Спосіб одержання амінокротонільних сполук
Номер патенту: 91006
Опубліковано: 25.06.2010
Автори: Кулінна Крістіан, Зойка Райнер, Шнаубельт Юрген, Ралль Вернер, Зігер Петер






Формула / Реферат
1. Спосіб одержання сполуки формули (VII)
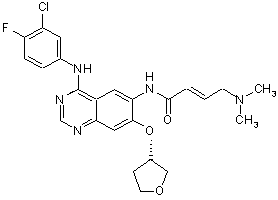 , (VII)
, (VII)
що включає наступні стадії синтезу
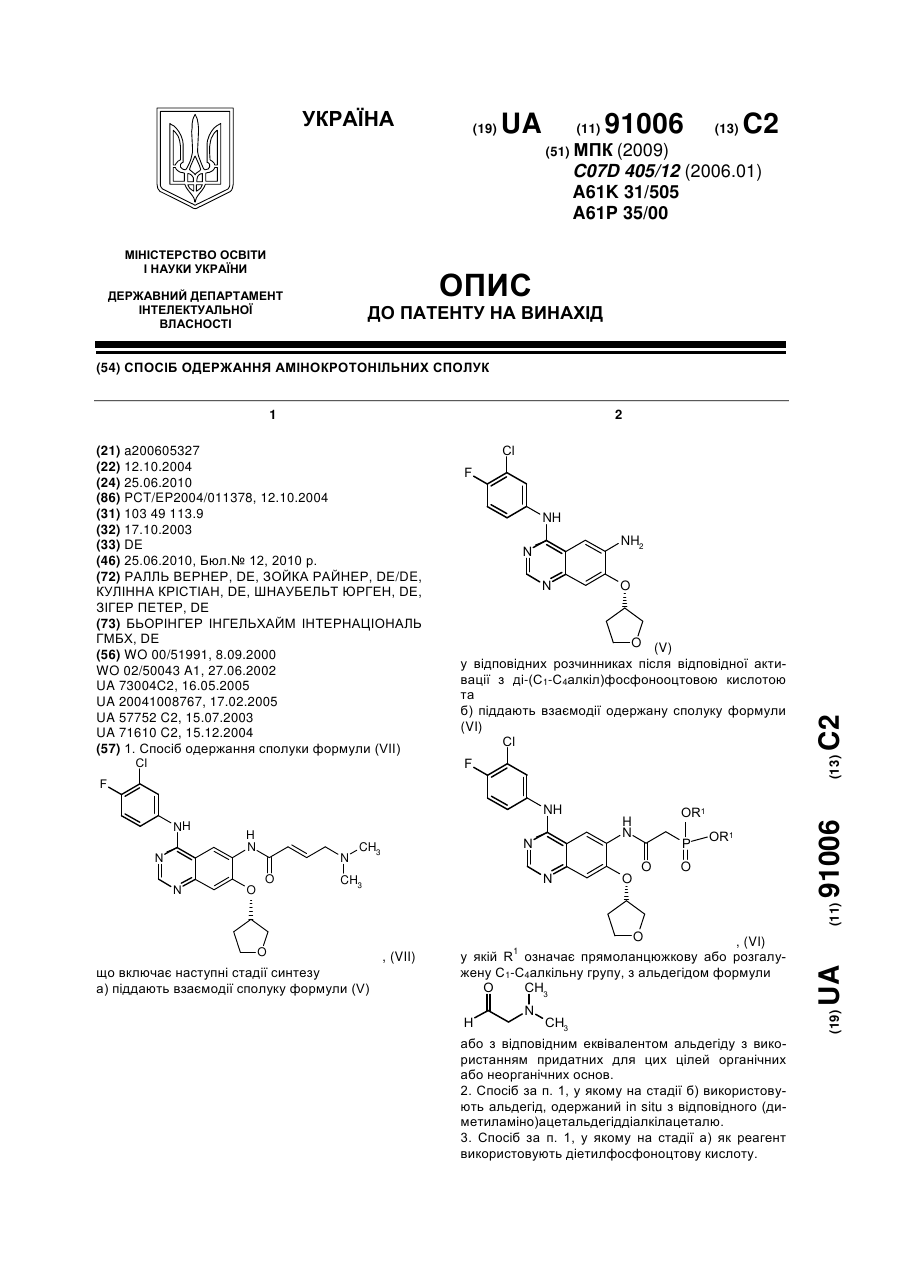
а) піддають взаємодії сполуку формули (V)
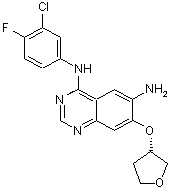 (V)
(V)
у відповідних розчинниках після відповідної активації з ді-(С1-С4алкіл)фосфонооцтовою кислотою та
