Спосіб отримання специфічних пептидів для терапевтичних цілей







Формула / Реферат
Спосіб одержання специфічних пептидів для терапевтичних цілей, при якому конструюють бібліотеку випадкових пептидів, виконують її скринінг та використовують методи для підтвердження специфічності зв'язуючих пептидів, який відрізняється тим, що отримують чисельну кількість бібліотек з правильно орієнтованими вставками у визначених видах експресійних векторів за допомогою фрагментації транскриптомів клітин-мішеней та клітин-елімінаторів людського походження, а скринінг виконують з використанням комбінацій клітин-мішеней та клітин-елімінаторів у декілька етапів для отримання груп специфічних пептидів, із яких виділяють та тестують індивідуальні пептиди з подальшим підтвердженням їх специфічності комбінованими тестами.
Текст
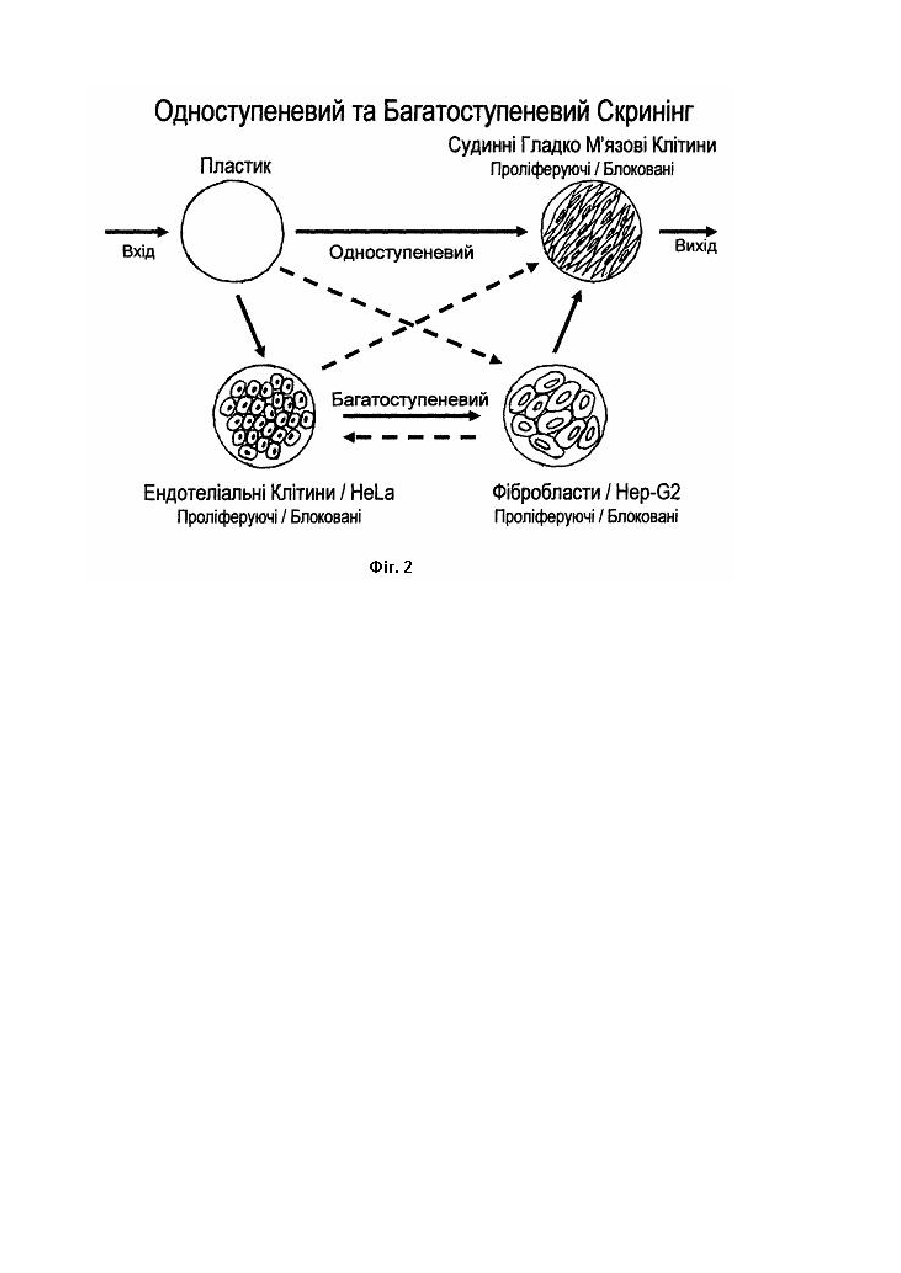
Винахід відноситься до генної інженерії, зокрема до способів отримання специфічних пептидів, які використовують у діагностиці, терапії та фармацевтичних застосуваннях. В наш час, однією з найголовніших проблем є підвищення рівня серцево-судинних, онкологічних та імунних захворювань. У більшості випадків, первинним джерелом таких порушень є швидкоплинна неконтрольована проліферація тканинно-клітинних стр уктур, яка утр уднює та ускладнює їх діагностику і терапію. Тому, першочерговою умовою стає своєчасне виявлення патологічного стану, розробка та доставка лікарських препаратів, які блокують клітинну активність у вогнищі захворювання. Відомі терапевтичні інгібітори судинних гладком'язових клітин, які використовують для пригнічення клітинної активності та/або знищення клітини шляхом націленої доставки терапевтичних агентів у вогнища активної проліферації (див. пат. США №6515009 від 04.02.2003 р., МПК А61К31/40). Для інгібування судинних гладком'язових клітин, згідно даного патенту, використовують терапевтичні кон'югати, які містять у собі лікарській препарат, зчеплений з пептидом чи білком, що специфічним чином зв'язується з клітинною мембраною судинних гладком'язових клітин (наприклад, моноклональні та поліклональні антитіла, фактори росту, поліпептидні гормони, цитокіни та подібні до них молекули) або з елементом внутрішньо - або зовнішньо клітинного матриксу (наприклад, макромолекули, що впізнають рецептори інтегринів та фібронектинів, а також епітопи колагену та позаклітинних протеогліканів) цих же клітин. Один з таких пептидів, наприклад, містить послідовність амінокислот, що специфічно впізнає хондроітін-сульфат протеоглікан, з наближеною молекулярною вагою біля 250 кілодальтон, що експресується на мембранах судинних гладком'язових клітин. Таку ж послідовність амінокислот знаходять у Fab та Fv фрагментах або CDR (регіони, що забезпечують компліментарність) моноклонального антитіла NR-AN-01 та/або його функціональних еквівалентів. Недоліками даного винаходу є використання пептидів, які впізнають білки з вираженою експресією в ряді інших функціонально вагомих клітин і тканин, це призводить до того, що даний терапевтичний кон'югат може зв'язуватися з іншими типами клітин не гладком'язового походження, як наприклад клітини нейроглії, кісткової тканини та інші, що призводить до небажаної акумуляції лікарського препарату в даних клітинах та побічній цитотоксичній дії (клітинної загибелі). Крім того, при використанні антитіл в якості ведучої складової кон'югатів, їх розмір, із-за досить високої щільності позаклітинного матриксу, може стати перешкодою при проникненні кон'югату в вогнище проліферації, при цьому, крос - реактивність антитіл до інших молекулярних структур знижує їх специфічність. Відомий також спосіб цілеспрямованої доставки пептидів до рестенозних судинних гладком'язових клітин in vitro та in vivo, з метою доставки лікарських препаратів до рестенозної бляшки за допомогою пептидів, які вбудовані в фатові частинки та зв'язуються з судинними гладком'язовими клітинами, що проліферують. Ці пептиди відбирають з бібліотеки синтетичних 15-ти членних пептидів, сконструйованої випадковим шляхом (див. Статтю Ingrid N. Michon et al. "Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo" в журналі " Biochimica at Biophysica Acta" вип. №1591, 2002p., стор.87-97). Відібрані пептиди шляхом багаторазового повторення одноступеневого скринінгу, подалі проходять підтвердження специфічності, виключно методом імуноферментного аналізу та візуалізуються за допомогою імунофлюоресценції, за якими виявляють два пептиду з консервативною амінокислотною послідовністю, такі як 5G6 (CNIWGVVLSWIGVFPEC) та 5E5(CESLWGGLMWTIGLSDC). Недоліками даного способу є використання однієї вихідної бібліотеки на основі синтетичних випадково орієнтованих пептидів, які не мають аналогів у природі, що визначає їх первинно низьку специфічність та селективність, а також високу імуногенність. Крім того, багаторазове повторення одноступеневого скринінгу призводить до послаблення специфічних та нарощуванню неспецифічних взаємодій. В основу заявленого винаходу поставлена задача, створити спосіб отримання природних високо специфічних пептидів людського походження з високою cтупінню селективності щодо заданого типу вибраних клітин-мішеней. Поставлена задача досягається шляхом здійснення нового способу отримання специфічних пептидів для терапевтичних цілей, при якому конструюють чисельну кількість бібліотек з правильно орієнтованими вставками в певних видах експресійних векторів, шляхом фрагментації транскриптомів клітин-мішеней та клітин-елімінаторів людського походження, а скринінг виконують з використанням комбінацій клітин-мішеней та клітин-елімінаторів в декілька етапів з отриманням груп специфічних пептидів, серед яких виділяють і тестують індивідуальні пептиди з подальшим підтвердженням їх специфічності з використанням комбінованих тестів. При здійсненні даного способу отримують високо специфічні пептиди з високою отупінню селективності, які можуть бути використанні для доставки лікарських препаратів у місце їх безпосередньої дії, з метою профілактики та/або терапії розладів, що пов'язані з порушенням функціонування судинних гладком'язових клітин, або для безпосереднього використання отриманих пептидів, як лікарської форми в терапевтичних застосуваннях. Крім того, використання специфічних пептидів та їх