 [M+H]. Проміжна сполука 13: 1,1-диметилетил N-{[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}-L-метіонінат O S O O O S N H N H O 25 30 35 40 Кашицю 1,1-диметилетил-L-метіонінату (218 мг, 0,902 ммоль) в ДХМ (3 мл) обробляли TEA (126 мкл, 0,902 ммоль) при температурі навколишнього середовища протягом 15 хвилин, після якого додавали (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбальдегід-1,1-діоксид (250 мг, 0,602 ммоль), та суміш струшували протягом 1 години. Додавали NaHB(OAc)3 (255 мг, 1,203 ммоль), та реакційну суміш струшували протягом ночі при температурі навколишнього середовища, після чого РХМС показала перетворення до бажаного продукту. Суміш розподіляли між ДХМ та сольовим розчином, та фази розділяли. Водну фазу екстрагували ДХМ, та об'єднані органічні фази сушили над MgSO 4, фільтрували та концентрували до прозорої олії. Залишок очищували на 40 г силікагелю, елююючи 20 до 60 % EtOAc/гексани, одержуючи 1,1-диметилетил N-{[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}-L-метіонінат у вигляді білої 1 твердої речовини (270 мг, 74 % вихід): H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,75 (т, J=7,0 Гц, 3 H) 0,80 (т, J=7,4 Гц, 3 H) 0,98-1,28 (м, 4 H) 1,36 (с, 9 H) 1,38-1,53 (м, 2 H) 1,65-1,85 (м, 3 H) 2,03 (с, 3 H) 2,03-2,13 (м, 1 H) 2,33-2,43 (м, 1 H) 2,53 (ш с, 2 H) 2,55 (д, J=4,3 Гц, 1 H) 3,03 (д, J=14,8 Гц, 1 H) 3,09-3,17 (м, 1 H) 3,43 (с, 3 H) 3,52 (д, J=14,8 Гц, 1 H) 3,62 (ш с, 2 H) 5,93 (д, J=9,6 Гц, 1 H) + 6,07 (с, 1 H) 7,25-7,36 (м, 1 H) 7,36-7,47 (м, 4 H) 7,94 (с, 1 H); РХ-МС (ES ) m/z 605.3 [M+H]. 18 UA 110338 C2 Проміжна сполука 14: (3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-карбальдегід-1,1-діоксид O O S H O N O OH 5 10 15 20 (3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-карбонітрил-1,1-діоксид (1,99 г, 4,64 ммоль), розчинені в ДХМ (25 мл), обробляли по краплям 1M DIBAL-H в толуолі (5,34 мл, 5,34 ммоль) при 0 °C зі струшуванням протягом 1 години. Додавали другу порцію 1M DIBAL-H (5,34 мл, 5,34 ммоль), та суміш струшували протягом наступних 2 годин при 0 °C, після чого РХМС показала перетворення до продукту. До суміші додавали 1N HCl (10mL), та суміш енергійно струшували протягом 16 годин. Органічну фазу віокремлювали, та водну фазу тричі екстрагували ДХМ. Органічні шари об'єднували, сушили над MgSO4, фільтрували та концентрували насухо. Залишок очищували на 40 г силікагелю, елююючи 10 до 50 % EtOAc/гексани, одержуючи (3R, 5R)-3-бутил-3-етил-4гідрокси-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-карбальдегід-1,1-діоксид 1 (1,42 г, 70,9 % вихід) у вигляді білої піни: H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,78 (т, J=7,1 Гц, 3 H), 0,85 (т, J=7,3 Гц, 3 H), 1,02-1,38 (м, 4 H), 1,38-1,52 (м, 1 H), 1,56-1,73 (м, 1 H), 1,78-1,91 (м, 1 H), 2,01-2,15 (м, 1 H), 3,39-3,51 (м, 2 H), 3,55 (с, 3 H), 6,31 (с, 1 H), 6,38 (с, 1 H), 7,32-7,75 (м, 5 H), + 8,20 (с, 1 H), 8,22 (с, 1 H), 10,26 (с, 1 H). РХ-МС (ES ) m/z 432.2 [M+H]. Проміжна сполука 15: {[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл]метил}амін O O S N N O 25 30 35 Суміш (3R, 5R)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8карбонітрил-1,1-діоксиду (468 мг, 1,134 ммоль) та 10 % паладію на вугіллі (60,4 мг, 0,567 ммоль) в етанолі (20 мл) додавали до гідрохлоридної кислоти (0,279 мл, 3,40 ммоль) та гідрогенізували при тиску 40 фунт на кв.дюйм протягом ночі, потім фільтрували та концентрували. Залишок очищували за допомогою ВЕРХ (елююючи MeCN/H2O з 0,05 % ТФО-H2O та 0,05 % ТФО-MeCN), 1 одержуючи названу сполуку (621 мг, 79 %, ТФО сіль) у вигляді білої твердої речовини: H ЯМР (CDCl3) м.