Похідні 1,4,10,13-тетраокса-7,16-діазациклооктадекану, динатрієва сіль n,n1 -біс(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-діазациклоокта-декан-кальцієвого комплексу та композиції на їх основі для виведення з ж
Номер патенту: 35547
Опубліковано: 16.04.2001
Автори: ЕМРІ Йожеф, КАНЯР Бейла, БРЮХЕР Ерньо, ДЕРІ Бейла, ВАРГА Ласло, КОВАЧ Золтан, КІРАЙ Йодьон, СТАНІК Ласло










Формула / Реферат
Спосіб самокорекції набутої патології міжхребцевих дисків, а також психофізичної реабілітації людини під час лікування і в післялікувальний період, здійнюваний шляхом виконання циклів вправ лікувальної фізкультури, що включають способи самомануальної терапії — самонадавлюють протирухом рук за допомогою кулаків або зчеплених кистей рук на хребет ззаду або самообертають тулуб в грудному відділі хребта, захоплюючи кистей руки ділянку або частину тіла і розгинають при цьому хребет спереду назад, вліво і вправо до максимально можливої межі, а також виконують нахили голови або тулуба вбік а також обертання їх навколо вертикальної осі, який відрізняється тим, що вправи групуються в цикли по дії на ділянки і частини тіла людини, склад вправ підбирають індивідуально під час сеансів мануальної терапії у відповідності зі станом опорно-рухового апарата, і змінюють цей склад відповідно до ступеня відновлення рухового стереотипу в післялікувальний період для його закріплення і підтримання, до того ж вправи погоджують з диханням, здійснюючи вдих при максимальному навантаженні, причому для обличчя, голови і шиї здійснюють самообертання голови при повертанні її навколо вертикальної осі в бік одного плеча, надавлюючи нижньою частиною долоні руки, відповідної цьому плечу, на протилежний край підборіддя, а другою рукою, охоплюючи потилицю, надавлюють на голову в ділянці вуха також в напрямку її плеча, для ділянок шиї здійснюють самофіксацію хребта, натискуючи на куприк кистями рук,захоплених за зап'ястя, при нахилах голови вбік, попередньо розправивши хребет, а також при крутінні головою навколо вертикальної осі в положенні розігнутого шийного відділу, здійснюють самонадавлювання протирухом рук або голови при нахилах голови вперед і вбік на підборіддя знизу за допомогою великих пальців кистей рук, стиснутих в кулаки, також при нахилах голови вперед до упирання підборіддям в груди на потилицю за допомогою рук із перехрещеними пальцями і попередньо зсунутими разом ліктями, також на м'язи над ключицями в середній їх частині за допомогою пучок великих пальців при згинанні шийного відділу хребта, а при його розгинанні чотирма пальцями кистей здійснюють в горизонтальному положенні обличчям вниз розгинання шийного відділу хребта спереду назад і в боки поперемінне з упором голови на підборіддя, для ділянок пояса верхніх кінцівок здійснюють розгинання в попере коболу і грудному відділах хребта вбік, самонадавлюючи плечем однієї руки на тулуб збоку при перетягуванні її другою рукою з захопленням за зап'ястя поперек хребта на рівні поперекової, а потім лопаткової ділянок спини, фіксуючи останнє положення з упором спини об стіну, здійснюють самообертання навколо вертикальної осі, перетягуючи на рівні грудей рукою, відповідного напрямку обертання, зчеплену з нею за допомогою кистей другу руку при обертанні тулуба вліво або вправо і одночасно виконують протирух головою вправо або вліво відповідно, здійснюють розгинання грудного відділу згинаючим моментом у фронтальній площині, для чого виконують різкий зустрічний рух кистей рук з витягнутими пальцями, причому одною на рівні потилочної ділянки, а другою на рівні поперекової ділянки спини, попередньо випрямивши хребет, повільно піднімаючи руки через боки вверх, здійснюють розгинання грудного відділу, для чого різко піднімають руки вгору-вбік, повертаючи долоні вгору із положення рук на рівні грудей і одночасно різко згинають шийний відділ вперед, нахиляючи голову до упора підборіддям в груди, здійснюють самофіксацію шийного відділу хребта взаємним протинадавлюванням кистей рук,стиснутих в кулаки на потилиці і голові,! різко піднімають вгору руки з витягнутими пальцями,фіксуючи верхній грудний відділ хребта, і одночасно різко згинають шийний відділ вперед, нахиляючи голову до упора підборіддям в груди, причому попередньо піднімають вгору руки з кистями, стиснутими в кулаки, у яких великі пальці, охоплюючи збоку, надавлюють на середні фаланги вказівного і середнього