Метансульфонат (е)-3-[2-н-бутил-1{(4-карбоксифеніл)метил}-1н-імідазол-5-іл]-2-(2-тієніл)метил-2-пропенової кислоти, спосіб його одержання, фармацевтична композиція на його основі











Формула / Реферат
1. Метансульфонат (Е)-3-[2-н-бутил-1-{(4-карбоксифенил)метил}-1н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропиновой кислоты.
2. Соединение по п.1, обладающее активностью антагониста ангиотензина II.
3. Соединение по п.1, обладающее антигипертензивной активностью.
4. Фармацевтическая композиция, обладающая активностю антагониста ангиотензина II, отличающаяся тем, что в качестве активного ингредиента содержит эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
5. Способ получения соединения по п.1, отличающийся тем, что осуществляют обработку (Е)-3-[2-н-бутил-1-{(4-карбоксифенил)метил}-1н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропиновой кислоты в 2-пропаноле метансульфоновой кислотой приблизительно при 8°С.
Текст
1. Метансульфонат (Е)-3-[2-н-бутил-1-{(4-карбоксифенил)метил}-1н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропеновой кислоты. 40587 предположить, что ингибирование ренин-ангиотензивной системы может быть успешным в предотвращении или замедлении прогрессирования хронической почечной недостаточности (Anderson S. и др. (1985), J. Clin. Invest., 76, 612). Также, недавно поданная Патентная Заявка (Патентная Заявка Южн. Африки № 87/01, 653) утверждает, что антагонисты АII полезны в качестве агентов, способных снижать и регулировать повышенное внутриглазное давление, особенно глаукому, у млекопитающих. Соединения данного изобретения ингибируют, блокируют и противодействуют действию гормона АII и, следовательно, являются полезными в регулировании и смягчении вызванной ангиотензином артериальной гипертензии, застойной сердечной недостаточности, почечной недостаточности и других расстройств, обусловленных действием АII. При приеме соединений данного изобретения животными повышенное кровяное давление, обусловленное AII, снижается, а другие проявления, основанные на действии AII, сводятся к минимуму и регулируются. Можно ожидать также, что соединения данного изобретения проявляют диуретическую активность. Признание важности блокирующего и ингибирующего действия AII стимулировали и другие усилия, относящиеся к синтезу антагонистов AII. Следующие ссылки раскрыли имидазоловые производные, которые характеризуются как обладающие блокирующей активностью в отношении AII и которые являются полезными в качестве гипотензивных агентов. Furukawa и др., Пат. США 4340598 описывает имидазол-5-ил-уксусные кислоты и имидазол-5-ил-пропановые (пропионовые) кислоты. В частности, автор изобретения отмечает 1-бензил2-н-бутил-5- хлоримидазол-4-уксусную кислоту и 1-бензил-2-фенил-5хлоримидазол-4-пропионовую кислоту. Furukawa и др., Пат. США 4356040 описывает замещенные производные имидазол-5-уксусной кислоты. В частности, раскрывается соединение 1-(2-хлорбензил)-2-н-бутил-4-хлоримидазол-5уксусная кислота. Carini и др. в Европ. пат. 253310 описывают определенные имидазолилпропеновые кислоты. Два промежуточных соединения, описанные в этом патенте, представляют собой этил-3-[1-(4нитробензил)-2-бутил-4-хлоримидазол-5-ил] пропеноат и этил-3-[2- бутил-4-хлор-1-(4-аминобензил)имидазол-5-ил]пропеноат. Кроме того, Wareing в РСТ/Евр. пат. 86/00297 раскрывает в качестве промежуточных соединений определенные имидазолилпропеноаты. На странице 62 формула (СХ) представляет собой 3-[1-(4-фторфенил)-4-изопропил-2-фенил1Н-имидазол-5-ил]-2- пропеноат. Описание изобретения Соединения представленного изобретения, являющиеся блокаторами рецепторов ангиотензина II, представляют собой: (Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-пропеновая (акриловая) кислота и (Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-про пеновая (акриловая) кислота, этиловый эфир, или фармацевтически приемлемая соль этого соединения и (Е)-3-[2-н-бутил-1-{(4-карбоксифенил)метил}-1Н-имидазол-5-ил]-2-(2-тиенил)метил2-пропеновая кислота-метансульфонат. Изобретение также относится к фармацевтическим композициям, включающим фармацевтический носитель и эффективное количество перечисленного выше соединения. В данное изобретение включаются также способы противодействия рецепторам ангиотензина II, которые состоят в приеме субъектом необходимого эффективного количества перечисленного выше соединения. В данное изобретение включены также способы лечения артериальной гипертензии, застойной сердечной недостаточности, глаукомы и почечной недостаточности приемом этих соединений. Соединения данного изобретения получаются в соответствии с описанными здесь методиками и иллюстрируются примерами. Реагенты, защитные группы и структура имидазола и других фрагментов молекулы должны быть составлены в соответствии с предложенными химическими превращениями. Этапы в синтезе должны быть согласованы с функциональными группами и защитными группами имидазола и других частей молекулы. Исходный материал, 2-н-бутилимидазол, является известным (J. Org. Chem., 45:4038, 1980) или синтезируется с использованием известных методик. Например, имидазол превращается в 2н- бутилимидазол реакцией имидазола с триэтилортоформиатом и п-толуолсульфоновой кислотой с образованием 1-диэтоксиортоамидимидазола и затем обработкой н-бутил литием с образованием 2-литиевого производного ортамида и алкилированием н-бутил иодидом в соответствующем растворителе, таком, как тетрагидрофуран (ТГФ). 1-замещенная нафтильная или бензильная группа вводится в 2-н- бутилимидазол известными методами, например, реакцией с замещенным нафтильным или бензильным галогенидом, мезилатом или ацетатом, таким, как 2-хлорбензилбромид или 4-карбометоксибензилбромид в соответствующем растворителе, таком как диметилформамид (ДМФ) в присутствии подходящего кислотного акцептора, такого как алкилат натрия, карбонат калия или натрия или гидрида металла, предпочтительно гидрида натрия при температуре реакции приблизительно от 25оС до 100оС, предпочтительно при 50оС. Образующийся 1-замещенный нафтильный или бензил-2-н-бутилимидазол представляет собой гидроксиметилированный в положении 5 продукт, например, реакцией с формальдегидом в присутствии ацетата натрия в уксусной кислоте с образованием 1-замещенного нафтильного или бензил-2-н-бутил-5-гидроксиметилимидазольного промежуточных соединений. Или же, полученные выше 5-гидроксиметилимидазольные промежуточные соединения получаются реакцией имидоэфира, такого, как метиловый эфир валерамидина с дигидроксиацетоном в жидком аммиаке под давлением с образованием 2-н-бутил-5-гидроксиметилимидазола. Это промежуточное соединение взаимодействует с ангидри 2 40587 дом уксусной кислоты с образованием 1-ацетил-5ацетоксиметил-2-н- бутилимидозола. Диацетатное промежуточное соединение представляет собой N-алкилат, например, при использовании 2-хлорбензилтрифлоата или 4-карбометоксибензилтрифлоата, и образующийся 1-замещенный-2-н-5ацетоксиметилимидазол обрабатывается водным основанием, таким как 10%-ый раствор гидроокиси натрия, с образованием 5-гидроксиметилимидазольных промежуточных соединений, описанных ранее. Гидроксиметильная группа полученного ранее промежуточного соединения оксиляется до альдегидной действием соответствующего реагента, такого, как безводная хромовая кислота-силикагель в тетрагидрофуране или, предпочтительно, активированной двуокисью марганца в подходящем растворителе, таком как бензол или толуол, или, предпочтительно, метиленхлорид при температуре от 25оС до 140оС, предпочтительно при 25оС. Имидазол-5-карбоксиальдегиды обрабатываются соответствующим фосфонатом, таким как триметил-3-(2-триенил)-2-фосфонопропионат. Фосфонаты получаются, например, из триалкилфосфоноацетатов алкилированием