Пірольні сполуки, спосіб їх одержання та фармацевтична композиція, що їх містить (варіанти)
Номер патенту: 115773
Опубліковано: 26.12.2017
Автори: Жан-Мішель Анлан, Янош Татаі, Джеймс Брук Маррей, Анн-Франсуаз Гійузік, Джеймс Едвард Пол Девідсон, Арнод Лє Тіран, Гійом Де Нантей, Жером-Бенуа Старк, Тьєррі Лє Дігуарер, Олівьє Генесте, Ай-Джен Чен, Міклош Ласло Ньєргеш, Імре Фейєш, Дідьє Дюран





































































































Формула / Реферат
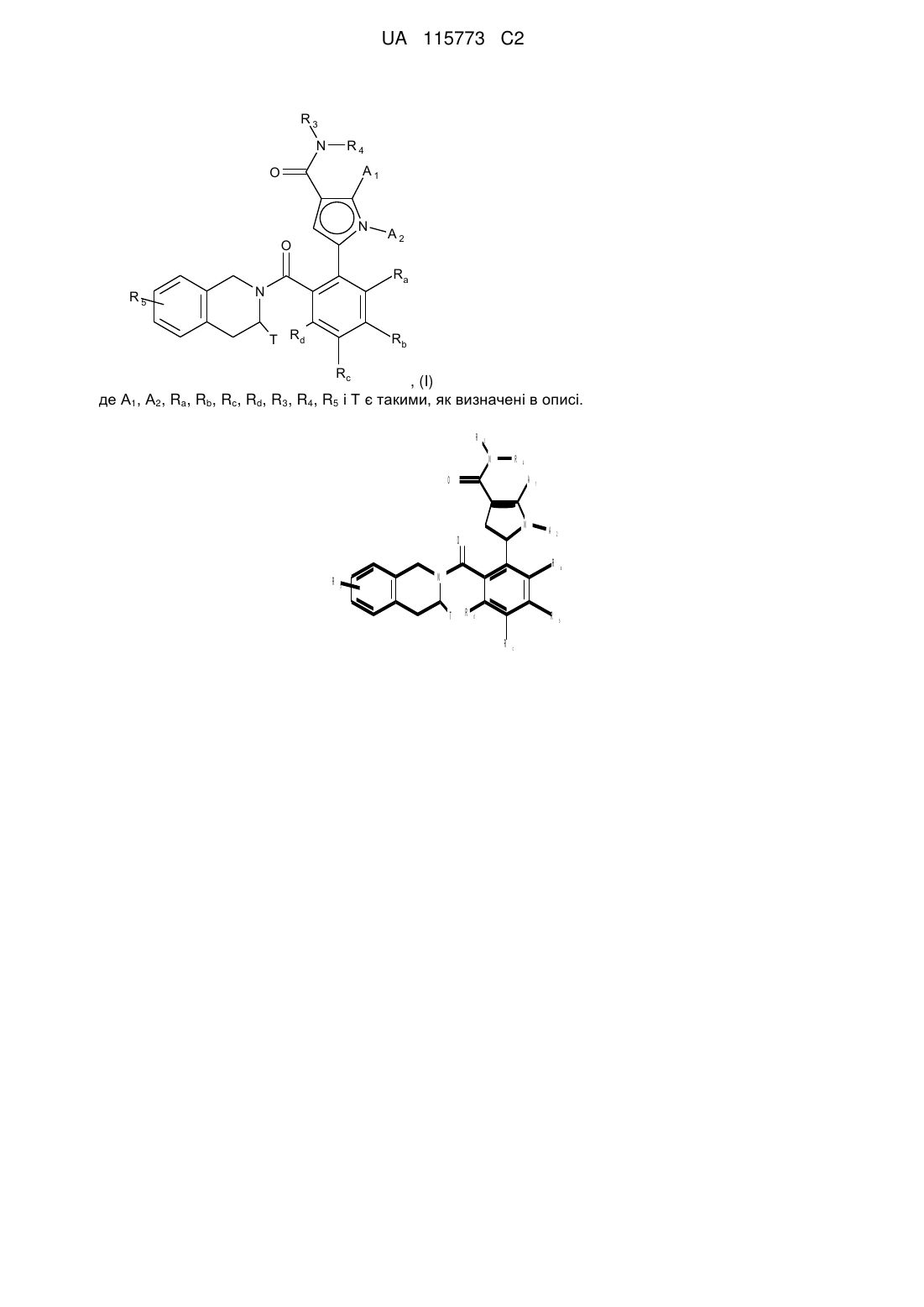
1. Сполука формули (І):
 , (I)
, (I)
де:
А1 і А2, кожен незалежно один від одного, являють собою атом водню або галогену, лінійну або розгалужену (С1-С6)полігалоалкільну групу, лінійну або розгалужену (С1-С6)алкільну групу або циклоалкільну групу,
Т являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу, необов'язково заміщену від одного до трьох атомів галогену, групу (С1-С4)алкіл-NR1R2 або групу (С1-С4)алкіл-OR6,
R1 і R2, кожен незалежно один від одного, являють собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу,
або R1 і R2 утворюють з атомом азоту, до якого вони приєднані, гетероциклоалкіл,
R3 являє собою лінійну або розгалужену (С1-С6)алкільну групу, лінійну або розгалужену (С2-С6)алкенільну групу, лінійну або розгалужену (С2-С6)алкінільну групу, циклоалкільну групу, (С3-С10)циклоалкіл-(С1-С6)алкільну групу, де алкільний фрагмент є лінійним або розгалуженим, гетероциклоалкільну групу, арильну групу або гетероарильну групу, слід розуміти, що один або більше атомів вуглецю з попередніх груп, або їх можливих замісників, можуть бути дейтеровані,
R4 являє собою арильну групу, гетероарильну групу, циклоалкільну групу або лінійну або розгалужену (С1-С6)алкільну групу, слід розуміти, що один або більше атомів вуглецю попередніх груп, або їх можливих замісників, можуть бути дейтеровані,
R5 являє собою атом водню або галогену, лінійну або розгалужену (С1-С6)алкільну групу або лінійну або розгалужену (С1-С6)алкоксигрупу,
R6 являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу,
Ra, Rb, Rc і Rd, кожен незалежно від інших, являють собою R7, атом галогену, лінійну або розгалужену (С1-С6)алкоксигрупу, гідроксигрупу, лінійну або розгалужену (С1-С6)полігалоалкільну групу, трифторметоксигрупу, -NR7R7', нітро, R7-CO-(C0-C6)алкіл-, R7-CO-NH-(С0-С6)алкіл-, NR7R7'-CO-(C0-C6)алкіл-, NR7R7'-CO-(C0-C6)алкіл-O-, R7-SO2-NH-(C0-C6)алкіл-, R7-NH-CO-NH-(C0-C6)алкіл-, R7-O-CO-NH-(C0-С6)алкіл-, гетероциклоалкіл, або замісники однієї з пар (Ra, Rb), (Rb, Rc) або (Rc, Rd) утворюють разом з атомами вуглецю, до яких вони приєднані, кільце, що складається з від 5 до 7 кільцевих членів, які можуть містити від одного до 2 гетероатомів, вибраних з кисню і сірки; слід також розуміти, що один або більше атомів вуглецю кільця, визначеного тут вище, можуть бути дейтеровані або заміщені від однієї до 3 групами, вибраними з галогену і лінійного або розгалуженого (С1-С6)алкілу,
R7 і R7', кожен незалежно один від одного, являють собою водень, лінійний або розгалужений (С1-С6)алкіл, лінійний або розгалужений (С2-С6)алкеніл, лінійний або розгалужений (С2-С6)алкініл, арил або гетероарил, або R7 і R7' разом з атомом азоту, до якого вони приєднані, утворюють гетероцикл, що складається з від 5 до 7 кільцевих членів,
причому слід розуміти, що:
"арил" означає фенільну, нафтильну, біфенільну або інденільну групу,
"гетероарил" означає будь-яку моно- або біциклічну групу, що складається з від 5 до 10 кільцевих членів, які мають принаймні один ароматичний фрагмент і містять від 1 до 4 гетероатомів, вибраних з кисню, сірки і азоту (в тому числі четвертинного азоту),
"циклоалкіл" означає будь-яку моно- або біциклічну, неароматичну, карбоциклічну групу, що містить від 3 до 10 кільцевих членів,
"гетероциклоалкіл" означає будь-яку моно- або біциклічну, неароматичну, конденсовану або спірогрупу, що складається з від 3 до 10 кільцевих членів і містить від 1 до 3 гетероатомів, вибраних з кисню, сірки, SO, SO2 та азоту,
причому для арильної, гетероарильної, циклоалкільної і гетероциклоалкільної груп, визначених таким чином, і груп алкілу, алкенілу, алкінілу і алкоксилу можливе заміщення від 1 до 3 групами, вибраними з необов'язково заміщеного лінійного або розгалуженого (С1-С6)алкілу, (С3-С6)спіро, необов'язково заміщеного, лінійного або розгалуженого (С1-С6)алкокси, (С1-С6)алкіл-S-, гідрокси, оксо (або N-оксиду, де це доречно), нітро, ціано, -COOR', -OCOR', NR'R", лінійного або розгалуженого (С1-С6)полігалоалкілу, трифторметокси, (С1-С6)алкілсульфонілу, галогену, необов'язково заміщеного арилу, гетероарилу, арилокси, арилтіо, циклоалкілу, гетероциклоалкілу, необов'язково заміщеного одним або кількома атомами галогену або алкільними групами, слід розуміти, що R' і R", кожен незалежно від іншого, являє собою атом водню або необов'язково заміщену, лінійну або розгалужену (С1-С6)алкільну групу,
її енантіомери і діастереоізомери та адитивні солі з фармацевтично прийнятною кислотою або основою.
2. Сполука формули (І) за п. 1, де А1 являє собою атом водню або метильну групу.
3. Сполука формули (І) за п. 1 або п. 2, де А2 являє собою лінійну або розгалужену (С1-С6)алкільну групу, необов'язково заміщену групою, вибраною з галогену, гідрокси, лінійного або розгалуженого (С1-С6)алкокси, NR'R" і морфоліну.
4. Сполука формули (І) за п. 1 або п. 2, де А2 являє собою лінійну або розгалужену (С1-С6)полігалоалкільну групу або циклопропільну групу.
5. Сполука формули (І) за будь-яким з пп. 1-3, де А1 і А2 обидва являють собою метильну групу.
6. Сполука формули (І) за будь-яким з пп. 1-5, де Т являє собою метил, амінометил, (морфолін-4-іл)метил, (4-метилпіперазин-1-іл)метил, 2-(морфолін-4-іл)етил, [2-(морфолін-4-іл)етокси]метил, гідроксиметил, [2-(диметиламіно)етокси]метил, гексагідропіразино[2,1-с][1,4]оксазин-8(1Н)-ілметил, 1-окса-6-азаспіро[3,3]гепт-6-ілметил, 3-(морфолін-4-іл)пропіл або трифторметильну групу.
7. Сполука формули (І) за будь-яким з пп. 1-6, де Ra і Rd кожен являє собою атом водню і (Rb, Rc), разом з атомами вуглецю, до яких вони приєднані, утворюють 1,3-діоксоланову групу або 1,4-діоксанову групу; або Ra, Rc і Rd кожен являє собою атом водню і Rb являє собою атом водню або галогену або метоксигрупу.
8. Сполука формули (І) за будь-яким з пп. 1-6, де Ra і Rd кожен являє собою атом водню, Rb являє собою атом водню або галогену і Rc являє собою гідрокси або метоксигрупу, або Ra і Rd кожен являє собою атом водню, Rb являє собою гідрокси або метоксигрупу і Rc являє собою атом галогену.
9. Сполука формули (І) за будь-яким з пп. 1-6, де Ra, Rb і Rd кожен являє собою атом водню і Rc являє собою групу, вибрану з R7-CO-NH-(C0-С6)алкілу-, R7-SO2-NH-(C0-C6)алкілy-, R7-NH-CO-NH-(C0-C6)алкілy- і R7-O-СО-NН-(С0-С6)алкілу.
10. Сполука формули (І) за будь-яким з пп. 1-9, де R4 являє собою феніл, 4-гідроксифеніл, 3-фтор-4-гідроксифеніл, 2-гідроксипіримідин або 3-гідроксипіридинову групу.
11. Сполука формули (І) за будь-яким з пп. 1-10, де R3 являє собою арильну або гетероарильну групу.
12. Сполука формули (І) за будь-яким з пп. 1-10, де R3 являє собою групу, вибрану з метилу, фенілу, 1Н-піразолу, -1H-індолу, 1Н-індазолу, піридину, піримідину, 1Н-піроло[2,3-b]піридину, 2,3-дигідро-1Н-піроло[2,3-b]піридину, 1Н-бензімідазолу, 1Н-піролу, 1Н-піроло[2,3-с]піридину, 1Н-піроло[3,2-b]піридину, 5Н-піроло[3,2-d]піримідину, тіофену, піразину, 1Н-піразоло[3,4-b]піридину, 1,2-оксазолу і 1Н-піразоло[1,5-а]піримідину, причому ці групи необов'язково мають один або кілька замісників, вибраних з галогену, лінійного або розгалуженого (С1-С6)алкілу, лінійного або розгалуженого (С1-С6)алкокси, ціано, циклопропілу, оксетану, тетрагідрофурану, -СО-О-СН3, тридейтеріометилу, 2-(морфолін-4-іл)етилу і 2-(морфолін-4-іл)етокси.
13. Сполука формули (І) за п. 10, де R3 являє собою лінійний або розгалужений (С1-С6)алкіл або гетероарил, необов'язково заміщений лінійним або розгалуженим (С1-С6)алкілом, і R4 являє собою 4-гідроксифенільну групу.
14. Сполука формули (І) за п. 1, вибрана з наступної групи:
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3R)-3-метил-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(піридин-4-іл)-1H-пірол-3-карбоксамід,
N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-5-(6-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1H)-іл]карбоніл}-1,3-бензодіоксол-5-іл)-1Н-пірол-3-карбоксамід,
N-(4-гідроксифеніл)-1,2-диметил-5-(6-{[(3R)-3-метил-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}-1,3-бензодіоксол-5-іл)-N-(піридин-4-іл)-1Н-пірол-3-карбоксамід,
5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піроло[2,3-b]піридин-5-іл)-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(піридин-4-іл)-1H-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(5-ціано-1-метил-1Н-пірол-3-іл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід,
N-(5-ціано-1-метил-1Н-пірол-3-іл)-5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(5-ціано-1,2-диметил-1Н-пірол-3-іл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-2,3-дигідро-1Н-піроло[2,3-b]піридин-5-іл)-1H-пірол-3-карбоксамід,
N-(5-ціано-1,2-диметил-1Н-пірол-3-іл)-5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід,
5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-[5-ціано-2-метил-1-(тридейтеріометил)-1Н-пірол-3-іл]-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід,
їх енантіомери, діастереоізомери та їх адитивні солі з фармацевтично прийнятною кислотою або основою.
15. Спосіб одержання сполук формули (І) за п. 1, який відрізняється тим, що як вихідний матеріал використовують сполуку формули (II):
 , (II)
, (II)
де Ra, Rb, Rc і Rd є такими, як визначені для формули (І),
де сполуку формули (II) піддають реакції Хека, у водному або органічному середовищі, в присутності паладієвого каталізатора, основи, фосфіну і сполуки формули (III):
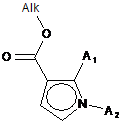 , (III)
, (III)
де групи А1 і А2 є такими, як визначені для формули (І), і Alk означає лінійний або розгалужений (С1-С6)алкіл,
для одержання сполуки формули (IV):
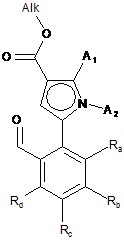 , (IV)
, (IV)
де А1, А2, Ra, Rb, Rc і Rd є такими, як визначені для формули (І), і Alk є таким, як визначено вище,
альдегідну функцію сполуки формули (IV) окислюють до карбонової кислоти для утворення сполуки формули (V):
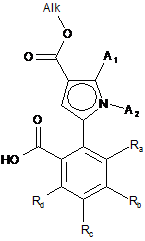 , (V)
, (V)
де А1, А2, Ra, Rb, Rc і Rd є такими, як визначені для формули (І), і Alk є таким, як визначено вище,
де сполуку формули (V) потім піддають пептидному зв'язуванню зі сполукою формули (VI):
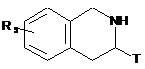 , (VI)
, (VI)
де Т і R5 є такими, як визначені для формули (І),
для одержання сполуки формули (VII):
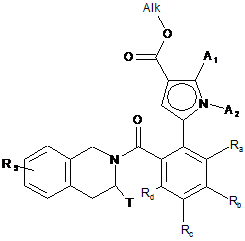 , (VII)
, (VII)
де A1, A2, Ra, Rb, Rc і Rd, T і R5 є такими, як визначені для формули (І), і Alk є таким, як визначено вище,
функцію складного ефіру сполуки формули (VII) гідролізують для одержання відповідної карбонової кислоти або карбоксилату, який може бути перетворений на похідну кислоти, таку як відповідний хлористий ацил або ангідрид, перед з'єднанням з аміном NHR3R4, де R3 і R4 мають такі ж значення, як у формулі (І), для одержання сполуки формули (І),
причому сполука формули (І) може бути очищена відповідно до звичайного методу сепарації, перетворена, якщо бажано, на її адитивні солі з фармацевтично прийнятною кислотою або основою і необов'язково поділена на її ізомери відповідно до звичайного методу сепарації,
слід розуміти, що в будь-який момент, який вважається доречним в ході описаного вище способу, певні групи (гідрокси, аміно…) реагентів або інтермедіатів синтезу можуть бути захищені, а потім позбавлені захисту відповідно до вимог синтезу.
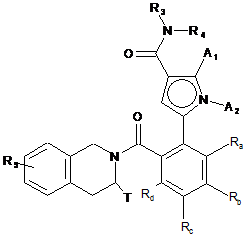
16. Спосіб одержання сполук формули (I'), які є окремими випадками сполук формули (І) за п. 1, як визначено далі:
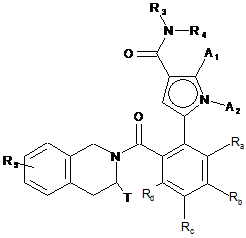 , (I')
, (I')
де:
A1, A2, Ra, Rd, R3, R4, T і R5 є такими, як визначені для формули (І),
Rb і Rc є такими, що один являє собою водень, а інший - групу, вибрану з R7-CO-NH-(C0-C6)алкілy-, R7-SO2-NH-(C0-C6)алкілy-, R7-NН-СО-NН-(С0-С6)алкілу- і R7-О-СО-NН-(С0-С6)алкілу-, R7 є таким, як визначено для формули (І),
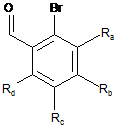
причому спосіб одержання використовує як вихідний матеріал сполуку формули (II'):
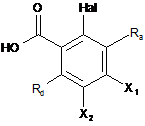 , (ІI')
, (ІI')
де:
Ra і Rd є такими, як визначені для формули (І),
Hal являє собою атом галогену,
Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2 групу,
тоді як інший являє собою атом водню,
де сполуку формули (II') потім піддають пептидному зв'язуванню зі сполукою формули (VI):
, (VI)
де Т і R5 є такими, як визначені для формули (І), для одержання сполуки формули (III'):
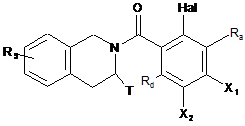 , (III')
, (III')
де:
Ra, Rd, T і R5 є такими, як визначені для формули (І),
Hal являє собою атом галогену,
Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2 групу, тоді як інший являє собою атом водню,
де сполуку формули (ІІI') піддають реакції Хека, у водному або органічному середовищі, в присутності паладієвого каталізатора, основи, фосфіну і сполуки формули (IV'):
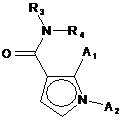 , (IV')
, (IV')
де A1, A2, R3 і R4 є такими, як визначені для формули (І), для утворення сполуки формули (V'):
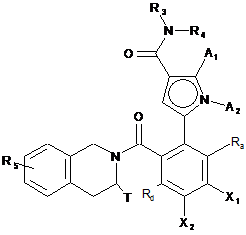 , (V')
, (V')
де:
A1, A2, Ra, Rd, R3, R4, T і R5 є такими, як визначені для формули (І),
Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2-групу, тоді як інший являє собою атом водню,
де сполуку формули (V') потім піддають реакції ацилювання або сульфонілування для одержання сполуки формули (I'),
причому сполука формули (I') може бути очищена відповідно до звичайного методу сепарації, перетворена, якщо бажано, на її адитивні солі з фармацевтично прийнятною кислотою або основою і необов'язково поділена на її ізомери відповідно до звичайного методу сепарації,
слід розуміти, що в будь-який момент, який вважається доречним в ході описаного вище способу, певні групи (гідрокси, аміно…) реагентів або інтермедіатів синтезу можуть бути захищені, а потім позбавлені захисту відповідно до вимог синтезу.
