Похідні пептидів, що є інгібіторами еластази лейкоцитів людини, спосіб їх отримання та фармацевтична композиція
Номер патенту: 39093
Опубліковано: 15.06.2001
Автори: СТЕЙН Марк Морріс, ШВАРЦ Джон Ентоні, ВІЛДОНГЕР Річард Алан, ШОУ Ендрю, ЕДВАРДС Філіп Д'юк, ВОЛАНІН Дональд Джон, ТРЕЙНОР Діана Емі, БЕРГЕСОН Скотт Хавен








Формула / Реферат
1. Производные пептидов общей формулы (I)
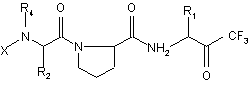 , (I)
, (I)
где
Х является группой, выбираемой из R3-А- или R3-АNНR6-СHR5-СО-,
А является группой, выбираемой из -NHCO-, -ОСО-, -СО-, -SO2-,
R1 является С1-С4-алкилом,
R2 выбирают из фенила, бензила, С1-С4-алкила, 2-(бензилоксикарбонил)этила, 4-(бензилоксикарбониламино)бутила, 4-(фенилсульфониламино)бутила и 2-[(фенил-сульфониламино)карбонил]этила,
R4 представляет собой водород,
R5 выбирают из бутила, 2-(бензилоксикарбонил)этила и 4-(бензил-оксикарбониламино)бутила,
R6 представляет собой водород,
и где R3 выбирают из числа следующих радикалов:
(1) С1-С6-алкил, необязательно замещенный С1-С4-алкоксикарбонилом, карбокси, 2-[(С1-С4)-алкокси]этокси, адамантилом, морфолинилом, пиридилом, тиенилом, оксопирролидинилом, фенилом, [(С1-С4)-алкоксикарбонил]фенилом, (карбокси)фенилом, [(С1-С4)-алкоксикарбонил]фенокси, (карбамоил)фенокси, (карбамоилметил)фенокси, (метансульфониламино)карбонилом, (бензолсульфониламино)карбонилом, (нафталинсульфониламино)карбонилом, (адамантансульфониламино)карбонилом или (хлорбензолсульфониламинокарбонил)фенилом;
(2) нафтил, аминохлорфенил, адамантил, фенил, необязательно замещенный гидроксигруппой, С1-С4-алкокси, С1-С4-алкоксикарбонилом, карбокси, галогеном, дигалогеном, ацетамидо, (метансульфониламино)карбонилом, (нафталинсульфониламино)карбонилом или [N-бензил-(4-бромбензолсульфонил)амино]карбонилом;
(3) группа формулы -W-SО2-NН-СО-фенил, в которой W представляет собой фенил, необязательно замещенный галогеном или нитрогруппой;
(4) циклопентил, замещенный С1-С4-алкокси, карбонилом или карбокси;
(5) 2-фенилвинил, замещенный в фенильном кольце группами С1-С4-алкоксикарбонил, карбокси или (хлорбензолсульфониламино)карбонил;
(6) 2-(уреидокарбонил)винил, которые являются ингибиторами эластазы лейкоцитов человека.
2. Способ получения производных пептидов общей формулы (I)
, (I)
где
X является группой, выбираемой из R3-A- или R3-АNНR6-СHR3-СО-,
А является группой, выбираемой из -NHCO-, -ОСО-, -СО-, -SO2-,
R1 является С1-С4-алкилом,
R2 выбирают из фенила, бензила, С1-С4-алкила, 2-(бензилоксикарбонил)этила,
4-(бензилоксикарбониламино)бутила, 4-(фенилсульфониламино)бутила и 2-[(фенил-
сульфониламино)карбонил]этила,
R4 представляет собой водород,
R5 выбирают из бутила, 2-(бензилоксикарбонил)этила и 4-(бензил-
оксикарбониламино)бутила,
R6 представляет собой водород,
и где R3 выбирают из числа следующих радикалов:
(1) С1-С6-алкил, необязательно замещенный С1-С4-алкоксикарбонилом, карбокси, 2-[(С1-С4)-алкокси]этокси, адамантилом, морфолинилом, пиридилом, тиенилом, оксопирролидинилом, фенилом, [(С1-С4)-алкоксикарбонил]фенилом, (карбокси)фенилом, [(С1-С4)-алкоксикарбонил] фенокси, (карбамоил)фенокси, (карбамоилметил)фенокси, (метансульфониламино)карбонилом, (бензолсульфониламино)карбонилом, (нафталинсульфониламино)карбонилом, (адамантансульфониламино)карбонилом или (хлорбензолсульфониламинокарбонил)фенилом;
(2) нафтил, аминохлорфенил, адамантил, фенил, необязательно замещенный гидроксигруппой, С1-С4-алкокси, С1-С4-алкоксикарбонилом, карбокси, галогеном, дигалогеном, ацетамидо, (метансульфониламино)карбонилом, (нафталинсульфониламино)карбонилом или [N-бензил-(4-бромбензолсульфонил)амино]карбонилом;
(3) группа формулы -W-SО2-NН-СО-фенил, в которой W представляет собой фенил, необязательно замещенный галогеном или нитрогруппой,
(4) циклопентил, замещенный С1-С4-алкокси, карбонилом или карбокси;
(5) 2-фенилвинил, замещенный в фенильном кольце группами С1-С4-алкоксикарбонил, карбокси или (хлорбензолсульфониламино)карбонил;
(6) 2-(уреидокарбонил)винил, отличающийся тем, что соединение общей формулы (II)
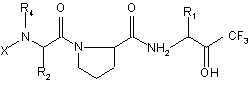 , (II)
, (II)
где R1, R2, R4 и Х имеют вышеуказанные значения,
окисляют с последующим, при необходимости, сульфонамидированием для тех
соединений, где R3 содержит карбокси- или этерифицированную карбоксигруппу, или, при
необходимости, гидролизом соединений, где R3 содержит этерифицированную
карбоксигруппу.
3. Фармацевтическая композиция, обладающая свойствами ингибитора эластазы лейкоцитов человека, содержащая активный компонент и фармацевтически приемлемый носитель, отличающаяся тем, что активным компонентом является производное пептидов общей формулы (I)
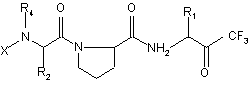 , (I)
, (I)
где
Х является группой, выбираемой из R3-А- или R3-АNНR6-СHR5-СО-,
А является группой, выбираемой из -NHCO-, -ОСО-, -СО-, -SO2-,
R1является С1-С4-алкилом,
R2 выбирают из фенила, бензила, С1-С4-алкила, 2-(бензилоксикарбонил)этила,
4-(бензилоксикарбониламино)бутила, 4-(фенилсульфониламино)бутила и 2-[(фенил-
сульфониламино)карбонил]этила,
R4 представляет собой водород,
R5 выбирают из бутила, 2-(бензилоксикарбонил)этила и 4-(бензил-
оксикарбониламино)бутила,
R6, представляет собой водород,
и где R3 выбирают из числа следующих радикалов:
(1) С1-С6-алкил, необязательно замещенный С1-С4-алкоксикарбонилом, карбокси, 2-[(С1-С4)-алкокси]этокси, адамантилом, морфолинилом, пиридилом, тиенилом, оксопирролидинилом, фенилом, [(С1-С4)-алкоксикарбонил]фенилом, (карбокси)фенилом, [(С1-С4)-алкоксикарбонил]фенокси, (карбамоил)фенокси, (карбамоилметил)фенокси, (метансульфониламино)карбонилом, (бензолсульфониламино)карбонилом, (нафталинсульфониламино)карбонилом, (адамантансульфониламино)карбонилом или (хлорбензолсульфониламинокарбонил)фенилом;
(2) нафтил, аминохлорфенил, адамантил, фенил, необязательно замещенный гидроксигруппой, С1-С4-алкокси, С1-С4-алкоксикарбонилом, карбокси, галогеном, дигалогеном, ацетамидо, (метансульфониламино)карбонилом, (нафталинсульфониламино)карбонилом или [N-бензил-(4-бромбензолсульфонил)амино]карбонилом;
(3) группа формулы W-SО2-NН-СО-фенил, в которой W представляет собой фенил, необязательно замещенный галогеном или нитрогруппой,
(4) циклопентил, замещенный С1-С4-алкокси, карбонилом или карбокси;
(5) 2-фенилвинил, замещенный в фенильном кольце группами С1-С4-алкоксикарбонил, карбокси или (хлорбензолсульфониламино)карбонил;
(6) 2-(уреидокарбонил)винил, в эффективном количестве.
Текст
39093 Изобретение относится к новым производным пептидов, применяемых в фармакологии и медицине в качестве ингибитора эластазы лейкоцитов человека, способу их получения, фармацевтической композиции. Некоторые ингибиторы эласта зы лейкоцитов че ловека, относящие ся к альдегидным производным пролинсодержащи х пептидов, раскрыты в Европейской заявке на патент, публикация № 0124317. Однако они проявляют невысокую активность в качестве ингибиторов эластазы лейкоцитов человека, кроме того, они недоста точно стабильны. Задача изобретения – поиск в ряду пепти дов новых производных пепти дов, обладающи х бо лее сильными ингибирующи ми свойствами по отношению к эластазе и более устойчивых, спо соба получения этих пептидов и фармацевтическая композиция на их основе, ингибирующая эластазу человеческих лейкоцитов. Поставленная задача достигается описываемыми производными пептидов общей формулы I: где Х является группой, выбираемой из R3-A- или R3-ANHR6-CHR5-CO-; А является группой, выбираемой из -NHCO-, -OCO-, -SO 2-, или -CO-; R1 является С1-С4-алкилом; R2 выбирают из фенила; бензила; С1-С4-алкила; 2-(бензилоксикарбонил)этила; 4-(бензилоксикарбониламино)бутила; 4-(фе нилсуль фониламино)бутила и 2-[(фе нилсуль фониламино)карбо-нил]этила; R4 представляет водород; R5 выбирают из бутила; 2-(бензилоксикарбонил)этила и 4-(бензилоксикарбониламино)бутила; и R6 представляет водород; и где R3 вы бирают из числа следующих радикалов; (1) С1-6-алкил, необязательно замещенный (С1-4)-алкоксикарбонилом, карбокси, 2-[(C1-4)-алкокси]этокси, адамантилом, морфо линилом, пиридилом, тиенилом, оксопирролидинилом, фенилом, [(C1-4)алкоксикарбонил]фенилом, (карбокси)фенилом, [(C1-4)алкоксикарбонил]фенокси, (карбамоил)фенокси, (карбамоилметил)фенокси, (метансульфо ниламино)карбонилом, (бензолсульфониламино)карбонилом, (нафталинсульфо ниламином)карбонилом, (адамантансульфониламино)карбонилом или (хлорбензолсульфониламинокарбонил)фенилом, (2) нафтил, аминохлорфе нил, адамантил, фенил, необязательно замещенный гидрокси, (С1-4)-алкокси), (С1-4)-алкоксикарбонилом, карбокси, галогеном, дигалогеном, ацетамидо, (метансульфониламино)карбонилом, (нафта линсульфо ниламино)карбонилом или [N-бензил-(4-бромбензолсульфонил)амино]карбонилом; (3) группа формулы -W-SO2-NH-CO-фенил, в которой W представляет собой фенил, необязательно замещенный галогеном или нитро; (4) циклопентил, замещен ный (С1-4)-алкокси, карбонилом или карбокси; (5) 2-фенилвинил, замещенный в фе нильном кольце группами (С1-4)-алкоксикарбонил, карбокси или (хлорбензолсульфо ниламино)карбонил; (6) 2-(уреидокарбонил)винил; способом получения производных пепти дов формулы I, заключающий ся в том, что производное пептида общей формулы II: значения радикалов определены выше, подвергают окислению с последующим, в случае необхо димости, сульфо намидированием соединения, где R3 - карбоксигруппы, или сложноэфирная группа, или превращением сложноэфирной группы карбоновой кислоты в соответствующую карбоксигруппу путем гидролиза; а также фармацевтической композицией, ингибирующей эластазу человеческих лейкоцитов, содержащей в качестве активного начала производные пептидов общей формулы I в количестве 100–250 мг на дозируемую единицу и фар мацевтически приемлемый носитель. В следующи х примерах и на всем протяжении описания изобретения используются следующие сокраще ния и условные обозначения: атм (атмосфер): т.кип. (температура кипения); оС (градус Цельсия), причем все температуры представляются в виде оС, если не отмечено особо; г (грамм); ч (час); мг (миллиграмм); мин (минута); мл (миллилитр); л (литр); моль (молей); ммоль (миллимоль); т.пл. (температура плавления); т.кип. (температура кипения); N (нормальный); нм (нанометр); нМ (наномолярный); х (раза); комнатная температура (20–23оС); ДЦК (1,3-дицилкогексилкарбодиимид); ДМФА (ди метилформамид); ДМСО (диметилсульфоксид); Et2O (диэти ловый эфир); EtOAc (эти лацетат); НОАс (ук сусная кислота); ОБТА (оксибензотриазол); МеОН (метиловый спирт); EtOH (этиловый спирт); Pd/C (катализатор – палладий на угле); пNA (пара-нитроанилид); ТГФ (тетрагидрофуран); КБЗ (бензилоксикарбонил); трет-БОК (третичный бутилоксикарбонил); ДМФА (диметилфор мамид); ТЭА (триэтиламин); ДЦК (1,3 дициклогексилкарбодиимид); АсОН 1 39093 (уксусная кислота); Исх.С. (исходное соединение); NMM (N-метилморфо лин); Ü (менее чем или равно); ТЭА (триэти ламин); ТФУК (трифторуксусная кислота); Ас2 О (ук сусный ангидрид); КДИМ (карбонилдиимидазол); ВРКДИМ (во дорастворимый карбодиимид); хлоргидрат 1 – этил-3-(3-диметиламинопропил)карбодиимида); ДМАП (4-диметиламинопиридин); периодинан (Десса-Мартина (1,1,1-триацетокси-2,1-бензоксиодол3(3Н)-он); газ. HCl (газообразный HCl), в других случаях НСl является водным раствором: Rh/C (катализатор – родий на угле); Ph (фе нильная группа); ТCХ (тон кослойная хроматография на силикагеле, если специально не указано); Rf (относительная подвижность в ТСХ); ЖХСД (жидкостная хроматография среднего давления); ЖХВД (жидкостная хроматография высокого давления); t12 (время удерживания в мин для ЖХВД); СП (скорость потока в мл/мин для ЖХВД); Col A (Зорбакс ОДS аналитическая колонка, 4,6 мм х 25 см); Col B (Феноменекс Зорбакс С-8 аналитическая колонка, 4,6 мм х 35 см); Col C (Альтекс Ультрасфера/Октил 10 мм 1 Д. х 25 см 5-микронная аналитическая и препаративная колонка); быстрая хроматография (быстрая колоночная хроматография на силикагеле, если не указано особо); поглоти тельная хроматография (поглотительная колоночная хроматография на силикагеле). Кроме того, С,Н,N и др. (обычные символы для элементов) используются в описании; 133,3 Па = 1 торр в качестве коэффи циента превраще ния с 760 торр = 14,7 фунтов на квадратный дюйм (рSi): 1Н ЯМР (ядерный магнитный резонанс), спектры получали, используя прибор на 80 МГц или на 250 МГц и тетраметилсилан (ТМС) в качестве внешнего стандарта (растворитель для конкретного примера отмечается в примере), d (части на миллион, слабое поле от ТМС); вместе с с. (синглет); д. (дублет), д.д. (дублет из дублетов); м. (мультиплет). Пример 1. Получение хло ристоводородной соли 2(RS),3(RS)-3-амино-4-метил-1,1,1-трифтор-2-пентанола. а. Синтез 2-метил-1-нитропропана. К заранее охлажденной (оС) суспензии AgNO2 (100,0 г, 0,65 моль) в Et2O (180 мл) по каплям добавляют 1-йод-2-метилпропан (94,0 г, 0,51 моль). Реакцию защища ли от света и перемешивали в те чение ночи, позволяя во время этой реакции подогреваться (доводиться) до комнатной температуры. Реакционную смесь фильтровали через целит. Фильтрат концентрировали под вакуумом и оста ток перегоняли под вакуумом (осторожно: потенциально взрывчатое вещество), что бы получить продукт (37,7 г, 0,366 моль); т. кип. 61–65оС при 6913,6 Па (52 мм рт.ст.). б. Синтез 2(RS),3(SR)-4-метил-3-нитро-1,1,1-трифтор-2-пентанола. 1-Нитро-2-метилпропан (37,7 г, 0,366 моль) из примера 1а, этиловый полуа цеталь трифторацетальдегида (58,5 г, 0,366 моль 90%-ной чистоты) и К2СО3 (3,4 г, 0,025 моль) смешивали и перемеши вали при 60оС в течение 3 ч, а затем при комнатной температуре в течение 3 дней. Солевой раствор (75 мл) и 1 N водный HCl (50 мл) добавляли и нижний органический слой разделяли. Водный слой экстраги ровали с помощью Et2O (дважды по 250 мл каждый раз) и объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали быстрой (flash) хроматографией на силикагеле с градиентным элюированием с помощью смеси СН 2Cl2 : гексан (50:50), СН2Cl2 : гексан (75:25), СН2Cl2 (100%) и МеОН:СН2Cl2 (5:95), что бы получить продукт (44,9 г), ТСХ, Rf = 0,65, силикагель, EtOAc : СНCl3 (5:95). в. Синтез хло ристоводородной соли 2(RS), 3(SR)-3-амино-4-метил-1,1,1-трифтор-2-пентанола. Раствор части продукта по примеру 1б (37,0 г, 0,184 моль) в Et2O (200 мл) по каплям добавляли к суспензии литийалюминийгидрида (22,0 г, 0,58 моля) в Et2O (800 мл). Реакционную смесь перемешивали в течение 45 мин, и осторожно добавляли насыщенный водный раствор Na 2SO4 (110 мл). Получившуюся суспензию фильтровали; фильтрат обрабатыва ли эфирным раствором НСl и концентрировали под вакуумом, чтобы получить продукт (37,6 г), который использовали без дальнейшей очистки. 1Н ЯМР данные (СД3СОСД3) (250 МГц) : 1,2 d, м, 6Н; 2,3 d, м, 1Н; 3,58 d, м, 1Н; 4,98 d, м, 2Н; 7,78 d, м, (NH 2). Пример 2. Получение 2(RS), 3(SR)-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-1-[(фенилметокси)-карбонил]-N-[3-(4-метил-1,1,1- трифтор-2-оксипентил)]-Lпролинамида. Раствор изобутилхлорформиата (11,01 г, 0,08 моль) в сухом ТГФ (30 мл по каплям добавляли в течение 5 мин к предварительно охлажденному раствору (-15оС) КБЗ-1-пролина (19,21 г, 0,077 моль) и N-метилморфолина (8,18 г, 0,081 моль) в ТГФ (300 мл) в атмосфере азота. Реакционную смесь перемешивали при -15оС в те чение 15 мин. Температуру реакции понижали затем до -40оС и раствор части продукта по примеру 1в (16,00 г, 0,077 моль) и N-метилморфолина (8,18 г, 0,081 моль) в ТГФ (200 мл) по каплям добавляли в реакционную смесь. Реакционную смесь перемешивали при -40оС в течение 1 ч, а затем позволяли постепенно нагреваться до комнатной температуры и перемеши вали дополнительно в течение 1 ч. Реакционную смесь фильтровали и концентрировали под вакуумом. Получающийся сироп растворяли в СНСl3 и промывали водной 20%-ной лимонной кислотой (дважды по 75 мл каждый раз). Органический слой концентрировали под вакуумом, чтобы получить сырой продукт в виде белого мутного сиропа. Сырой продукт растирали в порошок с помощью смеси эфира и гексана (1:2), чтобы получить три урожая продукта в виде белого порошка (17,11 г); ТСХ, Rf = 0,47, силикагель, МеОН : СHCl3 (3,97), т.пл. 152–154оС; ЖХВД, tR = 14,06, 16,63, 18,23, 19,00; Зорбакс ОДS аналитическая колонка. Н2О:СН3СН (70:30), скорость потока – 3 мл/мин. б. Синтез 2(RS), 3(SR)-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Продукт примера 2а (2,00 г, 4,97 ммоль) растворяли в абсолютном этаноле (50 мл), 10%-ном Pd/C (0,5 г) до бавляли и реакционную смесь гидрировали (310126.53 Па. 45 psi водорода) в те чение 3 ч при комнатной температуре. Реакционную смесь фильтровали через Целит и растворитель уда ляли под вакуумом, чтобы получить продукт (1,36 г), который использовали без дальнейшей очистки. 2 39093 да. Пример 3. Получение 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинами а. Синтез мети лового сложного эфи ра N-[(фенилметокси)-карбонил)-L-валил-L-пролина. 1-Оксибензотриазол (163,3 г, 1,2 моль) добавляли к предварительно охлажденному раствору N-бензилоксикарбонил-L-валина (151,8 г, 0,6 моль) в ДМФА (1,3 л) и перемеши вали в те чение 15 мин. Суспензию хлоргидрата метилово го сложного эфи ра L-пролина (100,0 г, 0,6 моль) и ТЭА (64,2 г, 0,63 моль) в ДМФА (0,7 л) добавляли, после чего добавляли ДЦК (137,1 г, 0,66 моль). Реакционную смесь перемешивали в течение 3 ч при 0оС, а затем при комнатной температуре в течение 3 дней. Реакционную смесь фильтровали и фильтрат концентрировали под вакуумом. Остаток смешивали с EtOAc (0,75 л) и фильтровали. Фильтрат промывали последовательно 20%-ной водной лимонной кислотой (0,75 л) насыщенной водной NaHCO3 и солевым раствором. Органическую фазу высуши вали над MgSO4, фильтровали и концентрировали под вакуумом, что бы получить сырой продукт (271,3 г). Продукт очи ща ли быстрой хроматографией на силикагеле, используя градиентное элюирование, начиная с СН2Сl2 и кончая с МеОН:СН2Cl2 (4:96), чтобы получить продукт (218,1 г): ТСХ, Rf = 0,48, силикагель, МеОН:СНСl3 (5:95). б. Синтез N-[(фенилметокси)-карбонил]-L-валил-L-пролина. К раствору части продукта примера 3а (158,8 г, 0,438 моль) в МеОН (1,6 л) добавляли 1 N водный NaOH (500 мл) и полученный раствор перемешива ли при комнатной температуре в течение 17 ч; 1 N водный NaOH (100 мл) добавляли и перемешивание продолжали в течение 5 ч. Дополнительное количество 1 N водного NaOH (50 мл) добавляли и реакционную смесь перемешивали в течение ночи. Реакционную смесь концентрировали под вакуумом, чтобы уда лить МеОН. До бавляли Н 2О (1,0 л) и водный раствор экстрагировали с помощью Et2O. Водный раствор подкисляли 1 N водным HCl (700 моль) и экстрагировали с EtOAc. Экстракты EtOAc промыва ли солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали под ва куумом, чтобы получить продукт (159,2 г), ТС Х, Rf = 0,34, силикагель, МеОН:СНСl3 : :AcOH (5:94:1). в. Синтез 2(RS), 3(SR)-[(фенилметокси)-кар-бонил]-L-валил-N-[3-(4-метил-1,1,1 -трифтор-2-оксипентил)]-L-пролинамида. Изобутилхлормиат (77,3 г, 0,566 моль) добавляли к предварительно охлажденному (-15оС) раствору N-метилморфо лина (59,25 г, 0,566 моль) и продукт примера 3б (197,2 г, 0,566 моль) в сухом ТГФ (2,5 л) и реакционную смесь перемешивали в течение 10 мин. Температуру понижали до -40оС и N-метилморфолин (59,25 г, 0,566 моль) добавляли, после чего по каплям добавляли раствор продук та по примеру 1в (117,5 г, 0,566 моль) в ТГФ (2,5 л). Реакционной смеси позволяли нагреваться до комнатной температуры и перемешивали в течение 3 дней. Реакционную смесь фильтровали и фильтрат концентрировали под вакуумом. Оста ток растворяли в EtOAc и промывали последовательно водой, 1 N водным HCl и солевым раствором. Органический слой высушива ли над Na2SO4, фильтровали и концентрировали, чтобы получить сырой продукт (267,8 г). Продукт очи щали быстрой хроматографией на силикагеле, используя гра диентное элюирование, начиная от смеси ТГФ : толуол (5:95) и кончая смесью ТГФ : толуол (25:75), чтобы получить продукт (183,8 г); ТСХ, Rf = 0,4, си ликагель, ТГФ : толуол (20:80). г. Синтез 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L- пролинамида. Смесь части продук та по примеру 3в (36,7 г, 0,073 моль) и 10%-ного Pd/C (10% на 50% увлажненный водой) в EtOH (0,6 л) гидрирова ли на трясучке Парра (303,924 Па, 3 атм Н2). После 1 ч реакционный сосуд откачивали и заново продавливали в него Н2. После встряхи вания дополнительно (0,5 ч) реакционную смесь фильтровали через целит и концентрировали под вакуумом, чтобы получить продукт (26,0 г); ТС Х, Rf = 0,16, силикагель, МеОН : СНСl3 (5:95). Пример 4. Получение хлористоводородной соли 2(RS), 3(SR)-3-амино-4-метил-1,1,1-трифтор-2-пентанола. а. Синтез 2-метил-1-нитропропана (альтернативный метод получения соединения примера 1а). Пятилитровую трехгорлую круглодонную колбу снабжали меха нической мешалкой, термометром, капельной воронкой и вводом N2. Колбу загружали с AgNO2 (1006,8 г, 6,54 моль) в Et2O (2,5 л), а йодистый изобутил (927,2 г, 5,03 моль) помеща ли в капельную воронку. Как колбу, так и капельную во ронку обвертывали алюминиевой фольгой, что бы защитить реакционную смесь от света. После это го перемеши ваемую суспензию охлаждали приблизительно до 5оС (ледяная баня) и начинали добавлять по каплям йод в течение 2 ч. Температуру реакции поддерживали при 5оС или менее в течение всего периода добавления. По завершении добавления реакционный сосуд ук ладывали в лед и позволяли медленно нагреваться до комнатной температуры в течение ночи. ЯМР-анализ аликвотной части, отобранной из реакционной смеси после 48 ч перемешивания, показывает, что весь йодистый изобутил расхо дуется. Реакционную смесь фильтровали через целит, чтобы удалить соли серебра и спекшуюся массу на фильтре промывали с помощью Et2O (3 х 500 мл). Объединенные фильтраты высуши вали MgSO4, фильтровали и концентрировали на роторном испарителе (температура бани 35оС) до объема примерно 600 мл. Фракционированная перегородка (атмосферное давление) (осторожно: потенциально взрывчатое вещество) дает очищенное нитросоединение (350,4 г, 68%-ный выход); т. кип. 135-142оС. б. Синтез 2(RS), 3(SR)-4-метил-3-нитро-1,1,1-трифтор-2-пентанола. Трехлитровую тре хгорлую круглодонную колбу, снабженную механической мешалкой и вводом N2, загружали с K2CO3 (470,0 г, 3,4 моль), продуктом по примеру 4а (350,0 г, 3,4 моль) и этиловым полуа цеталем трифторацетальдегида (708,0 г, 4,4 моль). Смесь энергично перемешивали при комнатной температуре в течение 76 ч, за время которого 1Н-ЯМР показывает почти полный расход нитроалкана. Реакционную смесь разбавляли с СН 2Сl2 и фильтровали. Фильтрат обрабаты вали водным НСl до тех пор, пока рН не 3 39093 достигнет 3. Слои разделяли и водный слой промывали с СН 2Сl2 (500 мл). Объединенные порции СН2Сl2 промывали с Н 2О (1 л) и со левым раствором (1 л). Высушивание (MgSO4) и концентрирование дает 854,6 г сырого продук та в ви де желтого масла. 