соответствующим галоидом, метилсульфонилатом или ацетатом в присутствии соответствующего основания, такого как гидрид натрия в подходящем растворителе, предпочтительно глиме, при температуре реакции приблизительно от 25оС до 110оС, предпочтительно при 55оС, с образованием соответствующего фосфоната. Взаимодействие имидазол5-карбоксиальдегидов с фосфонатами осуществляется в присутствии подходящего основания, такого как алкоголят металла, гидрид лития или, предпочтительно, гидрид натрия в соответствующем растворителе, таком как этанол, метанол, эфир, диоксан, тетрагидрофуран или, предпочтительно, глим, при температуре реакции приблизительно от 10оС до 50оС, предпочтительно при 25оС, с образованием непостоянной смеси транс и цис, например, (Е) и (Z), 1-замещенных-2-н-бутил5-СН [(2-тиенил)метил]-(СОО алкил)-имидазоС = лов. Эти изомеры легко разделяются хроматографированием через силикагель в соответствующих системах растворителей, предпочтительно в гексан / этилацетатных смесях. Эфиры гидролизуются до соответствующих кислотных соединений с использованием основания, такого, как гидроксид калия, гидроксид лития или гидроксид натрия, в соответствующей системе растворителя, такой как, например, водные спирты или диглим. Или же, 1-замещенные-2-н-бутилимидазол5-карбоксиальдегиды получаются следующим способом. Исходные 2–н-бутилимидазол-5- карбоксиальдегиды обрабатываются N-алкилирующим защитным агентом, таким как хлорметилпивалат (ПОМ-Сl) в присутствии основания, такого как карбонат калия, в соответствующем растворителе, таком, как диметилформамид, при температуре приблизительно от 20оС до 50оС, предпочтительно при 25оС, с образованием продуктов N-алкилирования (например, ПОМ-дериватизация) на наименее затрудненном атоме азота имидазольного ядра. 1-Замещенная-нафтильная или -бензильная группы объединяются в имидазол N-алкилированием приготовленного выше альдегида галоидме тилбензольными соединениями, такими как метил 4-бромметилбензоат или метил 4-бромметилнафталин-1-карбоксилат при температуре приблизительно от 80оС до 125оС, предпочтительно при 100оС. Защитная группа при 3-азоте имидазольного кольца удаляется с использованием основного гидролиза, например, при использовании двухфазной смеси этилацетата и водного карбоната натрия с образованием 1-замещенных-н-бутилимидазол-5-карбоксиальдегидных соединений. Соединения данного изобретения могут быть получены из этих 5-карбоксиальдегидных соединений с использованием описанных выше методов. Или же, 2-н-бутилимидазольные исходные вещества взаимодействуют с триметилсилилэтокси метил (СЭМ) хлоридом с образованием 1-( триметилсилил)-этоксиметил-2-н-бутилимидазола. Реакция проводится, например, в присутствии гидрида натрия в растворителе, таком, как диметилформамид. Производные 5-трибутилолова получаются обработкой литием, например, бутиллитием в соответствующем растворителе, предпочтительно в диэтиловое эфире, за которой следует обработка литийимидазольного производного галоидным трибутилоловом, предпочтительно хлоридом три-н-бутилолова приблизительно при температуре от –10оС до 35оС, предпочтительно при 25оС. 1-СЭМ-2-н-бутил-5- трибутилоловоимидазол соединяется с эфиром a, b-насыщенной кислоты, имеющим остаточную группу в b-положении, такую как галоидная или трифторметансульфонилокси группу, например, BrCR4 = C[(2-тиенил)метил](СОО алкил) в присутствии фосфинового лиганда, такого, как бис(дифенилфосфино)пропан или трифенилфосфин и соединения палладия (II), или предпочтительно тетракис(трифенилфосфин)палладия(О) в присутствии основания или без него – такого, как трибутиламин при температуре от 50оС до 150оС, предпочтительно при 120оС. Как (Е), так и (Z) олефиновые изомеры получаются по этой методике и изомерные эфиры легко разделяются хроматографированием с использованием силикагеля. 1-СЭМ группа (Е) и (Z) изомеров гидролизуется кислотой, например, водной хлористоводородной кислотой в подходящем спиртовом растворителе, таком как метанол или этанол, и 1-незамещенные имидазольные производные превращаются в 1-т-бутоксикарбонил (тБОК) имидазолы ди-т-бутилдикарбонатом (HoppeSeyler's Z. Physiol. Chem. (1976), 357, 1651). Т-БОК эфиры алкилируются и гидролизуются с использованием, например, 2-хлорбензилтрифлоата или 4карбометоксибензилтрифлоата в присутствии соответствующего основания, предпочтительно диизопропилэтиламина, в подходящем растворителе, предпочтительно метиленхлориде, образуя 1-замещенные имидазольные производные (эфиры). (Е) и (Z) изомеры гидролизуют в (Е) и (Z) кислоты с использованием описанного выше метода. Соединения данного изобретения получают также по следующей методике. 1-Замещенные-2н-бутилимидазол-5-карбоксиальдегиды, полученные, как описано выше, взаимодействуют с замещенными полукислотным, полуэфирным производным малоната, таким как этил-2-карбокбси-3(2-тиенил) пропионатом, в присутствии основания, такого как пиперидин в подходящем растворителе, 3 40587 рой R8 является водородом, получают известными способами из органических и неорганических оснований, включая нетоксичные основания щелочных и щелочноземельных металлов, например, гидроксидов кальция, лития, натрия и калия, гидроксида аммония и нетоксичных органических оснований, таких как триэтиламин, бутиламин, пиперазин, меглумин, холин, диэтаноламин и трометамин. Активность антагонистов ангиотензина II соединений формулы (I) оценивается по способам in vitro и in vivo. Антагонистическая активность in vitro определяется способностью соединений конкурировать с 125І-ангиотензином II в связывании рецепторов сосудистого ангиотензина II и их способностью противостоять сжимающей реакции ангиотензина II в выделенной аорте кролика. Активность in vivo оценивается эффективностью соединений в ингибировании прессорной реакции на экзогенный ангиотензин II у находящихся в сознании крыс и в снижении кровяного давления у крыс, моделирующих связанную с почками артериальную гипертензию. Связывание. Анализ связывания меченого лиганда представляет собой разновидность детально описанного ранее метода (Gunther и др., Circ. Res., 47:278, 1980). Особая фракция брыжжеечной артерии крысы термостатировалась в буфере Tris-a с рМ 80 125І-ангиотензина II в присутствии антагонистов ангиотензина II или без них в течение 1 часа при 25оС. Термостатирование заканчивается быстрой фильтрацией и связанный рецептор 125Іангиотензин II, осевший на фильтре, определяется количественно счетчиком гамма-излучения. Сила антагонистов ангиотензина II выражается как ІС50, которое представляет собой концентрацию антагониста, необходимую для замещения 50% общего в особенности связанного ангиотензина II. Примерное значение ІС50 соединений изобретения (Е изомеры) составляет приблизительно от 0,1 нМ до 30 мМ. Аорта. Возможность соединений противодействовать ангиотензину II, обусловливающему сужение кровеносных сосудов, определяется на аорте кролика. Из грудной аорты кролика вырезаются кольцевые сегменты и суспензируются в органных ваннах, соединяющих физиологический солевой раствор. Кольцевые сегменты укрепляются на металлических держателях и прикрепляются к измерительным датчикам смещения, связанным с записывающим устройством. Совокупные концентрационные кривые, отвечающие ангиотензину II, снимаются в отсутствии антагониста или через 30 минут после термостатирования с антагонистом. Константы диссоциации антагониста (КВ) рассчитываются по методу отношения доз с использованием значения эффективных концентраций. Примерными значениями КВ соединений изобретения (Е изомеры) являются значения от 0,1 нМ до 0,50 нМ. Ингибирование прессорной реакции на ангиотензин II у находящихся в сознании крыс. Готовятся крысы с постоянными бедренным артериальным и венозным катетерами и желудочной трубкой (Gellai и др., Kidney Int., 15:419, 1979). таком как толуол, при температуре от 80оС до 110оС, предпочтительно при 100оС. Образующиеся 1-замещенные-2-н-бутил- 5-СН=С(R5)СООалкилимидазолы гидролизуют до соответствующих соединений данного изобретения щелочным гидролизом, как описано выше. Или же соединения данного изобретения получают следующим образом. 