17. Спосіб за п. 15 або п. 16 одержання сполуки формули (І), де одна з груп R3 або R4 є заміщеною гідроксильною функцією, який відрізняється тим, що амін NHR3R4 піддають заздалегідь реакції захисту гідроксильної функції перед будь-яким з'єднанням з карбоновою кислотою, утвореною зі сполуки формули (VII), або з її відповідною похідною кислоти, причому одержана захищена сполука формули (І) надалі вступає в реакцію зняття захисту і потім необов'язково перетворюється на одну з її адитивних солей з фармацевтично прийнятною кислотою або основою.
18. Фармацевтична композиція, яка містить сполуку формули (І) за будь-яким з пп. 1-14 або її адитивну сіль з фармацевтично прийнятною кислотою або основою у поєднанні з одним або декількома фармацевтично прийнятними ексципієнтами.
19. Фармацевтична композиція за п. 18 для застосування як проапоптозного агента.
20. Фармацевтична композиція за п. 18 для застосування в лікуванні раку, аутоімунних захворювань і захворювань імунної системи.
21. Фармацевтична композиція за п. 18 для застосування в лікуванні раку сечового міхура, головного мозку, молочної залози і матки, хронічних лімфоїдних лейкозів, колоректального раку, раку стравоходу і печінки, лімфобластного лейкозу, неходжкінської лімфоми, меланоми, злоякісних захворювань крові, мієломи, раку яєчників, недрібноклітинного раку легенів, раку передміхурової залози та дрібноклітинного раку легенів.
22. Застосування фармацевтичної композиції за п. 18 у виробництві лікарського засобу для застосування як проапоптозного агента.
23. Застосування фармацевтичної композиції за п. 18 у виробництві лікарського засобу, призначеного для лікування раку, захворювань імунної системи і аутоімунних захворювань.
24. Застосування фармацевтичної композиції за п. 18 у виробництві лікарського засобу, призначеного для лікування раку сечового міхура, головного мозку, молочної залози і матки, хронічних лімфоїдних лейкозів, колоректального раку, раку стравоходу і печінки, лімфобластного лейкозу, неходжкінської лімфоми, меланоми, злоякісних захворювань крові, мієломи, раку яєчників, недрібноклітинного раку легенів, раку передміхурової залози та дрібноклітинного раку легенів.
25. Сполука формули (І) за будь-яким з пп. 1-14 або її адитивна сіль з фармацевтично прийнятною кислотою або основою для застосування в лікуванні раку сечового міхура, головного мозку, молочної залози і матки, хронічних лімфоїдних лейкозів, колоректального раку, раку стравоходу і печінки, лімфобластного лейкозу, неходжкінської лімфоми, меланоми, злоякісних захворювань крові, мієломи, раку яєчників, недрібноклітинного раку легенів, раку передміхурової залози та дрібноклітинного раку легенів.
26. Застосування сполуки формули (І) за будь-яким з пп. 1-14 або її адитивної солі з фармацевтично прийнятною кислотою або основою у виробництві лікарського засобу, призначеного для лікування раку сечового міхура, головного мозку, молочної залози і матки, хронічних лімфоїдних лейкозів, колоректального раку, раку стравоходу і печінки, лімфобластного лейкозу, неходжкінської лімфоми, меланоми, злоякісних захворювань крові, мієломи, раку яєчників, недрібноклітинного раку легенів, раку передміхурової залози та дрібноклітинного раку легенів.
27. Поєднання сполуки формули (І) за будь-яким з пп. 1-14 з протираковим агентом, вибраним з генотоксичних агентів, мітотичних отрут, антиметаболітів, інгібіторів протеасом, інгібіторів кінази і антитіл.
28. Фармацевтична композиція, яка містить поєднання за п. 27 у поєднанні з одним або декількома фармацевтично прийнятними ексципієнтами.
29. Поєднання за п. 27 для застосування в лікуванні раку.
30. Застосування поєднання за п. 27 у виробництві лікарського засобу для застосування в лікуванні раку.
31. Сполука формули (І) за будь-яким з пп. 1-14 для застосування у поєднанні з променевою терапією при лікуванні раку.
Текст
Реферат: Сполуки формули (І): UA 115773 C2 (12) UA 115773 C2 R3 N R4 A1 O N O R5 A2 Ra N T Rd Rb Rc , (I) де А1, А2, Ra, Rb, Rc, Rd, R3, R4, R5 і Т є такими, як визначені в описі. R 3 N R 4 A O N O 1 A 2 R R 5 a N T R R d R c b UA 115773 C2 5 10 15 20 25 30 Даний винахід стосується нових пірольних сполук, способу їх одержання та фармацевтичних композицій, що їх містять. Сполуки за даним винаходом є новими і мають дуже цінні фармакологічні характеристики у сфері апоптозу та онкології. Апоптоз, або запрограмована смерть клітин, є фізіологічним процесом, який має вирішальне значення для ембріонального розвитку і підтримки тканинного гомеостазу. Некроз клітин апоптозного типу включає в себе морфологічні зміни, такі як конденсацію ядра клітини, фрагментацію ДНК, а також біохімічні явища, такі як активацію каспаз, які спричиняють пошкодження ключових структурних компонентів клітини, таким чином, викликаючи її руйнування і смерть. Регулювання процесу апоптозу є складним і включає в себе активацію або пригнічення декількох внутрішньоклітинних сигнальних шляхів (Cory S. et al., Nature Review Cancer, 2002, 2, 647-656). Дерегулювання апоптозу бере участь у певних патологіях. Підвищений апоптоз пов'язаний з нейродегенеративними захворюваннями, такими як хвороба Паркінсона, хвороба Альцгеймера та ішемія. І навпаки, дефіцит реалізації апоптозу відіграє значну роль у розвитку ракових захворювань та їх хімічної стійкості, в аутоімунних захворюваннях, запальних захворюваннях і вірусних інфекціях. Відповідно, відсутність апоптозу є однією з фенотипічних ознак раку (Hanahan D. et al., Cell 2000, 100, 57-70). Антиапоптозні білки сімейства Всl-2 пов'язані з численними патологіями. Участь білків сімейства Всl-2 описана у численних типах раку, таких як колоректальний рак, рак молочної залози, рак легенів, дрібноклітинний рак легенів, недрібноклітинний рак легенів, рак сечового міхура, рак яєчників, рак передміхурової залози, хронічний лімфолейкоз, фолікулярна лімфома, мієлома тощо. Надекспресія антиапоптозних білків сімейства Всl-2 застосовується в онтогенезі, стійкості до хіміотерапії і клінічному прогнозі пацієнтів, які страждають на рак. Таким чином, існує терапевтична потреба у сполуках, які інгібують антиапоптозну активність білків сімейства Всl-2. Крім новизни, сполуки за даним винаходом мають проапоптозні властивості, які дозволяють їх використання у патологіях, що мають дефект апоптозу, таких як, наприклад, при лікуванні раку, аутоімунних захворювань і захворювань імунної системи. Даний винахід стосується особливо сполук формули (І): R3 N R4 A1 O N O R5 A2 Ra N T Rd Rb Rc 35 40 45 , (I) де: - А1 і А2, кожен незалежно один від одного, являють собою атом водню або галогену, лінійну або розгалужену (С1-С6)полігалоалкільну групу, лінійну або розгалужену (С 1-С6)алкільну групу або циклоалкільну групу, - Т являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу, необов'язково заміщену від одного до трьох атомів галогену, групу (С 1-С4)алкіл-NR1R2 або групу (С1-С4)алкілОR6, - R1 і R2, кожен незалежно один від одного, являють собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, або R1 і R2 утворюють з атомом азоту, до якого вони приєднані, гетероциклоалкіл, - R3 являє собою лінійну або розгалужену (С1-С6)алкільну групу, лінійну або розгалужену (С 2С6)алкенільну групу, лінійну або розгалужену (С2-С6)алкінільну групу, циклоалкільну групу, (С3С10)циклоалкіл-(С1-С6)алкільну групу, де алкільний фрагмент є лінійним або розгалуженим, гетероциклоалкільну групу, арильну групу або гетероарильну групу, слід розуміти, що один або більше атомів вуглецю з попередніх груп, або їх можливих замісників, можуть бути дейтеровані, 1 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 - R4 являє собою арильну групу, гетероарильну групу, циклоалкільну групу або лінійну або розгалужену (С1-С6)алкільну групу, слід розуміти, що один або більше атомів вуглецю попередніх груп, або їх можливих замісників, можуть бути дейтеровані, - R5 являє собою атом водню або галогену, лінійну або розгалужену (С 1-С6)алкільну групу або лінійну або розгалужену (С1-С6)алкоксигрупу, - R6 являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, - Ra, Rb, Rc і Rd, кожен незалежно від інших, являють собою R7, атом галогену, лінійну або розгалужену (С1-С6)алкоксигрупу, гідроксигрупу, лінійну або розгалужену (С 1С6)полігалоалкільну групу, трифторметоксигрупу, -NR7R7', нітро, R7-CO-(CO-C6)алкіл-, R7-CONH-(С0-С6)алкіл-, NR7R7'-CО-(C0-C6)алкіл-, NR7R7'-CО-(C0-C6)алкіл-O-, R7-SO2-NH-(C0-C6)алкіл-, R7-NH-CO-NH-(C0-C6)алкіл-, R7-O-CO-NH-(C0-С6)алкіл-, гетероциклоалкіл, або замісники однієї з пар (Ra, Rb), (Rb, Rc) або (Rc, Rd) утворюють разом з атомами вуглецю, до яких вони приєднані, кільце, що складається з від 5 до 7 кільцевих членів, які можуть містити від одного до 2 гетероатомів, вибраних з кисню і сірки; слід також розуміти, що один або більше атомів вуглецю кільця, визначеного тут вище, можуть бути дейтеровані або заміщені від однієї до 3 груп, вибраних з галогену і лінійного або розгалуженого (С1-С6)алкілу, -R7 і R7', кожен незалежно один від одного, являють собою водень, лінійний або розгалужений (С1-С6)алкіл, лінійний або розгалужений (С2-С6)алкеніл, лінійний або розгалужений (С2-С6)алкініл, арил або гетероарил, або R7 i R7' разом з атомом азоту, до якого вони приєднані, утворюють гетероцикл, що складається з від 5 до 7 кільцевих членів, причому слід розуміти, що: - "арил" означає фенільну, нафтильну, біфенільну або інденільну групу, - "гетероарил" означає будь-яку моно- або біциклічну групу, що складається з від 5 до 10 кільцевих членів, які мають принаймні один ароматичний фрагмент і містять від 1 до 4 гетероатомів, вибраних з кисню, сірки і азоту (в тому числі четвертинного азоту), - "циклоалкіл" означає будь-яку моно- або біциклічну, неароматичну, карбоциклічну групу, що містить від 3 до 10 кільцевих членів, - "гетероциклоалкіл" означає будь-яку моно- або біциклічну, неароматичну, конденсовану або спірогрупу, що складається з від 3 до 10 кільцевих членів і містить від 1 до 3 гетероатомів, вибраних з кисню, сірки, SO, SO2 та азоту, причому для арильної, гетероарильної, циклоалкільної і гетероциклоалкільної груп, визначених таким чином, і груп алкілу, алкенілу, алкінілу і алкоксилу можливе заміщення від 1 до 3 групами, вибраними з необов'язково заміщеного лінійного або розгалуженого (С 1-С6)алкілу, (С3-С6)спіро, необов'язково заміщеного, лінійного або розгалуженого (С 1-С6)алкокси, (С1С6)алкіл-S-, гідрокси, оксо (або N-оксиду, де це доречно), нітро, ціано, -COOR', -OCOR', NR'R", лінійного або розгалуженого (С1-С6)полігалоалкілу, трифторметокси, (С1-С6)алкілсульфонілу, галогену, необов'язково заміщеного арилу, гетероарилу, арилокси, арилтіо, циклоалкілу, гетероциклоалкілу, необов'язково заміщеного одним або кількома атомами галогену або алкільними групами, слід розуміти, що R' і R", кожен незалежно від іншого, являє собою атом водню або необов'язково заміщену, лінійну або розгалужену (С 1-С6)алкільну групу, їх енантіомерів і діастереоізомерів та адитивних солей з фармацевтично прийнятною кислотою або основою. Серед фармацевтично прийнятних кислот можливо відзначити, але без будь-яких обмежень, соляну кислоту, бромистоводневу кислоту, сірчану кислоту, фосфонову кислоту, оцтову кислоту, трифтороцтову кислоту, молочну кислоту, піровиноградну кислоту, малонову кислоту, бурштинову кислоту, глутарову кислоту, фумарову кислоту, винну кислоту, малеїнову кислоту, лимонну кислоту, аскорбінову кислоту, щавлеву кислота, метансульфонову кислоту, камфорну кислоту, тощо. Серед фармацевтично прийнятних основ можливо відзначити, але без будь-яких обмежень, гідроксид натрію, гідроксид калію, триетиламін, трет-бутиламін тощо. Переважно А1 являє собою атом водню або метильну групу. Крім того, А2 переважно являє собою лінійну або розгалужену (С 1-С6)алкільну групу, необов'язково заміщену групою, вибраною з галогену, гідрокси, лінійного або розгалуженого (С 1С6)алкокси, NR'R" і морфоліну. В іншому варіанті здійснення даного винаходу, А2 являє собою лінійну або розгалужену (С1С6)полігалоалкільну групу або циклопропільну групу. Ще більш переважно, А1 і А2 обидва являть собою метильну групу. У переважному варіанті здійснення даного винаходу, Т являє собою лінійний або розгалужений (С1-С6)алкіл. В іншому переважному варіанті, Т являє собою групу алкіл(С 1-С4) 2 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 NR1R2 і, зокрема, групу алкіл(С1-С4)-NR1R2, де R1 і R2 утворюють з атомом азоту, до якого вони приєднані, гетероциклоалкіл. У переважних сполуках за даним винаходом, Т являє собою метил, амінометил, (морфолін4-іл)метил, (4-метилпіперазин-1-іл)метил, 2-(морфолін-4-іл)етил, [2-(морфолін-4іл)етокси]метил, гідроксиметил, [2-(диметиламіно)етокси]метил, гексагідропіразино[2,1с][1,4]оксазин-8(1Н)-ілметил, 1-окса-6-азаспіро[3,3]гепт-6-илметил, 3-(морфолін-4-іл)пропіл або трифторметільну групу. Ще більш переважно, Т являє собою (морфолін-4-іл)метил, метил або 3-(морфолін-4-іл)пропільну групу. Переважно Ra і Rd являє собою атом водню і (Rb, Rc), разом з атомами вуглецю, до яких вони приєднані, утворюють 1,3-діоксоланову групу або 1,4-діоксанову групу; або Ra, Rc і Rd кожен являє собою атом водню і Rb являє собою атом водню або галогену або метоксигрупу. Ще більш переважно, Ra і Rd кожен являє собою атом водню і (Rb, Rc), разом з атомами вуглецю, до яких вони приєднані, утворюють 1,3-діоксоланову групу; або Ra, Rc і Rd кожен являє собою атом водню і Rb являє собою галоген, переважно атом хлору або фтору. В іншому варіанті даного винаходу Ra і Rd кожен являє собою атом водню, Rb являє собою атом водню або галогену і Rc - гідрокси або метоксигрупу, або Ra і Rd кожен являє собою атом водню, Rb являє собою гідрокси або метоксигрупу і Rc - атом галогену. Крім того, Ra, Rb і Rd переважно кожен являє собою атом водню і Rc являє собою групу, вибрану з R7-CO-NH-(C0-C6)алкілу-, R7-SO2-NH-(C0-С6)алкілу-, R7-NH-CO-NH-(C0-C6)aлкілy- і R7O-CO-NH-(C0-C6)алкілy-. Для цих конкретних сполук, R3 переважно являє собою лінійний або розгалужений (С1-С6)алкіл або гетероарил, необов'язково заміщений лінійним або розгалуженим (С1-С6)алкілом, і R4 являє собою 4-гідроксифенільну групу. Ще більш переважно, R3 являє собою метил. Переважні групи R4 є наступними: феніл; 4-гідроксифеніл; 3-фтор-4-гідроксифеніл, 2гідроксипіримідин; 3-гідроксипіридин. Ще більш переважно, R4 являє собою 4-гідроксифенільну групу. У переважних сполуках за винаходом, R3 являє собою лінійну або розгалужену (С1С6)алкільну групу (переважно метил), арил або гетероарил, усі з яких необов'язково заміщені. Ці групи арилу і гетероарилу є особливо переважними. Нарешті, R3 переважно являє собою групу, вибрану з фенілу, 1Н-піразолу, 1Н-індолу, 1Н-індазолу, піридину, піримідину, 1Н-піроло[2,3b]піридину, 2,3-дигідро-1Н-піроло[2,3-b]піридину, 1Н-бензімідазолу, 1Н-піролу, 1Н-піроло[2,3с]піридину, 1Н-піроло[3,2-b]піридину, 5Н-піроло[3,2-d]піримідину, тіофену, піразину, 1Нпіразоло[3,4-b]піридину, 1,2-оксазолу і 1Н-піразоло[1,5-а]піримідину, причому ці групи необов'язково мають один або кілька замісників, вибраних з галогену, лінійного або розгалуженого (С1-С6)алкілу, лінійного або розгалуженого (С1-С6)алкокси, ціано, циклопропілу, оксетану, тетрагідрофурану, -СО-О-СН3, тридейтеріометилу, 2-(морфолін-4-іл)етилу і 2(морфолін-4-іл)етокси. Більш переважно, R3 являє собою групу 1-метил-1Н-піразол-4-ілу, піридин-4-ілу, 1-метил-1Н-піроло[2,3-b]піридин-5-ілу, 5-ціано-1-метил-1Н-пірол-3-ілу, 5-ціано-1,2диметил-1Н-пірол-3-ілу, 1-метил-2,3-дигідро-1Н-піроло[2,3-b]піридин-5-ілу, 5-ціано-2-метил-1(тридейтеріометил)-1Н-пірол-3-ілу. Переважні сполуки за даним винаходом включені до наступної групи: - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-1Н-пірол-3-карбоксамід, 5-(5-хлор-2-{[(3R)-3-метил-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4гідроксифеніл)-1,2-диметил-N-(піридин-4-іл)-1Н-пірол-3-карбоксамід, N-(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-5-(6-{[(3S)-3-(морфолін-4ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}-1,3-бензодіоксол-5-іл)-1Н-пірол-3-карбоксамід, N-(4-гідроксифеніл)-1,2-диметил-5-(6-{[(3R)-3-метил-3,4-дигідроізохінолін-2(1Н)іл]карбоніл}-1,3-бензодіоксол-5-іл)-N-(піридин-4-іл)-1Н-пірол-3-карбоксамід, - 