Пептиди-агоністи crhr2 і їх використання
Номер патенту: 104877
Опубліковано: 25.03.2014
Автори: Шенклі Найджел П., Морено Вероніка, Свансон Роналд В., Дженго Пітер



























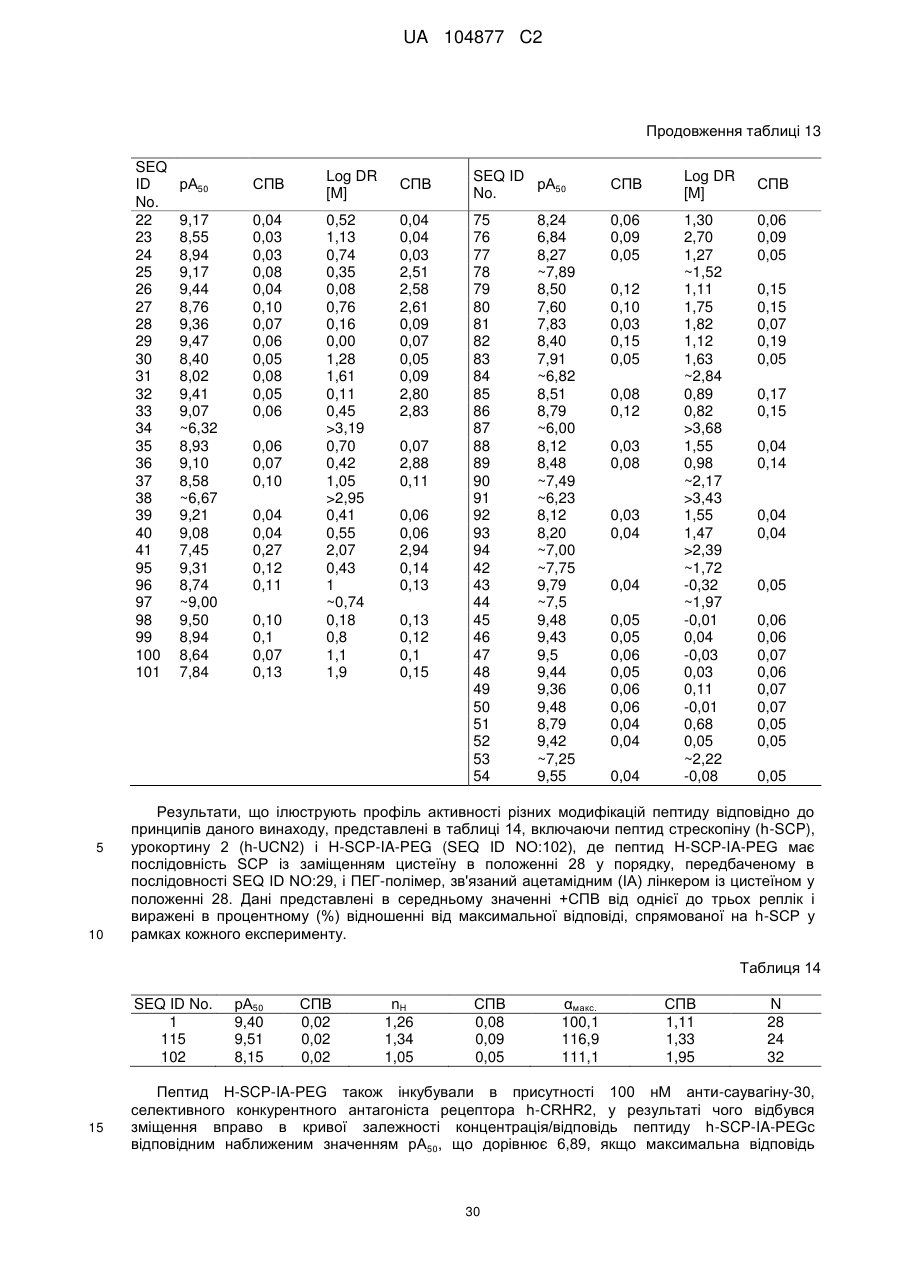





























































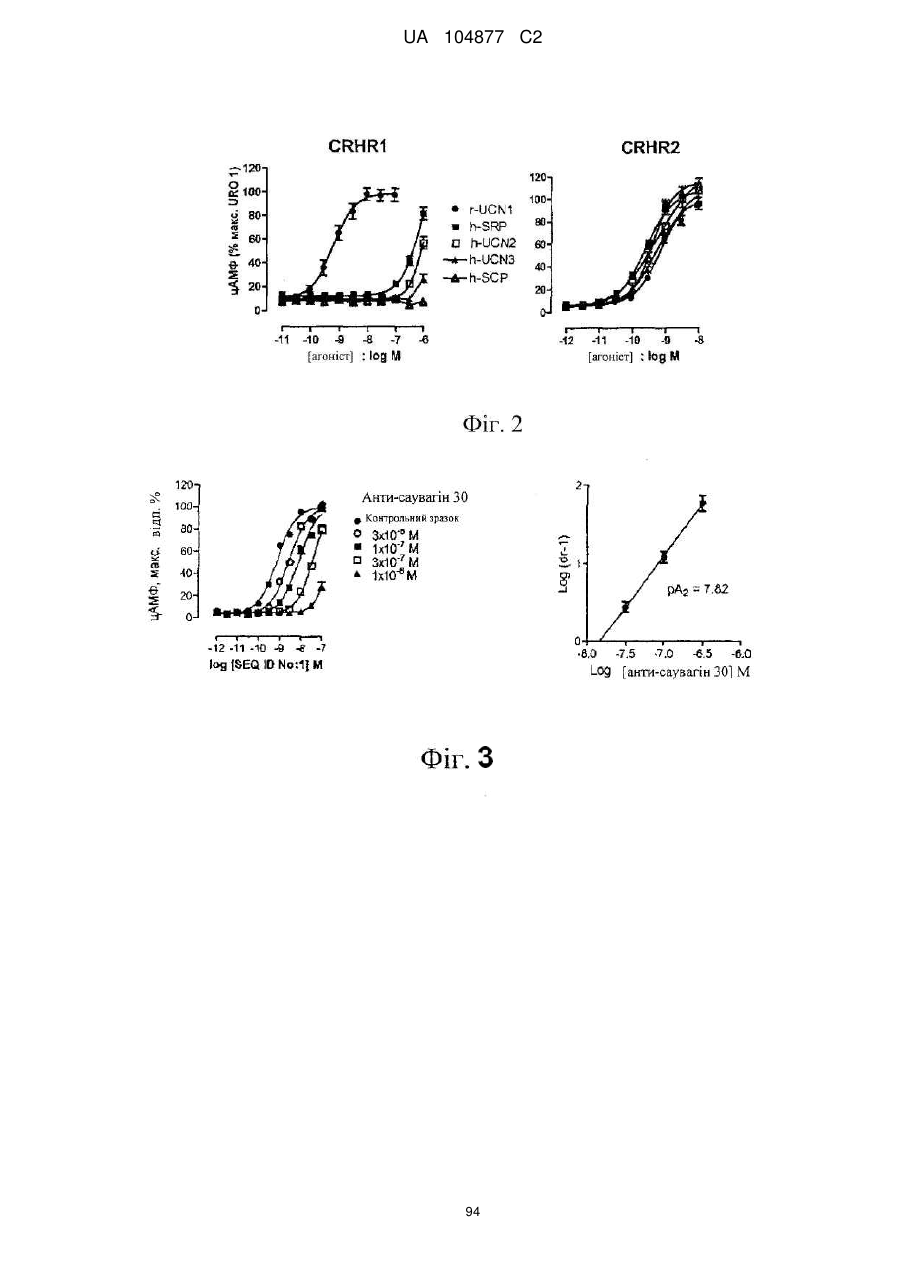

Формула / Реферат
1. Пептид, який має агоністичну активність відносно рецептора кортикотропін-рилізинг-гормону 2 типу (CRHR2), де вказаний пептид має амінокислотну послідовність:
![]()
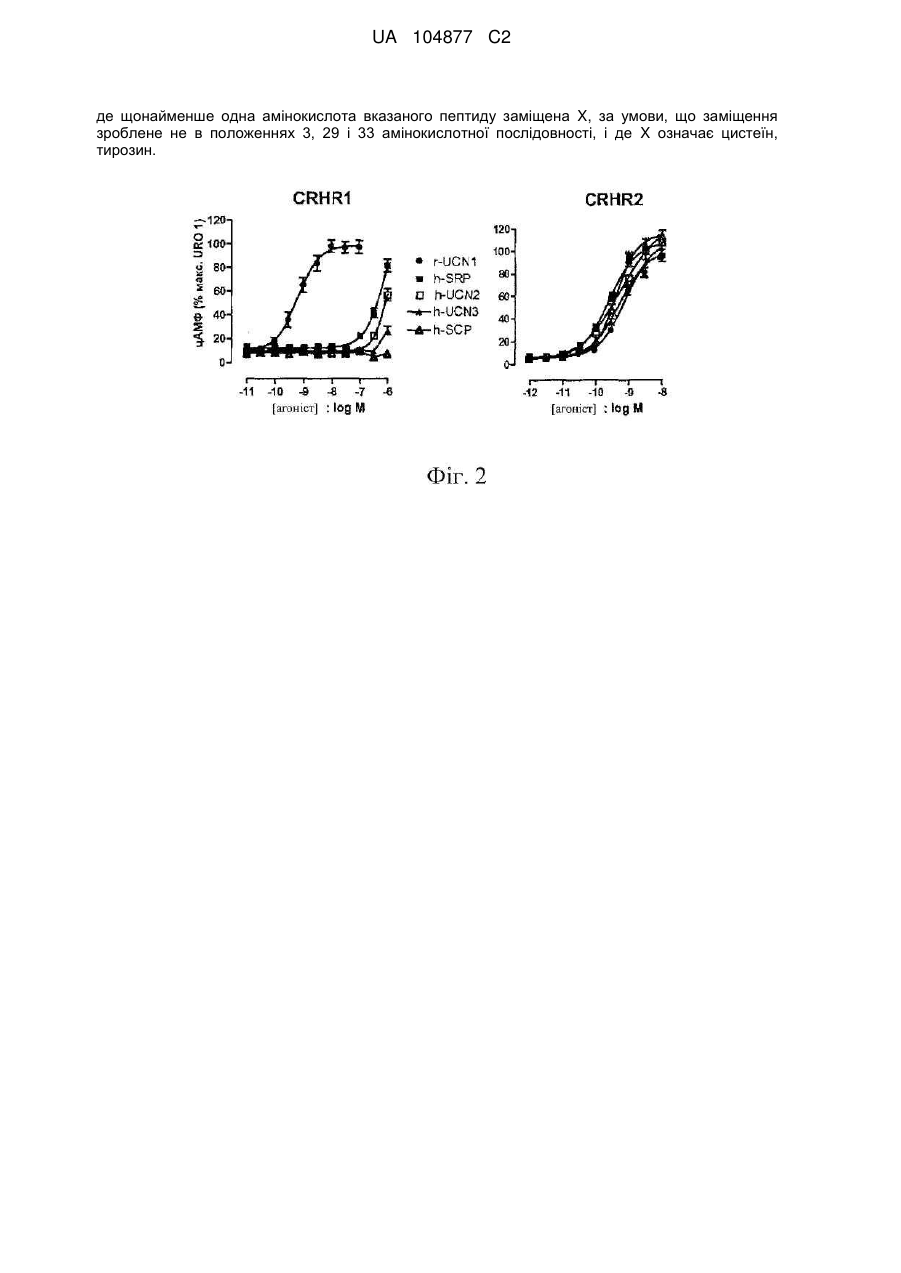
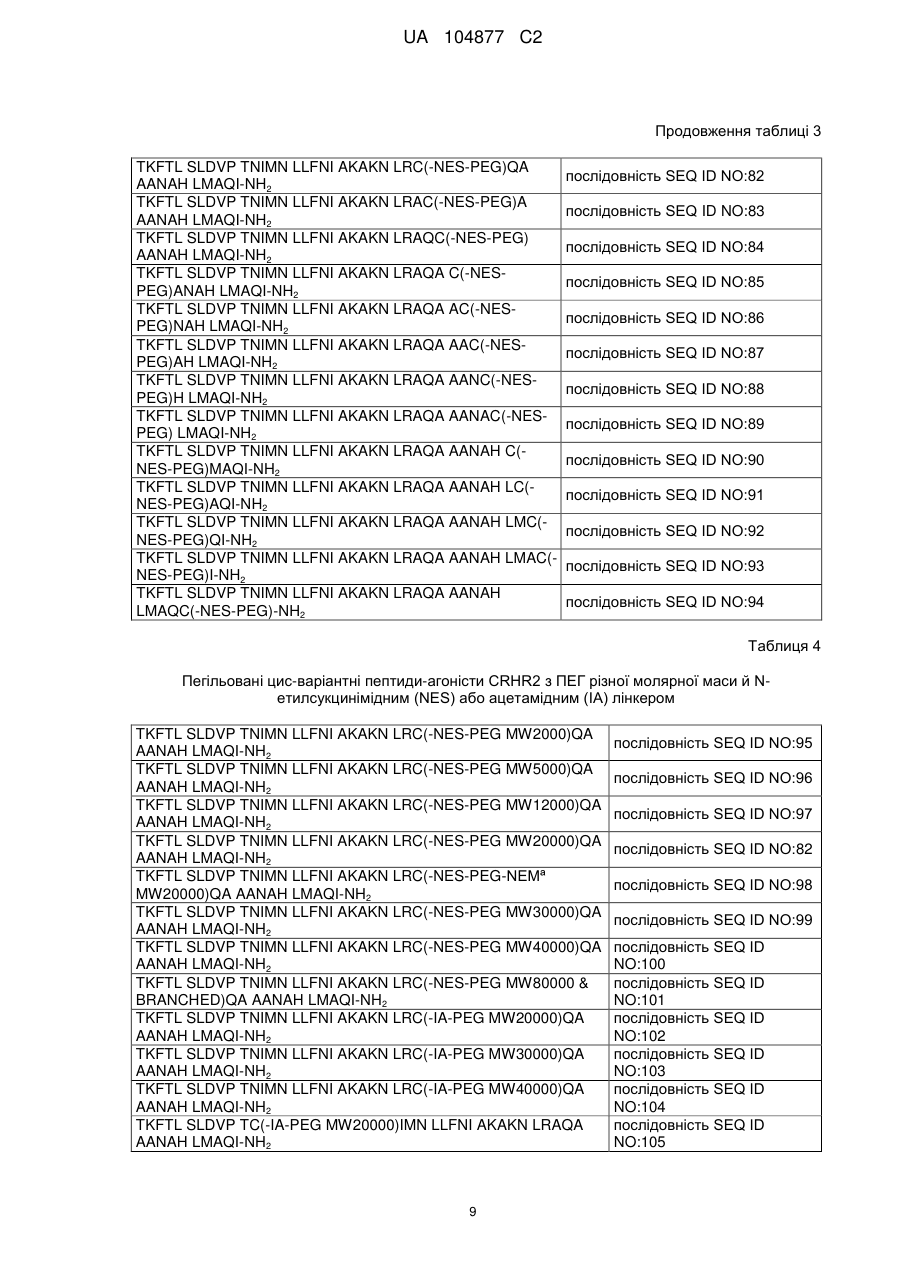
де щонайменше одна амінокислота вказаного пептиду заміщена X, за умови, що заміщення зроблене не в положеннях 3, 29 і 33 амінокислотної послідовності, і де X означає цистеїн або тирозин;
або їх фармацевтично прийнятні солі або аміди.
2. Пептид за п. 1, у якому заміщення стосується наступної групи заміщень:
X на L у положенні 1;
X на S у положенні 2;
X на D у положенні 4;
X на V у положенні 5;
X на Р у положенні 6;
X на Т у положенні 7;
X на N у положенні 8;
X на І у положенні 9;
X на М у положенні 10;
X на N у положенні 11;
X на L у положенні 12;
X на L у положенні 13;
X на F у положенні 14;
X на N у положенні 15;
X на І у положенні 16;
X на А у положенні 17;
X на K у положенні 18;
X на А у положенні 19;
X на K у положенні 20;
X на N у положенні 21;
X на L у положенні 22;
X на R у положенні 23;
X на А у положенні 24;
X на Q у положенні 25;
X на А у положенні 26;
X на А у положенні 27;
X на А у положенні 28;
X на А у положенні 30;
X на Н у положенні 31;
X на L у положенні 32;
X на А у положенні 34;
X на Q у положенні 35; і
X на І у положенні 36.
3. Пептид за п. 1, де вказаний пептид вибраний із групи, яка складається з:

4. Пептид за п. 1, де амінокислотна послідовність вибрана із групи, яка складається з послідовностей SEQ ID NO: 17, 18, 20, 21, 25, 26, 27, 29, 32, 33, 34, 36, і 41; а X є цистеїном.
5. Кон'югат, що містить пептид, за п. 1 і лінкер, приєднаний до X вказаного пептиду.
6. Кон'югат за п. 5, у якому X є цистеїном.
