Спосіб одержання похідних 1-бензил-3-оксиметиліндазолу або їх солей з фармацевтично прийнятними основами







Формула / Реферат
(57) 1. Способ получения производных 1-бензил-3-оксиметилиндазола формулы (I)
A-CH2-O-CRR|-COOR||| (|)
где А представляет собой 1 -бензилиндазол-З-ил,
R и R' могут быть одинаковыми или
различными и представляют собой С1-С4-алкил,
R'" представляет собой Н, или их солей с фармацевтически приемлемыми основаниями, отличающийся тем, что соединение формулы (На)
(||а)
A-CН2-Y,
где Y представляет собой ОН, подвергают взаимодействию с кетоном формулы RCOR1,
где R и R' имеют вышеуказанные значения,
и хлороформом в присутствии гидроксида щелочного металла и, при необходимости, получают соль кислоты формулы (I) с фармацевтически приемлемыми основаниями.
2. Способ по п. 1,отличающи-й с я тем, что реакцию проводят при температуре кипения реакционной смеси и времени реакции от 30 мин до 12 ч
Текст
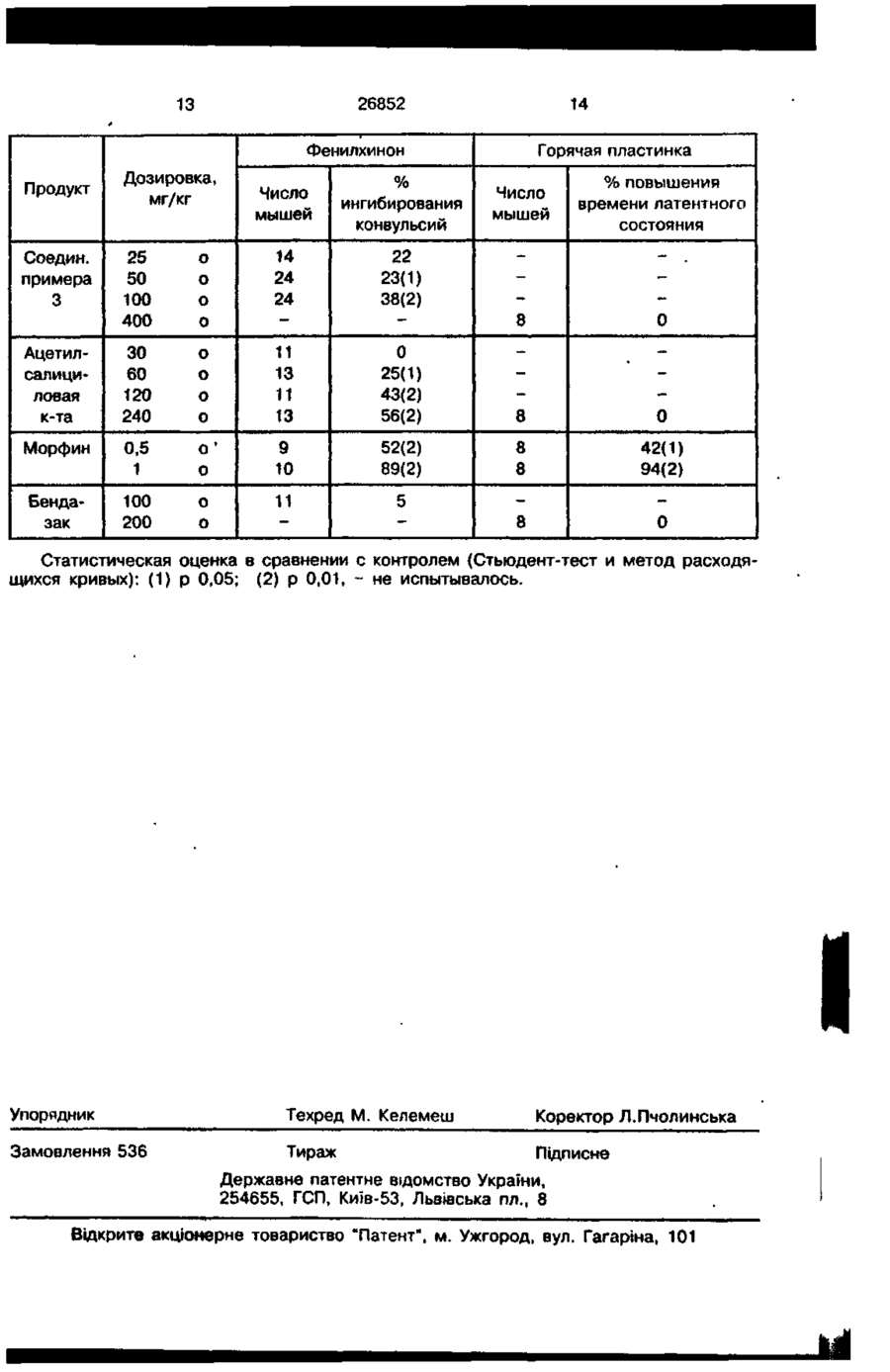
1. Способ получения производных 1бензил-3-оксиметилиндазола формулы (I) различными и представляют собой С,-С4алкил, R'" представляет собой Н, или их солей с фармацевтически приемлемыми основаниями, о т л и ч а ю щ и й с я тем, что соединение формулы (На) A-CH2-O-CRRf-COOR"\ (Ї) где А представляет собой 1 -бензилиндазол-З-ил, R и R' могут быть одинаковыми или A-CK-Y, (На) где Y представляет собой ОН, подвергают взаимодействию с кетоном формулы RCOR1, где R и R' имеют вышеуказанные значения, и хлороформом в присутствии гидроксида щелочного металла и, при необходимости, получают соль кислоты формулы (I) с фармацевтически приемлемыми основаниями. 2. Способ по п. 1 , о т л и ч а ю щ и й с я тем, что реакцию проводят при температуре кипения реакционной смеси и времени реакции от 30 мин до 12 ч. ОО ил К) Изобретение относится к простым эфирам 1-бензил-З-гидроксиметилиндазола с алифатическими 2-гидроксикислотами, к их солям с фармацевтически приемлемыми основаниями, к промежуточным соединениям и способам их получения, а также к содержащим эти эфиры фармацевтическим композициям. Более конкретно, первой целью настоящего изобретения является получение соединения формулы: A-CH2-O-CRR'-COORM (і) где А представляет ядро 1-бензилиндазол-3-ила формулы: 26852 R и R' могут быть одинаковы или различны и представляют Н или С^С^алкил, R'" предсатвляет Н или остаток алифатического насыщенного спирта с 1-4 атомами углерода; и если R'" - Н, соли соединения (I) с фармацевтически приемлемыми основаниями. Совершенно очевидно, что когда R и R' отличны друг от друга, соединение формулы (I) может существовать в виде единственного энантиомера или в виде рацемической смеси. Таким образом, настоящим изобретением охватываются как рацемические смеси, так и индивидуальные энантиомеры, полученные либо выделением обычными способами из рацемических смесей, либо стереоспецифичным синтезом. При отсутствии особых указаний в приведенных примерах исходные соединения, имеющие асимметрический атом углерода, используют в виде рацемических смесей. Известно соединение бендазак формулы: А-О-СН2-СООН (Bz) , где А принимает вышеуказанное значение, наделенное противовоспалительной акивностью (Патент США № 3470194). Непрекращавшиеся несколько лет исследования этого соединения показали, что бендазак и его соли с фармацевтически приемлемыми основаниями проявляет активность при лечении некоторых дислепимий (Патент США № 4352813), пигментозного