пальців, для ділянок верхніх кінцівок здійснюють самонадавлювання великими пальцями, стискуючи в кулаки кисті витягнутих вперед рук з фіксацією в цьому положенні і періодично різко розсувають пальці в боки також з фіксацією або між пальцями кистів у їх основі по черзі, або їх медіальними краями на дистальні суглоби середнього і безіменного пальців рук при обертанні кистей то в один, то в інший бік, для ділянок нижніх кінцівок, живота здійснюють розгинання хребта в бік однієї тазової кістки і одночасно надавлюють пучкою великого пальця синхронно нахилу на точки, розташовані по зовнішньому контуру клубового гребня цієї тазової кістки від точки у фронтальній площині до куприка, саморозгинаючи хребет, здійснюють самообертання кульшового суглоба, для чого роблять крок вперед однією ногою, а підняте коліно другої ноги захоплюють двома руками і притискають його до грудей, здійснюють саморозгинання поперекового відділу хребта в положенні лежачи на спині, для чого стискають кисті рук в кулаки і підкладають їх під крижі, притискують великі пальці один до одного, витягують ноги/складені ралом, а потім згинають поперековий відділ, для чого піднімають ноги вгору в напрямку до голови до повного відриву їх від рук, стиснутих в кулаки, здійснюють самообертання колінного і кульшового суглобів, для чого піднімають коліно однієї ноги вгору, притискають передню площину стегна до живота, охоплюють кистею руки, відповідної томуж бону тіла, передню ділянку коліна, а кистею другої руки-підошвену ділянку ступні, обертають гомілку у фронтальній площині, надавлюючи на коліно вниз, а на ступню вгору, здійснюють обертання в кульшовому суглобі в положенні лежачи на спині, для чого піднімають ноги по черзі вбік-вгору, до того ж їх попередньо витягують, здійснюють обертання кульшового суглоба, для чого самообертають хребет в грудному відділі, відтягуючи в бік обертання тулуба відповідною рукою зчіплену з нею другу руку і одночасно в тому ж напрямі переміщують центр ваги тіла, приставляючи до опорної ноги другу ногу, якою потім роблять різкий рух вбік-вгору, для ділянок нижніх кінцівок здійснюють самонадавлювання, різко встаючи зі стільця або на пальці ніг, для чого попередньо надавлюють ними на підлогу, напружуючи м'язи ніг, або на ступні, для чого попередньо випрямляють хребет і спираються на коліна руками, здійснюють вібрацію хребта, для чого в положенні стоячи на пальцях ніг, попередньо розслабивши м'язовий корсет, випрямляють хребет, напружуючи м'язи ніг і фіксують це положення, а потім різко падають на п'яти, здійснюють розгинання хребта в поперековому і нижнєгрудному відділах в положенні лежачи на животі, для чого різко згинають ноги в колінах і наносять удар по сідничній ділянці п'ятами, причому попередньо фіксують шийний і верхнєгрудний відділи, для чого упирають підборіддя в підлогу, а руки витягують вперед, для всіх ділянок і частин тіла здійснюють згинання хребта вперед, для чого ковзають долонями в напрямку переміщення із положення лежачи на животі в положення сидячи на розставлених в боки колінах, торкаючись лобом підлоги, причому попередньо розгинають хребет спереду назад, випрямляючи руки з упором долонями на рівні грудей, потім випрямляють хребет, для чого ковзають в зворотному напрямку, попередньо піднявши голову і пальці рук вгору, падаючи на живіт з витягнутими вперед руками, здійснюють самонадавлювання на плечовий суглоб опорної руки трьома пальцями другої руки в положенні стоячи боком на віддалі кроку від стінки і одночасно виконують коливні рухи тулубом до стінки до упора передпліччям, періодично переміщуючи кисть опорної руки вгору на розмір долоні із положення на рівні плеча до повністю випрямленої вгору руки, здійснюють самонадавлювання на колінні суглоби, для чого згинають хребет вперед і повільно переміщують долоні рук по ногах вниз, потім вгору до колін, різко повторюють два рази, випрямляють хребет і повільно присідають до кута 90° в колінному суглобі, не відриваючи ступні ніг і витягнувши вперед руки, здійснюють самообертання тулуба, для чого випрямляють хребет, витягуючи руки вгору, обертають тулуб навколо вертикальної осі хребта то в один, то в інший бік і одночасно виконують в шийному відділі повороти в тому ж напрямку, здійснюють самозгинання хребта, для чого глибоко присідають, не відриваючи ступні ніг, з витягнутими вперед випрямленими руками, охоплюють обома руками коліна, притискають їх до грудної ділянки і фіксують це положення.