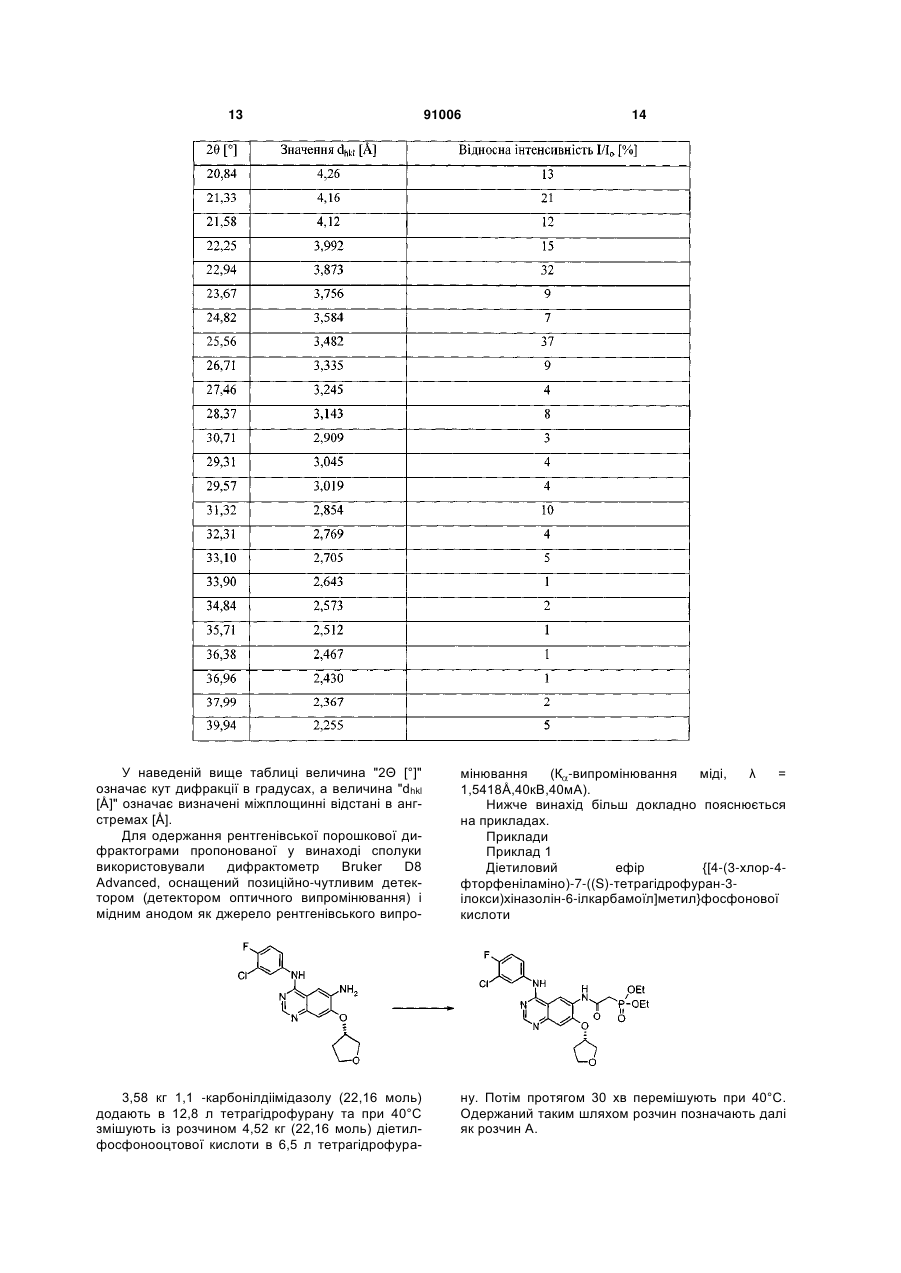
б) піддають взаємодії одержану сполуку формули (VI)
 , (VI)
, (VI)
у якій R1 означає прямоланцюжкову або розгалужену С1-С4алкільну групу, з альдегідом формули
![]()
або з відповідним еквівалентом альдегіду з використанням придатних для цих цілей органічних або неорганічних основ.
2. Спосіб за п. 1, у якому на стадії б) використовують альдегід, одержаний in situ з відповідного (диметиламіно)ацетальдегіддіалкілацеталю.
3. Спосіб за п. 1, у якому на стадії а) як реагент використовують діетилфосфоноцтову кислоту.