ч, 8,07-8,46 (ш с, 2H), 7,98 (с, 1H), 7,31-7,57 (м, 5H), 6,27 (с, 1H), 6,17 (с, 1H), 4,124,27 (м, 1H), 3,98-4,12 (м, 1H), 3,55 (с, 3H), 3,38-3,48 (м, 1H), 3,29 (д, J=15,0 Гц, 1H), 2,14-2,38 (м, 1H), 1,80-1,99 (м, 1H), 1,44-1,75 (м, 2H), 1,14-1,38 (м, 3H), 0,97-1,15 (м, 1H), 0,90 (т, J=7,2 Гц, 3H), + 0,82 (т, J=6,8 Гц, 3H); ЕС-РХМС m/z 417 (M+H) . Проміжна сполука 16: N-{[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}-2-хлорацетамід O Cl O O S N N O 19 UA 110338 C2 5 10 До охолодженого кригою розчину {[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}аміну (50 мг, 0,120 ммоль) в ДХМ (6 мл) додавали піридин (0,097 мл, 1,20 ммоль) та хлорацетилхлорид (0,048 мл, 0,60 ммоль). Реакційну суміш струшували при кімнатній температурі протягом ночі, потім концентрували при зниженому тиску, одержуючи світло-жовту олію. Сиру названу сполуку (55 мг, 92 %) 1 використовували без додаткової очистки: H ЯМР (CDCl3) м.ч. 7,27-7,45 (м, 5H), 6,91-7,13 (м, 1H), 6,17 (с, 1H), 6,05 (с, 1H), 4,44 (д, J=6,1 Гц, 2H), 4,05 (д, J=7,0 Гц, 3H), 3,53 (с, 3H), 3,37 (с, 1H), 3,02 (с, 2H), 2,09-2,24 (м, 1H), 1,69-1,90 (м, 1H), 0,99-1,60 (м, 6H), 0,86 (т, J=7,4 Гц, 3H), 0,80 + (т, J=7,0 Гц, 3H); ЕС-РХМС m/z 494 (M+H) . Проміжна сполука 17: етил (2E)-3-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]-2-пропеноат O EtO2 C O S N O 15 20 25 В запаяній трубці, розчин (3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл-трифлуорметансульфонату (3,5 г, 6,53 ммоль) в ДМФ (30 мл) обробляли триетиламіном (4,55 мл, 32,7 ммоль), після чого етилакрилатом (3,56 мл, 32,7 ммоль) та біс(трифенілфосфін)паладій(II) хлоридом (0,459 г, 0,653 ммоль). Реакційну суміш струшували при 120 °C протягом ночі, охолоджували до кімнатної температури та розподіляли між H2O та EtOAc. Органічний шар промивали насиченим сольовим розчином, сушили (Na2SO4), фільтрували та концентрували при зниженому тиску. Очистка на силікагелі (EtOAc: гексани= 1:6 до 2:1) призводила до одержання названої сполуки (3,06 г, 94 %) у вигляді 1 білої твердої речовини: H ЯМР (CDCl3): м.ч. 8,21 (с, 1H), 7,85 (д, J=16,2 Гц, 1H), 7,30-7,48 (м, 5H), 6,56 (д, J=16,0 Гц, 1H), 6,19 (с, 1H), 6,06 (д, J=6,8 Гц, 1H), 4,18-4,30 (м, 2H), 3,55 (с, 3H), 3,44 (д, J=14,9 Гц, 1H), 3,02 (д, J=14,9 Гц, 1H), 2,08-2,26 (м, 1H), 1,76-1,93 (м, 1H), 1,38-1,54 (м, 3H), 1,28-1,35 (м, 3H), 1,03-1,21 (м, 2H), 0,88 (т, J=7,4 Гц, 3H), 0,81 (т, J=7,0 Гц, 3H); ЕС-РХМС m/z + 486 (M+H) . Проміжна сполука 18: діетил 3-амінопентандіоат O 40 O EtO 30 35 N OEt До розчину β-глутамінової кислоти (500 мг, 3,40 ммоль) в EtOH (10 мл) додавали по краплям тіонілхлорид (0,992 мл, 13,59 ммоль). Реакційну суміш струшували при кімнатній температурі протягом ночі та концентрували при зниженому тиску. Залишок розподіляли між ДХМ та насиченим розчином карбонату калію. Органічний шар промивали насиченим сольовим розчином, сушили (Na2SO4), фільтрували та концентрували при зниженому тиску, одержуючи 1 названу сполуку (702 мг, 97 %) у вигляді безбарвної олії: H ЯМР (CDCl3) м.ч. 4,04-4,27 (м, 4H), 3,46-3,74 (м, 1H), 2,24-2,60 (м, 4H), 1,09-1,29 (м, 6H). Проміжна сполука 19: (3R, 5R)-8-(3-бромпропіл)-3-бутил-3-етил-7-(метилокси)-5-феніл2,3,4,5-тетрагідро-1,4-бензотіазепін 1,1-діоксид O O S Br N O 20 UA 110338 C2 5 10 До розчину 3-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро1,4-бензотіазепін-8-іл]-1-пропанол (84,2 мг, 0,189 ммоль) в ТГФ (5 мл) додавали трифенілфосфін (99 мг, 0,378 ммоль) та карбону тетрабромід (125 мг, 0,378 ммоль). Реакційну суміш струшували при кімнатній температурі протягом ночі та розподіляли між H2O та ДХМ. Органічний шар промивали сольовим розчином, сушили (Na 2SO4), фільтрували та концентрували при зниженому тиску. Очистка з використанням силікагелю (EtOAc:гексани = 10:90 до 50:50) призводила до одержання названої сполуки (78 мг, 80 %) у вигляді прозорої олії: + ЕС-РХМС m/z 508 (M+H) . Проміжна сполука 20: 3-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл]пропілметансульфонат O O S O O S O N O 15 20 25 До охолодженого кригою розчину 3-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]-1-пропанолу (250 мг, 0,561 ммоль) в ДХМ (10 мл) додавали триетиламін (0,235 мл, 1,683 ммоль) та метансульфонілхлорид (0,048 мл, 0,617 ммоль). Реакційну суміш струшували при кімнатній температурі протягом ночі та розподіляли між H2O та ДХМ. Органічний шар промивали насиченим сольовим розчином, сушили (Na2SO4), фільтрували та концентрували при зниженому тиску, одержуючи названий продукт (280 мг, 1 91 %) у вигляді світло-жовтої твердої речовини: H ЯМР (CDCl3) м.