2. Спосіб по п. 1, який відрізняється тим, що вправи в наборі містять елементи відпочинку.
3. Спосіб по п. 1, який відрізняється тим, що вправи в наборі містять підготовчі елементи.
4. Спосіб по п. 1, 2, 3, який відрізняється тим, що залежно від ступеня відновлення рухового стереотипе в набір включають вправи в повному або неповному обсязі.
Текст
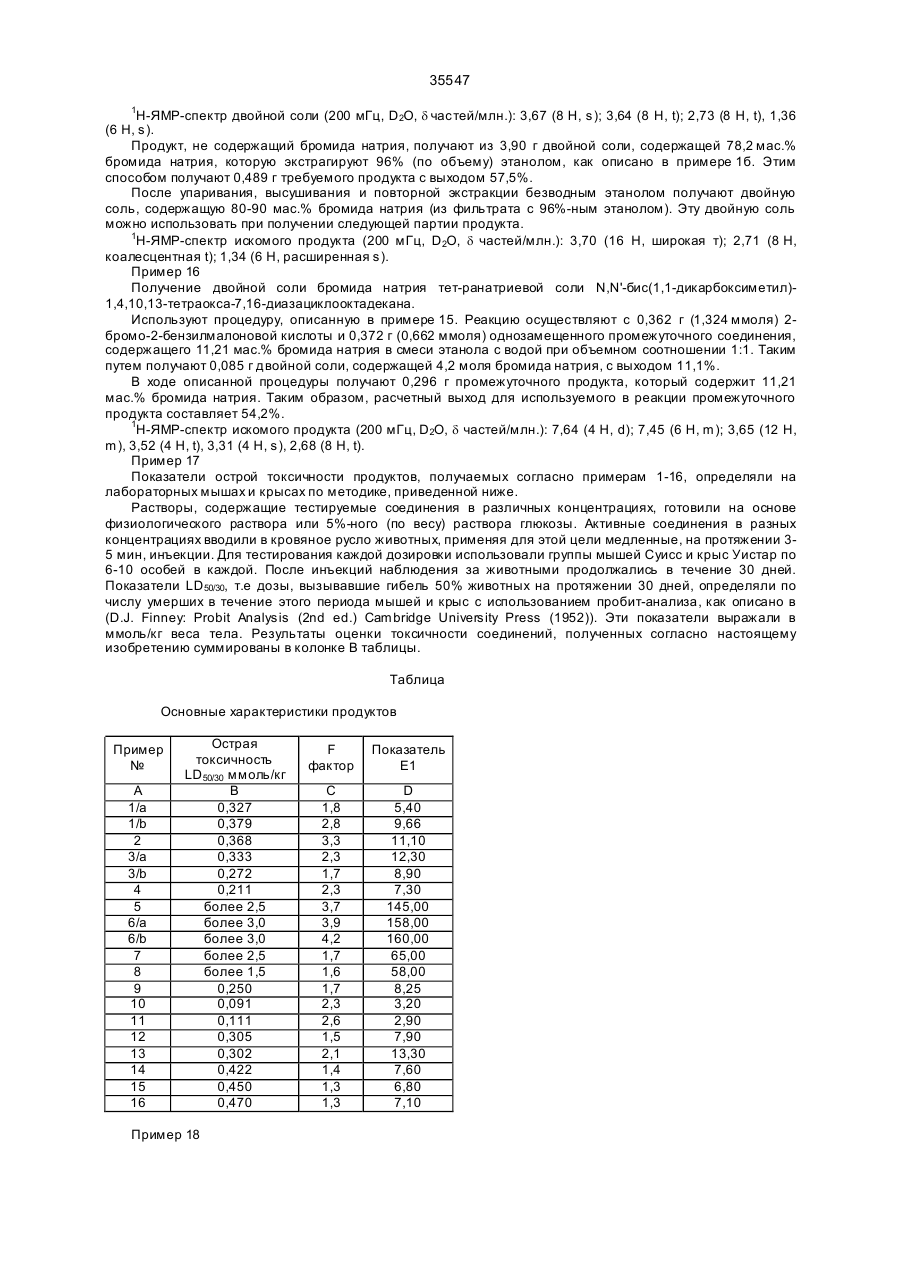
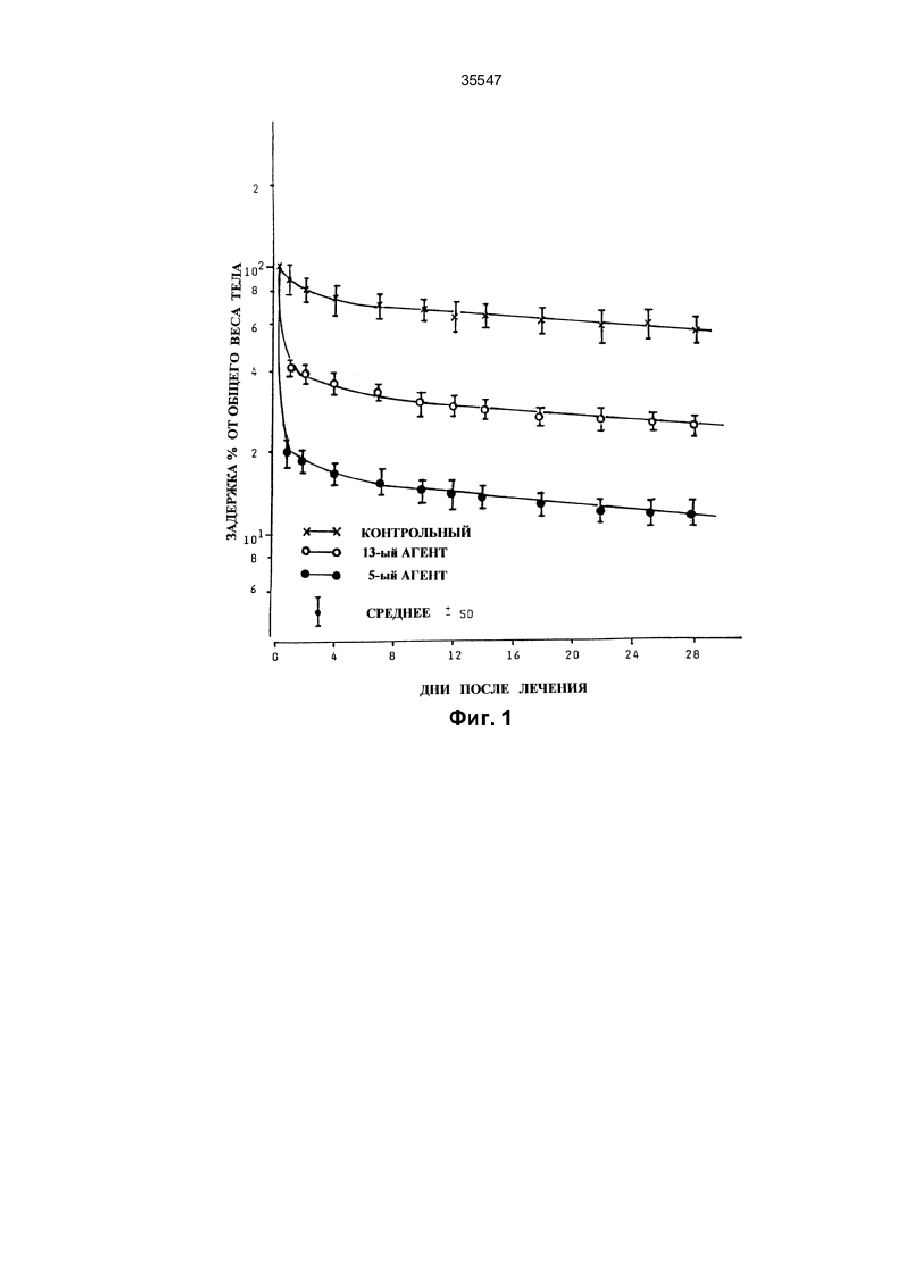
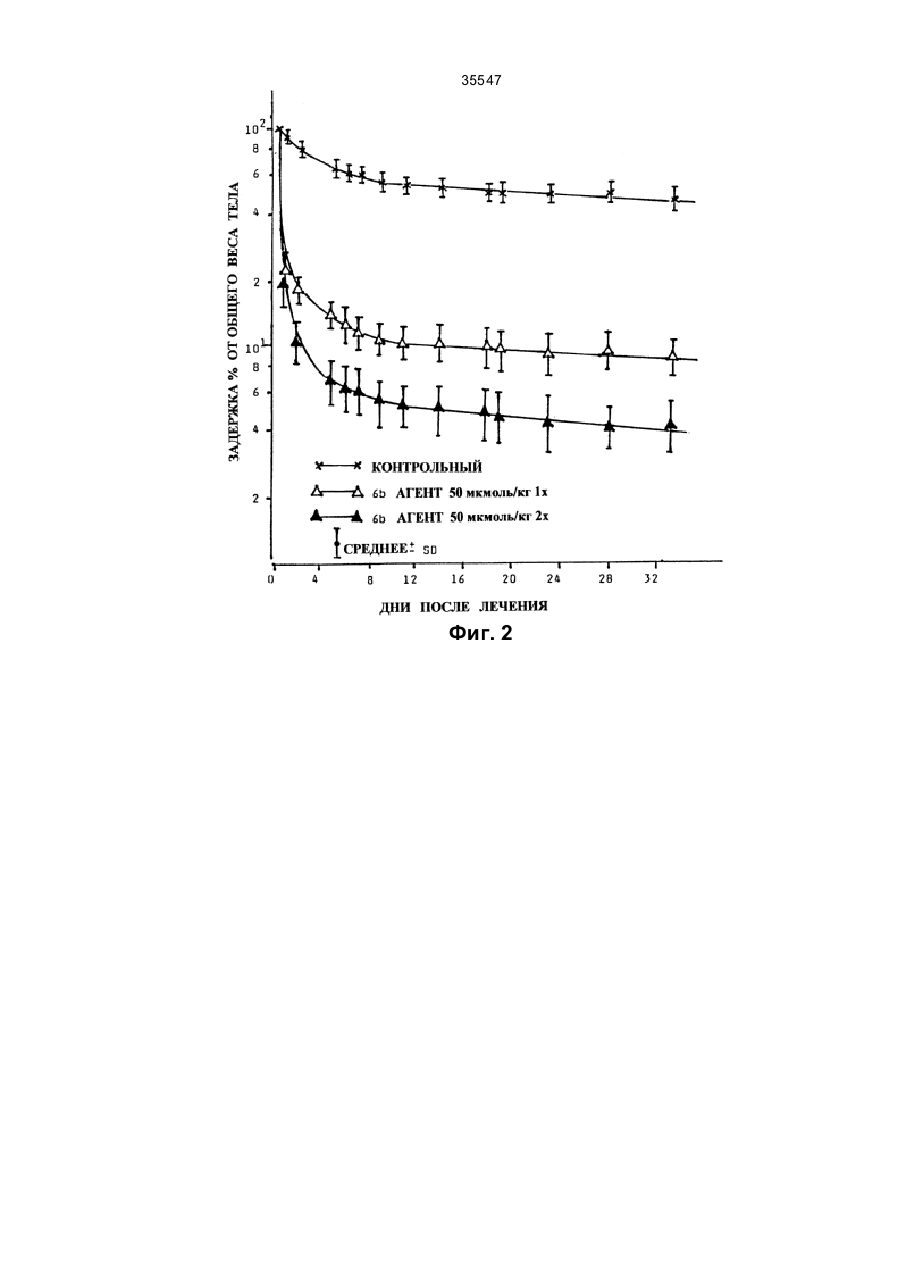
35547 Изобретение относится к области химии, в частности - к гетероциклическим соединениям, содержащим ядра только с атомами азота и кислорода в качестве гетероатомов, и может быть использовано в медицине в составе лекарственных средств для выведения из живого организма поражающих его ионов металлов, особенно радиоактивных изотопов. Известно, что при ядерных взрывах или при авариях на ядерных реакторах в а тмосферу поступает чрезвычайно опасные радиоактивные изотопы, такие как йод-131 (131І), стронций-89 и –90 (89Sr и 90Sr), а 134 137 141 144 также цезий-134 и –137 ( Cs и Cs) и церий-141 и –144 ( Ce и Се) (см.: Nuclear and Radiochemistry. John Wiley and Sons, p. 158-166, 1981). При попадании этих изотопов в легкие в процессе дыхания или в пищеварительный тракт с загрязненными пищей и жидкостями, а также в кровеносную и лимфатическую систему, в результате ресорбции через кожные покровы они откладываются и накапливаются в тканях, что, в конечном счете, приводит к тяжелым последствиям для здоровья (см.: Summery Report on the Post Acci-dent Review Meetinq on the Chernobyl Accident. - Safety Series No. 75, ІАЕА, Vienna, 1986). В случае радиоактивною поражения стронций уже через несколько часов обнаруживается в костной ткани, и его удаление из организма не представляется возможным, что чрезвычайно затрудняет защиту организма. Единственная возможность такой защиты состоит в предотвращении фиксации стронция в ткани, прежде всего костной, посредством введения в организм соответствующи х соединений, образующи х специфические комплексы со стронцием. Таким путем осуществляется связывание изотопа, присутствующего в крови или во внеклеточном пространстве в стабильной форме, и выведение его из организма. Решение этой проблемы усложняется тем, что известные комплексообразующие соединения, например, этилендиаминчетырехуксусная кислота или диэтилентриаминпентауксусная кислота, образуют с кальцием значительно более стабильные комплексы, чем со стронцием (см.: A. Catsch. Radioactive Metal Mobilization in Medicine. - Ed. Charles C. Thomas, Springfield, Illinois, 1964; A. Catsch. Dekorporierung radioaktiver und stabiler Metallionen. - Therapeutische Grundlagen, Ed. Thiemig, Munich, 1968; A. Catsch. Renoval of Tran-suranium Element by Chelating Agent // Diagnosis and Treament of Incorporated Radionuclides, IAEA Publication No. SII/PUB/411, IAEA, Vienna, p. 295, 1976). Новые перспективы для исследования в этом направлении открылись после синтеза кронэфи-ров и криптандовых молекул. В этом случае механизм комплексообразования отличается от известного ранее, так как благодаря структуре новых комплексирующи хся молекул ионы металлов локализуются в ячейках строго определенного размера, в связи с чем стабильность комплексов зависит прежде всего от размеров данного типа ионов. Первые обнадеживающие результаты были получены при исследованиях 4,7,13,16,21,24-гексаокса1,10-диазабицикло(8,8,8)-гексакозана, образующего со стронцием комплекс, показатель стабильности которого на несколько порядков выше, чем у комплекса его с кальцием (см.: Coordination Chemistry of Macrocyclic Compound. Ed. G. A. Melson, Plenum Press, 1979). Однако при изучении этого соединения в экспериментах на животных удалось доказать лишь то, что образующийся вне организма комплекс с лигандой не подвергался диссоциации после его введения в организм. В то же время не были получены данные о возможности выведения радиоактивного стронция из организма в форме стабильного комплекса с лигандой. Кроме того, ли-ганда оказалась очень токсичной (см.: W.H. Muller. Naturwiss, 57, 248 (1970); W.H. Muller and W.A. Muller. Naturwiss, 61, 455, 1974; W.H. Muller et al. Naturwiss, 64, 96, 1977; J. Knajfl el al. 12th Ann. Meeting of ESRB, Budapest, 1976; J. Batsch et al. Nukleonika 23, 305, 1978). F. de Jong et аl. предложили способ получения