1НЯМР показывает два диастереомерных нитроспирта (присутствуют в соотноше нии примерно 3:1, как это количественно определено путем интегрирования протонов спиртов), которые последовательно появляются в пределах 6,0-6,5 при проведении исследования в дейтерированном ацетоне – d6, загрязненном растворите лями и малыми количествами исходных ве ществ. Перегонка при пониженном давлении дает следующие фракции: А Б В Г Д Вес 191,7 г 34,8 г 213,6 г 337,8 г 114,0 г Т.кип. 42–50оС/атм И.С. (исход. соедин.) + растворители 35о/1 торр - 45о/0,5 то рр 45о/0,5 торр - 95о/1,5 то рр 95о/1,5 торр - 105о/2 торр летучие продукты в ло вушке Для упроще ния очисток в последующих синтетических стадиях были сделаны усилия с целью, чтобы получить основную диастереомерную пару, в основном в чистом состоянии, и что бы работать только с этим веществом в последующи х реакциях. Основная диастереомерная пара кристаллизуется из смеси диасте реомеров, а также из холодного пентана с образованием бесцветных игл. Та ким образом, фракции В из указанной выше перегонки позволяли кристаллизоваться в течение ночи в холодильнике. Продукт собирали, промывали холодным пентанолом и высуши вали в течение нескольких часов в ва куумной печи, чтобы получить 52,0 г по существу чистого вещества. Фракции, которые, как известно (по данным ЯМР), содержат значительные количества целевого изомера, повторно обрабатывали таким образом (и повторно перегоняли, что бы получить новые фракции, дополнительно обогащенные целевым диастереомером), чтобы в конечном счете получить в общем 197,7 г в основном чисто го нитроспирта. Это количество представляет тип проведенной обработки, но не отражает верхнего предела выхо да целевого продукта. в. Синтез хло ристоводородной соли 2(RS), 3(SR)-3-амино-4-метил-1,1,1-трифтор-2-пентанола. Безводный EtOH (232 мл) добавляли к 10%-ному ката лизато ру Pd/C (2,30 г) под атмосферой N 2. (Менее активные катализаторы (например, 10%-ный Pd/BaSO4, влажный 10%-ный Pd/C) или недостаточные продолжительности реакции могут приводить к получению одного или более побочных продук тов). Продукт по примеру 4б (22,93 г, 0,144 моль) добавляли и полученную смесь помеща ли в аппарат гидрирования Парра (примерно 480 000 Па, 55 psi H 2) и оставляли на ночь. Катализатор удаляли фильтрованием через целит. Спекшуюся массу на фильтре промывали затем с помощью EtOH. Газообразный НСl пробулькивался через объединенные фильтраты до тех пор, пока приблизительно 8 г (примерно 0,22 моль) не поглотится. Раствор концентрировали на роторном испарителе и полученный остаток концентрировали несколько раз из Et2O, чтобы получить белое твердое вещество. Твердое вещество промывали с помощью Et2O и высушивали в течение ночи в вакуумной печи, что бы получить 20,79 г (88%) хлоргидрата мина. Для определения т.пл.: при медленном нагревании вещество размягчается при 90оС и плавится при 118–120оС. Когда образец погружается в ба ню, предварительно нагретую до 110оС, образец плавится мгновенно. Пример 5. Получение 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L -пролинамида. а. Синтез мети лового сложного эфи ра N-[(фенилметокси)-карбонил]-L-валил-L-пролина. Раствор КБЗ-L-валина (100,0 г, 0,40 моль) в ДМФА (1 л, высушенный над ситами), добавляли в 3-литровую трехгорлую круглодонную колбу, снабженную механической мешалкой, вводом N 2 и термометром. Реакционную смесь охлаждали до 0оС и добавляли ОБТА-гидрат (108,1 г, 0,80 моля). Приблизительно 15минутное перемешивание проводили до добавления ДМФА (500 мл) суспензии хлоргидрата метилового сложного эфи ра L-пролина (66,2 г, 0,40 моль) и ТЭА (41,8 г, 0,42 моль) сразу в одну порцию. Дополнительное количество ДМФВ (500 мл) использовали, чтобы завершить полный перенос глинообразной массы. ДНК (90,8 г, 0,44 моль) добавляли в реакцию и промывали с помощью ДМФА (100 мл). Реакционную смесь перемеши вали в течение 3 ч при 0оС перед тем, как позво лить ей нагреться до комнатной температуры и перемеши ваться в течение 3 дней. Затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Спекшуюся массу на фильтре промывали с помощью EtOAc (3 х 1 л) концентрирование полученного фильтра приводило к продук ту, который объединяли с остатком из концентрирования раствора ДМФА. Объединенную смесь продуктов (примерно 2,5 л) разбавляли с помощью Et2O (2 л) и хранили в холодильнике в те чение ночи. Вы павший осадок удаляли фильтрованием. Затем фильтрат промывали с помощью 1N HCl (1 л), при этом образовавшееся дополнительное количество осадка удаляли фильтрованием. Затем фильтрат промывали с помощью 1N HCl (1 л), Н2О (0,5 л), насыщенного NaHCO3 (2 х 1 л) и солевого раствора (0,5 л). После высушивания с помощью MgSO4 и концентрирования сырой продукт ве сом 587,2 г подвергали быстрой хроматографии на силикагеле (3,5 кг) с помощью градиентного элюирования от CH2Cl2 до 5% МеОН : СН2Cl2 (5:95). Смешанные фракции подвергали повторной хроматографии, чтобы уда лить примеси. Объединение фракций, содержащи х це левой продукт, дает 500,7 г (87%) продукта, загрязненного малым количеством примесей с низким значением Rf; TCX, Rf = 0,37, силикагель, Et2O : гексан (3:1); R f = 0,53, си ликагель, MeOH : СНCl3 (5:95). б. Синтез N-[(фенилметокси)-карбонил]-L-валил-L-пролина. 4 39093 Ме танольный (4 л) раствор продукта по примеру 5а (500,7 г, 1,38 моль) добавляли в 12-литровую трехгорлую круглодонную колбу, снабженную меха нической мешалкой и вводом N 2. К перемешиваемому раствору добавляли 1 N NaOH (1,4 л), доводя рН приблизительно до 13. После перемешивания реакционной смеси в течение 3 ч рН падает до 11. Дополнительное количество 1 N NaOH (0,1 л) использовали, чтобы довести рН до 12 и реакционную смесь перемеши вали в те чение ночи при комнатной температуре. МеОН уда ляли из реакционной смеси путем концентрирования на роторном испарителе. В те чение процесса удаления растворителя добавляли в воду, общее количество которой составляет 1 л, чтобы понизить концентрацию основания. Водный раствор промывали с помощью Et2O до подкисления с помощью 1N HCl (1,5 л) до рН приблизительно равного 3,5. Слои разделяли и водный слой экстрагировали с помощью EtOАc (3x1 л). Объединенные органические продукты промывали солевым раствором (1 л), высушивали (MgSO4) и концентрировали, чтобы получить 493,0 г (100%) продукта в ви де белого твердого вещества; ТСХ, Rf = 0,51, си ликагель МеОН : СHCl3 (5 : 96) вместе с до бавленным АсОН. в. Синтез 2(RS), 3(SR)-[(фенилметокси)-карбонил]-L-валил-L-[3-(4-метил-1,1,1 -трифтор-2-оксипентил)]-L-пролинамида. Продукт по примеру 5б (105,3 г, 0,302 моль) растворяли в сухом ТГФ (1,5 л) в атмосфе ре N2 в 3-литровой трехгорлой круглодонной колбе, снабженной меха нической мешалкой, термометром, вводом N2 и капельной воронкой. Раствор охлаждали до -35о и обрабатывали с помощью первого эквивалента (34 мл, 0,309 моль) NMM. Изобутилхлорформиат (39 мл, 0,377 моль) добавляли по каплям в течение 20 мин, поддерживая температуру -35оС. После завершения добавления реакционную смесь перемешивали в течение 1 ч при -35оС. Второй эквивалент NMM (34 мл) добавляли. Продукт по примеру 4в (62,8 г), 0,302 моль в ТГФ (300 мл) добавляли затем с такой скоростью, чтобы поддерживать температуру -35оС. После завершения добавления температуру поддерживали равной -35оС в течение 1 ч прежде чем позволить смеси нагреться до комнатной температуры при перемешивании в те чение ночи. Реакционную смесь фильтровали и спекшуюся массу на фильтре промывали с помощью ТГФ (1л). Объединенные фильтраты концентрировали, чтобы получить 189 г сырого продукта. Это вещество подвергали быстрой хроматографии на силикагеле (5000 мл) и элюировали с помощью ТГФ: толуол (1:9). Как только первый продукт начинает элюировать, полярность растворителя изменяют в градиентном состоянии: ТГФ:толуол (15–85); ТГФ:толуол (20:80); и наконец МеОН:ТГФ:то луол (2,5:30:70) (использова ние МеОН минимизировали, чтобы предотвратить элюирование примесей с низкой величиной Rf. Концентрирование фракций с колонки с последующим высушиванием под вакуумом в те чение ночи дает 12,0 г (8%) незначительно загрязненного продукта и 131,6 г (87%) по существу чистого продукта. (Растворы этого вещества, взятого до завершения высуши вания, дают пену, которая в конечном счете затвердевает при продолжительной вакуумной обработке. Следует завершать эту операцию довольно в большой колбе, что бы разместить расширение пены). ТСХ, Rf = 0,25, силикагель, МеОН:CHCl (5:95); Rf=0,37, силикагель, ТГФ:толуол (20:80). Когда продукт хроматографи руется на пятна медленно, то два изомера разделяются, чтобы получить пятна при Rf=0,37 и Rf=0,46, силикагель, ТГФ:толуол (20:80), (поддержание внешних температур, описываемых в этой методике, по-видимому, является крити ческим для получения по существу чистого продукта). г. Синтез 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L -пролинамида. Продукт примера 5в (131,6 г, 0,262 моль) растворяли в EtOH (750 мл) и объединяли с 10%-ным катализатором Pd/C (560% Н2О) (13,0 г) в атмосфе ре N2 в большом сосуде для гидрирования по Парру. Реакционную смесь встряхи вали под давлением Н 2 480000 Па, 55 psi в аппарате Парра. Повторное продувание Н2 продолжали до тех пор, пока не прекратится дальнейшее поглоще ние Н 2. Исследование реакционной смеси с помощью ТСХ показывает полный расход исходного продукта. Реакционную смесь фильтровали через целит и концентрировали до пенообразования. Этот продукт растирали в порошок с помощью Et2O, фильтровали и высушивали, что бы получить 81,4 г (84%) светло-зеленого твердого вещества; ТСХ, Rf= = 0,41 силикагель, СНСl3:MeOH (10:1). Пример 6. Получение 3S (или R)-фенилметоксикарбонил-L-валил- N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пропиламида (фор мула 1b, R1 – CH(CH3)CH3, R2 - CH(CH3)CH3, R3 – PhCH 2, R4 – H, A – OCO, n = 1). a. Синтез D-виннокислой соли 2R, 3S-3-амино-1,1,1-трифтор-4-метил-2-пента нола. Хлоргидрат амина (20 г), получаемый, как это описано в примере 4д, растворяли в Н2 О и нейтрализовали с помощью твердого NaHCO3. Водный раствор экстрагировали несколько раз с помощью СН 2Сl2. Объединенные экстракты высуши вали (Na2SO4) и концентрировали для получения свободного аминового основания (14,04 г) в ви де белого твер дого ве щества. Это продукт со четали с D-винной кислотой (12,31 г) в кипящем безводном EtOH (100 мл), и полученный мутный раствор фильтровали в горячем состоянии через фильтровальную бумагу. Раствор сначала охлаждали медленно до комнатной температуры в течение ночи, а затем помещали в холодильник на несколько часов. Вы павший осадок собирали на фильтре Шотта, промывали хо лодным EtOH и высушива ли в те чение ночи в ва куумной печи при 40оС. Образец высушенного белого твердого вещества (4,56 г) плавится при 127–130оС. Большую часть этого продукта (4,05 г) растворяли в кипящем EtOH (20 мл) и раствор медленно охлаждали до комнатной температуры. Белое гелеобразное твердое вещество, которое выпадало в осадок, собирали в фильтре Шотта и промывали несколькими порциями EtOH. Высуши вание в вакуумной печи при 40оС в течение нескольких часов дает белое твердое вещество, т. пл. 132–134оС. 6. Синтез 2S, 3S-[(фенилметокси)-карбо-нил]-L-валил-N-[3-(1,1,1-трифтор-2-окси -4-метил-пентил]]-Lпролинамида (формула VIIb, R 1 – CH(CH3)-CH3, R2 – CH(CH3)CH3, R3 – PhCH2, R4 – H, A – OCO, n=1). 5 39093 Кислоту, полученную по примеру 5б (1,00 г, 2,87 ммоль), растворяли в сухом ТГФ (16 мл) в атмосфере N2 в 50-миллилитровой трехгорлой круглодонной колбе, снабженной термометром, вводом N 2, магнитной мешалкой и железной полоской для перемеши вания магнитной мешалкой. NMM до бавляли (0,34 мл, 3,09 ммоль) и получающийся перемешиваемый раствор охлаждали до температуры (внешней) -35оС. Изобутилхлорфор миат (0,37 мл, 2,85 ммоль) добавляли в течение 2 мин, никогда не позво ляя повышению внешней температуры выше -35оС. Реакционную смесь перемешивали в течение 1 ч при температурах от -45 до -35оС. Соль D-винной кислоты из примера 6а (0,92 г, 2,86 ммоль) в смеси ТГФ (5 мл) и ДМСО (2 мл) обрабатывали с помощью NMM (0,68 мл) и мутный раствор добавляли в реакционную смесь с такой скоростью, чтобы поддерживать температуру ниже -40оС. Реакционную смесь перемеши вали при температурах от -45 до -15оС в течение 1 ч, прежде чем ей позволяли нагреваться до комнатной температуры в течение ночи. Смесь разбавляли с СНСl3, а затем промыва ли (Н2О, насыщенным водным NaHCO3), вы сушивали (Na2SO4) и концентрировали, что бы получить названный выше продукт (1,15 г, 80%) в виде белого твердого вещества. 1Н-ЯМР-спектр этого продукта в ДМСО-d6 проявляет дублет при 86,43, который является соответствующим хи мическим сдвигом спиртового прото на в продукте с приписанной относительной конфи гурацией. в. Синтез 3S (или R)-фенилметоксикарбонил-L-валил-N-[3-(1,1,1-трифтор-4-метил -2-оксопентил)]-Nпролинамида (формула 1b, R1 – СН(СН3)СН3, R2 – CH(CH3)CH3, R3 – PhCH2, R4 – H, A – OCO, n = 1). Часть спирта, получаемого в примере 6в (0,25 г, 0,5 ммоль), растворяли в СН2Сl2 и обрабатывали периодинаном Десса-Марти на (0,42 г, 0,99 ммоль) в виде одной порции. Добавляли ТФУК (0,08 мл, 1,04 ммоль) и незначительно мутной смеси позволяли перемешиваться в течение ночи. В реакционной смеси образуется белая суспензия. Как показано с помощью ТСХ, исходное вещество в основном отсутствует. Воду, со держащую Na2S 2O3 (0,78 г) и NaHCO3 (0,42 г), до бавляли и перемешивали вместе с реакционной смесью. Когда органический слой в конечном счете становится полностью белой твердой суспензией, его разделяли от водной фазы. Органический слой промывали (насыщенный водный NaHCO3), вы сушивали (Na2SO4) и концентрировали до получения масла. Этот продукт заново растворяли и концентрировали из EtO/гексана, чтобы получить белое твердое вещество (0,21 г, 84%-ный выход). Перекристаллизация из горячей смеси Et2O/гексан дает по существу чистый образец названного соединения в виде в основном чистого изомера, который проявляется в виде единственного пика на ЖХВД со временем удерживания, идентичным таковому идентифицированного образца названного продук та, как это описано в примере 117; ЖХВД, tR = 5,65, Co1 A, H 2O : CH 3OH (55:45), СП = 2,0. Пример 7. Получение 1-[2-(трицикло[3.3.13.7]-дец-1-ил)этоксикарбонил]-N-[3-(RS)-3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 4-нитрофенил-2-[трицикло-(3.3.1.13.7)-дец-1-ил]-этилкарбоната. К раствору паранитрофе нилхлорформиата (1,17 г, 5,82 ммоль) в Et2O (25 мл) при 0оС добавляли пиридин (5 мл), после чего прибавляли 2-(1-адамантил)-этанол (1,00 г, 5,54 ммоль) в Et2O (20 мл) по каплям в течение 1 ч. Полученную смесь перемешивали при комнатной температуре в течение 12 ч и она распределялась между Н 2О Et2O. Эфирный слой промывали с водным НСl, фосфатным буфе ром с рН 7,0, высушивали над MgSO4, фильтровали и растворители удаляли под вакуумом. Сырой продукт очища ли с помощью быстрой хроматографии на силикагеле, элюируя с помощью EtOАc: гексан:смеси (5:95) для получения продукта (1,10 г) в ви де белого порошка; ТСХ, Rf = 0,29, силикагель, EtOAc:гексан (5:95). б. Синтез 1-[2-(трицикло-[3.3.1.13.7 ]-дец-1-ил)-этоксикарбонил]-N- [2(RS), 3(SR)-3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Раствор продукта по примеру 7а (731,0 мг, 2,98 ммоль), продукта, полученного при использовании методики примера 2б (500 мг, 1,80 ммоль), и К2СО3 (2,58 г, 18,6 ммоль) в ДМФА (50 мл) перемеши вали при комнатной температуре в течение 18 ч, фильтровали и растворитель удаляли под вакуумом. Остаток растворяли в EtOAc, промывали тремя порциями 10%-ного водного раство ра NaOH, высуши вали над твердой смесью К2СО3: Na2SO4 (10:90), фильтровали и растворитель удаляли при пониженном давлении. Сырой продукт хроматографировали на силикагеле, элюируя с помощью смеси МеОН:CHCl3 (1:99). Получающееся твердое вещество промывали гексаном, чтобы получить продукт (340 мг) в виде белого твердого вещества; ЖХВД, tR = 5,86; 6,38; Зорбакс ОДS аналитическая колонка; скорость потока – 2 мл/мин, СН3CN:H2O : ТФУК (70:30:0,1). в. Синтез 1-[2-(трицикло-[3.3.1.13.7]-дец-1-ил)-этоксикарбонил]-N-3(RS)- [3(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. К раствору хлористого оксалила (1,09 г, 8,60 ммоль) в сухом СН 2Сl2 (15 мл), охлажденному до -43оС, добавляли ДМСО (1,37 г, 17,3 ммоль) в сухом СН 2Сl2 (10 мл) по каплям в течение 15 мин. Раствор перемеши вали в течение 10 мин и продукт по примеру 7б (340 мг, 0,72 ммоль) добавляли таким же образом в течение 30 мин. После перемешивания раствора при -43оС в течение еще 1 ч медленно добавляли ТЭА (4,80 мл, 34,5 ммоль) и раствору позволяли нагреваться медленно до комнатной температуры, который затем перемеши вали в течение 2 ч. Раствор разбавляли с СН2Сl2, промывали с 10%-ным водным HCl, 5%-ным водным NaOCl, высушива ли над твердой смесью К2СО3:Na2SO 4 (10:90), фильтровали и растворитель удаляли под вакуумом. Сырой продукт очища ли двумя последовательными проведениями быстрой хроматографии на силикагеле, элюируя с помощью МеОН:CHCl3 (0,1:99,9) и EtOAc:гексан (1:5) соответственно, чтобы получить продукт (130 мг) в виде белого твердого вещества; ТСХ, Rf = 0,50, силикагель, MeOH:CHCl 3 (5:95), ЖХВД, tR = 5,31, Зор бакс ОДS аналитическая колонка, скорость потока – 2 мл/мин, СН3СN:H2O:ТФУК (70:30:0,01). 