5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піразол-4-іл)-1H-пірол-3-карбоксамід, - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N(4-гідроксифеніл)-1,2-диметил-N-(1-метил-1Н-піроло[2,3-b]піридин-5-іл)-7Н-пірол-3-карбоксамід, - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N(4-гідроксифеніл)-1,2-диметил-N-(піридин-4-іл)-1Н-пірол-3-карбоксамід, - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1H)-іл]карбоніл}феніл)-N(5-ціано-1-метил-1Н-пірол-3-іл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід, N-(5-ціано-1-метил-1Н-пірол-3-іл)-5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-1H-пірол-3карбоксамід, 3 UA 115773 C2 5 10 15 -5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]-карбоніл}феніл)-N(5-ціано-1,2-диметил-1Н-пірол-3-іл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3-карбоксамід, - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1H)-іл]карбоніл}феніл)-N(4-гідроксифеніл)-1,2-диметил-N-(1-метил-2,3-дигідро-1H-піроло[2,3-b]піридин-5-іл)-1Н-пірол-3карбоксамід, N-(5-ціано-1,2-диметил-1Н-пірол-3-іл)-5-(5-фтор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол-3карбоксамід, - 5-(5-хлор-2-{[(3S)-3-(морфолін-4-ілметил)-3,4-дигідроізохінолін-2(1Н)-іл]карбоніл}феніл)-N[5-ціано-2-метил-1-(тридейтеріометил)-1H-пірол-3-іл]-N-(4-гідроксифеніл)-1,2-диметил-1Н-пірол3-карбоксамід, їх енантіомери, діастереоізомери та адитивні солі з фармацевтично прийнятною кислотою або основою. Винахід стосується також способу одержання сполук формули (І), причому цей спосіб характеризується тим, що як вихідний матеріал використовують сполуку формули (II): O Br Ra Rb Rd Rc , (II) де Ra, Rb, Rc і Rd є такими, як визначені для формули (І), де сполуку формули (II) піддали реакції Хека, у водному або органічному середовищі, в присутності паладієвого каталізатора, основи, фосфіну і сполуки формули (III): Alk O A1 O N 20 A2 , (III) де групи А1 і А2 є такими, як визначені для формули (І), і Alk позначає лінійний або розгалужений (С1-С6)алкіл, для одержання сполуки формули (IV): Alk O A1 O N O A2 Ra Rb Rd Rc 25 , (IV) де А1, А2, Ra, Rb, Rc і Rd є такими, як визначені для формули (І), і Alk є таким, як визначено вище, альдегідну функцію сполуки формули (IV) окислили до карбонової кислоти для утворення сполуки формули (V): 4 UA 115773 C2 Alk O A1 O N A2 O Ra HO Rb Rd Rc , (V) де A1, A2, Ra, Rb, Rc і Rd є такими, як визначені для формули (І), і Alk є таким, як визначено вище, де сполуку формули (V) потім піддали пептидному зв'язуванню зі сполукою формули (VI): R5 5 NH T , (VI) де Т і R5 є такими, як визначені для формули (І), для одержання сполуки формули (VII): Alk O A1 O N O R5 A2 Ra N T Rd Rb Rc 10 15 20 25 , (VII) де A1, А2, Ra, Rb, Rc і Rd, T і R5 є такими, як визначені для формули (І), і Аlк є таким, як визначено вище, функцію складного ефіру сполуки формули (VII) гідролізували для одержання відповідної карбонової кислоти або карбоксилату, який може бути перетворений на похідну кислоти, таку як відповідний хлористий ацил або ангідрид, перед з'єднанням з аміном NHR 3R4, де R3 і R4 мають такі ж значення, як у формулі (І), для одержання сполуки формули (І), причому сполука формули (І) може бути очищена відповідно до звичайного методу сепарації, перетворена, якщо бажано, на її адитивні солі з фармацевтично прийнятною кислотою або основою і необов'язково поділена на її ізомери відповідно до звичайного методу сепарації, слід розуміти, що в будь-який момент, який вважається доречним в ході описаного вище способу, певні групи (гідрокси, аміно…) реагентів або інтермедіатів синтезу можуть бути захищені, а потім позбавлені захисту відповідно до вимог синтезу. Більш конкретно, коли одна з груп R3 або R4 аміну NHR3R4 є заміщеною гідроксильною функцією, остання може бути піддана заздалегідь реакції захисту перед будь-яким з'єднанням з карбоновою кислотою, утвореною зі сполуки формули (VII), або з її відповідною похідною кислоти, причому одержана захищена сполука формули (І) надалі вступає в реакцію зняття захисту і потім необов'язково перетворюється на одну з її адитивних солей з фармацевтично прийнятною кислотою або основою. 5 UA 115773 C2 Даний винахід стосується також альтернативного способу одержання сполук формули (I'), які є окремими випадками сполук формули (І), як визначено вище: R3 N R4 A1 O N O Ra N R5 A2 T Rd Rb Rc 5 , (I') де: A1, A2, Ra, Rb, Rc і Rd, T і R5 є такими, як визначені для формули (І), Rb і Rc є такими, що один являє собою водень, а інший групу, вибрану з R7-CO-NH-(C0C6)алкілу-, R7-SO2-NH-(C0-C6)алкілу-, R7-NН-СО-NН-(С0-6алкілу- і R7-O-CO-NH-(C0-C6)алкілу-, причому спосіб одержання використовує як вихідний матеріал сполуку формули (II'): O Hal Ra HO X1 Rd X2 10 15 , (ІI') де: Ra і Rd є такими, як визначені для формули (І), Hal являє собою атом галогену, Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2-групу, а другий являє собою атом водню, де сполуку формули (ІI') потім піддають пептидному зв'язуванню зі сполукою формули (VI): R5 NH T , (VI) де Т і R5 є такими, як визначені для формули (І), для одержання сполуки формули (III'): O R5 Hal Ra N T Rd X1 X2 20 25 , (III') де: Ra, Rd, T і R5 є такими, як визначені для формули (І), Hal являє собою атом галогену, Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2-групу, а другий являє собою атом водню, де сполуку формули (III') піддають реакції Хека, у водному або органічному середовищі, в присутності паладієвого каталізатора, основи, фосфіну і сполуки формули (IV'): 6 UA 115773 C2 R3 N O R4 A1 N A2 , (IV') де А1, А2, R3 і R4 є такими, як визначені для формули (І), для утворення сполуки формули (V'): R3 N R4 A1 O N O R5 A2 Ra N T Rd X1 X2 5 10 15 20 25 30 35 , (V') де: A1, A2, Ra, Rd, R3, R4, T і R5 є такими, як визначені для формули (І), Х1 і Х2 є такими, що один являє собою (С0-С6)алкіл-NН2-групу, а другий являє собою атом водню, де сполуку формули (V') потім піддають реакції ацилювання або сульфонілування для одержання сполуки формули (I'), причому сполука формули (I') може бути очищена відповідно до звичайного методу сепарації, перетворена, якщо бажано, на її адитивні солі з фармацевтично прийнятною кислотою або основою і необов'язково поділена на її ізомери відповідно до звичайного методу сепарації, слід розуміти, що в будь-який момент, який вважається доречним в ході описаного вище способу, певні групи (гідрокси, аміно…) реагентів або інтермедіатів синтезу можуть бути захищені, а потім позбавлені захисту відповідно до вимог синтезу. Сполуки формул (II), (III), (II'), (IV'), (VI) і амін NHR 3R4 або є комерційно доступними, або можутьбути одержані фахівцем у даній галузі з використанням звичайних хімічних реакцій, описаних у літературі. Фармакологічне дослідження сполук за винаходом засвідчило, що вони мають проапоптозні властивості. Здатність реактивувати процес апоптозу в ракових клітинах представляє значний терапевтичний інтерес при лікуванні раку, аутоімунних захворювань і захворювань імунної системи. Більш конкретно, сполуки за даним винаходом будуть корисними при лікуванні хіміорезистентних та радіорезистентних ракових захворювань, злоякісних захворювань крові і дрібноклітинного раку легенів. Серед видів раку, щодо яких передбачена терапія, можуть бути згадані, але не обмежуючись, рак сечового міхура, головного мозку, молочної залози і матки, хронічні лімфоїдні лейкози, колоректальний рак, рак стравоходу і печінки, лімфобластний лейкоз, неходжкінська лімфома, меланоми, злоякісні захворювання крові, мієломи, рак яєчників, недрібноклітинний рак легенів, рак передміхурової залози та дрібноклітинний рак легенів. Як неходжкінська лімфома можуть бути згадані більш переважно фолікулярні лімфоми, лімфоми з клітин мантійної зони, дифузні В-великоклітинні лімфоми, дрібноклітинні лімфоцитарні лімфоми та лімфоми з В-клітин маргінальної зони. Даний винахід також стосується фармацевтичних композицій, які містять принаймні одну сполуку формули (І) у поєднанні з одним або декількома фармацевтично прийнятними ексципієнтами. 7 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 Серед фармацевтичних композицій за даним винаходом можуть бути згадані, зокрема, ті, які є придатними для перорального, парентерального, назального, підшкірного або черезшкірного, ректального, сублінгвального, очного або респіраторного введення, зокрема таблетки або драже, сублінгвальні таблетки, саше, пакети, капсули, таблетки для повільного розчинення під язиком, пастилки, супозиторії, креми, мазі, шкірні гелі та питні або ін'єкційні ампули. Дозування варіюється залежно від статі, віку і ваги пацієнта, шляху введення, характеру терапевтичного показання або будь-яких супутніх видів лікування і коливається від 0,01 мг до 1 г на добу за один або декілька прийомів. Крім того, даний винахід також стосується поєднання сполуки формули (І) з протираковим агентом, вибраним з генотоксичних агентів, мітотичних отрут, антиметаболітів, інгібіторів протеасом, інгібіторів кінази і антитіл, а також фармацевтичних композицій, які містять цей тип поєднання, та їх використання при виготовленні лікарських засобів для застосування при лікуванні раку. Сполуки за даним винаходом можуть також використовуватися у поєднанні з променевою терапією при лікуванні раку. Нарешті, сполуки за даним винаходом можуть бути пов'язані з моноклональними антитілами або їх фрагментами або пов'язані з каркасними білками, які можуть бути пов'язані чи ні з моноклональними антитілами. Фрагменти антитіл слід розуміти як фрагменти типу Fv, ScFv, Fab, F(ab') 2, F(ab'), ScFv-Fc або діатіла, які зазвичай мають таку ж специфічність зв'язування, що і антитіло, від якого вони походять. Відповідно до даного винаходу, фрагменти антитіл за даним винаходом можуть бути одержані, виходячи з антитіл, способами, такими як перетравлювання ферментами, такими як пепсин або папаїн, та/або шляхом розщеплення дисульфідних містків хімічним відновленням. В іншому способі, фрагменти антитіл, що розкриваються в цьому винаході, можуть бути одержані за допомогою способів генетичної рекомбінації, також добре відомих фахівцям у даній галузі техніки, або шляхом синтезу пептидів за допомогою, наприклад, автоматичних синтезаторів пептидів, таких, які постачаються компанією "Applied Biosystems", тощо. Каркасні білки, які можуть бути пов'язаними або не пов'язаними з моноклональними антитілами, слід розуміти як такі, що означають білок, який містить або не містить укладку ланцюга імуноглобуліну і забезпечує здатність зв'язування, аналогічну до моноклонального антитіла. Фахівцям в даній галузі техніки відомо, як вибирати каркас білка. Зокрема, відомо, що для вибрання, наприклад, такий каркас повинен забезпечувати кілька функцій, таких як (Skerra A., J. Моl Recogn, 13, 2000, 167-187): належну філогенетичну консервативність, надійну архітектуру з добре відомою тривимірною молекулярною організацією (такі як, наприклад, кристалографія або ЯМР), малий розмір, не мати або мати тільки низький ступінь посттрансляційних модифікацій, бути простим для одержання, експресії і очищення. Такий каркас білка може бути, але без обмеження, структурою, вибраною з групи, що включає в себе фібронектин і переважно десятий подібний до фібронектину III домен (FNfn10), ліпокалін, антикалін (Skerra A., J. Biotechnol., 2001, 74 (4): 257-75), похідну білка Z від домену В стафілококового білка А, тиоредоксин А або будь-який білок з повторним доменом, таким як "анкіриновий повтор" (Kohl et al., PNAS, 2003, vol. 100, No. 4, 1700-1705), "повторний білок броненосця", "багатий на лейцин повтор" або "тетратрикопептидний повтор". Також можливо зазначити каркасні похідні від токсинів (таких як, наприклад, токсинів скорпіона, комах, рослин або молюсків) або інгібіторів білка нейрональної синтази оксиду азоту (PIN). Наступні Приготування і Приклади ілюструють винахід, жодним чином не обмежуючи його. Приготування 1: 4-хлор-2-[4-(етоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Етап А: етил-1,2-диметил-1Н-пірол-3-карбоксилат До розчину 10 г етил-2-метил-1Н-пірол-3-карбоксилату (65,3 ммоль) і 8,95 мл (130,6 ммоль) йодистого метилу в 70 мл диметилформаміду, розміщеного при 0 °C, додали трьома частинами 2,61 г (65,3 ммоль) гідриду натрію (NaH) 60 %. Суміш потім перемішували при 0 °C протягом 1 години. Потім, реакційну суміш гідролізували шляхом додавання 420 мл льодяної води. Потім реакційну суміш розбавили етилацетатом, послідовно промили 0,1 М водним розчином хлористоводневої кислоти (НСl), насиченим водним розчином LiCl і потім сольовим розчином. Органічну фазу потім висушили над MgSO4, фільтрували, концентрували досуха і очистили за допомогою хроматографії на силікагелі (петролейний ефір/етилацетат (AcOEt) в градієнті). 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 6,65 (д, 1Н-пірол); 6,3 (1д, 1Н-пірол); 4,1 (1к, 2Н, ОСН2СН3); 3,5 (с, 3Н N-пірол); 2,4 (с, 3Н-пірол); 1,5 (1т, 3Н ОСН2СН3), -1: -1 ІЧ: :>C=O: 1688 см : С-О-С: 1172 см . Етап В: етил-5-(5-хлор-2-формілфеніл)-1,2-диметил-1Н-пірол-3-карбоксилат 8 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 До розчину 10,5 г сполуки, одержаної на етапі А (62,8 ммоль), в 65 мл N,Nдиметилацетаміду послідовно додали 15,2 г 2-бром-4-хлорбензальдегіду (69 ммоль), 12,3 г ацетату калію (125,6 ммоль), і потім суміш перемішували в атмосфері аргону протягом 20 хвилин. Потім додали 2,2 г паладієвого каталізатора PdCl2(PPh3)2 (3,14 ммоль). Реакційну суміш потім нагрівали при 130 °C протягом ночі. Суміші дали охолонути до кімнатної температури і потім її розбавили дихлорметаном. Додали активоване вугілля тваринного походження (2 г на г продукту) і суміш перемішували при кімнатній температурі протягом 1 години, а потім фільтрували. Органічну фазу потім промили водою, висушили над MgSO 4 і концентрували досуха. Сирий продукт, одержаний таким чином, очистили за допомогою хроматографії на силікагелі (петролейний ефір/етилацетат в градієнті). Зазначений у заголовку продукт одержали у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 9,8 (с, 1Н, форміл); 7,91-7,69-7,61 (д, 3Н, ароматичні Hs); 6,5 (с, 1Н-пірол); 4,2 (к, 2Н, ОСН2СН3); 3,4 (с, 3Н, СН3-N-пірол); 2,55 (с, 3Нпірол); 1,28 (т, 3Н, ОСН2СН3). Етап С: 4-хлор-2-[4-(етоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Приготували розчин, який містить 12,85 г сполуки, одержаної на етапі В (42 ммоль), і 35,7 мл (336 ммоль) 2-метил-2-бутену, в суміші, що містить 20 мл ацетону і 20 мл тетрагідрофурану. Додали по краплях 200 мл водного розчину, що містить суміш 13,3 г хлориту натрію (NaClO 2) (147 ммоль) і 14,5 г гідрофосфату натрію (NaHPO 4) (105 ммоль). Потім суміш енергійно перемішували при кімнатній температурі протягом 7 годин. Реакційну суміш потім концентрували для видалення ацетону. Додали етилацетат і органічну фазу промили водою, висушили над MgSO4 і потім концентрували насухо. Залишок потім розчинили у мінімальній кількості діетилового ефіру. Потім одержану тверду речовину фільтрували, промили ефіром і потім сушили in vacuo при 40 °C протягом ночі. Зазначений у заголовку продукт одержали у формі твердої речовини, яку потім використовували без будь-якого іншого очищення. 1 Н-ЯМР (400 МГц, ДMCO-d6; 300К) м.