1-Замещенные-2-нбутилимидазол-5-карбоксиальдегиды, полученные, как описано здесь выше, обрабатывают литиевым производным замещенного этилового или метилового эфира. Эти литиевые производные получаются взаимодействием литий диизопропиламида в подходящем растворителе, предпочтительно тетрагидрофуране, с кислым эфиром, таким, как ROOC-CH2-CH2-(2-тиенил) с образованием a-литиевых производных, при температуре от –78оС до –10оС, предпочтительно при –78оС, которые обрабатывают имидазолкарбоксиальдегидом. Промежуточная b-гидрокси группа эфира имидазола превращается в метилсульфонилат или ацетат и метилсульфонилат или, предпочтительно, ацетат, нагревается в соответствующем растворителе, таком как толуол с одним или двумя эквивалентами 1,8-диазобицикло [5,4,0] ундек7-ена при температуре от 50 до 110оС, предпочтительно при 80оС с образованием эфиров 3-(имидазол-5-ил)-2-(2-тиенил)метил-2-пропеновой кислоты. (Е) изомер является преобладающим олефиновым изомером. Кислоты получаются из эфиров по описанному выше способу. Соединения данного изобретения, в которых заместитель в положении 1 имидазольного кольца представляет собой замещенный карбоксил, образуются из соединений, в которых эта группа является замещенным СО2С1-С4 алкилом, с использованием основного гидролиза, такого как водные натриевый или калиевый гидроксид в метаноле или этаноле, или с использованием кислотного гидролиза, такого как водная хлористоводородная кислота. Фармацевтически приемлемые кислотные аддитивные соли соединений формулы (I) образуются с использованием соответствующих органических или неорганических кислот и известных способов. Например, основание взаимодействует с соответствующей неорганической или органической кислотой в смешивающемся с водой растворителе, таком как этанол, с выделением соли удалением растворителя, или в смешивающемся с водой растворителе, когда кислота является растворимой в нем, таком как этиловый эфир или хлороформ, с непосредственным отделением нужной соли или с выделением ее удалением растворителя. Характерными примерами соответствующих кислот являются малеиновая, фумаровая, бензойная, аскорбиновая, pamoic, янтарная, бисметиленсалициловая, метансульфоновая, этандисульфоновая, уксусная, пропионовая, винная, салициловая, лимонная, глюконовая, аспартиковая, стеариновая, пальмитиновая, итаконовая, гликолевая, n-аминобензойная, глутаминовая, бензолсульфоновая, хлористоводородная, бромистоводородная, серная, циклогексилсульфаминовая, фосфорная и азотная кислоты. Фармацевтически приемлемые основные аддитивные соли соединений формулы (I), в кото 4 40587 Спустя два-три дня после хирургической операции крысы помещаются в приспособление с устройством для фиксации конечностей и осуществляется постоянное наблюдение кровяного давления через артериальный катетер датчиком давления и запись на многоканальный самописец. Изменение в значении артериального давления в ответ на инъекции 250 мг/кг ангиотензина II сравнивается в различных временных точках до приема и после приема соединений внутривенно или орально при дозах от 0,1 до 300 мг/кг. Доза соединения, необходимая для создания 50% ингибирования контрольной реакции на ангиотензин II (ІС50), применяется в качестве оценки эффективности соединений. Активность в предотвращении артериальной гипертензии. Активность соединений против артериальной гипертензии оценивается их способностью в снижении значения артериального давления у находящихся в сознании крыс, у которых создано ренин-зависимое артериальное гипертензивное состояние перевязыванием левой почечной артерии (Cangiano и др., J. Pharmacol. Exp. Ther., 208:310, 1979). Крысы с перевязанной почечной артерией получаются введением постоянных катетеров, как описано выше. Спустя семь-восемь дней после перевязывания почечной артерии, в течение которых уровни ренина в плазме являются наиболее высокими, находящиеся в сознании крысы помещаются в приспособление с устройством для фиксации конечностей и осуществляется непрерывная запись значения артериального давления до внутривенного или орального приема соединений и после их введения. В качестве оценки эффективности используется доза соединения, необходимая для снижения значения артериального давления на 30 мм Hg (IC30). Эффекты по снижению внутриглазного давления, достигнутые в данном изобретении, могут быть оценены по методике, описанной в работе Watkins и др., J. Ocular Pharmacol., 1(2):161–168 (1985). Соединения данного изобретения включаются в подходящие дозированные формы, такие как препараты для инъекций или активные соединения для орального приема, капсулы или таблетки. Применяются твердые или жидкие фармацевтические носители. К твердым носителям относятся крахмал, лактоза, дигидрат сульфата кальция, терра альба, сахароза, тальк, желатин, агар, пектин, акация, стеарат магния и стеариновая кислота. Жидкие носители включают сироп, масло земляного ореха, оливковое масло, физиологический раствор и воду. Аналогично, носитель или разбавитель могут включать любое вещество, пролонгирующее выделение, такое как моностеарат глицерина или дистеарат глицерина – одного или в сочетании с парафином. Количество твердого носителя может меняться в широких пределах, но, предпочтительно, составляет от 25 мг до 1 г на дозированную единицу. При использовании жидкого носителя препарат может иметь вид сиропа, эликсира, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекций, такой как ампула, или водной или неводной жидкой суспензии. Для местного офтальмологического применения фармацевтические композиции применяют ся в виде растворов, суспензий, мазей и твердых составов. Типичными фармацевтически приемлемыми носителями являются, например, вода, смеси воды и смешивающихся с ней растворителей, таких как низшие спирты или растительные масла, и водорастворимые офтальмологически приемлемые нетоксичные полимеры, например, производные целлюлозы, такие как метилцеллюлоза. Фармацевтические препараты могут также содержать нетоксичные вспомогательные вещества, такие как эмульгирующие, консервирующие, увлажняющие и связующие агенты, как, например, полиэтиленгликоли, антибактериальные компоненты, такие как четвертичные аммониевые соединения, буферирующие ингредиенты, такие как хлорид щелочного металла, антиоксиданты, такие как метабисульфит натрия и другие стандартные ингредиенты, такие как монолаурат сорбитана. Кроме того, в качестве сред-носителей для данной цели могут быть использованы подходящие офтальмологические наполнители, содержащие стандартные фосфатные буферные системы наполнителей. Фармацевтический препарат также может существовать в виде твердого состава. Например, в качестве носителя лекарственного средства можно использовать твердый водорастворимый полимер. Могут быть использованы также твердые нерастворимые в воде составы, такие как составы, приготовленные из сополимера этилен-винилацетат. Фармацевтические препараты готовятся с использованием следующих стандартных для химиков-фармацевтов методик, включающих смешение, гранулирование и сжатие, когда необходимо для таблетированных форм, или смешение, наполнение и растворение ингредиентов, что применимо для получения нужных оральных, парентеральных или местных продуктов. Дозы соединений данного изобретения в фармацевтической дозированной единичной форме, как описано выше, представляют собой эффективное нетоксичное количество в интервале от 0,01 до 200 мг/кг активного соединения, предпочтительно 1–100 мг/кг. Выбранная доза принимается пациентом при необходимости противодействия рецептору ангиотензина II от 1 и 6 раз ежедневно орально, ректально, местно, путем инъекций или постоянно посредством вливания. Оральные дозированные единицы для приема человеком предпочтительно содержат от 1 до 500 мг активного соединения. Предпочтительно, более низкие дозировки используются для парентерального приема. Оральный прием може использоваться также и с более высокими дозировками, однако при условии безопасности и удобства для пациента. Составы для местного применения содержат активное соединение в количествах от 0,0001 до 0,1 (вес./объемн.