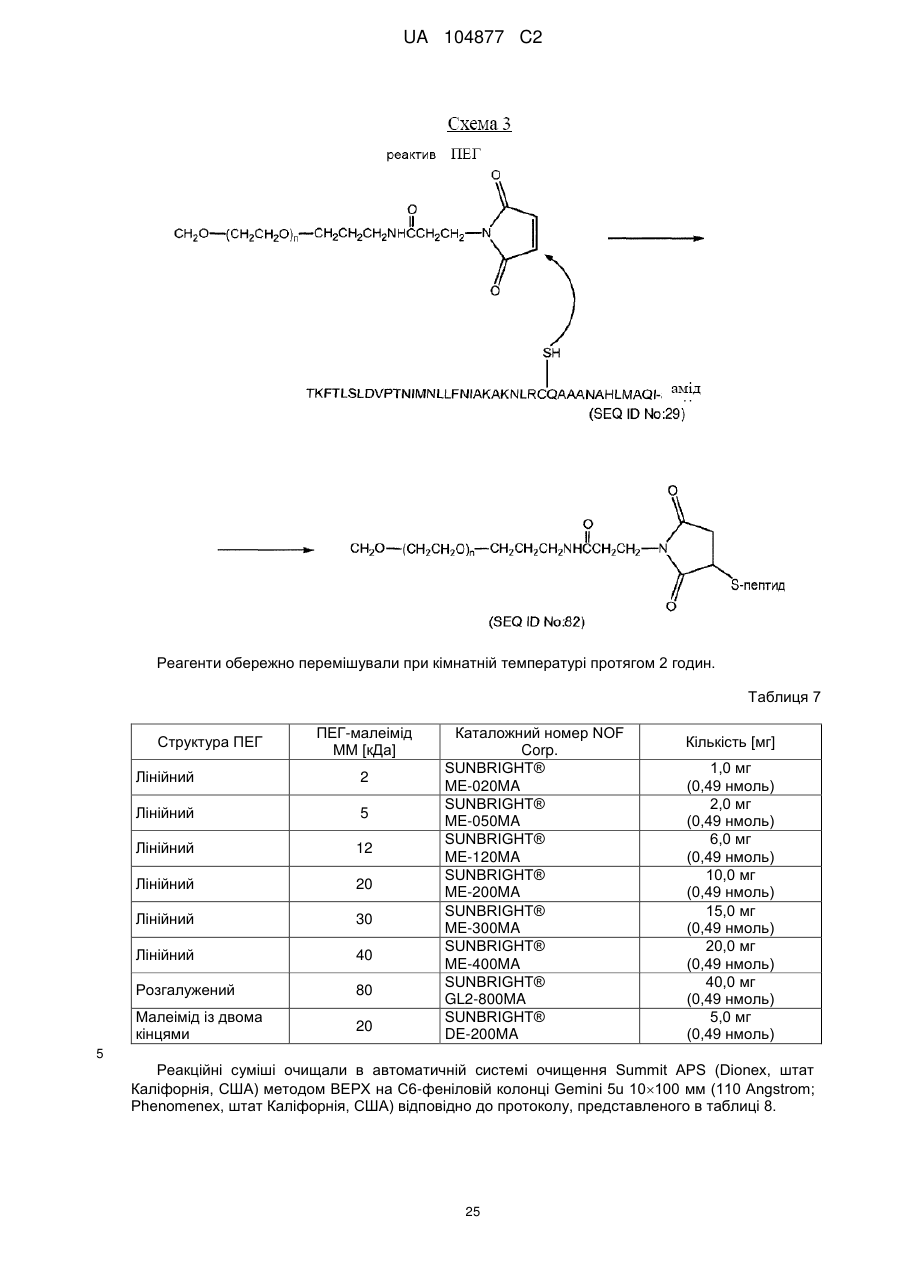
7. Кон'югат за п. 5, у якому вказаний лінкер представлений ацетамідом або N-етилсукцинімідом.
8. Кон'югат за п. 5, що додатково включає поліетиленгліколь (ПЕГ), приєднаний до вказаного лінкеру, де молекулярна маса ПЕГ не перевищує приблизно 80 кДа.
9. Кон'югат за п. 8, у якому вказаний лінкер представлений ацетамідом.
10. Кон'югат за п. 8, у якому вказаний ПЕГ має молекулярну масу, вибрану із групи молекулярних мас, що складається з приблизно 2 кДа, приблизно 5 кДа, приблизно 12 кДа, приблизно 20 кДа, приблизно 30 кДа або приблизно 40 кДа.
11. Кон'югат за п. 5, у якому вказаний ПЕГ має розгалужену або лінійну форму.
12. Кон'югат за п. 5, у якому вказаний ПЕГ додатково містить реакційноздатну групу.
13. Кон'югат за п. 12, у якому вказана реакційноздатна група є N-етилмалеїмідом.
14. Пептид за п. 1, у якому вказаний пептид має амінокислотну послідовність, вибрану із групи, яка складається з послідовностей SEQ ID NO: 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 35, 36, 37, 39, 40 і 41.
15. Кон'югат, що має формулу:
 ,
,
де R - пептид за п. 4, a S - атом сірки тіолової групи цистеїну X.
16. Кон'югат за п. 6, що має формулу:
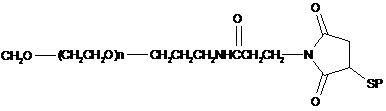 ,
,
де n - ціле число від приблизно 40 до приблизно 1900, R - пептид за п. 1, що має амінокислотну послідовність, вибрану із групи, яка складається з послідовностей SEQ ID NO: 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 35, 36, 37, 39, 40 і 41; a S - атом сірки тіолової групи цистеїну X.
17. Кон'югат за п. 6, що має формулу:
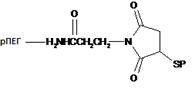 ,
,
де рПЕГ - розгалужений поліетиленгліколь з молекулярною масою приблизно 80 кДа, R - пептид за п. 1, що має амінокислотну послідовність SEQ ID NO: 29, a S - атом сірки тіолової групи цистеїну X.
18. Кон'югат за п. 6, що має формулу:
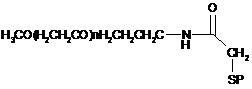 ,
,
де n - ціле число від приблизно 40 до приблизно 1900, R - пептид за п. 1, що має амінокислотні послідовності SEQ ID NO: 13, 29 і 36; a S - атом сірки тіолової групи цистеїну X.
19. Кон'югат за п. 16, у якому n - ціле число, що приблизно дорівнює 460, а R - пептид, що має амінокислотну послідовність SEQ ID NO: 29.
20. Кон'югат за п. 18, у якому n - ціле число, що приблизно дорівнює 460, а R - пептид, що має амінокислотну послідовність SEQ ID NO: 29.
21. Полінуклеотид, що кодує пептид, за п. 1.
22. Фармацевтична композиція, що включає (а) пептид за п. 1 і (b) фармацевтично прийнятний наповнювач.
23. Фармацевтична композиція, що містить (а) кон'югат за п. 5 і (b) фармацевтично прийнятний наповнювач.
24. Фармацевтична композиція, що містить (а) кон'югат за п. 19 і (b) фармацевтично прийнятний наповнювач.
25. Фармацевтична композиція, що містить (а) кон'югат за п. 20 і (b) фармацевтично прийнятний наповнювач.
26. Моноклональне антитіло, яке специфічно зв'язує пептид, що включає амінокислотну послідовність вказаного пептиду за п. 1.
27. Моноклональне антитіло за п. 26, у якому вказаний пептид є пегильованим.
28. Спосіб лікування пацієнта, що страждає на захворювання або має діагностоване захворювання, порушення або патологічний стан, опосередкований активністю кортикотропін-рилізинг-гормону 2 типу, вибраний із групи, яка складається з метаболічного захворювання, яке може бути зв'язане як з порушенням обміну речовин, так і із серцевою недостатністю, що включає введення пацієнту для здійснення такого лікування ефективної кількості пептиду за п. 1.
29. Спосіб за п. 28, у якому захворюванням, порушенням або патологічним станом є діабет.
30. Спосіб за п. 28, у якому захворюванням, порушенням або патологічним станом є серцева недостатність.
31. Пептид, що має амінокислотну послідовність SEQ ID NO: 29.
32. Кон'югат, що містить пептид, за п. 31 і лінкер, приєднаний до цистеїну в положенні 28 амінокислотної послідовності.
33. Кон'югат за п. 32, у якому вказаний лінкер представлений ацетамідом або N-ентилсукцинімідом.
34. Кон'югат за п. 32, що додатково включає поліетиленгліколь (ПЕГ), приєднаний до вказаного лінкеру, де молекулярна маса ПЕГ не перевищує приблизно 80 кДа.
35. Кон'югат за п. 34, у якому вказаний лінкер представлений ацетамідом.
36. Кон'югат, що має формулу:
,
де R - пептид за п. 33, a S - атом сірки тіолової групи цистеїну в положенні 28 амінокислотної послідовності.
37. Кон'югат за п. 32, що має формулу:
,
де n - ціле число від приблизно 40 до приблизно 1900, R - пептид за п. 33; S - атом сірки тіолової групи цистеїну в положенні 28 амінокислотної послідовності.
38. Кон'югат за п. 32, що має формулу:
,
де n - ціле число від приблизно 40 до приблизно 1900, R - пептид за п. 33; S - атом сірки тіолової групи цистеїну в положенні 28 амінокислотної послідовності.
39. Полінуклеотид, що кодує пептид, за п. 31.
40. Фармацевтична композиція, що містить пептид за п. 31 і фармацевтично прийнятний наповнювач.
41. Фармацевтична композиція, що містить пептид за п. 32 і фармацевтично прийнятний наповнювач.
42. Фармацевтична композиція, що містить пептид за п. 37 і фармацевтично прийнятний наповнювач.
43. Фармацевтична композиція, що містить пептид за п. 38 і фармацевтично прийнятний наповнювач.
Текст
Реферат: Даний винахід стосується пептиду, який проявляє агоністичну активність відносно рецептора кортикотропін-рилізинг-гормону 2 типу (CRHR2), де вказаний пептид має амінокислотну послідовність: UA 104877 C2 (12) UA 104877 C2 де щонайменше одна амінокислота вказаного пептиду заміщена X, за умови, що заміщення зроблене не в положеннях 3, 29 і 33 амінокислотної послідовності, і де X означає цистеїн, тирозин. UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 Перехресні посилання на суміжні винаходи Дана заявка вимагає пріоритет, заявлений у попередній заявці на одержання патенту США з реєстраційним № 61/111233 від 04 листопада 2008 року й № 61/178890 від 15 травня 2009 року. Галузь застосування винаходу У цілому, даний винахід стосується пептиду, що використовується як міметик стрескопіну для лікування порушень, викликаних активністю рецептора кортикотропін-рилізинг-гормону 2 типу, а також конструкцій для доставки пептидів, фармацевтичних композицій, що містять ці пептиди, і способів лікування пацієнта, у якого виявлено захворювання, порушення або патологічний стан, опосередкований кортикотропін-рилізинг-гормоном 2 типу. Передумови створення винаходу Серцева недостатність - розповсюджена патологія серцево-судинної системи. У Сполучених Штатах Америки і Європі вона вже досягла розмірів епідемії (Remme et al., Eur. Heart J., 2001, vol. 22, pp. 1527-1560). Чисельність госпіталізацій із приводу гострої серцевої недостатності тільки в США наближається до 1 мільйона в рік. У цей час частка повторної госпіталізації й смертності протягом 60 днів після виписки досягає 30-40% (Cuffee et al., JAMA, 2002, vol. 287(12), pp. 1541-7). При гострій серцевій недостатності, як правило, спостерігається погіршення гемодинаміки, зокрема, дуже високий кінцевий діастолічний тиск у лівому шлуночку. Сучасні методи лікування гострої серцевої недостатності є багатофакторними й найчастіше підбираються індивідуально. Хоча діуретики, вазодилататори й позитивні інотропи становлять основу терапії пацієнтів з гострою серцевою недостатністю, такі методи лікування пов'язані з високим рівнем смертності й повторної госпіталізації. Більше того, існуюча інотропна терапія (наприклад, терапія добутаміном) приводить до поліпшення серцевого викиду, але при цьому підвищується частота серцевих скорочень і споживання кисню міокардом. Інотропні препарати також мають проаритмічний потенціал для пацієнтів із серцевою недостатністю. Передбачається, що схильність до серцево-судинних захворювань пов'язана з витратою енергії й посиленням обміну кальцію, обумовленими прямою позитивною інотропною дією препарату. У спробі задовольнити наявні терапевтичні потреби, вивчення ряду нових підходів привело лише до невеликого успіху в питаннях забезпечення безпеки при поліпшенні гемодинамічного статусу й результатів лікування пацієнтів з таким синдромом. Одна з таких речовин - людський пептидний гормон урокортин 2 (h-UCN2) - пройшла дослідження на здорових індивідах і пацієнтах із серцевою недостатністю. Пептид показав здатність підвищувати фракцію викиду лівого шлуночка (ФВЛШ) і серцевий викид (СВ) у моделі вівці із серцевою недостатністю (Rademaker et al., Circulation, 2005, vol. 112, pp. 3624-3624). У наступних дослідженнях виконувалися внутрішньовенні уливання 8 здоровішим особинам (Davis et al., J. Am. Coll. Cardiol., 2007, vol. 49, pp. 461-471) і 8 пацієнтам із серцевою недостатністю (Davis et al., Eur. Heart J., 2007, vol. 28, pp. 2589-2597). При введенні обох досліджуваних доз у кожному із двох досліджень підвищення ФВЛШ і СВ супроводжувалося підвищенням частоти серцевих скорочень і зниженням артеріального тиску. Внутрішньовенні уливання гормону h-UCN2 протягом однієї години добре переносилися як здоровими особинами, так і пацієнтами із серцевою недостатністю. Людський стрескопін (h-SCP), 40-амінокислотний пептид, є спорідненим гормону h-UCN2, при цьому обидві речовини належать до класу пептидів сімейства кортикотропін-рилізинггормону (КРГ). Біологічна дія пептидів сімейства КРГ обумовлена двома 7-трансмембранними G-протеїнзв’язаними рецепторами, рецептором КРГ 1 типу (CRHR1) і рецептором КРГ 2 типу (CRHR2). Незважаючи на те, що для рецепторів характерна висока гомологія послідовності, різні пептиди сімейства КРГ демонструють істотні відмінності відносних значень спорідненості до зв'язування, ступеня активації рецептора й селективності до цих двох рецепторів. На відміну від багатьох пептидів сімейства КРГ, h-SCP демонструє більшу селективність до CRHR2 і виступає як посередник, що сприяє ослабленню процесу розвитку й підтримки фізіологічного стресу (Bale et al., Nat. Genet., 2000, vol. 24, pp. 410-414; Kishimoto et al., Nat. Genet., 2000, vol. 24, pp. 415-419). Крім очевидної участі у формуванні стану фізіологічного стресу, також є свідчення про те, що h-SCP викликає ряд інших фізіологічних впливів. Він впливає на ендокринну (Li et al., Endocrinology, 2003, vol. 144, pp. 3216-3224) систему, ЦНС, серцево-судинну систему (Bale et al., Proc. Natl. Acad. Sci., 2004, vol. 101, pp. 3697-3702; Tang et al., Eur. Heart J., 2007, vol. 28, pp. 2561-2562), бронхоелегеневу, шлунково-кишкову, ниркову, скелетно-м'язову системи й на розвиток запальних процесів (Moffatt et al., FASEB J., 2006, vol. 20, pp. 1877-1879). Крім того, активність CRHR2 відзначається при захворюванні, що викликає атрофію кістякових м'язів, таких як саркопенія (Hinkle et al., Endocrinology, 2003, vol. 144(11), pp. 4939 1 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 4946). CRHR2 також залучений до моторної діяльності і споживання їжі (Ohata et al., Peptides, 2004, vol. 25, pp. 1703-1709), бере участь у кардіозахисті (Brar et al., Endocrinology, 2004, vol. 145(1), pp. 24-35) і чинить бронхорелаксуючу й протизапальну дії (Moffatt et al., FASEB J., 2006, vol. 20, pp. E1181-E1187). Пегілювання - процес приєднання одного або декількох поліетиленгліколевих (ПЕГ) полімерів до молекул. Як правило, процесу пегілювання піддаються антитіла, пептиди й протеїни з метою поліпшення їх біофармацевтичних властивостей і подолання сприйнятливості сполук до дії протеолітичних ферментів, збільшення періоду напівжиття в кровотоці, збільшення строку придатності при зберіганні, підвищення розчинності, зниження ниркового кліренсу й потенціалу утворення антитіл до введеного препарату (Harris et al., Nature, 2003, vol. 2, pp. 214221; Hamidi et al., Drug Delivery, 2006, 3, pp. 399-409; Bailon et al., PSTT, 1998, vol. 1(8), pp. 352356). Недавно Управління по контролю над якістю харчових продуктів і лікарських засобів США (FDA) схвалило використання ПЕГ-полімерів як середовища або основи для продуктів харчування, косметичних і лікарських препаратів. У цілому, ПЕГ-полімери є відносно неімуногенними, мають низьку токсичність і виводяться в незміненому вигляді із сечею або калом. Ці властивості можуть давати лікарському препарату ряд клінічних переваг, якщо вдосконалити процес таким чином, щоб зберегти або поліпшити афінність, ефективність і фармакологічний профіль вихідної молекули. Короткий опис Винахід описує загальні й переважні варіанти здійснення винаходу, обумовлені, відповідно, незалежними й залежними пунктами формули винаходу, які додаються до даного документа й включаються в нього шляхом посилання. Переважні й зразкові властивості винаходу стають очевидними з докладного опису, представленого нижче з посиланням на супровідні малюнки. У конкретних варіантах здійснення винахід стосується пептиду, що має агоністичну активність відносно рецептора кортикотропін-рилізинг-гормону 2 типу (CRHR2), де зазначений пептид має амінокислотну послідовність LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I, де щонайменше одна амінокислота пептиду заміщена X, за умови, що заміщення зроблено не в положеннях 3, 29 і 33 амінокислотної послідовності, і де X означає цистеїн, тирозин або глутамінову кислоту, або їх фармацевтично прийнятні солі або аміди. Пептиди-агоністи CRHR2, що становлять предмет даного винаходу, містять амінокислотні послідовності, відносно схожі з послідовностями людського стрескопіну й людського урокортину III. В одному з варіантів здійснення заміщення амінокислоти вищевказаної амінокислотної послідовності стосується групи заміщень: X на L у положенні 1; X на S у положенні 2; X на D у положенні 4; X на V у положенні 5; X на P у положенні 6; X на T у положенні 7; X на N у положенні 8; X на I у положенні 9; X на M у положенні 10; X на N у положенні 11; X на L у положенні 12; X на L у положенні 13; X на F у положенні 14; X на N у положенні 15; X на I у положенні 16; X на A у положенні 17; X на K у положенні 18; X на A у положенні 19; X на K у положенні 20; X на N у положенні 21; X на L у положенні 22; X на R у положенні 23; X на A у положенні 24; X на Q у положенні 25; X на A у положенні 26; X на A у положенні 27; X на A у положенні 28; X на A у положенні 30; X на H у положенні 31; X на L у положенні 32; X на A у положенні 34; X на Q у положенні 35; і X на I у положенні 36. Винахід також стосується фармацевтично прийнятній солі або аміду пептидів-агоністів CRHR2 і, крім того, включає фармацевтичну композицію, що містить зазначені пептиди в комбінації з одним або декількома фармацевтично прийнятними наповнювачами. В іншому варіанті здійснення амінокислотна послідовність пептиду-агоніста CRHR2 вибрана із групи, що складається з: XLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI-NH2; XTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI-NH2; XFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI-NH2; і XKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2. У ще одному варіанті здійснення кон’югат містить амінокислотну послідовність і лінкер, приєднаний до X пептиду-агоніста CRHR2. Х, переважно, є цистеїном. У переважному варіанті лінкер представлений ацетамідом або N-етилсукцинімідом. В іншому варіанті здійснення кон’югат пептиду-агоніста містить поліетиленгліколь (ПЕГ), приєднаний до лінкеру, молекулярна маса якого не перевищує приблизно 80 кДа. У переважному варіанті ПЕГ має молекулярну масу, вибрану із групи молекулярних мас, що складається із приблизно 2 кДа, приблизно 5 кДа, приблизно 12 кДа, приблизно 20 кДа, приблизно 30 кДа або приблизно 40 кДа. 2 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 Лінкер забезпечує більш просте й селективне приєднання ПЕГ до пептиду з урахуванням положення в амінокислотній послідовності, а пегілювання пептиду дозволяє продовжити період напівжиття пегільованого пептиду й тим самим збільшити тривалість терапевтичного впливу в організмі пацієнта. Таким чином, переважним є таке заміщення на амінокислотну послідовність пептиду-агоніста CRHR2, у якому щонайменше одна амінокислота в послідовності є амінокислотою типу X. Це буде сприяти тому, що пегілювання пептиду буде спрямовано щонайменше на одне положення в послідовності. У деяких варіантах здійснення пептид-агоніст CRHR2 містить одну із зазначених амінокислотних послідовностей, що містяться в SEQ ID NO. 