4. Спосіб за п. 1 або 2, який відрізняється тим, що на стадії б) як основу використовують ДБН (1,5-діазабіцикло[4.3.0]нон-5-ен), гідроксид натрію або гідроксид калію.
5. Спосіб за п. 4, який відрізняється тим, що на стадії б) як основу використовують гідроксид калію.
Текст
1. Спосіб одержання сполуки формули (VII) O , (VI) у якій R означає прямоланцюжкову або розгалужену С1-С4алкільну групу, з альдегідом формули O CH3 H N CH3 або з відповідним еквівалентом альдегіду з використанням придатних для цих цілей органічних або неорганічних основ. 2. Спосіб за п. 1, у якому на стадії б) використовують альдегід, одержаний in situ з відповідного (диметиламіно)ацетальдегіддіалкілацеталю. 3. Спосіб за п. 1, у якому на стадії а) як реагент використовують діетилфосфоноцтову кислоту. UA що включає наступні стадії синтезу а) піддають взаємодії сполуку формули (V) , (VII) 1 (19) O 3 4. Спосіб за п. 1 або 2, який відрізняється тим, що на стадії б) як основу використовують ДБН (1,5-діазабіцикло[4.3.0]нон-5-ен), гідроксид натрію або гідроксид калію. Даний винахід стосується вдосконаленого способу одержання амінокротонільних сполук, таких, наприклад, як 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназолін, і їх фізіологічно сумісних солей, насамперед дималеату 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну, а також дималеату 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну та його застосування для одержання лікарських засобів. 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4-(N,Nдиметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((S)тетрагідрофуран-3-ілокси)хіназолін має наступну структурну формулу і вже відомий з заявки WO 02/50043, де описуються сполуки з цінними фармакологічними властивостями, до яких насамперед належать інгібуюча дія на опосередковувану тирозинкіназами трансдукцію сигналів та інгібуюча дія на опосередковувану рецептором епідермального фактора росту (EGF-R) трансдукцію сигналів. Тому сполуки такого типу придатні для лікування певних захворювань, насамперед для лікування онкологічних захворювань, захворювань легень і дихальних шляхів, так само як і захворювань шлунковокишкового тракту, а також жовчних шляхів і жовчного міхура. У заявці WO 02/50043 пропонується спосіб одержання амінокротонільних сполук (IV), таких, наприклад, як 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4(N,N-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7((S)-тетрагідрофуран-3-ілокси)хіназолін, який полягає у тому, що зазначені сполуки одержують за реакцією, яку проводять у одному апараті, з відповідного анілінового структурного фрагмента (II), бромкротонової кислоти (III), оксалілхлориду та вторинного аміну (див. схему 1) 91006 4 5. Спосіб за п. 4, який відрізняється тим, що на стадії б) як основу використовують гідроксид калію. Цей спосіб забезпечував максимум 50%-вий вихід продукту. Крім того, передбачалося, як правило, очищення за допомогою колоночної хроматографії. Тому можливість реалізації даного способу одержання 4-[(3-хлор-4-фторфеніл)аміно]-6{[4-(N,N-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}7-((S)-тетрагідрофуран-3-ілокси)хіназоліну в промисловому масштабі виключалася. Ще один недолік запропонованого підходу полягав у тому, що необхідна для його здійснення бромкротонова кислота не поставляється на ринок у великих кількостях, а відповідний метиловий ефір бромкротонової кислоти доступний лише з 80%-вим ступенем чистоти. Ці фактори також не дозволяють застосовувати спосіб одержання 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну в промисловому масштабі. З урахуванням вищеописаних недоліків відомого способу в основу даного винаходу було покладене завдання запропонувати спосіб, що забезпечував би одержання амінокротонілариламідів, насамперед 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4(N,N-диметиламіно)-1 -оксо-2-бутен- 1-іл]аміно} -7((S)-тетрагідрофуран-3-ілокси)хіназоліну, з використанням легкодоступних вихідних речовин, з високим ступенем чистоти та