%), предпочтительно от 0,0001 до 0,01. В качестве местной единичной дозировки для глаза человека применяется количество активного соединения от 50 нг до 0,05 мг, предпочтительно от 50 нг до 5 мг. При приеме соединений изобретения в соответствии с данным изобретением не наблюдается нежелательных токсикологических эффектов. 5 40587 Способ данного изобретения, состоящий в противодействии рецепторам ангиотензина II у млекопитающих, включая человека, включает прием нуждающегося в таком противодействии субъекта эффективного количества соединения данного изобретения. Способ данного изобретения, состоящий в активности в противодействии артериальной гипертензии и в лечении застойной сердечной недостаточности, глаукомы и почечной недостаточности, включает прием соединения данного изобретения нуждающимся в нем субъектом эффективного количества для создания названной активности. Предполагаемыми эквивалентами соединений данного изобретения являются соединения в остальном соответствующие им, в которые были введены заместители – в любое из незамещенных положений таких соединений, при условии, что такие соединения обладают фармацевтической полезностью, свойственной соединениям данного изобретения. Следующие примеры иллюстрируют препараты соединений и фармацевтические композиции данного изобретения. Предполагается, что примеры не ограничивают объем притязаний данного изобретения, как это определено выше и как заявлено ниже. Пример 1. (Е)-3-[2-н-Бутил-1-{(2-хлорфенил)метил}-1Нимидазол-5-ил]-2-(2-тиенил)метил-2-пропеновая кислота. (i) 2-н-Бутил-1-(2-хлорфенил)метил-1Н-имидазол. Имидазол превращается в 1-диэтоксиортоамидное производное по методу Curtis и Brown, J. Org. Chem., (1980), 45, 20. Имидазол (12,8 г, 0,19 мол) и 118,4 г (0,8 мол) триэтилортоформиат взаимодействовали в присутствии 1г п-толуолсульфоновой кислоты с образованием 20,6 (61%), с т. кип. 65–70оС (0,1 мм) 1-диэтоксиортоамида имидазола. Этот продукт (24,0 г, 0,14 мол) растворяли в сухом тетрагидрофуране (250 мЛ), охлаждали до –40оС и добавляли н-бутиллитий (0,14 мол, 56,4 мЛ 2,5 М-го раствора в гексане) при температуре от – 40оС до –35оС. Спустя 15 минут добавляли н-бутил иодид (31,1 г, 0,169 мол) при –40оС, и реакционная смесь перемешивалась в течение ночи при комнатной температуре. Реакционная смесь разделялась между эфиром и 0,3N хлористоводородной кислотой, а органический слой повторно экстрагировался разбавленной хлористоводородной кислотой. Объединенные водные экстракты нейтрализовались раствором бикарбоната натрия, экстрагировались метиленхлоридом, высушивались над сульфатом магния и концентрировались. Однократное равновесное испарение на приборе Kugelrohr дало 14,8 г (85%) 2-н-бутилимидазола. 2-н-Бутилимидазол (9,7 г, 0,078 мол) растворялся в метаноле (50 мЛ) и по каплям добавлялся к раствору метилата натрия (из гидрида натрия (2,31 г, 0,0934 мол) в метаноле (250 мЛ). Через час раствор выпаривали до сухого состояния и натриевую соль растворяли в сухом диметилформамиде (150 мЛ) и добавляли 2-хлорбензилбромид (16,3 г, 0,079 мол). Смесь нагревали в течение 17 часов при 50оС в атмосфере аргона, выливали в ледяную воду, а продукт экстрагировали этилацетатом. Экстракт промывали, сушили и концентрировали, получая 18,5 г неочищенного продукта, который подвергали хроматографированию над силикагелем с использованием смеси 2:1 этилацетат/гексан, получив 11,9 г (61%) 2-н-бутил1-(2- хлорфенил)метил-1Н-имидазола в виде масла. Тонкослойная хроматография на силикагеле смесью 4:1 этилацетат/гексан дала значение Rf 0,59. (ii) 2-н-Бутил-1-(2-хлорфенил)метил-5-гидроксиметил-1Н-имидазол. Способ 1. Смесь 2-н-бутил-1-(2-хлорфенил)метил-1Нимидазола (95,5 г, 0,384 мол), 37% формальдегида (500 мЛ), ацетата натрия (80 г) и уксусной кислоты (60 мЛ) нагревалась до температуры дефлегмации в течение 40 часов в атмосфере аргона. Реакционная смесь концентрировалась в вакууме, а остаток перемешивался с 500 мЛ 20% раствора гидроксида натрия в течение 4 часов, разбавлялся водой и экстрагировался метиленхлоридом. Экстракт промывался, сушился и концентрировался. Неочищенный продукт (117 г) подвергался флешхроматографированию над 600 г силикагеля с градиентом этилацетата к 10% метанолу в этилацетате с образованием 8,3 г исходного вещества, 24,5 г смеси исходного вещества и продукта и 44 г (41%) 2-н-бутил-1-(2-хлорфенил)-метил-5-гидроксиметил-1Н- имидазол, т.пл. 86–88оС (из этилацетата). Дальнейшее элюирование дало бис(4,5-гидроксиметил) производное, т.пл. 138–140оС (из этилацетата). Способ 2. Смесь гидрохлорида метилового эфира валерамидина (250 г, 1,66 мол) и дигидроксиацетона (150 г, 0,83 мол), растворенную в жидком аммиаке, оставляли на ночь при комнатной температуре в аппарате высокого давления, а затем нагревали в течение 4 часов при 65оС при 375 psi (26,36 кг/см2). Аммиак испаряли, а остаток растворяли в метаноле (3 Л). Образующееся пастообразное вещество подвергали ректификации с добавлением ацетонитрила (1 Л). Раствор декантировали в горячем виде из твердого хлорида аммония. Эту процедуру повторяли и объединенные ацетонитрильные экстракты обрабатывали активированным древесные углем, фильтровали в горячем виде, а фильтрат концентрировали в вакууме с образованием темного масла – 2-н-бутил-5-гидроксиметилимидазола (253 г, 1,63 мол, 98%). Этот неочищенный спирт (253 г) обрабатывали уксусным ангидридом (400 мЛ) при –15оС, затем давали ему нагреваться при перемешивании до комнатной температуры, а затем дополнительно перемешивали в течение 19 часов. Уксусный ангидрид испаряли при пониженном давлении, остаток растворяли в метиленхлориде и промывали органическую фазу 5%-ым раствором бикарбоната натрия и водой. Экстракт сушили над сульфатом натрия и концентрировали, получая 323 г (83%) 1ацетил-4-ацетоксиметил-2-н-бутилимидазола. Этот диацетат был подвергнут N-алкилированию по следующей методике. К раствору ангидрида трифловой кислоты (трифторзамещенный) (120 мЛ, 0,71 мол) в метиленхлориде (200 мЛ) при –78оС в атмосфере аргона добавлялся раствор диизопропилэтиламина (128 мЛ, 0,73 мол) и 2 6 40587 хлорбензиловый спирт (104 г, 0,72 мол) в метиленхлориде (350 мЛ) в течение 20 минут. После дополнительного перемешивания в течение 20 минут при –78оС этот раствор затем взаимодействовал с 1-ацетил-4-ацетоксиметил-2- н-бутилимидазолом (146 г, 0,61 мол), растворенным в метиленхлориде (300 мЛ) в течение 20 минут. Затем смесь перемешивалась в течение 18 часов при комнатной температуре, и растворители испарялись. Оставшийся 2-н-бутил-5-ацетоксиметил-1(2-хлорфенил)метил-1Н-имидазол был использован без очистки для гидролиза ацетатной группы. Раствор неочищенного 2-н-бутил-5-ацетоксиметил-1-(2-хлорфенил)метил-1Н-имидазола (250 г) в метаноле (200 мЛ) обрабатывался 10%ым раствором гидроксида натрия (700 мЛ) и смесь нагревалась на паровой бане в течение 4 часов. После охлаждения добавлялся метиленхлорид, органическая фаза отделялась, промывалась водой, сушилась и концентрировалась. Остаток растворяли в эфире, охлаждали и затравляли с образованием