ретинита (Европейский патент В 131317) и катаракта (Патент США № 4451477), а также было обнаружено, что бендазак и его соли могут предотвращать помутнение контактных линз (Европейский патент А 255967). Настоящее изобретение основано на открытии того, что введение метиленовой группы (-СН2-) между ядром А бензилиндазол-3-ила (II) и боковой цепью ( - 0 СН2-СООН) меняет фармакологические свойства бендазака, в отличие от которого соединения формулы (I) наделены обезболивающей активностью (пример 5). Второй целью настоящего изобретения является создание способа получения соединения формулы (I), и такой способ включает: !а) реакцию по обычным методикам соединения формулы: A-CH2-Y, • (На) где А принимает вышеуказанное значение и Y - гидроксил, с щелочным металлом или его приемлемым производным с образованием алкоголята формулы: А-СН г ОМе, (lib) где А принимает вышеуказанное значе 5 10 15 20 25 30 ние и Me - атом щелочного металла, с последующей реакцией соединения (II) с соединением формулы: X-CRR1 -COOR", (Ilia) где R и R' принимают вышеуказанные значения, X представляет отходящую группу, выбранную из галоидов и радикалов формулы: -Z-SO 2 -O-, где Z - арил или алкил, R1 -С^Сд-алкил, с образованием простого эфира формулы: A-CH2-O-CRR' -COOR", (la) где A, R, R\ R" принимают вышеуказанные значения, или Ib) реакцию по обычной методике соединения формулы: ("с) А-СН,-Х, где А и X принимают вышеуказанные значения, с алкоголятом формулы: MeO-CRR' -COOR", (IHb) где R, R\ R" и Me принимают вышеуказанные значения, с образованием простого эфира формулы (la) или Ic) реакцию по обычной методике соединения формулы (На) с кетоном и хлороформом в присутствии щелочи согласно следующему уравнению реакции: A -CH2-Y СНСІ R-CO-R* (Па) -» A-CH2-O-CRR' -СООН, (D 35 40 45 50 55 где А и Y принимают вышеуказанные значения, R и R" могут быть одинаковы или различны, и представляют С,-С5-алкил, II) гидролиз при желании сложного эфира (la) с образованием соответствующей кислоты формулы (I) по обычной методике и III) получения по обычным методикам и при желании (А) соли кислоты формулы (I) с фармацевтически приемлемым основанием или (В) эфира кислоты формулы (I) с насыщенным алифатическим спиртом с 1-4 атомами углерода. Стадии (la) и (Ib) направлены на способ получения асимметричных простых эфиров по Уильямсону (March I. Advanced Organic Chemistry. 3-е изд., с. 342-344, реакции 0-14 и 0-16), который рекомендуется осуществлять в присутствии приемлемого растворителя при температуре от комнатной до температуры кипения реакционной смеси и в течение времени от 18 минут до 48 часов. Примерами приемлемых растворителей являются апротонные растворители. Типичные примеры рекомендуемых растворителей включают: тетрагидрофуран, диметилформамид, толуол и их смеси. Алкоголяты (lib) и (Illb) рекомендуется получать в реакции с металлическим натрием, металлическим калием или гидра 26852 том натрия в присутствии приемлемого растворителя при температуре от комнатной до температуры кипения реакционной смеси в течение времени от 15 минут до 48 часов. Примером рекомендуемых растворителей являются апротонные растворители. Типичные примеры рекомендуемых растворителей включают тетрагидрофуран, диметилформамид, толуол и их смеси. Рекомендуемые значения для X - хлор, бром и Z-SO 2 -O-, где Z предсталяет п-метилфенил, фенил и метил. Стадию (Ic) рекомендуется проводить при температуре