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 35, 36, 37, 39, 40 і 41. В іншому аспекті даний винахід стосується способу лікування пацієнта, що страждає на або має діагностоване захворювання, порушення або патологічний стан, опосередкований активністю кортикотропін-рилізинг-гормону 2 типу, вибраний із групи, що складається з метаболічного захворювання й серцевої недостатності. Спосіб передбачає введення пацієнтові ефективної кількості пептиду-агоніста CRHR2 даного винаходу, що становить предмет, для терапії захворювання. Додаткові варіанти здійснення й переваги винаходи стануть очевидні з докладного опису, малюнків, прикладів і формули винаходу, представлених нижче. Короткий опис малюнків На фіг. 1A і 1B представлений слідовий аналіз методом ВЕРХ пептиду-агоніста з послідовністю SEQ ID NO:102, дериватизованого йодоацетамід-ПЕГ, після реакції тривалістю 2 години й після очищення, відповідно. На фіг. 1C представлений графік мас-спектроскопії пептиду-агоніста з послідовністю SEQ ID NO:102, дериватизованого йодоацетамід-ПЕГ. На фіг. 2 представлений агоністичний потенціал і селективність пептидів-агоністів до людських CRHR1 і CRHR2, відповідно. На фіг. 3 представлений ефект конкурентного антагонізму між пептидом-агоністом з послідовністю SEQ ID NO:1 і антисаувагіном-30 (SEQ ID NO:118). На фіг. 4 представлені криві агоністичного концентраційного ефекту різних пептидівагоністів, отримані шляхом вимірювання рівня цАМФ в SK-N-MC клітинах, трансфікованих hCRHR2. На фіг. 5 представлені криві концентраційного ефекту агоніста h-SCP (SEQ ID NO:1), отримані шляхом вимірювання рівня цАМФ в SK-N-MC клітинах, трансфікованих h-CRHR2 за відсутності й у присутності 10 мкм пептидів-агоністів CRHR2 з послідовністю SEQ ID NO:110, SEQ ID NO:111 і SEQ ID NO:112, відповідно. На фіг. 6 зображене розслаблення попереднє звуженої ізольованої аорти щура під дією пептидів-агоністів з послідовністю SEQ ID NO:1 і SEQ ID NO:115 (h-UCN2). На фіг. 7 представлені частота серцевих скорочень, тиск лівого шлуночка й зміни коронарного перфузійного тиску в моделях серця кролика, перфузованого по методу Лангендорфа, у присутності пептиду-агоніста з послідовністю SEQ ID NO:1 і плацебо у вигляді контрольного середовища. Докладний опис винаходу Даний винахід стосується нових пептидів, а саме - пептидів-агоністів рецептора CHRH2 і композицій для лікування, що їх містять, зниження інтенсивності або інгібування патологічних станів серцево-судинної системи, включаючи, крім іншого, серцеву недостатність і порушення обміну речовин. В одному з варіантів здійснення винаходу спосіб лікування або поліпшення стану пацієнта при серцевій недостатності в зазначених цілях включає введення пацієнтові терапевтично ефективної кількості щонайменше одного пептиду-агоніста CRHR2. У деяких варіантах здійснення пептид CRHR2 є пептидом ссавця, а саме миші, щура, морської свинки, кролика, собаки, кішки, коня, корови, свині або примата, або їх модифікаціями. У переважному варіанті здійснення пептид є модифікованим пептидом людини. Приклади пептидів-агоністів CRHR2 у рамках даного винаходу докладно описані нижче. Інший варіант здійснення винаходу включає реакційноздатну групу, ковалентно зв'язану з пептидом-агоністом. Реакційноздатна група вибрана за здатність утворювати стабільний ковалентний зв'язок з полімером або іншими хімічними частинками, які можуть продовжити період напівжиття пептиду в кровотоці пацієнта. В одному з варіантів здійснення як такий полімер використовується поліетиленгліколевий полімер (ПЕГ-полімер), який продовжує тривалість дії пептиду в кровотоці пацієнта до виведення. У такій формі реакційноздатна група виступає як лінкер між пептидом і полімером, взаємодіючи, з одного боку, з однією або 3 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 декількома амінокислотами пептиду, а з іншого боку - з полімером. В альтернативному варіанті здійснення реакційноздатна група початково, до утворення хімічного зв'язку з пептидом, пов'язана з ПЕГ. У переважному варіанті здійснення модифікованих пептидів лінкерна група представлена сукцинімідом, більш конкретно –N-етилсукцинімідом або ацетамідом. Крім того, лінкером може бути вінілсульфон або ортопіридилу дисульфід. Переважно, хімічні модифікації виконані на ізольованих пептидах, наприклад, для підвищення реакційної ефективності. Лінкери, використовувані для зв'язування пептиду й ПЕГ-частинок, повинні передавати хазяїнові мінімальну імуногенність і токсичність. Приклади таких лінкерів можна знайти в роботі Bailon et al., PSTT, 1998, vol. 1(8), pp. 352-356 або Roberts et al., 2002, Adv. Drug Del. Rev., vol. 54, pp. 459-476. Приклади прийнятних хімічних частинок, зокрема, ПЕГ або еквівалентних полімерів, описані в роботі Greenwald et al., 2003, Adv. Drug Del. Rev., vol. 55, pp. 217-250. Наприклад, неокарциностатин, співполімер стиролу й малеїнового ангідриду, співполімер (гідроксипропіл)метакриламіду, декстран, поліглутамінова кислота, гідроксіетиловий крохмаль і поліаспарагінова кислота можуть використовуватися як полімерні системи для здійснення доставки й мають фармакокінетичні характеристики, аналогічні ПЕГ-системі. У деяких варіантах здійснення винаходу пептид-агоніст CRHR2 містить амідіваний С-кінець. Така модифікація може бути виконана на ізольованому очищеному пептиді або, у випадку твердофазного синтезу, у процесі синтезу. Такі процеси докладно розглянуті Ray et al., Nature Biotech., 1993, vol. 11, pp. 64-70; Cottingham et al., Nature Biotech., 2001, vol. 19, pp. 974-977; Walsh et al., Nature Biotech., vol. 24, pp. 1241-1252, а також у патенті США № 2008/0167231. У конкретному варіанті здійснення винаходу композиція включає пептид амінокислотної послідовності в порядку, передбаченому в SEQ ID NO:82 або SEQ ID NO:102, що містить CONH2 на карбокси-кінці й лінкер, зв'язаний із цистеїновими залишками в положенні 28 амінокислотної послідовності, де як лінкер виступає N-етилсукцинімід або ацетамід, і лінкер приєднаний до ПЕГ-полімеру з молекулярною масою приблизно 20 кДа. Крім того, один з варіантів здійснення даного винаходу характеризує спосіб лікування пацієнта, що страждає на або має діагностоване захворювання, порушення або патологічний стан, опосередкований активністю CHRH2, що припускає введення пацієнтові терапевтично ефективної кількості щонайменше одного пептиду-агоніста CRHR2. Інший варіант здійснення даного винаходу також включає спосіб лікування або вповільнення прогресування одного або декількох патологічних станів, захворювань або порушень, опосередкованих CHRH2, при цьому зазначений спосіб включає введення пацієнтові в терапевтичних цілях фармацевтично ефективної кількості щонайменше одного пептиду-агоніста CRHR2. У додатковому варіанті здійснення винаходу передбачений процес виготовлення фармацевтичної композиції, що включає додавання одного з пептидів-агоністів CRHR2 і фармацевтично прийнятного носія. Терміни й визначення Для кращого розуміння даного винаходу в документі приводяться наступні визначення, малюнки й опису варіантів здійснення. Нижче зазначені абревіатури, що іноді зустрічаються в даному описі: розрахункова кількість (pА50) або розрахункова концентрація впливу (pЕС50)=кологарифм по основі 10 концентрації агоніста, необхідної для досягнення половини максимального ефекту; СПС=стандартна погрішність середнього; Log КД=логарифм по основі 10 коефіцієнта дозування агоніста; ММ=молекулярна маса; цАМФ=аденозин 3’,5’-циклічний монофосфат; кДНК=комплементарна ДНК; т. п. р.=тисяча пар нуклеотидів (1000 пар нуклеотидів); кДа=кілодальтон; АТФ=аденозин 5’-трифосфат; нт=нуклеотид; п. о.=пара основ; ПААГ=електрофорез у поліакриламідному гелі; ПЛР=полімеразна ланцюгова реакція, нм=наномолярний. Терміни «що складається», «що містить» і «що включає» використовуються в даному документі в їхньому відкритому, необмеженому значенні. Терміни «приймання» або «уведення» означають доставку в організм пацієнта лікарського препарату будь-яким способом, відомим у фармакології. Термін «композиція» означає продукт, що містить сполуку, яка становить предмет даного винаходу (наприклад, продукт, що містить зазначені інгредієнти в зазначених кількостях, а також будь-який продукт, отриманий напряму або опосередковано в результаті комбінування зазначених інгредієнтів у зазначених кількостях). Терміни «сполука» або «лікарський препарат» означають пептид-агоніст CRHR2 або його фармацевтично прийнятні форми. Термін «кон’югат» означає хімічну сполуку, отриману шляхом приєднання двох або більше сполук. Термін «лікарська форма» призначений для позначення одного або декількох сполук у 4 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 середовищі, носії, розчиннику або пристрій, прийнятний для введення пацієнтові. Термін «пероральна лікарська форма» призначений для позначення лікарської форми, придатної для перорального введення. Термін «доза» означає одиницю лікарського препарату. Як правило, доза обумовлена лікарською формою. Уведення дози лікарського препарату проводиться відповідно до різних режимів дозування. Найпоширеніші режими дозування включають приймання препарату перорально один раз на день (qd), перорально два рази на день (bid) і перорально три рази на день (tid). Термін «форми» включає різні ізомери й суміші одного або декількох пептидів-агоністів CRHR2. Термін «ізомер» означає сполуки, які мають однаковий хімічний склад і молекулярну масу, але відрізняються за фізичними і/або хімічними властивостями. Подібні сполуки мають однакові кількості атомів кожного типу, але відрізняються за структурою. Структурні відмінності можуть проявлятися в будові сполук (геометричні ізомери) або в здатності сполуки повертати площину поляризації падаючого світла (стереоізомери). Термін «стереоізомер» стосується ізомерів, що мають ідентичну будову, які відрізняються тільки розташуванням атомів у просторі. Енантіомери й діастереомери - це стереоізомери, у яких асиметрично заміщений атом вуглецю виступає як хіральний центр. Термін «хіральний» стосується молекули, яка не сполучається зі своїм дзеркальним відбиттям, припускаючи відсутність осі й площини або центру симетрії. Термін «лікарський препарат» означає продукт, використовуваний для профілактики, лікування або зниження інтенсивності прояву симптомів розладів, пов'язаних із прийманням речовин, наприклад, залежності, зловживання або порушень, викликаних прийманням препаратів, у пацієнта. Терміни «пацієнт», «індивід» або «особина» означають тварину, переважно, ссавця, більш переважно - людину, якій потрібне лікування. Термін «фармацевтично прийнятний» включає молекулярні суті й композиції, ступінь чистоти і якість яких дозволяють використовувати їх у складі композиції або лікарського препарату, що становить предмет даного винаходу. Оскільки об'ємом даного винаходу передбачене застосування препарату як у лікуванні людини (клінічному й безрецептурному), так і у ветеринарії, розробка композиції або лікарського препарату передбачає можливість застосування як при лікуванні людини, так і у ветеринарії. Термін «фармацевтично прийнятний наповнювач» означає речовину, яка є нетоксичною, біологічно стерпною і іншим способом біологічно прийнятною для призначення пацієнтові, таку як інертна речовина, додана у фармакологічну композицію або іншим способом використане як транспорт, носій або розріджувач для підвищення ефективності сполуки, що становить предмет винаходу, і сумісна із цією сполукою. Приклади наповнювачів включають карбонат кальцію, фосфат кальцію, різні цукри й типи крохмалів, похідні целюлози, желатин, рослинні олії й поліетиленгліколі. Термін «фармацевтично прийнятна сіль» означає кислоту або основну сіль сполук, що становлять предмет даного винаходу, ступінь чистоти і якість яких дозволяють використовувати їх у складі композиції або лікарського препарату, що становить предмет даного винаходу, і є добре стерпною й нетоксичною в тій мірі, яка дозволяє використовувати її у фармацевтичних препаратах. Відповідні фармацевтично прийнятні солі включають солі приєднання кислоти, які, наприклад, можуть бути отримані шляхом змішування розчину сполуки з розчином фармацевтично прийнятної кислоти, такої як соляна кислота, сірчана кислота, фумарова кислота, малеїнова кислота, бурштинова кислота, оцтова кислота, бензойна кислота, лимонна кислота, винна кислота, вугільна кислота або фосфорна кислота. Термін «пептид-агоніст CRHR2» означає пептид або його похідні, що демонструє агоністичну активність до рецептора кортикотропін-рилізинг-гормону 2 типу (CRHR2). Переважно, пептид-агоніст CRHR2 є похідним або варіантом стрескопіну, який також має активність до рецептора кортикотропін-рилізинг-гормону 1 типу (CRHR1). Пептид-агоніст CRHR2, як правило, являє собою селективний агоніст CRHR2 з меншою активністю до CRHR1. Таким чином, під селективністю до рецептора розуміється потенціал пептиду викликати відповідну активну реакцію в рецепторі, відносно якого проявляється селективність пептиду, на відміну від інших рецепторів, у яких пептид також може викликати відповідну активну реакцію, але з меншим потенціалом. Визначення пептиду-агоніста CRHR2 не обмежується тільки агоністом, а також поширюється на часткові агоністи. Активність пептиду-агоніста CRHR2 до CRHR1 і CRHR2 можна оцінити, наприклад, за допомогою проби з 3’,5’-циклічним аденозинмонофосфатом (цАМФ), активність якого аналогічна активності стрескопіну (h-SCP) стосовно CRHR2. Термін «терапевтично ефективна кількість» означає таку кількість сполуки, яка викликає 5 UA 104877 C2 5 10 15 20 25 30 35 біологічно або медично значиму відповідь із боку системи тканин організму тварини або людини, очікуваний дослідником, ветеринаром, лікарем або іншим фахівцем, і який викликає полегшення симптоматики захворювання або патологічного стану, лікування якого здійснюється. Термін «лікувати» у контексті даного документа, якщо не зазначено інше, означає викликати зворотний розвиток, полегшення протікання, уповільнення розвитку або здійснювати профілактику порушень або патологічних станів, до яких застосовується термін, або одного або більше симптомів такого порушення або патологічного стану. Термін «лікування» у контексті даного документа, якщо не зазначено інше, означає виконання лікувальних заходів. Сполуки Даний винахід стосується наступних пептидів і їх модифікацій. Сполуки, що становлять предмет даного винаходу, також включають нові й селективні пептиди-агоністи CRHR2 і їх модифікації. Крім того, сполуки, що становлять предмет даного винаходу, включають пептидні частинки, які зв'язують або утворюють комплекси з CRHR2, такі як h-SCP або пептиди-міметики h-SCP. Переважними сполуками є пептиди, що мають агоністичну активність до рецепторів CRHR2, наприклад, згідно із пробою із цАМФ, з pА50, що відповідає значенню в межах приблизно від 7,5 і вище, або pКi (кологарифм KI), що виміряний у пробі з радіологічним зв'язуванням і лежить у межах від приблизно 7,5 і вище. Поряд з рецепторзв’язувальною активністю пептиди-агоністи CRHR2 повинні частково активувати рецептор. У цьому випадку переважними є пептиди, гомологічні h-SCP, тому що ці пептиди мають схожі природні фізичні й хімічні властивості. Члени сімейства кортикотропін-рилізинг факторів мають помірковано короткий період напівжиття. Пептиди-агоністи CRHR2 мають унікальний терапевтичний профіль. Для лікування розладів, опосередкованих CRHR2, включаючи, крім іншого, серцево-судинні захворювання й порушення обміну речовин, один з варіантів здійснення даного винаходу передбачає форму пептиду-агоніста тривалої дії. Пептид-агоніст CRHR2 тривалої дії забезпечує певні переваги при лікуванні хронічних розладів, коли тривалий терапевтичний вплив і дотримання хворим режиму й схеми лікування викликають труднощі. У зв'язку із цим, один з варіантів здійснення даного винаходу в цілому допускає зміну(-и) послідовності h-SCP, сайти специфічної послідовності й ураховує просторовий або стеричний вплив для збереження бажаного терапевтичного профілю й/або залежність структурної активності відносно CRHR2. Приклади пептидів-агоністів CRHR2, які при необхідності можуть бути амідівані на C-кінцях, наведено в таблицях 1-5. Як реакційноздатна групи або лінкер, переважно, використовуються сукцинімід або ацетамід. Модифіковані пептиди при необхідності можуть містити ПЕГ-групу. ПЕГ варіюється по довжині й масі, переважно, має молекулярну масу приблизно 20 кДа. При необхідності можливо використовувати декілька реакційноздатних груп, однак переавжним є наявність однієї реакційноздатної групи. Таблиця 1 Людський стрескопін з амідіваним C-кінцем і цис-варіантними пептидами-агоністами CRHR2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 CKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TCFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKCTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFCL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTC SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL CLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SCDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLCVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDCP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVC TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP CNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TCIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNCMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNICN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMC LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN CLFNI AKAKN LRAQA AANAH LMAQI-NH2 6 послідовність SEQ ID NO:1 послідовність SEQ ID NO:2 послідовність SEQ ID NO:3 послідовність SEQ ID NO:4 послідовність SEQ ID NO:5 послідовність SEQ ID NO:6 послідовність SEQ ID NO:7 послідовність SEQ ID NO:8 послідовність SEQ ID NO:9 послідовність SEQ ID NO:10 послідовність SEQ ID NO:11 послідовність SEQ ID NO:12 послідовність SEQ ID NO:13 послідовність SEQ ID NO:14 послідовність SEQ ID NO:15 послідовність SEQ ID NO:16 послідовність SEQ ID NO:17 UA 104877 C2 Продовження таблиці 1 TKFTL SLDVP TNIMN LCFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLCNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFCI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNC AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI CKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI ACAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKCKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKACN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKC LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN CRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LCAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRCQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRACA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQC AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA CANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA ACNAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AACAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANCH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAC LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH CMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LCAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMCQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMACI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQC-NH2 послідовність SEQ ID NO:18 послідовність SEQ ID NO:19 послідовність SEQ ID NO:20 послідовність SEQ ID NO:21 послідовність SEQ ID NO:22 послідовність SEQ ID NO:23 послідовність SEQ ID NO:24 послідовність SEQ ID NO:25 послідовність SEQ ID NO:26 послідовність SEQ ID NO:27 послідовність SEQ ID NO:28 послідовність SEQ ID NO:29 послідовність SEQ ID NO:30 послідовність SEQ ID NO:31 послідовність SEQ ID NO:32 послідовність SEQ ID NO:33 послідовність SEQ ID NO:34 послідовність SEQ ID NO:35 послідовність SEQ ID NO:36 послідовність SEQ ID NO:37 послідовність SEQ ID NO:38 послідовність SEQ ID NO:39 послідовність SEQ ID NO:40 послідовність SEQ ID NO:41 Таблиця 2 Цис-варіантний пептид стрескопіну з N-етилсукцинімідною (NES) реакційноздатною групою TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQC(NES)-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAC(-NES) LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AAC(-NES)AH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AC(-NES)NAH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA C(-NES)ANAH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES)QA AANAH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKN C(-NES)RAQA AANAH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAKC(-NES) LRAQA AANAH LMAQINH2 TKFTL SLDVP TNIMN LLFNI AKAC(-NES)N LRAQA AANAH LMAQINH2 TKFTL SLDVP TNIMN LLFNC(-NES) AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFC(-NES)I AKAKN LRAQA AANAH LMAQINH2 TKFTL SLDVP TNIMN LC(-NES)FNI AKAKN LRAQA AANAH LMAQINH2 TKFTL SLDVP TNIMN C(-NES)LFNI AKAKN LRAQA AANAH LMAQINH2 7 послідовність SEQ ID NO:42 послідовність SEQ ID NO:43 послідовність SEQ ID NO:44 послідовність SEQ ID NO:45 послідовність SEQ ID NO:46 послідовність SEQ ID NO:47 послідовність SEQ ID NO:48 послідовність SEQ ID NO:49 послідовність SEQ ID NO:50 послідовність SEQ ID NO:51 послідовність SEQ ID NO:52 послідовність SEQ ID NO:53 послідовність SEQ ID NO:54 UA 104877 C2 Таблиця 3 Пегільовані цис-варіантні пептид-агоністи CRHR2 з N-етилсукцинімідним (NES) лінкером і ПЕГ масою приблизно 20 кДа C(-NES-PEG)KFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TC(-NES-PEG)FTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKC(-NES-PEG)TL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFC(-NES-PEG)L SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTC(-NES-PEG) SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL C(-NES-PEG)LDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SC(-NES-PEG)DVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLC(-NES-PEG)VP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDC(-NES-PEG)P TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVC(-NES-PEG) TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP C(-NES-PEG)NIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TC(-NES-PEG)IMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNC(-NES-PEG)MN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIC(-NES-PEG)N LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMC(-NES-PEG) LLFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN C(-NES-PEG)LFNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LC(-NES-PEG)FNI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLC(-NES-PEG)NI AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFC(-NES-PEG)I AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNC(-NES-PEG) AKAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI C(-NES-PEG)KAKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AC(-NES-PEG)AKN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKC(-NES-PEG)KN LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAC(-NES-PEG)N LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKC(-NES-PEG) LRAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN C(-NES-PEG)RAQA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LC(-NES-PEG)AQA AANAH LMAQI-NH2 8 послідовність SEQ ID NO:55 послідовність SEQ ID NO:56 послідовність SEQ ID NO:57 послідовність SEQ ID NO:58 послідовність SEQ ID NO:59 послідовність SEQ ID NO:60 послідовність SEQ ID NO:61 послідовність SEQ ID NO:62 послідовність SEQ ID NO:63 послідовність SEQ ID NO:64 послідовність SEQ ID NO:65 послідовність SEQ ID NO:66 послідовність SEQ ID NO:67 послідовність SEQ ID NO:68 послідовність SEQ ID NO:69 послідовність SEQ ID NO:70 послідовність SEQ ID NO:71 послідовність SEQ ID NO:72 послідовність SEQ ID NO:73 послідовність SEQ ID NO:74 послідовність SEQ ID NO:75 послідовність SEQ ID NO:76 послідовність SEQ ID NO:77 послідовність SEQ ID NO:78 послідовність SEQ ID NO:79 послідовність SEQ ID NO:80 послідовність SEQ ID NO:81 UA 104877 C2 Продовження таблиці 3 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAC(-NES-PEG)A AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQC(-NES-PEG) AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA C(-NESPEG)ANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AC(-NESPEG)NAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AAC(-NESPEG)AH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANC(-NESPEG)H LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAC(-NESPEG) LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH C(NES-PEG)MAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LC(NES-PEG)AQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMC(NES-PEG)QI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAC(NES-PEG)I-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQC(-NES-PEG)-NH2 послідовність SEQ ID NO:82 послідовність SEQ ID NO:83 послідовність SEQ ID NO:84 послідовність SEQ ID NO:85 послідовність SEQ ID NO:86 послідовність SEQ ID NO:87 послідовність SEQ ID NO:88 послідовність SEQ ID NO:89 послідовність SEQ ID NO:90 послідовність SEQ ID NO:91 послідовність SEQ ID NO:92 послідовність SEQ ID NO:93 послідовність SEQ ID NO:94 Таблиця 4 Пегільовані цис-варіантні пептиди-агоністи CRHR2 з ПЕГ різної молярної маси й Nетилсукцинімідним (NES) або ацетамідним (IA) лінкером TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW2000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW5000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW12000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW20000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG-NEMª MW20000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW30000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW40000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-NES-PEG MW80000 & BRANCHED)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-IA-PEG MW20000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-IA-PEG MW30000)QA AANAH LMAQI-NH2 TKFTL SLDVP TNIMN LLFNI AKAKN LRC(-IA-PEG MW40000)QA AANAH LMAQI-NH2 TKFTL SLDVP TC(-IA-PEG MW20000)IMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 9 послідовність SEQ ID NO:95 послідовність SEQ ID NO:96 послідовність SEQ ID NO:97 послідовність SEQ ID NO:82 послідовність SEQ ID NO:98 послідовність SEQ ID NO:99 послідовність SEQ ID NO:100 послідовність SEQ ID NO:101 послідовність SEQ ID NO:102 послідовність SEQ ID NO:103 послідовність SEQ ID NO:104 послідовність SEQ ID NO:105 UA 104877 C2 Продовження таблиці 4 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAC(-IA-PEG MW20000) LMAQI-NH2 послідовність SEQ ID NO:106 ªNES-PEG-NEM - лінійний ПЕГ із двома кінцями, на незчепленому кінці ланцюжка ПЕГ – Nетилмалеімідний кеп. Таблиця 5 Пептиди-агоністи CRHR2 з укороченою амінокислотною (аа) послідовністю в порівнянні з пептидом SEQ ID NO:1 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 KFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI-NH2 TLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI-NH2 LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I-NH2 SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI-NH2 LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI-NH2 DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI-NH2 FTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI-NH2 5 10 15 20 25 30 40aa послідовність SEQ ID NO:1 39aa послідовність SEQ ID NO:107 37aa послідовність SEQ ID NO:108 36aa послідовність SEQ ID NO:109 35aa послідовність SEQ ID NO:110 34aa послідовність SEQ ID NO:111 33aa послідовність SEQ ID NO:112 h-UCN3 послідовність SEQ ID NO:116 До лікарських сполук, що становлять предмет даного винаходу, також належать суміші стереоізомерів або кожний окремий чистий або переважно чистий ізомер. Наприклад, дана сполука може додатково мати один або декілька асиметричних центрів на атомі вуглецю, що несуть будь-який із заміснтків. Таким чином, сьогодення сполука може існувати у формі енантіомера або діастереомера, або їх суміші. Якщо дана сполука містить подвійний зв'язок, то вона може існувати у формі геометричної ізомерії (цис-сполука, транс-сполука), а якщо дана сполука містить такий ненасичений зв'язок, як, наприклад, карбонільний, дана сполука може існувати у формі таутомера, і до даної сполуки також належать зазначені ізомери і їх суміші. У процесі одержання даної сполуки може використовуватися вихідна сполука у формі рацемічної суміші, енантіомера або діастереоізомера. Якщо дана сполука отримана у формі діастереоізомера або енантіомера, їх можна розділити традиційним способом, наприклад, хроматографією або дробною кристалізацією. Крім того, до даної сполуки належать його міжмолекулярні солі, гідрати, сольвати або її поліморфні варіанти. Крім того, прийнятними є такі лікарські сполуки, які виявляють місцеву фізіологічну або системну дію або після проникнення через слизову, або - у випадку перорального введення після доставки зі слиною в шлунково-кишковий тракт. Лікарські форми, приготовлені на основі композицій, що становлять предмет даного винаходу, особливо придатні для створення лікарських сполук, що проявляють активність протягом тривалого періоду часу, зокрема, для лікарських препаратів, період напівжиття яких становить щонайменше кілька годин. Методи синтезу і очищення «Ізольований» пептид - це пептид, переважно вільний або відділений від клітинного матеріалу або інших забруднюючих протеїнів клітини або тканини, з яких витягнутий або ізольований пептид, або переважно вільний від хімічних вихідних речовин або інших хімічних сполук, якщо пептид синтезований хімічним шляхом. Наприклад, протеїн, переважно вільний від клітинного матеріалу, може включати препарати протеїну, що містять менше 30%, переважно 20%, більш переважно - 10%, найбільш переважно - 5%, ще більш переважно - 1% (по вазі в сухому стані) забруднюючих протеїнів. У переважних варіантах здійснення ізольований пептид є переважно чистим. Таким чином, якщо пептид отриманий рекомбінантним шляхом, він, переважно, не містить поживного 10 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 середовища для культивування, наприклад, поживного середовища для культивування, що становить менше 20%, більш переважно - 10%, більш переважно - 5 %, ще більш переважно 1% від об'єму протеїнового препарату. Якщо протеїн отриманий шляхом хімічного синтезу, він, переважно, не містить хімічних вихідних речовин або інших хімічних сполук, тобто, він відділений від хімічних вихідних речовин або інших хімічних сполук, що беруть участь у синтезі протеїну. Таким чином, препарати пептиду містять менше 30%, переважно - 20%, більш переважно - 10%, ще більш переважно - 5%, найбільш переважно - 1% (по вазі в сухому стані) хімічних вихідних речовин або сполук, відмінних від необхідного пептиду. Рекомбінантне продукування Експресія пептиду в клітинному середовищі досягається шляхом застосування ізольованих полінуклеотидів. «Ізольований» полінуклеотид - це полінуклеотид, переважно, відділений або вільний від молекул нуклеїнової кислоти з різними послідовностями нуклеїнової кислоти. Варіанти ізольованих полінуклеотидних молекул включають кДНК, геномні ДНК, РНК і антисмислові РНК. Переважні варіанти полінуклеотидів виділені з біологічних зразків, отриманих від людини, наприклад, з гістологічного препарату тканини. Для доставки й репродукції полінуклеотидів, що кодують пептид послідовності SEQ ID NO:1, можуть використовуватися вектори. Уведення таких векторів у клітину-хазяїна сприяє продукуванню кодованої мРНК або пептиду-міметика стрескопіну. В альтернативному варіанті експресуючі вектори можуть використовуватися в комбінації з очищеними елементами, включаючи, крім іншого, фактори транскрипції, РНК-полімеразу, рибосоми й амінокислоти для здійснення ефективних реакцій транскрипції/трансляції в позаклітинних умовах. Пептидиміметики стрескопіну, експресовані в результаті реакцій, можуть бути ізольовані для наступного очищення, модифікації й/або використання для приготування лікарської композиції. Термін «вектор» означає молекулу нуклеїнової кислоти, здатну транспортувати іншу нуклеїнову кислоту, з якою вона зв'язана. Прикладом вектора може служити плазміда, кругла доспіральна петля ДНК, у яку можуть бути додані додаткові сегменти ДНК. Іншим прикладом вектора є вірусний вектор, у який можуть бути додані додаткові сегменти ДНК. Деякі вектори здатні до незалежної реплікації в клітині-хазяїні, у яку введені (наприклад, бактеріальні вектори, що мають бактеріальну природу реплікації й епісомні вектори ссавця). Вектори іншого типу (наприклад, неепісомні вектори ссавця) інтегруються в геном клітини-хазяїна після введення в неї, і в такий спосіб реплікуються разом з геномом хазяїна. Крім того, деякі - експресуючі вектори здатні спрямовувати експресію генів, з якими функціонально зв'язані. Вектори, використовувані в технології рекомбінантних ДНК, можуть бути представлені у вигляді плазмід. В альтернативному варіанті здійснення фахівець може вибрати як прийнятні для застосування по призначенню інші форми векторів, такі як вірусні вектори (наприклад, ретровіруси з дефективною реплікацією, аденовіруси й аденоасоційовані віруси), що виконують аналогічні функції. Під «клітиною-хазяїном» розуміється клітина, у якій молекула ДНК або міститься на векторі, або інтегрована в клітинну хромосому. Клітина-хазяїн може бути як нативною клітиноюхазяїном, що містить молекулу ДНК усередині себе, так і рекомбінантною клітиною-хазяїном. Прикладом клітини-хазяїна може служити рекомбінантна клітина-хазяїн, що представляє собою клітину, змінену й трансфіковану екзогенною послідовністю ДНК. Трансформація клітини обумовлена екзогенною ДНК, якщо така екзогенна ДНК уводиться всередину клітинної мембрани. Екзогенна ДНК може, хоча й не обов'язково, інтегруватися (за допомогою ковалентного зв'язку) у хромосомну ДНК, що складає геном клітини. Наприклад, у прокаріотів і дріжджових грибків екзогенна ДНК може втримуватися на епісомному елементі, такому як плазміда. В еукаріотичних клітинах стабільно змінена або трансфікована клітина - це така клітина, у якій екзогенна ДНК інтегрується в хромосому й успадковується дочірніми клітинами шляхом реплікації хромосом. Така стабільність виражається здатністю еукаріотичної клітини утворювати клітинні лінії або створювати клони, що становлять популяцію дочірніх клітин, які містять екзогенну ДНК. Під терміном «клон» розуміється популяція клітин, утворених шляхом поділу від однієї клітини або загального попередника. Під терміном «клітинна лінія» розуміється клон первинної клітини, здатний до стабільного росту in vitro протягом багатьох поколінь. Як рекомбінантні клітини-хазяїни можуть виступати прокаріотичні або еукаріотичні клітини, включаючи бактерії, такі як E. coli, грибкові клітини, такі як дріжджі, клітини ссавців, такі як клітинні лінії людини, бика, свині, мавпи, гризуна, а також клітини комах, такі як клітини дрозофіли або шовковичного шовкопряда. Термін «рекомбінантна клітина-хазяїн» належать не тільки до конкретної клітини, але також до похідних або потенційних похідних цієї клітини. Зокрема, у наступних поколіннях можуть виникнути певні модифікації внаслідок мутацій або впливу середовища. Таке потомство, можливо, буде відрізнятися від материнської клітини, 11 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 однак воно також охоплюється зазначеним терміном. Приклади векторів, що відповідають принципам даного винаходу, також включають спеціальні експресуючі системи, що дозволяють ДНК курсувати між клітинами-хазяїнами, наприклад, між бактеріальною й дріжджовою клітинами, клітинами бактерії й тварини, клітинами бактерії й грибка або клітинами бактерії й безхребетного. Фахівцям у даній галузі відома множина клонуючих векторів, і вибір прийнятного клонуючого вектора входить у компетенцію відповідного фахівця. Інформація про інші прийнятні експресуючі системі як для прокаріотичних, так і для еукаріотичних клітин, представлена, наприклад, у гл. 16 і 17 публікації Maniatis et al., (1990), Molecular Cloning: A Laboratory Manual, vol. 2:16.3-16.81. Для досягнення високорівневої експресії клонованого гена або нуклеїнової кислоти, такої як кДНК, що кодує пептид-міметик стрескопіну, нуклеотидна послідовність, що відповідає послідовності пептиду-міметика стрескопіну, переважно, субклонується в експресуючий вектор, що містить сильний промотор прямої транскрипції, термінатор