неочищенного продукта. Перекристаллизация из этилацетата дала 176 г 2-н-бутил-1- (2хлорфенил)метил-5-гидроксиметил-1Н-имидазола, т. пл. 86–88оС. Это вещество было во всех отношениях идентично продукту, полученному по способу 1. (iii) 2-н-Бутил-1-(2-хлорфенил)метил-1Н-имидазол-5- карбоксиальдегид. Раствор 2-н-бутил-1-(2-хлорфенил)метил-5гидроксиметил-1Н- имидазола (5,4 г, 0,0194 мол) в толуоле (25 мЛ) добавляли к суспензии активированной двуокиси марганца (27 г) в метиленхлориде (325 мЛ). Суспензия перемешивалась при комнатной температуре в течение 17 часов. Твердые вещества отфильтровывались, а фильтрат концентрировался и подвергался флеш-хроматографированию над силикагелем смесью 6:4 гексан/этилацетат, давая 4,16 г (78%) 2-н-бутил-1-(2хлорфенил)метил-1Н-имидазол-5-карбоксиальдегида в виде масла. ЯМР и ИК совпадали со строением. (iv) (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}1Н-имидазол-5-ил]- 2-(2-тиенил)метил-2-пропеновая кислота. Способ А. (а) Триметил 3-(2-тиенил)-2-фосфонопропионат. К раствору 2-тиофенметанола (2,28 г, 0,02 мол) в четыреххлористом углероде (25 мЛ) добавлялся трифенилфосфин (6,81 г, 0,026 мол) и раствор подвергался дефлегмированию в течение 3 часов. Охлажденная реакционная смесь разбавлялась гексаном (60 мЛ), вымораживалась и фильтровалась. Концентрированный фильтрат (4,6 г) подвергался флеш-хроматографированию над силикагелем с использованием смеси гексан/этилацетат 7:3, образуя 2-хлорметилтиофен (1,52 г, 57%) в виде масла. Суспензия гидрида натрия (0,271 г, 11,3 ммол) в сухом глиме (40 мЛ) в атмосфере аргона обрабатывалась по каплям триметилфосфоноацетатом (1,87 г, 10,3 мол) в глиме (5 мЛ). Образующаяся смесь перемешивалась при комнатной температуре в течение 1,5 часа. Затем добавлялся 2хлорметилтиофен (1,5 г, 11,3 ммол) и смесь перемешивалась при 65оС в течение 18 часов. Реак ционная смесь разделялась между водой и этилацетатом и органический слой промывался водой и раствором хлористого кальция, сушился безводным сульфатом магния и концентрировался до 1,9 масла. Вещество хроматографировалось над силикагелем с использованием смеси этилацетат/гексан 4:1, образуя 800 мг (28%) триметил 3(2-тиенил)-2-фосфонопропионата. (b) Метил (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-пропеноат. К суспензии гидрида натрия (69 мг, 2,87 ммол) в глиме (5 мЛ) по каплям добавляется раствор триметил 3-(2-тиенил)-2- фосфонопропионата в глиме (3 мЛ) в атмосфере аргона. Когда выделение газа прекращено, смесь нагревают до 50оС в течение 15 минут. Добавляют раствор 2-н-бутил-1(2-хлорфенил)-метил-1Н-имидазол-5-карбоксиальдегида (0,53 г, 1,98 ммол) в глиме (3 мЛ) и смесь перемешивалась при 60–65оС в течение 5 часов. Охлажденная реакционная смесь разделялась между водой и этилацетатом и органический слой промывался водой, высушивался, концентрировался и подвергался флеш-хроматографированию над силикагелем с образованием 336 мг (41%) метил (Е)-3-[2-н-бутил-1-{(2- хлорфенил)метил}-1Нимидазол-5-ил]-2-(2-тиенил)метил-2-пропеноата в виде масла, ЯМР которого полностью совпадает с транс- или Е-формой олефина. (с) (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-пропеновая кислота. Раствор метил (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}- 1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-пропеноата (336 мг, 0,783 ммол) в этаноле (10 мЛ) обрабатывался 10%-ым раствором гидроксида натрия (4 мЛ), после чего раствор перемешивали в течение 3 часов при 25оС. рН доводилось до 5 и выделялся твердый осадок. Смесь разбавлялась водой, охлаждалась и фильтровалась, образуя 309 мг твердого вещества. Кристаллизация из этилацетата дала 195 мг (60%) (Е)-3-[2-н-бутил1-{(2-хлорфенил)метил}-1Н-имидазол-5-ил]-2-(2тиенил)метил-2-пропеновой кислоты, т.пл. 177– 179оС. Способ В. (а) Метил 3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5-ил]-3-гидрокси-2-(2-тиенил)метилпропаноат. К раствору диизопропиламина (1,96 г, 0,0194 мол) в сухом тетрагидрофуране (40 мЛ), поддерживаемому при температуре –78оС в атмосфере аргона, добавлялся н-бутиллитий (7,3 мЛ, 0,0183 мол 2,5 М раствора в толуоле) и смесь перемешивалась в течение 10 минут. Затем добавлялся метил 3-(2-тиенил)пропаноат (2,83 г, 0,0166 мол) в тетрагидрофуране (2 мЛ) и смесь перемешивалась в течение 30 минут при –78оС. Добавлялся раствор 2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-карбоксиальдегида (3 г, 0,0111 мол) в тетрагидрофуране (4 мЛ) и образующаяся смесь перемешивалась при –78оС в течение 30 минут. Реакционная смесь разделялась между насыщенным раствором хлорида аммония и эфиром, органический экстракт промывался раствором хлористого кальция, сушился над безводным сульфатом магния и концентрировался до 7 40587 образования 6,67 г неочищенного продукта. Этот продукт подвергался флеш-хроматографированию через 70 г силикагеля с использованием смеси этилацетат/гексан 4:1, образуя 4,03 г (81%) метил 3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-ил]-3-гидрокси-2-(2-тиенил)метилпропаноата. (b) Метил 3-ацетокси-3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5- ил]-2-(2-тиенил)метилпропаноат. Раствор метил 3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-ил]-3-гидрокси-2-(2-тиенил)метилпропаноата (4,03 г, 9,02 ммол) в метиленхлориде (100 мЛ) взаимодействовал с 4-диметиламинопиридином (0,386 г, 3,16 ммол). Затем к перемешиваемой смеси по каплям добавлялся уксусный ангидрид (8,5 мЛ, 9,02 ммол). Смесь перемешивалась в течение 18 часов, добавлялась вода (35 мЛ), смесь перемешивалась в течение 1 часа и затем разбавлялась эфиром и насыщенным раствором бикарбоната натрия. Эфирный слой промывался раствором хлористого кальция, сушился безводным сульфатом магния и выпаривался, давая озаглавленное 3-ацетокси производное в виде масла (4,37 г, 99%). (с) метил (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5- ил]-2-(2-тиенил)метил-2-пропеноат. Смесь метил 3-ацетокси-3-[2-н-бутил-1-(2хлорфенил)метил-1Н-имидазол-5ил]-2-(2-тиенил)метилпропаноата (4,36 г, 8,92 ммол) в сухом толуоле (80 мЛ) взаимодействовала с 1,8-диазабицикло [5.4.0] ундек-7-ен (ДБУ) (3,2 мЛ, 21,4 ммол) и образующийся раствор нагревался в течение 3 часов при 80оС в атмосфере аргона. Растворитель испаряли, остаток растирали в порошок с эфиром и добавляли активированный древесный уголь. После фильтрования фильтрат концентрировали до образования 6,29 г масла, которое подвергали хроматографированию над силикагелем с использованием смеси гексан/этилацетат 65:35, получая 2,89 г (76%) метил (Е)3-[2-н-бутил-1-[(2-хлорфенил)метил]-1Н-имидазол-5- ил]-2-(2-тиенил)метил-2пропеноата, ЯМР которого и ТСХ (50% этилацетата в гексане на силикагеле) были идентичны продукту, полученному по способу А. (d) (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}1Н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропеновая кислота. Основной гидролиз этого эфира (2,88 г, 6,71 ммол) в соответствии со способом А (iii) дал 2,59 г (93%) (Е)- 3-[2-н-бутил-1-[(2-хлорфенил)метил]-1Нимидазол-5-ил]-2-(2-тиенил)метил-2-пропеновой кислоты, т.пл. 175–177оС, что было идентичным продукту, полученному в способе А. Пример 2. (Е)-3-[2-н-бутил-1-{(4-карбоксифенил)метил}-1Н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропеновая кислота. (i) По методике примера 1 [(ii) способ 2, (iii) и (iv) способ В] с использованием 4-карбометоксибензилового спирта вместо 2-хлорбензилового спирта было получено озаглавленное соединение, т.