ч. 7,82 (с, 1H), 7,27-7,52 (м, 5H), 6,11 (с, 1H), 6,01 (д, J=8,0 Гц, 1H), 4,04-4,26 (м, 2H), 3,47 (с, 3H), 3,31-3,43 (м, 1H), 2,85-3,07 (м, 4H), 2,46-2,78 (м, 4H), 1,73-2,27 (м, 4H), 1,35-1,61 (м, 4H), 0,97-1,34 (м, 8H), 0,86 (т, J=7,4 Гц, 3H), 0,80 (т, J=6,8 Гц, 3H). Проміжна сполука 21: 4-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5тетрагідро-1,4-бензотіазепін-8-іл]бутан нітрил O O S NC N O 30 35 40 До розчину 3-[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро1,4-бензотіазепін-8-іл]пропілметансульфонату (90 мг, 0,172 ммоль) в ДМСО (5 мл) додавали ціанід натрію (16,84 мг, 0,344 ммоль). Реакційну суміш струшували при 60 °C протягом вихідних та розподіляли між H2O та EtOAc. Органічний шар промивали насиченим сольовим розчином, сушили (Na2SO4), фільтрували та концентрували при зниженому тиску. Очистка з використанням силікагелю (EtOAc:гексани= 10:90 до 1:1) призводила до одержання названої 1 сполуки (76 мг, 95 %) у вигляді білої твердої речовини: H ЯМР (CDCl3) м.ч, 7,81 (c, 1H), 7,267,48 (м, 5H), 6,12 (c, 1H), 6,02 (c, 1H), 3,40 (д, J=14,8 Гц, 1H), 3,01 (д, J=14,6 Гц, 1H), 2,64-2,80 (м, 2H), 2,29 (т, J=7,2 Гц, 2H), 2,08-2,21 (м, 1H), 1,75-1,98 (м, 3H), 1,37-1,51 (м, 2H), 0,99-1,35 (м, 4H), + 0,86 (т, J=7,4 Гц, 3H), 0,80 (т, J=7,0 Гц, 3H); ЕС-РХМС m/z 455 (M+H) . Проміжна сполука 22: (3R, 5R)-8-(бромметил)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3дигідро-1,4-бензотіазепін-4(5H)-ол-1,1-діоксид 21 UA 110338 C2 O O S Br N O OH 5 10 15 (3R, 5R)-8-(бромметил)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-1,1-діоксид (627 мг, 1,305 ммоль), розчинений в ДХМ (20 мл), обробляли m-CPBA (292 мг, 1,305 ммоль) зі струшуванням при 0 °C протягом 1 години, після чого РХМС показала завершене перетворення до продукту. Реакційну суміш обробляли 10 % Na2SO3 з енергійним струшуванням протягом 15 хвилин, розбавляли ДХМ, та органічну фазу відокремлювали, сушили над MgSO4, фільтрували та концентрували насухо. Залишок очищували на 40 г силікагелю, елююючи ДХМ, одержуючи (3R, 5R)-8-(бромметил)-3-бутил-3-етил-7-(метилокси)-5феніл-2,3-дигідро-1,4-бензотіазепін-4(5H)-ол-1,1-діоксид (621 мг, 96 % вихід) у вигляді білої піни: 1 H ЯМР (400 МГц, ДМСО-d6) м.ч. 0,79 (т, J=7,1 Гц, 3 H), 0,85 (т, J=7,3 Гц, 3 H), 1,04-1,37 (м, 4 H), 1,37-1,49 (м, 1 H), 1,58-1,71 (м, 1 H), 1,77-1,89 (м, 1 H), 2,01-2,14 (м, 1 H), 3,36-3,44 (м, 2 H), 3,45-3,55 (м, 3 H), 4,69 (с, 2 H), 6,15 (с, 1 H), 6,35 (с, 1 H), 7,30-7,72 (м, 5 H), 7,98 (с, 1 H), 8,10 (с, + 1 H); РХ-МС (ES ) m/z 496,1, 498,1 [M+H]. Проміжна сполука 23: діетил {[(3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-1,1-діоксидо5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}фосфонат O O P O O O S N O OH 20 25 30 (3R, 5R)-8-(бромметил)-3-бутил-3-етил-7-(метилокси)-5-феніл-2,3-дигідро-1,4-бензотіазепін4(5H)-ол-1,1-діоксид (171 мг, 0,344 ммоль), розчинений в толуолі (10 мл), обробляли триетилфосфітом (1,189 мл, 6,80 ммоль), потім нагрівали при кіп'ятінні зі зворотним холодильником протягом 16 годин. Реакційну суміш промивали H 2O, сушили над MgSO4, фільтрували та концентрували насухо. Залишок очищували за допомогою ОФ-ВЕРХ (з оберненою фазою) (30 × 150 мм, C18 5мкм H 2O Sunfire колонка; MeCN+0,05 % ТФО та H2O+0,05 % ТФО використовували як систему розчинників; 15 до 100 %), одержуючи діетил {[(3R, 5R)-3-бутил-3-етил-4-гідрокси-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,41 бензотіазепін-8-іл]метил}фосфонат (48,5 мг, 25,4 % вихід) у вигляді прозорої олії: H ЯМР (400 МГц, ДМСО-d6) δ м.