N,N1-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7, 16диазоциклооктадекана в форме четырехли-тиевой соли (см.: Rec. Trav. Chim. Pays-Bas, 102, 164, 1983). В соответствии с этой методикой, требуемый криптанд реагирует с метил-2-бро-момалонатом, и полученное эфирное производное подвергается гидролизу до образования литиевой соли с выходом не более 15%. Известно использование в нефтеперерабатывающей промышленности литиевой соли в форме композиции, повышающей растворимость сульфата бария (см.: Патент Великобритании № 2024822, 1980). Там же указана четырехнатриевая соль, хотя ее применение не описано в специальных примерах. Изобретение не дает никаких способов получения производных бис(дикарбоксилметила) или их солей и комплексов, которые имеют отличие среди соединений подобного типа за счет их способности отделять ионы металла, опасные для живого организма. Наиболее близким к изобретению является описанная в заявке Венгрии № 2614/89 двойная четырехнатриевая соль с бромидом натрия, а также использование ее в терапии. Однако пример 1 в указанной заявке не удобен для получения чистой тетранатриевой соли Na 4DMK,здесь можно получить только двойную соль Na4DMK·2,5NaBr, где NaBr составляет 30-60%. Эти два продукта качественно отличаются, поскольку Na4DMK легко растворяется в воде и в 70-процентном спирте. В 90-процентном спирте она растворяется только в нагретом состоянии, а в 97-процентном спирте не растворяется вообще. В противоположность этому, Na4DMK·2,5NaBr растворяется в чистом спирте, при разбавлении водой ее растворимость снижается, а в 96-про-центном спирте она практически не растворяется, что делает возможным получение чистого NaBr без тетранатриевой соли, и при этом комплексная двойная натриевая + кальциевая соль (Na2DMКCa) биологично более активна. Данная соль не позволяет эффективно вывести из живого организма радиоактивные изотопы или предотвратить накопление вредных для него ионов металлов. 35547 В основу изобретения поставлена задача получения соединений, образующих комплексы и способных отделять вредные для организма ионы металлов, а также получения композиций на основе этих соединений для выведения из живых организмов поражающих их ионов металлов, особенно радиоактивных изотопов, таких как стронций или цезий, при их внутреннем или местном, накожном и ректальном введении. Поставленная задача решается получением производных 1,4,10,13-тетраокса-7,16-диазациклооктадекана формулы (I) (І) где Q1 и Q2 означает водород или группу формулы (III) COO C R (III) COO при условии, что, как минимум, один из них не водород и R означает водород, С1-5-алкильную гр уппу с прямой или разветвленной цепью, С2-5-алкинильную групп у с прямой или разветвленной цепью, фенильную или фенил-С 2-5-алкильную группу или бензил. Из числа заместителей в соединениях формулы (I) R в качестве С 1-5-алкильной группы может быть представлен, например, метильной, этиль-ной, н-пропильной, изопропильной, н-бутильной, сексбутильной, трет-бутильной, н-пентильной или изопентильной группой; Me означает щелочной металл, кроме лития, или щелочноземельный металл, кроме бария, или ион переходного металла. В качестве переходного металла Me может быть представлен ионом металла, принадлежащего к 3-й, 4-й или 5-й группе, предпочтительно ионом двухвалентного железа или цинка. q - это 0 или 1; M и N - независимо друг от др уга означают водород или щелочной металл, или щелочноземельный металл, или, при необходимости, замещенный ион аммония. Замещенный ион аммония содержит 1, 2, 3 или 4 указанных алкильных, фенильных или фенилалкильных группы. m, n и р - эквиваленты зарядов соответственно N, М или Me. s, r и q независимо друг от друга равны 0, 1, 2, 3 при условии, что p·q+n·r+m·s=4". Поставленная задача решается также получением динатриевой соли N,N1-бис(дикарбоксиме-тил)1,4,10,13-тетраокса-7,16-диазациклооктаде-кан-кальциевого комплекса. Поставленная задача решается и получением композиции, усиливающей выведение из живого организма поражающих его ионов металлов, особенно радиоактивных изотопов, содержащей активный ингредиент и фармацевтически приемлемые разбавители или носители, которая, согласно изобретению, в качестве активного ингредиента содержит эффективное количество не менее одного соединения формулы (I) (І) где Q1 и Q2 означает водород или группу формулы (III) COO C R (III) COO при условии, что как минимум один из них не водород и R означает