кипения реакционной смеси в течение времени от 30 мин до 12 ч. Стадию (И) рекомендуется проводить в водном или водно-спиртовом растворе щелочи при температуре от комнатной до температуры кипения и времени реакции 1-48 часов. Типичные примеры фармацевтически приемлемых неорганических оснований, приемлемых для использования на стадии (На), включают щелочные или щелочноземельные металлы, более конкретно натрий, калий и кальций. Типичными примерами органических оснований являются метиламин, изопропиламин, гексиламин, диэтиламин, этаноламин, 2-гидроксиметил-2-амино-1,3-пропандиол, глюкамин, глицин, аланин, валин, лейцин, изолейцин, серии, треонин, аспаратиновую кислоту, глутаминовую кислоту, аргинин, лизин, цистин, метионин, фенилаланин, тирозин, триптофан и гистидин. К типичным представителям спиртов, рекомендуемых для использования на стадии (ШЬ). относятся спирты с прямой цепью. Еще одной целью настоящего изобретения является получение промежуточных соединений формулы: A-CH 2 -W, (II) где А представляет ядро 1-бензилиндазол-3-ила и W представляет ОН, ОМе (Me - атом щелочного металла) или отходящую группу, выбранную из галоидов или радикалов формулы: Z~SO 2 -Oгде Z - арил или алкил. К рекомендуемым отходящим группам относятся бром, хлор и Z-SO 2 -O-, где Z - п-метилфенил, фенил или метил. Спирт формулы (И)(А-СН 2 -ОН) может быть получен восстановлением кислоты формулы: А-СООН (IV), где А принимает вышеуказанное значение, или ее алифатического эфира по обычной методике. Восстановление сложного эфира рекомендуется проводить использованием 5 10 15 •20 25 30 35 40 45 50 55 приемлемого восстановителя, такого как литийалюминийгидрид, натрийбис(2-метокси-этокси)алюминийгидрид (70% в толуоле) или кальцийтетра (изопропокси)аланат (70% в толуоле) в присутствии приемлемого растворителя при температуре от 0*С до температуры кипения реакционной смеси и времени реакции от 30 минут до 12 часов. Примеры приемлемых растворителей включают диэтиловый эфир, тетрагидрофуран, толуол и их смеси. Соответствующие алкоголяты (W = =ОМе), галоидпроизводные (W = галоид), и сульфоновые эфиры (W = O-SO 2 -Z) могут быть легко получены известными способами. В практической медицине соединения настоящего изобретения и их фармацевтически приемлемые соли могут быть введены как таковые, но рекомендуется их вводить в виде фармацевтических композиций. Такие композиции являются еще одной целью настоящего изобретения, и они содержат эффективное количество одного или нескольких соединений формулы (I) или их солей с фармацевтически приемлемыми органическими или неорганическими основаниями в смеси с жидкими или твердыми фармацевтическими наполнителями, пригодными для системного введения, например перорально, ректально и парентерально или местно в виде аэрозоля или офтальмическим введением. Фармацевтические комопзиции настоящего изобретения могут быть твердыми, например, иметь вид таблеток, пилюль, капсул и медленно действующих препаратов или быть жидкими, т.