фрагментів, або модифікованого вектора з таким пептидом, дозволить діагностува ти порушення функціонування судинних гладком'язових клітин і прогнозувати їх лікування. Заявлений спосіб дозволяє отримувати високо специфічні та високо селективні пептиди до певного типу клітин-мішеней. Спосіб отримання специфічних пептидів пояснюється схемами та ілюстраціями, де: Фіг.1 - показано синтез кДНК і конструювання бібліотек; Фіг.2 - зображена схема одно - та багатоступеневого скринінгу. Спосіб отримання специфічних пептидів здійснюють таким шляхом. Спочатку, проводять відбір, і розподіл клітинних популяцій на типи: клітини-мішені та клітини-елімінатори. В якості перших, звичайно використовують клітини, що є безпосередньо домінуючими (ключовими) у розвитку патологічного процесу та, відповідно, потребують чіткого впізнання отриманими пептидами, тобто, висока специфічність до яких, є необхідною умовою. В якості клітин-елімінаторів використовують клітини, які можуть бути втягнуті в патологічний процес або здатні впливати на його течію, діагностику та терапію, і відповідно, пептиди, які отримують, не повинні бути специфічними до даного типу клітин, так як це може привести до перекручування результатів діагностики, ярко вираженої побічної дії та низької ефективності терапії. Потім конструюють серії бібліотек кДНК в експресійному векторі вірусного типу (бактеріофаги). Спочатку виділяють сумарну РНК загальновідомими методами в адаптаційних модифікаціях, з наступною, у випадку необхідності, очисткою її та виділенням фракції матричної РНК. Якість такої РНК аналізують і в подальшому, використовують РНК в реакціях оберненої транскрипції, що направляють праймерами, як випадкового походження, так і комплементарними до поли А+ ділянки матричних РНК. На цьому етапі здійснюють первинну фрагментацію транскриптому шляхом контрольованого синтезу фрагментів кДНК обмежених в розмірі та гетерогенності, що досягається використанням різних концентрацій іонів в реакційній суміші, комбінацією праймерів, кількістю вихідного матеріалу та режимів температури. Вторинну фрагментацію транскриптома здійснюють на етапі фракціонування та очистки отриманих молекул кДНК, для чого використовують гельфільтраційну хроматографію так, що після елюції пул молекул кДНК складається з двох частин: фракції молекул з кількістю пар нуклеотидів менше 500 та фракції, що складається з молекул розміром більше 500 пар нуклеотидів. Такий розподіл дозволяє, в першому випадку, створення бібліотек спрямованих на визначення точних місць взаємодії молекули пептиду з молекулами на клітинах - мішенях, а в др угому - ідентифікувати фрагмент нуклеїнової кислоти, який відповідає за синтез даного пептиду або білкового фрагмента. Далі, молекули кДНК легують з олігонуклеотидними лінкорами, які синтетично синтезовані і містять місця впізнання для рестрикційних ендонуклеаз (рестриктаз), які використовують для обробки та підготовки вектора для клонування. Для цього, кінці кДНК спочатку обробляють за допомогою полімерази і тільки потім, такі фрагменти легують з лінкерами та інкубують з рестриктазами. Використовуючи данні лінкери, стає можливим отримати фрагменти кДНК, які містять на різних кінцях місця впізнання для різних рестриктаз, в такому положенні, що можуть бути безперешкодно клоновані у вектор в певній орієнтації (смислове клонування). Одночасно з цим, експресійний вектор обробляють рестрикційними ендонук-леазами, з метою отримання фрагментів придатних для легування з синтезованими фрагментами кДНК. Отриманні в результаті легування фрагментів кДНК та молекул вектора, рекомбінантні молекули, упаковують in vitro та ініціюють експресію даного фрагмента шляхом одного циклу реплікації. Потім, визначають кількість рекомбінантів в бібліотеці та абсолютне число частинок. Проводять скринінг таких бібліотек, використовуючи комбінації клітин - мішеней та клітин-елімінаторів. При цьому, скринінг здійснюють багаторазовим повторенням раундів інкубації з клітинами, з наступними відмивками фагови х частинок, які не зв'язалися з клітинами, розчинами різної іонної сили та розчинами різної здатності руйнува ти білок-білкові взаємодії, з наступною елюцією та ампліфікацією шуканих фагови х частинок. Причому, у випадку прямого скринінгу, певну кількість фагових частинок у буфері, спочатку інкубують з матеріалом, який використовують для культивування клітин, а потім залишкову фракцію (супернатант), що містить вільні фати, переносять на клітини - мішені. У випадку багатоступеневого скринінгу, а саме, використання декількох типів клітин-елімінаторів, супернатант після інкубації з матеріалом, спрямовують на клітиниелімінатори, після яких супернатант спрямовують на клітини-мішені або на слідуючий тип клітин-елімінаторів, з наступною інкубацією на клітинах-мішенях. Аналогічно діють при використанні клітин - мішеней та клітинелімінаторів в різних структурно-функціональних станах. Таким чином, результатом багатоступеневого скринінгу є гр упа пептидів у складі фагови х частинок, яка має виражену специфічність у відношенні до клітин-мішеней. Для отримання кінцевих пептидів, з вищевказаних груп виділяють індивідуальні фатові частинки та тестують їх. В першому випадку, пептид, що тестують, напрацьовують (нарощують) в необхідній кількості, очищують та досліджують як первинне антитіло в прямому імуноферментному аналізі (ІФА), де антигеном виступають культивовані клітини вибраного функціонально стану. При дослідженні великої кількості фагових частинок, проводять мікроселекцію. Для чого, ви хідну груп у розсівають до отримання індивідуальних бляшок, елюцію яких проводять буфером для інкубації в ІФА та використовують безпосередньо, як джерело первинних антитіл в ІФА, без попереднього нарощування та очистки. У другому випадку, індивідуальний клон аналогічно першому, ампліфікують, очищують і інкубують з клітинами-мішенями та клітинами-елімінаторами одночасно. Після чого, клітини відмивають, проводять елюцію зв'язаних частинок та визначають їх титр, потім порівнюють його з аналогічним для іншого типу клітин. У третьому випадку, в аналізі специфічності пептидів використовують полімеразну ланцюгову реакцію (ПЛР), при цьому, декілька фатів очищують та змішують в однакових кількостях, потім інкубують з клітинами-мішенями та