пл. 250–253оС. (ii) Получение монометансульфоната. Озаглавленное соединение, 3600 г, добавлялось к 2-пропанолу (54 Л) в 20-галлонном (75,707 л-вый) футерованном стеклом реакторе. Перемешанная суспензия охлаждалась приблизительно до 8оС. К тщательно перемешанной суспензии быстро добавлялась метансульфокислота (2448 г). Исходное вещество быстро растворяли с получением прозрачного раствора в течение двух минут. Наблюдалась небольшая экзотермия приблизительно до 11оС. В течение следующих трех минут начал выделяться тонкий белый твердый осадок. Суспензия перемешивалась в течение 5,5 часа при температуре 3оС и твердое вещество было собрано центрифугированием. После промывания 10 Л 2-пропанола продукт высушивался в вакууме при 45оС до постоянного веса 4,21 кг (выход 94%, неподготовленный для анализа). Неочищенный продукт (4,20 кг) в твердом виде добавлялся к 12,6 Л перемешиваемой ледяной уксусной кислоте в 10-галлонном (37,85-литровом) футерованном стеклом реакторе. Пастообразная смесь нагревалась до 80оС, образуя гомогенный раствор. Раствор фильтровался в теплом виде через проходной фильтр, и реактор и линейный фильтр промывались дополнительной порцией (4,2 Л) уксусной кислоты. Объединенные растворы уксусной кислоты перемешивались при медленном охлаждении до 25оС в отдельном 10ти галлонном (37,85-литровом) футерованном стеклом реакторе. Выделение такого осадка началось приблизительно при 45оС. Через 2,5 часа суспензия разбавлялась 42 Л этилацетата, добавленным в виде двух равных порций с часовым интервалом между добавками. Для полного осаждения суспензия перемешивалась в течение дальнейших 18 часов. Твердый продукт собирался центрифугированием и промывался 10 Л этилацетата. После высушивания до постоянного веса в вакууме при 40оС выход составил 3,80 кг продукта, т.пл. 251–252оС (90,4%, неподготовленный для анализа). Пример 3. (Е)-3-[2-н-Бутил-1-{(2-хлорфенил)метил}-1Нимидазол-5- ил]-2-(4-пиридил)метил-2-пропеновая кислота. (i) Метил 3-[2-н-бутил-1-(2-хлорфенил)метил1Н-имидазол-5-ил]-3-гидрокси-2-(4-пиридил)метилпропаноат. К раствору диизопропиламина (3,58 мЛ, 25,6 ммол) в высушенном тетрагидрофуране (50 мЛ), выдержанному при –78оС в атмосфере аргона, добавлялся н-бутиллитий (10,2 мЛ, 26,6 ммол 2,5 Мго раствора в толуоле) и смесь перемешивалась в течение 10 минут. Затем добавлялся метил 3-(4пиридил)пропаноат (4,22 г, 25,6 ммол) (полученный взаимодействием 4-пиридин-карбоксиальдегида с триметилфосфоноацетатом в присутствии гидрида натрия в этиленгликольдиметиловом эфире с последующей каталитической гидрогенизацией двойной связи 10% палладием на углероде в этилацетатном растворе (98%) при давлении водорода 3 атмосферы с образованием насыщенного эфира) в тетрагидрофуране (40 мЛ) и эта смесь перемешивалась в течение 30 минут при –78оС. Добавлялся раствор 2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5- карбоксиальдегид (5,9 г, 21,3 ммол) в тетрагидрофуране (10 мЛ) и перемешивание при –78оС продолжали в течение 30 минут. Реакционная смесь разделялась между 8 40587 насыщенным раствором хлорида аммония и эфиром, органический экстракт промывался раствором хлористого кальция, сушился над сульфатом магния, концентрировался и подвергался флешхроматографированию через силикагель с использованием 5%-го метанола в этилацетате, образуя 3,32 г (30%) метил 3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-ил]-3-гидрокси-2-(4-пиридил)метилпропаноата. ТСХ на силикагеле с использованием 5% метанола в этилацетате выявляет гомогенный продукт с Rf 0,79. (ii) Метил 3-ацетокси-3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-ил]-2-(4-пиридил)пропаноат. Раствор метил 3-[2-н-бутил-1-(2-хлорфенил)метил-1Н-имидазол-5-ил]-3-гидрокси-2-(4-пиридил)метилпропаноата (3,32 г, 7,5 ммол) в метиленхлориде (50 мЛ), 4-диметиламинопиридин (150 мг, 1,3 ммол) и уксусный ангидрид (7,1 мЛ,(75 ммол) перемешивались при комнатной температуре в течение 18 часов. Добавлялась вода (5 мЛ), смесь перемешивалась в течение 2 часов и затем разбавлялась метиленхлоридом и 5% раствором бикарбоната натрия. Органическая фаза промывалась 5% раствором бикарбоната натрия и хлористого кальция, сушилась и концентрировалась, образуя 4 г неочищенного озаглавленного продукта. ТСХ на силикагеле с использованием 5% метанольного этилацетата обнаруживает в основном материал одной пробы с Rf 0,86. Исходный материал не был обнаружен. Этот материал в дальнейшем не подвергался очистке. (iii) Метил (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5-ил]-2-(4-пиридил)метил-2-пропеноат. Смесь метил 3-ацетокси-3-[2-н-бутил-1-(2хлорфенил)метил-1Н-имидазол-5-ил]-2-(4-пиридил)-пропеноата (7,5 ммол), толуола (50 мЛ) и 1,8диаза-бицикло [5,4,0]-ундек-7-ена (ДБУ) (3,4 мЛ), 22,5 ммол) нагревалась при 90оС в течение 18 часов в атмосфере аргона. Охлажденная смесь разбавлялась эфиром и промывалась раствором хлористого кальция, сушилась и концентрировалась до 3,1 г (97%) озаглавленного соединения. ЯМР показал, что основным продуктом был транс- или Е-изомер. (iv) (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}1Н-имидазол-5-ил]-2-(4-пиридил)-метил-2-пропеновая кислота. Раствор метил (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5-ил]-2-(4-пиридил)-метил2-пропеноата (3,1 г, 7,3 ммол) в этаноле (16 мЛ) взаимодействовал с 10%-ым раствором гидроксида натрия и смесь перемешивалась в течение 18 часов при 25оС. Раствор концентрировался в вакууме, добавлялась вода, рН доводили до 6,5 и образующееся твердое вещество отфильтровывали, промывали водой и кристаллизовали из смеси метанол/эфир с получением 0,48 г (Е)-3-[2-н-бутил-1-{(2-хлорфенил)метил}-1Н-имидазол-5-ил]-2-(4-пиридил)метил-2пропеновой кислоты, т. пл. 178–182оС (d). Пример 4. (Е)-3-[2-н-Бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропеновая кислота. (i) 2-н-бутил-5-гидроксиметил-4-йодоимидазол. N-йодосукцинимид (148,75 г, 0,661 мол) добавляли к перемешиваемому раствору 2-н-бутил4-гидроксиметилимидазолу (100,78 г, 0,652 мол) в 500 мЛ абсолютного этанола. Через 20 минут раствор нагревали при 40–45оС в течение 45 минут, разбавляли 2,5 литрами воды и быстро охлаждали. Кристаллический продукт собирали фильтрованием, промывали водой и сушили с получением 174,5 г (95%) кристаллического вещества, т.пл. 166–166,5оС. (ii) 2-н-Бутил-4-йодимидазол-5-карбоксиальдегид. Перемешиваемая смесь 174,1 г (0,62 мол) 2н-бутил-5- гидроксиметил-4-йодимидазола и 360 г (4,14 мол) двуокиси марганца в 3 литрах метиленхлорида кипятились в течение 24 часов с обратным холодильником с использованием ловушки для удаления воды. Горячая реакционная смесь фильтровалась через Celiteâ, который затем промывался 4,5 литрами кипящего метиленхлорида. Соединенные фильтраты концентрировались до сухого состояния, остаток растворялся дважды в 150 мЛ метанола и раствор концентрировался до сухого состояния. Остаток растворялся в 130 мЛ метанола и быстро охлаждался. Послу осуществления кристаллизации медленно добавлялось 700 мЛ воды. Смесь быстро охлаждалась, твердое вещество собиралось фильтрацией и промывалось водой, образуя 145,2 г (84%) продукта, т.пл. 104–105оС. (iii) Метил 4-[(2-н-бутил-5-формил-4-йод-1Нимидазол-1- ил)метил]нафталин-1-карбоксилат. Суспензия 29,53 г (0,214 мол) 2-н-бутил-4йодимидазол-5-карбоксиальдегида и 65,68 г (0,235 мол) метил 4-бромметилнафталин-1-карбоксилата (E.A. Dixon, A. Fischer и F.P. Robinson, Can. J. Chem., 59, 2629, (1981) в 600 мЛ диметилформамида перемешивалась в течение 5 часов в атмосфере аргона при 70оС. Дополнительно добавлялось 6,56 г (0,0235 мол) бромметилового эфира и суспензия перемешивалась еще 15 часов при 70оС. Реакционная смесь выливалась в воду и образующееся твердое вещество собиралось фильтрацией, промывалось водой и несколько раз растиралось с 250 мЛ кипящего метанола, образуя 86,8 г (85%) твердого вещества, т.