водород, C1-5-алкильную группу с прямой или разветвленной цепью, С2-5-алкинильную групп у с прямой или разветвленной цепью, фенильную или фенил-С 2-5-алкильную группу или бензил; Me означает щелочной металл, кроме лития, или щелочноземельный металл, кроме бария, или ион переходного металла; q - это 0 или 1; М и N независимо друг от др уга означают водород или щелочной металл, или щелочноземельный металл, или, 35547 при необходимости, замещенный ион аммония; m, n и р - эквиваленты зарядов соответственно N, М или Me; s, r и q независимо друг от др уга равны 0, 1, 2, 3 при условии, что p·q+n·r+m·s=4''. Композицию выполняют в виде таблеток, драже, капсул, свечей, растворов для инъекций, порошка или жидкого аэрозоля либо накожного пластыря. В другом варианте исполнения композиция, усиливающая выведение из организма поражающих его ионов металлов, особенно радиоактивных изотопов, содержащая активный ингредиент и фармацевтически приемлемые разбавители или носители, согласно изобретению, в качестве активного ингредиента содержит динатриевую соль N,N1-бис (дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диазациклооктадеканкальциевого комплекса. Соединения формулы (І) обладают специфическими комплексообразующими свойствами, которые позволяют им связывать и выводить ионы металлов, опасные для организма, особенно радиоактивный стронций и цезий, проникающие в живой организм и поступающие в кровяное русло и/или во внеклеточное пространство. При введении человеку или животным фармацевтических композиций, содержащих в качестве активных ингредиентов соединение формулы (I), удается предотвратить накопление в тканях радиоактивного стронция, что, в свою очередь, позволяет избежать или уменьшить тяжелое вредоносное действие радиоактивной нагрузки на состояние здо-ровья. Растворимые в воде соли и комплексы формулы (I) можно получить по аналогии с реакцией, предполагающей взаимодействие соответствующей галогенизированной дикарбоксильной кислоты формулы (II) COOH X C R (ІІ) COOH где R определен выше, а Х - галоген, предпочтительно бром, или реактивное производное (допускается использование эфира) с 1,4,10,13-тетраок-са-7,16-диазациклооктадеканом в среде органиче-ского растворителя с последующим гидролизом полученного продукта сильным основанием, например, гидроокисью натрия или смесью в требуемом молярном соотношении сильного основания с гидроокисью или солью, предпочтительно галидом комплексообразующего металла. По другому способу растворимые в воде соли и комплексы формулы (I) могут быть синтезированы посредством реакции между с 1,4,10,13-тетра-окса-7,16-диазациклооктадеканом и 2-галодикар-боксильной кислотой формулы (II), предпоч-тительно 2-бромомалоновой кислотой, в водной среде с рН от 6 до 13 в присутствии гидроокиси щелочного или щелочноземельного металла со-ответствующей соли, которую требуется получить. Соединения формулы (І), в которой q=1, можно получить посредством реакции соли щелочного металла формулы (I), в которой q=0, a M и N - соли щелочного металла, предпочтительно четырехнатриевой соли, с эквивалентным количеством комплексообразующего галида, допустимо хлорида металла. Действие соединений формулы (I), которое проявляется в усилении выведения ионов металлов, поражающих живой организм, изучали в условиях загрязнения мышей линии Суисс и крыс линии Уистер обоего пола ионами радиоактивных стронция или бария. На фиг. 1, 2, 3 изображены кривые задержки радиоактивности в зависимости от количества дней после введения препарата. На фиг. 1: 1 – задержка радиоактивности, в %, 2 – дни после введения, 3 – контроль, 4 – 13-й препарат, 5 – 5-й препарат, 6 – среднее ±S.Д. На фиг. 2: 1 – общая задержка радиоактивности, в %, 2 – дни после введения, 3 – контроль, 4 – 6-й препарат, 50 мкмоль/кг (однократно), 5 – 6-й препарат, 50 мкмоль/кг (двукратно), 6 – среднее ±S.