клітинами-елімінаторами, проводять елюцію та розсівають до окремих бляшок, які відбирають парами, виділяють ДНК і використовують її в якості матриці для ПЛР. Так визначають кількісний розподіл і ступінь специфічності пептидів. Також визначають стабільність і здатність проникати в клітину, які перевіряють, інкубуючи фат з клітинамимішенями при різних режимах температури та часу, з наступною елюцією життєздатних фагів. Приклади конкретного виконання заявленого способу. Приклад 1. Вибір клітинних популяцій. Для отримання високо специфічних пептидів до судинних гладком'язових клітин (СГМК), їх використовують в якості клітин-мішеней, а клітини, що взаємодіють з ними, як у складі стінки кровоносної судини (ендотеліальні) так і ті, що переносяться потоком крові (фібробласти), використовують в якості клітин-елімінаторів. Клітини карциноми цервікального каналу - HeLa, та клітини карциноми печінки - Нер-G2 , також використовують як клітини - елімінатори. Культуру СГМК ініціюють з магістральних кровоносних судин пацієнтів з патологіями серцево-судинної системи. Цілий ряд клітинних ліній також доступний у вигляді комерційних препаратів. Наприклад, паралельно культурам клітин, отриманим при обробці артерій і вен пацієнтів, використовували СГМК коронарної артерії (Cell Systems GmbH, Lot # 17151, Catalog # CC-2583). Препарати вихідної культури СГМК є первинними в своєму походженні та мають обмежену кількість дуплікацій, що лімітує кількість їх наступних пересівів. Тому, в усіх випадках, для подальшого дослідження ініціюють культури, використовуючи зразки клітин, які мають не більш ніж шість поділів. Для культивування використовують пластикові флакони, виробленні виключно для роботи з культурами тканин, загальною площею поверхні для росту від 25 до 182 квадратних сантиметрів (наприклад, флакони типів - Т25, Т75,Т182). Судинні гладком'язові клітини культивують, використовуючи поживне середовище, яке складається на 1/3 з кожного наступного компоненту - Waymouth (GIBCO BRL Cat # 31220-023), F12 (GIBCO BRL Cat. # 31220-024), SMCBM (Promocell Cat. # C-22260). Таке середовище доповнюють, попередньо інактивованою інкубацією протягом 30 хвилин при температурі 56°С на водяній бані, сироваткою плоду корови (GIBCO BRL Cat. # СС-4102) до кінцевої концентрації - 15%, а також розчином пеніциліну та стрептоміцину (GIBCO BRL Cat. # 15140-114) до кінцевої концентрації - 1,5%. Середовище, що містить в своєму складі сироватку плоду корови, називають збагаченим. Судинні гладком'язові клітини, які блоковані на стадії росту, культивують, використовуючи збіднене поживне середовище, тобто без додавання сироватки плоду корови, але з додаванням розчину пеніциліну - стрептоміцину у ви щевказаній концентрації. Для блокування судинних гладком'язових клітин на стадії росту, їх спочатку культивують, доки розмір моно шару не досягне 90% від всієї площі для росту, використовуючи збагачене поживне середовище, і тільки потім, проводять заміну збагаченого поживного середовища на збіднене і продовжують інкубацію протягом 72 годин, здійснюючи заміну застарілого поживного середовища на нове, кожні 24 години. Ендотеліальні клітини культивують, використовуючи поживне середовище - EGM-2 (Clonetics Company, Cat. # CC-3202) доповнене пакетом комплементації, що додається - EGM-2 MV (Clonetics Company, Cat. # CC-4147), який містить фактори росту, гормони та інактивовану сироватку плоду корови, кінцева концентрація якої в середовищі після комплементації складає 10%. Ендотеліальні клітини ініціюють з препарату (Promocell Cat. # CC2519). Блокують ендотеліальні клітини на стадії росту аналогічно тому, як це роблять у випадку судинних гладком'язових клітин. Фібробласти культивують, використовуючи поживне середовище, основою якого є стандартне середовище DMEM (GIBCO BRL) доповнене інактивованою сироваткою плоду корови до кінцевої концентрації 10%, та розчином пеніциліну-стрептоміцину до кінцевої концентрації 1,5%. Ініціюють культур у фібробластів із препаратів пацієнтів з патологіями серцево-судинної системи та блокують також, як це описано для судинних гладком'язових клітин. Клітини HeLa 3Т3 та клітини Hep-G2 являють собою імморталізовані лінії, тому практично любий доступний зразок клітин використовують для ініціації культури, однак, намагаються використовувати клітини, що пройшли не більш 100 поділів. Для культивування цих клітин використовують поживне середовище на основі будь-якої з MEM- композицій (GIBCO BRL), доповнену сироваткою плоду корови в кінцевій концентрації 15-20% (що залежить від бажаних темпів росту) та розчином пеніциліну-стрептоміцину до кінцевої концентрації 1,5%. Для отримання достовірних результатів скринінгу дані типи клітин також піддають культивуванню з використанням збідненого поживного середовища, імітуючи блокування росту подібне з тим, що спостерігається для первинних типів клітин (СГМК, ендотеліальні клітини та фібробласти). Приклад 2. Виділення РНК та реакції оберненої транскрипції (див. Фіг.1) На основі первинного відбору клітинних популяцій виділяють РНК, як з клітин-мішеней, так і з клітинелімінаторів. Клітини всіх типів нарощують протягом 5-7 діб, використовуючи флакони типу Т75 та 18мл поживного середовища, зміну якого проводять кожну добу. Судинні гладком'язові клітини культивують у 40 флаконах, з рахунку 3,5*105 клітин на флакон. Ендотеліальні клітини культивують в 20 флаконах, з розрахунку 3,5*105 клітин на флакон. Клітини HeLa та Hep-G2 культивують в 20 флаконах, з розрахунку 1,6*105 клітин на флакон. Клітини інтенсивно промивають 2 рази, використовуючи 12мл буфера HBSS (GIBCO BRL) кожний раз, далі обробляють протягом 3 хвилин використовуючи 6мл розчину, що містить 0,05% трипсину та 0,53мМ ЕДТА (етілендіамінтетраацетат), та нейтралізують додаванням 9мл буферу HBSS. Збирають клітини центрифугуванням протягом 10 хвилин на 3000*g, та промивають 3 рази, використовуючи 20мл буфера HBSS. Число клітин визначають за допомогою камери для рахунку клітин, для чого змішують 50мкл суспензії клітин з рівним об'ємом 15%-ого розчину трипанового блакитного, що є вітальним барвником. Число живих клітин фіксують та розраховують загальне число клітин в суспензії. У випадку використання методів жорсткої екстракції гуанідінізотіоцианатом, як з наступним осадженням