ч. 0,79 (т, J=6,6 Гц, 3 H), 0,82-0,93 (м, 3 H), 1,04-1,71 (м, 12 H), 1,91-2,02 (м, 1 H), 2,02-2,13 (м, 1 H), 3,13-3,28 (м, 2 H), 3,29-3,40 (м, 2 H), 3,41 (с, 3 H), 3,95 (м, J=7,2, 7,2, 7,2, 7,2, 2,5 Гц, 4 H), 6,12 (с, 1 H), 6,28-6,38 (м, 1 H), 7,31-7,39 (м, 1 H), 7,42 (т, J=7,4 Гц, 2 H), 7,46+ 7,60 (м, 2 H), 7,82 (д, J=1,8 Гц, 1 H), 7,99-8,08 (м, 1 H); РХ-МС (ES ) m/z 554,31 [M+H]. Проміжні сполуки 24: тетраетил [({[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}іміно)диметандііл]біс(фосфонат) 35 O O P O O O O O S N P O N H O 22 UA 110338 C2 5 10 15 {[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-іл]метил}амін (155 мг, 0,37 ммоль) змішували з тозилатною кислотою (каталітична кількість) в толуолі (20 мл) та концентрували насухо, азеотропно видаляючи будьяку залишкову H2O. Додавали параформальдегід (11,2 мг, 0,37 ммоль) та толуол (5 мл), та суміш нагрівали при 75 °C з енергійним струшуванням протягом 30 хвилин, після чого додавали діетилфосфіт (0,048 мл, 0,37 ммоль). Через 30 хвилин до суміші додавали ТГФ (20 мл), та одержану в результаті гомогенну реакційну суміш струшували протягом ночі при 75 °C. Суміш концентрували насухо та очищували за допомогою ОФ-ВЕРХ (30 × 150 Sunfire колонка в кислих умовах; MeCN+0,05 % ТФО та H2O+0,05 % ТФО використовували як систему розчинників; 30 % до 100 % понад 10 хвилин, 100 % до 100 % до 12 хвилин), одержуючи тетраетил [({[(3R, 5R)-3бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін-8іл]метил}іміно)диметандііл]біс(фосфонат) (49,7 мг, 18,6 % вихід) у вигляді жовтої олії: РХ-МС + (ES ) m/z 717,4 [M+H]. Проміжна сполука 25: діетил [({[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл2,3,4,5-тетрагідро-1,4-бензотіазепін-8-іл]метил}аміно)метил]фосфонат O O S O O P O N H N H O 20 25 30 35 {[(3R, 5R)-3-бутил-3-етил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-іл]метил}амін (45,8 мг, 0,110 ммоль) змішували з параформальдегідом (3,47 мг, 0,110 ммоль) та діетилфосфітом (0,014 мл, 0,11 ммоль) в ТГФ (4 мл) та струшували протягом 16 годин при 75 °C, після чого весь розчинник видалявся, та РХМС показала 95 %. Стадія 4: Очищений етил 2-бутил-N-(фенілметиліден)норлейцинат (112,3 г, 370 ммоль) обробляли 1N HCl (444 мл, 444 ммоль) та струшували при 25 °C протягом 30 хвилин. Реакційну суміш промивали гексанами (2 × 200 мл), потім водний шар охолоджували до 0 °C та регулювали pH до 12 поєднанням 3N та 6N NaOH. Водну суміш екстрагували MTBE (4x), потім об'єднані органічні шари промивали сольовим розчином, сушили (MgSO 4), фільтрували та концентрували в вакуумі, одержуючи етил 2-бутилнорлейцинат (74 г, 93 % вихід) у вигляді жовтої олії. Стадія 5: Розчин етил 2-бутилнорлейцинату (37 г, 172 ммоль) в ТГФ (275 мл) при 25 °C обробляли 2M LAH (86 мл, 172 ммоль) через додаткову воронку протягом понад 30 хвилин. Реакційну суміш нагрівали до 65 °C та струшували протягом 2 годин, потім давали досягнути температури навколишнього середовища, в той же час струшуючи протягом ночі. Реакційну суміш охолоджували до 0 °C, потім послідовно обробляли H20 (7,82 мл), 15 % NaOH (7,82 мл) та H2O (23,5 мл). Суміш струшували протягом 2 годин, потім фільтрували, промиваючи ТГФ. Фільтрат концентрували в вакуумі, одержуючи 2-аміно-2-бутил-1-гексанол (29,8 g, >95 % вихід) у вигляді густої жовтої олії, яка затвердівала при стоянні. Стадія 6: Розчин 2-аміно-2-бутил-1-гексанолу (62,8 г, 362 ммоль) в EtOAc (454 мл) при 25 °C обробляли хлорсульфоновою кислотою (29,1 мл, 435 ммоль). Реакційну суміш нагрівали до 40 °C та струшували протягом 3 годин, потім охолоджували до 25 °C. Густу суміш ставили в холодильник на всю ніч, потім фільтрували, промиваючи холодним EtOAc. Тверду речовину сушили, одержуючи 2-аміно-2-бутилгексанезилгідрогенсульфат у вигляді білої твердої речовини 1 з виходом >50 %: H ЯМР (D2O) δ м.