6 39093 Анализ, вычисленный для С 24Н35N2F3 O4 x х 0,25 H 2O: C 60,43, H 7,50, N 5,87. Найдено: C 60,50, H 7,45, N 5,74. Пример 8. Получение 3(RS)-1-(4-фенилбутилкарбонил)-N-[3-(4-метил-1,1,1-трифтор-2- оксопентил)]L-пролинамида. а. Синтез 2(RS), 3(SR)-1-(4-фенилбутилкарбонил)-N-[3-(4-метил-1,1,1-трифтор- 2-оксипентил)]-L-пролинамида. Раствор 5-фенилвалериановой кислоты (0,330 г, 1,86 ммоль) и N-метилморфо лина (0,280 г, 2,79 ммоль) в ТГФ (100 мл) охлаждали до -15оС. Раствор изобутилхлорформиата (0,280 г, 2,05 ммоль) в ТГФ (5 мл) добавляли по каплям и смесь перемеши вали при -15оС в течение 10 мин, после чего температуру понижали до -40оС и раствор продукта, полученного при использовании методики примера 2б (0,500 г, 1,86 ммоль) в ТГФ (25 мл), добавляли по каплям. Смесь перемешива ли в течение 1 ч при -40оС и в течение ночи при комнатной температуре. Смесь фильтровали и фильтрат концентрировали под вакуумом. Сырой продукт очища ли с помощью быстрой хроматографии на силикагеле, элюируя с помощью МеОН:СНCl3 (3:97), чтобы получить продукт (0,60 г) в ви де белого твердого вещества, ТС Х, Rf = 0,40–0,51, силикагель, МеОН : CHCl3 (3:97). б. Синтез 3(RS)-1-(4-фенилбутилкарбонил)-N-[3-(4-метил-1,1,1-трифтор- 2-оксoпентил)]-L-пролинамида. Раствор ДМСО (4,22 г, 54,0 ммоль) в сухом СН 2Сl2 (80 мл) добавляли по каплям в течение 10 мин в заранее охлажденный (-60оС), перемеши ваемый раствор хло ристо водородного оксалила (3,43 г, 27,0 ммоль) в СН2Сl2 (10 мл) под атмосферой азота. Температура не должна превышать -55оС в течение добавления. Смесь перемешивали при -60оС в течение 15 мин и раствор продукта по примеру 8а (0,580 г, 1,35 ммоль) в СН2Сl2 (100 мл) добавляли по каплям в течение 10 мин при -60оС. Реакционную смесь перемешивали в течение 1 ч, во время которого она нагревалась до комнатной температуры. Реакционную смесь промывали последовательно двумя порциями 1N водного раствора HCl и солевым раствором и концентрировали под вакуумом, чтобы получить сырой продукт (0,85 г) в виде сиропа оранжевого цве та. Сырой продукт очи щали тремя последовательными проведениями быстрой хроматографии на силикагеле, элюируя соответственно с 1) МеОН:CHCl3 (3:97) 2) MeOH:CHCl3 (3:97) и 3) Et2O : гексан (90:10), чтобы получить продукт (319 мг) в виде пены белого цвета; ТСХ, Rf = 0,33 – 0,40, силикагель, Et2O:гексан (90:10), ЖХВД, tR=17,93, 18,55, Зорбакс ОДS аналитическая колонка, Н2О:CH3CN (55:45), скорость потока – 2 мл/мин. Анализ, вычисленный для С 22H29N2O 3F3 x х1,25 H2O: C58,85, H 7,07, N 6,24. Найдено: C 58,91, H 6,83, N 6,13. Пример 9. Получение 3(RS)-1-[(фенилметокси)-карбонил]-N-[3-(4-метил-1,1,1-трифтор- 2-оксопентил)]-L-пролинамида. Раствор диметилсульфоксида (0,890 г, 11,4 ммоль) и сухо го хло ристого метилена (5 мл) добавляли по каплям к перемешиваемому раствору хло ристого оксалила (0,75 г, 5,9 ммоль) и сухо го хлористого метилена (5 мл) при -60оС в атмосфере азота. Реакционной смеси позволяют нагреваться до -25оС, затем добавляли раствор продукта, получаемого при использовании способа примера 2а (0,200 г, 0,497 ммоль), и сухо го хлористо го метилена (5 мл). Получившуюся смесь перемешивали при -25оС в течение 0,5 ч. Триэтиламин (1,94 г, 19,2 ммоль) добавляли и реакционной смеси позволяли нагреваться до комнатной температуры. Реакционную смесь фильтровали. Фильтрат выпаривали. Остаток растворяли в хлороформе. Хлороформный раствор промывали последовательно 1N раствором водного НСl, затем солевым раствором и высушивали над Na2SO4. Раствор фильтровали. Растворитель удаляли под вакуумом, чтобы получить сырой продукт (0,147 г). Сырой продукт очища ли путем быстрой хроматографии на силикагеле с помощью элюента из смеси СНСl3:MeOH (97:3), чтобы получить продукт (0,11 г); ТСХ, Rf = 0,25, CHCl 3:EtOAc (90:10). Анализ, вычисленный для С 19Н23F3N2 O4 x х 1,5 H 2 O: C 53,39, H 6,13, N 6,55. Найдено: С 53,55, Н 5,78, N 6,56. Пример 10. Получение 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L -пролинамида. а. Синтез мети лового сложного эфи ра N-[(фенилметокси)-карбонил]-L-валил-L-пролина. Ме тодику примера 3а повторяли. б. Синтез N-[(фенилметокси)-карбонил]-L-валил-L-пролина. Ме тодику примера 3б повторяли, используя продукт по примеру 10а. в. Синтез 2-метил-1-нитропропана. Ме тодику примера 1а повторяли. г. Синтез 2(RS), 3(SR)-4-метил-3-нитро-1,1,1-трифтор-2-пентанола. Ме тодику примера 1б повторяли, используя продукт по примеру 10в. д. Синтез хлористоводородной соли 2(RS), 3(SR)-3-амино-4-метил-1,1,1-трифтор-2-пентанола. Ме тодику примера 1в повторяли, используя продукт по примеру 10г. е. Синтез 2(RS), 3(SR)-[(фенилметокси)-карбонил]-L-валил-N -[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Способ примера 3в повторяли, используя соединения по примерам 10б и 10д. ж. Синтез 2(RS), 3(SR)-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)] -L-пролинамида. Способ примера 3г повторяли, используя соединение по примеру 10е. 7 39093 Пример 11. Получение 3(RS)-[(фенилметокси)-карбонил]-L-валил -N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а-е. Стадии а-е повторяли, как это описано в примерах 10а-е. ж. Синтез 3(RS)-[(фенилметокси)-карбонил]-L-валил-N-[3-(4-метил-1,1,1 -трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (12,46 г, 159,50 ммоль) в сухом СН 2Сl2 (12 мл) добавляли по каплям в течение 10 мин в предварительно охлажденный (-60оС), перемешиваемый раствор хлористого оксалила (10,12 г, 79,75 ммоль) в СН2Сl2 (160 мл) в атмосфере азота. Температура реакционной смеси никогда не превышает -50оС во время добавления. Раствор спирта в примере 11е (2,00 г, 3,99 ммоль) в СН2Сl2 (160 мл) добавляли по каплям в течение 10 мин при 60оС. Реакционную смесь перемешивали при -60оС в течение 1 ч. Диизопропилэтиламин (20,62 г, 159,50 ммоль) добавляли по каплям в течение 10 мин при -60оС. Реакционную смесь перемешивали в те чение 1 ч, в то вре мя как она нагревалась до комнатной температуры. Реакционную смесь промывали 1N водным раствором HCl, затем солевым раствором, после этого концентрировали под вакуумом, чтобы получить сырой продукт в ви де сиропа оранжевого цвета (2,76 г). Сырой продукт очи ща ли тремя последовательными проведениями быстрой хроматографии на силикагеле, элюируя соответственно с 1) эфир:гексан смесью (80:20), 2) МеОН:CHCl3 (2,5:97,5), 3) MeOH : CHCl 3 (2,5 : 97,5), чтобы получить продукт в ви де пены белого цвета (0,88 г); ТС Х, Rf=0,45, силикагель, МеОН:CHCl 3 (3:97), ЖХВД, tR = 6,45, 11,10 Зорбакс ОДS аналитическая колонка Н2О:CH3CN (55:45) вместе с 0,1% трифто руксусной кислотой, скорость потока – 2 мл/мин. Анализ, вычисленный для С 24H32N3O5 F3 х x 0,5 H 2 O: C 56,68, P 6,54, N 8,26. Найдено: C 56,58, H 6,52, N 8,21. Пример 12. Получение 3(RS)-[2-(2-оксопирролидинил)-этоксикарбонил]-L-валил-N- [3-(4-метил-1,1,1трифтор-2-оксипентил)]-L-пролинамида. а. Синтез 4-нитрофе нил-2-(2-оксопирролидинил)-этилкарбоната. N-(2-Оксиэтил)-пирролидон (3,00 г, 23,2 ммоль) растворяли в диэти ловом эфире (20 мл) под атмосфе рой азота и перемешивали при комнатной температуре. Раствор паранитрофе нилхлорформиата (4,68 г, 23,2 ммоль) в диэтиловом эфире (25 мл) добавляли по каплям к смеси в те чение 2 ч. Смесь перемешивали в те чение дополнительно 2 ч при комнатной температуре. Смесь концентрировали под вакуумом, чтобы получить сырой продукт (7,90 г). Сырой продукт очища ли с помощью быстрой хроматографии на силикагеле с МеОН:CHCl3 (5:95), что бы получить продукт в ви де белого порошка (4,62 г); ТСХ, Rf = 0,51, си ликагель, NeOH:CHCl3 (3:97). б) Синтез 2(RS),3(SR)-[2-(2-оксопирролидинил)-этоксикарбонил]-L-валил-N-[3-(4 -метил-1,1,1-трифтор- 2-оксипентил)]-L-пролинамида. Карбонат калия (2,820 г, 40,80 ммоль) добавляли к раствору этил-2-пирролидон-паранитрофе нилкарбоната (1,20 г, 4,08 ммоль) и продукта, полученного при использовании методики примера 3 г (1,50 г, 4,08 ммоль) в ДМФА (100 мл) при комнатной температуре под атмосфе рой азота. Реакционную смесь перемешивали в те чение ночи. Реакционную смесь разбавляли с помощью EtOAc и избыток К2СО3 фильтровали. Фильтрат концентрировали под вакуумом, чтобы получить остаток, который раство ряли в EtOAc и промывали последовательно водным 10%-ным раствором NaHCO3, во дой, водной 5%-ной лимонной кислотой и солевым раствором. Органическую фа зу высуши вали над Na2SO4, фильтрова ли и концентрировали под вакуумом, что бы получить сырой спирт. Спирт очищали, используя быструю хроматографию на силикагеле с помощью МеОН:CHCl3 (5:95), чтобы получить продукт (0,72 г); ТС Х (Rf = 0,46, силикагель, МеОН: НСl3 (7:93). в. Синтез 3(RS)-[2-(2-оксопирролидинил)-этоксикарбонил]-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Раствор ДМСО (4,310 г, 55,20 ммоль) в сухом СН 2Сl2 (6 мл) добавляли по каплям в течение 10 мин в предварительно охлажденный (-60оС), перемешиваемый раствор хло ристого оксалила (3,500 г, 27,60 ммоль) в СН2Сl2 (80 мл) под атмосферой азота. Во время добавления температура реакционной смеси никогда не превышала -50оС. Раствор спирта из примера 12б (0,720 г, 1,38 ммоль) в СН2Сl2 (80 мл) добавляли по каплям в течение 10 мин при -60оС. Реакционную смесь перемешивали при -60оС в течение 1 ч. Раствор диизопропилэтиламина (7,13 г, 55,2 ммоль) в СН2Cl2 (50 мл) добавляли по каплям в течение 10 мин при -60оС. Реакционную смесь перемешивали в те чение 1 ч, по мере того как она нагревалась до комнатной температуры. Реакционную смесь промывали 1 N водным раствором НCl и солевым раство ром, а затем концентрировали под вакуумом, чтобы получить сырой продукт в виде сиропа оранжевого цве та. Сырой продукт очищали быстрой хроматографией на силикагеле, используя МеОН:CHCl3 (5:95) для получения продукта в виде пены белого цвета (0,43 г); ТСХ, Rf = 0,33, силикагель, МеОН:CHCl 3 (5:95), ЖХВД, tR = 3,50, 4,63, Зорбакс ОДS аналити ческая колонка, Н2О:СН3СN (55:45), скорость потока – 1 мл/мин. Анализ, вычисленный для С 23H35N4O6 F3 x х H2 O: C 51,29 H, 6,92 N, 10,40. Найдено: С 51,20, Н 6,86, N 10,30. Пример 13. Получение 3(RS)-[2-метоксикарбонил)-этилкарбонил]-L-валил-N-[3-(4-метил-1,1,1-три-фторметил- 2-оксопентил)]-L-пролинамида. а. Синтез трет-бутилово го сложного эфи ра N-бензилоксикарбонил-L-валил-L-пролина. 8 39093 Раствор N-бензилоксикарбонил-L-валина (56,25 г, 0,244 моль) и ОБТА (60,67 г, 0,45 моль) в ДМФА (565 мл) охлаждали до 5оС. ДЦК (50,89 г, 0,247 моль) добавляли в виде одной порции. Смесь перемеши вали еще 15 мин при 5оС, а затем добавляли трет-бутиловый сложный эфир L-пролина (38,36 г, 0,224 моль). Смесь перемеши вали еще 2 ч при 5 оС, затем в течение 48 ч при комнатной температуре. Смесь фильтровали и концентрировали под вакуумом. Маслообразный остаток растворяли в EtOAc (1 л) и промывали последовательно 20%-ной водной лимонной кислотой, насыщенным водным раствором NaHCO3 и солевым раствором. Органическую фа зу высуши вали над Na2SO4, фильтровали и концентрировали под вакуумом до получения продук та (92,0 г) в ви де пены белого цвета; ТС Х, Rf= 0,9, си ликагель, СНCl3: EtOAc (85:15). б. Синтез трет-бутилового сложного эфира L-валил-L-пролина. Смесь продукта примера 13а (92,0 г, 0,227 моль) и 10%-ного Pd/С (10 г) в EtOH (1 л) гидрировали в трясучке Парра в течение 6 ч, при давлении и (60 psi) при комнатной температуре. Смесь фильтровали через целит и концентрировали под вакуумом для получения продук та (62 г) в ви де вязкого желтого масла; ТСХ, Rf = 0,3, силикагель, МеОН:CHCl 3 (10:90). в. Синтез 1,1-диметилэтилового сложного эфира N-[2-(метоксикарбонил)-этилкарбонил]-L-валил-L-пролина. 1N Водный раствор NaOH (8,0 мл) добавляли в предварительно охлажденный (0оС) раствор продукта примера 13б (2,1 г, 7,8 ммоль) в СН2Сl2 (60 мл). Смесь перемешивали и добавляли 3-карбометоксипропионил хлористый (0,96 мл, 7,8 ммоль). Раствор убирали из ледяной бани, разбавляли водой (30 мл) и подкисляли 1N водным раствором НСl. Органический слой разделяли и водный слой экстрагировали хлористым мети леном. Органические слои объединяли, высушивали над Na2SO4, фильтрова ли и концентрировали под вакуумом. Получающий ся остаток очищали с помощью быстрой хроматографии на колонке с силикагелем, элюируя с помощью Et2O, а затем с EtOAc, что бы получить продукт (2,68 г) ТСХ, Rf = 0,28, силикагель, Et2O. г. Синтез N-[2-(метоксикарбонил)-этилкар-бонил]-L-валил-L-пролина. Три фтор уксусную кислоту (11 ,0 мл, 143 ммоль) добавляли к раство ру продукта из примера 13в (2,68 г, 6,98 ммоль) в СН2Сl2 (11 мл). Смесь перемеши вали в те чение 4 ч и концентрирова ли под вакуумом, чтобы по лучи ть продукт (2 ,13 г), ко торый использова ли без да льнейшей о чистки . д. Синтез 2(RS), 3(SR)-[2-(метоксикарбонил)-этилкарбонил]-L-валил-N-[3 -(4-метил-1,1,1-трифтор-2оксипентил)]-L-пролина амида. Изобутилхлорформиат (0,85 мл, 6,5 ммоль) добавляли к предварительно охлажденному (-15оС) раствору N-метилморфолина (0,71 мл, 6,5 ммоль) и продукта из примера 13 г (2,13 г, 6,5 ммоль) в ТГФ (50 мл). Реакционную смесь перемеши вали в течение 10 мин и температуру реакции понижали до 50оС. Суспензию N-метилморфо лина (0,71 мл, 6,5 ммоль) и продукта, полученного при использовании методики примера 1в (1,39 г, 6,5 ммоль) в ТГФ (50 мл), добавляли в виде одной порции и реакционную смесь перемеши вали в течение ночи, по мере того как она нагревалась до комнатной температуры. Реакционную смесь фильтровали и фильтрат концентрировали под вакуумом. Остаток растворяли в EtOAc и промывали последовательно 1N водным раствором НСl и солевым раствором. Органический слой высушива ли над Na2SO4, фильтровали и концентрировали под вакуумом. Получающийся остаток очищали с помощью быстрой хроматографии на силикаге ле, элюируя с помощью МеОН : CHCl3 (5:95), чтобы получить продукт (1,92 г); ТСХ, Rf = 0,24, силикагель, МеОН : CHCl 3 (5:95). е. Синтез 3(RS)-[2-(метоксикарбонил)-этил-карбонил]-L-валил-N-[3- (4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (11,2 мл, 0,158 моль) в СН2Сl2 (12 мл) медленно добавляли к предварительно охлажденному (-60оС) раствору хлористого оксалила (6,9 мл, 0,79 моль) в СН2Сl2 (160 мл). Раствор продукта по примеру 13д (1,90 г, 3,985 ммоль) в СН2Сl2 (160 мл) добавляли в реакционную смесь и перемешивали в течение 1 ч при -60оС. Диизопропилэтиламин (28,0 мл, 0,158 моль) медленно добавляли и реакционной смеси позволяли нагреваться до температуры окружающей среды. Раствор промыва ли 1N водным раствором HCl (2 х 80 мл) и солевым раствором. Органический слой высушивали над Na2SO4, фильтровали и концентрировали, чтобы получить сырой продукт (3,5 г). Продукт очища ли фильтрованием через силикагель вместе с EtOAc с последующей быстрой хроматографией на силикагеле, используя МеОН:CHCl3 (5:95), чтобы получить продукт (1,49 г); ТС Х, Rf = 0,31, си ликагель, МеОН :CHCl 3 (5:95). Диагностические 1Н-ЯМР сдвиги (CD 3SOCD3, 250 МГц) 0,9, м, 12Н; 3,54, с, 3Н; 3,54-3,68, м, 2Н; 4,34, м, 1Н; 4,40, м, 1Н; 4,49 д.д., 1/2H; 4,58 д.д., 1/2Н; 8,1, д, 1Н; 8,58, д.д.; 1Н. Пример 14. По лучение 3(RS)-[(2-кар-боксиэтил)-карбонил]-L-валил -N-[3-(4-метил-1,1,1-трифтор-2оксопентил)]-L-пролинамида. 1N Водный раствор NaOH (0,92 мл) добавляли к раствору продук та примера 13е (0,20 г, 0,42 ммоля) в МеОН (10 мл) и реакционную смесь перемешивали в течение ночи при температуре окружающей среды. 1N Водный раствор НСl (1 мл) добавляли и реакционную смесь концентрировали под вакуумом, чтобы удалить MeOH. Остающий ся водный раствор экстрагировали с EtOAc и органический слой промывали солевым раствором, высуши вали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить продукт (0,18 г); ТСХ, Rf = 0,2, силикагель, МеОН : CHCl3: ТФУК (5:94:1); ЖХВД, tR = 2,9, 5,86. Применяется колонка с Сайенс Абсорбосфе ре С8; 4,6 мм х 10 см, СН3CN : H2O : ТФУК (20:80:0,1), скорость потока – 1,6 мл/мин. 9 39093 Пример 15. По лучение 3(RS)-[4-(этоксикарбонил)-фенил)-аминокарбонил]-L-валил-N-[3- (4-метил1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-[(4-этоксикарбонилфе нил)-аминокарбонил]- L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Раствор эти лового эфира 4-изоцианато бензойной кислоты (0,86 г, 4,5 ммоль) в СНСl3 (2 мл) добавляли по каплям к раствору продукта, получающе гося при использовании методики примера 3 г (1,65 г, 4,5 ммоль) в СНСl3 (20 мл) и реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь концентрировали под вакуумом, можно получить сырой продукт. Продукт очища ли быстрой хроматографией на силикагеле, используя МеОН:CHCl3 (2,5:97,5) и МеОН:CHCl3 (5:95) в качестве элюентов, чтобы получить продукт (1,76 г); ТСХ, Rf = 0,34, силикагель, МеОН:CHCl 3 (5:95). б. Получение 3(RS)-[(4-этоксикарбонилфе нил)-аминокарбонил]-L- валил-N]-3-(4-метил-1,1,1-трифтор2-оксопентил)-L-пролинамида. Раствор ДМСО (4,5 мл, 63 ммоль) в СН2Сl2 (5 мл) медленно добавляли к предварительно охлажденному (-60оС) раствору хло ристо го оксалила (2,75 мл, 31,5 ммоль) в СН2Сl2 (60 мл). Реакционную смесь перемеши вали в течение 15 мин при -60оС. Раствор продукта примера 15а (1,76 г, 3,15 ммоля) в СН2Сl2 (60 мл) добавляли к реакционной смеси и перемешива ли в течение 1 ч при -60оС. Диизопропилэтиламин (11 мл, 63 ммоль) медленно добавляли и реакционной смеси позволяли нагреваться до температуры окружающей среды. Раствор промывали 1N водным раствором HCl (2 х 60 мл) и солевым раствором. Органический слой высуши вали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить сырой продукт (2,3 г). Продукт очища ли быстрой хроматографией на силикагеле, используя стадийный градиент СНСl3, MeOH:CHCl3 (2,5 : 97,5) и МеОН:CНСl3 (5:95), что бы получить продукт (0,91 г); ТС Х, Rf = 0,42, силикагель, МеОН : CHCl3 (5:95); ЖХВД, tR = 6,77, 11,27, Зорбакс ОДS аналитическая колонка, СН3CN : H2O (45:55), скорость потока – 2 мл/мин. Пример 16. Получение 3(RS)-[(4-карбоксифенил)-аминокарбонил]-L-валил-N-[3-(4-метил- 1,1,1-трифтор-2-оксопентил)]-L-пролинамида. 1N Водный раствор NaOH (1 мл) добавляли к раство ру продук та примера 15б (0,48 г, 0,86 ммоль) в МеОН:H2O (8 мл : 7 мл). Реакционную смесь перемешивали в течение 3 ч, добавляли дополнительное количество 1 N водного раствора NaOH (1 мл) и реакционную смесь перемешивали в те чение ночи при температуре окружающей среды. Добавляли 1N водный HCl метанол удаляли под вакуумом. Н2О (10 мл) добавляли к остатку и экстрагировали с EtOAc (2 х 50 мл). Объединенные органические слои промывали солевым раствором, высуши вали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить продукт (0,42 г); ТС Х, Rf = 0,17, силикагель, МеОН:CHCl 3 : AcOH (5:94:1); ЖХВД, tR = 2,38; 2,78, Зорбакс ОДS аналитическая колонка, СН3CN:H2O (30:70), скорость потока – 1 мл/мин. Пример 17. Получение 3(RS)-[(4-фенилбутил)-карбонил]-L-валил- L-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-[(4-фенилбутил)-карбонил] -L-валил- N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. ДЦК (0,906 г, 4,4 ммоль) добавляли к раствору 5-фенилвалериановой кислоты (0,728 г, 4,08 ммоль), продукта, получающе гося при использовании методики примера 3г (1,5 г, 4,08 ммоль) и ОБТА (1,19 г, 8,8 ммоль) в ТГФ (75 мл) при 0оС. Смесь перемешивали и позволяли медленно нагреваться до комнатной температуры в течение ночи. Смесь концентрировали под вакуумом и получающийся остаток растворяли в СНСl3 (60 мл) и промывали последовательно 20%-ным водным раствором лимонной кислоты (30 мл) Н2О (30 мл), 5%-ным водным раствором NaHCO3 (30 мл) и солевым раствором (30 мл). Органическую фа зу собирали, высушивали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить сырой продукт (2,0 г). Очистка путем быстрой хроматографии на силикагеле, элюируя с помощью МеОН:CHCl3 (5:95) дает продукт (1,0 г) в виде пены белого цве та; ТС Х, Rf = (0,5–0,55), силикагель, МеОН:CHCl 3 (5:95). б. Синтез 3(RS)-[(4-фенилбутил)-карбонил]-L-ва лил-N-[3 -(4-ме ти л-1 ,1 ,1-три фто р-2-ок сo пентил)]-Lпролинамида. Раствор ДМСО (3,4 мл, 48 ммоль) в сухом СН 2Сl2 (4 мл) добавляли по каплям к перемешиваемому раствору хлористого оксалила (2,10 мл, 24 ммоль) в сухом СН 2Сl2 (50 мл), охлажденному до -60оС под атмосферой азота. Раствор перемеши вали при -60оС в течение 15 мин. Раствор продукта по примеру 17а (1,00 г, 1,89 ммоль) в сухом СН 2Сl2 (30 мл) добавляли медленно, поддерживая температуру раствора ниже -50оС. Смесь перемеши вали при (-50оС) в течение 1 ч. Диизопропилэтиламин (8,48 мл, 48 ммоль) добавляли по каплям и реакционной смеси позволяли нагреваться медленно до комнатной температуры. Реакционную смесь промывали последовательно 1N водным раствором НСl и солевым раство ром. Органическую фа зу собирали, вы сушивали с помощью Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить сырой кетон. Кетон очища ли прове дением трех последовательных быстрых хро матографией с помощью силикагеля и элюентов (МеОН:CHCl 3 (5:95) – CHCl3 (100%) – MeOH:CHCl3 (3:97) и Et2O (100%) – МеОН:Et 2O (10:90), чтобы получить конечный продукт (0,2 г) в виде белого воскообразного твердого вещества; ЖХВД, tR = 6,80, 8,90, Зорбакс ОДS колонка; Н2О:CH3CN: ТФУК (40:60:0,1), скорость потока – 0,75 мл/мин. Анализ, вычисленный для С 27N3O4F 3H38 х x 0,5 H 2 O: C 60,65, H 7,35, N 7,85. 