транскрипції/трансляції і, у випадку нуклеїнової кислоти, що кодує пептид, рибосомозв’язувальна ділянка ініціації трансляції. Прийнятні бактеріальні промотори відомі в даній галузі техніки й описані, наприклад, у публікації Sambrook et al., (1989), Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York and Macrides, 1996, Microbiol. Rev. 60(3):512-38. Бактеріальні експресуючі системи для експресії протеїнів-міметиків стрескопіну, описані в рамках даного винаходу, присутні, наприклад, у бактеріях E. coli, Bacillus sp. і Salmonella (Palva et al., 1983, Gene, 22:229-235; Mosbach et al., 1983, Nature, 302:543-545). Комплекти таких експресуючих систем доступні в продажу. Еукаріотичні експресуючі системи для клітин ссавців, дріжджових клітин і клітин комах відомі в даній галузі техніки й також доступні в продажу. У прикладах здійснення як еукаріотичний експресуючий вектор використовується бакуловірусний вектор, аденовірусний вектор, вектор на основі аденоасоційованого вірусу або ретровірусний вектор. Промотор - регуляторна послідовність ДНК, що бере участь у зв'язуванні РНК-полімерази з метою ініціації транскрипції гена. Промотори, як правило, розташовуються вище (тобто, 5’) сайту ініціації транскрипції гена. Ген - сегмент ДНК, що бере участь у продукуванні пептиду, пептид або протеїн, включає кодуючу область, некодуючі області, що передують (5'-кінцева нетрансльована область) і наступні (3'-кінцева нетрансльована область) за кодуючою областю, а також проміжні некодуючі послідовності (інтрони) між окремими кодуючими ділянками (екзонами). Кодування - визначення специфікації конкретних амінокислот або термінація сигналів у триосновних триплетах (кодонах) ДНК і мРНК. Промотор, що використовується для прямої експресії полінуклеотида, може бути вибраний за стандартною методикою відповідно до конкретного застосування. За необхідності промотор розміщується приблизно на однаковій відстані як від сайту ініціації гетерологічної транскрипції, так і від сайту ініціації транскрипції в природному середовищі. Однак для фахівця очевидно, що відстань може до деякої міри варіюватися без шкоди для промоторної функції. Крім промотору, експресуючий вектор може включати одиницю транскрипції або полігенний експресуючий кластер, який містить усі додаткові елементи, необхідні для експресії полінуклеотиду, що кодує міметик стрескопіну в клітинах-хазяїнах. Приклад експресуючого кластера містить промотор, функціонально зв'язаний з полінуклеотидною послідовністю, що кодує пептид-міметик стрескопіну, і сигнали, необхідні для ефективного поліаденілування транскрипта, рибосомозв’язувальних ділянок і термінації транскрипції. Полінуклеотидна послідовність, що кодує пептид-міметик стрескопіну собаки, може бути зв'язана з розчіплюваною послідовністю сигнального пептиду для активізації секреції кодованого протеїну трансфікованою клітиною. Приклади сигнальних пептидів включають сигнальні пептиди тканинного активатора плазміногена, інсуліну й фактора росту нейронів, а також естерази ювенільного гормону Heliothis virescens. До додаткових елементів кластера належать підсилювачі й, у випадку, якщо геномна ДНК використовується як структурний ген, інтрони з функціональними донорським і акцепторним сайтами сплайсингу. Крім промоторної послідовності, експресуючий кластер також може містити ділянку термінації транскрипції, розташовану після структурного гена, для забезпечення ефективної термінації. Ділянка термінації може бути виділена з того ж гена, що й промоторна послідовність, з гена стрескопіну людину або з різних генів. В ілюстративних варіантах здійснення допускається використання кожного з векторів, придатних для експресії в еукаріотичних і прокаріотичних клітинах, відомих у даній галузі техніки. Приклади бактеріальних експресуючих векторів включають плазміди, наприклад, плазміди на основі pBR322, pSKF, pET23D і фузійні експресуючі системи, такі як GST і LacZ. Прикладами експресуючих векторів ссавця можуть бути, наприклад, pCDM8 (Seed, 1987, Nature, 12 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 329:840) і pMT2PC (Kaufman et al., 1987, EMBO J., 6:187-195). У продажу доступні експресуючі вектори ссавця, придатні для рекомбінантної експресії пептидів, що становлять предмет винаходу, до яких належать, наприклад, pMAMneo (Clontech, Маунтин-В'ю, штат Каліфорнія), pcDNA4 (Invitrogen, Карлсбад, штат Каліфорнія), pCiNeo (Promega, Медісон, Вісконсин), pMC1neo (Stratagene, Ла Джолла, штат Каліфорнія), pXT1 (Stratagene, Ла Джолла, штат Каліфорнія), pSG5 (Stratagene, Ла Джолла, штат Каліфорнія), EBO-pSV2-neo (ATCC 37593), pBPV-1(8-2) (ATCC 37110), pdBPV-MMTneo (342-12) (ATCC 37224), pRSVgpt (ATCC 37199), pRSVneo (ATCC 37198), pSV2-dhfr (ATCC 37146), pUCTag (ATCC 37460) і lZD35 (ATCC 37565). Для забезпечення прийнятних способів ізолювання до рекомбінантних протеїнів також можуть бути додані епітопні кінці, наприклад, c-myc, гемагглютинін (HA)-tag, 6-Нis tag, мальтозозв’язувальний протеїн, VSV-G tag, anti-FLAG tag і інші відомі в даній галузі техніки. Експресуючі вектори, що містять регуляторні елементи еукаріотичних вірусів, можуть використовуватися в еукаріотичних експресуючих векторах, наприклад, вектори SV40, вектори вірусу папіломи й вектори, похідні від вірусу Епштейна-Барра. Інші приклади еукаріотичних векторів включають pMSG, pAV009/A+, pMTO10/A+, pMAMneo 5, бакуловірус pDSVE і інші вектори, що дозволяють експресувати протеїни під керівництвом ЦМВ-промотору, раннього промотору SV40, пізнього промотору SV40, металотіонеїнового промотору, промотору на основі мишачого вірусу пухлини молочної залози, промотору на основі вірусу саркоми Рауса, полігедринового промотору або інших промоторів, що довели ефективність в експресії еукаріотичних клітин. Деякі експресуючі системи мають маркери, що забезпечують ампліфікацію гена, наприклад, неоміцин, тимідинкіназа, гігроміцин-В-фосфотрансфераза й дигідрофолат редуктази. В альтернативному варіанті здійснення також допускається використання високопродуктивних експресуючих систем, що не включають ампліфікацію гена, наприклад, використання бакуловірусних векторів у клітинах комах з послідовністю, що кодує пептид-міметик стрескопіну під керівництвом полігедринового промотору або інших сильних бакуловірусних промоторів. До елементів, які можуть міститися в експресуючих векторах, також належать реплікон, що функціонує в E. coli, ген, що кодує резистентність до антибіотиків, забезпечуючи селекцію бактерій, що містять рекомбінантні плазміди, а також специфічні сайти рестрикції в другорядних областях плазмід, що забезпечують контрольоване введення еукаріотичних послідовностей. Конкретний ген резистентності до антибіотиків може бути вибраний з множини генів резистентності, відомих у даній галузі техніки. Прокаріотичні послідовності можуть бути вибрані таким чином, щоб вони не перешкоджали реплікації ДНК в еукаріотичних клітинах, якщо це необхідно або переважно. Відомі способи трансфекції можуть використовуватися для продукування клітинних ліній бактерій, ссавців, дріжджів або комах, експресуючих велику кількість міметика стрескопіну, який потім очищається стандартними методиками, наприклад, селективним осадженням такими речовинами, як сульфат амонію, хроматографія на колонках і імуносорбційне очищення. Трансформація еукаріотичних і прокаріотичних клітин може бути виконана стандартними методиками (наприклад, Morrison, 1977, J Bact., 132:349-351; Clark-Curtiss & Curtiss, Methods in Enzymology, 101:347-362). Кожна зі стандартних методик, придатних для введення чужорідних нуклеотидних послідовностей у клітини-хазяїни, може застосовуватися для введення експресуючого вектора. Такі методики мають на увазі використання реактивів, наприклад, Superfect (Qiagen), ліпосом, трансфекції фосфатом кальцію, полібрену, злиття протоплазми, електропорації, мікроін'єкції, плазмідних векторів, вірусних векторів, біолістичене прискорення частинок («генний пістолет») або будь-яких інших відомих способів уведення клонованої геномної ДНК, кДНК, синтетичної ДНК або чужорідного генетичного матеріалу в клітину-хазяїна (наприклад, Sambrook et al., supra). Вибрана методика генної інженерії повинна забезпечувати успішне введення щонайменше одного гена в клітину-хазяїна, здатну експресувати РНК, мРНК, кДНК або ген міметика стрескопіну. Як стане очевидно фахівцям, при стабільній трансфекції клітин ссавців, залежно від експресуючого вектора й використовуваної методики трансфекції, лише невелика фракція клітин здатна інтегрувати чужорідну ДНК у геном. Для того, щоб визначити й вибрати ці інтегранти, ген, що кодує селектований маркер (наприклад, резистентність до антибіотиків), може вводитися в клітину-хазяїна разом із цільовим геном. До прикладів селектованих маркерів належать маркери з резистентністю до лікарських препаратів, таких як G-418, пуроміцин, генетицин, гігроміцин і метотрексат. Клітини, стабільно трансфіковані введеною нуклеїновою кислотою, можуть бути селектовані й ідентифіковані шляхом лікарської селекції (наприклад, клітини, що інкорпорували ген селектованого маркера, виживуть, тоді як інші клітини загинуть). 13 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 Гетерологічний регуляторний елемент може бути введений у стабільну клітинну лінію або клонований мікроорганізм, у якому він функціонально зв'язується з ендогенними генами й активує їхню експресію, такими способами, як спрямована гомологічна рекомбінація, наприклад, відповідно до патенту США № 5272071 і міжнародної публікації № WO 91/06667. Після введення експресуючого вектора трансфіковані клітини, переважно, культивують в умовах, сприятливих для експресії пептиду-міметика стрескопіну, які виділяють із культури стандартними способами, зазначеними нижче. Способи культивування прокаріотичних або еукаріотичних клітин відомі в даній галузі техніки (наприклад, Sambrook et al., supra; Freshney, 1993, Culture of Animal Cells, 3rd ed). Як альтернатива використання клітинних систем для продукування пептиду допускається використання безклітинних систем, що мають здатність експресувати і синтезувати ген у прокаріотичних (Zubay G., Annu Rev Genet., 1973, 7:267-287) і еукаріотичних системах (Pelham et al., Eur J Biochem., 1976, 67:247-256; Anderson et al., Meth Enzymol., 1983, 101:635-644). У таких системах у реакціях синтезу пептиду можуть використовуватися як нуклеотиди мРНК, так і нуклеотиди ДНК. У переважному способі для безклітинного продукування пептиду використовуються лізат ретикулоцитів, РНК-полімераза, нуклеотиди, солі й інгібітор рибонуклеази в сполученій реакції транскрипції/трансляції (TNT®, Promega, Медісон, штат Вісконсин, США). Синтетичне продукування Пептиди, що становлять предмет винаходу, можуть бути отримані шляхом синтезу у твердій фазі. Спочатку ця методика була описана Merrifield, in J. Am. Chem. Soc., 15:2149-2154 (1963). Опис інших методик синтезу пептидів можна знайти, наприклад, у публікації M. Bodanszky et al., (1976) Peptide Synthesis, John Wiley & Sons, 2d Ed.; Kent and Clark-Lewis in Synthetic Peptides in Biology and Medicine, p. 295-358, eds. Alitalo, K., et al. Science Publishers, (Amsterdam, 1985), а також в інших довідкових виданнях, відомих фахівцям у даній галузі техніки. Короткий опис методики синтезу пептидів можна знайти в публікації J. Stuart і J. d. Young, Solid Phase Peptide Synthelia, Pierce Chemical Company, Rockford, III. (1984), включеній в даний документ шляхом посилання. Також допускається синтез пептидів шляхом розчинення, як описано в The Proteins, Vol. II, 3d Ed., p. 105-237, Neurath, H. et al., Eds., Academic Press, New York, N.Y. (1976). Захисні групи, прийнятні для використання в синтезі, описані в перерахованих вище публікаціях, а також у публікації J. f. W. Mcomie, Protective Groups in Organic Chemistry, Plenum Press, New York, N.Y. (1973), включеній в даний документ шляхом посилання. Як правило, такі методи синтезу мають на увазі послідовне додавання одного або декількох амінокислотних залишків у зростаючий пептидний ланцюг. Як правило, як аміногрупа, так і карбоксильна група першого амінокислотного залишку захищена прийнятною захисною групою, що селективно видаляється. Інша захисна група, що селективно видаляється, використовується для амінокислот, що містять реакційноздатну бічну групу, таку як лізин. Крім того, як у методі синтезу пептидів у твердій фазі, так і в методі синтезу пептидів розчиненням може застосовуватися методика блокового синтезу. Замість послідовного додавання окремих амінокислотних залишків використовуються попередньо сформовані блоки, що складаються із двох або більше амінокислотних залишків у послідовності, або як пускові підодиниці, або як одиниці, що послідовно додаються, а не у вигляді окремих амінокислотних залишків. За аналогією із синтезом у твердій фазі, захищена або похідна амінокислота приєднується до інертної твердої підкладки за допомогою незахищеної карбоксильної або аміногрупи. Захисна група аміногрупи або карбоксильної групи потім селективно видаляється, і додається наступна амінокислота в послідовності, що має комплементарну групу (аміногрупу або карбоксильну групу), захищену відповідним чином, яка вступає в реакцію із залишком, уже приєднаним до твердої підкладки. Захисна група аміногрупи або карбоксильної групи потім видаляється з нового доданого амінокислотного залишку, після чого додається наступна амінокислота (захищена відповідним чином) і т. д. Після зв'язування всіх необхідних амінокислот у прийнятну послідовність усі захисні групи, що залишилися, термінальної й бічної груп (і тверда підкладка) видаляються послідовно або одночасно, утворюючи шуканий пептид. Пептиди, що становлять предмет даного винаходу, переважно, позбавлені бензилованих або метилбензилованих амінокислот. Такі частини захисних груп можуть використовуватися в процесі синтезу, однак перед використанням пептиду вони видаляються. Як описано в літературі, можуть знадобитися додаткові реакції для формування міжмолекулярних зв'язків з метою зміцнення структури. Синтез у твердій фазі може здійснюватися за допомогою автоматичного синтезатора протеїну (Protemist®, Cellfree Sciences, Мацуяма, Ехіме 790-8577, Японія, Symphony SMPS-110, 14 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 Rainin, Уобурн, штат Массачусетс, США, синтезатор пептидів ABI 433A, Applied Biosystems, ФостереСіті, штат Каліфорнія, США). Такі синтезатори можуть здійснювати автоматизовані протеїнові реакції, що дозволяє забезпечити гарний контроль і оптимізацію процесу синтезу. Очищення Існує ряд методик для ізолювання або очищення пептидів відповідно до принципів даного винаходу. Наприклад, для очищення пептидів на основі фізичних властивостей, тобто, гідрофобності, може застосовуватися колонкова хроматографія. В іншому випадку протеїни, відомі здатністю до молекулярної адгезії, можуть реверсивно зливатися, утворюючи пептид відповідно до принципів даного винаходу. При наявності прийнятного ліганду злитого протеїну, пептид-міметик стрескопіну може селективно адсорбуватися колонкою очищення, після чого вивільнятися із колонки в переважно очищеному вигляді. Потім злитий протеїн може бути видалений шляхом регуляції ферментативної активності. Альтернативна стратегія колонкового очищення може включати використання антитіл, індукованих пептидом-міметиком стрескопіну. Такі антитіла можуть бути кон’юговані в матриці імуноафінних колонок, за допомогою яких проходять очищення пептиди. Рекомбінантні протеїни можуть бути ізольовані від реакцій клітини-хазяїна за допомогою відповідного методу поділу, наприклад, фракціонування солями. Цей спосіб може використовуватися для відділення небажаних протеїнів клітини-хазяїна (або протеїнів, отриманих із середовища для культивування клітин) від шуканого рекомбінантного протеїну. Як правило, як сіль використовується сульфат амонію, який осаджує протеїни, ефективно скорочуючи кількість води в суміші протеїнів (після чого вони осаджуються залежно від розчинності). Чим вища гідрофобність протеїну, тим вища ймовірність його осадження сульфатом амонію при меншій концентрації. Приклад протоколу виділення включає додавання насиченого розчину сульфату амонію до розчину протеїну, у результаті чого концентрація сульфату амонію становить 20-30%, для осадження найбільш гідрофобних протеїнів. Потім фазу, що виділилася, відкидають (за винятком випадків, коли протеїн є гідрофобним) і додають у супернатант сульфат амонію в концентрації, що дозволяє осадити шуканий протеїн. Фазу, що виділилася, розчиняють у буферному розчині, а надлишок солі видаляють для досягнення бажаного ступеня чистоти, наприклад, шляхом діалізу або діафільтрації. Для фракціонування суміші комплексних протеїнів також можуть застосовуватися інші відомі способи, основані на розчинності протеїнів. В інших прикладах способів виділення або очищення молекулярна маса пептиду відповідно до принципів даного винаходу може використовуватися для виділення пептиду із суміші протеїнів більшого або меншого розмірів шляхом ультрафільтрації через мембрани з порами різних розмірів (наприклад, мембрани Amicon або Millipore). Спочатку суміш протеїнів піддається ультрафільтрації через мембрану, розмір пор якої дозволяє пропускати протеїни з молекулярними масами меншими молекулярної маси шуканого протеїну. Після цього матеріал ультрафільтрації піддають повторній ультрафільтрації через мембрану, розмір пор якої більший молекулярної маси шуканого протеїну. Рекомбінантний протеїн проходить через мембрану й потрапляє у фільтрат, після чого фільтрат