солями літію, так і без осадження ними, клітини концентрують центрифугуванням, супернатант видаляють, а осад ресуспендують інтенсивно в буфері для лізису (5,25М гуанідінізотіоцианату; 50мМ Трис-СІ, рН 6,4; 20мМ ЕДТА; 1% Тритон-Х100; 0,1М 2-меркаптоетанол) з розрахунку 1мл на 107 клітин. До даного розчину додають 0,1 об'єму 2М р-ну ацетату натрію, 1 об'єм р-ну водного фенолу, 0,3 об'єму суміші хлороформ-ізоаміловий спирт (у співвідношенні 24:1) та інтенсивно перемішують протягом 30-60 секунд. Інкубують отриману суміш 15 хвилин при температурі 0°С, після чого центрифугують протягом 30 хвилин на 8000*g при температурі 4°С, з метою розділення водної та органічної фаз. Водну фазу після осадження переносять в нову пробірку та додають до неї 0,6 об'єму ізопропілового спирту, попередньо охолодженого до температури -20°С, перемішують та поміщують на не менш 60 хвилин у о холоджувач з температурою -70°С. Після інкубації розчин центрифугують 30 хвилин на 12000*g при температурі 4°С, супернатант ретельно видаляють, а осад промивають декілька разів центрифугуванням по 5 хвилин у присутності 1мл 70%-ого р-ну етилового спирту. Отриманий осад розчиняють в 0,4мл води обробленої діетилпірокарбонатом (ДЕПК). У випадку застосування методів м'якої екстракції з використанням протеїнази К, клітини концентрують центрифугуванням, супернатант видаляють, а осад ресуспендують інтенсивно в попередньо охолодженому буфері для лізису (0,15М хлориду натрію; 10мМ Трис - СІ, рН 8,5; 0,05% NP-40; 0,01% 3-карбоксиаурінової кислоти; 0,1М 2-меркаптоетанолу) з розрахунку 1мл на 5*107 клітин. Потім, суміш центрифугують 2 хвилини на 12000*g та температурі 4°С, супернатант переносять в нову пробірку і додають рівний об'єм буфера для протеїнази К (0,2М Трис - Ацетат; 0,3М натрію ацетат; 0,05 М ЕДТА, рН 7,5; 2% (в/о) натрію додецилсульфат), що містить 400 мг/мл протеїнази К, та інкубують 60 хвилин при температурі 37°С. Далі, додають 1 об'єм суміші фенол-хлороформ-ізоаміловий спирт (у співвідношенні 25:24:1) та інтенсивно перемішують 5 хвилин при кімнатній температурі. Після чого, суміш центрифугують 2 хвилини на 12000*g при кімнатній температурі, отриману водну фазу переносять в нову пробірку та повторюють екстракцію сумішшю фенол-хлороформізоаміловий спирт (у співвідношенні 25:24:1) декілька разів. До водної фази, що отримана в результаті екстракції, додають 2,5 об'єму абсолютного етилового спирту, інтенсивно перемішують, і осаджують РНК, розміщуючи зразок на 30 хвилин у охолоджувач з температурою -70°С. Надалі розчин, що містить РНК, центрифугують 10 хвилин на 12000*g при температурі 4°С, та отриманий осад промивають 3 рази центрифугуванням 5 хвилин на 12000*g, у присутності 700мкл 70%-ого розчину етилового спирту. Осад висушують 2,5 хвилини під вакуумом і розчиняють в 0,4мл води обробленої ДЕПК. Отриманий в результаті первинної екстракції розчин сумарної РНК, піддають обробці дезоксирибонуклеазой першого типу, з метою видалення фрагментів ДНК, здатних потрапити у розчин РНК в процесі ЇЇ екстракції і здатних утр удняти подальше фракціонування такої РНК та її використання в реакціях оберненої транскрипції. Для цього, до 0,4мл розчину, що містить 4 мг/мл РНК, додають 12мкл (480 одиниць) інгібітору рибонуклеаз (Invitrogen, Cat. # 10777-019), 50мкл 10-ти кратного стандартного буфера для дезоксирибонуклеази та 50мкл (500 одиниць) дезоксирибонуклеази першого типу (GIBCO BRL). Потім, отриману суміш інкубують 2 години при температурі 37°С, та зупиняють реакцію одночасним додаванням 0,1 об'єму 3М р-н у ацетату натрію (рН 5,2), 1,1 об'єму розчину водного фенолу, 0,3 об'єму суміші хлороформ-ізоаміловий спирт (у співвідношенні 24:1). Розчин інтенсивно перемішують протягом 1 хвилини, інкубують 15 хвилин при температурі 0°С та центрифугують 10 хвилин на 12000*g при температурі 4°С. Водну фазу переносять в нову пробірку і осаджують РНК за допомогою абсолютного етилового спирту, як описано вище. Для виділення фракції матричної РНК (мРНК), 1-2мг сумарної РНК розчиняють в 5мл буфера для нанесення (додається до колонки), і наносять на колонку, що містить Оліго - (дТ) - целюлозу (Sigma, Cat # 03131), попередньо промиту 3 рази 1мл буфера для нейтралізації. Після екстракції колонку промивають 5 разів, використовуючи 1мл буфера для нейтралізації, та проводять елюцію зв'язаної мРНК 1,5мл буфера для елюції. Надалі, мРНК, що міститься в отриманому розчині, осаджують додаванням 150мкл 3М р-ну ацетату натрію (рН 5,2) та 3,75мл абсолютного етилового спирту. Приклад 3. Синтез кДНК і фрагментація транскриптома. З метою отримання молекул кДНК для клонування, сумарну РНК або/та мРНК використовують в реакціях оберненої транскрипції, що направляються РНК - залежною - ДНК - полімеразою. Для цього, змішують 0,001-5,000мг сумарної РНК та 1мкл праймера (500мкг оліго - (дТ) або 0,050-0,250мкг праймера, що випадково зв'язується), до яких додають 1мкл 10мМ р-ну дезоксирибонуклеотидів (10мМ кожного у нейтральному розчині) тадоводять об'єм суміші до 12мкл, використовуючи подвійно дистильовану воду оброблену ДЕПК. Отриману суміш інкубують 5 хвилин при температурі 65°С, швидко охолоджують і додають 4 мкл 5-ти кратного буферу для синтезу первинного ланцюгу кДНК (250мМ Трис -СІ, рН 8,3; 375мМ хлориду калію; 15мМ хлориду магнію), 2мкл 0,1 М р-ну дитіотриетолу (ДТТ), 1мкл (40 одиниць) інгібітору рибонуклеаз. Вміст пробірки інтенсивно перемішують та інкубують 2 хвилини при температурі 42°С, після чого, додають 1мкл (200 одиниць) Superscript II РНК - залежної - ДНК - полімерази (GIBCO BRL, Cat. # 18064-014) інтенсивно перемішують та інкубують 60 хвилин при температурі 42°С. Реакцію оберненої транскрипції зупиняють шляхом інактивації ферменту, для чого суміш інкубують 15 хвилин при температурі 70°С. З метою видалення фрагментів РНК компліментарних новосинтезованим молекулам кДНК та здатним утруднювати наступн у ампліфікацію кДНК за допомогою ПЛР, до розчину додають 1мкл (2 одиниці) рибонуклеази Н та інкубують 20 хвилин при температурі 37°С . На цьому етапі здійснюють первинну фрагментацію транскриптома шляхом контрольованого синтезу фрагментів кДНК обмежених у розмірі та гетерогенності, що досягається використанням різних концентрацій іонів у реакційній суміші, комбінацій праймеров, кількістю вихідного