10 39093 Найдено: 60,68, 7,30, 7,67. Пример 18. Получение 3(RS)-2-[2-(трицик-ло[3.3.1.13.7]-дец-1-ил) -этоксикарбонил]-L-валил- N-[3-(4-метил1,1,1-трифтор-2-оксoпентил)]-пролинамида. а. Синтез 4-нитрофенил 2-(трицикло-[3.3.1.13.7]-дец-1-ил)-этилкарбоната. Продукт был получен с использованием способа примера 7а. б. Синтез 2(RS), 3(SR)-[2-(трицикло[3.3.1.13.7]-дец-1-ил) -этоксикарбонил]-L-валил- N-[3-(4-метил-1,1,1трифтор-2-оксипентил)]-L-пролинамида. Раствор продукта по примеру 18а (0,758 г, 2,19 ммоль) продукта, получающегося при использовании методики примера 3г (0,768 г, 2,09 ммоль), и К2СО3 (2,89 г, 20,9 моль) в ДМФА (75 мл) перемешивали при комнатной температуре в течение 18 ч, фильтровали и растворители уда ляли под вакуумом. Оста ток разбавляли эти лацетатом, промывали тремя порциями 10%-ного водного раствора NaOH и солевым раствором, высушивали над твердой смесью К2СО3 : Na2SO4 (10:90), фильтровали и растворители удаляли при пониженном давлении. Сырой продукт очи щали с помощью быстрой хроматографии на силикагеле, элюируя с помощью МеОН:CHCl3 (2,98), чтобы получить продукт (0,965 г) в ви де пены белого цве та; ТС X Rf = 0,14 и 0,18, силикагель, МеОН:CHCl3 (2:98). в. Синтез 3(RS)-[2-(трицикло[3.3.1.13.7]-дец-1-ил)-этоксикарбонил]-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксoпентил)]-L-пролинамида. К раствору хлористого оксалила (2,14 г, 16,8 ммоль) в сухом СН 2Сl2 (30 мл), охлажденному до -43оС, по каплям добавляли ДМСО (2,66 г, 33,6 ммоль) в СН2Сl2 (20 мл) в течение 1 ч, затем до бавляли продукт примера 18б (0,965 г, 1,68 ммоль) тем же путем, в течение 30 мин. После перемешивания раствора при -43оС еще в те чение 1 ч добавляли триэтиламин (8,50 г, 84,0 ммоль) и раствору позволяли медленно нагреваться до комнатной температуры. Раствор разбавляли с СН 2Сl2, промывали 5%-ным водным раствором НСl, 5%-ным водным раствором NaOCl, высуши вали над MgSO4, фильтрова ли и растворители удаляли под вакуумом. Сырой продукт очищали с помощью быстрой хроматографии на силикагеле, элюируя с помощью СНСl3, после предварительной обработки силикагеля с МеОН:CHCl3 (1:99), чтобы получить продукт (410 мг) в ви де пены белого цвета; ТС Х, Rf = 0,39, си ликагель, МеОН:CHCl3 (2:98); ЖХВД, tR = 8,12 и 10,75, Зорбакс ОДS колонка, скорость потока – 1,5 мл/мин, СН3СN : H2O : ТФУК (70:30:0,1). Анализ, вычисленный для С 29Н44N3F3 O5 х x 0,75 H 2 O: C 59,52, H 7,83, N 7,18. Найдено: C 59,48, H 7,70, N 7,17. Пример 19. Получение 3(RS)-[(2-меток-сиэтокси)-этоксикарбонил]-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2-(2-метоксиэтокси)-этил-4-нитрофе нилкарбоната. К раствору паранитрофенилового эфи ра хлормуравьиной кислоты (2,00 г, 9,92 ммоль) в Et2O (50 мл) при 0оС добавляли пиридин (8 мл), затем добавляли 2-(2-метоксиэтокси)-этанола (1,14 г, 9,45 ммоль) в Et2O (25 мл) по каплям в течение 1 ч. Получившуюся смесь перемешивали при комнатной температуре в течение 12 ч и разделяли между Н 2О и Et2O. Эфирный слой промывали 5%-ным водным раствором НСl, фосфатным буфе ром с рН 7,0, высуши вали над NgSO4, фильтровали и растворите ли удаляли под вакуумом. Сырой продукт очищали с помощью быстрой хроматографии на силикагеле, элюируя с ЕtOAc : гексан (30:70) чтобы получить продукт (1,30 г) в ви де прозрачного бесцветного масла: ТСХ, Rf = 0,11, силикагель, EtOAc:гексан (30:70). б.Синтез 2(RS), 3(SR)-[(2-метоксиэтокси-этокси )-ка рбон ил]-L-вали л-N-[3 -(4 -мети л-1,1,1-трифтор2-оксипентил)]-L-пролинамида. Раствор продук та из примера 19а (1,11 г, 3,90 ммоля), продукта, получаемого при использовании методики примера 3 г, (1,37 г, 3,72 ммоль) и К2СО3 (5,14 г, 37,2 ммоль) в ДМФА (100 мл) перемешивали при комнатной температуре в течение 18 ч, фильтровали и растворите ли удаляли под вакуумом. Остаток растворяли в этилацета те, промывали тремя порциями 10%-ного водного раствора NaOH и солевым раствором, высуши вали над твердой смесью К2СО3 : Na2SO4 (10:90), фильтровали и растворители удаляли под вакуумом. Сырой продукт очища ли с помощью быстрой хроматографии на силикагеле, элюируя с EtOAc после предварительной обработки силикагеля с ТЭА:гексан (1:9), чтобы получить продукт (1,13 г) в виде прозрачной бесцветной стеклообразной массы: ТС Х, Rf = 0,43 и 0,48, си ликагель, МеОН : СНСl3 (1:9). Анализ, вычисленный для С 22Н38N3O 7F3 : C 51,45, H 7,45, N 8,18. Найдено: C 51,48, H 7,35, N 8,01. в. Синтез 3(RS)-[(2-метоксиэтокси)-этоксикар-бонил]-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]L-пролинамида. К раствору хлористого оксалила (3,26 г, 2,57 ммоль) в сухом СН 2Сl2 (70 мл), охлажденному до -43оС, добавляли ДМСО (4,07 г, 51,4 ммоль) в СН2Сl2 (20 мл) по каплям в течение 1 ч, затем добавляли продукт примера 19б (1,10 г, 2,14 ммоль) в СН2Cl2 (25 мл) таким же образом в течение 30 мин. После перемешивания раствора при -43оС в течение еще 1 ч добавляли ТЭА (10,80 г, 107,0 ммоля) и раствору позволяли медленно нагреваться до комнатной температуры. Раствор разбавляли с СН 2Cl2, промывали с 5%ным водным раство ром НСl, 5%-ным водным раствором NaOCl, высуши вали над MgSO4, фильтровали и растворители удаляли под вакуумом. Сырой продукт очища ли с помощью быстрой хроматографии на силикагеле, элюируя с МеОН:CHCl 3 (2:98), чтобы получить продукт (420 мг) в виде прозрачной светло-желтой сиропообразной массы; ТСХ, Rf = 0,32 и 0,37, силикагель, МеОН : CHCl3 (5:95); ЖХВД, 11 39093 tR = 7,38 и 9,55, Зорбакс ОДS аналитическая колонка, скорость потока – 0,5 мл/мин, СН3CN : H2O : ТФУК (50:50:0,1). Анализ, вычисленный для С 22Н36N3O7 F3 х x 0,75 H 2 O: C 50,33, H 7,20, N 8,00. Найдено: C 50,34, H 7,21, N 7,58. Пример 20. Получение 3 (RS)-[(4-метоксифе нил)-карбонил]-L-валил-N-[3-(4-метил-1,1,1-три-фтор-2оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-[4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. К раствору продукта, полученного при использовании методики примера 3г, (1,50 г, 4,08 ммоль) и ТЭА (2,06 г, 20,4 ммоль) в СНСl3 (50 мл), охлажденному до 0оС, добавляли 4-метоксибензоил хлористый (0,766 г, 4,49 ммоль) в СНСl3 (40 мл) по каплям в течение 1 ч и раствор перемеши вали при комнатной температуре в течение ночи. Растворители удаляли под вакуумом и оста ток растворяли в этилацетате, промывали 5%-ным водным раствором НСl, 20%-ным водным раствором NaOH, высушивали над твердой смесью К2СО3: NaSO4 (10:90), фильтровали и растворители удаляли под вакуумом. Сырой продукт очищали с помощью быстрой хроматографии на силикагеле, элюируя с МеОН : CНCl3 (5:95), чтобы получить продукт (1,84 г) в виде пены белого цве та; ТС Х, Rf = 0,33, силикагель, MeOH:CHCl 3 (5:95). Анализ, вычисленный для С 24Н34О5N3 F3 х x 0,3 H 2 O: C 56,86, H 6,88, N 8,29. Найдено: C 56,80, H 6,88, N 8,07. б. Синтез 3(RS)-[(4-метоксифенил)-карбонил]-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. К раствору хлористого оксалила (3,79 г, 29,9 ммоль) в сухом СН 2Сl2 (50 мл), охлажденному до -43оС, добавляли ДМСО (4,73 г, 59,8 ммоль) в СН2Сl2 (20 мл) по каплям в течение 40 мин, затем добавляли продукт по примеру 20а (1,50 г, 2,99 ммоль) в СН2Cl2 (20 мл) таким же образом в те чение 30 мин. После перемеши вания раство ра при -43оС в те чение еще 1 ч добавляли ТЭА (15,10 г, 149,5 ммоль) и раствору позволяли медленно нагреваться до комнатной температуры. Раствор разбавляли с СН2Сl2, промывали 5%-ным водным раствором НСl, 5%ным водным раствором NaOCl, высушивали над MgSO4, фильтровали и растворители уда ляли под вакуумом. Сырой продукт очи щали с помощью быстрой хроматографии на силикаге ле, элюируя с EtOAc:гексан (1:1) после предва рительной обработки силикагеля с ТЭА: гексан (10:90), чтобы получить продукт (489 мг) в виде пены белого цвета: ТС Х, Rf = 0,15 и 0,19, силикагель, МеОН:CHCl3 (2:98); ЖХВД, tR = 6,62 и 9,72, Зорбакс ОДS колонка, скорость потока – 1 мл/мин, СН3СN : H2O : ТФУК (50:50:0,1). Анализ, вычисленный для С 24Н32О 5N3F3 : C 57,71, H 6,46, N 8,41. Найдено: C 57,39, H 6,67, N 8,18. Пример 21. Получение 3(RS)-N 2, N6-ди-[(фе нилметокси)-карбонил]-L-лизил-N[3-(4-метил-1,1,1 -трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-N-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Получен продукт при использовании способа, описанного в примере 2б. б. Синтез 2(RS), 3(SR)-N 2, N6-ди-[(фе нилметокси)-карбонил]-L-лизил-N[3-(4-метил-1,1,1–триф-тор-2оксипентил)]-L-пролинамида. ДЦК (0,84 г, 4,09 ммоль) добавляли к перемешиваемому раствору (N2, N6-ди бензилоксикарбонил)-Lлизина (1,54 г, 3,72 моль), продук та по примеру 21а (1,0 г, 3,72 ммоль), 1-оксибензотриазола (1,01 г, 7,44 ммоль) и сухо го ТГФ (70 мл) при 0 оС в атмосфере азота. Реакционную смесь перемешивали при 0оС в течение 1 ч, и ей позволяли медленно нагреваться до комнатной температуры в течение ночи. Реакционную смесь фильтровали, растворитель удаляли под вакуумом и остаток растворяли в СНСl3. Хлороформный раствор промывали 20%-ным водным раствором лимонной кислоты, органический слой высуши вали над Na2SO4 и фильтровали. Растворитель удаляли под вакуумом, чтобы получить сырой продукт (3,14 г). Продукт очи щали с помощью быстрой хроматографии на силикагеле, элюируя с СНСl3 : MeOH (97:3), что бы получить 1,50 г конечного продукта, Rf = 0,33-0,45, CHCl3 : MeOH (95:5), силикагель. с. Синтез 3(RS)-N 2, N6-ди-[(фе нилметокси)-карбонил]-L-лизил-N-[4-метил- 1,1,1-трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (3,56 г, 46 ммоль) и сухого СН 2Сl2 (30 мл) добавляли к перемеши ваемому раствору хло ристого оксалила (3,04 г, 24 ммоль) и сухо го СН 2Сl2 (50 мл) при -60оС в атмосфере азота. Реакционную смесь перемеши вали при -60оС в течение 5 мин и позволяли ей нагреваться до -30оС. Раствор продукта по примеру 21б (1,32 г, 2,0 ммоль) и сухо го СН 2Сl2 (30 мл) добавляли по каплям. Полученную реакционную смесь перемешивали при -25оС в течение 1 ч, добавляли ТЭА (7,8 г, 77,4 ммоль) и реакционной смеси позволяли медленно нагреваться до комнатной температуры. Реакционную смесь промывали 1 N водным раствором HCl и солевым раствором. Органический слой высушива ли над Na2SO4, фильтровали и растворитель удаляли под вакуумом, чтобы получить продукт (2,6 г). Продукт очи щали с помощью быстрой хроматографии (силикагель, СНСl3 : MeOH, 97:3, чтобы получить продукт (0,99 г), ТСХ, Rf = 0,4–0,52, CHCl3: MeOH (95:5), силикагель. Пример 22. Получение 3(RS)-[(фенилметокси)-карбонил]-L-фенилаланил-N- [3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-[(фенилметокси)-карбонил]-L-фе ни лала нил-N-[3-(4-ме тил-1,1,1-трифтор-2оксипентил)]-L-пролинамида. 12 39093 ДЦК (1,01 г, 4,92 ммоль) добавляли к перемеши ваемому раствору части продук та по примеру 21а (1,41 г, 4,47 ммоль), N-(бензилоксикарбонил)-L-фенилаланина (1,20 г, 4,47 ммоль), 1-оксибензотриазола (1,21 г, 8,94 ммоль) и сухо го ТГФ (75 мл) при 0оС в атмосфере азота. Реакционную смесь перемешивали при 0оС в течение 1 ч и позволяли ей нагреваться до комнатной температуры и перемешивали в те чение ночи. Реакционную смесь фильтровали и растворитель удаляли под вакуумом, чтобы получить сырой остаток, который растворяли в СНСl3. Хло роформный раствор промывали 20%-ным водным раствором лимонной кислоты и солевым раствором и высуши вали над Na2SO4. Хлороформный раствор фильтровали и растворитель удаляли под вакуумом, чтобы получить сырой продукт (3,57 г). Продукт очища ли с помощью быстрой хроматографии на силикагеле, элюируя с СНСl3 : MeOH (97:3), чтобы получить продукт (1,45 г) в виде белой пены; ТСХ, Rf = 0,39–0,60, СHСl3:MeOH (95:5), силикагель, ЖХВД, Зорбакс ОДS аналитическая колонка, СН3СN : H2O (50:50), скорость потока – 2,5 мл/мин, tR = 6,47 и 7,63. б. Синтез 3(RS)-[(фенилметокси)-карбонил]-N-фенилаланил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (2,33 г, 29,9 ммоль) и сухо го СН 2Сl2 (40 мл) добавляли по каплям к перемешиваемому раствору хло ристого оксалила (1,89 г, 14,9 ммоль) и сухого СН 2Сl2 (40 мл) при -60оС в атмосфере азота. Реакционную смесь перемешивали при -60оС в течение 0,5 ч. Раствор продук та по примеру 22а (1,43 г, 2,49 ммоль) и сухого СН2Cl2 (40 мл) добавляли при -50оС. Полученную смесь перемеши вали при -60оС в течение 1 ч. Диизопропилэтиламин (7,70 г, 59,7 ммоль) добавляли и реакционной смеси позволяли нагреваться до комнатной температуры. Смесь промывали дважды 1N водным раствором НСl, а затем солевым раствором. Органический слой высушивали над Na2SO4, фильтровали и растворитель удаляли под вакуумом, чтобы получить сырой продукт (1,87 г). Продукт очи щали с помощью быстрой хроматографии на силикагеле, СНСl3 : MeOH (98:2), чтобы получить продукт (0,77 г) в виде белой пены; ТСХ, Rf = 0,62–0,69, СНСl3 : MeOH (95:5), силикагель; ЖХВД, Зорбакс ОДS аналитическая колонка, С3CN: H2O (50:50), скорость потока – 2,5 мл/мин, tR = 6,11 и 6,21. Анализ, вычисленный для С 28H32F 3N3O5 : C 61,41, H 5,84, N 7,67. Найдено: С 61,53, Н 5,82, N 7,67. Пример 23. Получение 3(RS)-[2-(метоксикарбонил)-этилкарбонил]-L-норлейцил-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез N-[2-(метоксикарбонил)-этилкар-бонил]-L-норлейцина. Раствор 1N гидроокиси натрия (100 мл, 100 ммоль) добавляют по каплям к энергично перемешиваемой смеси 1-норлейцина (6,55 г, 50 ммоль) и хлористого метилена (250 мл) при 0 оС в атмосфе ре азота. 3Карбометоксипропионил хлорид (7,52 г, 50 ммоль) добавляли по каплям. Полученную реакционную смесь перемеши вали при 0оС в течение 15 мин. Охлаждающую баню уби рали и добавляли воду (100 мл). С помощью 3 N водной НСl рН доводили до 1. Добавляли этилацетат (200 мл) и органический слой разделяли. Водный слой экстраги ровали этилацета том и органические слои объединяли, промывали с солевым раствором и высушивали над Na2SO4. Раствор фильтровали. Растворитель удаляли под вакуумом, чтобы получить сырой продукт (10,73 г). Часть сырого продукта (6,47 г, 26,4 ммоль) очища ли с помощью быстрой хроматографии на силикагеле СНСl3:MeOH (97:3), чтобы получить продукт (5,31 г) ТСХ, Rf = 0,45, силикагель, СНСl3:MeOH : AcOH (95:4,75:0,25). б. Синтез 2(RS), 3(SR)-[2-(метоксикарбонил)-этилкарбонил]-L-норлейцил-L-валил-N- [3-(4-метил1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Дициклогексилкарбодиимид (3,46 г, 16,8 ммоль) добавляли к предварительно охлажденному (0оС), раствору продукта, получаемого при использовании методики примера 3г (5,60 г, 15,3 ммоль), продук та примера 23а м (3,75 г, 15,3 ммоль) и 1-оксибензотриазола (4,13 г, 30,6 ммоль) в ТГФ (70 мл). Получающемуся раствору позволяли нагреваться медленно до комнатной температуры и перемеши вали в течение ночи. Реакционную смесь фильтровали и концентрировали под вакуумом. Оста ток разбавляли этилацетатом и получающийся раствор промывали насыщенным водным раствором NaHCO3 и солевым раство ром, высушивали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить сырой продукт. Продукт очища ли с помощью быстрой хроматографии на колонке с силикаге лем, используя в качестве элюента градиент Et2O (100%), Et2O : EtOAc (90:10), Et2O : EtOAc (75:25), Et2O : EtOAc (50:50), чтобы получить продукт (5,6 г); ТСХ, Rf = 0,45, силикагель, MеOH : CHCl 3 (1:9). в. Синтез 3(RS)-[2-(метоксикарбонил)-этил-карбонил]-L-норлейцил-L -валил-N-[3-(4-мети л-1,1,1трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (27,0 мл, 0,378 моль) в СН2Сl2 (27 мл) медленно добавляли к предва рительному охлажденному (-65оС) раствору хло ристого оксалила (16,5 мл, 0,189 моль) в СН2Сl2 (350 мл). Получающийся раствор перемешивали в течение 15 мин и добавляли раствор продук та по примеру 23б (5,60 г, 0,00943 моль) в СН2Сl2 (250 мл). Реакционную смесь перемеши вали в те чение 1 ч при -65оС и по каплям добавляли диизопропилэтиламин (67,0 мл, 0,378 моль). Реакционной смеси позволяли нагреваться до комнатной температуры, а затем промывали ее 1 N водным раствором НСl и солевым раствором, высушива ли над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить сырой продукт. Сы рой продукт очищали с помощью быстрой хроматографии на колонке с силикагелем, элюируя с помощью поэтапного градиента Et2O (100%), Et2O : EtOAc (50:50), EtОAc (100%), что бы получить частично очищенный продукт, который дополнительно очища ли с помощью быстрой хроматографии на силикагеле, элюируя с помощью поэтапного градиента СНСl3 (100%), МеОН:СHCl3 (2,5:97,5) и MeOH : CHCl3 (5:95), чтобы получить конеч 13 39093 ный продукт (3,24 г); ЖХВД, tR = 6,80 и 12,98, Зорбакс ОДS аналити ческая колонка, Н2О : СН3СN (65:35), скорость потока – 2 мл/мин. Приме р 24. По лучение 3(RS)-[(2-карбокси-эти л)-кар бони л]-L -но рле йци л-L -валил-N -[3-(4-метил1,1,1-три фтор-2-ок сопентил)]-L-про линамида. Добавляли 1N водный раствор NaOH (9,5 мл) к раствору продук та по примеру 23в (2,60 г, 4,39 ммоль) в МеОН (95 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре и 1 N водный раствор HCl (10,5 мл) добавляли. Реакционную смесь концентрировали под вакуумом и добавляли Н 2О (35 мл). С успензию экстрагировали эти лацетатом и органический слой промывали солевым раствором, высуши вали над Na2SO4, фильтровали и концентрировали под вакуумом, чтобы получить продукт (2,1 г), ЖХВД, tR = 12,63 и 19,05, Зорбакс ОДS аналитическая колонка, Н2О : CH3CN (65:35), скорость потока – 0,5 мл/мин. Пример 25. Получение 3(RS)-[(фенилметокси)-карбонил]-L-альфа-глутамил-L-валил-N- [3-(4-метил1,1,1-трифтор-2-оксопентил)]-L-пролиламида фенилметилового сложного эфи ра. а. Синтез фенилметилового сложного эфи ра 2(RS), 3(SR)-[(фенилметокси)-карбонил]-L-альфа-глутамил-L-валил-N- [3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. Изобутилхлорформиат (0,53 мл, 4,1 ммоль) добавляли к предварительно охлажденному (-15оС) раствору альфа-бензилового сложного эфи ра N-бензилоксикарбонил-L-глутаминовой кислоты (1,52 г, 4,1 ммоль) и N-метилморфо лина (0,45 мл, 4,1 ммоль) в ТГФ (30 мл). Реакционную смесь перемешивали в течение 10 мин, а затем охлаждали до -40оС. Добавляли по каплям раствор продукта по примеру 3г (1,5 г, 4,1 ммоль) в ТГФ (30 мл) и реакционную смесь перемешивали в течение ночи и позволяли ей медленно нагреваться до комнатной температуры. Реакционную смесь фильтровали и концентрировали под вакуумом. Остаток растворяли в этилацета те, промывали 1N водным раствором НСl и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали, что бы получить сырой продукт. Продукт очи щали с помощью быстрой хроматографии на колонке с силикагелем, используя в качестве элюента градиент СНСl3 (100%), MeOH:CHCl3 (2,5:97,5) и MeOH:CHCl3 (5:95), чтобы получить продукт (2,13 г); ТС Х, Rf = 0,43, силикагель, MeOH:CHCl3 (5:95). б. Синтез фенилметилового сложного эфи ра 3(RS)-[(фенилметокси)-карбонил]-L-альфа-глутамил-Lвалил-N- [3-(4-метокси-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (8,40 мл, 0,118 моль) в СН2Сl2 (8 мл) осторожно добавляли к предварительному о хлажденному (-65оС) раствору хло ристого оксалила (5,2 мл, 0,059 моль) в СН2Сl2 (100 мл). Раствор перемешивали в течение 15 мин и раствор продукта по примеру 25а (2,13 г, 2,96 ммоля) в СН2Сl2 (100 мл) добавляли по каплям. Реакционную смесь перемешивали в течение 1 ч при -60оС и N,N-диизопропилэтиламин (20,9 мл, 0,118 моль) добавляли по каплям. Реакционной смеси позволяли нагреваться до комнатной температуры, промывали ее 1N водным раствором НСl и солевым раствором, высушивали над NaSO4, фильтровали и концентрировали, что бы получить сырой продукт. Продукт частично очищали с помощью быстрой хроматографии на колонке с силикагелем, используя в качестве элюента поэтапный градиент Et2O (100%), Еt2O : EtOAc (50:50) и EtOAc (100%). Продукт в конечном счете очища ли с помощью быстрой хроматографии на колонке с силикагелем, используя в качестве элюента поэтапный градиент СНСl3 (100%), МеОН:CHCl3 (1:99), MeOH:CHCl3 (2:98), MeOH:CHCl3 (3:97) и MeOH:CHCl3 (5:95), чтобы получить продукт (1,35 г); ЖХВД, tR = 7,2 и 11,5, Зорбакс ОДS аналитическая колонка, Н2О : CH3CN (50:50), скорость пото ка – 2 мл/мин. Пример 26. Получение 3(RS)-N 2-[2-(метоксикарбонил)-этилкарбони л]-N 6-[(фе нилметок си)– карбонил]-L-ли зил-L-ва лил-N -[3-(4 -ме тил-1,1,1-трифтор-2 -оксопентил)]-L-пролинамида. а. Синтез N2-[(2-метоксикарбонил)-этилкар-бонил]-N6-фенилметоксикарбониллизина. 1N Водный раствор NaOH (43 мл) добавляли к предварительно охлажденному раствору (0оС) N-бензилоксикарбонил-L-лизина (6,06 г, 0,0216 моль) в CH2Cl2 (160 мл). Реакционную смесь энергично перемешивали и добавляли 3-карбоксиметоксипропионил хлористый (2,66 мл, 0,0216 моль). Реакционную смесь энергично перемешивали в течение 15 мин при 0оС. Воду (100 мл), 1 N водный HCl (25 мл) и этилацетат (500 мл) последовательно добавляли и слои разделяли. Органический слой промывали солевым раствором, высуши вали над Na2SO4, фильтровали и концентрирова ли, чтобы получить продукт (6,78 г). Продукт использовали без дальнейшей очистки. б. Синтез 2(RS), 3(SR)-N 2-[2-(метоксикарбонил)-L-лизил-L-валил-N6-[3-(4-метил-1,1,1-трифтор-2-оксипентил)]-L-пролинамида. ДЦК (2,85 г, 13,9 ммоль) добавляли к смеси продукта, получающе гося при использовании методики примера 3г (4,63 г, 12,6 ммоль), продукта по примеру 26а (5,00 г, 12,6 ммоль) и ОБТА (3,76 г, 27,8 ммоль) в ТГФ (65 мл), предварительно охлажденного до 0 оС. Смесь в течение 1 ч нагревали до комнатной температуры и, в те чение 1 ч, нагревали до комнатной температуры и перемеши вали при 0оС в течение ночи. Растворитель удаляли под вакуумом, остаток разбавляли этилацетатом и промывали последова тельно насыщенным раствором NaHCO3 и солевым раствором. Органическую фа зу высушивали над твердой смесью К2СО3 : Na2SO4 (10:90), фильтровали и раство ритель удаляли под вакуумом, чтобы получить сырой продукт. Очистка с помощью быстрой хроматографии на силикагеле с элюентом МеОН : CHCl 3 (1:99) дает продукт (6,22 г) в ви де белой пены; ТСХ, Rf = 0,40, силикагель, МеОН : CHCl 3 (5:95). в. Синтез 3(RS)-N2-[2-(метоксикарбонил)-этилкарбонил]-N 6-[(фе нилметокси)-карбонил]-L-лизил-Lвалил-N-[3-(4-метил-1,1,1-трифтор -2-оксопентил)]-L-пролинамида. 