Д. На фиг. 3: 1 – общая задержка радиоактивности, в %, 2 – дни после введения, 3 – контроль, 4 – 6-й препарат (б), 50 мкмоль/кг (однократно), 5 – 6-й препарат (б), 50 мкмоль/кг (двукратно). Выведение радиоизотопов изучали после их введения разными способами в различные ткани и участки тела, в частности в кровяное русло, брюшную полость, легкие, мышцы или подкожную соединительную ткань подопытных животных. Соединение, способствующее выведению радиоактивных изотопов, вводили ежедневно один или два раза в день в виде порошка, жидкого аэрозоля или пластыря. После этого определяли общую радиоактивность организма и получали кривые задержки, которые затем сравнивали с аналогичными параметрами, полученными на контрольных животных. Кривые задержки анализировали с помощью компьютера, используя для этой цели программу, известную как "Nonlinear Regression by the Code of BMDP-3R" BMDP Statistical Software Manual, UCLA, Los Angeles, 1990, Chief Ed. W. J. Dixon. На основании полученных результатов можно сделать вывод, что кривые описываются двухкомнонентной нисходящей экспоненциальной функцией. Для расчета эффективности использовали два показателя. Одним из них служил так называемый Е фактор, характеризующий усиление выведения радиоактивности в сопоставлении с наблюдавшимся у контрольных животных, который отражает увеличение выведения радиоизотопа под воздействием тестируемого соединения по сравнению с его интенсивностью у животных, не получавши х этого соединения (колонка С в таблице). Для более наглядной сравнительной характеристики соединений, являющихся предметом настоящего изобретения, приведены так называемые показатели Е1, которые получали посредством перемножения интенсивности выведения радиоизотопа в процентах к наблюдавшемуся в контрольной группе 35547 (эффективность Е) показателя острой токсичности (ЛД50/30, безвредный, 1), приведенного в колонке D таблицы. Хотя показатель Е1 в цифровом выражении и не идентичен показателю терапевтической безопасности соединения, он во всех случаях характеризует слабую или высокую активность тестируемого продукта. Важным экспериментальным результатом считают отсутствие определяемого с помощью данного метода радиоактивного стронция в костях животных, которым вводили активное соединение, согласно настоящему изобретению. В мягких тканях и печени животных остаточная радиоактивность составляла 510% ее общей задержки, тогда как у контрольных животных основная часть задержки была обусловлена накоплением радиоактивности в костях (65-70%). Сходные результаты были получены при различных путях введения тестируемых соединений в организм животных. Показано, что очень высокой эффективностью обладали соединения общей формулы (I). Из числа этих соединений особенно хорошими свойствами обладал N 1-бис(дикарбоксиметил)-1,4,10,13-тетраокса7,16-диазациклооктадекан-кальциевый комплекс. Ценность высокоэффективных соединений формулы (I) обусловлена также их отличными показателями терапевтической безопасности. Особенно предпочтительна динатриевая соль кальциевого комплекса благодаря ее низкой токсичности и выраженной способности усиливать выведение радиоактивных изотопов из живого организма. Соединения формулы (І) можно использовать в составе лекарственных средств в комбинации с обычными носителями и другими хорошо известными вспомогательными материалами. Подходящие для этой цели носители и другие добавки описаны в многочисленных руководства х по данному вопросу. Изучение эффективности соединений, описанных в примерах 1-16, показало, что после их введения происходит абсорбция активного ингредиента, который затем обеспечивает выведение радиоактивных изотопов. Введение осуществляют в составе инъекцируемых растворов или в виде таблетки, помещаемой под язык, драже, капсул, таблеток для приема во внутрь, порошков, жидких аэрозолей и накожных повязок. Эффективные дозы составляют от 1,0 до 200 мкмоль веса тела, предпочтительно 10-100 мкг/кг. Введение производится в виде одной или нескольких частей данной общей дозировки, желательно в виде двух субдозировок. Лекарственные средства, содержащие в качестве активного ингредиента соединения формулы (I), пригодны также для предотвращения накопления в живом организме вредных для него ионов металлов. Более подробно изобретение иллюстрируется следующими, не ограничивающими его, примерами. Пример 1 Получение тетранатриевой соли N,N1-бис (дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадена а) 2,74 г (14,98 ммоля) 2-бромомалоновой кислоты нейтрализуют в 1,0 мл воды, добавляя в нее раствор гидроокиси натрия в концентрации 7,410 моль/л в присутствии индикатора фенолфталеина. Затем в полученную смесь добавляют 1,75 г (3,69 ммоля) N-дикарбоксиметил-1,4,10,13-тетраокса-7,16диазациклооктадекана (двунатрие-вой соли), содержащего 14,10 мас.