е. в виде растворов, суспензий и эмульсий. Кроме традиционных наполнителей композиции могут включать приемлемые в фармацевтических целях добавки, такие как консерванты, стабилизаторы, эмульгаторы, регулирующие осмотическое давление соли, буферные системы, отдушки и красители. В случае необходимости при конкретном лечении композиции настоящего изобретения могут включать другие совместимые активные компоненты, совместное введение которых приносит лечебную пользу. В лечебной практике эффективное количество вводимого соединения настоящего изобретения может меняться в широком интервале в зависимости от известных факторов, таких как конкретный вид лечения, вида фармацевтической композиции, пути введения и эффективности конкретного применяемого соединения изобретения. Однако оптимальное эффек 26852 тивное количество может быть легко определено обычным способом. Фармацевтические композиции могут быть приготовлены обычными способами из практики химика-фармацевта с приме- 5 нением смешивания, гранулирования и прессования, когда это необходимо, или смешивания и растворения различным образом компонентов с получением целевого конечного продукта. 10 В целом, в случае системного введения ежедневная дозировка соединения (I) предпочтительно задается таким образом, чтобы его уровень в тканях достигал 105—1 *3 М, и такой уровень обычно 15 достигается при дозах в 0,5-100 мг/кг. В свою очередь в случае местного употребления рекомендуется использовать фармацевтическую композицию (примочки для глаз, кремы, мази и т.п.), содержащие 20 0,1-5 мае. % соединения формулы (I) или соответствующее количество его фармацевтически приемлемой соли. И, наконец, еще одной целью изобретения является создание способа лече- 25 ния, заключающегося в введении нуждающему в лечении больному эффективного количетсва соединения формулы (I) или его фармацевтически приемлемой соли. С целью дальнейшей иллюстрации 30 изобретения ниже приводятся следующие примеры. П р и м е р 1. a) 1 - Бензил-3-гидроксиметилиндазол. В суспензию 2 г литийалюминийі ид- 35 рида в 50 мл диэтилового эфира прикапывают с перемешиванием этиловый эфир 1 -бензил-3-индазолкарбоновой кислоты (Von Auwers Schaich. Chem. Ber., 1921, 54, 1756) в ЗО мл безводного тетрагидро- 40 фурана. По окончании прикапывания реакционную смесь кипятят 90 мин. После охлаждения реакционную смесь обрабатывают обычным образом, образо- 45 вавшийся осадок отфильтровывают и перекристаллизацией остатка, полученного испарением растворителя, из изопропилового спирта получают 1-бензил-З-гидрокеиметилиндазол (соединение (Иа), т.пл. 50 85-86*С. b) Простой эфир 1-бензил-3-гидроксиметилиндазола с гликолевой кислотой. К раствору всего количества 1-бензил-3-гидроксиметилиндазола, полученно- 55 го вышеописанным способом, в 70 мл тетрагидрофурана добавляют 2,4 г гидрида натрия (60%-ная суспензия в масле) и реакционную смесь кипятят в токе инертного газа (азот). Затем добавляют раст 8 вор 3,5 г бромуксусной кислоты в 40 мл тетрагидрофурана и реакционную смесь кипятят 90 минут. После охлаждения реакционную смесь обрабатывают обычным способом и подкисляют. Перекристаллизацией полученного продукта из изопропанола получают простой эфир 1 -бензил3-гидроксиметилиндазола с гликолевой кислотой (соединение (I), R = R' = R" = =Н), т.пл. 136-138'С. П р и м е р 2. a) 1-Бензил-З-хлорметилиндазол. Раствор 1 -бензил-3-гидроксиметилендазола (11 г, получение см. пример 1а) и 11,9 г тионилхлорида в 100 мл толуола кипятят 4 часа. После испарения растворителя получают твердый остаток сырого 1 -бензил-3-хлорметилиндазола (соединение (II), W = СІ), который применяют на следующей стадии (Ь) без дополнительной очистки. Образец, перекристаллизованный из гексана, имеет т.