ч. 3,92 (с, 2H), 1,42-1,65 (м, 4H), 1,02-1,25 (м, 8H), 0,72 (т, J=6,7 Гц, 6H). Проміжна сполука 29: S-[5-бром-4-(метилокси)-2-(фенілкарбоніл)феніл] діетилтіокарбамат O Br N S O O 45 50 Стадія 1: До розчину 2-бром-1,4-біс(метилокси)бензолу (203 г, 934 ммоль) та бензоїлхлориду (125 мл, 1074 ммоль) в ДХМ (800 мл) при 5ºC додавали трифлатну кислоту (83 мл, 934 ммоль) протягом 1 години. Реакційній суміші давали нагрітися до температури навколишнього середовища та потім повільно обережно нагрівали до кипіння зі зворотнім холодильником та струшували протягом 48 годин. Реакційну суміш охолоджували, та додавали MeOH (20 мл), та струшування продовжували протягом 30 хвилин. Реакційну суміш виливали в крижану H2O та струшували протягом 1 години. Шари розділяли та органічну фазу промивали H2O, сушили над MgSO4 та концентрували до оранжевої твердої речовини. Додавали MTBE (500 мл), та суміш струшували протягом ночі. Однржану в результаті тверду речовину 25 UA 110338 C2 5 10 15 20 25 30 35 40 45 фільтрували, промивали сумішшю 1:1 MTBE/гексани та сушили на повітрі, одержуючи [4-бром2,5-біс(метилокси)феніл](феніл)метанон (210,6 г, 70 %) у вигляді світло-сірої твердої речовини: 1 H ЯМР (ДМСО-d6) δ м.ч. 7,68 (д, J=8,0 Гц, 2H), 7,62 (т, J=8,0 Гц, 1H), 7,48 (т, J=7,4 Гц, 2H), 7,42 (с, 1H), 7,07 (с, 1H), 3,76 (с, 3H), 3,59 (с, 3H). Стадія 2: Розчин [4-бром-2,5-біс(метилокси)феніл](феніл)метанону (290 г, 0,903 моль) в ДХМ (1,5 л) по краплям додавали протягом понад 1 години до розчину 1M BCl3 в ДХМ (1,13 л, 1,13 моль), що струшували, в той же час підтримуючи температуру реакції нижче 5 °C. Реакційну суміш струшували при або нижче 0 °C протягом 30 хвилин та потім гасили повільним додаванням MeOH (500 мл) протягом 30 хвилин при 10 °C. Додавали 2N HCl (1 л) при 15 °C протягом понад 30 хвилин. Шари розділяли, та органічну фазу концентрували, використовуючи роторний випарювач до об'єму близько 500 мл, потім розбавляли гексанами. Одержані в результаті жовті кристали збирали фільтрацією та сушили на повітрі, одержуючи [4-бром-21 гідрокси-5-(метилокси)феніл](феніл)метанон (258 г, 93 %): H ЯМР (CDCl3) δ м.ч. 11,67 (с, 1H), 7,64-7,72 (м, 2H), 7,59 (т, J=7,4 Гц, 1H), 7,51 (т, J=7,2 Гц, 2H), 7,33 (с, 1H), 7,02 (с, 1H), 3,71 (с, 3H) Стадія 3: Твердий калію трет-бутоксид (116 г, 1,03 моль) додавали порціями до розчину [4бром-2-гідрокси-5-(метилокси)феніл](феніл)метанону (253,33 г, 0,825 моль) в ДМФ (800 мл), що струшували, в той же час підтримуючи температуру всередині нижче 20ºC. Струшування підтримували з охолодженням протягом 15 хвилин до досягнення температури всередині приблизно 0ºC. Охолоджуючу баню видаляли, та струшування продовжували протягом 30 хвилин з нагріванням до 6ºC температури всередині. Потім повільним струменем додавали розчин N, N-діетилтіокарбамоїлхлориду (150 г, 0,99 моль) в ДМФ (300 мл) протягом понад 5 хвилин. Далі одержану в результаті темну суміш нагрівали до 60ºC та витримували протягом 3 годин. Суміш розбавляли MTBE (1,5 л) та H2O (1,5 л), потім швидко струшували. Шари розділяли, та водну фазу екстрагували MTBE (1,5 л). Об'єднані органічні шари промивали 0,2N NaOH (2 × 1,5 L) та один раз сольовим розчином (1 л). Темно-червону органічну фазу сушили над MgSO4, фільтрували та концентрували до 1 л густої кашиці, відганяючи гептан. Тверді речовини збирали фільтрацією, промиваючи гептаном, та сушили на повітрі, одержуючи O-[5бром-4-(метилокси)-2-(фенілкарбоніл)феніл]діетилтіокарбамат (301,3 г, 86 %) у вигляді світло1 оранжевої твердої речовини: H ЯМР (CDCl3) δ м.ч. 7,81 (д, J=8,0 Гц, 2H), 7,52 (т, J=7,6 Гц, 1H), 7,36-7,45 (м, 3H), 7,00 (с, 1H), 3,86 (с, 3H), 3,63 (кв, J=7,0 Гц, 2H), 3,22 (кв, J=7,2 Гц, 2H), 1,06 (кв, 6H). Стадія 4: Суміш O-[5-бром-4-(метилокси)-2-(фенілкарбоніл)феніл]діетил-тіокарбамату (301 г, 0,71 моль) в дифеніловому етері (1 л), що струшували, поступово нагрівали до 215ºC температури всередині протягом понад 1 години та підтримували дану температуру протягом 3,5 годин. Темний розчин охолоджували до 100ºC та обробляли Darco G-60 (20 г). Далі суміш охолоджували до 70ºC зі струшуванням та фільтрували через целіт, промиваючи гептаном. Фільтрат розбавляли гептаном (4 л) з охолодженням до 5ºC та витримували при 0ºC протягом понад 30 хвилин. Супернатант видаляли декантуванням, та тверді речовини, що залишились, розтирали з гексанами. Тверді речовини, що залишились, розчиняли в ДХМ та концентрували в вакуумі, до твердої речовини, яку розтирали з гексанами та збирали фільтрацією. Речовину сушили під високим вакуумом, одержуючи S-[5-бром-4-(метилокси)-21 (фенілкарбоніл)феніл]діетилтіокарбамат (224 г, 74 %) у вигляді темно-сірої твердої речовини: H ЯМР (CDCl3) δ м.