14 39093 Раствор ДМСО (15,9 г, 100 ммоль) в сухом СН 2Сl2 (50 мл) добавляли по каплям к перемешиваемому раствору хло ристого оксалила (8,8 мл, 200 ммоля) в сухом СН 2Сl2 (150 мл), охлажденному до -43оС в атмосфере азота. Раствор продук та по примеру 26б (6,22 г, 8,37 ммоль) в СН2Сl2 (60 мл) добавляли таким же образом. Реакционную смесь перемеши вали при -20оС в течение 1 ч и ТЭА (70 мл, 400 ммоль) добавляли по каплям. Смеси позволяли медленно нагреваться до комнатной температуры, перемешивали в те чение еще 1 ч, а затем разбавляли с СН 2Сl2, промывали 5%-ным водным раствором NaOCl, высушивали над твердой смесью К2СО3 : Na2SO4 (10:90), фильтровали и растворитель удаляли под вакуумом, чтобы получить сырой продукт. Очистка с помощью быстрой хроматографии на си ликагеле с элюентом МеОН:СHСI3 (1:99) дает продукт (4,5 г) в ви де светло-желтой пены; ТСХ, R f = 0,51, силикагель, MeOH:CHCl 3 (1:9), ЖХВД, tR = 6,99 и 12,01, скорость потока – 1 мл/мин, Зорбакс ОДS, аналитическая колонка, Н2О : СН3СN : ТФУК (50:50:0,1). Пример 27. Получение 3(RS)-N 2-[(2-кар-боксиэтил)-карбонил]-N6- [(фенилметокси)-карбо-нил]-L-лизил-L-валил-N-(4-метил-1,1,1- трифтор-2-оксопентил)]-L-пролинамида. Раствор продукта по примеру 26в (2,0 г, 2,7 ммоль) в МеОН (60 мл) и 1 N NaOH (5,4 мл, 5,4 ммоль) перемеши вали при комнатной температуре в течение 12 ч, а затем доводили до рН 7 с помощью 1 N водного раствора НСl (6,0 мл, 6,0 ммоль). Под вакуумом удаляли МеОН, получающийся остаток растворяли в этилацетате, промыва ли солевым раствором, высушива ли над MgSO4, фильтровали и растворитель удаляли под вакуумом, чтобы получить сырой продукт. Очистка с помощью быстрой хроматографии на силикагеле (рН 5,5 фирмы Бейкер) с хлороформом дает продукт (1,7 г) в виде белой пены; ЖХВД, tR = 4,06 и 5,56, скорость потока – 1 мл/мин, Зорбакс ОДS аналитическая колонка, Н2О:CH3CN:ТФУК (50:50:0,1). Анализ, вычисленный для С 34Н48N5O9F 34 x x 1,75 H 2 O: C 53,78, H 6,83, N 9,22. Найдено: C 53,46, H 6,39, N 9,03. Пример 28. Получение 3S (или R)-N2, N6-ди-[(фе нилметокси)-карбонил]-L-лизил-L-валил-N-[3- (4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. а. Синтез 2(RS), 3(SR)-N 2, N6-ди-[(фенилметокси)-карбонил]-L-лизил-L-валил-N-[3-(4-метил-1,1,1трифтор-2-оксипентил)]-L-пролинамида. ДЦК (0,93 г, 4,49 ммоль) добавляли к перемешиваемому раствору N2, N6-бензилоксикарбонил-L-лизина (1,69 г, 4,08 ммоль), продукта, получающе гося при использовании методики примера 3г (1,50 г, 4,08 ммоль), 1-оксибензотриазола (1,10 г, 8,16 ммоль) и сухого ТГФ (75 мл) при 0оС в атмосфе ре азота. Реакционную смесь перемешивали при 0оС в те чение 1 ч, затем позволяли ей нагреваться до комнатной температуры и перемешивали в те чение ночи. Реакционную смесь фильтровали. Фильтрат выпарива ли под вакуумом. Оста ток растворяли в СН 2Сl2 и раствор промыва ли 1N водным раствором НСl и солевым раствором, а затем высушивали над Na2SO4. Отфильтровывали Na2SO4 и фильтрат концентрировали под вакуумом, чтобы получить сырой продукт (3,94 г), который очищали с помощью быстрой хроматографии на силикагеле, СНСl3:MeOH (95:5), что бы получить 2,48 г продук та, ТСХ, Rf = 0,36–0,56, СНСl3:MeOH (95:5), силикагель, ЖХВД, Зорбакс ОДS аналитическая колонка, скорость потока – 1,5 мл/мин, СН3CN:H2O (50:50), tR = 18,33, 14,99. Анализ, вычисленный для С 33H41F 3N4O7 : C 59,01, H 6,24, N 8,45. Найдено: C 58,89, H 6,33, N 7,89. б. Синтез 3S (или R)-N2, N6-ди-[(фе нилметокси)-карбонил]-L-лизил-L-валил-N-[3-(4-метил-1,1,1-трифтор-2-оксопентил)]-L-пролинамида. Раствор ДМСО (2,8 г, 36,13 ммоль) и сухого СН 2Сl2 (40 мл) добавляли в перемешиваемый раствор хло ристого оксалила и сухого СН 2Сl2 (40 мл) при -60оС в атмосфере азота. Затем раствор продукта по примеру 28а (2,30 г, 3,01 ммоль) и сухо го СН 2Сl2 (40 мл) добавляли в реакционную смесь при -50оС. Полученную реакционную смесь перемешивали при -60оС в течение 1 ч. Триэтиламин (7,290 г, 72,26 ммоль) добавляли и реакционной смеси позволяли нагреваться до комнатной температуры. Смесь промывали дважды 1N водным раствором НСl, затем солевым раствором и высуши вали над Na2SO4×Na2SO4, отфильтровывали и фильтрат концентрировали под вакуумом, чтобы получить сырой продукт (2,67 г). Продукт очи щали с помощью быстрой хроматографии на силикагеле с элюентом СНСl3 : MeOH (97:3), чтобы получить 64 мг продукта; ТС Х, Rf = 0,6, CHCl3 : MeOH (95:5), ЖХВД, Зорбакс ОДS аналитическая колонка, СН3CN : H2O (60:40), скорость потока – 1,5 мл/мин, tR = 5,29. Анализ, вычисленный для С 38Н50F3N5 O8 x x H2 O: C 58,53, H 6,72, N 8,98. Найдено: C 58,95, H 6,59, N 8,74. Пример 29. Получение 3(RS)-1-(12-метокси-12-оксододе цилокси)-кар бонил-N-[3-(1 ,1,1 -три фтор-4метил-2-оксопентил)]-L-пролинамида. (формула Ia, R1 = CH(CH3)CH3, R3 = CH 3OCO(CH2)11, A = OCO, n=1). а. Синтез мети лового эфира 12-оксидодекановой кислоты. Смесь 1-оксидодекановой кислоты (4,0 г, 18,5 ммоль), МеОН (450 мл), концентрированную Н2SO 4 (2,5 мл) и молекулярных сит ти па 3А (3 мл) перемешивали при температуре кипения с обратным холодильником в те чение 16 ч. Смесь нейтрализовали насыщенным водным раствором NaHCO3, концентрировали под вакуумом и разделяли между Et 2O и водой. Эфирный раствор промывали (водой, насыщенным водным раствором NaHCO3, со левым раствором), высушивали (Na2SO4), фильтровали и концентрировали под ва 15 39093 куумом, чтобы получить продукт (3,94 г) в виде белого твердого вещества; ЯМР (ДМСО-d6) 83,65 (3Н, с); 1,7 –1,0 (22 Н, м). б. Синтез N-метоксикарбонилундецил-4-нитрофенилкарбоната. Используя метод примера 7а, продукт по примеру 29а превраща ли в названное соединение и очищали с помощью быстрой хроматографии (EtOAc : гексан (1:9)), чтобы получить названное соединение с 59%ным выхо дом; ТСХ, Rf = 0,20, EtOAc : гексан (1:9). в. Синтез 2(RS), 3(SR)-1-(12-метокси-12-оксододецилокси)-карбонил- N-[3-(1,1,1-трифтор-2-окси-4метилпентил)]-L-пролинамида. (формула VIIa, R1 = CH(CH3)–CH3, R3 = CH 3OCO(CH2)11, A = OCO, n=1). Используя мето дику примера 7б, продукт по примеру 29б подверга ли взаимодействию с соединением, полученным при использовании методики примера 2б, чтобы обеспечить после очистки с помощью быстрой хроматографии (ацетон:гексан (3:7)) выше названный продукт (45%); ЖХВД, tR = 4,43, Col A, CH 3CN : H2O (35:65), СП = 2,0. г. Синтез 3(RS)-1-(12-метокси-12-оксододецилокси)-карбонил-N- [3-(1,1,1-трифтор-4-метил-2-оксопентил)]-1-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = CH 3OCO-(CH2)11, A = OCO, n = 1). К продук ту по примеру 29в (1,1 ммоль) добавляли ДМСО (65 мл) и Ас2О (50 ммоль). Получающийся раствор перемешивали 18 ч при комнатной температуре и разбавляли эфиром. Органический раствор промывали (насыщенным водным NaHCO3 три раза, водой и солевым раствором), высушивали (Na2SO4), фильтровали, концентрировали под вакуумом и очища ли с помощью быстрой хроматографии (эфир:гексан (1:1)), чтобы получить названный выше продукт (100%), ЖХВД, tR = 12,73, Col A, CH 3CN : H 2O (60:40), CП = 2,0. Анализ, вычисленный для С 22Н28F3N3 O5 x x 0,4 H 2 O: C 55,91, H 8,00, N 5,21. Найдено: С 56,05, Н 8,00, N 5,19. Пример 30. Получение 3(RS)-1-(12-окси-12-оксододецилокси)-карбонил-N-[3-(1,1,1-трифтор-4-метил2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = HOCO(CH 2)11 , A = OCO, n = 1). Используя мето дику примера 14, продукт по примеру 29г превраща ли в названное выше соединение, очищали с помощью быстрой хроматографии (этилацетат:гексан (1:1)) и получали с 10%-ным выхо дом; ЖХВД, tR = 4,55, Сol A, CH 3CN : H2O (60:40), СП = 2,0. Анализ, вычисленный для С 24Н39F3N2 O6 x x 0,1 H 2 O: C 56,48, H 7,74, N 5,49. Найдено: C 56,48, N 7,96, N 5,23. Пример 31. Получение 3(RS)-1-[1-оксо-5-(фенилметоксикарбониламино)-пентил]-N-[3-(1,1,1-трифтор-4метил-2-оксопентил)]-L-пролинамида (формула Ia, R 1 = CH(CH3)CH3, R3 = PhCH2OCONH-(CH 2)4, A = CO, n=1). а. Синтез 5-(фе нилметоксикарбониламино)-валериановой кислоты. К перемешиваемому охлажденному раствору (0оС) 5-аминовалериановой кислоты (5,00 г, 42,68 ммоль) и 2 N NaOH (32,0 мл, 32,0 ммоль) добавляли одновременно бензиловый эфир хлормуравьиной кислоты (7,65 г, 6,40 мл, 44,81 ммоль) и 2 N NaOH (32,0 мл, 32,0 ммоль). После перемешивания при 0оС в течение 0,5 ч, раствор промыва ли эфиром. Эфирный слой подкисляли до рН 2,0 с помощью 6 N HCl, приводящего к выпадению в осадок продукта из раствора. Названное выше соединение фильтровали, промывали (Н2О) и высуши вали (вакуумная печь), что бы получить чистый продукт в виде белого твердого ве щества (8,55 г, 80,0%), т.пл 104–105оС; ТСХ, R5 = 0,48, MeOH : CHCl 3 : AcOH (3:97:0,1). б. Синтез 2(RS), 3(SR)-1-[1-оксо-5-(фенилметоксикарбониламино)-пентил]- N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (фор мула VIIa, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)4, A = CO, n = 1). К перемешиваемому охлаждаемому раствору (0оС) продукту по примеру 31а (0,47 г, 1,86 ммоль), ОБТА (0,50 г, 3,72 ммоль) и ДЦК (0,40 г, 1,95 ммоль) в СНCl3 (50 мл) добавляли продукт, по лученный при использовании методики примера 2б (0,50 г, 1,86 ммоль). После перемешивания реакционной смеси в те чение ночи при комнатной температуре ее отфильтровывали и концентрировали, что бы получить сироп, который частично растворяется в этилацетате. Нерастворимый продукт фильтровали из этилацетатного раствора до его промывания (насыщенным водным раствором NaHCO3, 5%-ной водной лимонной кислотой и солевым раство ром), высушивания (Na2SO4) и концентрирования до смеси, которую очища ли с помощью быстрой хроматографии (МеОН:CHCl3 (4:96)), чтобы получить названное выше соединение в виде белой пены (0,78 г, 84%); ТСХ, Rs = =0,4, MeOH:CHCl 3 (4:94). в. Синтез 3(RS)-1-[1-оксо-5-(фенилметоксикарбониламино)-пентил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3, R3 = PhCH 2O-CONH(CH2)4, A = CO, n=1). К продукту по примеру 31б (1 ммоль) добавляли ДМСО (85 ммоль) и Ас2О (64 ммоль). Получающийся раствор перемеши вали в течение 18 ч при комнатной температуре, выливали в ледяную во ду (50 мл) и перемеши вали в течение 1 до 4 ч. Сырой продукт экстрагировали этилацета том и этилацетатный раствор промывали (насыщенным водным NaHCO3 и солевым раствором), высушива ли (Na2SO4), фильтровали и концентрировали под вакуумом, прежде чем продукт очищали с помощью быстрой хроматографии (СHСl3 : MeOH (97:3)), чтобы получить продукт (58%); ЖХВД, tR = =6,56 и 7,79, Col A, H 2O : CH 3CN (60:40), СП = 2,0. 16 39093 Анализ, вычисленный для С 24H32F3N3 O5 x x 1,5 H 2 O: C 54,74, H 6,69, N 7,98. Найдено: С 54,87, Н 6,20, N 8,02. Пример 32. Получение 3(RS)-1-(1-оксо-4-феноксибутил)-N-[3-(1,1,1-трифтор-4-метил- 2-оксопентил)]L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhO(CH 2)3, A = CO, n=1). а. Синтез 2(RS), 3(SR)-1-(1-оксо-4-феноксибутил)-N-[3-(1,1 ,1-три фтор-2 -окси-4 -мети лпентил)]-Lпролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhO(CH 2)3, A = CO, n=1). К 0,25 молярному раствору 4-фе ноксибутановой кислоты в ТГФ добавляли молярное эквивалентное количество КДИМ в ви де одной порции. После перемеши вания реакционной смеси в течение 1 ч при комнатной температурe в виде одной порции добавляли молярное эквивалентное количество продукта, полученного при использовании методики примера 2б. После перемеши вания реакционной смеси в течение ночи добавляли избыток насыщен ного водного раствора NaHCO3 и смесь экстрагировали этилацетатом. Этилацетатные экстракты промывали (в НСl, солевым раствором), высушивали (MgSO 4), фильтровали и концентрировали под вакуумом, чтобы получить названный выше продукт (88%); ТСХ, Rs = 0,53 и 0,61, МеОН:CH2Cl2 (1:9). б. Синтез 3(RS)-1-(1-оксо-4-феноксибутил)-N-[3-(1,1,1-трифтор-4-метил -2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhO(CH 2)3, A = CO, n=1). По методике примера 31в продукт по примеру 32а превраща ли в названное выше соединение с 39%ным выхо дом после кристаллизации из воды; ТСХ; Rf = 0,68 и 0,64, СН2Cl2 : MeOH (9:1). Анализ, вычисленный для С 21H27F3O 4 x x 1,25 H 2 O: C 55,93, H 6, 69, N 6,21. Найдено: С 55,88, Н 6,67, N 6,15. Пример 33. По лучение 3(RS)-1-[2-(4-морфо лин)-этоксикарбонил]-N-[3-(1,1,1-трифтор-4 -метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = 4-морфолинил-(CH2)2, A = OCO, n=1). а. Синтез хлор гидрата 2-(4-морфолинил)-этил-4-нитрофе нилкарбоната. Используя методику примера 7а, но без использования пиридина (и кислотной промывки) 2-(4-морфо линил)-этанол обрабатывали с 4-нитрофе ниловым эфиром хлормуравьиной кислоты. Сырой продукт фильтровали, промывали эфи ром и высуши вали под вакуумом. Полученный продукт (91%) использовали для последующей реакции без дальнейшей идентифи кации. б. Синтез 2(RS), 3(SR)-1-[2-(4-морфо линил)-этоксикарбонил]-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIa, R1 = СH(CH3)CH3, R3 = 4-морфо линил-(CH2)2, A = OCO, n=1). Используя методику примера 7б, продукту по примеру 33а позволяли реагировать с продуктом, полученным при использовании методики примера 2б, чтобы обеспечить после очистки с помощью быстрой хроматографии (МеОН:CHCl3 (1:99)) названный выше продукт (68%); ТС Х; Rs = =0,34, MeOH:CHCl3 (5:95), СП = 2,0. в. Синтез 3(RS)-1-[2-(4-морфолинил)-этокси-карббонил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопе-нтил)]L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = 4-морфолинил-(СН2)2, А = ОСО, n=1). К продукту по примеру 33б (1,1 ммоль) добавляли ДМСО (65 ммоль) и Ас2О (50 ммоль). Получающийся раствор перемешивали в течение 18 ч при комнатной температуре и разбавляли с СНСl2. Органический раствор промывали (насыщенным водным NaHCO3 3 раза, водой и солевым раствором), высуши вали (Na2SO4), фильтровали и концентрирова ли под вакуумом до проведения очистки с помощью быстрой хроматографии (МеОН : CHCl3 (2:98)), чтобы получить названный выше продукт (37%), ЖХВД, tR = 8,44 и 9,88, Сol A, CH3CN : H2O (60:40), СП = 2,0. Анализ, вычисленный для С 18Н28F3N3 O5 x x 1,0 H 2 O: C 48,97, H 6,85, N 9,51. Найдено: 48,97, 6,61, 9,73. Пример 34. Получение 3(RS)-1-[1-оксо-6-[2-(2-пири дил)-этокси ]-карбони ламиногексил]-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)] -L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = (2-пиридил)-(СН2)2ОСОNH-(CH2)5, A = CO, n=1). а. Синтез 4-нитрофе нил-2-(2-пиридил)-этилкарбоната. Раствор 2-пиридинэтанола (1,38 г, 11 ммоль) в Et2O (20 ммоль) добавляли в те чение 1 ч к перемешиваемому раствору паранитрофе нилового эфи ра хлормуравьиной кислоты (2,26 г, 11 ммоль) при 0 оС в атмосфере азота. Получающуюся смесь перемеши вали в течение 1 ч при 0 оС до образования осадка, который собирали под атмосфе рой азота и перекристаллизовывали из абсолютного EtOH, чтобы получить 1,53 г (58%) названного выше соединения в виде не совсем белых кристаллов, т.пл. 125,127оС. б. Синтез 2(RS), 3(SR)-1-[1-оксо-6-[2-(2-пиридил)-этокси]-карбониламиногексил]-N -[3 -(1 ,1,1-трифтор-2-окси-4-метилпентил)] -L-пролинамида (формула VIIa, R1 = CH(CH3)CH3, R3 = (2-пиридил)-(СН2)2ОСОNH(CH2)5, A = CO, n=1). Раствор аминного продукта по примеру 50б (0,75 г, 1,8 ммоль), продукта по примеру 34а (0,675 г, 1,8 ммоль), ТЭА (0,52 мл, 3,6 ммоль), СH3CN (25 мл) и воды (25 мл) перемешивали при комнатной температу 17 39093 ре в те чение 2 дней, прежде чем удалить растворитель под вакуумом, чтобы получить сырой продукт, который очищали с помощью быстрой хроматографии (СН3ОН : CHCl3 (2,5:97,5)) для получения продукта (1,13 ммоль, 60%) в виде бледно-желтого твер дого ве щества; ТС X, Rf = 0,5, CH 3OH : CHCl3 (5:95)). в. Синтез 3(RS)-1-[1-оксо-6-[2-(2-пиридил)-этокси]-карбониламиногексил] -N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = (2-пиридил)-(СН2)2ОСОNH(CH2)5, A = CO, n=1). Используя мето дику примера 31в, продукт по примеру 34в превращали в названный выше продукт, чтобы получить после очистки с помощью быстрой хроматографии (МеОН : CH2Cl2 (3:97)) названный выше продукт с 10%-ным выхо дом; ЖХВД, tR = 1,84, Col A, H 2 O; CH3CN (60:40), СП = =2,0. Анализ, вычисленный для С 25Н35F3N4 O5 x x 0,5 H 2 O: C 55,85, H 6,75, N 10,40. Найдено: C 56,08, H 6,82, N 10,43. Пример 35. Получение 3(RS)-1-[2-фенилметокси-1-(фенилметоксиметил)-этоксикарбонил]-N- [3(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = (PhCH 2O-CH2)2CH, A = OCO, n=1). a. Синтез 4-нитрофе нил-2-фенилметокси-1-(фенилметоксиметил)-этилкарбоната. ТЭА (0,74 г, 7,34 ммоль) добавляли по каплям к перемешиваемому раствору па ранитрофе нилового эфи ра хлормуравьиной кислоты (1,48 г, 7,34 ммоль) и Et2O (30 мл) при температуре 0–5оС. К полученной выше реакционной смеси добавляли при температуре 0–5оС раствор 1,3-дибензилглицерина (2,0 г, 7,34 ммоль) и эфира (20 мл) и получающуюся смесь перемешивали в те чение 2 ч при 0–5оС до ее нагревания до комнатной температуры и перемешивания в течение ночи. Реакционную смесь фильтровали и концентрировали под вакуумом, получая в остатке 3,6 г желтого масла, которое очищали с помощью быстрой хроматографии (гексан:эфир (9:2)), чтобы получить 2,19 г (68%) названного выше соединения в виде прозрачного масла; ТСХ, Rf = 0,33, гексан:эфир (7:3). б. Синтез 2(RS), 3(SR)-1-[2-фенилметокси-1-(фенилметоксиметил)-этоксикарбонил]-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = (PhCH2OCH2)CH, A = OCO, n=1). Используя методику примера 7б, продукт по примеру 35а подвергали реакции с продуктом, полученным при использовании методики примера 2б, что бы получить после очистки с помощью быстрой хроматографии (СHCl 3:EtOAc (95:5)) названное выше соединение (62%): ЖХВД, tR = 5,81 и 6,29; Col A, H2O : CH3CN (40:60), СП = 2,0. в. Синтез 3(RS)-1-[2-фенилметокси-1-(фенилметоксиметил)-этоксикарбонил]-N-[3-(1,1,1-трифтор-4метил-2-оксопентил)]-L-про линамида (формула Ia, R1 = CH(CH3)CH3, R3 = (PhCH 2OCH2)2CH, A = OCO, n=1). Используя методику примера 31в, продукт по примеру 35б окисляли, чтобы получить после очистки с помощью быстрой хроматографии (СНСl3 : EtOAc (98:2)) названное выше соединение (13%); ЖХВД, tR = 5,62, Col A, H2O : CH3CN (40:60), СП = 2,0. Анализ, вычисленный для С 29Н35F3N2 O6 x x 0,25 H 2 O: C 61,20, H 6,28, N 4,92. Найдено: C 61,28, H 6,34, N 5,15. Пример 36. Получение 3(RS)-1-[1-оксо-4-(1-оксо-2-феноксиэтиламино)-бутил]-N-[3-(1,1,1-три-фтор-4метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2CONH(CH2)3, A = CO, n=1). а. Синтез эти лового эфи ра 4-(1-оксо-2-феноксиэтиламино)-бутановой кислоты. К перемешиваемой смеси хлоргидрата этилового эфи ра 4-аминобутановой кислоты (3,4 г) и хлористого фе ноксиацети ла (2,76 мл) в 50 мл эфира и 50 мл воды добавляли в виде одной порции NaHCO3 (4,2 г). После 2 ч слои разделяли и органическую фазу промывали (1N HCl, солевым раствором), высушивали (MgSO4) и фильтровали. Выпаривание растворителя под вакуумом дает 3,1 г (53%) названное выше соединение в виде масла). б. Синтез 4-(1-оксо-2-феноксиэтиламино)-бутановой кислоты. Смесь продукта по примеру 36а (3,1 г) в 1 N NaOH (15 мл) перемешивали в течение 6 ч при комнатной температуре. Получающийся раствор подкисляли с помощью 2N HCl. Твердое вещество, вы павшее в осадок, собирали, промыва ли водой и высушивали под высоким вакуумом. Получено 2,5 г (95%) названного выше соединения в виде белого твердого вещества, т.