пл. 89-9VC. b) Простой эфир 1-бензил-З-гидроксиметилиндазола с молочной кислотой и ее этиловым эфиром. К кипящему раствору сырого 1-бензил-3-хлорметилиндазолз, полученного вышеописанным способом, и 52 г этил лактата в 100 мл диметилформамида в течение примерно 60 минут порциями прибавляют 2,7 г гидрида натрия (60%-ная суспензия в масле). По окончании прибавления реакционную смесь кипятят еще 30 минут, затем охлаждают, разбавляют водой и отделившееся масло экстрагируют этилацетатом. Полученный после испарения растворителя остаток представляет собой сырой этиловый эфир (соединение (1), R = Н, R' = =СН3, R'" = С2Н5), который растворяют в растворе 560 г водно-спиртовой смеси (1:1), содержащей 3,4 г NaOH. После кипячения в течение четырех часов большую часть спирта испаряют, оставшийся водный раствор подкисляют и перекристаллизацией образовавшегося твердого вещества из смеси гексана с этилацетатом получают простой эфир 1-бензил-З-гидроксиметилиндазола с молочной кислотой (соединение (I), R = R'"= H, R1 = =СН3), т.пл. 126-128'С. Или NaOH добавляют при комнатной температуре и по окончании прибавления реакционную смесь нагревают при 40-50'С. По методике примера 2Ь, но использованием вместо этиллактата метил-2-гидроксибутирата, метил-2-этил-2-гидрокси.бутирата и метил-2-гидроксикапроата могут быть получены соединения формулы (I) со значением R, R' и R"\ приведенными ниже. 26852 R = H, R' = C2H5, R f l t = CH3 (эфир) и Н (кислота), R = С2Н5, R1 = С2Н5, R"' = СН3 (эфир) и Н (кислота), R = Н, R1 = С4Н9, R'" = СН3 (эфир) и Н (кислота). П р и м е р 3. Простой эфир 1бензил-3-гидроксиметилиндазола с 2-гидрокси-2-метилпропионовой кислотой. В круглодонную колбу, снабженную мощной мешалкой, последовательно вносят 1,9 г NaOH, 10 г ацетата и 1-бензил3-гидроксиметилиндазола, полученного вышеописанным способом. Затем добавляют 1,6 г хлороформа (экзотермическая реакция) и смесь нагревают на водяной бане два часа. Добавляют воду, реакционную смесь промывают этилацетатом и подкисляют. Перекристаллизацией полученного в результате продукта из смеси гексана с этилацетатом (1:1) получают простой эфир-1 -бензил-3-гидроксиметилиндазола с 2-гидрокси~2-метилпропионовой кислотой (соединение (I), R = R' = СН3, R m = = Н), т.пл. 132-134'С. П р и м е р 4. Простой эфир 1бензил-3-гидроксиметилиндазола с 2-гидрокси-2-этилпропионовой кислотой. К суспензии 5,9 г 1-бензил-3-гидроксиметилиндазола, полученного вышеописанным способом, 12 NaOH и 35 мл метилэтилкетона по каплям в течение примерно 30 минут прибавляют раствор 6 мл хлороформа и 6,8 мл метилэтилкетона. По окончании прибавления реакционную смесь кипятят 60 мин. Затем охлаждают, добавляют воду, водную фазу отделяют и подкисляют. Образовавшееся масло экстрагируют диэтиловым эфиром, и после испарения растворителя получают масло, которое затвердевает и перекристаллизацией которого из смеси гексана с этилацетатом (1:1) получают простой эфир 1бензил-2-гидроксиметилиндазола с 2-гидрокси-2-этилпропионовой кислотой (соединение (I), R = СН_, R' = C2HS, R'" = =Н), т.