хроматографують. Модифікації хімічної структури Пептид відповідно до принципів даного винаходу може піддаватися прямим модифікаціям хімічної структури, таким як малеімідне кепування, приєднання поліетиленгліколю (ПЕГ), малеїдифікація, ацилування, алкілування, естерифікація й амідифікація, для продукування структурних аналогів пептиду. Фахівець у даній галузі техніки зможе оцінити застосовність різноманіття способів і агентів для модифікації хімічної структури, описаних, наприклад, у патентах США № 5554728, 6869932, 6828401, 6673580, 6552170, 6420339, а також у публікації патенту США № 2006/0210526 і міжнародній заявці на патент США № WO 2006/136586. У переважному варіанті здійснення модифікації хімічної структури виконуються на ізольованому пептиді, наприклад, з метою підвищення ефективності реакції. У деяких варіантах здійснення винаходу пептид відповідно до принципів даного винаходу містить амідований C-кінець. Такі модифікації хімічної структури пептиду можуть виконуватися на ізольованому очищеному пептиді або, у випадку синтезу у твердій фазі, можуть виконуватися в процесі синтезу. Огляд методик представлений в Ray et al., Nature Biotechnology, 1993, vol. 11, pp. 64-70, Cottingham et al., Nature Biotechnology, 2001, vol. 19, pp. 974-977, Walsh et al., Nature Biotechnology, 2006, vol. 24, pp. 1241-1252, а також у публікації заявки на патент США № 2008/0167231. Пептиди відповідно до принципів даного винаходу можуть містити проміжні лінкери, що зв'язують пептид і ПЕГ-групу. Такі лінкери мають мінімальну імуногенність і токсичність для клітини-хазяїна. Приклади таких лінкерів представлені в публікації Bailon et al., PSTT, 1998, vol. 15 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 1(8), pp. 352-356. У деяких варіантах здійснення винахід стосується кон’югату, що включає ізольований пептид, який переважно складається з послідовності SEQ ID NO:29, що включає CONH 2 на карбоксильному кінці й проміжний лінкер, кон’югований із цистеїновим залишком у положенні 28 амінокислотної послідовності SEQ ID NO:29. У деяких варіантах здійснення як проміжний лінкер використовується N-етилсукцинімід. У додаткових варіантах здійснення як проміжний лінкер може використовуватися вінілсульфон. У додаткових варіантах здійснення як проміжний лінкер може використовуватися ацетамід. У деяких варіантах здійснення як проміжний лінкер може використовуватися ортопіридил дисульфід. У додаткових варіантах здійснення винахід стосується кон’югату, що включає пептид з амінокислотною послідовністю SEQ ID NO:29 з CONH2 на карбоксильному кінці, Nетилсукцинімідним лінкером, кон’югованим із цистеїновим залишком у положенні 28 послідовності SEQ ID NO:29, де N-етилсукцинімідний лінкер також зв'язаний з ПЕГ-групою. У деяких варіантах здійснення молекулярна маса ПЕГ-групи може перебувати в діапазоні від приблизно 2 кДа до приблизно 80 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 20 кДа. У переважних варіантах здійснення пептид-агоніст CRHR2 містить пептид з послідовністю SEQ ID NO:82 або SEQ ID NO:102. У деяких варіантах здійснення маса ПЕГ становить приблизно 5 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 12 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 20 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 30 Так. У деяких варіантах здійснення маса ПЕГ становить приблизно 40 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 80 кДа. У деяких варіантах здійснення ПЕГ має лінійну форму. В інших варіантах здійснення ПЕГ має розгалужену форму. ПЕГ-частинки можуть бути синтезовані способами, відомими фахівцям у даній галузі техніки. Як альтернатива ПЕГ-частинки доступні в продажу, наприклад, SUNBRIGHT® ME-020MA, SUNBRIGHT® ME-050MA і SUNBRIGHT® ME-200MA (NOF corp., Японія, Sigma Aldrich, Сент-Луїс, штат Міссурі, США). Винахід також стосується фармацевтично прийнятних солей пептиду відповідно до принципів даного винаходу й способів застосування таких солей. Термін «фармацевтично прийнятна сіль» означає сіль вільної кислоти або основи пептиду, яка є нетоксичною, біологічно стерпною або іншим способом біологічно прийнятною для введення пацієнтові. Див., загалом, S.M. Berge, et al., Pharmaceutical Salts J. Pharm. Sci., 1977, 66:1-19, і довідник Handbook of Pharmaceutical Salts, Properties, Selection, and Use, Stahl and Wermuth, Eds., Wiley-vch and VHCA, Zurich, 2002. Переважними фармацевтично прийнятними солями є фармакологічно ефективні солі, які можуть контактувати із тканинами пацієнта, не викликаючи неприйнятної токсичності, роздратування або алергійної реакції. Пептид може мати в достатній мірі кислу групу або в достатній мірі основну групу, або обидва типи функціональних груп, і відповідним чином реагувати з рядом неорганічних або органічних основ, а також неорганічних або органічних кислот, з утворенням фармацевтично прийнятних солей. Приклади фармацевтично прийнятних солей включають наступні солі: сульфати, піросульфати, бісульфати, сульфіти, бісульфіти, фосфати, моногідрогенфосфати, дигідрогенфосфати, метафосфати, пірофосфати, хлориди, броміди, йодиди, ацетати, пропіонати, деканоати, каприлати, акрилати, формати, ізобутирати, капроати, гептаноати, пропіолати, оксалати, малонати, сукцинати, суберати, себакати, фумарати, малеати, бутин-1,4-діоати, гексин-1,6-діоати, бензоати, хлорбензоати, метилбензоати, динітробензоати, гідроксибензоати, метоксибензоати, фталати, сульфонати, ксиленсульфонати, фенілацетати, фенілпропіонати, фенілбутирати, цитрати, лактати, γгідроксибутирати, гліколяти, тартрати, метансульфонати, пропансульфонати, нафталін-1сульфонати, нафталін-2-сульфонати й манделати. Пептид відповідно до принципів даного винаходу містить азотисту основа, для одержання бажаної фармацевтично прийнятної солі може використовуватися будь-який прийнятний спосіб, відомий у даній галузі техніки, наприклад, обробка вільної основи неорганічною кислотою, такою як соляна кислота, бромистоводнева кислота, йодистоводнева кислота, перхлорна кислота, сірчана кислота, сульфамінова кислота, азотна кислота, борна кислота, фосфорна кислота й т. п., або органічною кислотою, такою як оцтова кислота, трифтороцтова кислота, фенілоцтова кислота, пропіонова кислота, стеаринова кислота, молочна кислота, аскорбінова кислота, малеїнова кислота, гідроксималеїнова кислота, яблучна кислота, памова кислота, ізетіонова кислота, бурштинова кислота, валеріанова кислота, фумарова кислота, сахаринова кислота, малонова кислота, піровиноградна кислота, щавлева кислота, гліколева кислота, саліцилова кислота, олеїнова кислота, пальмітинова кислота, лауринова кислота, піранозидильною кислотою, такою як глюкуронова кислота або галактуронова кислота, альфа-оксикислотою, такою як мигдальна кислота, лимонна кислота або винна кислота, амінокислотою, такою як 16 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 аспарагінова кислота або глютамінова кислота, ароматичною кислотою, такою як бензойна кислота, 2-ацетоксибензойна кислота, нафтойна кислота або корична кислота, сульфоновою кислотою, такою як лаурилсульфонова кислота, бензолсульфонова кислота, 2нафталінсульфонова кислота, патолуолсульфонова кислота, метансульфонова кислота, етансульфонова кислота, гідроксіетансульфонова кислота, циклогексансульфаміновою кислотою, а також сумішшю будь-яких сумісних кислот, наприклад, із числа тих, які наведені як приклад у даному документі, і будь-якою іншою кислотою й сумішшю кислот, розглянутих як еквіваленти або прийнятні замінники у світлі загальноприйнятої практики в даній галузі. Якщо пептид відповідно до принципів даного винаходу містить кислотну групу, таку як карбонова кислота або сульфонова кислота, то бажана фармацевтично прийнятна сіль може бути приготована за будь-якою відповідною методикою, наприклад, обробкою вільної кислоти неорганічною або органічною основою, такою як амін (первинний, вторинний або третинний), гідроксид лужного металу, гідроксид лужноземельного металу, будь-яка сумісна суміш основ, таких як наведені в прикладах даного документа, і будь-яка інша основа й суміші основ, які розглядаються як еквівалентні або прийнятні замінники, відомі будь-якому фахівцеві в даній галузі. Приклади прийнятних солей включають органічні солі, отримані з амінокислот, таких як гліцин і аргінін, аміак, карбонати, бікарбонати, первинні, вторинні й третинні аміни, і циклічні аміни, такі як бензиламіни, піролідини, піперидини, морфолін, і піперазин, і неорганічні солі, похідні натрію, кальцію, калію, магнію, марганцю, заліза, міді, цинку, алюмінію й літію. До типових органічних і неорганічних основ також належать бензатин, хлоропрокаїн, холін, діетаноламін, етилендіамін, меглумін і прокаїн. Даний винахід також стосується фармацевтично прийнятних проліків сполук і способів лікування з використанням подібних фармацевтично прийнятних проліків. Термін «проліки» означає попередник певного сполуки, який після введення пацієнтові дає сполуку in vivo у результаті хімічного або фізіологічного процесу, такого як сольволіз або ферментативне розщеплення, або при фізіологічних умовах. «Фармацевтично прийнятні проліки» означає проліки, які є нетоксичними, біологічно стерпними і іншим способом біологічно прийнятними для призначення пацієнтові. Ілюстративні процедури вибору й приготування припустимих похідних проліків описані, наприклад, у публікації Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985. Приклади проліків включають сполуки, що мають амінокислотний залишок або поліпептидний ланцюжок із двох або більше (наприклад, два, три або чотири) амінокислотних залишків, ковалентно зв'язаних по амідному або ефірному зв'язку з вільною аміно-, гідрокси-, або карбоксигрупою сполуки. Приклади амінокислотних залишків включають двадцять існуючих у природі амінокислот, які звичайно позначаються трьома буквами, а також 4-гідроксипролін, гідроксилізин, демозин, ізодемозин, 3-метилгістидин, норвалін, бета-аланін, гаммааміномасляна кислота, цитрулін гомоцистеїн, гомосерин, орнітин і метіонін сульфон. Додаткові типи проліків можуть бути отримані, наприклад, при використанні вільних карбоксильних груп структур сполуки для одержання похідних, амідів або алкілефірів. Приклади амідів включають похідні від амонію, первинні C 1-6алкіламіни й вторинні ді(C1-6алкіл) аміни. Вторинні аміни включають 5- або 6-членні циклічні гетероциклоалкільні або гетероарильні фрагменти. Приклади амідів включають похідні амонію, C 1-3алкіл первинні аміни й ді(C12алкіл)аміни. Приклади ефірів винаходу включають C 1-7алкіл, C5-7циклоалкіл, феніл, і феніл(C 16алкіл) ефіри. Переважні ефіри включають метилові ефіри. Проліки також можуть бути приготовлені шляхом використання вільних гідроксильних груп для одержання похідних з використанням груп, включаючи гемисукцинати, фосфатні ефіри, диметиламіноацетати й фосфорилоксиметилоксикарбоніли, дотримуючись процедур, таких як описані в публікації Fleisher et al., Adv. Drug Delivery Rev. 1996, 19, 115-130. Для одержання проліків можуть також використовуватися карбаматні похідні гідроксильних і аміногруп. Карбонатні похідні, сульфонатні ефіри й сульфатні ефіри гідроксигруп можуть також давати проліків. Також для одержання проліків можливе використання гідроксильних груп для приєднання (алкокси)метил- і (ацилоксі)етил- функціональних груп з утвором ефірів, де ацильна група може бути алкілефіром, необов'язково заміщеним одним або більше ефірними, аміно- або карбоксильними функціональними групами, або де зазначена ацильна група являє собою ефір амінокислоти, як зазначено вище. Проліки цього типу можуть бути приготовлені згідно з описом, представленим у публікації Greenwald, et al., J Med Chem. 1996, 39, 10, 1938-40. Вільні аміни також можуть бути отримані у вигляді амідів, сульфонамідів або фосфонамідів. Усі перераховані фрагменти проліків можуть мати у своїй структурі додаткові функціональні групи, такі як ефірні, аміно- і карбоксильні групи. Даний винахід також стосується фармацевтично активних метаболітів сполук і можливостей використання подібних метаболітів у способах, що становлять предмет даного винаходу. 17 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 «Фармацевтично активний метаболіт» означає фармакологічно активний продукт метаболізму в складі сполуки або її солі. Проліки й активні метаболіти сполуки можуть бути визначені за допомогою звичайних способів, відомих і доступних фахівцям у даній галузі. Див., наприклад, публікацію Bertolini, et al., J Med Chem. 1997, 40, 2011-2016; Shan, et al., J Pharm Sci. 1997, 86 (7), 765-767; Bagshawe, Drug Dev Res. 1995, 34, 220-230; Bodor, Adv Drug Res. 1984, 13, 224-331; Bundgaard, Design of Prodrugs (Elsevier Press, 1985), а також Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen, et al., eds., Harwood Academic Publishers, 1991). Фармацевтичні композиції У деяких варіантах здійснення винаходу пептиди-агоністи CRHR2 використовуються самі по собі або в комбінації з одним або декількома інгредієнтами, що складають фармацевтичні композиції. Фармацевтична композиція містить ефективну кількість щонайменше однієї сполуки відповідно до принципів даного винаходу. У деяких варіантах здійснення фармацевтична композиція містить пептид, що має амінокислотну послідовність SEQ ID NO:29, де пептид містить CONH 2 на карбоксильному кінці, а також включає N-етилсукцинімідний або ацетамідний лінкер, зв'язаний із цистеїновим залишком у положенні 28, при цьому зазначений лінкер також зв'язаний з ПЕГ-групою. Пегчастинки класифікуються по молекулярній масі й фізичних характеристиках. Вони можуть бути лінійними й розгалуженими, а також можуть містити одну або декілька частинок лінкеру, використовуваного для зв'язку ПЕГ з пептидним субстратом. У деяких переважних варіантах здійснення пептид містить один або два зазначені лінкери. У деяких варіантах здійснення фармацевтична композиція, що містить ПЕГ-групу, може містити ПЕГ-групу, молекулярна маса якої може варіюватися в діапазоні від приблизно 2 кДа до приблизно 80 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 2 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 5 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 12 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 20 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 30 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 40 кДа. У деяких варіантах здійснення маса ПЕГ становить приблизно 80 кДа. Подібні композиції додатково можуть містити фармацевтично прийнятний наповнювач. В описі також представлені композиції (у тому числі фармацевтичні композиції), що містять сполуку або описані в даному документі похідні, а також один або декілька фармацевтично прийнятних носіїв, наповнювачів і розчинників. У деяких варіантах здійснення винаходу композиція також може містити незначну кількість зволожувачів, емульгуючих речовин або pНбуферних речовин. В окремому варіанті здійснення фармацевтична композиція є фармацевтично прийнятною для введення людині. У деяких варіантах здійснення фармацевтична композиція містить терапевтично або профілактично ефективну кількість сполуки або похідного, описаного в даному документі. Кількість сполуки або похідних даного винаходу, що становить предмет, яка буде мати терапевтичний або профілактичний ефект, можна визначити, керуючись стандартною клінічною практикою. Зразкові ефективні кількості більш докладно будуть розглянуті нижче. У деяких варіантах здійснення винаходу композиція також може містити стабілізатор. Стабілізатор являє собою сполуку, яка знижує швидкість хімічного розпаду модифікованого пептиду, що входить до складу композиції. Прийнятні стабілізатори включають, крім іншого, антиоксиданти, такі як аскорбінова кислота, pН-буферні розчини або сольові буфери. Фармацевтичні композиції можуть бути виконані в будь-якій формі, прийнятній для введення пацієнтові, переважно людині. У деяких варіантах здійснення композиції виконані у формі розчинів, суспензій, емульсії, таблеток, пігулок, капсул, порошків і композицій з уповільненим вивільненням. Композиції також можуть бути представлені у вигляді конкретних стандартних лікарських форм. Приклади стандартних лікарських форм включають, крім іншого, наступні : таблетки, каплети, капсули, такі як м'які еластичні желатинові капсули, крохмальні капсули, пастилки, таблетки для розсмоктування, дисперсні системи, супозиторії, мазі, припарки (гарячі компреси), пасти, порошки, пов'язки, креми, гіпс, розчини, пластири, аерозолі (наприклад, назальні спреї або інгалятори), гелі, рідкі лікарські форми, прийнятні для перорального й черезслизового введення пацієнтові, включаючи суспензії (наприклад, водні або безводні рідкі суспензії, емульсії типу олія-в-воді, або рідкі емульсії типу вода-в-ілії), розчини, і еліксири; рідкі лікарські форми, прийнятні для парентерального введення пацієнтові, і стерильні тверді форми (наприклад, кристали або аморфні тверді речовини), відновлювані в рідкі лікарські форми, придатні для парентерального введення пацієнтові. У конкретному варіанті здійснення пацієнтом є ссавець, такий як корова, кінь, вівця, свиня, 18 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 птах, кішка, собака, миша, щур, кролик або морська свинка. У переважному варіанті здійснення пацієнтом є людина. Переважно, фармацевтична композиція підходить для використання у ветеринарії й для лікування людини. Відповідно до цього варіанта здійснення термін «фармацевтично прийнятний» означає схвалений контрольним органом федерального уряду або уряду штату або внесений у Фармакопею США або іншу загальновизнану