пл. 91–94оС. в. Синтез 2(RS), 3(SR)-1-[1-оксо-4-(1-оксо-2-феноксиэтиламино)-бутил]-N- [3-(1,1,1-трифтор-2-оксо-4метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3), R3 = PhOCH 2CONH(CH2)3, A = CO, n=1). Используя методику примера 32а, продукт по примеру 36б подвергали взаимодействию с продуктом, полученным при использовании методики примера 2б, чтобы получить после очистки путем обработки – промывки кислотой и основанием – названный выше продукт (92%); ТСХ, Rf = =0,43 и 0,48, MeOH:CH2Cl2 (1:9). г. Синтез 3(RS)-1-[1-оксо-4-(1-оксо-2-фенок-сиэтиламино)-бутил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhOCH 2CONH(CH2)3, A = CO, n=1). Используя методику примера 61в, продукт по примеру 36г окисляли, что бы получить после очистки с помощью быстрой хроматографии 18 39093 (МеОН:CH2Cl2 (3:97)) названный выше продукт (75%), ЖХВД, tR = 4,62 и 6,02, Col A, CH3CN : H2O (65:35), СП = 2,0. Анализ, вычисленный для С 23Н30F 3N5O5 : C 56,26, H 6,28, N 8,56. Найдено: C 56,28, H 6,40, N 8,30. Пример 37. Получение 3(RS)-1-(4-метокси-1,4-диоксобутил)-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = CH3OCO(CH2)2, A = CO, n=1). а. Синтез 2(RS), 3(SR)-4-(4-метокси-1,4-ди-оксобутил)-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-Lпролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = CH 3OCO(CH2)2 , A = CO, n=1). К перемеши ваемой смеси продук та, полученного при использовании методики примера 2б, (1,34 г) в СН2Cl2 (50 мл) и 1N NaOH (6 мл), охлажденной в бане с ледяной водой, добавляли по каплям 3-карбометоксипропионил хлористый (0,75 г). После 1 ч слои разделяли и органическую фазу вы сушивали (Na2SO4), фильтровали и выпарива ли, чтобы получить 1,1 г (58%) названного выше соединения в виде белого порошка; ТСХ; Rf = =0,57, MeOH:CH 2Cl2 (1:9). б. Синтез 3(RS)-1-(4-метокси-1,4-диоксобутил)-N-[3-(1,1,1-трифтор -4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1=CH(CH3)CH3, R3 = CH 3OCO(CH2)2 , A = CO, n=1). Используя методику примера 61в, продукт по примеру 37а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CH2Cl2 (2:98)) названный выше продукт (71%); ТС Х, Rf = 0,58, MeOH : CH 2Cl2 (1:9). Анализ, вычисленный для С 16Н23F3N2 O5 x x 0,75 H 2 O: C 48,79, H 6,27, N 7,11. Найдено: C 49,04, H 6,12, N 6,83. Пример 38. Получение 3(RS)-1-[3-(1,1,1-ди-метилэтоксикарбонил)-амино-1-оксопропил]-N- [3-(1,1,1трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = (CH 3)3COCONH(CH2)2, A = CO, n=1). а. Синтез 2(RS), 3(SR)-1-[3-((1,1-диметил-этоксикарбонил)амино-1-оксопропил]-N -[3-(1,1,1-трифтор2-окси-4-метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = (CH3)3COCONH(CH2)2, A = CO, n=1). Используя методику примера 32а, 3-(БОК-амино)-пропановой кислоте позволяли реагировать с продуктом, полученным при использовании методики примера 2б, для получения названного выше продук та (80%): ТСХ, Rf = 0,35, MeOH:CH 2Cl2 (1:9). б. Синтез 3(RS)-1-[3-(1,1-диметилэтоксикарбонил)-амино-1-оксопропил] -N-[3-(1,1,1-трифтор-4-метил2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = (CH 3)3COCONH(CH2)2, A = CO, n=1). Используя методику примера 61в, продукт по примеру 38а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (MeOH:CH2Cl2 (2:98)) названное выше соединение (61%); ТСХ, Rf = 0,46, MeOH : CH 2Cl2 (1:9). Анализ, вычисленный для С 19H30F3N3 O5 x x 0,75 H 2 O: C 50,83, H 6,62, N 9,39. Найдено: С 51,18, Н 7,00, N 9,28. Пример 39. Получение 3(RS)-1-(3-бензоиламино-1-оксопропил)-N -[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCONH(CH2)2, A = CO, n=1). а. Синтез 2(RS), 3(SR)-1-(3-бензоиламино-1-оксопропил)-N-[3-(1,1,1-трифтор-2-окси-4-метилпе-нтил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhCONH(CH2)2, A = C O, n=1). Используя методику примера 32а, 3-(бензоиламино)-пропановой кислоте позволяли реагировать с продуктом, полученным при использовании мето дики примера 2б, чтобы получить названный выше продукт (83%); ТС Х, Rf = 0,39 и 0,42, МеОН:CH2Cl2 (1:9). б. Синтез 3(RS)-1-(3-бензоиламино-1-оксопропил)-N-[3-(1 ,1,1 -три фтор -4-ме тил-2-оксопентил)]-Lпролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCONH(CH2)2, A = CO, n=1). Используя методику примера 61в, продукт по примеру 39а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CH2Cl2 (2:98)) названный выше продукт (48%); ТС Х, Rf = 0,54, MeOH:CH 2Cl2 (1:9). Анализ, вычисленный для С 21H26F 3N3O4 : C 57,14, H 5,94, N 9,52. Найдено: C 57,12, H 6,95, N 9,45. Пример 40. Получение 3(RS)-1-[3-(1-оксо-2,2-дифенилэтил)-амино]-1-оксобутил-N -[3-(1,1,1-трифтор4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = Ph2CHCONH(CH2)3, A = CO, n=1). а. Синтез эти лового эфи ра 3-(1-оксо-2,2-дифенилэтил)-аминобутановой кислоты. К перемешиваемому раствору хлоргидрата этилового эфи ра 4-аминобутановой кислоты (2,51 г) и дифе нилацетил хло рида (3,46 г) в 50 мл СH2Cl2 добавляли 50 мл воды, а затем добавляли в виде одной пор 19 39093 ции NaHCO3 (3,4 г). После 2 ч слои разделяли и органическую фазу вы сушивали (Na2SO4), фильтрова ли и выпаривали. Получали 3,5 г (72%) названного выше соединения в виде белого твердого вещества; ТСХ, Rf = 0,71, MeOH:CH 2Cl2 (5:95). б. Синтез 3-(1-оксо-2,2-дифенилэтил)-аминобутановой кислоты. Смесь продукта по примеру 40а (3,5 г) в 1N NaOH (30 мл) и EtOH (10 мл) перемешивали в те чение 10 ч. Раствор экстрагировали затем эфиром. Водную фазу подкисляли с помощью 1 N HCl; и выпавшее в осадок твердое вещество собирали, промывали водой и высушивали под высоким вакуумом. Получали 2,9 г (93%) названного вы ше соединения в ви де белого порошка. в. Синтез 2(RS), 3(SR)-1-[3-(1-оксо-2,2-дифенилэтил)]-амино-1-оксобутил-N -[3-(1,1,1-три-фтор-2окси-4-метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = Ph2CHCONH(CH2)3, A = CO, n=1). Используя методику примера 32а, продукту по примеру 40б позволяли реаги ровать с продуктом, полученным при использовании методики примера 2б, чтобы получить названный выше продукт (81%): ТСХ, Rf = 0,13, MeOH:CH 2Cl2 (5:95). г. Синтез 3(RS)-1-[3-(1-оксо-2,2-дифенил-этил)]-амино-1-оксобутил-N-[3-(1 ,1,1-три фтор-4-метил-2оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = Ph2CHCONH(CH2)3, A = CO, n=1). Используя методику примера 61в, продукт по примеру 40в окисляли, чтобы получить после очистки с помощью быстрой хроматографии (MeOH:CH2Cl2 (2:98)) названный выше продукт (27%); ТС Х, Rf = 0,24, MeOH:CH2Cl2 (5:95). Анализ, вычисленный для С 29H34F3N3 O4 x x 1,0 H 2 O: C 61,31, H 6,48, N 7,39. Найдено: С 61,58, H 6,77, N 7,43. Пример 41. Получение 3(RS)-1-[2-(2-ме-токсиэтокси)-этоксикарбонил] -N-3-(1,1,1-трифтор-4-метил-2оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = CH3O(CH2)2O(CH2)2 , A = OCO, n=1). а. Синтез 2(RS), 3(SR)-1-[2-метоксиэтокси)-этоксикарбонил]-N-[3-(1,1,1-трифтор-2-окси-4-ме-тилпентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = CH3O(CH2)2O(CH2)2 , A = OCO, n=1). Используя методику примера 7б, продукту по примеру 19а позволяли реагировать с продуктом, полученным при использовании методики примера 2б, чтобы получить после очистки с помощью сухой колоночной быстрой хроматографии над силикаге лем, используя градиентное элюирование со смесями ацетон:гексан от (10:90) до (50:50), названный выше продукт (75%); ТСХ, R1 = =0,30 и 0,35, ацетон:гексан (40:60). б. Синтез 3(RS)-1-[2-(2-метоксиэтокси)-эток-сикарбонил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = CH3O(CH2)2O(CH2)2 , A = OCO, n=1). Используя методику примера 61в, продукт по примеру 41а окисляли для получения очистки с помощью быстрой хроматографии (ацетон:CHCl3 (1:3) названного выше продукта (42%); ЖХВД, tR = =5,69, Col A, H2O:CH3CN (75:25), СП=2,0. Анализ, вычисленный для С 17Н27F 3N2O6 : C 49,39, H 6,83, N 6,78. Найдено: 49,27, 6,80, 6,48. Пример 42. По лучение 3(RS)-1-[1,4-диоксо-4-(фенилметиламино)-бутил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)3CH3, R3 = PhCH 2NHCO(CH2)2, A = CO, n=1). a. Синтез 4-оксо-4-(фенилметиламино)-бутановой кислоты. Смесь бензиламина (10,7 г) и янтарного ангидрида (10 г) перемешивали в ТГФ (1 л) в те чение 2 дней. Твердое вещество фильтровали и растворяли в 1 N NaOH (110 мл). Водную фа зу промыва ли эфиром, а затем подкисляли с концентрированной НСl, охлаждая при этом в бане с ледяной водой. Твердое вещество собирали, промывали во дой и высушивали под высоким вакуумом. Получали 10,9 г (53%) названное выше соединение в виде белого порошка; т.пл. 137,5–138оС. б. Синтез 2(RS), 3(SR)-1-[1,4-диоксо-4-(фенилметиламино)-бутил]-N-[3-(1,1,1-трифтор-2-окси-4метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhCH 2-NHCO(CH2)2, A = CO, n=1). Используя мето дику примера 32а, продукт по примеру 42а позволяли реагировать с продуктом, полученные при использовании методики примера 2б, что бы получить названное выше соединение (74%); ТСХ, Rf = 0,48, MeOH:CH 2Cl2 (1:9). в. Синтез 3(RS)-1-[1,4-диоксо-4-(фенилмети ламино)-бутил] -N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2NHCO(CH2)2, A = CO, n=1). Используя методику примера 61в, продукт примера 42б окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CH2Cl2 (2:98)) названный выше продукт (40%); ТС Х, Rf = 0,56, MeOH : CH 2Cl2 (1:9). Анализ, вычисленный для С 22H28F 3N3O4 : C 58,10, H 6,20, N 9,23. 20 39093 Найдено: С 57,80, Н 6,36, N 9,27. Пример 43. Получение 3(RS)-1-[1-оксо-3-(фен и лметоксик арбон иламин о)-пр опи л]-N -[3-(1,1,1трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)2, A = CO, n=1). а. Синтез 2(RS), 3(SR)-1-[1-оксо-3-(фенилметоксикарбониламино)-пропил] -N-[3-(1,1,1-трифтор-2окси-4-метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)2, A = CO, n=1). Используя методику примера 33а, КБЗ-бета-аланину позволяли реагировать с продуктом, полученным при использовании мето дики примера 2б, чтобы получить названный выше продукт (82%); ТСХ, Rf = 0,59, MeOH : CH 2Cl2 (1:9). б. Синтез 3(RS)-1-[1-оксо-3-(фенилметоксикарбониламино)-пропил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)2, A = CO, n=1). Используя методику примера 61в, продукт по примеру 43а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CH2Cl2 (2:98)) названный выше продукт (35%); TC Х, Rf = 0,23, МеОН:CH2Cl2 (5:95). Анализ, вычисленный для С 22H28F3N3 O5 x x 0,4 H 2 O: C 50,20, H 6,06, N 8,77. Найдено: C 55,28, H 6,24, N 8,55. Пример 44. Получение 3(RS)-1-[1-оксо-4-(фенилметоксикарбониламино)-бутил] -N-[3-(1,1,1-трифтор4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)3, A = CO, n=1). a. Синтез 2(RS), 3(SR)-1-[1-оксо-4-(фенилметоксикарбониламино)-бутил]-N-[3-(1 ,1,1 -три фтор-2окси-4-метилпентил)]-L-пролинамидa (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)3, A = CO, n=1). Используя методику примера 32а, КБЗ-4-аминобутановой кислоте позволяли реагировать с продуктом, полученным при использовании методики примера 2б, чтобы получить названный выше продукт (72%), ТСХ, Rf = 0,47, Et 2O : EtOAc (1:1). б. Синтез 3(RS)-1-[1-оксо-4-(фенилметоксикарбониламино)-бутил]-N -[3-(1,1 ,1-три фтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)3, A = CO, n=1). Используя методику примера 61в, продукт по примеру 44а окисляли, чтобы получить после очистки с помощью препаративных ТСХ (МеОН:CHCl3 (2,5:97,5), названный выше продукт (32%); ТС Х, Rf = 0,65 и 0,68. МеОH:CHCl 3 (1:9). Пример 45. Получение 3R (или S)-1-[1-оксо-4-фенокси-2-(2-феноксиэтил)-бутил]-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = [PhO(CH 2)2 ]CH, A = CO, n=1). а. Синтез диэтилового эфира 2,2-ди-(2-феноксиэтил)-малоновой кислоты. К перемешиваемому раствору натрия (2,3 г) в абсолютном EtOH (50 мл) добавляли диэтиловый эфир малоновой кислоты (15,2 мл), после чего добавляли 2-феноксиэтил хлористый (15,7 г). Реакционную смесь кипятили затем с обратным холодильником в течение 12 ч EtOH вы паривали под вакуумом и смесь разбавляли водой (40 мл). Водную фа зу экстрагировали эфи ром. Объединенные эфирные экстракты промывали (солевым раствором), высушивали (MgSO4), фильтровали и выпаривали. Перегонка из сосуда в сосуд приводит к 12,2 г (27%) названному выше сложному диэфиру в ви де прозрачной жидкости; т.кип. 155–175оС (106 Па 0,8 торр); ТСХ, Rf = 0 ,34, СН2Cl2. б. Синтез 4-фенокси-2-(2-феноксиэтил)-бута новой кислоты. Смесь продукта по примеру 45а (10,0 г) и гидроокиси калия (17,7 г) в воде (22 мл) кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали и подкисляли с помощью концентрированной НСl. Выпадающее в осадок твердое вещество собирали, промывали водой и высуши вали воздухом. Полученное твердое вещество (8,44 г) нагревали при 170оС в течение 2 ч, а затем охлаждали. Перекристаллизация твердого вещества и циклогексана дает 4,1 г (93%) названного выше соединения в виде тонких белых игл; т.пл. 85–86оС. Анализ, вычисленный для С 18Н20О 4: С 71,98, Н 6,71. Найдено: С 71,92, Н 6,71. в. Синтез 4-фенокси-2-(2-феноксиэтил)-бутаноила хло ристого. Смесь продукта по примеру 45б (1,5 г) и хло ристого тионила (0,73 мл) нагревали на паровой бане в течение 1 ч. Реакционную смесь затем откачивают. Названный выше продукт - хлорангидрид кислоты – получали в ви де прозрачного масла с количественным выхо дом и был использован непосредственно. г. Синтез 2(RS), 3(SR)-1-[1-оксо-4-фенокси-2-(2-феноксиэтил)-бутил]-N-[3-(1,1,1-трифтор-2-ок-си-4метилпентил)]-L-пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = [PhO(CH 2)2 ]2CH, A = CO, n=1). Используя методику примера 37а, продукту по примеру 45в позволяли реагировать с продуктом, полученным при использовании методики примера 2б, чтобы получить названный выше продукт (95%, ТСХ, Rf = 0,47 и 0,54, Et2O. д. Синтез 3R (или S)-1-[1-оксо-4-фенокси-2-(2-феноксиэтил)-бутил]-N-[3-(1,1 ,1-три фтор-4-метил-2оксопентил)]-L-пролинамида 21 39093 (формула Ia, R1 = CH(CH3)CH3, R3 = [PhO(CH 2)2]CH, A = CO, n=1). Используя методику примера 61в, продукт по примеру 45г окисляли, чтобы получить сырой продукт в виде смеси диастереомеров, кото рую разделяли с помощью быстрой хроматографии (эфир:гексан, градиентное элюирование, от 60:40 до 75:25). Более быстро элюирующийся диастереомер является названным выше соединением, полученным с 27,7%-ным выхо дом; ЖХВД, tR = =6,94, Col A, CH3СN : H2O (65:35), СП = 2,0. Анализ, вычисленный для С 29H35F 3N2O5 : C 63,49, H 6,43, N 5,11. Найдено С 63,39, H 6,47, N 5,07. Пример 46. Получение 3S (или R)-1-[1-оксо-4-фенокси-2-(2-феноксиэтил)-бутил]-N-[3-(1 ,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = [PhO(CH 2)2]CH, A = CO, n=1). Из данных по разделению диастереомеров из сырого продукта, описанного в примере 45д, более медленно элюирующий диасте реомер является названным выше соединением данного примера, полученным с 28,3%-ным выхо дом; ЖХВД, tR = 5,04, Col A, СH3CN : H 2 O (65:35), СП = =2,0. Анализ, вычисленный для С 29H35F 3N2O5 : C 63,49, H 6,43, N 5,11. Найдено: C 63,50, Н 6,45, N 5,26. Пример 47. Получение 3(RS)-1-[6-[(4-метоксикарбонилфе нил)-аминокарбониламино] -1-оксо]-гексилN-[3 -(1 ,1 ,1-три фтор-4-ме ти л-2-оксо пентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = 4-(CH3CH2OCO)-PhNHCONH(CH2)5, A = CO, n=1). а. Синтез 2(RS), 3(SR)-1-[6-[(4-этоксикарбонилфе нил)-аминокарбониламино]-1-оксо] -гексил-N-[3(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIa, R1 = CH(CH3)CH3, R3 = 4-(CH3CH2OCO)-PhNHCONH(CH2)5, A = CO, n=1). Этиловый эфир параизоцианатобензойной кислоты (0,288 г, 1,5 ммоль) добавляли к перемешиваемому раствору продукта по примеру 50б (0,6 г, 1,5 ммоль), ТЭА (0,15 г, 1,5 ммоль) и ДМФА (20 мл) в атмосфе ре азота при комнатной температуре. Получающуюся смесь перемешивали при комнатной температуре в течение ночи, после чего ее концентрировали под вакуумом, получая янтарный остаток, который растворяли в эти лацета те. Этилацетатный раствор промывали (1N HCl), высушивали (MgSO 4), фильтровали и концентрировали под вакуумом, получая 1,15 г маслянистого остатка. Этот остаток очища ли с помощью быстрой хроматографии (СНCl3 : CH3OH (97:3)), чтобы получить 0,62 г (70%) названного выше соединения в ви де белого твер дого ве щества; ТC Х, Rf = =0,28 и 0,35, СНСl3: CH3OH (95:5). б. Синтез 3(RS)-1-[6-[(4-этоксикарбонилфе нил)-аминокарбониламино]-1-оксо]-гек сил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = 4-(CH3CH2OCO)PhNHCONH(CH2)5, A = CO, n=1). Используя методику примера 61в, продукт по примеру 47а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (СНСl3 : MeOH (97:3)) названное выше соединение (43%); ЖХВД, tR = 12,39 и 15,79, Сol A, H 2 O : :CH 3CN (65:35), СП = 2,0. Анализ, вычисленный для С 27H37F3N4 O6 x x 1,0 H 2 O: C 55,09, H 6,68, N 9,51. Найдено: C 54,75, H 6,63, N 9,29. Пример 48. Получение 3(RS)-1-[6-(фенилметоксикарбониламино)-1-оксогексил] -N-[3-(1,1,1-трифтор4-метил-2-оксопентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)5, A = CO, n=1). Используя методику примера 31в, продукт по примеру 50а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (CHCl3 : MeOH (97:3)) названный выше продукт (31%); ЖХВД, tR = 4,06, Сol A, CH3CN : H 2 O (1:1), CП = 2,0. Анализ, вычисленный для С 25H34F3N3 O5 x x 0,5 H 2 O: C 57,46, H 6,75, N 8,04. Найдено: С 57,87, H 6,24, N 7,86. Пример 49. Получение 3(RS)-1-[6-[(4-оксикарбонилфенил)-аминокарбониламино]-1-оксогек-сил]-N-[3(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = 4-(HOCO)-PhNHCONH(CH 2)5, A = CO, n=1). Используя методику примера 14а, продукт по примеру 47б превращали в названный выше продукт с 38%-ным выхо дом; ЖХВД, tR = 6,24 и 8,00, Сol A, H 2O : CH3CN (75:25), СП = 2,0. Анализ, вычисленный для С 25Н33F3N4 O6 x x 2,5 H 2 O: C 51,1, H 6,50, N 9,50. Найдено: C 51,34, H 5,93, N 8,95. Пример 50. Получение 3(RS)-1-(6-фенилсульфониламино-1-оксогексил)-N-[3-(1,1,1-трифтор-4-метил2-оксопентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhS(O2)NH(CH2)5, A = CO, n=1). Синтез 2(RS), 3(SR)-1-[6-(фенилметоксикарбониламино)-1)-оксогексил] -N-[3-(1,1,1-трифтор-2-окси-4метилпентил)]-L -пролинамида 22 39093 (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)5, A = CO, n=1). ДЦК (6,35 г, 30,8 ммоль) добавляли к перемеши ваемому раствору N-КБЗ-аминокапроновой кислоты (6,84 г, 25,7 ммоль), продукта, полученного по методике примера 2б (6,89 г, 25,7 ммоль), ОБТА (6,94 г, 51,4 ммоль) и сухо го ТГФ (250 мл) при 0оС в атмосфе ре азота. По лучающуюся реакционную смесь перемеши вания при 0оС в течение 1 ч, допускали к нагреванию до комнатной температуры и перемеши вали в течение ночи, после чего ее фильтровали. Фильтрат концентрировали под вакуумом до образования коричневого остатка, который раство ряли в СНСl3, и хлороформный раствор промывали (20%-ным раствором лимоннойкислоты), высуши вали (Na2SO4), фильтровали и концентрировали под вакуумом. Оста ток очищали и с помощью быстрой хроматографии (СHCl3 : MeOH (97:3)), чтобы получить 8,0 г (61%) названного выше соединения в виде воскообразного твер дого ве щества; ТС Х, Rf = 0,35, СНСl3 : MeOH (95:5). б. Синтез 2 (RS), 3(SR)-1-(6-амино-1-оксогексил)-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L -пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = H2N(CH2)5, A = CO, n=1). Смесь продук та по примеру 50а (2,06 г, 3,99 ммоль), EtOH (100 мл) и 10%-ного Pd/C (0,3 г) загружали в трясучку Парра под давлением (45 psi) H2 в те чение 3 ч. Смесь фильтровали через целит и спекшуюся массу на целите промывали с EtOH. Спиртовые промывки и указанный выше фильтрат объединяли и концентрировали под вакуумом, чтобы получить 1,36 г (86%) названного вы ше соединения в виде бледно-зеленого воскообразного масла; ТС Х, Rf = 0,2, СHСl3:MeOH (85:15). в. Синтез 2(RS), 3(SR)-1-(6-фенилсуль фониламино-1-оксогексил)-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L -пролинамида (формула VIIa, R 1 = CH(CH3)CH3, R3 = PhS(O2)NH-(CH2)5, A = CO, n=1). Бензолсульфонилхлористый (0,6 г, 1,5 ммоль) добавляли к перемешиваемому раствору продукта по примеру 50б (0,26 г, 1,5 ммоль), ТЭА (0,3 г, 3,0 ммоль) и сухого ДМФА (20 мл) в атмосфере азота при комнатной температуре и получающуюся смесь перемешивали при комнатной температуре в течение ночи. ДМФА удаляли под вакуумом до образования коричневого остатка, который растворяли в этилацетате. Этилацетатный раствор промывали (1 N HCl), высуши вали (MgSO 4) и фильтровали. Фильтрат концентрировали под вакуумом до образо вания остатка, который очищали с помощью быстрой хроматографии (СHCl 3 : MeOH (97:3)), чтобы получить 0,48 г (60%) названного выше соединения в виде белого порошка; ТСХ, Rf = 0,30 и 0,40 CHCl 3 : MeOH (95:5). г. Синтез 3(RS)-1-(6-фенилсульфо ниламино-1-оксогексил) -N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Ia, R1 = CH(CH3)CH3, R3 = PhS(O2)NH(CH2)5, A = CO, n=1). Используя методику примера 8б, продукт по примеру 50в окисляли, чтобы получить после очистки с помощью быстрой хроматографии (Et 2O:гексан (3:1)), после чего на второй колонке использовали СНСl3 : MeOH (97:3)), названный выше продукт (36%); ЖХВД, tR = 8,48 и 10,33, СН3CN : H 2O (35:65), СП = 2,0. Пример 51. Получение 3(RS)-(1-нафтилкарбонил)-L-валил -N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iб, R1 = CH(CH3)CH3, R3 =СН(CH3)2, R3 = 1-нафтил, R4 = H, A = CO, n=1). а. Синтез 2(RS), 3(SR)-(1-нафтилкарбонил)-L-валил-N-[3-(1,1 ,1-три фтор-2-окси-4-ме тилпентил)]-L