пл. 177,5– 179оС. (iv) Метил 4-[(2-н-бутил-5-формил-1Н-имидазол-1- ил)метил]нафталин-1-карбоксилат. Суспензия 40,0 г (83,9 ммол) метил 4-[(2-нбутил-5-формил-4-йод- 1Н-имидазол-1-ил)метил]нафталин-1-карбоксилата, 9,07 г (92,4 ммол) ацетата калия и 6,0 г 10% палладия на угле в 1,2 литра этилацетата подвергалась гидрогенизации в течение 2 часов. Твердое вещество удалялось фильтрацией и дополнительно добавлялось 8,0 г 10% палладия на угле и 9,01 г (92,4 ммол) ацетата калия. После гидрогенизации реакционной смеси еще в течение 2 часов, твердые вещества удаляли фильтрацией, а раствор концентрировали приблизительно до 1/3 объема. Этилацетатный раствор промывали водным раствором карбоната натрия, сушили над сульфатом магния и концентрировали в вакууме с образованием масла, которое подвергали кристаллизации. Перекристаллизация из смеси метиленхлорид/гексан дала 25,77 г (87,6%) бесцветных кристаллов, т.пл. 95,5–97оС. 9 40587 (v) метил (Е)-3-[2-н-бутил-1-{(4-карбоксинафт1-ил)метил}-1Н-имидазол-5-ил]-2-(2-тиенил)метил-2пропеноат. Озаглавленное соединение было получено из 25,0 г метил 4-[(2-н-бутил-5-формил-1Н-имидазол-1-ил)метил]нафталин-1-карбоксилата последовательными процедурами примера 3 с образованием 22,12 г (56%) продукта в виде хлористоводородной соли, т.пл. 217–218оС. (vi) (Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол-5-ил]-2-(2-тиенил)метилпропеновая кислота. Паста, содержащая 14,46 г (26,14 ммол) метил(Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол- 5-ил]-2-(2-тиенил)метил-2-пропеноата, 8,38 г (2,09 ммол) гидроксида калия в смеси 165 мЛ этанола и 85 мЛ воды перемешивалась в течение 18 часов при комнатной температуре. Концентрирование в вакууме и разбавление водой дали 400 мЛ прозрачного раствора. При доведении рН до 4,03 хлористоводородной кислотой были получены кристаллы, которые при перекристаллизации из метанола образовали 9,89 г (80%) бесцветных кристаллов, т.пл. 218–219оС в виде частичного гидрата. Пример 5. (Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н-имидазол-5-ил]-2-[(2-тиенил)метил]-2-пропеновая кислота. Этиловый эфир. Раствор 5,0 г (14,27 мол) метил 4-[(2-бутил5-формил-1Н-имидазол-1-ил)метил]нафталин-1карбоксилата в 60 мл этанола взаимодействовал с раствором, содержащим 2,0 г (50 ммол) гидроксида натрия в 30 мл воды. После перемешивания в течение 18 часов при 25оС реакционная смесь концентрировалась в вакууме, разбавлялась водой до 50 мл, рН доводилось до 3,15 с помощью 12N хлористоводородной кислоты. Фильтрация быстро охлажденной смеси дала 4,71 г белых кристаллов, т.пл. 183–184оС. Перекристаллизация из этилацетата дала другую кристаллическую форму, т.пл. 134–135оС. К раствору, содержащему 27,2 г (0,119 мол) этил 2-карбокси-3- (2-тиенил)пропионата в 250 мл бензола, добавлялось 4,71 г (14 ммол) вышеприведенной альдегидо-кислоты, 3,58 г (42 ммол) пиперидина и 10 мл пиридина и раствор кипятился с обратным холодильником в течение 18 час. С использованием ловушки для удаления воды. Затем летучие вещества удалялись с использованием вакуума, добавлялся толуол, а летучие вещества вновь удалялись. Остаток обрабатывался 2,5%ым раствором бикарбоната натрия и гексаном, который вызывал отделение масла. Добавление этилацетата дало две фазы. Водная фаза отфильтровывалась, рН ее доводилось 12N хлористоводородной кислотой до 3,86 и экстрагировалась этилацетатом. Этот этилацетатный раствор сушился над сульфатом магния и концентрировался в вакууме, образуя смолу, которая растворялась в эфире и затем подкислялась эфирным раствором HCl. Растирание образующейся смолы с эфиром дало 5,32 г хорошо отделенных белых кристаллов, т.пл. 180–181,5оС, размягчающихся при 176оС (гидрохлоридная соль). Пример 6. Оральная дозированная форма для приема оральным способом активного соединения формулы (I) производится просеиванием, смешиванием и наполнением в твердые желатиновые капсулы ингредиентов в пропорциях, например, показанных ниже: Ингредиенты Количества Метансульфонат (Е)-3-[2-н-бутил-1{(4-карбоксифенил) метил}-1Н- имидазол-5-ил]-2-(2-тиенил) метил-2-пропеновой кислоты 100 мг Стеарат магния 10 мг Лактоза 100 мг Пример 7. Сахароза, дигидрат сульфата кальция и орально активные соединения формулы (I) смешивались и гранулировались с 10% желатиновым раствором. Влажные гранулы просеивались, сушились, смешивались с крахмалом, тальком и стеариновой кислотой и уплотнялись в таблетки. Ингредиенты Количества (Е)-3-[2-н-бутил-1{(4-карбоксинафт-1-ил) метил}-1Н- имидазол5-ил]-2-(2-тиенил)метил2-пропеновая кислота 75 мг Сульфат кальция дигидрат 100 мг Сахароза 15 мг Крахмал 8 мг Тальк 4 мг Стеариновая кислота 2 мг Пример 8. (Е)-3-[2-н-бутил-1-{(4-карбоксинафт-1-ил)метил}-1Н- имидазол-5-ил]-2-(2-тиенил)метил-2-пропеновая кислота, этиловый эфир, 50 мг, диспергируется в 25 мл нормального солевого физиологического раствора для получения препарата для инъекций. Пример 9. Местный офтальмологический раствор для применения соединений формулы (I) производится смешением в стерильных условиях ингредиентов в пропорциях, например, показанных ниже. Ингредиенты Количества (Е)-3-[2-н-бутил-1{(4-карбоксифенил) метил}-1Н-имидазол5-ил]-2-(2-тиенил)метил2-пропеновая кислота метансульфонат 1,0 Двухосновный фосфат натрия 10,4 Одноосновный фосфат натрия 2,4 Хлорбутанол 5,0 Гидроксипропанолметилцеллюлоза 5,0 Стерилизованная вода достаточное количество до 1,0 мЛ 1,0 N гидроксид натрия достаточное количество до рН 7,4 Понятно, что изобретение не ограничивается иллюстрированными выше осуществлениями, и 10 40587 следующие далее притязания сохраняют права как на проиллюстрированные осуществления, так и на все модификации, входящие в объем притязаний. Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03 11
ДивитисяДодаткова інформація
Назва патенту англійськоюMethanesulfonate (e)-3-[2-n-butyl-1-{(4-carboxyphenyl)methyl}-1n-imidazole-5-yl]-2-(2-thienyl)methyl-2-propionic acid, process for obtaining thereof, pharmaceutical composition based thereon
Автори англійськоюKEENAN RICHARD MCCULLOCH, WEINSTOCK JOSEPH
Назва патенту російськоюМетансульфонат (е)-3-[2-н-бутил-1{(4-карбоксифенил)метил}-1н-имидазол-5-ил]-2-(2-тиенил)метил-2-пропионовой кислоты, способ его получения, фармацевтическая композиция на его основе
Автори російськоюРичард МакКаллок Кинен, Джозеф Вайншток
МПК / Мітки
МПК: A61K 31/415, A61P 13/02, A61P 9/08, C07D 409/06, A61P 9/10, A61P 15/00, A61P 9/12
Мітки: кислоти, композиція, фармацевтична, основі, е)-3-[2-н-бутил-1{(4-карбоксифеніл)метил}-1н-імідазол-5-іл]-2-(2-тієніл)метил-2-пропенової, одержання, метансульфонат, спосіб
Код посилання
<a href="https://ua.patents.su/11-40587-metansulfonat-e-3-2-n-butil-14-karboksifenilmetil-1n-imidazol-5-il-2-2-tiehnilmetil-2-propenovo-kisloti-sposib-jjogo-oderzhannya-farmacevtichna-kompoziciya-na-jjogo-osnovi.html" target="_blank" rel="follow" title="База патентів України">Метансульфонат (е)-3-[2-н-бутил-1{(4-карбоксифеніл)метил}-1н-імідазол-5-іл]-2-(2-тієніл)метил-2-пропенової кислоти, спосіб його одержання, фармацевтична композиція на його основі</a>
Похідні 5-аміно-8-метил-7-піролідинілхінолін-3-карбонової кислоти, спосіб їх одержання (варіанти), фармацевтична композиція, спосіб лікування інфекційних захворювань, проміжна сполука

Номер патенту: 39859
Опубліковано: 16.07.2001
Автори: Іто Яшіо, Ямамото Йоічі, Кадо Норіюкі, Като Хідео, Йошіда Тошіхіко, Ясуда Сінго
МПК: C07D 215/56, C07D 401/04
Мітки: одержання, кислоти, лікування, фармацевтична, сполука, проміжна, варіанти, похідні, 5-аміно-8-метил-7-піролідинілхінолін-3-карбонової, спосіб, захворювань, інфекційних, композиція
Формула / Реферат:
1. Производные 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты общей формулы (I):, (I)гдеR1 выбирают из группы, включающей атом водорода и низший алкил,R2 выбирают из группы, включающей атом водорода, низший алкил, низший алканоил, галогенированный низший алканоил и остаток эфира карбоновой кислоты, R3 выбирают из группы,...