матеріалу та режимами температури. Синтез 2-ого ланцюгу кДНК здійснюють за допомогою полімеразної ланцюгової реакції (ПЛР), для чого використовують не більш 10% кінцевого продукту після обробки рибонуклеазою Н. Збирають суміш послідовним додаванням 5мкл 10-ти кратного буферу для ПЛР (200мМ Трис -СІ, рН 8,4; 500мМ хлориду калію), 1,5мкл 50мМ р-ну хлориду магнію, 1мкл 10мМ р-ну дезоксирибонуклеотидів, по 1мкл (10мкмоль) кожного з праймеров для ампліфікації, 0,5мкл (2,5 одиниці) Taq - ДНК - по-лімерази, 2мкл р-ну кДНК (кінцевий продукт реакції синтезу первинного ланцюгу кДНК), 38,1мкл води обробленої ДЕПК. Розчин інтенсивно перемішують та інкубують протягом 2 хвилин при температурі 94°С. Потім проводять 15-40 стандартних циклів ампліфікації ПЛР. З метою отримання фрагментів придатних для клонування, кінці молекул кДНК обробляють та легують з синтетичними олігонуклеотидними лінкерами, які містять в своєму складі місце впізнання для рестрикційної ендонуклеази - Eco R1 та частково місце впізнання для рестрикційної ендонуклеази - Hind III. Це дозволяє отримати після обробки таких лінкерів відповідними рестрикційними ендонуклеазами, молекули кДНК, що здатні ефективно легуватись з 5'-кінці лише з Eco R1 обробленою ДНК, та з 3'-кінця лише з Hind ІІІ обробленою ДНК. Для цього до 20мкл (не більш 10мкг) розчину молекул кДНК додають 3мкл 10-ти кратного буферу для Т4 - ДНК поліме-рази (200мМ Трис - СІ, рН 8,4; 500мМ хлорид калію), 1,5мкл 100мМ р-ну ДТТ, 3мкл 1 мМ р-ну дезоксирибонуклеотидів, та 1мкл (1,5 одиниць) Т4 - ДНК - полімерази, та доводять кінцевий об'єм розчину до 30мкл, використовуючи подвійно дистильовану воду. Розчин повільно перемішують та інкубують протягом 20 хвилин при температурі 11°С. Реакцію зупиняють додаванням рівного об'єму суміші фенол-хлороформізоаміловий спирт (у співвідношенні 25:24:1) і потім ДНК, що міститься у водній фазі, осаджують додаванням розчинів ацетату натрію та абсолютного етилового спирту, як описано вище. Отриманий осад, що містить молекули кДНК, розчиняють в 10мкл подвійно дистильованої води. Олігонуклеотидні лінкери фосфорилюють шляхом інкубації з Т4 - полінкулеотидкіназою, безпосередньо перед використанням в реакціях легування. Для цього, збирають суміш послідовним додаванням до 10мкл р-ну молекул кДНК (з попереднього етапу), 2мкл 10-ти кратного буферу для легування (10х=200мМ Трис - СІ, рН 7,6; 50мМ хлорид калію), 2мкл 1мМ р-ну АТФ, 2мкл (100 пікомоль) р-ну олігонуклеотидних лінкерів, 2мкл 100 мМ р-ну ДТТ, 2,0мкл (5 одиниць) Т4-полінуклеотидкінази. Розчин перемішують та інкубують протягом 5 хвилин при температурі 37°С. Потім додають 6-8 одиниць Т4 - ДНК лігази та інкубують протягом 6-20 годин при температурі 16°С. Далі молекули кДНК розщеплюють за допомогою 2-х рестриктаз - Hind III та Eco R1. Для цього, до розчину молекул кДНК, додають 10мкл 10-ти кратного буфера для рестрикційної ендонуклеази Hind III, та 10мкл (100 одиниць) рестрикційної ендонуклеази Hind III (Invitrogen), об'єм такої суміші доводять до 100мкл за допомогою подвійно дистильованої води. Отриману суміш інкубують протягом 2 годин при температурі 37°С. Потім додають 10мкл 10-ти кратного буфер у для рестрикційної ендонуклеази Eco R1 та 10мкл (100 одиниць) рестрикційної ендонуклеази Eco R1 (Invi-trogen), та знов інкубують протягом 4 годин при температурі 37°С. Реакцію зупиняють додаванням рівного об'єму суміші фенол-хлороформ-ізоаміловий спирт (у співвідношенні 25:24:1) і потім ДНК, що міститься у водній фазі, осаджують додаванням розчинів ацетату натрію та абсолютного етилового спирту, як описано вище. Вторинну фрагментацію транскриптома здійснюють на етапі фракціонування та очистки отриманих молекул кДНК, для чого застосовують гельфільтраційну хроматографію. Хроматографічну колонку, що містить частинки сефарози певного розміру (Novagen) промивають 5 разів, використовуючи 1мл буфера для нейтралізації кожний раз. Наносять матеріал, що необхідно розділити, популяцію молекул кДНК, в об'ємі 1мл та збирають фракції по 25мкл. Приклад 4. Клонування та конструкція бібліотек. Створення бібліотек всіх органів проводять за однією схемою, як для клітин-мішеней так і для клітин-елімінаторів. Для цього використовують ДНК бактеріофагу Т7 (No vagen, T7 Select 10-3 вектор), оброблену з використанням 2-ох рестрикційних ендонуклеаз - Eco R1 та Hind III, тобто, того ж типу, що використовувались для обробки кінців кДНК. Розчин молекул ДНК вектора та розчин молекул кДНК змішують у співвідношенні 1:3, у перерахунку на моль речовини, додають еквівалентну кількість Т4 - ДНК - лігази (GIBСО BRL) та інкубують не більш ніж 28 годин при температурі 16°С. Реакцію зупиняють додаванням рівного об'єму суміші фенол - хлороформ - ізоаміловий спирт (у співвідношенні 25:24:1), і потім ДНК, що міститься у водній фазі, осаджують додаванням розчинів ацетату натрію та абсолютного етилового спирту, як описано вище. Отримані рекомбінантні молекули ДНК, в кількості не більш 1мкг, змішують з 250мкл р-ну пакувальних білків бактеріофагу та інкубують протягом 5 хвилин при температурі 0°С. Потім додають поживне середовище та продовжують інкубацію ще 60 хвилин при температурі 37°С. Приклад 5. Ампліфікація бактеріофагів. Бактеріофаг Т7 культивують, використовуючи штам E.coli BLT 5403 в якості хазяїна. При цьому, ампліфікацію проводять кількома шляхами. В одному випадку, мікро-ампліфікація, одиничну бляшку або елюат після скринінгу, абу будь-яку іншу малу кількість бактеріофагів, розчиняють в 1мл поживного середовища, виготовленого за методом Луріа-Бертані. Потім додають 1мл свіжо приготовленої культури штаму-хазяїна з оптичною густиною 0,4-0,6 при довжині хвилі 600 нм (OГ 600=0,4-0,6), яку готують нарощуванням 0,5мл нічної культури в 50мл поживного середовища протягом 2-3 годин. Суміш фагів і бактерій загальним об'ємом 2мл інкубують на шейкері при температурі 37°С та 200 обертів за хвилину протягом 3-4 годин, спостерігаючи повний лізис, що виражається просвітленням вмісту пробірки. В другому випадку, макро-ампліфікацію проводять, використовуючи колби, шляхом інокуляції необхідної кількості фагів в 135мл свіжо приготовленої денної культури штаму-хазяїну (OГ 600=0,4-0,6) з наступним ростом протягом 4-5 годин до моменту повного лізису. До лізованих культур додають 