пролинамида (формула VIIб, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 1-нафтил, R4 = H, A = CO, n=1). Согласно методике примера 20а вещество, полученное по методике примера 3 г, допускали к взаимодействию с 1-нафталинкарбонилом хлористым, что бы получить названное выше соединение, выделенное с 38%-ным выхо дом после очистки с помощью препаративной ТСХ (гексан:Et2O (4:6)); ТС Х, Rf = 0,46 и 0,41, МеОН : CHCl 3 (5:95). Анализ, вычисленный для С 27H32F 3N3O4 : C 60,32, H 6,37, N 7,82. Найдено: С 60,89, Н 6,21, N 7,68. б. Синтез 3(RS)-(1-нафтилкарбонил)-L-валил -N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iб, R1 = CH(CH3)CH3, R2 = СH(CH3)2, R3 = 1-нафтил, R4 = H, A = CO, n=1). Раствор ДМСО (29,7 г, 380 ммоль) в СН2Сl2 (81 мл) медленно добавляли к предварительно охлажденному раствору (-43о) хло ристого оксалила (24,0 г, 190 ммоль) в СН2Сl2 (350 мл) и получающийся раствор перемешивали в течение 15 мин, после чего раствор продукта по примеру 51б (9,4 ммоль) в СН2Cl2 (83 мл) добавляли в смесь. После перемешивания реакционной смеси в те чение 1 ч при -30оС по каплям добавляли диизопропилэтиламин (48,9 г, 380 ммоль) и реакционной смеси позволяли нагреваться до комнатной температуры, после чего ее промывали (1 N HCl, 5%-ным водным NaOCl, солевым раствором), высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (гексан : Et2O (40:60)), чтобы получить названный выше продукт (38%); ЖХВД, tR = 5,21 и 7,31, Col A, СН3CN : H 2 : ТФУК (50:50:1), СП = 1,5. Анализ, вычисленный для С 27H32F 3N3O4 x х0,75 H2O: C60,32, H 6,37, N 7,82. Найдено: С 60,69, Н 6,37, N 7,68. Пример 52. Получение 3(RS)-[4-(метилсульфо ниламинокарбонил)-фениламинокарбонил] -L-валилN-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iб, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 4-[CH 3S(O2) NHCO]-Ph, R 4 = H, A = NHCO, n=1). Используя методику примера 89, соединение, полученное по методике примера 16, подвергали взаимодействию с метансульфо намидом, чтобы получить после очистки с помощью быстрой хроматографии 23 39093 над силикагелем Бейкера с рН 5,0 (градиент, СНСl3 : MeOH (97:3) – (90:10)) названный выше продукт (59%), ЖХВД, tR = 2,60 и 3,33 Сol C, H 2O : CH3CN (60:40), СП = 6.0. Анализ, вычисленный для С 25Н34F3N5 O7S x x 0,5 H 2 O: C 48,85, H 5,74, N 11,39. Найдено: С 49,03, Н 5,74, N 10,36. Пример 53. Получение 3(RS)-[2-(4-морфо линил)-этоксикарбонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 4-морфолинил-(CH2)2, R4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-[2-(4-морфолинил)-этоксикарбонил]-L-валил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L -пролинамида (формула VIIб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 4-морфолинил-CH2(CH3)2, R4 = H, A = OCO, n=1). Используя методику примера 7б, соединению, полученному по методике примера 3г, позволяли реагировать с соединением, полученным по методике примера 33а, чтобы получить названное выше соединение, выделенное с 55%-ным выходом после очистки с помощью быстрой хроматографии (МеОН : CHCl3 (2,5 : 97,5)); ЖХВД, tR = 4,62 и 5,85, Col A, CH 3CN : H 2O (1:1), СП = 2,0. Анализ, вычисленный для С 23Н39F3N4 O6 x x H2O: C 50,91, H 7,61, N 10,32. Найдено: С 50,95, Н 7,20, N 10,02. б. Синтез 3(RS)-[2-(4-морфо линил)-этокси-карбонил]-L- валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 4-морфолинил-CH2СН2, R4 = H, A= OCO, n=1). Раствор ДМСО (29,7 г, 380 ммоль) в СН2Сl2 (135 мл) добавляли к предварительно охлажденному (43оС) раствору хлористого оксалила (24,0 г, 190 ммоль) в СН2Cl2 (350 мл) и получающий ся раствор перемешивали в течение 15 мин, после чего добавляли раствор продукта по примеру 53а (9,4 ммоль) в СH2Сl2 (125 мл). Реакционной смеси позволяли нагреваться от -43оС до -20оС во время перемешивания ее в течение 1 ч. Затем по каплям добавляли диизопропилэтиламин (48,9 г, 380 ммоль) и реакционную смесь подвергали нагреванию до комнатной температуры, после чего ее разбавляли с СН 2Cl2, промывали (водный NaOH, pH 10), высушивали (К2СО3/Na2SO4) и концентрировали под вакуумом. Остаток очища ли с помощью быстрой хроматографии (МеОН:CHCl3 (2:98)), что бы получить названный выше продукт (18%); ЖХВД, tR = 2,00 и 2,60, Col A, H 2O : CH 3CN (1:1), СП = 2. Анализ, вычисленный для С23Н37F3N4 O6 x x 1,5 H 2 O: C 50,26, H 7,34, N 10,19. Найдено: C 50,49, H 6,96, N 9,96. Пример 54. Получение 3(RS)-[(2,4-дихлорфенил)-карбонил)]-L- валил-N-[3-(1,1,1-трифтор-4-метил-2оксопентил)]-L -пролинамида (формула Iб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2,4-дихлорфе нил, R4 = H, A = CO, n=1). а. Синтез 2(RS), 3(SR)-[(2,4-дихлорфенил)-карбонил)]-L-валил-N-[3-(1,1,1-трифтор-2-оксо-4-метилпентил)]-L -пролинамида (формула VIIб, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2,4-дихлорфенил, R4 = H, A = CO, n=1). Используя мето дику примера 20а, соединение, полученное по методике примера 3г, подвергали взаимодействию с 2,4-ди хлорбензилом хлористым, чтобы получить названное выше соединение, выделенное с 98% выхо дом; ТСХ, Rf = 0,54, МеОН:CHCl 3 (5:95). б. Синтез 3(RS)-[(2,4-дихлорфе нил)-карбонил)]-N- валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]L -пролинамида (формула Iб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2,4-дихлорфе нил, R4 = H, A = CO, n=1). Раствор ДМСО (29,7 г, 380 ммоль) в СН2Сl2 (27 мл) медленно добавляли к предварительно охлажденному раствору (-65оС) хло ристого оксалила (24,0 г, 190 ммоль) в СН2Сl2 (350 мл) и получающий ся раствор перемешивали в течение 15 мин, после чего добавляли раствор продукта, полученного по методике примера 54а (9,4 ммоль) в СН2Сl2 (250 мл). После перемешивания реакционной смеси в те чение 1 ч при 65оС по каплям добавляли диизопропилэтиламин (48,9 г, 380 ммоль). Реакционной смеси позволили нагреваться до комнатной температуры, после чего ее промывали (1N HCl, солевым раствором), высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали с помощью быстрой хроматографии (МеОН:CHCl3 (3:97)), чтобы получить названный выше продукт (15%); ЖХВД, tR = =17,93 и 18,55, Col A, H2O:CH3CN (55:45), СП = 2,0. Анализ, вычисленный для С 23Н28Cl2F3N3 O4 x xH2O: С 49,65, H 5,43, N 7,55 . Найдено: C 49,95, Н 5,31, N 7,35. Пример 55. Получение 3(RS)-феноксикарбонил-L- валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]L -пролинамида (формула Iб, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = Ph, R 4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-феноксикарбонил-L- валил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L -пролинамида (формула VIIб, R1 = CH(CH3)CH3, R2 = (CH(CH), R 3 = Ph, R 4 = H, A = OCO, n=1). Используя методику примера 20а, соединению, полученному при методике примера 3г, позволяли реагировать с фениловым эфиром хлормуравьиной кислоты, что бы получить названное выше соединение, выделяемое с 61%-ным выхо дом после очистки с помощью быстрой хроматографии (МеОН : CHCl3 (5:95); ТСХ, Rf = 0,31 и 0,36 МеОН : CHCl 3 (3:97). 24 39093 мида б. Синтез 3(RS)-феноксикарбонил-L- валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролина (формула Iб, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = Ph, R 4 = H, A = OCO, n=1). Используя методику примера 54б, продукт по примеру 55а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (гексан:Et2O) (15:85)) названный выше продукт (37%): tR = 2,72 и 3,55, Col A, H 2O : CH 3CN (1:1), СП = 2,0. Анализ, вычисленный для С 23Н30F3N3 O5 x x 0,5 H 2 O: C 55,86, H 6,32, N 8,50. Найдено: C 56,07, H 6,30, N 8,48. Пример 56. Получение 3(RS)-[2-(2-пиридил)-этоксикарбонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2оксопентил)]-L-пролинамида (формула Iб, R 1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2-пиридил-(СН2)2, R4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-[2-(2-пиридил)-эток-сикарбонил]-L- валил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2-пиридил-СН 2CH2, R4 = H, A = О, n=1). Используя методику примера 34б, соединению, полученному по методике примера 3г, позволяли реагировать с соединением, полученным по методике примера 34а, чтобы получить названное выше соединение, выделенное с 50%-ным выхо дом после очистки с помощью быстрой хроматографии (МеОН:CHCl3) (4:96)); ТС Х, Rf = 0,30 и 0,34, СНСl3 : MeOH (95:5). б. Синтез 3(RS)-[2-(2-пиридил)-этоксикар-бонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2-пиридил-СН 2СН2, R4 = H, A = OCO, n=1). Используя методику примера 54б, за исключением промывки кислотой, продукт по примеру 56а окисляли, чтобы получить после очистки с помощью быстрой хроматографии дважды (гексан:эфир (1:1)), затем МеОН:CHCl3 (5:95)), названный выше продукт (19%); ЖХВД, tR = 9,52 и 14,58, Соl A, H2 O: CH3CN (60:40), CП = 1,0. Анализ, вычисленный для С 24Н33F3N4 O5 x x 0,75 H 2 O : C 54,59, H 6,58, N 10,61. Найдено: C 54,63, H 6,47, N 10,55. Пример 57. Получение 3(RS)-[(4-фторфе нил)-аминокарбонил)]-L-ва лил-N-[3-(1 ,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R 3 = 4-FPh, R 4 = Н, A = NHCO, n=1). а. Синтез 2(RS),3(SR)-[(4-фторфенил)-аминокарбонил)]-L-валил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L -пролинамида (формула VIIв, R1 = CH(CH3)CH, R 2 = CH(CH3)2, R3 = 4-FPh, R 4 = Н, A = NHCO, n=1). Используя мето дику примера 15а, соединение, полученное по методике примера 3г, подвергали взаимодействию с 4-фторфе нилизоцианатом, чтобы получить названное выше соединение, выделяемое с 84%-ным выхо дом после очистки с помощью быстрой хроматографии (градиент, МеОН:CHCl3 от (2,5:97,5) до (5:95); ТС Х, Rf = 0,37, МеОН : СНСl3 (5:95). б. Синтез 3(RS)-[(4-фторфенил)-аминокарбонил)]-L- валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R 3 = 4-FPh, R 4 = Н, A = NHCO, n=1). Используя методику примера 54б, продукт по примеру 57а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CHCl3 (3:97)) названный выше продукт (42%); ЖХВД, tR = 8,87 и 12,10, Сol A, H2O : CH3CN (60:40), СП = 1,0. Анализ, полученный для С 23Н30F 4N4O4 : C 54,97, H 6,02, N 11,15. Найдено: С 55,18, Н 6,15, N 11,08. Пример 58. Получение 3(RS)-[4-(фенилсульфониламинокарбонил)-фениламинокарбонил]-L-валил-N-[3(1,1,1-трифтор -4-ме тил-2-ок сопенти л)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 4-[PhS(O 2)NHCO]Ph, R 4 = Н, A = NHCO, n=1). Используя методику примера 89, соединению, полученному по методике примера 16, позволяли реагировать с бензолсульфо намидом, чтобы получить после очистки с помощью быстрой хроматографии на силикагеле Бейкера с рН 5,0 (СНСl3:MeOH (97:3)) названный выше продукт (42%); ЖХВД, tR = 4,05 и 5,93, Сol H2O : CH3CN (60:40), CП = 6,0. Анализ, вычисленный для С 30Н36F3N5 O7 x x 0,5 H 2 O: C 53,25, H 5,51, N 10,34. Найдено: C 53,38, H 5,61, N 10,02. Пример 59. Получение 3(RS)-[2-(3-тиофенил)-этоксикарбонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 3-тио фенил-СН 2СН2, R4 = Н, A = ОCO, n=1). а. Синтез 4-нитрофенил-2-(3-тиофенил)-этилкарбоната. Используя методику примера 7а, 3-тиофенэтанол обрабатывали с 4-нитрофе ниловым эфиром хлормуравьиной кислоты, что бы получить после очистки с помощью быстрой хроматографии (EtOAc:гексан (1:9)) названный выше продукт (56%); ТС Х, Rf = 0,25 Et 2Oc : гексан (1:9). 25 39093 б. Синтез 2(RS), 3(SR)-[2-(3-тиофе нил)-этоксикарбонил]-L-валил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH(CH 3)2, R3 = 3-тиофенил-СН 2СН2, R4 = Н, A = О, n=1). Используя методику примера 7б, соединению, полученному по методике примера 3г, позволяли реагировать с продуктом по примеру 59а, что бы получить названное выше соединение, выделенное с 58%ным выхо дом после очистки с помощью быстрой хроматографии (ацетон:гексaн (3:7)); ТС Х, Rf = 0,23 и 0,27, МеОН:CHCl 3 (5:95). Анализ, вычисленный для С 24Н34F3N3 O5S : C 52,96, H 6,57, N 8,06. Найдено: C 53,28, H 6,46, N 7,77. в. Синтез 3(RS)-[2-(3-тиофе нил)-этоксикар-бонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 3-тио фенил-СН 2СН2, A = ОCO, n=1, R4 = Н). Раствор ДМСО (29,7 г, 380 ммоль) в СН2Сl2 (135 мл) медленно добавляли в предварительно охлажденный (-43оС) раствор хлористого оксалила (24,0 г, 190 ммоль) в СН2Cl2 (350 мл). Получающийся раствор перемеши вали 15 мин, после чего добавляли раствор соединения, полученного по мето дике примера 59б (9,4 ммоль) в СН2Сl2 (125 мл) и реакционную смесь перемешивали при -43оС в течение еще 1 ч. Диизопропилэтиламин (48,9 г, 380 ммоль) добавляли по каплям и реакционной смеси позволяли нагреваться до комнатной температуры, после чего ее промывали (1N водным НСl, 5%ным водным NaOCl, солевым раствором), высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Очистка остатка с помощью быстрой хроматографии (ацетон:гексан (1:4)) приводит к названному выше продукту (23%); ЖХВД, tR = 5,09 и 7,61, Сol A, H 2O : CH 3CN (1:1), СП = 2,0. Анализ, вычисленный для С 24H32F 3N3O5S: C 53,17, H 6,21, N 8,09. Найдено: C 52,92, H 6,26, N 8,09. Пример 60. Получение 3(RS)-[(1,1-диме-тилэтоксикарбонил)]- L-альфа-аминобутирил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]- L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = СН3С(СН3)2, R4 = Н, A = ОCO, n=1). а. Синтез 2(RS), 3(SR)-[(1,1-диметилэтоксикарбонил)]-L-альфа-аминобутирил-N-[3-(1,1,1-три-фтор-2окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = СН3С(СН3)2 , R4 = Н, A = ОCO, n=1). ДЦК (5,90 г, 28,7 ммоль) добавляли к перемеши ваемому раствору ОБТА (7,76 г, 57,4 ммоль), БОКальфа-аминобутановой кислоты (5,60 г, 27,4 ммоль), и соединения, полученного по мето дике примера 2б (7,00 г, 26,1 ммоль) в сухом ТГФ (130 мл) при 0оС в атмосфере азота. После перемеши вания реакционной смеси при 0оС в течение 1 ч ей позволяли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали под вакуумом до образования остатка, кото рый повторно растворяли в этилацетате. Получающийся раствор промывали насыщенным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4), фильтровали и концентрировали под вакуумом до образования остатка, который очища ли с помощью быстрой хроматографии (EtOAc : :CH2Cl2 (1:3)), чтобы получить названное выше соединение (95%); ТСХ, Rf = 0,29, EtOAc : CH 2Cl2 (3:7). б. Синтез 3(SR)-(1,1-диметилэтоксикарбонил)-L-альфа-аминобути рил-N-[1,1,1-трифтор-4-метил-2-оксопентил)]- L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = СН3С(СН3)2, R4 = Н, A = ОCO, n=1). Используя методику примера 61в, продукт по примеру 60а, окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CHCl3) (1,5:98,5) названный выше продукт (47%); ЖХВД, tR = 10,21 и 14,54, Сol A, H2O: :CH3CN (65:35), СП = 2,0. Анализ, вычисленный для С 20H32F 3N3O5 : C 53,21, H 7,14, N 9,31. Найдено: C 53,65, H 7,21, N 9,51. Пример 61. Получение 3(RS)-[1-оксо-2-(2-тиофе н и л)-эти л]-L -а льфа -ам инобутир ил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]- L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = (2-тио фенил)-СН3, R4 = Н, A = CO, n=1). а. Синтез 2(RS), 3(SR)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида, соль трифто руксусной кислоты (формула IVв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R4 = Н, n=1). Раствор соединения, полученного по методике примера 60а, (4,0 г, 8,84 ммоль) и ТФУК (32 мл, 415 ммоль) в СН2Cl2 (32 мл) перемеши вали при комнатной температуре в течение 22 ч, после чего растворители удаляли при пониженном давлении, чтобы получить сырой продукт (5 г, более 100%) в виде бесцветной стеклообразной массы, которую использовали без дальнейшей очистки или идентификации. б. Синтез 2(RS), 3(SR)-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)] -L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = (2-тиофе нил)-СН2, R4 = H, A = CO, n=1). ДЦК (0,315 г, 1,53 ммоль) добавляли к перемеши ваемому раствору ОБТА (0,413 г, 3,06 ммоль) 2-тиофе нуксусной кислоты (0,222 г, 1,53 ммоль), NMM (0,154 г, 1,53 ммоль) и соединения, полученного по методике примера 61а (0,650 г, 1,39 ммоль) в сухом ТГФ (20 мл) при 0 оС в атмосфе ре азота. После перемешивания реакционной смеси при 0оС в те чение 1 ч ей позволяли нагреваться до комнатной температуры и перемеши вали в течение ночи. Реакционную смесь фильтрова ли и фильтрат концентрировали под вакуумом 26 39093 до образования остатка, который растворяли в этилацетате. Получающийся раствор промывали (солевым раствором), высуши вали (Na2SO4), фильтровали и концентрировали под вакуумом до образования остатка, который очищали с помощью быстрой хроматографии (ацетон:гексан (1:1)), чтобы получить названное выше соединение (33%); ТСХ, Rf = 0,40 и 0,44, МеОН : CHCl 3 (1:9). Анализ, вычисленный для С 21Н30F 3N3O4 : C 52,82, H 6,33, N 8,80. Найдено: C 52,43, H 6,53, N 8,08. в. Синтез 3(RS)-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = (2-тио фенил)-СН2, R4 = H, A = CO, n=1). К суспензии СrO3 (0,84 г, 8,4 ммоль) в сухом СН2Cl2 (50 мл) до бавляли сухой пиридин (1,36 мл, 17 ммоль) и смесь перемеши вали при комнатной температуре в течение 30 мин. К получающей ся суспензии, окрашенной в бургун ди-цвет (темно-красный), добавляли 1 г целита, за кото рым следуе т добавление продукта по примеру 61б (0,20 г, 0,42 ммоль) в СН2Cl2 (5 мл). Смесь перемешивали до те х пор, пока ТСХ не будет показывать, что весь спирт израсхо дован. Смесь фильтровали затем через слой силикагеля с помощью смеси метанол:хлороформ (1:9) и растворите ли, уда ляли из фильтрата под вакуумом. Сырой продукт очи ща ли с помощью препаративной ТСХ (Ме ОН:CHCl3 (5:95)), что бы получить продукт (150 мг) в ви де белого твер дого ве щества; ЖХВД, tR = 4,18 и 5,65, Col A, H 2O : CH 3CN (60:40), СП = 2 ,0. Анализ, вычисленный для С 21Н28F3N3 O4S x x 0,5 H 2 O: C 52,06, H 6,03, N 8,67. Найдено: С 52,03, Н 6,19, N 8,38. Пример 62. Получение 3(RS)-(фенилметоксикарбонил)-L-альфа-аминобути рил-N-[3 -(1,1 ,1-трифтор4-метил-2-оксопентил)] -L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = PhCH 2, R 4 = H, A = OCO, n=1). а. Cинтез 2(RS), 3(SR)-(фенилметоксикарбонил)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-2-окси-4метилпентил)] -N-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = PhCH 2, R4 = H, A = OCO, n=1). Используя методику примера 20а, соединению, полученному по методике примера 61а, позволяют реагировать с 1,5 эквивалента ми бензилового эфира хлормуравьиной кислоты в одной четвертой части количества СНСl3, используемого в методике примера 20а, чтобы получить названное выше соединение, выделяемое с 51%-ным выхо дом после очистки с помощью быстрой хроматографии (ацетон:гексан (1:4)); ТСХ, Rf = 0,38 и 0,43, ацетон:гексан (40:60). б. Синтез 3(RS)-(фенилметоксикарбонил)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)] -L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = PhCH 2, R 4 = H, A = OCO, n=1). Используя методику примера 61в, продукт по примеру 62а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (ацетон:гексан (30:70)), названный выше продукт (64%); ЖХBД, tR = 5,69 и 7,75, Col A, H 2 O : CH 3CN (55:45), СП = 2,0. Анализ, вычисленный для С 23Н30F3N3 O5 x x 0,8 H 2 O: C 55,26, H 6,37, N 8,40. Найдено: C 55,10, H 6,19, N 8,77. Пример 63. По лучение 3(RS)-(феноксикарбонил)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)] -L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = Ph, R 4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-(фе ноксикарбонил)-L-альфа-аминобути рил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)] -L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = Ph, R 4 = H, A = OCO, n=1). Используя методику примера 20а, но используя NMM вместо ТЭА, сое динению, полученному по методике примера 61а, позволяли реагировать с 1,5 эквивалента ми фенилового эфи ра хлормуравьиной кислоты, что бы получить названное выше соединение, выделяемое с 18%-ным выходом после очистки с помощью препаративной ТСХ (ацетон:гексан (3:7)); ТСХ, Rf = 0,32 и 0,37, ацетон:гексан 3:7). б. Синтез 3(RS)-(феноксикарбонил)-L-альфа-аминобути рил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)] -L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = Ph, R 4 = H, A = OCO, n=1). Используя методику примера 61в, продукт по примеру 63а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (EtOAc : гексан (1:9)), названный выше продукт (60%); ЖХВД, tR = 4,57 и 6,51, Сol A, H 2 O : CH 3CN (55:45). Анализ, вычисленный для С 22Н28F 3N3O5 : C 56,04, H 5,98, N 8,90. Найдено: C 56,04, H 6,18, N 8,85. Пример 64. Получение 3(RS)-[4-(1-оксоэтиламино)-фенилсульфони л]-L -ва лил-N-[3-(1 ,1,1-трифтор4-метил-2-оксопентил)]-N-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)CH3, R3 = 4-(CH 3CONH)-Ph, R 4 = H, A = S(O2), n=1). а. Синтез 2(RS), 3(SR)-[4-(1-оксоэтиламино)-фенилсульфонил]-L-валил-N-[3-(1,1,1-трифтор-2-окси-4метилпентил)]- L -пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH(CH3)CH3, R3 = 4-(CH3CONH)-Ph, R 4 = H, A = S(O 2), n=1). 