ч.: 13 (м, 1Н СООН); 7,85-7,6-7,41 (д, дд, ушир, д, 3Н, ароматичні Hs); 6,3 (с, 1Н, Н-пірол); 4,15 (к, 2Н, ОСН2СН3); 3,25 (с, 3Н, СН3-N-пірол); 2,5 (с, 3Н, СН3-пірол); 1,25 (т, 3Н, ОСН2СН3), -1 -1 ІЧ: :-OH 3100-2500 см кислота; :>C=O: 1681 см ефір + кислота. Приготування 2: 2-[4-(етоксикарбоніл)-1-метил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи, з одного боку, етил-2-метил-1Нпірол-3-карбоксилат, який використовували на етапі А, на етил 1Н-пірол-3-карбоксилат і, з іншого боку, 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 2-бромбензальдегід. Приготування 3: 4-хлор-2-[4-(етоксикарбоніл)-1-метил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи етил-2-метил-1Н-пірол-3карбоксилат, який використовували на етапі А, на етил 1Н-пірол-3-карбоксилат. Приготування 4: 6-[4-(етоксикарбоніл)-1-метил-1Н-пірол-2-іл]-1,3-бензодіоксол-5-карбонова кислота Процедура відповідає способу Приготування 1, замінюючи, з одного боку, етил-2-метил-1Нпірол-3-карбоксилат, який використовували на етапі А, на етил 1Н-пірол-3-карбоксилат і, з іншого боку, 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3бензодіоксол-5-карбальдегід. -1 -1 ІЧ: :-OH 3500-2300 см кислота; :>C=О: 1688-1670 см ефір + кислота. Приготування 5: 4-хлор-2-[4-(етоксикарбоніл)-1-етил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на етапі А, з одного боку, етил-2метил-1Н-пірол-3-карбоксилат на етил 1Н-пірол-3-карбоксилат і, з іншого боку, йодистий метил на йодистий етил (див. протокол, описаний в US 6.258.805 В1). Приготування 6: 4-хлор-2-[1-циклопропіл-4-(етоксикарбоніл)-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на етапі А, з одного боку, етил-2метил-1Н-пірол-3-карбоксилат на етил 1Н-пірол-3-карбоксилат і, з іншого боку, йодистий метил на циклопропілборну кислоту (див. протокол, описаний у Benard S. et al, Journal of Organic Chemistry 73(16), 6441-6444, 2008). Приготування 7: 4-хлор-2-[4-(етоксикарбоніл)-1-(пропан-2-іл)-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на етапі А, з одного боку, етил-2метил-1Н-пірол-3-карбоксилат на етил 1Н-пірол-3-карбоксилат і, з іншого боку, йодистий метил на йодистий ізопропіл (див. протокол, описаний у Okada E. et al, Heterocycles 34(7), 1435-1441, 1992). Приготування 8: 4-фтор-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота 9 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 Процедура відповідає способу Приготування 1, замінюючи на етапі А, етил-2-метил-1Нпірол-3-карбоксилат на метил 2-метил-1Н-пірол-3-карбоксилат, а також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-4-фторбензальдегід. -1 -1 ІЧ: :-OH 2727-2379 см кислота; >C=O: 1687 см . Приготування 9: 6-(1-[2-(бензилоксі)етил]-4-(етоксикарбоніл)-5-метил-1H-пірол-2-іл)-1,3бензодіоксол-5-карбонова кислота Етап А: етил-1-[2-(бензилоксі)етил]-2-метил-1Н-пірол-3-карбоксилат Процедура відповідає способу Етапу А Приготування 1, замінюючи йодистий метил, який використовували як алкілуючий агент, на бензил-2-брометиловий ефір. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,32 (т, 2Н, ароматичні Hs, H-мета-бензиловий ефір); 7,3 (т, 1Н, ароматичні Н, Н-пара-бензиловий ефір); 7,23 (д, 2Н, ароматичні Hs, Н-ортобензиловий ефір); 6,72 (д, 1Н, Н-пірол); 6,35 (д, 1Н, Н-пірол); 4,48 (с, 2Н, аліфатичні Hs, O-CH2Ph);4,15 (к, 2Н, аліфатичні Hs, О-СН2-СН3); 4,1 (т, 2Н, аліфатичні Hs, CH2-O-CH2-Ph); 3,7 (т, 2Н, аліфатичні Hs, CH2-CH2-O-CH2-Ph); 2,45 (с, 3Н, СН3-пірол); 1,25 (т, 3Н, аліфатичні Hs, O-CH2CH3), -1 ІЧ: >C=O: 1689 см . Етап В: 6-{1-[2-(бензилоксі)етил]-4-(етоксикарбоніл)-5-метил-1Н-пірол-2-іл}-1,3-бензодіоксол5-карбонова кислота Процедура відповідає способам Етапів В і С Приготування 1, замінюючи 2-бром-4хлорбензальдегід на 6-бром-1,3-бензодіоксол-5-карбальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,35 (с, 1Н, ароматичні Н, Н-бензодіоксол); 7,30 (м, 3Н, ароматичні Hs, Н-бензиловий ефір); 7,25 (с, 1Н, ароматичні Н, Н-бензодіоксол); 7,10 (д, 2Н, ароматичні Hs, H-орто-бензиловий ефір); 12,55 (ушир, с, 1Н, СООН); 6,75 (с, 1Н, Н-пірол); 6,15 (ушир, с, 2Н, аліфатичні Hs, О-СН2-О); 4,30 (с, 2Н, аліфатичні Hs, O-CH2-Ph); 4,15 (к, 2Н, аліфатичні Hs, О-СН2-СН3); 3,9 (м, 2Н, аліфатичні Hs, СН2-СН2-O-CH2-Ph); 3,40 (т, 2Н, аліфатичні Hs, CH2-CH2-O-CH2-Ph); 2,50 (с, 3Н, СН3-пірол); 1,25 (т, 3Н, аліфатичні Hs, O-CH2CH3), -1 -1 ІЧ: :-OH 3200-2300 см кислота; >C=O: 1687 см кислота. Приготування 10: 2-{1-[3-(бензилокси)пропіл]-4-(етоксикарбоніл)-5-метил-1Н-пірол-2-іл}-4фторбензойна кислота Процедура відповідає способу Приготування 9, замінюючи бензил-2-брометиловий ефір, який використовували на етапі А, на бензил 3-бромпропіловий ефір, а також 6-бром-1,3бензодіоксол-5-карбальдегід, який використовували на етапі В, на 2-бром-4-фторбензальдегід. -1 -1 ІЧ: :-OH: 3200-2305 см кислота; :>C=O: 1690 см кислота. Приготування 11: 6-[4-(етоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає способу Приготування 1, замінюючи 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5-карбальдегід. Приготування 12: 4-метокси-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи етил-2-метил-1Н-пірол-3карбоксилат, який використовували на етапі А, на метил 2-метил-1Н-пірол-3-карбоксилат, а також 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 2-бром-4метоксибензальдегід. -1 -1 ІЧ: :-OH: 3000-2500 см кислота; :>C=O: 1693+1670 см кислота + ефір. Приготування 13: 7-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]-2,3-дигідро-1,4бензодіоксин-6-карбонова кислота Процедура відповідає способу Приготування 1, замінюючи на Етапі А етил-2-метил-1Нпірол-3-карбоксилат на метил 2-метил-1Н-пірол-3-карбоксилат і також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 7-бром-2,3-дигідро-1,4-бензодіоксин-6карбальдегід. -1 -1 ІЧ: :-OH: 3100-2500 см кислота; :>C=O: 1690+1674 см кислота + ефір. Приготування 14: 4-фтор-5-метокси-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на етапі А етил-2-метил-1Нпірол-3-карбоксилат на метил 2-метил-1Н-пірол-3-карбоксилат і також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-4-фтор-5-метоксибензальдегід. Приготування 15: 4-фтор-5-гідрокси-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2іл]бензойна кислота 10 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 Процедура відповідає способу Приготування 1, замінюючи на етапі А етил-2-метил-1Нпірол-3-карбоксилат на метил-2-метил-1Н-пірол-3-карбоксилат і також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-4-фтор-5-гідроксибензальдегід. Приготування 16: 6-[4-(етоксикарбоніл)-1-метил-5-трифторметил-1Н-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Процедура відповідає способу Приготування 1, замінюючи на етапі А етил-2-метил-1Нпірол-3-карбоксилат на етил 2-(трифторметил)-1Н-пірол-3-карбоксилат, а також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5-карбальдегід. Приготування 17: 6-[4-(етоксикарбоніл)-1-етил-5-метил-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає способу Приготування 11, замінюючи йодистий метил на йодистий етил (див. протокол, описаний в US 6.258.805 В1). Приготування 18: 6-[4-(етоксикарбоніл)-1-метил-5-(трифторметил)-1Н-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Етап А: етил-1-метил-2-(трифторметил)пірол-3-карбоксилат Етил-4,4,4-трифтор-3-оксобутаноат (29 мл, 0,219 ммоль) додали до метиламіну (40 % розчин у воді) (50 мл, 0,580 ммоль), охолодженого до 10 °C; утворюється білий осад. 1,2Диброметилацетат (одержаний відповідно до Molecules, 16, 9368-9385; 2011) додали по краплях. Потім реактор герметизували і нагрівали при 70 °C протягом 45 хвилин. Реакційну суміш охолодили, потім екстрагували етилацетатом, висушили над сульфатом натрію (Na2SO4) і випарили насухо. Одержаний сирий продукт реакції очистили за допомогою хроматографії на силікагелі з використанням гептану і етилацетату як елюентів. Одержали очікувану сполуку у формі кристалів. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,11 (д, 1Н), 6,52 (д, 1Н), 4,21 (кв, 2Н), 3,8 (с, 3Н), 1,27 (т, 3Н), 19 F-ЯМР (400 МГц, ДМСО-d6) м.ч.: -53,9, -1 ІЧ (ATR) см : 3145+3128 -CH, 1711 >C=O, 1183+1117+1078 -CF3. Етап В: 6-[4-(етоксикарбоніл)-1-метил-5-(трифторметил)-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає протоколу, описаному на етапах В і С Приготування 1, замінюючи 2бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5карбальдегід. Приготування 19: 6-[1-(2-бензилоксіетіл)-4-етоксикарбоніл-5-метил-1Н-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Етап А: етил-1-(2-бензилоксіетил)-2-метилпірол-3-карбоксилат Етил-2-метил-1H-пірол-3-карбоксилат (10 г, 65,3 ммоль) розчинили у 100 мл диметилформаміду в атмосфері аргону, охолодженого до 0 °C, і потім додали 2брометоксиметилбензол (28,1 г, 130,6 ммоль) увесь відразу. Реакційну суміш помістили при перемішуванні. Потім до неї додавали при 0 °C, трьома частинами, NaH (1,72 г, 71,83 ммоль) протягом 15 хвилин. Реакційну суміш перемішували протягом 15 хвилин при 0 °C і потім протягом 15 годин при кімнатній температурі. Потім її вилили у льодяну баню і екстрагували 3 рази етилацетатом. Органічну фазу промили 3 рази насиченим водним розчином хлориду літію, висушили над MgSO4, фільтрували і потім випарили насухо. Одержаний таким чином залишок очистили за допомогою хроматографії на силікагелі з використанням петролейного ефіру і етилацетату як елюентів. Одержали очікувану сполуку у формі олії. 1 Н-ЯМР (400 МГц, ДMCO-d6) м.ч.: 7,32 (т, 2Н), 7,3 (т, 1Н), 7,23 (д, 2Н), 6,72 (д, 1Н), 6,35 (д, 1Н), 4,48 (с, 2Н), 4,15 (кв, 2Н), 4,1 (т, 2Н), 3,7 (т, 2Н), 2,45 (с, 3Н), 1,25 (т, 3Н), -1 IЧ (ATR) cм : 1689 -C=O. Етап В: етил-1-(2-бензилоксіетил)-5-(6-форміл-1,3-бензодіоксол-5-іл)-2-метилпірол-3карбоксилат Процедура відповідає способу Етапу В Приготування 1, замінюючи 2-бром-4хлорбензальдегід на 6-бром-1,3-бензодіоксол-5-карбальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 9,5 (с, 1Н), 7,3 (с, 1Н), 7,25 (м, 3Н), 7,05 (м, 2Н), 7 (с, 1Н), 6,4 (с, 1Н), 6,2 (ушир, с, 2Н), 4,25 (с, 2Н), 4,2 (кв, 2Н), 4,05 (м, 2Н), 3,4 (м, 2 Н), 2,55 (с, 3Н), 1,25 (т, 3Н). Етап С: 6-[1-(2-бензилоксіетил)-4-етоксикарбоніл-5-метил-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає способу Етапу С Приготування 1. 1 H-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,55 (ушир, с, 1Н), 7,35 (с, 1Н), 7,3 (м, 3Н), 7,2 (м, 3Н), 6,75 (с, 1Н), 6,15 (с, 2Н), 4,3 (с, 2Н), 4,15 (кв, 2Н), 3,9 (м, 2Н), 3,4 (т, 2Н), 2,5 (с, 3Н), 1,25 (т, 3Н), 11 UA 115773 C2 ІЧ (ATR) см :3200-2300 -OH, 1687 (+ плече) -C=O карбонова кислота + кон'югований ефір. Приготування 20: 6-[4-(етоксикарбоніл)-1-етил-5-метил-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає протоколу, описаному у Приготуванні 11, замінюючи йодистий метил на йодистий етил. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,49 (ушир, с, 1Н), 7,33 (с, 1Н), 6,89 (с, 1Н), 6,17 (с, 2Н), 6,13 (с, 1Н), 4,15 (к, 2Н), 3,69 (к, 2Н), 2,51 (с, 3Н), 1,24 (т, 3Н), 1,01 (т, 3Н). Приготування 21: 6-[4-(етоксикарбоніл)-1-(2-фторетил)-5-метил-1Н-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Процедура відповідає способу Приготування 11, замінюючи йодистий метил на 1-бром-2фторетан. 1 Н-ЯМР (400 МГц, ДМСО-d6, 300К) м.ч.: 12,53 (ушир, с, 1Н), 7,34 (с, 1Н), 6,9 (с, 1Н), 6,16 (с, 1Н), 6,16 (с, 2Н), 4,4 (д, 2Н), 4,15 (к, 2Н), 4,01 (м, 2Н), 2,51 (с, 3Н), 1,24 (т, 3Н), 19 F-ЯМР (400 МГц, ДМСО-d6, 300К) м.ч.: -222, -1 -1 -1 ІЧ: -OH: 3700-2400 см кислота; :> С=О: 1689 см кислота; :> CF:1213 cм . Приготування 22: 6-{4-(етоксикарбоніл)-1-метил-5-[2-(морфолін-4-іл)етил]-1Н-пірол-2-іл}-1,3бензодіоксол-5-карбонова кислота Етап А: 3,3-діетоксипропанова кислота До розчину 25 г етил-3,3-діетоксипропіонату (131 ммоль) у 79 мл метанолу додали 13,1 мл водного розчину 35 % гідроксиду натрію (452 ммоль). Реакційну суміш перемішували при кімнатній температурі протягом 3 годин. Реакційну суміш потім концентрували для видалення метанолу. Після розчинення нерозчиненого матеріалу шляхом додавання води, 5N водного розчину НСl додали для одержання рівня рН 5. Додали дихлорметан і потім органічну фазу промили насиченим сольовим розчином. Після сушіння над MgSO 4 і концентрування досуха, зазначений у заголовку продукт одержали у формі олії, яку використовували на наступному етапі без будь-якого іншого очищення. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,2 (с, 1Н), 4,8 (т, 1Н), 3,58/3,47 (2м, 4Н), 2,5 (д, 2Н), 1,09 (т, 6Н). Етап В: етил-5,5-діетоксі-3-оксопентаноат До розчину 16 мл 3-етоксі-3-оксомалонової кислоти (135 ммоль) у 40 мл тетрагідрофурану додали в атмосфері аргону 21,9 г порошкоподібного магнію (90,4 ммоль). Одержану суміш потім нагрівали при 80 °C протягом 7 годин. Після охолодження до кімнатної температури, цю суміш перенесли за допомогою канюлі до розчину 10 г сполуки, одержаної на Етапі А (61,7 ммоль), у 64 мл тетрагідрофурану, до якої раніше додали, частинами, 11 г карбонілдіімідазолу (66 ммоль). Реакційну суміш перемішували протягом 3 днів при кімнатній температурі. Після концентрування залишок розчинили в суміші етилацетату і водного розчину бісульфату натрію (NaHSO4). Суміш енергійно перемішували до припинення виділення газу. Після сепарації фаз, органічну фазу промили послідовно водою, насиченим водним розчином NaHCO 3 і, нарешті, сольовим розчином. Після сушіння над MgSO 4 і концентрування досуха, зазначений у заголовку продукт одержали у формі олії, яку використовували на наступному етапі без будь-якого іншого очищення. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 4,84 (т, 1Н), 4,08 (к, 2Н), 3,59 (с, 2Н), 3,56/3,46 (2м, 4Н), 2,8 (д, 2Н), 1,18 (т, 3Н), 1,09 (т, 6Н). Етап С: етил-2-(2,2-діетоксіетил)-1-метилпірол-3-карбоксилат До розчину 11,8 г сполуки, одержаної на етапі В (50,8 ммоль), у 76 мл води, додали по краплях при 0 °C 6,6 мл водного 40 % розчину метиламіну (76,2 ммоль). Реакційну суміш перемішували і обережно нагрівали до кімнатної температури протягом 5 годин. Після охолодження до 0 °C, додали 8,8 мл водного 40 % розчину метиламіну (102 ммоль) і потім 16,6 мл водного 50 % розчину хлорацетальдегіду (127 ммоль), кожен по краплях, при температурі менше 10 °C. Реакційну суміш перемішували при кімнатній температурі протягом 16 годин і потім розбавили етилацетатом. Органічну фазу промили сольовим розчином, висушили над MgSO4 і концентрували досуха. Сирий продукт, одержаний таким чином, очистили за допомогою хроматографії на силікагелі з використанням петролейного ефіру і етилацетату як елюентів. Зазначений у заголовку продукт одержали у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 6,7 (д, 1Н), 6,33 (д, 1Н), 4,55 (т, 1Н), 4,15 (к, 2Н), 3,6 (м, 2 Н), 3,6 (с, 3Н), 3,3 (м, 2Н), 3,15 (д, 2Н), 1,25 (т, 3Н), 1,05 (т, 6Н). Етап D: етил-1-метил-2-(2-морфоліноетил)-пірол-3-карбоксилат До розчину 3,8 г сполуки, одержаної на Етапі С (14,05 ммоль), у 28 мл тетрагідрофурану, додали 58 мл 10 % водного розчину сірчаної кислоти. Реакційну суміш перемішували при кімнатній температурі протягом 2 годин і потім розбавили сумішшю етилацетату і води. Після -1 5 10 15 20 25 30 35 40 45 50 55 60 12 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 сепарації, органічну фазу промили сольовим розчином, висушили над MgSO 4 і концентрували досуха. До розчину залишку, одержаного таким чином, у 70 мл дихлоретану додали розчин 13,5 мл морфоліну (14,5 ммоль) у суміші, що складається з 30 мл дихлоретану і 3,6 мл 4N водного розчину НСl у діоксані (14,5 ммоль), а потім 7,4 г триацетоксиборгідриду натрію (NaBH(OAc) 3) (35,13 ммоль). Реакційну суміш перемішували при кімнатній температурі протягом 3 годин, а потім розбавили сумішшю дихлорметану і насиченого водного розчину NaHCO 3. Після сепарації фаз та екстракції водної фази дихлорметаном, органічні фази висушили над MgSO 4 і концентрували досуха. Сирий продукт, одержаний таким чином, очистили за допомогою хроматографії на силікагелі з використанням дихлорметану та метанолу як елюентів. Зазначений у заголовку продукт виділили у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 6,66 (д, 1Н), 6,31 (д, 1Н), 4,14 (к, 2Н), 3,58 (с, 3Н), 3,57 (м, 4Н), 3,05 (м, 2Н), 2,45-2,38 (м, 6Н), 1,24 (т, 3Н). Етап Е: 6-{4-(етоксикарбоніл)-1-метил-5-[2-(морфолін-4-іл)етил]-1Н-пірол-2-іл}-1,3бензодіоксол-5-карбонова кислота Процедура відповідає Етапам В і С Приготування 1, замінюючи 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5-карбальдегід. Приготування 23: 6-(4-(етоксикарбоніл)-1-метил-5-[2-(морфолін-4-іл)метил]-1Н-пірол-2-іл)1,3-бензодіоксол-5-карбонова кислота Етап А: етил-4,4-діетоксі-3-оксобутаноат До розчину 40 г етилдіетоксіацетату (227 ммоль) у 67 мл етилацетату додали в атмосфері аргону, частинами, 6 г натрію (261 ммоль). Реакційну суміш потім перемішували при кімнатній температурі протягом 48 годин. Додали 10 мл метанолу, і потім суміш гідролізували з 65 мл води. Рівень рН реакційної суміші довели до рН 6 шляхом додавання 1N водного розчину НСl. Суміш відокремили і потім екстрагували етилацетатом. Органічні фази об'єднали, промили сольовим розчином, висушили над MgSO4, фільтрували і концентрували досуха. Зазначений у заголовку продукт одержали у формі олії, яку використовували на наступному етапі без будьякого іншого очищення. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 3,6 (с, 2Н), 1,28 (т, 9Н), 3,7-3,6 (2м, 4Н), 4,7 (с, 1Н), 4,2 (к, 2Н). Етап В: етил-2-(2,2-діетоксиметил)-1-метилпірол-3-карбоксилат Зазначену в заголовку сполуку одержали відповідно до способу, описаного на етапі С Приготування 22, виходячи з етил-4,4-діетоксі-3-оксобутаноату, одержаного на попередньому етапі. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 6,73 (д, 1Н), 6,32 (д, 1Н), 6,23 (с, 1Н), 4,17 (к, 2Н), 3,7 (с, 3Н), 3,68/3,43 (2м, 4Н), 1,26 (т, 3Н), 1,13 (т, 6Н). Етап С: етил-1-метил-2-(2-морфолінометил)-пірол-3-карбоксилат Зазначену в заголовку сполуку одержали відповідно до способу, описаного на Етапі D Приготування 22, виходячи з етил-2-(2,2-діетоксиметил)-1-метилпірол-3-карбоксилату, одержаного на попередньому етапі. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 6,74 (д, 1Н), 6,32 (д, 1Н), 4,15 (к, 2Н), 3,8 (с, 2Н), 3,65 (с, 3 Н), 3,5 (м, 4Н), 2,32 (м, 4Н), 1,22 (т, 3Н). Етап D: 6-{4-(етоксикарбоніл)-1-метил-5-[2-(морфолін-4-іл)метил]-1Н-пірол-2-іл}-1,3бензодіоксол-5-карбонова кислота Процедура відповідає протоколу, описаному на Етапах В і С Приготування 1, замінюючи 2бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5карбальдегід. Приготування 24: 6-[4-(метоксикарбоніл)-5-(2-метоксіетил)-1-метил-1H-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Етап А: метил-2-(2-метоксіетил)-1-метилпірол-3-карбоксилат Зазначену в заголовку сполуку одержали відповідно до протоколу Етапу С Приготування 22, використовуючи метил-5-метоксі-З-оксовалерат. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 6,69 (ушир, д, 1Н), 6,31 (ушир, д, 1Н), 3,69 (с, 3Н), 3,59 (с, 3Н), 3,49 (т, 2Н), 3,2 (с, 3Н), 3,11 (т, 2Н). Етап В: 6-[4-(метоксикарбоніл)-5-(2-метоксіетил)-1-метил-1Н-пірол-2-іл]-1,3-бензодіоксол-5карбонова кислота Процедура відповідає протоколу, описаному на Етапах В і С Приготування 1, замінюючи 2бром-4-хлорбензальдегід, який використовували на етапі В, на 6-бром-1,3-бензодіоксол-5карбальдегід. Приготування 25: 2-[4-(етоксикарбоніл)-1,5-диметил-1H-пірол-2-іл]бензойна кислота 13 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 Процедура відповідає способу Приготування 1, замінюючи 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 2-бромбензальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,81 (м, 1Н), 7,84 (м, 1Н), 7,59 (м, 1Н), 7,51 (м, 1Н), 7,35 (м, 1Н), 6,23 (с, 1Н), 4,16 (к, 2Н), 3,24 (с, 3Н), 2,5 (с, 3Н), 1,25 (т, 3Н). Приготування 26: 5-фтор-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на Етапі А етил-2-метил-1Нпірол-3-карбоксилат на метил-2-метил-1Н-пірол-3-карбоксилат, а також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-5-фторбензальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,6 (дд, 1Н), 7,42 (м, 2Н), 6,22 (с, 1Н), 4,15 (к, 2Н), 3,21 (с, 3Н), 2,5 (с, 3Н), 1,23 (т, 3Н), 19 F-ЯМР (400 МГц, ДМСО-d6) м.ч.: -113. Приготування 27: 2-[4-(етоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]-5-фтор-4метоксибензойна кислота Процедура відповідає способу Приготування 1, замінюючи на Етапі А етил-2-метил-1Нпірол-3-карбоксилат на метил-2-метил-1Н-пірол-3-карбоксилат, а також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-4-метокси-5-фторбензальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 9,65 (д, 1Н), 7,67 (д, 1Н), 7,24 (д, 1Н), 6,48 (с, 1Н), 4,19 (к, 2Н), 3,97 (с, 3Н), 3,38 (с, 3Н), 2,56 (с, 3Н), 1,26 (т, 3Н). Приготування 28: 2-(4-етоксикарбоніл-1,5-диметил-1Н-пірол-2-іл)-4-фтор-5-метоксибензойна кислота Процедура відповідає Приготуванню 1, замінюючи 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 4-фтор-5-метоксибензальдегід. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,9 (м, 1Н), 7,6 (д, 1Н), 7,22 (д, 1Н), 6,23 (с, 1Н), 4,2 (к, 2Н), 3,95 (с, 3Н), 3,2 5 (с, 3Н), 2,5 (с, 3Н), 1,25 (т, 3Н). Приготування 29: 5-хлор-2-[4-(етоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи 2-бром-4-хлорбензальдегід, який використовували на етапі В, на 2-бром-5-хлорбензальдегід. Приготування 30: 2-{1-[2-(бензилоксі)етил]-4-(етоксикарбоніл)-5-метил-1Н-пірол-2-іл}-4хлорбензойна кислота Процедура відповідає способу Приготування 19, замінюючи 6-бром-1,3-бензодіоксол-5карбальдегід на 2-бром-4-хлорбензальдегід. Приготування 31: 5-метокси-2-[4-(метоксикарбоніл)-1,5-диметил-1Н-пірол-2-іл]бензойна кислота Процедура відповідає способу Приготування 1, замінюючи на Етапі А етил-2-метил-1Hпірол-3-карбоксилат на метил-2-метил-1Н-пірол-3-карбоксилат, а також 2-бром-4хлорбензальдегід, який використовували на етапі В, на 2-бром-5-метоксибензальдегід. 1 Н-ЯМР (400 МГц, ДMCO-d6) м.ч.: 12,8 (ушир, с, 1Н), 7,34 (ушир, д, 1Н), 7,26 (д, 1Н), 7,15 (дд, 1Н), 6,19 (с, 1Н), 3,84 (с, 3Н), 3,69 (с, 3Н), 3,22 (с, 3Н), 2,5 (с, 3Н). Приготування 32: 6-[1-(2,2-дифторетил)-4-(етоксикарбоніл)-5-метил-1Н-пірол-2-іл]-1,3бензодіоксол-5-карбонова кислота Процедура відповідає протоколу Приготування 19, замінюючи 2-брометоксиметилбензол, який використовували на етапі А, на 2-бром-1,1-дифторетан. Приготування 1': (3R)-3-метил-1,2,3,4-тетрагідроізохіноліну гідрохлорид Етап А: {(3S)-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін-3-іл}метил-4метилбензолсульфонат До розчину 30,2 г [(3S)-1,2,3,4-тетрагідроізохінолін-3-іл]метанолу (185 ммоль) в 750 мл дихлорметану послідовно додали 91,71 г тозилхлориду (481 ммоль) і потім, по краплях, 122,3 мл N,N,N-триетиламіну (740 ммоль). Реакційну суміш потім перемішували при кімнатній температурі протягом 20 годин. Потім розбавили дихлорметаном, промили послідовно 1М розчину НСl, насиченим водним розчином NaHCO 3 і потім сольовим розчином до нейтральної реакції. Органічну фазу потім висушили над MgSO 4, фільтрували і концентрували досуха. Одержану тверду речовину потім розчинили в мінімальному об'ємі дихлорметану, потім додавали циклогексан, поки не утворився осад. Цей осад потім фільтрували і промили циклогексаном. Після сушіння одержали зазначений у заголовку продукт у формі кристалів. 1 Н-ЯМР (400 МГц, ДMCO-d6; 300К) м.ч.: 7,75 (д, 2Н, ароматичні Hs, орто-О-тозил); 7,6 (д, 2Н, ароматичні Hs, орто-N-тозил); 7,5 (д, 2Н, ароматичні Hs, мета-О-тозил); 7,3 (д, 2Н, ароматичні Hs, мета-N-тозил); 7,15-6,9 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,4-4,15 (дд, 2Н, аліфатичні Hs, тетрагідроізохінолін); 4,25 (м, 1Н, аліфатичні Н, тетрагідроізохінолін); 4,0-3,8 14 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 (2дд, 2Н, аліфатичні Hs, СН2-О-тозил); 2,7 (2дд, 2Н, аліфатичні Hs, тетрагідроізохінолін); 2,45 (с, 3Н, O-SO2-Ph-CH3); 2,35 (с, 3Н, N-SO2-Ph-CH3), -1 ІЧ: :-SO2: 1339-1165 см . Етап В: (3R)-3-метил-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін До суспензії 8,15 г (214,8 ммоль) алюмогідриду літію (LіАlН 4) у 800 мл метил-третбутилового ефіру (МТБЕ), додали 101,2 г сполуки дитосилу, одержаної на Етапі А (214,8 ммоль), розчиненої у 200 мл МТБЕ. Суміш потім нагрівали при 50 °C протягом 2 годин. Її залишили охолонути і помістили при 0 °С, а потім додали по краплях 12 мл 5N розчину NaOH. Суміш перемішували при кімнатній температурі протягом 45 хвилин. Тверду речовину, одержану таким чином, потім фільтрували і промили МТБЕ, а потім дихлорметаном. Потім фільтрат концентрували насухо. Зазначений у заголовку продукт нарешті одержали у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,70 (д, 2Н, ароматичні Hs, орто-N-тозил); 7,38 (д, 2Н, ароматичні Hs, мета-N-тозил); 7,2-7,0 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,4 (м, 2Н, аліфатичні Hs, тетрагідроізохінолін); 4,3 (м, 1Н, аліфатичні Н, тетрагідроізохінолін); 2,852,51 (2дд, 2Н, аліфатичні Hs, тетрагідроізохінолін); 2,35 (с, 3Н, N-SO2-Ph-СН3); 0,90 (д, 3Н, тетрагідроізохінолін-СН3), -1 ІЧ: :-SO2: 1332-1154 см . Етап С: (3R)-3-мeтил-1,2,3,4-тетрагідроізохінолін До розчину 31,15 г (103,15 ммоль) сполуки монотозилу, одержаної на Етапі В, у 500 мл безводного метанолу, додали порціями 3,92 г (161 ммоль) магнієвих стружок. Суміш перемішували у присутності ультразвуку протягом 96 годин. Реакційну суміш потім фільтрували і тверду речовину кілька разів промили метанолом. Потім фільтрат концентрували насухо. Після очищення за допомогою хроматографії на силікагелі з використанням дихлорметану та аміаку в етанолі як елюентів, зазначений у заголовку продукт одержали у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,05 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 3,90 (м, 2Н, аліфатичні Hs, тетрагідроізохінолін); 2,85 (м, 1Н, аліфатичні Н, тетрагідроізохінолін); 2,68-2,4 (2дд, 2Н, аліфатичні Hs, тетрагідроізохінолін); 1,12 (д, 3Н, тетрагідроізохінолін-СН3); 2,9-2,3 (ушир, м, 1Н, HN (тетрагідроізохінолін)), -1 ІЧ: :-NH: 3248 см . Етап D: (3R)-3-мeтил-1,2,3,4-тетрагідроізохінолін До розчину 14,3 г (97,20 ммоль) сполуки, одержаної на етапі С, у 20 мл безводного етанолу, додали по краплях 100 мл 1М розчину НСl в ефірі. Суміш перемішували при кімнатній температурі протягом 1 години і потім фільтрували. Одержані при цьому кристали промили діетиловим ефіром. Після сушіння одержали зазначений у заголовку продукт у формі кристалів. 1 + Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 9,57 (ушир, м, 2Н, NH2 (тетрагідроізохінолін)), 7,22 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,27 (с, 2Н, аліфатичні Hs, тетрагідроізохінолін); 3,52 (м, 1Н, аліфатичні Н, тетрагідроізохінолін), 3,03-2,85 (2дд, 2Н, аліфатичні Hs, тетрагідроізохінолін), 1,39 (д, 3Н, тетрагідроізохінолін-СН3), + -1 -1 ІЧ: :-NH2 : 3000-2300 см ; : ароматичний -СН: 766 см . Приготування 2': тpeт-бyтил-[(3S)-1,2,3,4-тетрагідроізохінолін-3-ілметил]карбамат Етап А: бензил-(3S)-3-(гідроксиметил)-3,4-дигідро-1Н-ізохінолін-2-карбоксилат Цю сполуку одержали з використанням протоколу згідно з літературою (Kawthekar R. В. et al. South Africa Journal of Chemistry 63, 195, 2009), починаючи з 15 г (3S)-1,2,3,4тетрагідроізохінолін-3-ілметанолу (91,9 ммоль) у присутності бензилхлорформіату і триетиламіну у розчині в дихлорметані. Після очищення за допомогою хроматографії на силікагелі з використанням петролейного ефіру і етилацетату як елюентів, одержали зазначений у заголовку продукт у формі олії. 1 Н-ЯМР (300 МГц, ДМСО-d6; 300К) м.ч.: 7,33 (м, 5Н, ароматичні Hs, О-бензил); 7,15 (с, 4Н, ароматичні Hs, Н-тетрагідроізохінолін); 5,13 (с, 2Н, СН2-Ph); 4,73 (д, 1Н, Н-тетрагідроізохінолін); 4,47 (м, Н, СН2ОН); 4,36 (м, 1Н, Н-тетрагідроізохінолін); 4,28 (д, 1Н, Н-тетрагідроізохінолін); 3,39 (дд, 1Н, СН2ОН); 3,23 (дд, 1Н, СН2ОН); 2,93 (дд, 1Н, Н-тетрагідроізохінолін); 2,86 (дд, 1Н, Нтетрагідроізохінолін), -1 -1 -1 ІЧ: : ОН: 3416 см ; : СН: 754 см . Етап В: бензил-(3S)-3-(азидометил)-3,4-дигідроізохінолін-2(1Н)-карбоксилат Цю сполуку одержали з використанням протоколу згідно з літературою (Page D. et al. J. Med. Chem, 44, 2387, 2001), починаючи з 23 г сполуки, одержаної на Етапі А (77,3 ммоль), у присутності дифенілфосфорилазиду і трифенілфосфіну в розчині у ТГФ. Після очищення за 15 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 допомогою хроматографії на силікагелі з використанням петролейного ефіру і етилацетату як елюентів, одержали зазначений у заголовку продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,36 (м, 5Н, ароматичні Hs, О-бензил); 7,19 (м, 4Н, ароматичні Hs, Н-тетрагідроізохінолін); 5,16 (с, 2Н, СН2-Ph); 4,76 (д, 1Н, Н-тетрагідроізохінолін); 4,53 (м, 1Н, Н-тетрагідроізохінолін); 4,30 (м, 1Н, Н-тетрагідроізохінолін); 3,28 (м, 2Н, CH2N3); 3,06 (дд, 1Н, Н-тетрагідроізохінолін); 2,78 (дд, 1Н, Н-тетрагідроізохінолін), -1 -1 -1 ІЧ: : N3: 2095 см ; : СН: 754 см . Етап С: бензил-(3S)-3-(амінометил)-3,4-дигідроізохінолін-2(1Н)-карбоксилат До розчину 20,9 г (64,5 ммоль) сполуки азидо, одержаної на Етапі В, у 650 мл ТГФ, послідовно додали 25,5 г (97,2 ммоль) трифенілфосфіну і 157 мл води. Суміш нагрівали зі зворотним холодильником протягом 2 годин 30 хвилин. Реакційну суміш потім концентрували досуха і залишок олії помістили в ізопропіловий ефір. З'явився білий осад, його фільтрували і промили ізопропіловим ефіром. Потім фільтрат концентрували досуха і очистили за допомогою хроматографії на силікагелі з використанням дихлорметану та метанолу як елюентів. Зазначений у заголовку продукт одержали у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,40 (м, 5Н, ароматичні Hs, О-бензил); 7,20 (м, 4Н, ароматичні Hs, Н-тетрагідроізохінолін); 5,15 (с, 2Н, СН2-Ph); 4,75-4,3 (м, 2Н, Нтетрагідроізохінолін); 4,30 (д, 1Н, Н-тетрагідроізохінолін); 2,90 (м, 2Н, CH2NH2); 2,45 (м, 2Н, Нтетрагідроізохінолін); 1,40 (м, 2Н, NH2), -1 -1 ІЧ: : NH2: 3400-3300 см ; : С=О: 1694 см ; :> С-О-С СН-Аr: 751; 697 см . Етап С: (3S)-3-(4-морфолінілметил)-1,2,3,4-тетрагідроізохінолін До розчину 4,9 г сполуки Етапу В (13,4 ммоль) у 67 мл етанолу додали 0,980 г дигідроксиду паладію (20 % ваг.) при кімнатній температурі. Реакційну суміш тримали під тиском 1,2 бара водню при кімнатній температурі протягом 4 годин. Потім пропустили через фільтр Ватмана і потім промили паладій кілька разів етанолом. Фільтрат випарювали досуха. Зазначений у заголовку продукт одержали у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,12-7,0 (недозволений пік, 4Н, ароматичні Hs); 3,92 (с, 2Н-тетрагідроізохінолін); 3,60 (т, 4Н-морфолін); 2,98 (м, 1Н-тетрагідроізохінолін); 2,68 (дд, 1Н-тетрагідроізохінолін); 2,5-2,3 (недозволений пік, 8Н, 1Н-тетрагідроізохінолін, 6Нморфолін, 1Н-NH), -1 -1 -1 ІЧ: :> NH: 3322 см ; :> C-O-C CH-Ar: 742 см . Приготування 4': (3S)-3-[(4-метил-1-піперазиніл)метил]-1,2,3,4-тетрагідроізохінолін Процедура відповідає способу Приготування 3', замінюючи морфолін, який використовували на етапі А, на 1-метилпіперазин. Приготування 5': (3S)-3-[2-(морфолін-4-іл)етил]-1,2,3,4-тетрагідроізохіноліну гідрохлорид Етап А: трет-бутил-(3S)-3-(2-морфоліно-2-оксоетил)-3,4-дигідро-1Н-ізохінолін-2-карбоксилат До розчину 3 г (10,30 ммоль) [(3S)-2-(трет-бутоксикарбоніл)-1,2,3,4-тетрагідроізохінолін-3іл]оцтової кислоти у 100 мл дихлорметану додали по краплях 1,10 мл (11,32 ммоль) морфоліну, також по краплях 4,3 мл (30,9 ммоль) триетиламіну, 2,20 г (12,40 ммоль) EDC і 1,70 г (1,68 ммоль) HOBt (гідроксибензотриазол). Суміш перемішували при кімнатній температурі протягом 15 годин. Реакційну суміш потім розбавили дихлорметаном, промили послідовно 1М розчину НСl, насиченим водним розчином NaHCO3 і потім сольовим розчином до нейтральної реакції. Органічну фазу потім висушили над MgSO4, фільтрували і концентрували досуха. Після очищення за допомогою хроматографії на силікагелі з використанням дихлорметану та метанолу як елюентів, одержали зазначений у заголовку продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,20-7,10 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,70 (м, 1Н, аліфатичні Hs, CH-тетрагідроізохінолін); 4,75-4,20 (2м, 2Н, аліфатичні Hs, CH2-альфа до N-тетрагідроізохіноліну); 3,60 (м, 8Н, аліфатичні Hs, морфолін); 3,00 і 2,70 (2дд, 2Н, аліфатичні Н, тетрагідроізохінолін); 2,50-2,20 (2д, 2Н, аліфатичні Hs, т СН2СО); 1,40 (с, 9Н, Вu), -1 ІЧ: : C=O: 1687; 1625 см . Етап В: 1-(морфолін-4-іл)-2-[(3S)-1,2,3,4-тетрагідроізохінолін-3-іл]етанону гідрохлорид До розчину 2,88 г (7,18 ммоль) сполуки, одержаної на Етапі А, в 16 мл дихлорметану, додали по краплях 80 мл (80 ммоль) 1М розчину НСl в ефірі. Суміш перемішували при кімнатній температурі протягом 15 годин, а потім суспензію фільтрували і осад промили ефіром. Після сушіння одержали зазначений у заголовку продукт у формі твердої речовини. 