без значних виробничих витрат. Таким чином, новий спосіб призначається для здійснення синтезу в промисловому масштабі та тим самим для одержання комерційно доступних продуктів. Зазначене завдання вирішується за допомогою пропонованого у винаході способу одержання 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4(N,Nдиметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((S)тетрагідрофуран-3-ілокси)хіназоліну та інших амінокротонільних сполук. Поряд із промисловою застосовністю пропонованого у винаході синтезу, що забезпечує високий вихід, до його інших переваг варто віднести винятково високу хімічну чисто 5 91006 6 ту одержуваних продуктів і низький вміст цисізомерів, що становить менше 0,1%. Відповідно до пропонованого у винаході способу відповідну аміноарильну сполуку формули (V) піддають взаємодії з ді-(С1С4алкіл)фосфонооцтовою кислотою, переважно з діетилфосфонооцтовою кислотою, у придатних для цих цілей розчинниках, після відповідної активації, краще 1,1-карбонілдіімідазолом, 1,1карбонілдитриазолом або ангідридом пропанфосфонової кислоти, особливо бажано 1,1карбонілдіімідазолом, як це показано на схемі 2. Як розчинники можуть використовуватися, наприклад, тетрагідрофуран (ТГФ), диметилформамід (ДМФ) та етилацетат. Активацію можна проводити з використанням всіх звичайних методів амідного приєднання, тобто, наприклад, за допомогою 1,1карбонілдіімідазолу, 1,1-карбонілдитриазолу, ДЦК (Ν,Ν-дициклогексилкарбодііміду), ДЕК (N’(диметиламінопропіл)-N-етилкарбодііміду), ТБТУ (тетрафторборату О-(бензотриазол-1 -іл)- N,N, N’,N’ -тетраметилуронію), тіазолідин-2-тіону або переведенням у відповідний хлорангідрид кислоти, наприклад, за допомогою тіонілхлориду. За певних умов активацію проводять з використанням органічних основ, таких як триетиламін або піридин, при цьому додатково можна додавати ДМАП (диметиламінопіридин). Як розчинники в цих випадках прийнятні ДМФ, ТГФ, етилацетат, толуол, хлоровані вуглеводні або їх суміші. У наведені нижче формулах замісники мають наступні значення: X означає метинову групу або атом азоту, Ra означає бензильну, 1-фенілетильну або 3хлор-4-фторфенільну групу та R1 означає С1-С4алкільну групу з прямим або розгалуженим ланцюгом. Пропонований у винаході спосіб краще використовувати для одержання сполук, у яких X означає атом азоту, Ra означає 3-хлор-4-фторфенільну групу та R1 означає етильну групу. а) Ді-(С1-С4алкіл)фосфонооцтова кислота, активатор Одержаний описаним шляхом з високим виходом і високим ступенем чистоти ари л амід формули (VI) піддають за реакцією Віттига-ХорнераЕммонса взаємодії з 2-аміноацетальдегідом з використанням відповідних органічних або неорганічних основ (схема 3). Цю реакцію можна проводити безпосередньо після одержання сполуки формули (VI) або після її виділення, наприклад шляхом осадження додаванням, наприклад, третбутилметилового ефіру. До придатних для зазначеної мети основ належать серед інших ДБН (1,5діазабіцикло[4.3.0]нон-5-ен), гідроксид натрію та гідроксид калію, кращими з яких є гідроксид натрію та гідроксид калію, особливо кращий гідроксид калію. Замість альдегіду можна використовувати також відповідний еквівалент, наприклад гідрат або ацеталь, з якого альдегід вивільняють (попередньо або in situ). б) Альдегід, основа, ТГФ/вода Як ацеталі можуть використовуватися, наприклад, сполуки наступної загальної формули: у якій кожний з R2-R5 означає прямоланцюжкову або розгалужену С1-С4алкільну групу, при цьому такі залишки можуть бути ідентичними або різними. У кращому варіанті кожний з R3 та R4 означає метильну групу, а кожний з R2 та R5 означає етильну групу. Одержаний описаним шляхом амінокротонілариламід формули (VII), наприклад, 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо 7 91006 2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназолін формули (І), можна потім за відомою методикою переводити в його солі, насамперед у його фізіологічно сумісні солі. Краще переведення у фумарати, тартрати або малеати. Особливо кращим є дималеат 