27 39093 К перемеши ваемому раствору соединения, полученного по мето дике примера 3г (1,00 г, 2,72 ммоль) и NMM (0,28 г, 0,30 мл, 2,80 ммоль) в СН2Cl2 (50 мл) в атмосфере азота добавляли 4-ацетамидобензолсульфо нилхлорид (0,64 г, 2,72 ммоль). После перемеши вания реакционной смеси в течение ночи при комнатной температуре раствор промывали (5%-ным водным раствором лимонной кислоты, со левым раствором), высушивали (Na2SO4), фильтровали и концентрировали до образования сиропа оранжевого цвета, который подвергали быстрому хроматографи рованию над силикагелем (350 г), (МеОН : CHCl3 (7:93)), чтобы получить названный выше продукт (810 мг, 52,6%); ТС Х, Rf = 0,34 и 0,45, МеОН :CHCl 3 (7:93). б. Синтез 3(RS)-[4-(1-оксоэтиламино)-фе-нилсульфонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-N-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)CH3, R3 = 4-(CH 3CONH)Ph, R 4 = H, A = S(O2), n=1). Используя методику примера 31в, продукт по примеру 64а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CHCl3 (5:95)) названный выше продукт (84%); ЖХВД, tR = 3,22 и 4,58, Сol A, CH 3CN : H 2O (40:60), СП = 2,0. Анализ, вычисленный для С 24Н33F3N4 O6 x x 5 H2 O: C 49,65, H 6,08, N 9,65. Найдено: C 49,73, H 5,86, N 9,53. Пример 65. Получение 3(RS)-N 2-(1,1-диметилэтоксикарбонил)-N6-фенилметоксикарбонил-L-лизил-N[3-(1,1 ,1-три фтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R3 = PhCH 2OCONH(CH2)4, R2 = C(CH3)3, R4 = H, A = OCO, n=1) а. Синтез 2(RS), 3(SR)-N 2-(1,1-диметилэтоксикарбонил)-N6-фенилметоксикарбонил-L-лизил-N-[3(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = PhCH 2OCONH(CH2)4, R3 = C(CH3)3С, R4 = H, A = OCO, n=1). Используя методику примера 84б, N2-БОК-N6-КБЗ-L-лизину позволяли реагировать с соединением, полученным по методике примера 2б, чтобы получить после очистки с помощью быстрой хроматографии (градиентное элюирование, МеОН:CHCl 3 от (2,5:97,5) до (5:95) названный выше продукт (73%); ТСХ, R f = 0,57, МеОН:СHCl3 (5:95). б. Синтез 3(RS)-N 2-(1,1-диметилэтоксикарбонил)-N6-фенилметоксикарбонил-L-лизил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]-N-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = PhCH 2OCONH(CH2)4, R3 =(CH3)3C, R4 = H, A = OCO, n=1) Используя методику примера 31в, продукт по примеру 65а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (градиентное элюирование, МеОН:CHCl3 от (2,5:97,5) до (5:95)) названный выше продукт (65%); ЖХВД, tR = 4,15 и 5,14, Col A, H 2 O : CH 3CN (45:55), СП = =2,0. Анализ, вычисленный для С 30H43F3N4 O7 x x 0,5 H 2 О: C 56,51, H 6,95, N 8,79. Найдено: C 56,45, H 6,58, N 8,42. Пример 66. Получение 3(RS)-[(2-амино-5-хлорфе нил)-карбонил]-L -ва лил-N-[3-(1 ,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R 3 = 2-NH2-S-Cl-Ph, R 4 = H, A = CO, n=1). а. Синтез 2(RS), 3(SR)-[(2-амино-5-хлорфенил)-карбонил]-L-валил-N-[3-(1,1,1-трифтор-2-ок-си-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R3 = 2-NH 2-5-Cl-Ph, R 4 = H, A = CO, n=1). Раствор соединения, полученного по примеру 3г (1 г, 2,7 ммоль) в СН2Cl2 (50 мл) обрабатывали 5хло ризотоевым ангидридом (0,54 г, 2,7 ммоль) при комнатной температуре и смесь перемеши вали в течение ночи, после чего ее промывали (5%-ным водным NaHCO, солевым раствором), высушивали (Na2SO4), концентрировали под вакуумом и очища ли с помощью быстрой хроматографии (МеОН:CHCl3 (3:97)), чтобы получить названный выше продукт (1,4 г, 78%) в виде белой пены; ТСХ, Rf = 0,30 и 0,25, МеОН:СHСl3 (5:95). б. Синтез 3(RS)-[(2-амино-5-хлорфенил)-карбонил]-L-валил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH(CH3)2, R 3 = 2-NH2-5-Cl-Ph, R 4 = H, A = CO, n=1). Используя методику примера 31в, продукт по примеру 66а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (МеОН:CHCl3 (3:97)) названный выше продукт (76%); ЖХВД, tR = 3,41 и 4,62, Col A, H 2O : CH3CN (50:50), СП = 2,0. Анализ, вычисленный для С 23H30F 3N4O4 x xH2 O: C 51,45, H 6,01, N 10,43. Найдено: C 51,65, H 5,74, N 9,68. Пример 67. Получение 3(RS)-(4-метоксифенилкарбонил)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = 4-(CH3O)-Ph, R 4 = H, A = CO, n=1). а. Синтез 2(RS), 3(SR)-(4-метоксифенилкарбонил)-L-альфа-аминобути рил-N-[3-(1 ,1,1 -трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = 4-(CH 3O)-Ph, R 4 = H, A = CO, n=1). Используя методику примера 20а, соединению, полученному по мето дике примера 61а, позволяли реагировать с 4-метоксибензоилом хлористым, что бы получить названное выше соединение, выделяемое 28 39093 с 34%-ным выхо дом после очистки с помощью быстрой хроматографии (EtOAc : :гексан (60:40), ТСХ, Rf = 0,71 и 0,73, МеОН : CHCl 3 (1:9). Анализ, вычисленный для С 23Н32F 3N3O4 x x0,3 H 2 O: C 56,04, H 6,67, N 8,52. Найдено: C 56,06, H 6,60, N 8,14. б. Синтез 3(RS)-(4-метоксифе нилкарбонил)-L-альфа-аминобутирил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R 3 = 4-(CH3O)-Ph, R 4 = H, A = CO, n=1). Используя методику примера 61в, за исключением того, что спирт растворяли в пятикратном объеме растворителя, продукт по примеру 67а окисляли, чтобы получить после очистки с помощью препаративной ТСХ (МеОН : CHCl3 (1:9)) названный выше продукт (40%) ЖХВД, tR = 8,89 и 11,75, Col A, H2 O : CH3CN (70:30), СП = 2,0. Анализ, вычисленный для С 23Н30F 3N3O5 x xH2 O: C 54,86, H 6,41, N 8,35. Найдено: C 54,89, H 6,38, N 7,48. Пример 68. Получение 3(RS)-[2-(трицикло-[3,3,1,13.7]-дец-1-и л)-этоксикарбони л]-L-аль фа-аминобутирил-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH3CH2-, R3 = (1-адамантил)-СН2СН2, R4 = H, A = CO, n=1). а. Синтез 2(RS), 3(SR)-[2-(трицикло-[3,3,1,13.7]-дец-1-ил)-этоксикарбонил]-L-альфа-аминобутирил-N-[3(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = (1-ада мантил)-СН2СН2, R4 = H, A = ОCO, n=1). Используя методику примера 34б, соединению, полученному по мето дике примера 61а, позволяли реагировать с соединением, полученным по методике примера 7а, чтобы получить названное выше соединение, выделяемое с 42%-ным выхо дом после очистки с помощью быстрой хроматографии (EtOAc : гексан( 20:80), затем (50:50)), ТСХ, Rf = 0,33 и 0,44, МеОН:CHCl 3 (5:95). б. Синтез 3(RS)-[2-(трицикло-[3,3,1,13.7]-дец-1-ил)-этоксикарбонил]-L-альфа-аминобутирил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]-L -пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = (1-адамантил)-СН2СН2, R4 = H, A = ОCO, n=1). Используя методику примера 61в, продукт по примеру 68а окисляли, чтобы получить после отгонки с помощью быстрой хроматографии (эфир-гексан (50:50), затем (90:10)) названный выше продукт (66%); ЖХВД, tR = 4,64 и 5,63, Col A, H 2O: CH 3CN (25:75), СП = 2,0. Анализ, вычисленный для С 28Н42F 3N3O5 x x0,15 H2O : C 60,01, H 7,60 . Найдено: С 59,76, H 7,65. Пример 69. Получение 3(RS)-N2-(1,1-ди-метилэтоксикарбонил)-N6-[фенилсульфонил-L-лизил- N-[3(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = PhS(O 2)NH(CH2)4, R3 = (CH 3)3C, R4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-N 2-(1,1-диметилэтоксикарбонил)-L-лизил-N-[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = H2N(CH2)4, R3 = (CH3)3C, R 4 = H, A = OCO, n=1). К раствору соединения, полученного по методике примера 65а (3,0 г, 4,8 ммоль) в абсолютном EtOH (60 мл) добавляли 10%-ный Pd на углероде (0,6 г). Полученную суспензию перемешивали в течение ночи в атмосфе ре водорода. Дополнительное количество 10%-ного Pd на углероде (0,3 г) добавляли и перемешивание продолжали в те чение нескольких часов. Реакционную смесь фильтровали через целит и концентрировали под вакуумом, что бы получить продукт (2,48 г), который использовали непосредственно. б. Синтез 2(RS),3(SR)-N 2-(1,1-диметилэтоксикарбонил)-N6-фе нилсуль фо нил-L-ли зил-N -[3-(1,1,1трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = PhS(O2)NH(CH2)4, R3 = (CH3)3C, R 4 = H, A = OCO, n=1). Используя методику примера 72б, за исключением того, что NMM использовали вместо ТЭА, продукт по примеру 69а обрабаты вали с бензолсульфо нилхлористым, чтобы получить названный выше продукт, который очищали с помощью быстрой хроматографии (СНСl3 : MeOH (95:5)), с 69%-ным выхо дом; ТСХ, Rf = 0,29, МеОН : CHCl 3 (95:5)). в. Синтез 3(RS)-N-(1,1-диметилэтоксикарбонил)-N6-[фе нилсуль фо нил-L-ли зил-N-[3-(1 ,1,1-трифтор4-метил-2-окси)]-N-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = PhS(O 2)--NH(CH 2)4, R3 = (CH 3)3C, R4 = H, A = OCO, n=1). Используя методику примера 31в, продукт по примеру 69б окисляли, чтобы получить после очистки с помощью быстрой хроматографии (градиентное элюирование, МеОН : CHCl3 от (0:100) до (2,5:97,5) названный выше продукт (57%); ЖХВД, tR = 7,48 и 9,11, Col A, CH 3CN : H 2O (1:1), СП = 1,0. Анализ, вычисленный для С 28H16F 3N3O7S x xH2 O: C 51,52, H 6,64, N 8,58. Найдено: C 51,47, H 6,46, N 7,80. Пример 70. Получение 3(RS)-(4-этоксикарбонилфенил)-аминокарбонил-L-альфа-аминобутаноил-N[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = 4-[CH 3CH2OC(O)]-Ph, R 4 = H, A = NHCO, n=1). а. Синтез 2(RS), 3(SR)-(4-этоксикарбонилфе нил)-аминокарбонил-L-альфа-аминобутироил- N-[3(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIIв, R1 = CH(CH3)CH3, R2 = CH 2CH3, R3 = 4-[CH 3CH2OC(O)]-Ph, R 4 = H, A = NHCO, n=1). 29 39093 Используя методику примера 15а, соединению, полученному по мето дике примера 61а, позволяли реагировать с этиловым эфиром 4-изоцианатобензойной кислоты, что бы получить названное выше соединение, выделяемое с 46%-ным выхо дом; ТСХ, Rf = 0,53, MeOH : CHCl 3 (5:95). б. Синтез 3(RS)-(4-этоксикарбонилфе нил)-аминокарбонил-L -аль фа-ами нобутири л-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = 4-[CH 3CH2OC(O)]-Ph, R 4 = H, A = NHCO, n=1). Используя методику примера 61в, продукт по примеру 70а окисляли, чтобы получить после очистки с помощью быстрой хроматографии (СНСl3 : MeOH (97:3)) названный выше продукт (68%); ЖХВД, tR = 6,35 и 8,70, Col A, H 2 O : CH 3CN (60:40), СП = 2,0. Анализ, вычисленный для С 25Н33F3N4 O6 x x 0,5 H 2 O: C 54,44, H 6,21, N 10,15. Найдено: C 54,76, H 6,13, N 10,27. Пример 71. По лучение 3(RS)-(4-оксикарбонилфе нил)-аминокарбонил-L-альфа-аминобути рил- N-[3(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = CH2CH3, R3 = 4[HOC(O)]-Ph, R 4 = H, A = NHCO, n=1). Используя методику примера 14 соединение, полученное по методике примера 70б, превраща ли в названный выше продукт и выделяли с помощью быстрой хроматографии на силикаге ле Бейкера с рН 5,0 (СНCl3 : MeOH (97:3)), c 61%-ным выхо дом; ЖХВД, tR = 3,68 и 4,69, Col A, H2 O: :CH3CN (3:1), СП = 2,0. Анализ, вычисленный для С 23Н29F 3N4O6 x xH2 O: C 51,88, H 5,86, N 10,52. Найдено: C 51,98, H 5,69, N 10,19. Пример 72. По лучение 3(RS)-N 6-фенилметоксикарбонил-N2-фенилсульфонил-L-лизил-N-[3-(1,1,1трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = (CH2)4NHCOOCH2Ph, R 3 = Ph, R 4 = H, A = S(O)2, n=1). а. Синтез 2(RS), 3(SR)-N 6-фе нилмето ксикарбон ил-L-лизи л-N-[3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида, соли трифторуксусной кислоты (формула IVв, R1 = CH(CH3)CH3, R2 = PhCH 2OCONH(CH2)4-, R4 = H, n=1). К раствору продук та, полученного по методике примера 65а (2,75 г, 4,4 ммоль) в СН2Сl2 (7 мл), добавляли ТФУК (10,4 г, 90 ммоль) и раствор перемешива ли при комнатной температуре в те чение 1 ч. Добавляли то луол (10 мл) и реакционную смесь концентрировали под вакуумом, чтобы получить названный выше продукт (3,4 г), который использова ли без дальнейшей очистки. б. Синтез 2(RS), 3(SR)-N 6-фен илметоксикарбони л-N 2-фени лсульфо ни л-L -лизи л-N -[3-(1,1,1-трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = CH(CH3)CH3, R2 = (CH 2)4NHC(O)OCH2Ph, R 3 = Ph, R 4 = H, A = S(O2), n=1). К раствору продукта по примеру 72а (0,83 г, 3,9 ммоль) и бензолсульфонила хлористого (0,25 г, 1,4 ммоль) и реакционную смесь перемешивали в те чение ночи при комнатной температуре. После концентрирования реакционной смеси под вакуумом оста ток растворяли в эти лацетате. Эти лацетатный раствор фильтровали, концентрировали под вакуумом до образования остатка, который очища ли с помощью быстрой хроматографии (градиентное элюирование, МеОН : CHCl 3 от (2,5:97,5) до (5:95), что бы получить названный выше продукт (81%); ТСХ, Rf = 0,52, MeOH : CHCl 3 (5:95). в. Синтез 3(RS)-N 6-фенилметоксикарбонил-N2-фенилсуль фонил-L-лизил-N- [3-(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = CH(CH3)CH3, R2 = (CH2)4NHCO(O)ОCH2Ph, R 3 = Ph, R 4 = H, A = S(O2), n=1). Используя методику примера 31в, продукт по примеру 72б окисляли, чтобы получить после очистки с помощью быстрой хроматографии (градиентное элюирование от СНСl3 до СНCl3 : MeOH (97,5:2,5) названный выше продукт (63%); ЖХВД, tR = 5,0 и 6,3, Col A, CH 3CN : H 2O (3:2), СП = 1,0. Анализ, вычисленный для С 31Н39F 3N4O7S x xH2 O: C 54,22, H 6,02, N 8,16. Найдено: С 54,17, Н 5,80, N 7,86. Пример 73. Получение 3(RS)-[2-(2-ме-токсиэтокси)-этоксикарбонил]-L-альфа-аминобутирил -N-[3(1,1,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = СH(CH3)CH3, R2 = CH3CH2-, R3 = CH 3O(CH2)2 O(CH2)2, R4 = H, A = OCO, n=1). а. Синтез 2(RS), 3(SR)-[2-(2-метоксиэтокси)-этоксикарбон ил]-L -аль фа-ами нобути рил-N -[3 -(1,1,1трифтор-2-окси-4-метилпентил)]-L-пролинамида (формула VIIв, R1 = СH(CH3)CH3, R2 = CH3CH2-, R 3 = CH 3O(CH2)2O(CH2)2, R4 = OCO, n=1). Используя методику примера 34б, соединению, полученному по мето дике примера 61а, позволяли реагировать с соединением, полученным по методике примера 19а, чтобы получить названное выше соединение, выделенное с 75%-ным выхо дом после очистки с помощью быстрой хроматографии (градиентное элюирование, ацетон:гексан от (1:9) до (7:3H)), ТСХ, Rf = 0,30 и 0,35, ацетон:гексан (40:60). Анализ, вычисленный для С 17H29F 3N2O6 x x0,5 H 2 O: C 48,22, H 7,14, N 6,61. Найдено: C 48,13, H 6,90, N 6,07. б. Синтез 3(RS)-[2-(2-метоксиэтокси)-этокси-карбонил]-L-альфа -ам инобути р ил-N -[3 -(1 ,1 ,1-трифтор-4-метил-2-оксопентил)]-L-пролинамида (формула Iв, R1 = СH(CH3)CH3, R2 = CH3CH2-, R3 = CH 3O(CH2)2 O(CH2)2, R4 = OCO, n=1). 30
ДивитисяДодаткова інформація
Назва патенту англійськоюDerivatives of peptides being elastase inhibitors of humans leukocytes, method for obtaining thereof and pharmaceutical composition
Автори англійськоюBERGESON Scott Haven, EDWARDS Philipe Duke, SCHWARtZ John Anthony, SHAW Andrew, STANiE Mark Morris, TRAiNOR Diane Amy, WILDONGER Richard Alan, VOLANIN Donald John
Назва патенту російськоюПроизводные пептидов, которые являются ингибиторами эластазы лейкоцитов человека, способ их получения и фармацевтическая композиция
Автори російськоюБЕРГЕСОН Скотт Хавен, ЭДВАРДС Филип Дьюк, ШВАРЦ Джон Энтони, ШОУ Эндрю, СТЕЙН Марк Моррис, ТРЕЙНОР Диана Эми, ВИЛДОНГЕР Ричард Алан, ВОЛАНИН Дональд Джон
МПК / Мітки
МПК: A61P 29/00, A61K 31/535, A61K 31/401, C07D 403/12, C07K 5/08, A61K 31/4418, C07K 5/04, C07C 205/00, C07K 5/093, C07D 333/00, A61K 38/00, A61P 43/00, C07D 401/12, A61K 31/44, A61K 31/40, A61K 31/4433, A61K 31/4025, C07K 1/113, C07D 213/26, A61P 9/10, A61K 31/4427, C07K 5/06, C07K 14/81, C12N 9/99, A61K 38/55, C07D 409/12, C07D 207/16, A61K 31/4409, A61P 11/00
Мітки: отримання, пептидів, лейкоцитів, композиція, еластази, інгібіторами, людини, фармацевтична, спосіб, похідні
Код посилання
<a href="https://ua.patents.su/52-39093-pokhidni-peptidiv-shho-eh-ingibitorami-elastazi-lejjkocitiv-lyudini-sposib-kh-otrimannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Похідні пептидів, що є інгібіторами еластази лейкоцитів людини, спосіб їх отримання та фармацевтична композиція</a>
Похідні піримідину, що мають активність антагоністів ангіотензину іі, спосіб їх отримання та фармацевтична композиція

Номер патенту: 27748
Опубліковано: 16.10.2000
Автори: Бажлі Жак Фрамрос, Нікайдо Маделен, Еллінгбой Джон Ватсон
МПК: C07D 471/04, C07D 487/04
Мітки: активність, іі, похідні, отримання, композиція, мають, спосіб, ангіотензину, піримідину, антагоністів, фармацевтична
Текст:
...Результаты анализа из расчеты на формулу 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 минут после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение, в соответствии с примером 1, при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от А II в среднем на 41 % в...
Сірковмісні похідні імідазолу, що мають гіпотензивну активність, спосіб їх отримання, проміжні сполуки і фармацевтична композиція

Номер патенту: 26130
Опубліковано: 07.06.1999
Автори: Вевер Жан-Поль, Амон Жіль, Фортен Мішель, Кай Жан-Клод, Корб'є Ален, Жукей Сімон
МПК: A61K 31/415, A61P 43/00, A61K 31/4164, A61P 21/00, A61P 9/10, C07D 403/10, A61P 9/00, C07D 233/84, C07D 233/94, C07D 233/90, C07D 401/12, A61K 31/4178, A61P 13/02, C07D 403/12, A61P 15/00, A61P 9/12
Мітки: гіпотензивну, мають, проміжні, отримання, композиція, похідні, сполуки, спосіб, сірковмісні, фармацевтична, активність, імідазолу
Формула / Реферат:
1. Серосодержащие производные имидазола общей формулы (I)где R1 - алкил;R2 - водород, гидроксиметил, карбоксигруппа, группа формулы -S(=O)2-A, где A - низший алкил или фенил,- группа формулы -S(=O)2(CH2)k-B, где k = 1 и 2, B - низшая алкокси- или гидроксигруппа,- группа -S-D, где D - незамещенный фенил или замещенный галогеном, низшей алкоксигруппой низший алкил, возможно замещенный галогеном, и...
Похідні азолів, їх фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб їх отримання, фармацевтична композиція
Номер патенту: 27343
Опубліковано: 15.09.2000
Автори: Бейкер Реймонд, Матасса Віктор Г., Стріт Леслі Дж.
МПК: A61K 31/4433, C07D 409/04, A61K 31/445, A61K 31/425, C07D 413/06, C07D 413/14, C07D 403/14, A61K 31/4427, C07D 521/00, C07D 409/06, A61K 31/41, C07D 417/14, C07D 409/14, A61K 31/42, C07D 403/06, A61K 31/415, A61P 25/04, C07D 417/06, C07D 401/14, C07D 403/04
Мітки: рецепторів, азолів, 5-нt1-подібних, композиція, проліки, солі, прийнятні, агоністами, фармацевтично, спосіб, похідні, отримання, фармацевтична
Текст:
...2-15-(1-бензилтетразол-5-илметил)-1Н-индол-3ил]этиламин М,М-диметил-2-{5-(2-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(1-метилтетразол-5-илметил)1 Н-индол-3-ил]этиламин М,М-диметил-2-[5-(тетрээол-2-ил метил )• 1 Н-индол3-ил]этиламин N,N-димeтил-2-[5-(тeтpaзoл-1-илмeтил)-1 Н-индол3-ил]этиламин N,N-flHMeTHn-2-tS-{1 -метил-1,2,4-триазол-5-и л метил) 1Н-индол-3-ил]этиламин...
Похідні 7-(2-аміноетил)бензотіазолону, що проявляють агоністичну активність по відношенню до бета2-адренорецепторів, фармацевтична композиція, спосіб їх отримання та проміжні сполуки

Номер патенту: 27901
Опубліковано: 16.10.2000
Автори: Чешир Дейвід Рейнулф, Інсе Френсіс, Боннет Роджер Віктор, Браун Роджер Чарльз
МПК: A61P 27/02, A61K 31/425, A61P 9/00, A61P 11/00, C07D 417/12, A61P 43/00, A61P 29/00, C07D 277/68, A61P 37/08, A61P 25/02, A61P 27/14, A61K 31/44, A61K 31/428
Мітки: агоністичну, проявляють, композиція, спосіб, активність, відношенню, похідні, сполуки, проміжні, отримання, фармацевтична, бета2-адренорецепторів, 7-(2-аміноетил)бензотіазолону
Текст:
...общей формулы I показаны к применению при лечении ряда состояний, известных как обратимое обструктивное заболевание дыхательных путей. Специалистам выражение "обратимое обструктивное заболевание дыхательных путей" должно быть хорошо понятно. Под ним подразумевается астма, в том числе бронхиальная астма, аллергическая астма, наследственная, приобретенная, пылевая астма, особенно хроническая или застарелая астма (например, поздняя астма и...
Похідні триазолопіримідину, які мають антагоністичну активність до рецепторів ангіотензину іі, та фармацевтична композиція, що їх містить

Номер патенту: 26627
Опубліковано: 11.10.1999
Автори: Ніколя Ерік, Брю-Маньє Ніколь, Тьолон Жан-Марі
МПК: A61K 31/519, C07D 487/04, A61P 43/00, A61P 9/00, A61P 9/04, A61K 31/505, A61P 9/10, A61P 9/12
Мітки: іі, композиція, мають, антагоністичну, фармацевтична, похідні, триазолопіримідину, містить, активність, рецепторів, ангіотензину
Формула / Реферат:
1. Производные триазолопиримидина общей формулы lгде R1 является низшим алкилом с 1 - 6 атомами углерода или C3-C7-циклоалкилом;R2 является атомом водорода, низшим алкилом с 1 - 6 атомами углерода, группой NH-NH2, группой (CH2)mOR4, (CH2)mSR4;R4 является атомом водорода, низшим алкилом с 1 - 6 атомами углерода и m равно целому числу от 0 до 2;группа является радикалом, выбранным среди следующих...
Попередній патент: Спосіб одержання тетрахлориду титану
Випадковий патент: Спосіб видобування металофази з природних і техногенних руд