ч. 7,72-7,82 (м, 3H), 7,52 (т, J=7,4 Гц, 1H), 7,39 (т, J=7,6 Гц, 2H), 6,90 (с, 1H), 3,87 (с, 3H), 3,15-3,29 (ш с, 2H), 3,02-3,15 (ш с, 2H), 0,68-1,09 (м, 6H). Проміжна сполука 30: [2-[(2-аміно-2-бутилгексанезил)тіо]-4-бром-5(метилокси)феніл](феніл)метанон Br O S NH2 O 50 Суспензію S-[5-бром-4-(метилокси)-2-(фенілкарбоніл)феніл]діетилтіо-карбамату (проміжна сполука 29) (15 г, 35,5 ммоль) в EtOH (75 мл) обробляли 6M KOH (23,68 мл, 142 ммоль), потім нагрівали при кип'ятінні зі зворотнім холодильником протягом 3 годин. Реакційну суміш 26 UA 110338 C2 5 10 15 розбавляли додатковою кількістю EtOH (200 мл), потім нагрівали при 70 °C протягом 15 годин. Реакційну суміш концентрували до одержання густої темно-червоної олії. В окремій колбі розчиняли 2-аміно-2-бутилгексанезилгідрогенсульфат (9,90 г, 39,1 ммоль) в H 2O (25 мл), потім нагрівали до 85 °C, після чого за допомогою піпетки додавали розчин тіофенолята в H 2O. Залишковий тіофенолят переносили з мінімальною кількістю EtOH та H 2O. Реакційну суміш струшували при 85 °C протягом 15 годин, потім охолоджували до температури навколишнього середовища. Реакційну суміш виливали в H2O та екстрагували ДХМ (3х). Об'єднані органічні шари промивали сольовим розчином, сушили (MgSO4), фільтрували та концентрували в вакуумі, одержуючи густу червону олію. Залишок очищували за допомогою хроматографії на силікагелі, використовуючи ДХМ/MeOH з 1 % NH4OH (градієнт від 100/0 % до 90:10 протягом понад 30 хвилин), одержуючи [2-[(2-аміно-2-бутилгексанезил)тіо]-4-бром-51 (метилокси)феніл](феніл)метанон (16,3 г, 96 % вихід) у вигляді густої червоної олії: H ЯМР (CDCl3) δ м.ч. 7,76-7,83 (м, 3H), 7,60 (т, J=7,4 Гц, 1H), 7,47 (т, J=7,8 Гц, 2H), 6,84 (c, 1H), 3,88 (c, + 3H), 2,81 (c, 2H), 0,78-1,55 (м, 18H); РХ-МС (ЕС ) m/z 478,2 [M+H], 480,2 [M+H], Проміжна сполука 31: [2-[(2-аміно-2-бутилгексанезил)тіо]-4-бром-5(метилокси)феніл](феніл)метанон S Br N O 20 25 30 Розчин [2-[(2-аміно-2-бутилгексанезил)тіо]-4-бром-5-(метилокси)феніл](феніл)метанону (16,3 г, 34.1 ммоль) в толуолі (341 мл) обробляли лимонною кислотою (0,327 г, 1,703 ммоль), потім нагрівали при кип'ятінні зі зворотнім холодильником протягом 15 годин. Реакційну суміш в ємності з насадкою Діна-Старка (Dean-Stark) потім нагрівали при 130 °C протягом 10 годин, після чого додавали додаткову кількість лимонної кислоти (325 мг). Струшування продовжували при 130 °C протягом 15 годин, потім додавали лимонну кислоту (100 мг) та продовжували струшування при 130 °C протягом 10 годин. Реакційну суміш концентрували в вакуумі, потім очищували, за допомогою хроматографії на SiO 2, використовуючи суміш гексани:EtOAc (100:0 до 80:20) як елюєнт, одержуючи 8-бром-3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4бензотіазепін (10 г, 63,7 % винахід) у вигляді олії, яка затвердівала у рудувато-коричневу тверду 1 речовину при стоянні: H ЯМР (CDCl3) м.ч. 7,79 (с, 1H), 7,53 (д, J=8,2 Гц, 2H), 7,30-7,44 (м, 3H), + 6,59 (с, 1H), 3,71 (с, 3H), 3,23 (с, 2H), 1,16-1,66 (м, 12H), 0,86 (т, J=7,2 Гц, 6H); РХ-МС (ES ) m/z 460,2 [M+H], 462,2 [M+H]. Проміжна сполука 32: метил 3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4бензотіазепін-8-карбоксилат 35 O S O N O 40 45 Монооксид карбону барботували через суміш 8-бром-3,3-дибутил-7-(метилокси)-5-феніл2,3-дигідро-1,4-бензотіазепіну (проміжна сполука 31) (10 г, 21,72 ммоль), Pd(OAc) 2 (0,488 г, 2,172 ммоль) та dppp (0,896 г, 2,172 ммоль) в ДМСО (71,6 мл) протягом 15 хвилин. Реакційну суміш обробляли MeOH (3,51 мл, 87 ммоль) та TEA (4,54 мл, 32,6 ммоль), потім нагрівали при 70 °C протягом 3 годин, після чого додавали додаткову кількість MeOH (3,51 мл, 87 ммоль). Нагрівання в атмосфері CO продовжували протягом 39 годин. Реакційну суміш розбавляли MTBE, потім фільтрували черех пластинку целіту, промиваючи MTBE. Фільтрат виливали в H2O, далі відокремлений водний шар екстрагували MTBE (1x). Об'єднані органічні шари промивали 27 UA 110338 C2 5 сольовим розчином, сушили (MgSO4), фільтрували та концентрували в вакуумі. Залишок очищували за допомогою хроматографії на SiO2, використовуючи суміш гексани:EtOAc (100:0 до 60:40) як елюєнт, одержуючи метил 3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,41 бензотіазепін-8-карбоксилат (86 % вихід) у вигляді оранжевої олії: H ЯМР (CDCl3) м.