% бромида натрия (промежуточного), полученного в предыдущей реакции, и 1,95 г (7,43 ммоля) 1,4,10,13-тет-раокса-7, 16диазациклооктадекана. Реакционную смесь оставляют при 60°С на 10-11 часов, добавляя в нее раствор гидроокиси натрия в концентрации 7,410 моля/л (14,98 ммоля) порциями по 0,1 мл общим количеством 2,02 мл. После завершения реакции смесь при необходимости отфильтровывают, добавляют бромид натрия, упаривают, высушивают при пониженном давлении и экстрагируют несколькими порциями метиленхло-рида, общий объем которого составляет 20-25 мл. Экстракт упаривают до сухого остатка, добавляют к последнему 10-15 мл петролейного эфира, а полученный после фильтрации осадок высушивают в токе азота. 2,27 г (4,82 ммоля) полученного таким образом продукта, содержащего 13,3 мас.% бромида натрия (промежуточного) используют при изготовлении следующей партии искомого про- дукта. Идентификационные показатели промежуточ-но: 1Н-ЯМР-спектр (200 мГц, D2О, d частей/млн.): 3,87 (1 Н, s), 3,67 (18 Н, m), 2,78-2,92 (8 Н, m). Остаток, образующийся после экстракции ме-тиленхлоридом, экстрагируют 60 мл безводного этанола до тех пор, пока в экстракте практически не останется твердого материала. Остаток после конечной экстракции разводят в 6-7 мл воды, и после добавления бромида натрия упаривают и высушивают до сухого остатка. Последний экстрагируют 30 мл безводного этанола, как описано выше. Оба этанольных экстракта объединяют и упаривают до сухого остатка, который содержит 4,89 г двойной соли, в которой имеется 2,71 моля бромида натрия. Выход для используемого мак-роцикла составляет 94,1%. Идентификационные показатели для двойной соли: ИК спектр (КВr, см -1): 2950, 2868 (m, g СН), 1605 (vs, g COO/as), 1430 (m, g СОО/as). Другие характерные, но не идентифицированные частоты: 1350 (s), 1320 (s), 1095 (s), 928 (w). 1 Н-ЯМР-спектр (200 мГц, D2O, d частей/млн.): 4,00 (2 Н, s); 3,70 (8 Н, s); 3,63 (8 Н, t); 2,92 (8 Н, t). б) Бромид натрия удаляют из двойной соли посредством экстракции 50 мл 95 мас.%-ного эта-нола. Образующийся остаток высушивают и освобождают от этанола упаривавшем при пониженном давлении. Получают 3,22 г конечного продукта. Выход для данного макроцикла составляет 93,2%. Идентификационные показатели для последнего: 1 Н-ЯМР-спектр (200 мГц, D2О, d частей/млн.): 3,95 (2 Н, s); 3,64 (8 Н, s); 3,60 (8 H, t); 2,85 (8 H, t). 35547 13 С-ЯМР-спектр (50 мГц, D20, d частей/млн.): 179,95 (C=0); 76,45 (N-CH-(COO)2); 71,66 и 70,84 (O-CH2); 54,06 (N-CH2). Пример 2 Получение тетранатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадена, содержащей двунатриевый оксималомат В соответствии с процедурой, описанной в примере 1, 2,84 г (15,54 ммоля) 2-бромома-лоновой кислоты, 2,02 г (4,11 ммоля) двунатрие-вой соли N-дикарбоксиметил-1,4,10,13-тетраокса-7,16диазациклооктадекана, содержащей 16,87 мас.% бромида натрия, и 2,01 г (7,66 ммоля) 1,4,10,13тетраокса-7,16-диазациклооктадена смешивают между собой в растворе, за исключением того, что реакционную смесь оставляют при 50°С, а гидроокись натрия отдельными порциями добавляют на протяжении 10 часов. В результате экстракции метиленхлоридом получают однозамещенное производное в количестве 2,17 г (4,41 ммоля). Оно содержит 16,93 мас.% бромида натрия. Получают 3,92 г конечного продукта. Выход 96,5% (при расчете на использованный мак-роцикл). Продукт содержит 1,6 мас.% динатриево-го оксималоната. 1 Н-ЯМР-спектр (200 мГц, D2O, d частей/млн.) соответствует спектру продукта, полученного по примеру 1, за исключением того, что он дает также резонансный сигнал при 4,31 (s), что характерно для оксималоната. Пример 3 Получение тетракалиевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана a) 1,32 г (7,22 ммоля) 2-бромомалоновой кислоты нейтрализуют в 1,0 мл воды, добавляя раствор гидроокиси калия в концентрации 5,760 моль/л в присутствии индикатора фенол-фталеина. В полученный таким образом раствор добавляют 1,25 г (4,77 ммоля) 1,4,10,13-тет-раокса-7,16диазациклооктадекана. Реакционную смесь нагревают до 50°С в течение 26 часов, добавляя в нее порциями эквивалентное количество раствора гидроокиси калия в концентрации 5,760 моль/л. После упаривания смеси твердый осадок высушивают при пониженном давлении, а затем экстрагируют метиленхлоридом общим объемом 20 мл в несколько приемов. После упа-ривания остаток высушивают. Получают 0,85 г (1,73 ммоля) дикалиевой соли N-дикарб-оксиметил-1,4,10,13-тераокса-7,16-диазациклооктадекана, содержащей 16,7 мас.