4-[(3-хлор-4фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1-оксо2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну структурної формули (Іа) і відповідно переведення 4-[(3-хлор-4-фторфеніл)аміно]6-{[4-(N,N-диметиламіно)-1-оксо-2-бутен-1іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну в його дималеат, як це показано на схемі 4. Із цією метою сполука формули (І) розчиняють у відповідному розчиннику, такому, на 8 приклад, як метанол, ізопропанол, н-бутанол або етанол, необов'язково з додаванням води, краще у етанолі, і при нагріванні змішують із кристалічною малеїновою кислотою або з розчином малеїнової кислоти. При використанні як розчинник етанолу краще працювати при температурі в інтервалі від 60 до 75°С з використанням розчину малеїнової кислоти в етанолі. Умови реакції доцільно вибирати з таким розрахунком, щоб забезпечити максимально швидке викристалізування необхідної солі. Краще використовувати приблизно 2 еквіваленти малеїнової кислоти. Після початку кристалізації суміш охолоджують до кімнатної температури, перемішують та відокремлюють кристалізат, що утворився, який являє собою сполуку формули (Іа) Схема 4 в) Малеїнова кислота, етанол Вихідну сполуку формули (V) можна одержувати, наприклад, відповідно до наступних методів, відомих з літератури. Хінолінові структурні фрагменти формули (V), у якій X означає СН, можна одержувати виходячи з доступного як комерційний продукт 3-фтору-6нітрофенолу формули (XIV) шляхом алкілування, обміну атома фтору на аміногрупу та взаємодією з ефірами етоксіакрилової кислоти, ефірами етоксиметиленціаноцтової кислоти або ефірами етоксиметиленмалонової кислоти (схема 5а). Потім одержану описаним шляхом сполуку формули (XVII) переводять у сполуку формули (XVIII), як це показано на схемі 6 на прикладі аналога хіназоліну. При одержанні сполуки формули (V), де X означає N, працюють у такий спосіб: Виходячи з доступної як комерційний продукт 4-хлорантранілової кислоти (формула VIII, X означає СІ), взаємодією з формамідин-ацетатом одер жують хіназолінон формули (IX), який потім нітрують з використанням сірчаної кислоти та концентрованої азотної кислоти (схема 5б). В іншому варіанті можна виходити з 4-фторантраніловоїкислоти. 9 а: X' означає СІ б: X' означає F г) формамідин-ацетат д) H2SO4, HNO3 концентрована є) SOCl2, ацетонітрил є) RaNH2 Одержану описаним шляхом сполуку формули (XII) взаємодією з (S)-(+)-3гідрокситетрагідрофураном перетворюють у спо 91006 10 Необхідний регіоізомер формули (X) одержаних описаним шляхом продуктів нітрування потім хлорують і продукт хлорування формули (XI) піддають in situ взаємодії з відповідним аміном (схема 6). луку формули (XIII). Наступним гідруванням сполуки формули (XIII), відповідно сполуки формули (XVIII) зі схеми 5а одержують вихідну сполуку формули (V) (схема 7). Схема 7 ж) (S)-(+)-3-гідрокситетрагідрофуран з)Н2 Ще одним об'єктом винаходу є дималеат 4-[(3хлор-4-фторфеніл)аміно]-6-{[4-(N,Nдиметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7-((S)тетрагідрофуран-3-ілокси)хіназоліну. Ця сіль особливо придатна для застосування у фармацевтиці, оскільки вона існує лише в одній кристалічній модифікації, яка крім іншого не містить води та відрізняється винятковою стабільністю. Діюча речовина для можливості її фармацевтичного застосування як лікарський засіб не тільки повинна мати необхідну дію, але й за багатьма критеріями повинна відповідати і іншим вимогам. Подібні критерії здебільшого пов'язані з фізикохімічними властивостями діючої речовини. Як не обмежуючі приклади подібних критеріїв можна назвати стабільне збереження вихідною речовиною своєї дії в різних навколишніх умовах, у процесі приготування фармацевтичного препарату та у складі кінцевих лікарських засобів. Тому лікарська діюча речовина, використовувана для приготування лікарських композицій, повинна мати високу стабільність, що зберігається навіть у різних навколишніх умовах. Обов'язкове додержання цієї вимоги обумовлено необхідністю виключити застосування лікарських