ч. 8,01 (с, 1H), 7,53 (д, J=6,8 Гц, 2H), 7,30-7,44 (м, 3H), 6,69 (с, 1H), 3,93 (с, 3H), 3,72 (с, 3H), 3,22 (с, 2H), + 1,14-1,65 (м, 12H), 0,86 (т, J=7,2 Гц, 6H); РХ-МС (ES ) m/z 440,3 [M+H]. Проміжна сполука 33: [3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4-бензотіазепін-8іл]метанол S HO N O 10 15 20 Розчин метил 3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4-бензотіазепін-8карбоксилату (8,2 г, 18,65 ммоль) в ТГФ (34,0 мл) при 25 °C по краплям обробляли 2M LAH в ТГФ (13,99 мл, 28,0 ммоль). Реакційну суміш струшували протягом 15 хвилин, потім гасили H2O (1,1 мл), 15 % NaOH (1,1 мл) та H2O (3,4 мл). Після струшування протягом 30 хвилин реакційну суміш фільтрували через прокладку целіту, промиваючи ТГФ. Фільтрат концентрували в вакуумі, одержуючи [3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4-бензотіазепін-81 іл]метанол (7,7 г, 100 % вихід): H ЯМР (CDCl3) м.ч. 7,49-7,59 (м, 3H), 7,30-7,42 (м, 3H), 6,58 (с, + 1H), 3,68 (с, 3H), 3,22 (с, 2H), 1,13-1,65 (м, 12H), 0,86 (т, J=7,2 Гц, 6H); РХ-МС (ES ) m/z 412,3 [M+H]. Проміжна сполука 34: [3,3-дибутил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4бензотіазепін-8-іл]метанол S HO N H O 25 30 35 Розчин [3,3-дибутил-7-(метилокси)-5-феніл-2,3-дигідро-1,4-бензотіазепін-8-іл]метанолу (7,7 г, 18,71 ммоль) в ТГФ (51,1 мл) при 25 °C обробляли по краплям 1M BH3.ТГФ (20,58 мл, 20,58 ммоль), потім реакційну суміш струшували протягом 2 годин. Реакційну суміш гасили MeOH (30 мл), далі концентрували при зниженому тиску, одержуючи сиру білу піну. Очистка залишку за допомогою хроматографії на SiO2, використовуючи градієнт 6:1/гексани:EtOAc до 2:1/гексани:EtOAc давала суміш непрореагувавшої вихідної речовини та продукту (приблизно 85:15, відповідно). Дану речовину знову піддавали ідентичним умовам та очищували подібним способом, одержуючи [3,3-дибутил-7-(метилокси)-5-феніл-2,3,4,5-тетрагідро-1,4-бензотіазепін1 8-іл]метанол (6,56 г, 85 % вихід), що містить 5-10 % іміну: H ЯМР (CDCl3) м.ч. 7,50 (с, 1H), 7,24-7,47 (м, 5H), 6,09 (с, 1H), 5,78 (с, 1H), 4,59 (д, J=3,1 Гц, 2H), 3,49 (с, 3H), 2,76 (д, J=14,2 Гц, 1H), 2,50 (д, J=14,2 Гц, 1H), 2,01-2,24 (м, 2H),,08-1,72 (м, 10H), 0,90 (т, J=6,9 Гц, 3H), 0,85 (т, + J=7,2 Гц, 3H); РХ-МС (ES ) m/z 414,3 [M+H]. Проміжна сполука 35: [3,3-дибутил-7-(метилокси)-1,1-діоксидо-5-феніл-2,3,4,5-тетрагідро1,4-бензотіазепін-8-іл]метанол 40 28
ДивитисяДодаткова інформація
Назва патенту англійськоюChemical compounds
Автори англійськоюAquino, Christopher Joseph, Collins, Jon Loren, Cowan, David John, Wu, Yulin
Автори російськоюАквино Кристофер Джозеф, Коллинз Джон Лорен, Кован Дэйвид Джон, Ву Юлинь
МПК / Мітки
МПК: C07D 281/10, A61K 31/554, A61P 5/00, A61P 3/00
Код посилання
<a href="https://ua.patents.su/76-110338-khimichni-spoluki.html" target="_blank" rel="follow" title="База патентів України">Хімічні сполуки</a>
Хімічні сполуки, що є інгібіторами ненуклеозидної зворотної транскриптази

Номер патенту: 98784
Опубліковано: 25.06.2012
Автори: Жанґ Гуічанґ, Піт Ендрю Джеймс, Себагар Паул Річард, Янґмен Майкл, Чонґ Пек Йок
МПК: A01N 43/50, A61K 31/415
Мітки: транскриптази, ненуклеозидної, зворотної, інгібіторами, сполуки, хімічні
Формула / Реферат:
1. Сполука формули (І):, (I) де m дорівнює 1, 2, 3 або 4; n дорівнює 1, 2, 3 або 4; кожен R1 незалежно є галогеном, -CN, С1-С5алкілом, С2-С5алкенілом, С2-С6алкінілом, С3-С7циклоалкілом, гідроксилом, С1-С8алкоксилом, -C(O)OR5, -C(O)N(R5)2, -OR5, -R3CN або -N(R5)2; кожен R2 незалежно є...