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Едді Жан Едгард Фрейн, Жерар Шарль Санз, Альфонс Герман Маргарета Реймакерс
МПК: A61K 31/47, A61P 43/00, A61K 31/4427, A61P 5/00, A61P 35/00, A61K 31/425, A61K 31/44, C07D 521/00, C07D 403/14, A61P 19/06, A61K 31/443, C07D 417/14, C07D 403/06, A61P 17/00, C07D 401/14, C07D 405/14, C07D 409/14, A61K 31/4433, A61K 31/415
Мітки: стереоізомерів, 1н-імідазол-1-ілметіл)замішаного, металів, спосіб, бензімідазола, похідних, солей, прийнятих, кислоти, фармацевтично, одержання
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Фармацевтична композиція, що призначена для одержання порошків або шипучих таблеток, яка містить ефективну кількість ібупрофену, фармацевтичний препарат на її основі та спосіб його одержання

Номер патенту: 27058
Опубліковано: 28.02.2000
Автори: Брю-Ман'єз Ніколь, Кордоліані Жан-Франсуа, Товєн Жерар, Друєн Жан Ів
МПК: A61K 31/19, A61K 9/46
Мітки: препарат, спосіб, яка, основі, містить, кількість, одержання, ібупрофену, фармацевтична, композиція, шипучих, призначена, ефективну, порошків, таблеток, фармацевтичний
Формула / Реферат:
1. Фармацевтическая композиция, предназначенная для получения порошков или шипучих таблеток, содержащая эффективное количество ибупрофена или одной из его фармацевтически приемлемых солей в качестве активного ингредиента, фармацевтически приемлемую систему газирования, содержащую, по крайней мере, один щелочной карбонат и, по крайней мере, одну органическую кислоту, отличающаяся тем, что она дополнительно содержит, по крайней мере, один...
Ефіри 2,6-диметил-1,4-дигідропіридин-3-карбонової кислоти, що є блокаторами кальцієвих каналів, спосіб їх одержання та фармацевтична композиція
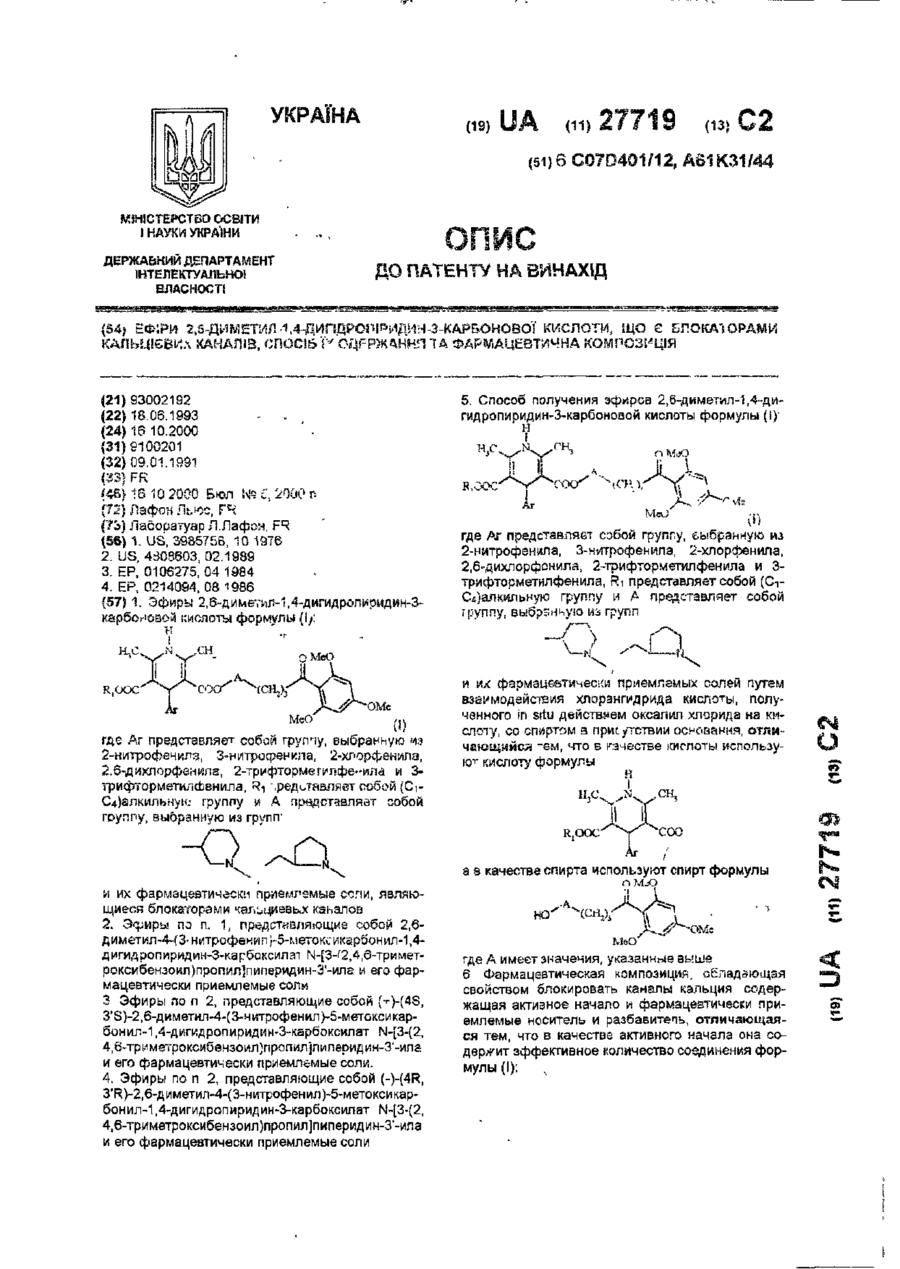
Номер патенту: 27719
Опубліковано: 16.10.2000
Автор: Лафон Льюс
МПК: A61P 9/12, A61K 31/4433, A61K 31/455, A61P 9/08, C07D 211/90, C07D 409/14, C07D 401/12, A61K 31/451, A61K 31/4427, A61K 31/435, A61K 31/445
Мітки: 2,6-диметил-1,4-дигідропіридин-3-карбонової, одержання, фармацевтична, кальцієвих, спосіб, ефіри, композиція, блокаторами, кислоти, каналів
Текст:
..."COOH в которой Аг и Ri имеют указанное выше значение, а в качестве спирта используют спир г формулы ОМеО Mel) где А имеет значение, указанное выше Аддитивные соли получают по классическим методикам реакцией соединения формулы (I) с фармацевтически приемлемой кислотой в подходящем растворителе И, напротив, основания могут быть получаны и; зддитизных солей обработкой их сильным основанием Кисго^ы формулы (ПІ моп.'т быть получше! классическим...
Похідне хіназоліну, спосіб його одержання, фармацевтична композиція на його основі

Номер патенту: 34426
Опубліковано: 15.03.2001
Автор: Баркер Ендрю Джон
МПК: C07D 239/94, C07D 491/04, C07D 491/056, C07D 403/04
Мітки: спосіб, основі, одержання, хіназоліну, фармацевтична, похідне, композиція
Текст:
...суль финильную груп пу,также можно использовать более слабый окислитель, например, метапериодат натрия или калия, обычно в полярном растворителе, таком как уксусная кислота или этанол. При необходимости получения соединения формулы I, содержащего /1-4С/алкилсульфонильную группу, его можно получить путем окисления соответствующего /1-4С/алкилсульфи нильного соединения, а также соответствующего /14С/алкилтиосоединения. (d) Для получе ния...
Попередній патент: Клиновий патрон
Наступний патент: Спосіб добування сірки підземним виплавленням із слабопроникних покладів
Випадковий патент: Спосіб вдосконалення визначення загального вмісту пігментів у баранині фотометричним методом