композицій, у яких поряд із по суті діючою речовиною присутні, наприклад, і продукти її розкладу. У такому разі вміст діючої речовини у фармацевтичних препаратах може виявитися меншим зазначеного виробником. У результаті абсорбції вологи вміст лікарської діючої речовини зменшується через обумовлене поглинанням вологи збільшення її маси. Тому лікарські засоби, що проявляють схильність до поглинання вологи, необхідно на час зберігання захищати від вологи, наприклад, за рахунок додавання прийнятних осушувачів або зберігання лікарського засобу в умовах, у яких він захищений від контакту з вологою. Крім цього поглинання вологи може привести до зменшення змісту лікарського діючої речовини в процесі виготовлення фармацевтичного препарату, якщо лікарська діюча речовина не захищена від контакту з вологою та безпосередньо піддається впливу факторів навколишнього середовища. Із цієї причини лікарська 11 діюча речовина бажано повинна мати лише мінімально можливу гігроскопічність. Оскільки для одержання лікарської форми з постійно відтворюваним вмістом у ній діючої речовини важливе значення має її кристалічна модифікація, спочатку необхідно одержувати максимально повну інформацію про будь-який можливий поліморфізм діючої речовини, представленої в кристалічній формі. Якщо діюча речовина може існувати в різних поліморфних модифікаціях, то необхідно впевнитися в тому, що певна кристалічна модифікація діючої речовини не зазнає ніяких змін у виготовленому пізніше на її основі лікарському препараті. У іншому випадку подібні поліморфні перетворення можуть негативно вплинути на ефективність медикаменту та його відтворюваності. З урахуванням сказаного вище кращі діючі речовини, що характеризуються лише мінімальною схильністю до поліморфних перетворень. Іншим критерієм, що за певних умов може мати особливо важливе значення залежно від вибраної лікарської форми або вибраної технології її приготування, є розчинність діючої речовини. Так, наприклад, при приготуванні лікарських форм у вигляді розчинів (наприклад, для інфузій) обов'язковою умовою є наявність у діючої речовини достатньої розчинності у фізіологічно сумісних розчинниках. Особливо важливе значення має й достатня розчинність діючої речовини, яка включається до складу лікарських засобів, які вводять в організм перорально. 91006 12 Відповідно до цього ще одне покладене в основу даного винаходу завдання полягало у одержанні лікарської діючої речовини, яка мала б не тільки високу фармакологічну ефективність, але й, крім того, у максимально можливому ступені задовольняла б за своїми фізико-хімічними властивостями розглянутим вище вимогам. Зазначене завдання вирішується за допомогою дималеату 4-[(3-хлор-4-фторфеніл)аміно] -6{[4-(N,N-диметиламіно)-1 -оксо-2-бутен-1 -іл]аміно} -7-((S)-тетрагідрофуран-3-ілокси)хіназоліну. Температура плавлення дималеату 4-[(3-хлор4-фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1оксо-2-бутен-1-іл]аміно}-7-((S)-тетрагідрофуран-3ілокси)хіназоліну дорівнює 178°С (див. представлені на фіг. 2 у графічному вигляді результати термоаналізу). Кристалічний дималеат 4-[(3-хлор4-фторфеніл)аміно]-6-{[4-(N,N-диметиламіно)-1 оксо-2-бутен- 1-іл]аміно} -7-((S)-тетрагідрофуран3-ілокси)хіназоліну більш детально аналізували за допомогою рентгенівської порошкової дифрактометрії. Отримана дифрактограма представлена на фіг. 1. У нижченаведеній таблиці наведені результати цього аналізу. Таблиця: Дифракційні максимуми (рефлекси) та їх інтенсивність (відносна), виявлені при аналізі дималеату 4-[(3-хлор-4-фторфеніл)аміно]-6-{[4(N,N-диметиламіно)-1-оксо-2-бутен-1-іл]аміно}-7((S)-тетрагідрофуран-3-ілокси)хіназоліну рентгенівською порошковою дифрактометрією 13 91006 14 У наведеній вище таблиці величина "2Θ [°]" означає кут дифракції в градусах, а величина "dhkl [Å]" означає визначені міжплощинні відстані в ангстремах [Å]. Для одержання рентгенівської порошкової дифрактограми пропонованої у винаході сполуки використовували дифрактометр Bruker D8 Advanced, оснащений