фармакопею, а також придатний для призначення тваринам, більш конкретно - людям. Фармацевтичні носії, придатні для використання в складі композицій, включають стерильні рідини, такі як вода й масла, включаючи нафтові масла, а також масла тваринного, рослинного й синтетичного походження. У конкретному варіанті здійснення як олії використовується арахісова, соєва олія, мінеральне масло або кунжутна олія. Вода є переважним носієм, у випадку, якщо фармацевтична композиція призначена для внутрішньовенного введення. Фізіологічні розчини, водний розчин глюкози й розчини гліцерину також можуть застосовуватися як рідкі носії, зокрема, у розчинах дляін'єкцій. У даній галузі техніки відомі й інші приклади прийнятних фармацевтичних носіїв, наприклад, описані в публікації E. W. Martin in Remington's Pharmaceutical Sciences (1990) 18th ed. (Mack Publishing, Easton Pa.). До наповнювачів, прийнятних для використання в складі композицій, належать крохмаль, глюкоза, лактоза, сахароза, желатин, солод, рис, борошно, крейда, силікагель, стеарат натрію, гліцеролмоностеарат, тальк, хлорид натрію, сухе зняте молоко, гліцерин, пропіл, гліколь, вода й етанол. Той факт, наскільки конкретний наповнювач підходить для включення до складу фармацевтичної композиції, залежить від ряду факторів, відомих у даній галузі техніки, включаючи, крім іншого, шлях уведення й конкретні активні інгредієнти в складі композиції. У деяких варіантах здійснення винаходу композиція є безводною. Безводні композиції виготовляють із безводних інгредієнтів або інгредієнтів з низьким вологовмістом, або в умовах низької вологості. Композиції, що містять модифіковані пептиди, які включають первинний або вторинний амін, переважно, є безводними, якщо під час виробництва, упакування й/або зберігання передбачається великий контакт із вологим середовищем і/або вологою. Виготовлення й зберігання безводної композиції повинно здійснюватися таким чином, щоб зберегти безводну природу композиції. Відповідно, безводні композиції, переважно, упаковують, використовуючи матеріали, які можуть перешкоджати взаємодії з водою. Такі матеріали включені у відповідні довідники. Приклади прийнятних пакувальних матеріалів включають, крім іншого, герметичні плівки, пластики й контейнери для одноразових доз (наприклад, ампули), блістери й контурні упаковки стрічкового типу. Фармацевтичні композиції, що містять сполуки або похідні, описані в даному документі, їх фармацевтично прийнятні солі й сольвати, складені відповідно до передбачуваного способу введення. Препарати, переважно, призначені для підшкірного введення, але також можуть бути призначені для введення іншим шляхом, наприклад, шляхом інгаляції або інсуфляції (вдмухування препарату в порожнину рота або носа), внутрішньошкірним, пероральним, букальним, парентеральним, вагінальним або ректальним шляхом. Переважно, композиції складають таким чином, щоб забезпечити підвищену хімічну стабільність у процесі зберігання й транспортування. Препарати можуть бути ліофілізованими або рідкими. В одному з варіантів здійснення сполуки або похідні призначені для внутрішньовенного введення. Препарати для внутрішньовенного введення можуть містити стандартні носії, такі як фізіологічні розчини. В іншому варіанті здійснення сполуки або похідні призначені для ін'єкцій. У переважному варіанті здійснення сполуки або похідні представлені в стерильній ліофілізованій формі, переважно вільній від забруднюючого клітинного матеріалу, хімічних сполук, вірусів і токсинів. У конкретному варіанті здійснення суміші й похідні представлені в рідкій формі. В іншому конкретному варіанті здійснення препарати для ін'єкцій упаковані в індивідуальні стерильні контейнери. У конкретному варіанті здійснення препарати для ін'єкцій упаковані в індивідуальні стерильні контейнери. Препарати можуть містити або не містити консервантів. Рідкі препарати можуть бути приготовлені у вигляді суспензій, розчинів або емульсій у масляному або водному носії, і можуть містити такі агенти, як суспендуючі, стабілізуючі і/або диспергуючі речовини. Способи введення Сполука або похідне, описане в даному документі, або їх фармацевтично прийнятні солі, переважно, вводяться у вигляді компонента композиції, яка при необхідності включає фармацевтично прийнятний носій. Сполука або похідне переважно вводиться підшкірно. Іншим переважним способом уведення є внутрішньовенна ін'єкція або тривале внутрішньовенне уливання сполуки або похідного. У переважному варіанті введення забезпечує стабільну концентрацію препарату в плазмі крові шляхом повільної системної адсорбції й кліренсу сполуки або похідного, або шляхом підтримки концентрації в плазмі крові у встановлених межах 19 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 протягом певного періоду часу. У деяких варіантах здійснення сполука або похідне вводяться будь-яким іншим традиційним шляхом, наприклад, шляхом уливання або болюсної ін'єкції, або шляхом адсорбції через епітеліальну або слизову оболонку (наприклад, слизова оболонка порожнини рота, ректальна слизова оболонка й слизова оболонка кишки). Способи введення включають, крім іншого, парентеральний, внутрішньошкірний, внутрішньом'язовий, внутрішньоочеревинний, внутрішньовенний, підшкірний, інтраназальний, епідуральний, пероральний, під'язичний, інтраназальний, внутрішньоцеребральний, інтравагінальний, черезшкірний, ректальний способи введення, уведення шляхом інгаляції, або місцеве введення, зокрема, у порожнину вуха, носа, очей або нанесення на шкіру. У більшості випадків уведення приводить до вивільнення сполуки або похідного в кровотік. Препарат може бути представлений у формі таблеток, капсул, саше, драже, порошків, гранул, пастилок, порошків для відновлення, рідких препаратів або супозиторіїв. Переважно, композиції складені для внутрішньовенного уливання або болюсної ін'єкції, підшкірного уливання або болюсної ін'єкції, або для внутрішньом'язової ін'єкції. Сполука, переважно, призначена для введення непероральним шляхом. Наприклад, композиції можуть бути виготовлені у вигляді супозиторіїв для ректального застосування. У випадку композицій для парентерального введення, включаючи внутрішньовенне, внутрішньом'язове, внутрішньоочеревинне або підшкірне введення, агенти відповідно до принципів даного винаходу можуть бути приготовлені у вигляді стерильних водних розчинів або суспензій з додаванням відповідних буферних розчинів до одержання необхідних значень pН і ізотонічності, або у вигляді парентерально прийнятного масла. Відповідні рідкі носії включають розчин Рінгера, розчин глюкози й ізотонічний розчин хлориду натрію. Подібні лікарські форми можуть бути приготовлені у вигляді однодозової форми, такої як ампула або одноразове пристосування для ін'єкцій, у вигляді багатодозової форми, такої як флакони, з яких може бути відібрана необхідна кількість препарату, або у твердій формі або у формі первинного концентрату, який може бути використаний для приготування сполук для ін'єкцій. Зразкові дози розчинів для уливань можуть призначатися протягом періоду часу з інтервалами від декількох хвилин до декількох днів. У ще одному варіанті здійснення ефективна кількість пептиду відповідно до принципів даного винаходу може бути нанесена у вигляді покриття на наночастинки або закладена в «депо», придатне для підшкірної доставки (Hawkins et al., Adv Drug Deliv Rev., 2008, vol. 60, pp. 876-885, а також Montalvo et al., Nanotechnology, 2008, vol. 19, pp. 1-7). Можливе введення активних речовин інгаляційними способами. Такі способи мають на увазі використання сухих порошків (Johnson K.A., Adv Drug Del Rev., 1997, vol. 26(1), pp. 3-15) і/або аерозолів (Sangwan et al., J Aerosol Med., 2001, vol. 14(2), pp. 185-195, міжнародна заявка на патент № WO2002/094342). У варіантах здійснення способів лікування відповідно до принципів даного винаходу терапевтично ефективна кількість щонайменше однієї активної речовини вводиться пацієнтові, що страждає або має такі захворювання, порушення або патологічні стани, як серцева недостатність, діабет, атрофія кістякових м'язів і саркопенія. До інших патологічних станів належать порушення рухової діяльності, апетиту або необхідність у кардіозахисних, бронхорелаксуючих і/або протизапальних заходах. Терапевтично ефективні кількості або дозування активних речовин, що становлять предмет даного винаходу, можуть бути визначені стандартними способами, такими як моделювання, дослідження зі збільшенням дози або клінічні випробування, а також шляхом прийняття в уваги стандартних факторів, таких як спосіб або шлях уведення або доставки ліків, фармакокінетика агента, ступінь тяжкості й характер перебігу хвороби, розлади або стани, поточна або попередня терапії пацієнта, стан здоров'я й реакція на ліки пацієнта, а також здоровий глузд лікаря. Зразкова швидкість внутрішньовенного введення дози становить від приблизно 0,2 нг до приблизно 52 нг стрескопін-асоційованої активної речовини на один кілограм ваги пацієнта за хвилину, переважно - від приблизно 0,2 нг/кг/хв. до приблизно 22 нг/кг/хв., або еквівалентно від приблизно 0,3 мкг/кг до приблизно 32 мкг/кг на день. У випадку болюсного уливання або підшкірної ін'єкції загальна доза вводиться разово або частинами (наприклад, два рази на день, три рази на день, чотири рази на день, двічі на тиждень, один раз на два тижні або один раз на місяць). Для людини вагою 70 кг зразковим прийнятним дозуванням є кількість від приблизно 1 мкг/доба до приблизно 1 мг/доба. Тижневий режим дозування може використовуватися як альтернатива щоденному введенню. В іншому переважному варіанті здійснення пептид-агоніст CRHR2 з послідовністю SEQ ID NO:102, що містить ацетамідний лінкер, який зв'язує ПЕГ масою приблизно 20 кДа із цистеїновим залишком у положенні 28 пептидної послідовності, уводиться пацієнтовів терапевтичних цілях у 20 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 55 60 дозуванні 10 мкг/кг шляхом підшкірної болюсної ін'єкції. Частота такого дозування може змінюватися - один раз на день або рідше, залежно від терапевтичних потреб пацієнта й інших клінічних факторів. При досягненні поліпшення стану пацієнта, захворювання або розладу дозування може бути відрегульовано для профілактичного або підтримуючого лікування. Наприклад, дозування або частота призначення, або й те й інше, можуть бути знижені, залежно від симптомів, до рівня, при якому підтримується бажаний терапевтичний або профілактичний ефект. Зрозуміло, у випадку полегшення симптомів до прийнятного рівня лікування можна припинити. Однак при наявності рецидивів пацієнтові може знадобитися довготривале періодичне лікування. У деяких варіантах здійснення сполука вводиться в комбінації з однією або декількома біологічно активними речовинами в рамках схеми лікування. У деяких варіантах здійснення сполука вводиться до, одночасно або після введення одного або декількох біологічно активних речовин. В одному з варіантів здійснення однієї або дкількох біологічно активних речовин уводять разом зі сполукою, описаною у даному документі, у складі однієї фармацевтичної композиції. В іншому варіанті здійснення одну або декілька біологічно активних речовин уводять окремо від сполуки, описаної в даному документі, у складі різних фармацевтичних композицій. Відповідно до даного варіанта здійснення, одна або декілька біологічно активних речовин можуть бути введені пацієнтові тим самим або різними способами введення із числа тих, які використовуються для введення сполуки. В іншому варіанті здійснення сполука вводиться в комбінації з однією або декількома сполуками або композицією для зниження ризику розвитку або для лікування серцево-судинних захворювань. Сполуки або композиції, які знижують ризик розвитку або спрямовані на лікування серцево-судинних захворювань, включають, крім іншого, протизапальні засоби, антитромботичні засоби, антиагрегантні засоби, фібринолітичні засоби, тромболітики, ліпідовідновлюючі засоби, прямі інгібітори тромбіну, інгібітори anti-Хa, інгібітори anti-IІa, інгібітори рецепторів глікопротеїну IІb/IІІa і прямі інгібітори тромбіну. Приклади препаратів, які можуть уводитися в комбінації зі сполукою, описаною у даному документі, включають бівалірудин, гірудин, гіруген, ангіомакс, аргатробан, PPACK, аптамери тромбіну, аспірин, інгібітори GpІlb/IІІa (наприклад, інтегрелін), інгібітори P2Y12, тієнопіридин, тиклопідин і клопідогріл. У варіантах здійснення сполука входить до складу лікарських форм, прийнятних для введення пацієнтові в терапевтичних цілях. Процес і обладнання для приготування лікарських частинок і частинок носія описані в публікаціях Pharmaceutical Sciences, Remington, 17th Ed., pp. 1585-1594 (1985); Chemical Engineers Handbook, Perry, 6th Ed., pp.21-13 to 21-19 (1984), Journal of Pharmaceutical Sciences, Parrot, Vol. 61, No. 6, pp. 813-829 (1974) і Chemical Engineer, Hixon, pp. 94-103 (1990). Кількість сполуки, включеної до складу лікарських форм відповідно до принципів даного винаходу, як правило, може варіюватися в межах від приблизно 10 до приблизно 90% по вазі від ваги композиції залежно від терапевтичних показань і періоду введення, наприклад, кожні 12 годин, кожні 24 години і т. д. Залежно від дози сполуки, яку необхідно ввести, допускається використання однієї або декількох лікарських форм. Залежно від рецептури композиції сполука переважно може бути представлена у вигляді солі соляної кислоти (HСl) або у вигляді вільної основи. Крім того, даний винахід також стосується фармацевтичної композиції або фармацевтичної лікарської форми, як зазначено вище в даному документі, призначеної для використання в діагностиці й лікуванні людини або тварини. Даний винахід також стосується фармацевтичної композиції, призначеної для використання фармацевтичної лікарської форми для перорального введення ссавцю в терапевтичних цілях, що й відрізняється тим, що зазначена лікарська форма призначена для введення зазначеному ссавцю в будь-який час дня, незалежно від прийому їжі. Даний винахід також стосується способу лікування й діагностики людини або тварини, який включає введення в зазначений організм терапевтично або діагностично ефективної дози фармацевтичної композиції, описаної в даному документі. Даний винахід також стосується фармацевтичної упаковки, прийнятної для продажу, що включає контейнер, лікарську форму, як описано в даному документі, а також супутні інструкції, що включають не тільки вказування, того, як приймати лікарську форму - натще або разом із прийомом їжі. Наступні приклади композицій наведені винятково в ілюстративних цілях і не обмежують об'єм винаходу. Приклади 21 UA 104877 C2 5 10 15 20 25 30 35 40 45 50 Приклад 1. Синтез і очищення пептиду Пептид послідовності SEQ ID NO:29 отриманий шляхом реакції синтезу у твердій фазі на синтезаторі пептидів Rainin Symphony Multiple Peptide Synthesizer (модель SMPS-110) з використанням програмного забезпечення версії 3.3.0. Смола (Novasyn TGR®, 440 мг, приблизно 0,1 ммоль, 0,23 ммоль/г заміщення, № партії A33379), використовувана для синтезу пептидамідів, була представлена у вигляді поліетиленгліколю й полістирену, функціоналізованих кислотно-нестійким модифікованим амідним лінкером Рінка. Амінокислоти, використовувані в синтезі, мають захисні групи Nα-9флуоренілметоксикарбонілу (Fmoc) на C-кінці й наступні захисні групи бічного ланцюга Arg(2,2,4,6,7-пентаметилдигідробензофуран-5-сульфоніл, pbf), Asp(третинний-бутокси, OtВu), Asn(тритил, Trt), Gln(Trt), Cys(Trt), His(Trt), Lys(t-бутоксикарбоніл, Boc), Ser(третинний-бутил, tВu) і Thr(tВu). Реакція зв'язування здійснена шляхом змішування смоли, попередньо розмоченої Nметилпіролідиноном (NMP) (0,1 ммоль), Fmoc-амінокислоти в диметилформаміді (ДМФА) в 5кратному молярному надлишку (2,5 мл) і гексафторфосфата (HBTU) в 5-кратному молярному надлишку з N-метилморфоліном (NMM) в 10-кратному молярному надлишку в ДМФА (2,5 мл), після чого реакція зв'язування протікала протягом 45 хвилин. Для видалення Fmoc проводили інкубацію реакції з 20%-ним розчином піперидину/ДМФА протягом 2 хвилин. Потім розчин зливали й додавали свіжий 20%-й розчин піперидину/ДМФА й інкубували протягом 18 хвилин. Після цього реагенти промивали N-метилпіролідиноном (NMP) і виконували послідовне додавання амінокислот, повторюючи етапи зв'язування. Для зв'язування C-кінця з Ile40, Gln39, Asn19, Asn12 і Val9, пронумерованих від N-кінця, етапи зв'язування виконували двічі. Відщіплення пептиду від смоли виконували за допомогою двогодинної програми розщеплення й інкубації з 9 мл розщеплюючого розчину, що складається із трифтороцтової кислоти (ТФК) (100 мл), 1,2-етандитіолу (ЕДТ) (20,0 мл), фенолу (7,5 г), тіоанізолу (5 мл), триізопропілсилану (ТІС) (5 мл) і води (5 мл). Розчин відщипленого пептиду поміщали в 50мілілітрову поліпропіленову центрифужну пробірку високої прозорості й осаджували пептид холодним діетиловим ефіром (40 мл). Суміш центрифугували, після чого діетиловий ефір декантували з пептиду. Додавали діетиловий ефір (40 мл), суміш перемішували на вортексі й центрифугували, після чого діетиловий ефір декантували. Ці етапи (додавання свіжого діетилового ефіру, перемішування на вортексі, центрифугування й декантування) повторювали ще два рази. Пептид висушували у вакуумі, одержуючи 408 мг (92% зібраної кількості) неочищеного продукту. Очищення пептиду виконувалося з використанням системи Waters для препаративної ВЕРХ (Waters, штат Массачусетс, США). Неочищений пептид (~100 мг) розчиняли в розчині оцтової кислоти/ацетонітрилу/води в пропорції 20/30/50, що містить 0,1% ТФК. Матеріал наносили на два стовпчики Vydac C-18 (10 мм, 2,525 див). Після нанесення для очищення пептиду використовували градієнт 0-45% розчинника B (розчинник B=80% ацетонітрил, що містить 0,1% ТФК) протягом 5 хвилин і 45-70% розчинника B протягом 60 хвилин зі швидкістю розтікання 6 мл/хв. Фракції збирали й аналізували методом аналітичної обернено-фазової рідинної хроматографії (RP-HPLC), матричної лазерної десорбційної часопролітної мас-спектрометрії (MALDI-TOF MS) і капілярного електрофорезу (CE). Найчистіші фракції об’єднували й ліофілізували, одержавши 23 мг продукту. Матрична лазерна десорбційна часопролітна мас-спектрометрія (MALDI-TOF MS) показала молекулярну масу продукту, еквівалентну 4400,5, що більше розрахункової молекулярної маси C195H326N56O53S3, еквівалентної 4399,2, на один атом водню. Ліофілізація виконана шляхом шокового заморожування рідини в ацетоновій бані із сухим льодом протягом приблизно 30 хвилин. Після заморожування продукт у відкритій посудині покривали фільтрувальним папером і піддавали впливу глибокого вакууму. Через 24 години впливу глибокого вакууму висушений зразок витягали з вакуумного приладу й поміщали в герметичний контейнер на зберігання. Приклад 2. Кон’югування пептиду з N-етилмалеімідом Сайт-спрямований кепінг цистеїнових залишків N-етилмалеімідом, як показано на схемі 1, виконували в наступних умовах: 22 UA 104877 C2 5 В 2,5-мілілітровому поліпропіленовому флаконі розчиняли 2,0 мг пептиду відповідно до принципів даного винаходу в 1,0 мл води. Потім відразу ж додавали двадцять мікролітрів 0,1M водного N-етилмалеіміду. Реагенти обережно перемішували при кімнатній температурі протягом 2 годин. Реакційні суміші очищали в автоматичній системі очищення Summit APS (Dionex, штат Каліфорнія, США) методом ВЕРХ на колонці Vydac C18 300 Angstrom, 10×250 мм (Grace Davison, Іллінойс, США) відповідно до протоколу, представленого в таблиці 6. Кінцеві фракції збирали, аналізували методом ВЕРХ, чисті фракції об’єднували й ліофілізували. 