0,1 об'єму 5М р-ну хлориду натрію, інтенсивно перемішують 30 секунд та центрифугують протягом 3 хвилин на 7000*g та при температурі 4°С. Супернатант переносять в нову пробірку та додають 1/6 об'єму розчину, що містить 20% поліетиленгліколю та 2,5М хлориду натрію, інтенсивно перемішують, та інкубують не менше 3 годин при температурі 0°С. Далі, розчин центрифугують 10 хвилин на 8000*g, а осад, що містить фаги, розчиняють в 1мл стандартного буфера TBS (Трис - Боратний Буфер, GIBCO BRL) та знов осаджують фагові частинки додаванням розчину поліетиленгліколю - хлористого натрію з наступною інкубацією та центрифугуванням протягом 10 хвилин на 12000*g та температурі 4°С. Отриманий осад розчиняють в 0,2мл буфера TBS. Приклад 6. Скринінг (див. Фіг.2) Скринінг бібліотек здійснюють, як багаторазовим повторенням одноступеневої процедури, так і за допомогою багатоступеневої комплексної процедури, що включає селекцію на комбінаціях різних типів клітин-елімінаторів. Для цього ініціюють культуру судинних гладком'язових клітин в чашках Петрі діаметром 60мм з розрахунку 64000 клітин на чашку та нарощують протягом 2-ох діб. Одночасно, в аналогічних чашках ініціюють культури ендотеліальних клітин, фібробластів, HeLa, Hep-G2, відповідно в кількостях - 48000,24000,16000,16000 клітин на чашку, та нарощують протягом 2-ох діб. Клітини промивають інтенсивно 5 разів по 5 хвилин, використовуючи 3мл буфера HBSS кожного разу. При використанні одноступеневої процедури, 1010 фагових частинок з будь-якої бібліотеки, розчиняють в 2мл буфера HBSS та інкубують 20 хвилин з преінкубаційним матеріалом (чашкою Петрі, в якій замість клітин містилось лише середовище, надалі - пластик). Після цього, верхню фазу, що містить не зв'язані з пластиком фаги, переносять на попередньо промиті судинні гладком'язові клітини та інкубують 40 хвилин при температурі 4°С. Після інкубації, з метою видалення незв'язаного матеріалу, клітини інтенсивно відмивають 5 разів по 5 хвилин, використовуючи кожного разу 3мл буфера HBSS, з наступною елюцією специфічно зв'язаних фагів за допомогою 1мл буфера для елюції, який містить в своєму складі не більш ніж 1,0% додецилсульфат натрію, або за допомогою 1мл свіжо виготовленої денної культури штаму хазяїна. Титр фагів в елюаті визначають та елюат ампліфікують для отримання як мінімум 1010 фагів, котрі інкубують з новою порцією преінкубаційного матеріалу та новою порцією судинних гладком'язових клітин, повторюючи селекційний цикл 3-5 разів. При використанні багатоступеневої процедури, 1010 фагових частинок з будь-якої бібліотеки, розчиняють в 2мл буфера HBSS та інкубують 20 хвилин з преінкубаційним матеріалом (пластик). Після цього, верхню фазу, що містить не зв'язані з пластиком фаги, переносять на попередньо промиті ендотеліальні клітини та інкубують протягом 40 хвилин при температурі 4°С, з наступним переносом верхньої фази на нову порцію ендотеліальних клітин та аналогічною інкубацією, що повторюють 2-3 рази і лише після цього, верхню фазу спрямовують на фібробласти та проводять схожу селекцію 2-3 рази. Або верхню фазу після інкубації на ендотеліальних клітинах, без додаткової селекції на фібробластах, переносять безпосередньо на судинні гладко м'язові клітини та інкубують протягом 40 хвилин при температурі 4°С. Відмивку та елюцію проводять, як описано вище. В іншому випадку, верхню фазу після інкубації з преінкубаційним матеріалом (пластик) переносять на попередньо промиті клітини HeLa та інкубують з ними протягом 40 хвилин при температурі 4°С, а далі, верхню фазу переносять на клітини Hep-G2 та інкубують з ними протягом 40 хвилин при температурі 4°С, після чого, верхню фаз у переносять на нову порцію клітин HeLa та клітин Hep-G2, повторюючи комбіновану селекцію 2-3 рази. Кінцеву верхню фазу переносять на судинні гладко м'язові клітини та інкубують протягом 40 хвилин при температурі 4°С. В іншому варіанті скринінгу, матеріал після селекції на імморталізованих типах клітин, переносять на ендотеліальні клітини та/або фібробласти, і лише потім на судинні гладком'язові клітини. Приклад 7. Виконання комбінованого тестування пептидів. А. Імуноферментний аналіз. Специфічність індивідуальних пептидів у складі фагових частинок , оцінюють, використовуючи їх, замість першого антитіла в прямому імуноферментному аналізі, де в якості антигену виступають клітини-мішені та клітини-елімінатори. Для цього, всі типи клітин, що використовуються, культивують в 96-лункових мікропланшетах, ініціюючи культур у з розрахунку 10000 клітин на лунку. Клітини нарощують протягом 2 діб, при цьому, проводять заміну поживного середовища кожні 24 години. Потім, мікропланшети промивають інтенсивно 5 разів по 5 хвилин, використовуючи кожного разу по 200мкл буфера HBSS. Далі інкубують 106-107 фагових частинок з клітинами протягом 1 години при температурі 4°С, з інтенсивною наступною відмивкою буфером HBSS, та інкубацією з другим антитілом, протягом 1 години при кімнатній температурі. Друге антитіло розпізнає специфічну амінокислотну послідовність на фаговій частинці та представляє собою кон'югат з пероксидазой хрону. Потім, планшети промивають знов буфером HBSS та проводять детекцію другого антитіла, використовуючи субстрат для пероксидази, розщеплення якого зупиняють додаванням 50мкл 1М р-ну сірчаної кислоти. Б. Аналіз на зв'язування. Зв'язування індивідуальних пептидів досліджують, використовуючи одноступеневу селекцію, при якій пептид, що аналізується, тестують паралельно, на зв'язування з клітинами-мішенями та клітинами-елімінаторами. Для цього ініціюють культуру судинних гладком'язових клітин в чашках Петрі діаметром 60мм з розрахунку 64000 клітин на чашку та нарощують протягом 2-ох діб. Одночасно, в аналогічних чашках ініціюють культури ендотеліальних клітин, фібробластів, HeLa, Hep-G2, відповідно в кількостях - 48000,24000,16000,16000 клітин на чашку, та нарощують протягом 2-ох діб. Використовують 109 фа гових частинок індивідуального фагу, які розчиняють в 2мл буфера HBSS. Такий розчин інкубують спочатку протягом 20 хвилин з преінкубаційним матеріалом (пластик) і потім переносять верхню фракцію на досліджувані клітини, попередньо промиті 5 разів буфером HBSS, тривалістю по 5 хвилин кожного разу. При цьому, процедуру паралельно виконують як для судинних