1 + Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 9,80-9,50 (м, 2Н, NH2 ); 7,30-7,10 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,30 (м, 2Н, аліфатичні Hs, CH 2-альфа до Nтетрагідроізохінолін); 3,80 (м, 1Н, аліфатичні Hs, CH-тетрагідроізохінолін); 3,70-3,40 (2м, 8Н, аліфатичні Hs, морфолін); 3,15 і 2,8 (м, 4Н, аліфатичні Н, СН2-тетрагідроізохінолін і СН2СО), + -1 -1 ІЧ: :-NH2 : 2800-1900 см ; : С=О 1620 см . Етап С: (3S)-3-[2-(морфолін-4-іл)етил]-1,2,3,4-тетрагідроізохіноліну гідрохлорид -1 5 10 15 20 25 30 35 40 45 50 55 17 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 Приготували розчин 2,2 г (7,44 ммоль) сполуки, одержаної на Етапі В, у 22 мл МТБЕ і 5 мл дихлорметану. Після охолодження у льодяній бані при 0 °C, до нього додали по краплях 15 мл (15 ммоль) 1М розчину LіАlН4 у тетрагідрофурані. Суміш потім перемішували при кімнатній температурі протягом 6 годин. Її тримали при 0 °C, і потім додали по краплях 1 мл 5N розчину NaOH. Суміш перемішували при кімнатній температурі протягом 45 хвилин. Потім тверду речовину фільтрували і промили МТБЕ, а потім дихлорметаном і фільтрат концентрували досуха. Одержану таким чином олію розбавили дихлорметаном і додали по краплях 6,3 мл 1М розчину НСl в ефірі. Суміш перемішували при кімнатній температурі протягом 1 години і потім фільтрували. Одержані таким чином кристали промили діетиловим ефіром. Після сушіння одержали зазначений у заголовку продукт у формі твердої речовини. 1 + + Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 11,35+9,80 (2 м, 2Н, NH2 ); 10,00 (м, Н, NH ); 7,20 (м, 4Н, ароматичні Hs, тетрагідроізохінолін); 4,30 (с, 2Н, аліфатичні Hs, СН 2-альфа до Nтетрагідроізохіноліну); 4,00+3,85 (2м, 4Н, аліфатичні Hs, CH2-альфа до N-морфоліну); 3,70 (м, 1Н, аліфатичні Hs, СН-тетрагідроізохінолін); 3,55-3,30 (м, 4Н, аліфатичні Hs, СН-альфа до Оморфоліну і СН2-морфоліну); 3,15 (дд, 1Н, аліфатичні Н, СН 2-тетрагідроізохінолін); 3,10 (м, 2Н, аліфатичні Н, СН-альфа до О-морфоліну); 2,90 (дд, 1Н, аліфатичні Н, СН2-тетрагідроізохінолін); 2,30+2,15 (2м, 2Н, аліфатичні Н, СН2-тетрагідроізохінолін), + + -1 -1 ІЧ: : NH /-NH2 : між 3500 і 2250 см ; : С=С: слабкий 1593 см ; : ароматичний С-Н: -1 765 см . Приготування 6': (3R)-3-[3-(морфолін-4-іл)пропіл]-1,2,3,4-тетрагідроізохінолін Етап А: {(3S)-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін-3-іл}метил-4метилбензолсульфонат Спосіб є таким же, що і на Етапі А Приготування 1. Етап В: трет-бутил-2-({(3R)-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін-3іл}метил)-3-(морфолін-4-іл)-3-оксопропаноат До суспензії 1 г NaH (60 %) (25,08 ммоль) у 30 мл МТБЕ додали по краплях розчин 5 г третбутил-3-морфоліно-3-оксопропаноату (21,81 ммоль) у 20 мл безводного МТБЕ. Цю суспензію перемішували при кімнатній температурі протягом 1 години, а потім додали сполуку, одержану на Етапі А, у формі порошку. Суміш перемішували при 60 °C протягом 30 годин. Додали 100 мл насиченого водного розчину хлориду амонію. Одержаний розчин екстрагували дихлорметаном. Органічну фазу потім висушили над MgSO4, фільтрували і концентрували досуха. Після очищення за допомогою хроматографії на силікагелі з використанням дихлорметану і МеОН як елюентів, одержали очікуваний продукт у формі олії. 1 Н-ЯМР (500 МГц, ДМСО-d6) м.ч.: 7,63/7,59 (2д, 2Н), 7,3/7,26 (2д, 2Н), 7,13 (м, 2 Н), 7,09/6,97 (2т, 2Н), 4,64/4,55/4,36/4,28 (2АВ, 2Н), 4,25/4,11 (2м, 1Н), 3,81 (м, 1Н), 3,73-3,48 (м, 4Н), 3,57-3,32 (м, 4Н), 2,51 (м, 2Н), 2,32/2,31 (2с, 3Н), 1,88/1,79 (2м, 2Н), 1,39/1,38 (2с, 9Н), -1 ІЧ (ATR) см ::> C=О: 1731 (ефір); :> C=О: 1644 (амід); :-SO2: 1334-1156; :> C-O-C CH-Ar: 815-746-709. Етап С: 2-({(3R)-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін-3-іл}метил)-3(морфолін-4-іл)-3-оксопропанова кислота До розчину 9,5 г (17,97 ммоль) сполуки, одержаної на Етапі В, у 40 мл діоксану, додали по краплях 20 мл 4М розчину НСl в діоксані. Суміш перемішували при кімнатній температурі протягом 48 годин, а потім розчин концентрували досуха. Після сушіння одержали очікуваний продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 12,75 (м, 1Н), 7,6 (2*д, 2Н), 7,3 (2*д, 2Н), 7,1/6,95 (2*м, 4 Н), 4,7-4,2 (д, 2Н), 4,25/4,12 (2*м, 1Н), 3,9-3,3 (м, 9Н), 2,55 (д, 2Н), 2,3 (2*с, 3Н), 1,8 (т, 2Н), -1 і ІЧ (ATR) см : -OH: 3500-2000 ; :> C=O: 1727 (кислота); :> C=O: 1634 (амід); :-SO2: 13301155. Етап D: 3-{(3R)-2-[(4-метилфеніл)сульфоніл]-1,2,3,4-тетрагідроізохінолін-3-іл}-1-(морфолін-4іл)пропан-1-он До розчину 7,80 г (16,51 ммоль) сполуки, одержаної на Етапі С, у 100 мл ДМСО, додали 1,16 г (19,8 ммоль) твердофазного хлориду натрію (NaCl), а потім по краплях 5 мл води. Суміш перемішували при 130 °C протягом 1 години, а потім розчин концентрували до ¾. Реакційну суміш потім розбавили дихлорметаном і промили послідовно насиченим водним розчином хлориду літію, а потім сольовим розчином. Органічну фазу потім висушили над MgSO 4, фільтрували і концентрували досуха. Після очищення за допомогою хроматографії на силікагелі з використанням циклогексану і етилацетату як елюентів, одержали очікуваний продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,65 (д, 2Н), 7,3 (д, 2Н), 7,15/7 (2м, 4Н), 4,6 (д, 1Н), 4,25 (д, 1Н), 4,2 (м, 1Н), 3,5 (м, 4Н), 3,4 (2м, 4Н), 2,6 (2дд, 2Н), 2,35 (с, 3Н), 2,3 (м, 2Н), 1,5 (кв, 2Н), 18 UA 115773 C2 ІЧ (ATR) см : :> C=O: 1639; :-SO2: 1331-1156; : > CH-Ar: 815-675. Етап Е: (3R)-2-[(4-метилфеніл)сульфоніл]-3-[3-(морфолін-4-іл)пропіл]-1,2,3,4тетрагідроізохінолін До розчину 6,0 г (14,0 ммоль) сполуки, одержаної на етапі D, у 60 мл МТБЕ і 14 мл дихлорметану, додали 1,06 г (28 ммоль) алюмогідриду літію (LAH) частинами протягом 5 хвилин. Суміш перемішували при кімнатній температурі протягом 15 годин. Додали по краплях 1,5 мл води і перемішування продовжили протягом 15 хвилин. Потім додали по краплях 1,5 мл 5М розчину гідроксиду натрію і перемішування продовжили протягом 15 хвилин. Реакційну суміш потім розбавили МТБЕ і дихлорметаном. Потім суспензію фільтрували і осад промили МТБЕ і дихлорметаном. Органічну фазу потім висушили над MgSO 4, фільтрували і концентрували досуха. Після очищення за допомогою хроматографії на силікагелі з використанням дихлорметану та аміаку в етанолі як елюентів, одержали очікуваний продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,68 (д, 2Н), 7,32 (д, 2Н), 7,1 (недозволений пік, 4Н), 4,65/4,23 (АВ, 2Н), 4,2 (м, 1Н), 3,55 (т, 4Н), 2,7/2,6 (АВХ, 2Н), 2,35 (с, 3Н), 2,25 (т, 4Н), 2,2 (т, 2Н), 1,4/1,3 (2м, 4Н), -1 ІЧ (ATR) см : :-SO2: 1333-1158. Етап F: (3R)-3-[3-(морфолін-4-іл)пропіл]-1,2,3,4-тетрагідроізохінолін До розчину 1,50 г (3,62 ммоль) сполуки, одержаної на етапі Е, у 20 мл безводного метанолу, додали 2,0 г (82,3 ммоль), частинами, магнієвих стружок. Суміш перемішували у присутності ультразвуку протягом 96 годин. Потім реакційну суміш фільтрували, тверду речовину промили кілька разів метанолом і фільтрат концентрували досуха. Після очищення за допомогою хроматографії на силікагелі з використанням дихлорметану та аміаку в етанолі як елюентів, одержали очікуваний продукт у формі олії. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,3 (д, 2Н), 7,1 (т, 2Н), 7,1 (д + т, 3Н), 7 (д, 2Н), 3,9 (с, 2Н), 3,55 (т, 4Н), 2,75 (м, 1Н), 2,72/2,45 (дд, 2Н), 2,35 (т, 4Н), 2,25 (т, 2Н), 1,6 (м, 2Н), 1,45 (м, 2Н), -1 ІЧ (ATR) см : :> NH2+/NH+: 3500-2300; :> C-O-C NH. Приготування 9': (3S)-3-[(9аR)-октагідропіперазино[2,1-с]морфолін-8-ілметил]-1,2,3,4тетрагідроізохіноліну тригідрохлорид Етап А: трет-бутил-(9аR)-4-оксооктагідропіперазино[2,1-с]морфолін-8-карбоксилат Синтез цієї сполуки описано в літературі (J. Med. Chem. 2012, 55, 5887). Етап В: (9аR)-октагідропіперазино[2,1-с]морфолін-4-ону гідрохлорид 4М розчину хлористого водню у діоксані (39 мл, 154 ммоль) додали до сполуки Етапу А (11,3 г, 44,1 ммоль). Потім розчин перемішували при кімнатній температурі протягом 5 годин, а потім знову додали 4М розчину НСl у діоксані (12 мл, 48 ммоль). Суміш перемішували протягом 16 годин. Потім розчин випарили досуха для одержання очікуваного продукту у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 2,79-2,93 (м, 2Н), 3,02 (тд, J=13,1, 2,7 Гц, 1Н), 3,22-3,34 (м, 2Н), 3,59-3,67 (м, 1Н), 3,86-3,96 (м, 1Н), 3,96-4,01 (м, 1Н), 4,05 (АВ кв, J=13,3 Гц, 2Н), 4,48 (дд, J=14,1, 2,5 Гц, 1Н), 9,71 (ушир, с, 1Н), 9,91 (ушир, с, 1Н). Етап С: трет-бутил-(3S)-3-[(9aR)-4-оксооктагідропіперазино[2,1-с]морфолін-8-карбоніл]1,2,3,4-тетрагідроізохінолін-2-карбоксилат EDC (5,17 г, 27,0 ммоль) додали до розчину сполуки попереднього етапу (4,0 г, 20,7 ммоль), (3S)-2-[(трет-бутокси)карбоніл]-1,2,3,4-тетрагідроізохінолін-3-карбоксилату (6,04 г, 21,8 ммоль), триетиламіну (11,6 мл, 83,1 ммоль) і HOBt (3,65 г, 27,0 ммоль) у дихлорметані (100 мл). Суміш перемішували при кімнатній температурі протягом 16 годин. Додали 1N водного розчину НСl (70 мл) і одержаний осад відфільтровували з використанням лійки Бюхнера. Фази фільтрату відокремили. Органічну фазу промили насиченим водним розчином карбонату калію і потім концентрували при зниженому тиску. Залишок попередньо абсорбували на силікагелі і очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 1,25-1,58 (м, 9Н), 2,50-2,76 (м, 2Н), 2,76-3,25 (м, 3Н), 3,37-3,76 (м, 2Н), 3,92-4,50 (м, 5Н), 4,06 (с, 2Н), 4,66 (д, J=15,6 Гц, 1Н), 4,76-5,28 (м, 1Н), 7,057,31 (м, 4Н). Етап D: (3S)-3-[(9aR)-октагідропіперазино[2,1-с]морфолін-8-ілметил]-1,2,3,4тетрагідроізохіноліну тригідрохлорид 4М розчин хлористого водню в діоксані (24,0 мл, 96,2 ммоль) додали до розчину сполуки Етапу С (8,00 г, 12,25 ммоль) у дихлорметані (25 мл). Суміш перемішували при кімнатній температурі протягом 16 годин і потім концентрували досуха. Одержаний сирий продукт додали до суспензії LiAIH4 (1,97 г, 51,91 ммоль) у тетрагідрофурані (140 мл). Суміш нагрівали зі зворотним холодильником до завершення реакції (контролювали способом РХ-МС) і потім охолоджували до 0 °C. Воду (2,5 мл) додали по краплях. Після перемішування протягом 10 хвилин додали по краплях 2М водного розчину гідроксиду натрію (5 мл). Знову додали воду (5 мл) після перемішування протягом 10 хвилин. Нарешті додали Целіт (4 г) і Na 2SO4 (12 г) після перемішування протягом додаткових 10 хвилин. Суспензію фільтрували через Целіт і фільтрат концентрували насухо. Сирий залишок, одержаний таким чином, розчинили у метанолі (80 мл), а потім додали 4М розчину НСl у діоксані (16,75 мл, 67,0 ммоль). Суміш перемішували при кімнатній температурі протягом 3 годин, а потім концентрували досуха. Залишок розчинили у мінімальній кількості теплого метанолу (70 мл), а потім МТБЕ (3-5 мл). Розчин охолоджували при 0 °C протягом 1 години на льодяній водяній бані, і продукт випадає в осад. Знову додали трохи МТБЕ (2-3 мл) і суміш витримали протягом ще 1 години при 0 °C. Одержану тверду речовину фільтрували через лійку Бюхнера і висушили in vacuo для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, CD3OD) м.ч.: 2,65 (т, J=11,1 Гц, 1Н), 2,74-2,97 (м, 4Н), 3,10 (д, J=12,5 Гц, 1Н), 3,19 (дд, J=17,6, 4,8 Гц, 1Н), 3,25-3,55 (м, 5Н), 3,59-3,74 (м, 2Н), 3,82-4,15 (м, 4Н), 4,45 (АВ кв, J=15,9 Гц, 2Н), 7,21-7,35 (м, 4Н), 13 С-ЯМР (100 МГц, CD3OD) м.ч.: 30,21, 45,67, 50,22, 51,99, 52,90, 53,58, 53,71, 59,25, 62,58, 65,30, 67,07, 127,76, 128,40, 129,08, 129,36, 130,16, 131,95, + MS (ESI): [М + Н] 288,2. Приготування 1": N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-метилпіразол-4-амін Етап А: 4-{[трет-бутил(диметил)силіл]оксі}анілін Зазначену в заголовку сполуку одержали, виходячи з 4-амінофенолу в ТГФ у присутності імідазолу і трет-бутил(диметил)силілхлориду відповідно до протоколу, описаного в літературі (Knaggs S. et al., Organic & Biomolecular Chemistry, 3(21), 4002-4010; 2005). 1 5 10 15 20 25 30 35 40 45 50 55 60 21 UA 115773 C2 H-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 6,45-6,55 (дд, 4Н, ароматичні Hs); 4,60 (м, 2Н, NH2Ph); 0,90 (с, 9Н, Si(CH2)2CH(CH3)2); 0,10 (с, 6Н, Si(CH2)2CH(CH3)2), + -1 ІЧ: :-NH2 : 3300-3400 см . Етап В: N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-метилпіразол-4-амін До розчину 30,8 г (0,137 моль) сполуки Етапу А у 525 мл безводного толуолу послідовно додали 29,8 г трет-бутилату натрію (0,310 моль), 4,55 г Pd 2(dba)3 (також відомого як трис(дибензиліденацетон)дипаладій(0)) (4,96 ммоль), 4,81 г 2-ди-трет-бутилфосфіно-2',4',6'триізопропіл-1,1'-біфенілу (9,91 ммоль) і 12,8 мл 4-бром-1-метил-1Н-піразолу (0,124 моль). Суміш дегазували в атмосфері аргону протягом 30 хвилин, а потім нагрівали зі зворотним холодильником протягом 3 годин. Її залишили охолонути. Реакційну суміш концентрували досуха і потім розчинили в дихлорметані, фільтрували через Целіт і потім знову концентрували досуха. Потім залишок очистили за допомогою хроматографії на силікагелі з використанням дихлорметану і етилацетату як елюентів для одержання очікуваного продукту у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,55 (с, 1Н, піразол); 7,23 (с, 1Н, піразол); 7,18 (ушир, с, 1Н, NH2-Ph); 6,64 (м, 4Н, ароматичні Hs); 3,77 (с, 3Н, СН3-піразол); 0,90 (с, 9Н, Si(CH2)2CH(CH3)2); 0,12 (с, 6Н, Si(CH2)2CH(CH3)2), + -1 -1 -1 -1 ІЧ: -NH : 3275 см ; Аr і C=N: 1577 і 1502 см ; -Si-C-: 1236 см ; -Si-O-: 898 см ; -1 -Si-C-: 828, 774 см . Приготування 2": 4-{[трет-бутил(диметил)силіл]окси}-N-феніланілін До розчину 12 г 4-анілінфенолу (64,7 ммоль) у 200 мл ацетонітрилу додали при кімнатній температурі 6,7 г імідазолу (97,05 ммоль) і 11,7 г трет-бутил(диметил)силілхлориду (77,64 ммоль). Суміш перемішували при 70 °C протягом 4 годин. Реакційну суміш потім вилили у воду і екстрагували ефіром. Органічну фазу потім висушили над MgSO 4, фільтрували і випарили насухо. Сирий продукт, одержаний таким чином, потім очистили за допомогою хроматографії на силікагелі з використанням петролейного ефіру і дихлорметану як елюентів. Зазначений у заголовку продукт одержали у формі порошку. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,84 (с, 1Н NH); 7,17 (т, 2Н-анілін); 6,98 (д, 2Нфенокси); 6,94 (д, 2Н-анілін); 6,76 (д, 2Н-фенокси); 6,72 (т, 1Н-анілін); 0,95 (с, 9Н-трет-бутил); 0,15 (с, 6Н-диметил), -1 -1 ІЧ: :> NH: 3403 см ; :> Ar: 1597 см . Приготування 3": трет-бутил-5-[(4-{[трет-бутил(диметил)силіл]окси}феніл)аміно]-1Н-індол-1карбоксилат Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на Етапі В, на трет-бутил-5-бром-1Н-індол-1-карбоксилат. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,85 (д, 1Н); 7,78 (с, 1Н); 7,55 (д, 1Н); 7,15 (д, 1Н); 6,95 (м, 3Н); 6,75 (д, 2Н); 6,58 (д, 1Н); 1,65 (с, 9Н); 1,00 (с, 9Н); 0,2 (с, 6Н). Приготування 4": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-метил-1Н-індол-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метил-1Н-індол. Приготування 5": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-метил-1Н-індазол-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метил-1Н-індазол. Приготування 6": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-3-фтор-4-метиланілін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 4-бром-2-фтор-1-метилбензол. Приготування 7": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-3-фторанілін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 1-бром-3-фторбензол. Приготування 8": 4-бензилокси-N-феніланілін До розчину 4-гідрокси-N-феніланіліну (30 г; 162 ммоль) в ацетонітрилі (400 мл) додали 58 г CS2CO3 (178 ммоль) і перемішування продовжували протягом 15 хвилин при кімнатній температурі. Потім додали по краплях бензилбромід (22,5 мл, 178 ммоль) і потім реакційну суміш нагрівали зі зворотним холодильником протягом 4 годин. Після фільтрації і промивання ацетонітрилом, фільтрат концентрували і очистили за допомогою хроматографії на силікагелі з використанням петролейного ефіру і етилацетату як елюентів. Зазначений у заголовку продукт потім одержали у формі безбарвної твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6; 300К) м.