Нові хімічні сполуки в ряду імідів перилентетракарбонової кислоти як органічні люмінофори червоного світіння та спосіб їх одержання

Номер патенту: 52316
Опубліковано: 16.12.2002
Автори: Гриньов Борис Вікторович, Шершуков Віктор Михайлович, Барикін Костянтин Едуардович
МПК: C07D 221/04, G02B 1/04, C07D 471/06, C09B 5/00
Мітки: імідів, перилентетракарбонової, хімічні, ряду, кислоти, світіння, червоного, люмінофори, одержання, спосіб, органічні, нові, сполуки
Формула / Реферат:
Пристрій виготовлено і випробувано. Автори Пухиря В. И. і Пухыря Л. С. щодня надягали пристрій на очі й дивилися телевізор, серіали протягом 4-х місяців. За час тренувань в авторів збільшилася гострота зору, і зникли чорні крапки в очах.
Сполуки бензимідазолу, фармацевтичні композиції, що містять ці сполуки, та спосіб лікування чутливих до позитивної модуляції гамк-рецепторних комплексів розладів цнс

Номер патенту: 54394
Опубліковано: 17.03.2003
Автори: САСАКІ Тосіро, УСІРОДА Осаму, Ветьєн Франк, ФУКУДА Йосімаса, ТЕУБЕР Лене
МПК: A61K 31/4439, C07D 417/10, A61P 43/00, C07D 405/10, C07D 409/10, A61K 31/00, A61P 25/00, A61K 31/505, C07D 401/10, A61P 25/04, A61K 31/425, A61K 31/4427, C07D 413/14, C07D 405/14, A61K 31/4184, C07D 417/14, A61K 31/506, C07D 235/06, A61K 31/4164, A61K 31/427, C07D 403/10, A61K 31/415, A61K 31/443, A61K 31/44, A61K 31/4433, C07D 521/00
Мітки: лікування, містять, чутливих, комплексів, бензимідазолу, композиції, позитивної, гамк-рецепторних, модуляції, цнс, спосіб, фармацевтичні, розладів, сполуки
Формула / Реферат:
1. Сполука формули:або її фармацевтично придатна сіль чи оксид, де R3 - ,де кожний А, В та D - СН, або один чи два з А, В та D - N, а інші - СН;R11 - феніл, бензимідазол або моноциклічний гетероарил, кожний з яких може бути заміщеним одним або більше замісниками,...
Спосіб одержання 4-арил-1,4,5,10-тетрагідропіразоло[4,3-b][1,5]бензодіазепінів

Номер патенту: 82264
Опубліковано: 25.03.2008
Автори: Лозинський Мирон Онуфрійович, Дзвінчук Ігор Борисович
МПК: A61K 31/5517, C07D 243/30
Мітки: одержання, спосіб, 4-арил-1,4,5,10-тетрагідропіразоло[4,3-b][1,5]бензодіазепінів
Формула / Реферат:
Спосіб одержання 4-арил-1,4,5,10-тетрагідропіразоло[3,4-b][1,5]бензодіазепінів загальної формули 1, (1)де при R=CH2CH2CN, Аr=4-NO2С6Н4 (а), 3-NO2С6Н4 (б), 4-СlС6Н4 (в), 4-FС6Н4 (г), 4-піридил (д), 3-піридил (е), 2-піридил (є);а при R-Ph Аr=4-NO2С6Н4 (ж), 3-NO2С6H4 (з), 4-СlС6Н4 (і), 4-FС6Н4 (й), 4-піридил (к), 2-піридил (л), який відрізняється тим, що...
Триазолопіримідинові сполуки, спосіб їх одержання (варіанти) та проміжні сполуки (варіанти)

Номер патенту: 73182
Опубліковано: 15.06.2005
Автори: Пальмгрен Андреас, Мусіль Тібор, Ларссон Ульф, Магнуссон Маттіас
МПК: C07D 239/56, C07D 487/04, C07D 317/44, C07B 61/00, C07D 239/47, C07D 405/12
Мітки: спосіб, варіанти, одержання, проміжні, сполуки, триазолопіримідинові
Формула / Реферат:
1. Сполука формули (І): (I).2. Спосіб одержання сполуки формули (І), що включає взаємодію сполуки формули (II): (II)з сіллю сполуки формули (III): (III).3. Спосіб одержання сполуки формули...
Попередній патент: Виготовлення капсул типу серцевина/оболонка, які мають різну геометричну форму, і їх подальша обробка
Наступний патент: Спосіб переробки целюлозної або лігноцелюлозної вихідної сировини
Випадковий патент: Секція вагонних тензометричних ваг