гладком'язових клітин (клітини-мішені), так і для ендотеліальних клітин, фібробластів, клітин HeLa та Hep-G2 ( клітини-елімінатори), використовуючи для всіх типів клітин 2 режими температури: 4°С та 37°С. Після інкубації, не зв'язані частинки, інтенсивно відмивають, використовуючи буфер HBSS, після чого проводять елюцію зв'язаних частинок, використовуючи 1мл буфера для елюції. Визначають титр фагів в елюаті та порівнюють одержані значення для всіх типів клітин. В. Використання полімеразної ланцюгової реакції (ПЛР). Специфічність індивідуальних пептидів тестують, використовуючи ПЛР для ідентифікації клонів, а також для аналізу їх кількісного розподілу при взаємодії з клітинами-мішенями. Для цього, ініціюють культур у судинних гладком'язових клітин в чашках Петрі діаметром 60мм з розрахунку 64000 клітин на чашку та нарощують протягом 2-ох діб. Одночасно, в аналогічних чашках ініціюють культури ендотеліальних клітин, фібробластів, HeLa, Hep-G2, відповідно в кількостях - 48000, 24000,16000, 16000 клітин на чашку, та нарощують протягом 2-ох діб. Фатові частинки змішують в еквівалентних кількостях, звичайно 107 частинок кожного типу, з розрахунку на кінцевий об'єм розчину 2мл. Отриманий розчин інкубують спочатку 20 хвилин з пре інкубаційним матеріалом (пластик) і потім переносять верхню фракцію на досліджувані клітини, попередньо промиті 5 разів буфером HBSS, тривалістю по 5 хвилин кожний раз. При цьому, процедуру одночасно проводять, як для судинних гладком'язових клітин (клітини-мішені), так і для ендотеліальних клітин, фібробластів, клітин HeLa та Нер-02 ( клітини-елімінатори), використовуючи для всіх типів клітин 2 режими температури: 4°С та 37°С. Після інкубації незв'язані частинки, інтенсивно відмивають, використовуючи буфер HBSS, після чого проводять елюцію зв'язаних фагів, використовуючи 1мл буфера для елюції. Потім визначають титр фагів в елюаті та розсівають рівні аліквоти елюату до отримання одиничних бляшок. Відбирають з кожної чашки 100 бляшок, розподіляють їх групами по дві і виділяють ДНК шляхом раптового лізису фагови х частинок. Для цього інкубують фатові частинки в 10мМ р-ні ЕДТА протягом 10 хвилин при температурі 65°С, с наступним охолодженням і центрифугуванням протягом 3 хвилин на 14000*g. Отриманий розчин, що містить ДНК бактеріофагу використовують в якості матриці для ПЛР за стандартною для даного вектора схемою, спрямованою праймерами до ділянок вектора або спеціально розробленими праймерами. Після цього визначають абсолютну частоту зустрічальності кожного з фагів в залежності від типу клітин. Г. Тест на стабільність та здатність проникати в клітину. Стабільність специфічного пептиду, тобто здатність максимально довго зберігати свою функціональну ефективність в клітині, до якої він є специфічним, а також в самому організмі хазяїна, прямо перетинається зі здатністю ефективно проникати в клітину, та зберігати активність і специфічно бомбардувати внутрішньоклітинні цілі. Для цього, ініціюють культур у судинних гладком'язових клітин в чашках Петрі діаметром 60мм з розрахунку 64000 клітин на чашку та нарощують протягом 2-ох діб. 1010 фагових частинок, що являють собою фракцію, отриману після елюції фагів в результаті 3-х раундів селекції багатоступеневим скринінгом, інкубують з судинними гладком'язовими клітинами при 2-ох режимах температури: 4°С та 37°С, та протягом різних періодів часу. Далі клітини відмивають та проводять їх лізис протягом 10 хвилин, використовуючи 2мл буфера для лізису (1мл 2%-ого р-ну дезоксихолієвої кислоти; 10мМ Трис; 2мМ ЕДТА, рН 8,0). Після нейтралізації, розчин, що містить фаги, розсівають до окремих бляшок, які потім відбирають з метою визначення їх нуклеотидноЇ первинної послідовності. Приклад 8. Визначення нуклеотидної первинної та кодованої амінокислотної послідовності та їх комп'ютерний аналіз. Послідовність ДНК ділянки геному вектора, що представляє собою вставку кДНК, обмежену праймерами вектора, ампліфікують, використовуючи ПЛР, та визначають її нуклеотидну первинну послідовність, за допомогою автоматичного секвенатора АВІ Prism 310 (Applied Biosystems Inc.), керуючись рекомендованими виробником протоколами проведення реакцій. Використовуючи специфічні сигнали розпізнавання у векторі, нуклеотидну первинну послідовність транслюють в послідовність амінокислот пептиду. Послідовність нуклеїнової кислоти та послідовність пептиду, перевіряють на наявність гомологій з зареєстрованими послідовностями, що знаходяться в базах даних міжнародних агентств. Для цього, використовують пакет програм комп'ютерного аналізу - GCG або подібний пакет для автоматичного аналізу послідовностей, що розповсюджується Національним Центром Біотехнологічної Інформації США.
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for the preparation of specific peptides for therapeutic purposes
Автори англійськоюRazumenko Mykhailo Viktorovych
Назва патенту російськоюСпособ получения специфических пептидов для терапевтических целей
Автори російськоюРазуменко Михаил Викторович
МПК / Мітки
МПК: G01N 33/15, A61K 38/00, C12N 15/09, C12N 15/12
Мітки: спосіб, цілей, специфічних, пептидів, терапевтичних, отримання
Код посилання
<a href="https://ua.patents.su/8-66110-sposib-otrimannya-specifichnikh-peptidiv-dlya-terapevtichnikh-cilejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання специфічних пептидів для терапевтичних цілей</a>
Нові солі врс-пептидів з органопроективною активністю, спосіб їх отримання і їх використання в терапії

Номер патенту: 61955
Опубліковано: 15.12.2003
Автори: Грабаревич Желько, Місе Степан, Дувняк Марко, Туркович Бранко, Зайверт Свен, Петек Маріян, Удовічич Іван, Ротквич Іво, Сікірич Предраг
МПК: A61K 38/00, A61P 43/00, C07K 14/47, A61P 39/00, C07K 7/06, C07K 7/08
Мітки: нові, використання, спосіб, терапії, активністю, врс-пептидів, солі, отримання, органопроективною
Формула / Реферат:
1. Сіль пептидної сполуки для захисту організму (ВРС), що містить 8 амінокислотних залишків, де аніон солі є негативно заряджений пептид, що має загальну формулу [Zaa Pro Pro Pro Xaa Yaa Pro Ala] (-)або(2-),деXaa є нейтральний аліфатичний амінокислотний залишок,Yaa є основний амінокислотний залишок, іZaa є кислий амінокислотний залишок,і де катіон солі є катіоном неорганічної або органічної...