позиційно-чутливим детектором (детектором оптичного випромінювання) і мідним анодом як джерело рентгенівського випро мінювання (К -випромінювання міді, λ = 1,5418Å,40кВ,40мА). Нижче винахід більш докладно пояснюється на прикладах. Приклади Приклад 1 Діетиловий ефір {[4-(3-хлор-4фторфеніламіно)-7-((S)-тетрагідрофуран-3ілокси)хіназолін-6-ілкарбамоїл]метил}фосфонової кислоти 3,58 кг 1,1 -карбонілдіімідазолу (22,16 моль) додають в 12,8 л тетрагідрофурану та при 40°С змішують із розчином 4,52 кг (22,16 моль) діетилфосфонооцтової кислоти в 6,5 л тетрагідрофура ну. Потім протягом 30 хв перемішують при 40°С. Одержаний таким шляхом розчин позначають далі як розчин А. 15 91006 16 6,39 кг (17,05 моль) N4-(3-хлор-4-фторфеніл)7-(тетрагідрофуран-3-ілокси)хіназолін-4,6-діаміну додають в 26,5 л тетрагідрофурану та при 40°С змішують із розчином А з наступним перемішуванням протягом 2 год при 30°С. До суспензії, що утворилася, додають 64 л трет-бутилметилового ефіру та після охолодження до 20°С осад відокремлюють центрифугуванням. На завершення спочатку промивають сумішшю з 16 л тетрагідрофу рану та 16 л трет-бутилметилового ефіру, а потім 32 л води і сушать при 50°С. Вихід: 6,58 кг (69,8%) білих кристалів, чистота згідно з даними РХВР (рідинна хроматографія високого розділення): 99,1%, що припадає на площу поверхні під піком. Приклад 2 [4-(3-хлор-4-фторфеніламіно)-7-((6г)тетрагідрофуран-3-ілокси)хіназолін-6-іл]амід(Е)-4диметиламінобут-2-енової кислоти До 4,4 л води додають 5,6 л 30%-вий розчин соляної кислоти (53,17 моль). Потім при 30°С продовж 20 хв по краплях додають 4,28 кг 95%-вого (диметиламіно)ацетальдегіддіетилацеталю (26,59 моль). Реакційну суміш перемішують протягом 8 год при 35°С, охолоджують до 5°С и в наступному зберігають в атмосфері аргону. Цей розчин означають далі як розчин Б. 4,55 кг (68,06 моль) гідроксиду калію розчиняють в 23,5 л води та охолоджують до -5°С. Цей розчин позначають далі як розчин В. 5,88 кг (10,63 моль) діетилового ефіру ((4-(3хлор-4-фторфеніламіно)-7-(тетрагідрофуран-3ілокси)хіназолін-6-ілкарбамоїл)метил)фосфонової кислоти та 0,45 кг хлориду літію (10,63 моль) додають в 23,5 л тетрагідрофурану та охолоджують до -7°С. Далі протягом 10 хв додають холодний розчин В. Потім при -7°С у продовж 1 год додають розчин Б. Після 1-годинного перемішування при 5°С реакційну суміш нагрівають до 20°С та змішують із 15 л води. Після охолодження до 3 °С суспензію піддають вакуум-фільтрації та залишок промивають водою й сушать. Вихід: 5,21 кг (100%) сирого продукту, вологовміст: 6,7%. Кристалізацію сирого продукту здійснюють із використанням бутилацетату/метилциклогексану. Вихід: 78%, чистота згідно з даним РХВР: 99,4%, що припадає на площу поверхні під піком, вологовміст: 5,4%. Приклад 3 Дималеат [4-(3-хлор-4-фторфеніламіно)-7-((S)тетрагідрофуран-3-ілокси)хіназолін-6-іл]аміду (Е)4-диметиламінобут-2-енової кислоти 6,0 кг (12,35 моль) [4-(3-хлор-4фторфеніламіно)-7-((S)-тетрагідрофуран-3ілокси)хіназолін-6-іл]аміду (Е)-4-диметиламінобут2-енової кислоти додають в 84 л етанолу, нагрівають до 70°С та змішують із розчином 2,94 кг (25,31 моль) малеїнової кислоти в 36 л етанолу. Після початку кристалізації суміш спочатку охолоджують до 20°С і перемішують протягом 2 год, а потім протягом 3 год при 0°С. Осад відокремлюють вакуум-фільтрацією, промивають 19 л етанолу та сушать у вакуумі при 40°С. Вихід: 8,11 кг(91,5%),tпл 178°С. 1H-ЯМР (CD3OD): δ = 2,47 + 2,27 (m+m, 2H), 2,96 (s, 6H), 4,03 (m, 2H), 4,07 + 3,92 (m+m, 2Н), 4,18 + 4,03 (m+m, 2Н), 5,32 (m, 1H), 6,26 (s, 4Н), 6,80 (m, 1Н), 6,99 (m, 1H), 7,27 (s, 1H), 7,30 (t, 1H), 7,66 (m, 1H), 7,96 (dd, 1H), 8,62 (s, 1H), 9,07 (s, 1H) част./млн. 17 Комп’ютерна верстка І. Скворцова 91006 Підписне 18 Тираж 28 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the production of amino crotonyl compounds
Автори англійськоюRall Werner, Soyka Rainer, Kulinna Christian, Schnaubelt Juergen, Sieger Peter
Назва патенту російськоюСпособ получения аминокротонильных соединений