пл. 115-116'С. По методике примера 4, но использованием 2- и 3-пентанона, 2- и 3-гексанона, 2-, 3- и 4-гептанона, 3-октанона, 5нонанона и 6-ундеканона вместо метилэтилкетона получают соединения формулы (!) со значениями R, R1 и R"\ приведенными ниже: R == Сп 3 ,, R === С-Н7,7, RR = =Н, Сп 3 R С-Н Н, R R R R R « = ~ = — С 2 Н 5 , R' СН 3 , R' C-Hgt R СН,, R' С2Н5, R = d2 H s , R"f = Н, d « С А , R'" = К. ~ G3H7, R — Н, = С5Н,,, R"* = И, == = С4Н9, R'* Н, 5 10 15 20 25 30 35 40 45 50 55 10 R = С3Н7, R' = С3Н7, R"' - Н, R = С2Н5, R' = C 5 H n , R'" =• Н, R = С4Н9, R' = С4Н9, R'" = Н, R = C 5 H n , R' = C 5 H n , R'" = Н. П р и м е р 5. Обезболивающая активность соединений настоящего изобретения может быть выявлена в испытании с горячей пластинкой, а также в испытании в вызванным фенилхиноном вытягиванием у мышей. А. Испытание с горячей пластинкой. Обезболивающую активность испытывают по методике Woolf и MacDonald (J.Pharmacol. Exp. Ther. 80, 300, 1944), Eddy и др. (J. Pharmacol. Exp. Ther 98, 121, 1950), Janssen и Jagencan un. (J.Pharm. Pharmacol. 9, 381, 1957), которая была модифицирована. 1. Оборудование для испытания с Т о рячей пластинкой", каталожный № фирмы Ugo Basile (Корнерио-Варез, Италия). Алюминиевую пластинку нагревают электричеством с помощью батареи по всей поверхности испытаний. Температурный регулятор чувствует изменение температуры пластинки и регулирует подачу напряжения с целью свести к минимуму перегрев. Потенциометр позволяет установить заданную температуру в интервале 45-62'С (±0,2'С). 2. Создание дискомфорта. Мышь (одну) помещают на пластинку, нагретую до 55±0,2'С. Для удерживания зверька в пределах "площади испытания" используют прозрачный цилиндр из перспекса диаметром 19 см и высотой 13 см. Зверек проявляет дискомфорт одной из следующих реакций (Eddy и др. J.Pharmacol. Exp. Ther. 98, 12, 1950): - отдергивание задних лапок (S) - выворачивание и облизывание подошв задних лапок (L), - танец около ограничивающего цилиндра (D) - подъем одной из задних лапок и прижимание ее к телу (А). Эта реакция обычно проявляется при убывании обезболивающего действия лекарства. - подпрыгивание с попыткой выйти из ограничивающего цилиндра (J). 3. Измерение продолжительности реакции. Время реакции определяют с помощью встроенного электронного таймера, отсчитывающего каждые 0,1 сек и приводимого в действие с помощью педального выключателя. Таймер включают в момент, когда мышь попадает на пластинку, и выключают, когда зверек проявляет од 11 26852 ну из вышеописанных реакций. Сразу же после этого зверька снимают с пластинки и отмечают время пребывания в секундах, соответствующие показаниям таймера и отмеченные символами (S, L, D, A, J) для каждого конкретного типа наблюдаемой реакции (см. пункт 2). 4. Отсчет времени. Базовый отсчет: два отсчета проводят до введения лекарства соответственно при 20 и 10 минутах. Среднее значение для этих двух отсчетов служит "нормальным временем реакции" (Janssen и Jagencan, J.Pharm.Pharmaco»., 9, 381, 1957). Отсчет после введения лекарства. Oreчет проводят при 10-20-30-40-50-60-90120 минутах после введения лекарства. Удлинение времени отсчета. Максимальное увеличение времени отсчета не должно превышать 30 секунд, чтобы избежать повреждения лап зверьков. После указанного времени при отсутствии реакции зверька снимают с пластинки и отмечают время реакции как "> ЗР\ цифра 30 используется при расчетах (Eddy и Jeimbach, J.Pharm. E.Th. 107, 385, 1953). 5. Положительные реакции. Данный параметр означает "конечную точку" для подсчета ЭД^, и определяется следующим образом (Janssen и Jagencan. J.Pharm. Pharmacol., 9, 381, 1957). Реакция считается положительной, если время реакции, по меньшей мере, единажды > 30 или е е л к в, по меньшей мере, 3 отсчетах время реакции в 3 или более раз превышает нормальное время реакции. 6.'