ч.: 7,80 (м, 1Н, NH); 7,45 (м, 2Н, арил); 7,40 (м, 2Н, арил); 7,30 (м, 1Н, арил); 7,15 (с, 2Н, арил); 7,05 (д, 2Н, арил); 6,9-7,0 (м, 4Н, арил); 6,70 (т, 1Н, арил); 5,05 (с, 2Н, бензил), 1 5 10 15 20 25 30 35 40 45 50 55 60 22 UA 115773 C2 ІЧ: :> NH: 3408 см . Приготування 9": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)піридин-4-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 4-бромпіридин. -1 -1 -1 ІЧ: -NH-: 3200 і 2500 см ; : -Si-O-: 902 см ; : -Si-C-: 820 см . Приготування 10": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-4-фторанілін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 1-бром-4-фторбензол. Приготування 11": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-метил-1Н-пірол[2,3b]піридин-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метил-1Н-пірол[2,3-b]піридин (одержаний відповідно до протоколу згідно з літературою: Heterocycles, 60(4), 865, 2003). -1 -1 ІЧ: : -NH-: 3278 см ; : ароматичні-С=С-фрагменти: 1605 см . Приготування 12": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-2-метоксипіримідин-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-2-метоксипіримідин. Приготування 13": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-метил-2,3-дигідро-1Нпіроло[2,3-b]піридин-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метил-2,3-дигідро-1Н-піроло[2,3-b]піридин. Приготування 14": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-метил-1Н-бензімідазол-5амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метил-1Н-бензімідазол. 42 2 Приготування 15": N (4-{[трет-бутил(диметил)силіл]окси}феніл)-N ,N -диметилпіридин-2,4діамін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 4-бром-N,N-диметилпіридин-2-амін. Приготування 16": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-піразоло[1,5-а]піримідин-6амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 6-бромпіразоло[1,5-а]піримідин. -1 -1 -1 ІЧ: -NH-: 3272 см ; -C=N-: 1634 см ; -C=C-: 1616 см . Приготування 17": N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-метилпіразоло[3,4b]піридин-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-метилпіразоло[3,4-b]піридин (одержаний відповідно до протоколу згідно з літературою: WO 2006/052568, починаючи з 2-метилпіразол-3-аміну і 2бромпропандіолу). Приготування 18": 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1,5-диметил-1Н-пірол2-карбонітрил Етап А: 4-бром-1,5-диметил-1Н-пірол-2-карбонітрил Розчин брому (6,58 мл, 0,13 моль) в оцтовій кислоті (60 мл) додали по краплях за допомогою крапельної лійки до розчину 1,5-диметил-1Н-пірол-2-карбонітрилу (15,0 г, 0,12 моль) в оцтовій кислоті (300 мл). Суміш перемішували при кімнатній температурі протягом 24 годин. Реакційну суміш потім вилили у хімічний стакан, що містить 300 мл води. Утворену тверду речовину фільтрували і промили водою. Потім розчинили у дихлорметані (300 мл) і органічну фазу промили сольовим розчином, висушили над Na2SO4, фільтрували і концентрували in vacuo для одержання очікуваного продукту у формі твердої речовини. 1 Н-ЯМР (CDCl3) м.ч.: 2,25 (с, 3Н), 3,67 (с, 3Н), 6,74 (с, 1Н). Етап В: 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1,5-диметил-1Н-пірол-2карбонітрил Розчин сполуки попереднього етапу (1,5 г, 7,53 ммоль), 4-[(третбутилдиметилсиліл)оксі]анілін (2,02 г, 9,04 ммоль), трет-бутилату натрію (1,45 г, 15,06 ммоль) і 2-ди-трет-бутилфосфіно-2',4',6'-триізопропілбіфенілу (0,13 г, 0,30 ммоль) в толуолі (20 мл) продували азотом. Додали трис(дибензиліденацетон)дипаладій(0) (0,28 г, 0,30 ммоль), а потім реакційну суміш нагрівали при 90 °C до завершення реакції (контролювали за допомогою ТШХ). Нагрівання припинили і суміш залишили охолоджуватися до кімнатної температури. Додали воду (75 мл) і суміш екстрагували етилацетатом (3×75 мл). Об'єднані органічні фази промили -1 5 10 15 20 25 30 35 40 45 50 55 60 23 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 сольовим розчином і потім концентрували. Сирий продукт абсорбували на силікагелі і очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів. Продукт, одержаний таким чином, розчинили у гептані в теплому стані і дозволили йому випасти в осад при перемішуванні при кімнатній температурі, а потім при 0 °C. Тверду речовину відфільтрували і операцію повторили на фільтраті для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,15 (с, 6Н), 0,97 (с, 9Н), 2,13 (с, 3Н), 3,66 (с, 3Н), 4,68 (ушир, с, 1Н), 6,49 (д, J=8,5 Гц, 2Н), 6,64 (с, 1Н), 6,66 (д, J=8,7 Гц, 2Н), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: 4,34, 9,72, 18,30, 25,88, 32,94, 101,27, 114,37, 114,70, 116,41, 120,73, 124,52, 131,23, 141,54, 148,27, + MC(ESI+): [M + Н] виміряно: 342,3. Приготування 19": 4-[(4-{[трет-бутил(диметил)силіл]окси}феніл)аміно]-1-метил-1Н-пірол-2карбонітрил Етап А: 1-метил-1Н-пірол-2-карбонітрил N,N-диметилформамід (3 мл) і 1,4-діазабіцикло[2,2,2]октан (0,49 г, 4,3 ммоль) додали до розчину пірол-2-карбонітрилу (4 г, 43,4 ммоль) у диметилкарбонаті (56 мл). Розчин перемішували при 90 °C протягом 15 годин, а потім нагрівали при 110 °C протягом 8 годин. Суміш охолодили до кімнатної температури, а потім додали етилацетат (80 мл). Фази відокремили і органічну фазу промили водою (2×80 мл) і 1N водного розчину НСl (1×80 мл). Об'єднані водні фази екстрагували ще раз етилацетатом (1×80 мл). Об'єднані органічні фази промили сольовим розчином (1×80 мл), висушили над MgSO 4, фільтрували і концентрували in vacuo для одержання очікуваного продукту у формі рідини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 3,78 (м, 2Н), 6,12-6,18 (м, 1Н), 6,74-6,82 (м, 1Н). Етап В: 4-бром-1-метил-1Н-пірол-2-карбонітрил N-бромсукцинімід (6,2 г, 34,9 ммоль) додали до розчину 1-метил-1Н-пірол-2-карбонітрилу (3,7 г, 34,9 ммоль) у N,N-диметилформаміді (150 мл). Розчин перемішували протягом 15 годин при кімнатній температурі. Додали іншу кількість N-бромсукциніміду (2,0 г, 11 ммоль) і суміш перемішували протягом 3 годин. Потім додали діоксид кремнію (7 г) і суспензію випарили насухо. Матеріал, попередньо абсорбований на діоксиді кремнію, помістили у колонку з силікагелем і продукт очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів для одержання очікуваного продукту у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 3,77 (с, 3Н), 6,75 (д, J=1,7 Гц, 1Н), 6,80 (д, J=1,7 Гц, 1Н). Етап С: 4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1-метил-1Н-пірол-2-карбонітрил Азот барботували через розчин 4-бром-1-метил-1Н-пірол-2-карбонітрилу (2,82 г, 15,2 ммоль) і 4-[(трет-бутилдиметилсиліл)оксі]аніліну (4,08 г, 18,3 ммоль) у толуолі (55 мл) протягом5 хвилин. Потім до реакційної суміші додали трет-бутилат натрію (2,92 г, 30,4 ммоль), трис(дибензиліденацетон)дипаладій(0) (556 мг, 0,6 ммоль) і 2-ди-трет-бутилфосфіно-2',4',6'триізопропілбіфеніл (255 мг, 0,6 ммоль). Суміш перемішували протягом 1 години при 80 °C в атмосфері азоту. Потім суспензію охолодили до кімнатної температури і фільтрували через Целіт. Целітний осад потім промили етилацетатом. Фільтрат промили водою, а потім сольовим розчином. Органічну фазу висушили над MgSO 4, фільтрували і концентрували in vacuo. Продукт очистили двічі за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів, а потім шляхом розтирання у порошок в гептані одержали очікуваний продукт у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,16 (с, 6Н), 0,97 (с, 9Н), 3,73 (с, 3Н), 6,57 (д, J=1,9 Гц, 1Н), 6,64-6,66 (м, 1Н), 6,70 (с, 4Н), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,48, 18,17, 25,72, 35,46, 103,01, 113,56, 113,69, 115,92, 119,55, 120,67, 129,04, 139,94, 148,85, + MC (ESI+): [М + Н] 328,25. Приготування 20": N-[4-[трет-бутил(диметил)силіл]окси-3-фторфеніл]-1-метил-1Н-піразол-4амін Процедура відповідає протоколу Приготування 1", замінюючи 4-амінофенол, який використовували на Етапі А, на 2-фтор-4-амінофенол. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,59 (ушир, с, 1Н), 7,39 (м, 1Н), 7,24 (д, 1Н), 6,74 (дд, 1Н), 6,52 (дд, 1Н), 6,42 (ддд, 1Н), 3,76 (с, 3Н), 0,92 (с, 9Н), 0,1 (д, 6Н). Приготування 21": 2-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)піридин-4-карбонітрил Розчин, що складається з 2-бром-4-піридинкарбонітрилу (5,00 г, 36,1 ммоль), 4-[(третбутилдиметилсиліл)оксі]аніліну (8,06 г, 36,1 ммоль), трет-бутилату натрію (4,50 г, 46,9 ммоль) і 2-ди-трет-бутилфосфіно-2',4',6'-триізопропілбіфенілу (0,458 г, 1,08 ммоль) у толуолі (50 мл) 24 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 продули азотом. Потім до реакційної суміші додали трис(дибензиліденацетон)паладій(0) (0,99 г, 1,08 ммоль), і потім суміш нагрівали при 50 °C протягом 1,5 години. Потім суміш залишили охолоджуватися до кімнатної температури. Додали воду і реакційну суміш екстрагували етилацетатом (3×20 мл). Об'єднані органічні фази промили сольовим розчином і потім концентрували при зниженому тиску. Сирий продукт абсорбували на силікагелі і очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів. Одержаний продукт розчинили в теплому стані у гептані і осадах при перемішуванні при кімнатній температурі, а потім при 0 °C. Після фільтрації, одержали очікувану сполуку у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,22 (с, 6Н), 1,00 (с, 9Н), 6,61 (ушир, с, 1Н), 6,81-6,84 (м, 2Н), 6,84-6,89 (м, 2Н), 7,12-7,17 (м, 2Н), 8,26 (дд, J=5,1, 0,9 Гц, 1Н), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,29, 18,31, 25,78, 109,11, 114,73, 117,23, 121,17, 121,74, 124,93, 132,12, 149,79, 153,45, 158,00, + МС (ESI+): [М + Н] 326,19. Приготування 22": N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-тетрагідрофуран-3ілпіразол-4-амін 4-[Трет-бутил(диметил)силіл]оксіанілін (0,92 г, 3,48 ммоль) і 4-йод-1-тетрагідрофуран-3ілпіразол (0,78 г, 3,48 ммоль), розчинений у безводному тетрагідрофурані (20 мл), перемішували протягом однієї години при кімнатній температурі в присутності трет-бутилату натрію (1,7 мл, 2М розчин у ТГФ) і хлор(2-ди-трет-бутилфосфіно-2',4',6'-триізопропіл-1,1'біфеніл)[2-(2-аміноетил)феніл]паладію(II) (84 мг, 0,122 ммоль). Реакційну суміш фільтрували через Целіт, потім випарили насухо. Залишок кристалізували з суміші гептану/етилацетату, фільтрували і промили гептаном, а потім очистили за допомогою хроматографії на силікагелі з використанням дихлорметану та метанолу як елюентів для одержання очікуваного продукту. 1 Н-ЯМР (500 МГц, ДМСО-d6, 300 К) м.ч.: 7,62 (д, 1Н, піразол-Н5), 7,29 (д, 1Н, піразол-Н3), 7,26 (с, 1Н, NH), 6,68 (д, 2Н, Аr-Н), 6,64 (д, 2Н, Аr-Н), 4,93 (м, 1Н, ТГФ-3'Н), 3,95 (м, 1Н, ТГФ-5'Н), 3,94 (м, 1Н, ТГФ-2'Н), 3,87 (м, 1Н, ТГФ-2'Н), 3,80 (м, 1Н, ТГФ-5'Н), 2,33 (м, 1Н, ТГФ-4'Н), 2,27 (м, т 1Н, ТГФ-4'Н), 0,93 (с, 9Н, Вu), 0,12 (с, 6Н, Me), -1 -1 -1 ІЧ: СН: 2857 см ; ароматичні: 1505 см ; Si-C: 1249 см . Приготування 23": 6-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)піридин-2-карбонітрил 4-Амінофенол (3,3 г, 30,2 ммоль) додали до розчину 6-хлорпіридин-2-карбонітрилу (3,5 г, 25,3 ммоль) у 1-метил-2-піролідиноні (70 мл). Реакційну суміш нагрівали при 140-150 °C протягом 16 годин у герметичній колбі. Потім суміш охолодили до кімнатної температури. Потім послідовно додали імідазол (3,4 г, 49,9 ммоль) і трет-бутил(диметил)силілхлорид (7,6 г, 50,4 ммоль) і суміш перемішували протягом 16 годин при кімнатній температурі. Суміш розбавили водою (140 мл) і продукт екстрагували етилацетатом (4×50 мл). Органічні фази об'єднали і промили водою (3×50 мл), а потім сольовим розчином (1×50 мл). Органічну фазу потім висушили над MgSO4, фільтрували і концентрували in vacuo. Сирий продукт очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів для одержання очікуваного продукту. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,20 (с, 6Н), 0,99 (с, 9Н), 6,74 (дд, J=7,6, 4,9 Гц, 1Н), 6,816,88 (м, 3Н), 7,36-7,42 (м, 2Н), 7,74 (дд, J=7,6, 1,9 Гц, 1Н), 8,34 (дд, J=4,9, 1,9 Гц, 1Н), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,38, 18,21, 25,73, 92,58, 113,50, 116,53, 120,30, 123,20, 131,97, 141,67, 152,42, 152,45, 156,51, + МС (ESI): [M + H] 326,24. Приготування 24": 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)піримідин-2-карбонітрил Етап А: N-{4-[(трет-бутилдиметилсиліл)окси]феніл}-2-хлорпіримідин-4-амін 4-Амінофенол (8,8 г, 80,6 ммоль) і триетиламін (18,6 мл, 133,4 ммоль) додали до розчину 2,4-дихлорпіримідину (10,0 г, 67,1 ммоль) в етанолі (150 мл). Реакційну суміш нагрівали при 150 °C протягом 14 годин у герметичній колбі. Суміш потім охолодили до кімнатної температури і розчинник випарили in vacuo. Дихлорметан (200 мл) додали до залишку, а потім додали імідазол (9,1 г, 133,7 ммоль) і трет-бутил(диметил)силілхлорид (12,1 г, 80,3 ммоль). Суміш перемішували протягом 15 годин при кімнатній температурі. Реакційну суміш розбавили водою (200 мл). Фази відокремили і органічну фазу промили сольовим розчином (1×100 мл). Потім висушили над MgSO4, фільтрували і концентрували in vacuo. Залишок очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів для одержання твердої речовини. Останню розтерли на порошок у гептані, фільтрували і промили гептаном для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 0,22 (с, 6Н), 0,99 (с, 9Н), 6,42 (д, J=5,9 Гц, 1Н), 6,81 (ушир, с, 1Н), 6,85-6,90 (м, 1Н), 7,13 (д, J=8,7 Гц, 2Н), 8,07 (д, J=5,9 Гц, 2Н). 25 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 Етап В: 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)піримідин-2-карбонітрил Безводний N,N-диметилформамід (10 мл) помістили в атмосфері азоту в колбу і потім додали сполуку Етапу А (670 мг, 2,0 ммоль). Послідовно додали ціанід цинку (468 мг, 4,0 ммоль) і тетракис(трифенілфосфін)паладій(0) (404 мг, 0,3 ммоль). Азот барботували через розчин протягом 5 хвилин, а потім реакційну суміш перемішували при 120 °C протягом 2 годин в атмосфері азоту. Реакція, контрольована за допомогою РХ-МС, була завершена. Суміш охолодили до кімнатної температури, а потім до неї додали воду (15 мл). Продукт екстрагували етилацетатом (3×25 мл). Органічні фази об'єднали і промили водою (4×25 мл), а потім сольовим розчином (1×25 мл). Органічну фазу висушили над MgSO4, фільтрували і концентрували in vacuo. Залишок очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,22 (с, 6Н), 0,99 (с, 9Н), 6,63 (д, J=6,1 Гц, 1Н), 6,86-6,92 (м, 2Н), 7,03 (ушир, с, 1Н), 7,17 (д, J=8,5 Гц, 2Н), 8,22 (д, J=6,1 Гц, 1H), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,51, 18,10, 25,55, 106,32, 115,92, 121,00, 125,22, 129,73, 144,55, 154,16, 156,07, 161,56. Приготування 25": N-{4-[(трет-бутилдиметилсиліл)окси]феніл}-1-тридейтеріометил-1Нпіразол-4-амін Етап А: 4-бром-1-тридейтеріометил-1Н-піразол 4-Бром-1Н-піразол (9,05 г, 61,6 ммоль) додали частинами до суспензії NaH (60 % в олії) (2,83 г, 70,8 ммоль) у тетрагідрофурані (90 мл), охолодженої на льодяній бані. Після видалення льодяної бані, розчин перемішували при кімнатній температурі протягом 0,5 години. Його знову охолодили у льодяній бані і додали йодометан-d3 (5,0 мл, 80,3 ммоль). Розчин перемішували при кімнатній температурі протягом 19 годин. Потім суспензію концентрували. Залишок після випарювання розтерли на порошок з МТБЕ (90 мл) і фільтрували. Фільтрат концентрували in vacuo для одержання очікуваної сполуки у формі олії. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 7,37 (с, 1Н), 7,43 (с, 1Н). Етап В: N-{4-[(трет-бутилдиметилсиліл)окси]феніл}-1-тридейтеріометил-1Н-піразол-4-амін 4-Бром-1-тридейтеріометил-1Н-піразол (9,6 г, 58,5 ммоль), 4-[(третбутилдиметилсиліл)оксі]анілін (14,4 г, 64,6 ммоль) і толуол (150 мл) додали у 500 мл тригорлій колбі. Розчин дегазували азотом протягом 15 хвилин, а потім послідовно додали трет-бутилат натрію (11,4 г, 0,12 моль), 2-ди-трет-бутилфосфіно-2',4',6'-триізопропілбіфеніл (0,77 г, 1,81 ммоль) і трис(дибензиліденацетон)паладій(0) (1,64 г, 1,79 ммоль). Суспензію нагрівали при 85 °C протягом 1,5 години. Реакційну суміш потім охолодили до кімнатної температури і додали воду (270 мл). Суміш перемішували протягом 30 хвилин. Потім додали Целіт (30 г) і суспензію фільтрували на шарі Целіту. Фази фільтрату відокремили і водну фазу екстрагували етилацетатом (3×200 мл). Об'єднані органічні фази висушили над Na 2SO4 і фільтрували. До фільтрату додали діоксид кремнію (36 г) і суміш випарювали досуха. Продукт очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів. Одержаний продукт рекристалізували з гептану (80 мл) для одержання очікуваної сполуки. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,16 (с, 6Н), 0,97 (с, 9Н), 4,92 (с, 1Н), 6,61-6,73 (м, 4Н), 7,25 (с, 1Н), 7,36 (с, 1Н), 13 1 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,37, 18,28, 25,86, 38,67 (септ, JCD=21,0 Гц), 115,12, 120,73, 123,76, 126,52, 134,74, 141,07, 148,43, + МС (ESI): [M + H] 307,08. Приготування 26": N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-(оксетан-3-іл)-1Н-піразол-4амін Етап А: 4-бром-1-(оксетан-3-іл)-1Н-піразол 4-Бром-1Н-піразол (1,53 г, 10,7 ммоль) розчинили у безводному диметилформаміді (15 мл). До нього послідовно додали 3-бромоксетан (2,0 г, 14,6 ммоль) і карбонат цезію (4,7 г, 14 ммоль). Реакційну суміш нагрівали протягом 8 годин при 130 °C у герметично закритій колбі. Наприкінці реакції розчинник випарили in vacuo і залишок очистили за допомогою хроматографії на силікагелі з використанням дихлорметану, що містить діетиламін та метанол як елюенти, для одержання очікуваної сполуки. Етап В: N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-(оксетан-3-іл)-1Н-піразол-4-амін 4-[Трет-бутил(диметил)силіл]оксіанілін (1,5 г, 7,2 ммоль) і 4-бром-1-(оксетан-3-іл)піразол (1,6 г, 7,2 ммоль), розчинений у безводному тетрагідрофурані (25 мл), перемішували протягом 3 годин при кімнатній температурі в присутності трет-бутилату натрію (3,7 мл, 2М розчин у ТГФ) і хлор(2-ди-трет-бутилфосфіно-2',4',6'-триізопропіл-1,1'-біфеніл)[2-(2-аміноетил)феніл]паладію(II) (101 мг, 0,145 ммоль). Реакційну суміш фільтрували через Целіт, потім випарили насухо. 26 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 Залишок очистили за допомогою хроматографії на силікагелі з використанням дихлорметану, що містить діетиламін і етилацетат як елюенти, для одержання очікуваного продукту. 1 Н-ЯМР (500 МГц, ДМСО-d6, 300 К) м.ч.: 7,76 (с, 1Н, піразол-5'Н), 7,40 (с, 1Н, піразол-3'Н), 7,34 (ушир, с, 1Н, NH), 6,70 (д, 2Н, Аr-Н), 6,65 (д, 2Н, Аr-Н), 5,49 (м, 1Н, оксетан), 4,89 (д, 4Н, т оксетан), 0,93 (с, 9Н, Вu), 0,13 (с, 6Н, Me), -1 -1 -1 ІЧ: : СН: 2955 см ; ароматичні: 1505 см ; Si-C: 1237 см . Приготування 27": суміш N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1,5-диметил-1Нпіразол-4-амін і N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1,3-диметил-1Н-піразол-4-амін Етап А: суміш 4-бром-1,5-диметил-1Н-піразолу і 4-бром-1,3-диметил-1Н-піразолу До суспензії 60 % NaH в олії (0,3 г, 7,45 ммоль) в тетрагідрофурані (150 мл) додали по краплях, при 10 °C, 4-бром-3-метил-1Н-піразол, розчинений у 15 мл тетрагідрофурану, протягом 15 хвилин. Після перемішування протягом 40 хвилин при кімнатній температурі додали по краплях йодометан (0,45 мл, 7,45 ммоль), а потім реакційну суміш перемішували протягом ночі. Після додавання води реакційну суміш випарили і розчинили у дихлорметані. Органічну фазу відокремили і висушили над MgSO4, фільтрували і концентрували досуха. Залишок очистили за допомогою хроматографії на силікагелі для одержання суміші зазначених у заголовку сполук (4бром-1,3-диметилпіразолу і 4-бром-1,5-диметилпіразолу відповідно у співвідношенні 4:6). 4-Бром-1,5-диметил-1Н-піразол: 1 Н-ЯМР (500 МГц, ДМСО-d6) м.ч.: 7,41 (с, 1Н), 3,76 (с, 3Н), 2,22 (с, 3Н). 4-Бром-1,3-диметил-1Н-піразол: 1 Н-ЯМР (500 МГц, ДМСО-d6) м.ч.: 7,81 (с, 1Н), 3,74 (с, 3Н), 2,09 (с, 3Н). Етап В: суміш N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1,5-диметил-1Н-піразол-4-амін і N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1,3-диметил-1Н-піразол-4-амін Процедура відповідає Етапу В Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол на суміш ізомерів Етапу А. Одержали суміш ізомерів у співвідношенні 4:6 (відповідно N-(4-{[третбутил(диметил)силіл]окси}феніл)-1,5-диметил-1Н-піразол-4-амін і N-(4-{[третбутил(диметил)силіл]окси}феніл)-1,3-диметил-1Н-піразол-4-амін). Приготування 28": N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-циклопропіл-1Н-піразол-4амін Етап А: 4-бром-1-циклопропіл-1Н-піразол 4-Бром-1Н-піразол (1,76 г, 12 ммоль) розчинили у безводному диметилформаміді (15 мл). До нього послідовно додали циклопропілбромід (2,9 мл, 36 ммоль) і карбонат цезію (7,8 г, 24 ммоль). Реакційну суміш нагрівали протягом 15 годин при 160 °C у герметично закритій колбі. Наприкінці реакції розчинник випарили in vacuo і залишок очистили за допомогою хроматографії на силікагелі з використанням гептану і дихлорметану як елюентів для одержання очікуваної сполуки. Етап В: N-[4-[трет-бутил(диметил)силіл]оксифеніл]-1-циклопропіл-1Н-піразол-4-амін 4-[Трет-бутил(диметил)силіл]оксіанілін (1,64 г, 7,3 ммоль) і 4-бром-1-циклопропіл-1Н-піразол (1,4 г, 7,3 ммоль), розчинені у безводному тетрагідрофурані (30 мл), перемішували протягом 3 годин при кімнатній температурі в присутності трет-бутилату натрію (3,7 мл, 2М розчину в ТГФ) і хлор(2-ди-трет-бутилфосфіно-2',4',6'-триізопропіл-1,1'-біфеніл)[2-(2-аміноетил)феніл]паладію(II) (101 мг, 0146 ммоль). Реакційну суміш фільтрували через Целіт, потім випарили насухо. Залишок очистили за допомогою хроматографії на силікагелі з використанням гептану і етилацетату як елюентів для одержання очікуваного продукту. 1 Н-ЯМР (500 МГц, ДМСО-d6, 300 К) м.ч.: 7,61 (с, 1Н, піразол-5'Н), 7,23 (с, 1Н, піразол-3'Н), 7,22 (ушир, с, 1Н, NH), 6,65 (д, 2Н, Аr-Н), 6,63 (д, 2Н, Аr-Н), 3,64 (м, 1Н, циклопропіл-Н), 1,00-0,91 т (м, 4Н, циклопропіл), 0,93 (с, 9Н, Вu),0,12(с, 6Н, Ме), -1 -1 -1 ІЧ: : СН: 2930 см ; ароматичні: 1504 см ; Si-C: 1237 см . Mac-спектрометрія високої роздільної здатності (ESI+): емпірична формула: C18H27N3OSi, + [М + Н] обчислено: 330,2003, + [М + Н] виміряно: 330,1989. Приготування 29": 1,5-диметил-4-(феніламіно)-1Н-пірол-2-карбонітрил Процедура відповідає протоколу Приготування 1", замінюючи 4-{[третбутил(диметил)силіл]оксі}анілін, який використовували на етапі В, на анілін. 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 7,19 (с, 1Н), 7,05 (т, 2Н), 6,79 (с, 1Н), 6,6 (м, 3Н), 3,61 (с, 3Н), 2,1 (с, 3Н). Приготування 30": 1-метил-N-феніл-1Н-пірол[2,3-b]піридин-6-амін 27 UA 115773 C2 5 10 15 20 25 30 35 40 45 50 55 60 Процедура відповідає протоколу Етапу В Приготування 1", використовуючи анілін і 5-бром-1метил-1Н-пірол[2,3-b]піридин (одержаний відповідно до протоколу згідно з літературою: Heterocycles, 60(4), 865, 2003). 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 8,1 (д, 1Н), 7,9 (с, 1Н), 7,7 (д, 1Н), 7,45 (д, 1Н), 7,17 (т, 2Н), 6,9 (д, 2Н), 6,7 (т, 1Н), 6,38 (д, 1Н), 3,8 (с, 3Н). Приготування 31": N-(4-{[трет-бутил(диметил)силіл]окси}феніл)-1-тридейтеріометил-1Нпіроло[2,3-b]піридин-5-амін Процедура відповідає способу Приготування 1", замінюючи 4-бром-1-метил-1Н-піразол, який використовували на етапі В, на 5-бром-1-(тридейтеріометил)-1Н-пірол[2,3-b]піридин (одержаний відповідно до протоколу згідно з літературою Heterocycles, 60(4), 865, 2003, замінюючи йодистий метил на тридейтерований йодистий метил). 1 Н-ЯМР (400 МГц, ДМСО-d6) м.ч.: 8,05 (д, 1Н), 7,6 (м + д, 2Н), 7,4 (д, 1Н), 6,85/6,7 (2d, 4Н), 6,3 (д, 1Н), 0,95 (с, 9Н), 0,15 (с, 6Н). Приготування 32": 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1-тридейтеріометил1Н-пірол-2-карбонітрил Етап А: 1-тридейтеріометил-1Н-пірол-2-карбонітрил NaH (60 % в олії) (2,61 г, 65,2 ммоль) суспендували у тетрагідрофурані (110 мл) при 0 °C. 1Н-пірол-2-карбонітрил (5 г, 54,3 ммоль) додавали по краплях протягом 10 хвилин. Реакційну суміш потім нагрівали до кімнатної температури протягом 30 хвилин. Її знову охолодили до 0 °C, а потім додали йодометан-d3 (10,23 г, 70,6 ммоль). Реакційну суміш перемішували протягом 16 годин в атмосфері азоту при кімнатній температурі. Реакційну суміш потім випарювали при зниженому тиску, а потім додали етилацетат (200 мл) і воду (200 мл). Фази відокремили і водну фазу екстрагували етилацетатом (2×200 мл). Об'єднані органічні фази промили сольовим розчином (1×80 мл), висушили над MgSO4, фільтрували і концентрували in vacuo. Залишок очистили за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів. Фракції об'єднали і випарили in vacuo для одержання очікуваної сполуки. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 6,13 (дд, J=2,7, 3,9 Гц, 1Н), 6,75 (дд, J=1,5, 4,1 Гц, 1Н), 6,79 (дд, J=1,7, 2,6 Гц, 1Н). Етап В: 4-бром-1-тридейтеріометил-1Н-пірол-2-карбонітрил N-бромсукцинімід (6,68 г, 37,5 ммоль) додали до розчину сполуки попереднього етапу (4,09 г, 37,5 ммоль) у N,N-диметилформаміді (188 мл). Розчин перемішували протягом 16 годин при кімнатній температурі. Залишок очистили за допомогою хроматографії з використанням етилацетату і гептану як елюентів для одержання очікуваної сполуки у формі твердої речовини. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 6,75 (д, J=1,7 Гц, 1Н), 6,80 (д, J=1,7 Гц, 1Н). Етап С: 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1-тридейтеріометил-1Н-пірол-2карбонітрил Азот барботували через розчин, що складається зі сполуки, одержаної на попередньому етапі (5,85 г, 31,1 ммоль), 4-[(трет-бутилдиметилсиліл)оксі]аніліну (8 г, 35,8 ммоль), третбутилату натрію (3,88 г, 40,4 ммоль) і 2-ди-трет-бутилфосфіно-2',4',6'-триізопропілбіфенілу (662 мг, 1,56 ммоль), у толуолі (260 мл) протягом 5 хвилин. Потім додали трис(дибензиліденацетон)паладій(0) (1,43 г, 1,56 ммоль). Суміш перемішували протягом 1 години при 70 °C в атмосфері азоту. Потім суспензію охолодили до кімнатної температури, розбавили етилацетатом і фільтрували через Целіт. Целітний осад потім промили етилацетатом. Фільтрат промили водою (3 рази), а потім сольовим розчином (один раз). Органічну фазу висушили над Na2SO4, фільтрували і концентрували in vacuo. Продукт очистили двічі за допомогою хроматографії на силікагелі з використанням етилацетату і гептану як елюентів, а потім за допомогою оберненофазової хроматографії з використанням метанолу та води як елюентів для одержання очікуваної сполуки у формі порошку. 1 Н-ЯМР (400 МГц, CDCl3) м.ч.: 0,17 (с, 6Н), 0,98 (с, 9Н), 4,96 (ушир, с, 1Н), 6,57 (д, J=1,9 Гц, 1Н), 6,64 (д, J=1,8 Гц, 1Н), 6,70 (ушир, с, 4Н), 13 С-ЯМР (100 МГц, CDCl3) м.ч.: -4,38, 18,26, 25,83, 34,83, 102,96, 113,69, 113,71, 115,95, 119,58, 120,75, 129,14, 140,10, 148,84, + MC (ESI):[M + H] 331,09. Приготування 33": 4-({4-[(трет-бутилдиметилсиліл)окси]феніл}аміно)-1-тридейтеріометил-5метил-1Н-пірол-2-карбонітрил Етап А: 5-метил-1Н-пірол-2-карбонітрил Сполуку одержали відповідно до протоколу, описаного у Heterocycles 2011, 82, 1503. Етап В: 1-тридейтеріометил-5-метил-1Н-пірол-2-карбонітрил Розчин 5-метил-1Н-пірол-2-карбонітрилу (0,30 г, 2,82 ммоль) у N,N-диметилформаміді (5 мл) охолодили до 0 °C. NaH (60 % в олії) (0,118 г, 2,96 ммоль) додали частинами і реакційну суміш 28
ДивитисяДодаткова інформація
Автори російськоюArnaud Le Tiran, Thierry Le Diguarher, Jerome-Benoit Starck, Jean-Michel Henlin, Anne-Francoise Guillouzic, Guillaume De Nanteuil, Olivier Geneste, Imre Fejes, Janos Tatai, Miklos Lazlo Nyerges, James Edward Paul Davidson, James Brooke Murray, I-Jen Chen, Didier Durand
МПК / Мітки
МПК: A61K 31/44, A01N 43/38, A01N 43/34
Мітки: містить, сполуки, варіанти, спосіб, одержання, пірольні, фармацевтична, композиція
Код посилання
<a href="https://ua.patents.su/103-115773-pirolni-spoluki-sposib-kh-oderzhannya-ta-farmacevtichna-kompoziciya-shho-kh-mistit-varianti.html" target="_blank" rel="follow" title="База патентів України">Пірольні сполуки, спосіб їх одержання та фармацевтична композиція, що їх містить (варіанти)</a>
Акрилоїлзаміщенa похідна дистаміцину, спосіб її одержання (варіанти) та фармацевтична композиція на її основі

Номер патенту: 61922
Опубліковано: 15.12.2003
Автори: Калдареллі Маріна, Кодзі Паоло, Бьясолі Джованні, Францетті Крістіна, Беріа Італо, Каполонго Лаура
МПК: A61P 35/00, A61K 31/40, C07D 207/34, A61P 31/12
Мітки: фармацевтична, похідна, варіанти, основі, спосіб, одержання, акрилоїлзаміщенa, дистаміцину, композиція
Формула / Реферат:
1. Акрилоїлзаміщена похідна дистаміцину формули:,де:n означає 2, 3 або 4;R1 або R2 вибирають, кожен незалежно, з: водню, галогену і С1-С4 алкілу;R3 означає водень або галоген;В вибирають з:де R4, R5, R6, R7 і R8 означають, кожен незалежно, водень або С1-С4 алкіл, за умови, що, принаймні, один з R4, R5 і R6 означає С1-С4 алкіл, або її фармацевтичнo прийнятна сіль.2. Сполука за п....
Гетероциклічні сполуки для пригнічення кислотності шлункового секрету, спосіб їх одержання (варіанти) та фармацевтична композиція, що їх містить (варіанти)

Номер патенту: 70932
Опубліковано: 15.11.2004
Автори: Старке Інгемар, Амін Косрат, Норберг Петер, Дальстрем Мікаель
МПК: A61P 1/04, A61K 31/437, A61K 31/5025, C07D 471/04, C07D 487/04, A61P 1/00, A61P 31/04, A61K 31/4985, A61K 31/519
Мітки: гетероциклічні, сполуки, варіанти, композиція, спосіб, містить, секрету, одержання, шлункового, фармацевтична, пригнічення, кислотності
Формула / Реферат:
1. Сполука формули (I)чи її фармацевтично прийнятна сіль, де R1 – С1–6алкіл, R2 – С1–6алкіл, R3 – гідроген чи галоген, ає заміщеним гетероциклом, а саме [1,2-а]піразином, формули,де R4 – Н, СН3, СН2ОН чи СН2СN,R5 – Н чи С1–6алкіл,Х – NН чи О.2. Сполука за п. 1 чи її фармацевтично прийнятна сіль, яка відрізняється тим, щоR1 та R2, незалежно, – СН3...
Сполуки, ефективні як інгібітори ксантиноксидази, спосіб одержання таких сполук (варіанти) та фармацевтична композиція, що містить такі сполуки

Номер патенту: 110197
Опубліковано: 10.12.2015
Автори: Парк Док Сонг, Кім Тае Хун, Парк Хеуі Сул, Кім Гин Тае, Сонг Йонг Ук, Артемов Василій, Йунг Чол Кю, Чоі Сунг Піл, Чоі Ин Сіл, Коо Кі Чул, Парк Ван Су, Парк Хюн Йюнг
МПК: A61K 31/40, A61P 3/10, C07D 403/02, C07D 209/04
Мітки: фармацевтична, спосіб, ксантиноксидази, інгібітори, композиція, такі, сполуки, таких, сполук, одержання, варіанти, ефективні, містить
Формула / Реферат:
1. Сполуки наступної формули (1):, (1) в якій: А вибирається з наступних заміщень А-і, A-iv, A-v, A-vi та A-vii: , ,
Сполуки індолу, спосіб їх одержання і фармацевтична композиція (варіанти), яка їх містить

Номер патенту: 91646
Опубліковано: 10.08.2010
Автори: Кеняр Даніель-Анрі, Делагранж Філіпп, Дюфло Мюріель, П'єсар Сільві, Бабоно Вінсан, Одіно Валері, Бутан Жан Альбер, Маршан Паскаль
МПК: C07D 209/42, A61P 3/04, A61P 35/00, A61P 15/00, A61P 25/00, A61K 31/404
Мітки: індолу, варіанти, одержання, фармацевтична, сполуки, спосіб, містить, яка, композиція
Формула / Реферат:
1. Сполука формули (І):, (I) в якій: R1 являє собою лінійну або розгалужену (С1-С6)алкільну групу, лінійну або розгалужену (С3-С8)циклоалкільну групу або (С3-С8)циклоалкіл(С1-С6)алкільну групу, в якій алкільна частина може бути лінійною або розгалуженою, R2 являє собою лінійну або розгалужену...
Сполуки азабіцикло[3.2.0]гепт-3-илу, спосіб їх одержання і фармацевтична композиція, яка їх містить (варіанти)

Номер патенту: 106721
Опубліковано: 10.10.2014
Автори: Пьєр Лєстаж, Фанні Панайї, Ан-Марі Шолє, Патрік Касара, Ален Дено
МПК: C07D 309/00
Мітки: сполуки, фармацевтична, яка, азабіцикло[3.2.0]гепт-3-илу, містить, спосіб, варіанти, композиція, одержання
Формула / Реферат:
1. Сполука формули (І):, (І)в якій:• ALK являє собою алкіленовий ланцюг,• W являє собою групу ,в якій R і R' являють собою, кожний незалежно один від одного, атом водню або лінійну або розгалужену (С1-С6)алкільну групу, необов'язково заміщену однією або більше групами,...
Попередній патент: Композиція програмованого нуклеопротеїнового молекулярного комплексу і спосіб модифікування заданої послідовності нуклеїнової кислоти-мішені
Наступний патент: Вузол кріплення навісного обладнання скребкового конвеєра
Випадковий патент: Двері коксової печі