Автори російськоюРалль Вернер, Зойка Райнер, Кулинна Кристиан, Шнаубельт Юрген, Зигер Петер
МПК / Мітки
МПК: A61K 31/505, C07D 405/12, A61P 35/00
Мітки: амінокротонільних, сполук, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/9-91006-sposib-oderzhannya-aminokrotonilnikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання амінокротонільних сполук</a>
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Еллегор Петер, Петерсен Ханс, Рок Майкл Харольд
МПК: C07D 307/87, C07C 253/30, C07C 255/56
Мітки: сполук, s-циталопраму, проміжні, кетони, рацемічних, циталопраму, варіанти, одержання, спосіб
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Спосіб одержання похідних 4-трифтрометилсульфінілпіразолу та способи одержання проміжних сполук

Номер патенту: 73752
Опубліковано: 15.09.2005
Автори: Клавель Жан-Луї, Шарро Філіпп, Ле Бар Сільві, Пельта Ізабелль
МПК: C07D 401/04, C07D 231/44
Мітки: одержання, похідних, спосіб, сполук, способи, 4-трифтрометилсульфінілпіразолу, проміжних
Формула / Реферат:
1. Спосіб (А) одержання сполуки формули (І):, (I)в якій W означає азот або -CR3; R1 означає галоген, галоалкіл, галоалкокси, R4S(O)n- або -SF5; R2 означає водень або галоген; R3 означає галоген; R4 означає алкіл або галоалкіл і n означає 0, 1 або 2, що включає окислення сполуки формули (II):
Стероїдні сполуки, застосування цих сполук для одержання лікарських засобів, які регулюють мейоз, і спосіб одержання цих сполук (варіанти)

Номер патенту: 75130
Опубліковано: 15.03.2006
Автори: Хегеле-Хартунг Хріста, Блюме Торстен, Есперлінг Петер
МПК: A61P 15/08, A61K 31/58, C07J 43/00, A61P 15/00, A61P 43/00, C07J 41/00
Мітки: цих, засобів, регулюють, сполук, мейоз, застосування, одержання, спосіб, варіанти, сполуки, лікарських, стероїдні
Формула / Реферат:
1. Стероїдна сполука, яка має загальну формулу Х:,де у фрагменті ХА:подвійний зв’язок присутній у 8 і/або 14 позиції;C3R3 означаєа) C=O,б) CH-OR3', де R3' вибирають із групи, яка включає водень, незаміщений або заміщений, лінійний або розгалужений...
Спосіб одержання сполук, які посилюють секрецію гормону росту
Номер патенту: 70306
Опубліковано: 15.10.2004
Автори: Пост Рональд Джеймс, Мелц Кліффорд Натаніель, Чіу Чарльз Квок-Фунг, Роуз Пітер Роберт, Буш Френк Роберт
МПК: A61K 38/00, C07K 5/02, C07B 57/00, A61P 19/10, A61P 5/00, C07K 5/06, C07D 471/04, A61K 31/437
Мітки: посилюють, гормону, сполук, секрецію, спосіб, одержання, росту
Формула / Реферат:
1. Спосіб одержання сполуки формули II , (ІІ) в якійR1 є -(С1-С10)алкілом, необов'язково, заміщеним до трьох атомів фтору,R2 є фенілметилом або 2-піридилметилом,R3 є -(С1-С5)алкіл-О-(С0-С5)алкілфенілом, в якому фенільний замісник необов'язково заміщений до трьох атомів фтору, таPrt - амінозахисна...
Азольні сполуки з протигрибковою активністю, спосіб одержання цих сполук, спосіб одержання проміжних сполук та фармацевтична композиція

Номер патенту: 55405
Опубліковано: 15.04.2003
Автори: Наполетано Мауро, Скіоппакассі Джованна, Альбіні Енріко, Фраіре Крістіна
МПК: A61K 31/41, A61K 31/4164, A61P 31/00, C07D 521/00, C07D 233/60, A61K 31/415, A61K 31/00, C07D 249/08, A61P 31/10, A61K 31/4196
Мітки: азольні, протигрибковою, проміжних, одержання, цих, спосіб, активністю, фармацевтична, композиція, сполук, сполуки
Формула / Реферат:
1. Сполука формули (ІІ-А), (ІІ-А)де:R1 являє собою хлор, фтор, бром або трифторметил;R2 являє собою водень, хлор, фтор, бром або трифторметил;Z являє собою СН або N;R3, R4, R5, які можуть бути однаковими або різними, являють собою водень або С1-С4 алкіл при умові, що R4 відрізняється від R5, якщо R3 - водень;Х являє собою О, S, SO або SO2;R6 являє собою С1-С5 поліфторалкільну групу, яка...
Попередній патент: Спосіб боротьби з мікроорганізмами та спосіб зниження рівня забруднювача з використанням силікатів перехідних металів
Наступний патент: Заміщені 6-циклогексилалкілом заміщені 2-хінолінони та 2-хіноксалінони як інгібітори полі-(адф-рибоза)полімерази
Випадковий патент: Спосіб визначення свіжості зрізаних квітів