Похідні пептидів, що є інгібіторами еластази лейкоцитів людини, спосіб їх отримання та фармацевтична композиція

Номер патенту: 39093
Опубліковано: 15.06.2001
Автори: ТРЕЙНОР Діана Емі, СТЕЙН Марк Морріс, БЕРГЕСОН Скотт Хавен, ЕДВАРДС Філіп Д'юк, ВОЛАНІН Дональд Джон, ШВАРЦ Джон Ентоні, ВІЛДОНГЕР Річард Алан, ШОУ Ендрю
МПК: A61K 31/4427, C12N 9/99, C07K 5/06, A61K 31/4025, A61K 31/4409, C07K 5/08, C07K 1/113, A61K 31/4418, A61K 31/535, C07K 14/81, C07D 213/26, A61K 31/44, A61P 11/00, C07K 5/04, A61P 29/00, C07D 409/12, A61K 38/00, A61K 31/4433, A61K 31/401, A61P 43/00, C07K 5/093, C07D 333/00, C07D 403/12, C07D 207/16, C07D 401/12, A61P 9/10, C07C 205/00, A61K 38/55, A61K 31/40
Мітки: лейкоцитів, похідні, людини, спосіб, фармацевтична, отримання, еластази, інгібіторами, композиція, пептидів
Формула / Реферат:
1. Производные пептидов общей формулы (I) , (I)гдеХ является группой, выбираемой из R3-А- или R3-АNНR6-СHR5-СО-,А является группой, выбираемой из -NHCO-, -ОСО-, -СО-, -SO2-,R1 является С1-С4-алкилом,R2 выбирают из фенила, бензила, С1-С4-алкила,...
Спосіб заглушування завад для доплеровської рлс з селекцією рухомих цілей, які викликані відбиттям електромагнітних хвиль від об’ємних розподілених цілей

Номер патенту: 17872
Опубліковано: 03.06.1997
Автори: Дубровський Сергій Єгорович, Рогов Петро Дмитрович, Касаткін Леонід Веніамінович, Феньов Дмитро Васильович
МПК: G01S 7/36, G01S 13/50
Мітки: хвиль, розподілених, викликані, рлс, завад, цілей, заглушування, доплеровської, спосіб, об'ємних, відбиттям, рухомих, селекцією, електромагнітних
Формула / Реферат:
Способ подавления помех для доплеровских РЛС с селекцией движущихся целей, вызванных отражениями электромагнитных волн от распределенных объемных целей - дождя, снега, града и т.п., заключающийся в выделении и усилении отраженных сигналов с доплеровскими поправками частоты, фильтрации сигналов доплеровских частот системой узкополосных фильтров (гребенкой фильтров) для определения скорости цели, индикации движущейся цели, отличающийся тем, что...
Спосіб експериментального використання тканинних пептидів для лікування аутоімунного діабету

Номер патенту: 60700
Опубліковано: 15.10.2003
Автори: Гейко Ольга Олексіївна, Кайдашева Ельвіра Ільїнічна, Кайдашев Ігор Петрович, Беркало Любов Володимирівна
МПК: A61K 38/00
Мітки: пептидів, експериментального, лікування, діабету, використання, тканинних, спосіб, аутоімунного
Формула / Реферат:
Спосіб експериментального використання тканинних пептидів для лікування аутоімунного діабету, що включає введення тканинних пептидів, який відрізняється тим, що як тканинні пептиди використовують пептиди, видобуті з підшлункової залози.
Спосіб перетворення пептидів класу ехінокандину у їх с4-гомотирозинові монодезоксі-аналоги
Номер патенту: 64787
Опубліковано: 15.03.2004
Автори: Яіванті Кенія, Кумар Ерра Котесвара Сатія Війая, Мукхопадхіаі Тріптікумар
МПК: C07K 7/56
Мітки: перетворення, ехінокандину, с4-гомотирозинові, пептидів, спосіб, класу, монодезоксі-аналоги
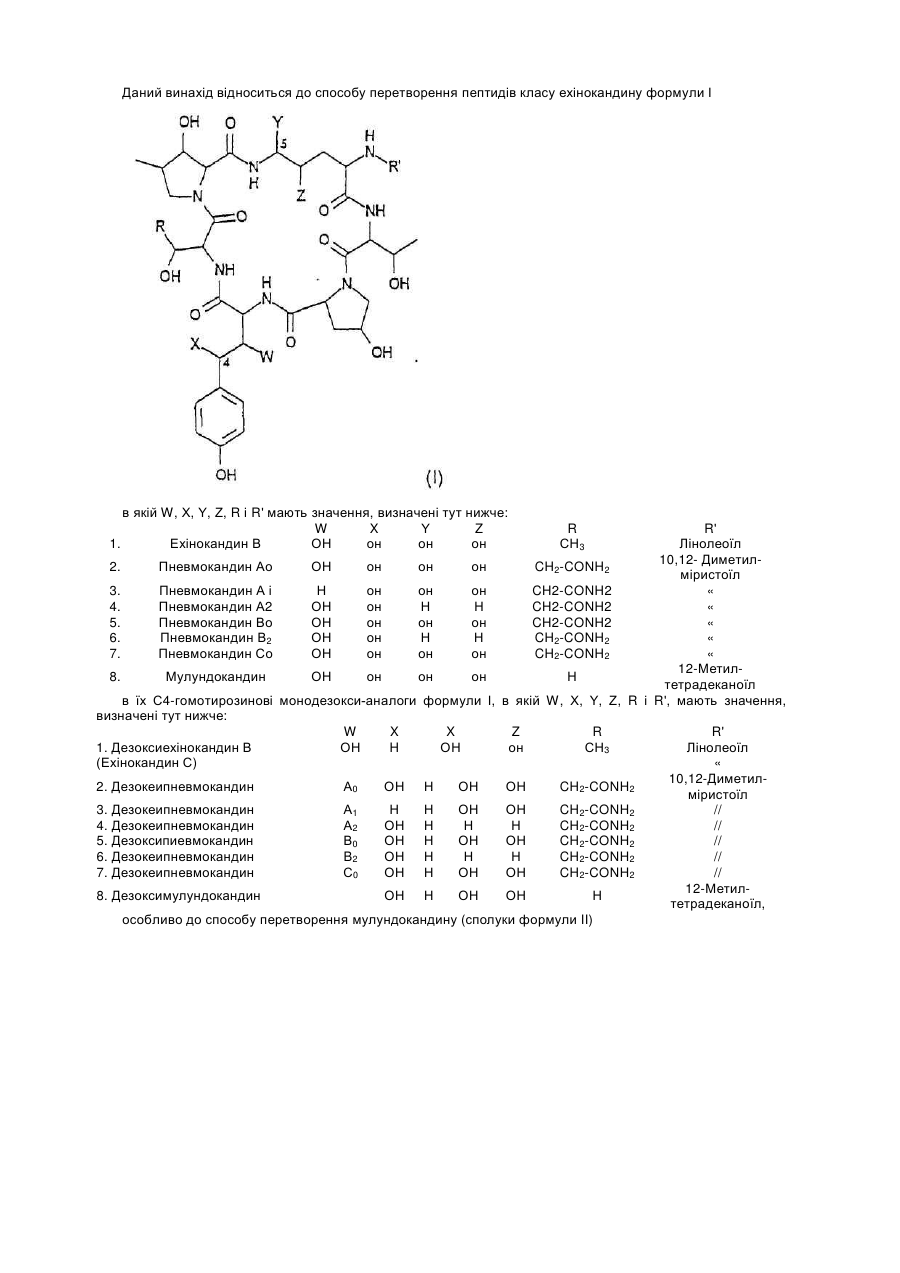
Формула / Реферат:
1. Спосіб перетворення пептидів класу ехінокандину формули (І)в якій W, X, Y, Z, R і R' мають значення, визначені нижче: W X Y Z R R' 1. Ехінокандин В OH OH OH OH ...