Подопытные группы и введение лекарства. Для каждого продукта и каждой дозы образуют группы из двух зверьков при максимальном количестве 14 штук. Лекарство в основном вводят внугрибрюшинно или подкожно. 12 Испытания проводят на мышах по модифицированной методике Henderson, Forsaith (J.PharmacoI.Exp.Ther. 125, 237, 1959). Вызывающее боль средство: 0,08% (20 5 мг) (20 мг, 20 мл) фенилхинона (2-фенил-1,4-бензхинона), суспендированного в кукурузном масле согласно Luo x, Smith, Aslem (Arzneim. Forsch. 28, 1644, 1978). Группы подопытных животных и вве10 дение фенилхинона. Образуют группы из 4 мышей (20-30 г) и каждую мышь метят пикриновой кислотой (насыщенный раствор в спирте). Каждому зверьку перентерально вводят фенилхинон (10 мл/кг для 15 каждого зверька весом выше 25 г и 0,25 мл для каждого зверькт весом ниже 25 г), помещают в прозрачную пластиковую клетку (23,5 х 13,7 х 13,1 см) и наблюдают в течение 20 минут после введения фенил20 хинона. 25 30 35 40 В. Испытание с вытягиванием под 45 действием фенилхинона. Подсчет вытягиваний и оценка. Наблюдатель регистрирует число вытягиваний для каждого зверька с помощью счетчика нажимного действия. Вытягивания классифицируют следующим образом. - полное - сокращение брюшной полости, периодическое искривление туловища и вытягивание задних лапок; - половинчатое - сокращение брюшной полости и некоторое искривление туловища. Каждые два вытягивания наблюдатель регистрирует как одно полное. Введение лекарства. Продукты вводят перорально (O s ) или подкожно (Sc) при - 3 0 или - 2 0 минутах после введения фенилхинона. Троим животным 8 каждой группе вводят различные продукты, четвертому животному вводят носитель. Действие соединения примера 3 и ссылочного лекарства на реакцию мышей в Испытаниях с применением фенилхинона и горячей пластинки приведены в нижеследующей таблице. 13 14 26852 Фенилхинон Продукт Дозировка, мг/кг Число мышей ингибирования конвульсий 14 24 24 Ацетилсалициловая к-та 30 60 120 240 о о о о 11 13 11 13 0 25(1) 43(2) 56(2) Морфин 0,5 1 о' о 9 10 52(2) 89(2) Бендазак 100 200 о о 11 5 22 23(1) 38(2) 1 о о о о % повышения времени латентного состояния I 25 50 100 400 Число мышей I Соедин. примера 3 Горячая пластинка 00 0 1 1 1 00 0 8 8 42(1) 94(2) 8 0 Статистическая оценка в сравнении с контролем (Стьюдент-тест и метод расходя щихся кривых): (1) р 0,05; (2) р 0,01, - не испытывалось. Упорядник Техред М. Келемеш Коректор Л.Пчолинська Замовлення 536 Тираж Підписне Державне патентне відомство України, 254655, ГСП, Київ-53, Львівська пл., 8 Відкрите акціонерне товариство "Патент", м. Ужгород, вул. Гагаріна, 101
ДивитисяДодаткова інформація
МПК / Мітки
МПК: A61P 27/02, A61P 25/04, C07D 231/56, A61K 31/415
Мітки: 1-бензил-3-оксиметиліндазолу, одержання, спосіб, прийнятними, фармацевтично, солей, основами, похідних
Код посилання
<a href="https://ua.patents.su/8-26852-sposib-oderzhannya-pokhidnikh-1-benzil-3-oksimetilindazolu-abo-kh-solejj-z-farmacevtichno-prijjnyatnimi-osnovami.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних 1-бензил-3-оксиметиліндазолу або їх солей з фармацевтично прийнятними основами</a>
Спосіб одержання похідних оксадіазолілалкілпурину або їх фармацевтично прийнятних кислих аддітивних солей

Номер патенту: 3533
Опубліковано: 27.12.1994
Автори: Йене Мартон, Еміль Мінкер, Каталін Мармароші, Шандор Віраг, Андреа Болеховські, Гергелі Хейа, Віра Гергелі, Ласло Тардош, Шандор Антуш, Золтан Варгаї, Габор Ковач, Петер Кьормеці, Лоранд Дебрецені, Дежьо Карбонітш, Габор Хорват, Агнеш Готтшеген