10 Таблиця 6 Стовпчик: Розчинники: УФ: Розтікання: Градієнт (%B) за час: 4,000 хв. 40,000 хв. 60,000 хв. 62,000 хв. 75,000 хв. 15 Vydac C18 300 Ångstrom (10250 мм) A: 0,1% ТФК у воді B: 0,1% ТФК із 80% ацетонітрил/вода (1) 214 нм (2) 280 нм 2,000 мл/хв. за 0,000 хв. 0,0% 100,0% 100,0% 0,0% 0,0% Приклад 3. Кон’югування пептиду з йодацетамідом-ПЕГ Взаємодія йодацетаміду-ПЕГ, лінійного ланцюга поліетиленгліколю масою 20 кДа з йодацетамідним кінцем, що присутній в обмежених кількостях у слабколужному pН з пептидом послідовності SEQ ID NO:29 привела до модифікації цистеїну в результаті реакції виключення, як показано на схемі 2. Цистеїновий тіол виступив як селективна точка приєднання йодацетаміду-ПЕГ. У результаті цього була отримана похідна ахіральна альфа сульфагідрилацетамідна структура. 23 UA 104877 C2 5 10 15 20 В 15-мілілітрову конічну колбу додавали 25 мг (5,68 ммоль, еквів. 1,0) пептиду послідовності SEQ ID NO:1. У ту ж колбу додавали 140 мг (6,82 ммоль, еквив. 1,2, 95% активн.) ПЕГ-20 йодацетаміду (№ партії M77592) виробництва Nippon, Oil and Fat (NOF) Corp. Потім додавали 10 мл води й перемішували розчин на вортексі до повного розчинення всіх твердих частинок. У мутний розчин додавали 50 мл піридину при pН розчину, що приблизно дорівнює 8,91. Через 2 години витягали аліквоту 20 мл зразка й аналізували методом зворотнофазної рідинної хроматографії із застосуванням C6-фенілової колонки Phenomenex з 0,1% розчином ТФК/ацетонітрилу як елюенту. Через 2 години зразок показав практично завершену реакцію (фіг. 1A). Реакційну суміш очищали безпосередньо способом ВЕРХ на С6-феніловій колонці Phenomenex 10×150 мм. Як елюенти для очищення використовували 0,1% водний розчин ТФК і 80% розчин ацетонітрилу в 0,1 водному розчині ТФК. Очищення проводили на пробах зразка 23 мл (фіг. 1B). Очищені фракції об’єднували й ліофілізували в 50-мілілітровій конічній колбі. Ліофілізовані тверді частинки розчиняли в 10 мл води й повторно ліофілізували. Приблизно 1 мг кінцевого продукту розчиняли в пропорції 1 мг/мл і піддавали мас-спектроскопії (фіг. 1C). Середня маса пегільованої сполуки послідовності SEQ ID NO:102 склала 25,449 Дальтона, частково внаслідок гетерогенності довжини ПЕГ-полімеру; сполука має вигляд білого аморфного твердого тіла. Приклад 4. Пегілювання пептиду з N-етилмалеімідним лінкером В 2,5-мілілітровому поліпропіленовому флаконі розчиняли 2,0 мг (~ 0,44 нмоль) пептиду в 2,5 мл води, потім відразу ж додавали активовані гліколі, похідні N-етилмалеіміду, з різними молекулярними масами, у кількостях, зазначених у таблиці 7. 24 UA 104877 C2 Реагенти обережно перемішували при кімнатній температурі протягом 2 годин. Таблиця 7 Структура ПЕГ ПЕГ-малеімід ММ [кДа] Лінійний 2 Лінійний 5 Лінійний 12 Лінійний 20 Лінійний 30 Лінійний 40 Розгалужений 80 Малеімід із двома кінцями 20 Каталожний номер NOF Corp. SUNBRIGHT® ME-020MA SUNBRIGHT® ME-050MA SUNBRIGHT® ME-120MA SUNBRIGHT® ME-200MA SUNBRIGHT® ME-300MA SUNBRIGHT® ME-400MA SUNBRIGHT® GL2-800MA SUNBRIGHT® DE-200MA Кількість [мг] 1,0 мг (0,49 нмоль) 2,0 мг (0,49 нмоль) 6,0 мг (0,49 нмоль) 10,0 мг (0,49 нмоль) 15,0 мг (0,49 нмоль) 20,0 мг (0,49 нмоль) 40,0 мг (0,49 нмоль) 5,0 мг (0,49 нмоль) 5 Реакційні суміші очищали в автоматичній системі очищення Summit APS (Dionex, штат Каліфорнія, США) методом ВЕРХ на С6-феніловій колонці Gemini 5u 10100 мм (110 Angstrom; Phenomenex, штат Каліфорнія, США) відповідно до протоколу, представленого в таблиці 8. 25 UA 104877 C2 Таблиця 8 Колонка: Розчинники: УФ: Розтікання: Градієнт (%B) за час: 2,500 хв. 40,000 хв. 45,000 хв. 52,000 хв. 54,000 хв. 60,000 хв. 5 10 15 20 25 30 35 40 Phenomenex Gemini 5u C6-феніл 110 Ångstrom (10×100 мм) A: 0,1% ТФК у воді B: 0,1% ТФК із 80% ацетонітрил/вода (1) 214 нм (2) 280 нм 4,000 мл/хв. за 0,000 хв. 0,0% 70,0% 100,0% 100,0% 0,0% Кінець Приклад 5. Агоністична активність CRHR2 і CRHR1 - цАМФ аналіз Оцінку агоністичної активності CRHR2 і CRHR1 сімейства кортикотропін-рилізинг гормонів (КРГ) проводили у двох лініях клітин SK-N-MC (людська нейробластома), трансфікованих або людським CRHR2, або людським CRHR1, шляхом аналізу із циклічним 3’,5’аденозинмонофосфатом (цАМФ). У даному аналізі h-SCP (SEQ ID NO:1) і h-UCN2 (SEQ ID NO:115) мали однакову силу, і h-SCP показав себе як більш селективний агоніст CRHR2 у сімействі КРГ (фіг. 2). Концентрація, необхідна для досягнення 50% максимального ефекту (A50), склала 0,4 нМ. Людський CRHR1 (номер доступу X72304) або CRHR2 (номер доступу U34587) клонують в експресуючий вектор pcDNA3.1/Zeo і стабільно трансфікують у клітини SK-N-MC шляхом електропорації. Клітини витримують у мінімальному підтримуючому середовищі, що складається з наступних компонентів: вода/солі Ерла з 10% ембріональної телячої сироватки (ЕТС), 50 МЕ пеніциліну, 50 мкг/мл стрептоміцину, 2 мМ L-глутаміну, 1 мМ пірувату натрію й 0,1 мМ несуттєвих амінокислот, 600 мкг/мл G418. Клітини культивували при температурі 37 °C в 5% CO2. Увечері попереднього дня клітини висівали на 96-ямкові мікропланшети (Biocoat виробництва BD Biosciences) у концентрації 50000 клітин/ямка. Клітини промивали фосфатносольовим буферним розчином (ФБР), потім ресуспендировали в модифікованому за способом Дульбекко середовищі Ігла F-12 без фенолового червоного, що містить 10 ммоль ізобутилметилксантину (ІБМК). Клітини інкубували з пептидами в концентрації від приблизно 1 М до приблизно 10 мкМ протягом 60 хвилин при температурі 37 °C. Для наступної оцінки будьякої антагоністичної активності тих пептидів, які не дали агоністичної відповіді, пептиди інкубували в концентрації 10 мкМ протягом 20 хвилин за 60 хвилин до введення h-SCP. Для позитивного контролю використовується форсколін (10 мкМ), прямий стимулянт аденілатциклази. Аналіз зупинено шляхом додавання 0,5 М HСl і перемішування орбітальним обертанням протягом 2 годин при температурі 4 °C. Для оцінки активності пептиду відповідно до принципів даного винаходу на CRHR2 використовується контрольне вимірювання міжклітинного цАМФ за допомогою радіоактивного аналізу електролітичного покриття (каталожний номер Cus56088; Perkin Elmer, штат Массачусетс, США). Увечері попереднього дня трансфіковані клітини SK-N-MC висівали на 96-лункові мікропланшети Biocoat (BD Biosciences, Сан-Хосе, штат Каліфорнія, США) у концентрації 50000 клітин/ямка. Спочатку клітини промивали ФБР, а потім суспендували в модифікованому за способом Дульбекко середовищі Ігла F-12 без фенолового червоного, що містить ммоль ізобутилметилксантину (ІБМК). Суспендовані клітини переносили на 96нямковий планшет з електролітичним покриттям з нанесеною сцинтиляційною рідиною. Клітини інкубували з пептидами в концентрації у від приблизно 1 пМ до приблизно 10 мкМ протягом 60 хвилин при температурі 37 °C. Для позитивного контролю використовували форсколін (10 мкМ). Після стимуляції лігандом клітини лізували шляхом додавання 0,5 М HСl і перемішування орбітальним обертанням протягом 2 годин при температурі 4 °C для вивільнення міжклітинного цАМФ у середовище. Середовище, яке містить внутрішньоклітинний цАМФ, що вивільнився, переносили на 96нямковий планшет з електролітичним покриттям і нанесеної сцинтиляційною рідиною, що 26 UA 104877 C2 125 5 10 містить анти-цАМФ антитіло. У даному аналізі внутрішньоклітинний цАМФ конкурує з Iміченим цАМФ, що зв'язується з антитілом. Для одержання стандартної кривої в експеримент 125 включений цАМФ із концентрацією від приблизно 2,5 до приблизно 250 пмоль/мл. [ I]-цАМФ визначений за допомогою сцинтиляційного лічильника TopСount (Perkin Elmer, штат Массачусетс, США). Індивідуальні характеристики кривої, що відображає співвідношення концентрації агоніста до відповіді, уводяться в рівняння Хіла, формула якого представлена нижче, у програмі Graphpad Prism (Graphpad Software, Ла-Джола, штат Каліфорнія, США), з метою оцінки концентрації агоніста, необхідної для формування половини максимальної відповідної реакції (A50) і забезпечення максимальної асимптоти (a) і параметрів відхилення Хіла (nН). У цьому рівнянні [A] - концентрація агоніста, а E - вимірюваний ефект: E 15 20 [ A ]nH n H [ A ]50 [ A ]nH , Для наочного відображення при побудові одиночної кривій E/[A] використані оцінки значень середніх підігнаних параметрів, накладені на середні експериментальні дані. Оціночні показники потенціалу агоністів pА50 виражені у вигляді кологарифма середньої точки кожної кривої й зазначені з урахуванням стандартної помилки вимірювання (СПВ). Логарифм по основі 10 значень коефіцієнта дозування агоніста (Log DR) обчислюється шляхом вирахування значення пробної сполуки pА50 з відповідного контрольного значення h-SCP (SEQ ID NO:1) pА50 у рамках однієї проби аналізу. Значення СПВ значень логарифма Log DR являють собою квадратний корінь суми значень СПВ значень контрольної й пробної сполуки h-SCP (SEQ ID NO:1) pА50 у квадраті. Таблиця 9 Пептид-антагоніст CRHR – анти-саувагін-30 FHLLR KMIEI EKQEK EKQQA ANNRL LLDTI-NH2 25 SV30 SEQ ID NO:118 CRHR 2-опосередкована відповідь цАМФ на h-SCP (SEQ ID NO:1) блокована селективним антагоністом CRHR2, анти-саувагіном-30 (SV30, SEQ ID NO:118, зазначеним у таблиці 9), залежно від концентрації й з урахуванням переборного конкурентного антагонізму (фіг. 3). Присутність анти-саувагіну-30 дає значення a pА2, що дорівнює 7,82, для сполуки з послідовністю SEQ ID NO:1. Таблиця 10 TKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI DDPPL SIDLT FHLLR TLLEL ARTQS QRERA EQNRI IFDSV-NH2 IVLSL DVPIG LLQIL LEQAR ARAAR EQATT NARIL ARV-NH2 HPGSR IVLSL DVPIG LLQIL LEQAR ARAAR EQATT NARIL ARV-NH2 неамідований послідовність SEQ ID NO:113 h-SCP r-UCN1 послідовність SEQ ID NO:114 h-UCN2 послідовність SEQ ID NO:115 h-SRP послідовність SEQ ID NO:117 30 35 Людські й щурячі пептиди (див. таблицю 10) використовувалися для стимуляції клітин hCRH1 або h-CRHR2, трансфікованих SK-N-MC в аналізі електролітичного покриття цАМФ. Пептиди інкубували протягом 1 години при температурі 37 °C. Криві розраховані з використанням нелінійного сигмоїдального аналізу залежності відповідної реакції від концентрації. Розрахунок проводився з використанням ПО GraphРad Prism. Отримані значення pА50 представлені в таблиці 11 на додаток до даних, опублікованих у літературі. 27 UA 104877 C2 Таблиця 11 Рецептор CRHR1 CRHR1 CRHR1 CRHR1 CRHR1 CRHR1 CRHR1 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 CRHR2 Опубліковано Пептид pА50 1 r- UCN1 9,82 3 h-SRP >7 3 h-SRP >7 3 h-SRP >7 h-UCN2 h-UCN3 h-SCP 2 r-UCN1 10,06 2/9 5 h-UCN2 9,37 ,12 2 h-UCN3 9,92 4 h-SCP ~9 4 h-SRP ~9 4 h-SCP ~9 4 h-SCP ~9 4 h-SCP ~9 2/9 5 h-UCN2 9,37 ,12 4 h-SRP ~9 4 h-SRP ~9 Отримано експериментальним шляхи СПВ nН СПВ αмаксимум СПВ 0,07 1,15 0,19 99,61 3,28 0,03 1,61 0,15 Немає 0,04 1,33 0,17 Немає 0,03 1,26 0,13 Немає 0,02 1,69 0,18 Немає N 12 20 11 17 15 0,05 0,05 0,05 0,06 0,05 0,03 0,04 0,02 0,04 0,05 0,03 pА50 9,19 6,34 6,2 6,28 6,02
ДивитисяДодаткова інформація
Назва патенту англійськоюCrhr2 peptide agonists and uses thereof
Автори російськоюSwanson, Ronald, V., Shankley, Nigel, P., Moreno, Veronica, Gengo, Peter
МПК / Мітки
МПК: C07K 16/18, A61K 38/22, C07K 14/575, A61P 9/00, C12N 15/12
Мітки: використання, пептиди-агоністи, crhr2
Код посилання
<a href="https://ua.patents.su/99-104877-peptidi-agonisti-crhr2-i-kh-vikoristannya.html" target="_blank" rel="follow" title="База патентів України">Пептиди-агоністи crhr2 і їх використання</a>
Коротколанцюгові пептиди як агоністи рецептора паратиреоїдного гормону (pth)

Номер патенту: 102016
Опубліковано: 27.05.2013
Автори: Джеін Мукул Р., Бахекар Раджеш, Пател Панкадж Р.
МПК: A61K 38/29, C07K 14/635
Мітки: пептиди, коротколанцюгові, гормону, pth, паратиреоїдного, рецептора, агоністи
Формула / Реферат:
1. Ізольований коротколанцюговий пептид, який має послідовність формули (І), включаючи його таутомери, сольвати,A-Z1-Z2-Z3-Z4-Z5-Z6-Z7-Z8-Z9-Z10-Z11-Z12-Z13-Z14-Z15-B, (І)у якійА позначає групи -NH-R1 або R3-CO-NH-, де R1 позначає водень, біотин або необов'язково заміщений лінійний чи розгалужений (C1-18)алкільний ланцюг, або амінокислоти, такі як піроглутамінова кислота (Pyr), Pro (P), альфа-метилпролін (αМе-Р),...
Пептиди, похідні від тромбоспондину-1, та способи лікування

Номер патенту: 94046
Опубліковано: 11.04.2011
Автор: Агах Рамтін
МПК: C07K 16/00, C07K 7/00, C07K 14/00, C07K 2/00, A61K 38/00, C07K 4/00, A61K 39/40, C07K 17/00
Мітки: пептиди, способи, тромбоспондину-1, лікування, похідні
Формула / Реферат:
1. Пептид, який включає послідовність Хаа1-Хаа2-Хаа3-Хаа4-Хаа5-Хаа6-Хаа7-Хаа8-Хаа9-Хаа10-Хаа11-Хаа12-Хаа13-Хаа14-Хаа15, де Хаа1, Хаа2 та Хаа4 вибрані з групи, що складається з основних, нециклічних амінокислот, включаючи Arg та Lys; Хаа3, Хаа9, Хаа11, Хаа12 та Хаа15 вибрані з групи, що складається знейтральних, неполярних, великих та циклічних амінокислот, включаючи Phe, Тrр та Pro; Хаа5 вибраний з групи, що складається з...
Алоферони-імуномодулюючі пептиди

Номер патенту: 69457
Опубліковано: 15.09.2004
Автори: Кім Су Ін, Бюлє Філіп, Бєккєр Гєрман Пєтровіч, Чєрниш Сєргєй Івановіч, Хоффман Жуль А., Махалдіані Наталя Борісовна
МПК: A61K 38/08, A61K 38/10, A61P 37/02, C07K 7/06, C07K 7/08
Мітки: пептиди, алоферони-імуномодулюючі
Формула / Реферат:
1. Алоферони - імуномодулюючі пептиди загальної формули (1):X1-His-Gly-X2-His-Gly-Val-X3 (1)або їх фармацевтично прийнятні солі або ефіри, або аміди, де:Х1 відсутній або містить не менше 1 амінокислоти,Χ2 містить не менше 1 амінокислоти або є пептидним зв'язком,Х3 відсутній або містить не менше 1 амінокислоти, причому зазначені амінокислоти вибрані з групи: аліфатичні, ароматичні або...
Пептиди, що біологічно не розкладаються; інгібітор ангіотензинперетворювального ферменту, лікарський засіб і функціональний харчовий продукт

Номер патенту: 87278
Опубліковано: 10.07.2009
Автори: Мізуно Сейіті, Ямамото Наоюкі, Готоу Таканобу, Мацуура Кейіті, Нісімура Сінго (помер)
МПК: A61P 9/12, C07K 5/062, A61P 43/00, C07K 5/072, C07K 5/068, C07K 5/083, A61K 38/01
Мітки: лікарський, ферменту, розкладаються, продукт, ангіотензинперетворювального, харчовий, засіб, інгібітор, пептиди, функціональний, біологічно
Формула / Реферат:
1. Пептид, що не перетравлюється in vivo, який має Pro на карбоксильному кінці, вибраний з групи, яка складається з Ile-Pro, Glu-Pro, Arg-Pro, Gln-Pro, Met-Pro і Ser-Pro-Pro.2. Інгібітор ангіотензинперетворювального ферменту, що включає як активний інгредієнт пептид, що не перетравлюється in vivo, який має Pro на карбоксильних кінцях, вибраний з групи, яка складається з Ile-Pro, Glu-Pro, Arg- Pro, Gln-Pro, Met-Pro i Ser-Pro-Pro або їх...
Очищені пептиди та сполуки, які їх містять

Номер патенту: 84432
Опубліковано: 27.10.2008
Автори: Коернер Штеффі К., Сан Веі-Чуан, Караван Пітер Д., Ніворожкін Алєксандр Л., Жанг Жаода, Ванг Ксіфанг, Дюма Стефан, Наір Шрікумар, Амедіо Джон С., Колодзєй Ендрю, Макмуррі Томас Дж.
МПК: C07K 7/00, C07K 5/00, A61B 5/055
Мітки: сполуки, містять, очищені, пептиди
Формула / Реферат:
1. Очищений пептид, що має наступну амінокислотну послідовність:P*-Y*-Х1*-L* (SEQ ID NO:1), в якійР* є проліном або його неприродним похідним;Y* є тирозином або його неприродним похідним;Х1* є гліцином або аспарагіновою кислотою, або неприродним похідним гліцину та аспарагінової кислоти;L* є лейцином або його неприродним похідним,де один з перерахованих Р*, Y*, Х1* і L* є неприродним похідним...
Попередній патент: Модулятори atф-зв’язувальних касетних транспортерів
Наступний патент: Спосіб лікування т-клітинних лімфом шкіри
Випадковий патент: Сенсорний пристрій