Мітки: похідних, кислих, одержання, фармацевтично, оксадіазолілалкілпурину, аддітивних, спосіб, солей, прийнятних
Формула / Реферат:
Ф о р м у л а и з о б р е т е н и яСпособ получения производных оксадиазолилалкилпурина общей формулыгде A=С1-С4-алкилен; R1=C1-С6-алкил, С4-С6-оксиалкил, галоидный С1-С6 - алкил, С1-С4 карбоксиалкил, циклогексил, аминоалкил общей формулы (СН2)nNR2R3, где n=1-3;R2 и R3 каждый С1-С4-алкил или вместе с атомом азота, с которым они связаны, образуют пиперидиновое или морфолиновое кольцо,или R1 – фенил, оксифенил,...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Шіпош Юдіт, Хермец Іштван, Пайор Аніко, Балог Марія, Хорват Агнеш, Керестурі Геза, Рітлі Петер, Вашварі Лелле
МПК: C07F 5/00, C07D 215/56, A61K 31/495, C07D 401/04, A61P 31/04
Мітки: кислоти, придатних, хінолінкарбонової, сполуки, похідних, солей, спосіб, одержання, проміжні, фармацевтично
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Спосіб одержання похідних 3 (2н) пірідазінона, або фармацевтично прийнятних солей

Номер патенту: 5591
Опубліковано: 28.12.1994
Автори: Мотоо Муцукадо, Ріозо Сакода, Кен-Іті Сікада, Кейзо Таніківа
МПК: C07D 237/22
Мітки: фармацевтично, солей, одержання, 2н, пірідазінона, прийнятних, похідних, спосіб
Формула / Реферат:
Способ получения производных 3(2Н)пиридазинона общей формулыгде R1- водород, метил, аллил, циклопентил, бензил, фенил, группа -(СН2)m, СО2R3, где R3 - водород или третбутил, m - целое число 1 или 2, группа -(СН2)nОН, где n = 2,3,-(СН2)2N(СН3)2 или группа -СН2ОF3;R2- хлор или бром;Y1 и Y2- одинаковые или различные, каждый независимо водород, С1-С3-алкил, С7-алкенил, галоген при условии, что Y1 и Y2 одновременно...
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Едді Жан Едгард Фрейн, Альфонс Герман Маргарета Реймакерс, Жерар Шарль Санз
МПК: C07D 521/00, A61K 31/47, A61P 19/06, C07D 405/14, A61K 31/425, A61K 31/4427, C07D 403/14, C07D 403/06, A61P 5/00, A61K 31/4433, C07D 409/14, C07D 417/14, A61P 35/00, A61P 17/00, A61P 43/00, C07D 401/14, A61K 31/415, A61K 31/443, A61K 31/44
Мітки: 1н-імідазол-1-ілметіл)замішаного, солей, бензімідазола, стереоізомерів, спосіб, одержання, фармацевтично, кислоти, металів, похідних, прийнятих
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Спосіб одержання похідних 2-тієнілоксіуксусної кислоти чи їх фармацевтично прийнятних солей

Номер патенту: 4227
Опубліковано: 27.12.1994
Автори: Франц Ровенсцкі, Дітер Біндер, Хуберт Петер Фербер
МПК: A61P 29/00, A61P 11/00, A61P 43/00, A61P 9/00, C07D 333/32, A61K 31/38, A61P 7/02, A61K 31/381
Мітки: одержання, кислоти, солей, прийнятних, фармацевтично, 2-тієнілоксіуксусної, похідних, спосіб
Формула / Реферат:
1. Способ получения производных 2-тиенил-оксиуксусной кислоты общей формулы где R - незамещенный или замещенный галогеном фенил, или их фармацевтически применимых солей, отличающийся тем, что соединение общей формулыгде R имеет указанные значения, подвергают окислению окисью серебра в водно-щелочной среде с последующим выделением целевого продукта в свободном виде или в виде фармацевтически применимой если....
Попередній патент: Калібрувальний пристрій до машини для зварювання поздовжніх швів обичайок
Наступний патент: Теплообмінна труба для опалювального котла
Випадковий патент: Пристрій захисту джерел живлення від перевантажень та коротких замикань