% бромида калия (промежуточного). Этот продукт может быть использован для приготовления следующей партии требуемого продукта. Остаток после экстракции метиленхлоридом экстрагируют 60 мл безводного этанола и после упаривания раствора остаток высушивают. Получают 2,18 г искомого продукта, т.е. вы ход - 94,0% при расчете на использованный макроцикл. Продукт представляет собой двойную соль с бромидом калия, которая содержит 29,97 мас.% бромида калия. 1 Н-ЯМР-спектр промежуточного соединения (200 мГц, D2O, d частей/млн.): 3,86 (1 Н, s); 3,63 (16 Н, m); 2,89 (4 H, t); 2,78 (4 H, m). 1 Н-ЯМР-спектр двойной соли, образованной бромидом калия (200 мГц, D2O, d частей/млн.): 3,99 (2 Н, s); 3,69 (8 Н, s); 3,63 (8 Н, t); 2,86 (8 Н, t). б) Чистый свободный от бромида калия продукт может быть получен, как описано выше, с применением 97% (по объему) этанола. В результате образуется 1,16 г искомого продукта, т.е. выход составляет 75,6% при перечете на использованный макроцикл. 1 Н-ЯМР-спектр искомого продукта (200 мГц, D2O, d частей/млн.): 4,00 (2 Н, широкий s); 3,70 (8 Н, широкий s); 3,65 (8 Н, широкий t); 2,88 (8 Н, широкий). Пример 4 Получение динатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан магниевого комплекса 0,30 г (1,46 ммоля) гексагидрата хлористого магния, разведенного в 2,0 мл воды, добавляют в раствор, содержащий 0,81 г (1,46 ммоля) продукта, полученного, как описано в примере 1б, в 3,0 мл воды. Спустя 30 мин раствор упаривают при пониженном давлении и остаток высушивают. Получают 0,93 г (97,8%) искомого продукта, содержащего 17,99 мас.% хлористого натрия. 1 Н-ЯМР-спектр искомого продукта (200 мГц, D2О в присутствии NaOD, d частей/млн.): 4,00 (2 Н, s); 3,67 (8 H, s); 3,62 (8 Н, широкая); 2,88 (8 Н, широкая). 13 Н-ЯМР-спектр (50 мГц, D2О, d частей/млн.): 179,95 (С=0); 71,88 и 71,05 (О-СН2); 54,51 (N-CH2). Примечание: резонансный сигнал СН(СОО)2 отсутствует из-за дейтерирования. Пример 5 Получение динатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан кальциевого комплекса, содержащей хлористый натрий Используют процесс, описанный в примере 4, с 0,92 г (1,65 ммоля) продукта, полученного в соответствии с примером 1б, и 0,25 г (1,65 ммоля) дигидрата хлористого кальция, что дает 1,08 г (98,2%) искомого продукта, содержащего 17,57 мас.% хлористого натрия. 1 Н-ЯМР-спектр продукта (200 мГц, D2O, d частей/млн.): 3,90 (4 Н, широкая); 3,53 (14 Н, широкая); 2,92 (4 Н, коалесцентная t); 2,72 (4 Н, коа-лесцентная t). 13 Н-ЯМР-спектр (50 мГц, D2О, d частей/млн.): 179,23 (С=0); 82,38 (CH(COOH)2), 71,62 (О-СН2); 55,63 (NCH2). Пример 6 35547 Получение динатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан кальциевого комплекса, содержащей динатриевый оксималонат и хлористый натрий 3,44 г (18,825 ммоля) 2-бромомалоновой кислоты нейтрализуют в 1,0 мл воды, добавляя раствор гидроокиси калия в концентрации 8,360 моль/л в присутствии индикатора фенол-фталеина. В полученный таким образом раствор добавляют 2,010 г (7,65 ммоля) 1,4,10,13-тет-раокса-7,16диазациклооктадекана. Реакционную смесь нагревают при 30-45°С в течение 43-45 часов, добавляя в нее 1,83 мл раствора гидроокиси калия, который необходим для образования двухзамещенного соединения. После этого реакционную смесь оставляют на 22-25 час при 60°С, добавляя отдельными порциями раствор гидроокиси натрия в количествах, необходимых для гидролизации непрореагированного 2броммало-ната. После окончания реакции раствор упаривают и в дальнейшем действуют, как описано в примере 1б. Получают 4,720 г сухого неочищенного продукта, который содержит 12 мас.% динатрие-вого малоната и практически не содержит бромида натрия. Затем полученный продукт используют для осуществления одной из следующи х процедур. а) Сырой продукт разводят в смеси из 3,0 мл воды и 7,5 мл раствора хлористого кальция, имеющего концентрацию 1000 моль/л. Добавляют 12,0 мл 99,7% (по объему) этанола и 0,85 мл хлористого кальция при непрерывном помешивании. После этого содержание этанола в смеси доводят до 90%, добавляя 105 мл 99,7% этанола. Полученную суспензию энергично перемешивают в течение 30-60 мин при нагревании, после чего отфильтровывают твердые частицы. Фильтрат упаривают при пониженном давлении и наполовину сухой продукт продолжают подсушивать при 75-85°С и пониженном давлении до получения 4,48 г (90,6%) требуемого вещества, содержащего 14,0 мас.% хлористого натрия и 1,08 мас.% динатриевого оксималоната. 1 Н-ЯМР-спектр полученного продукта (200 мГц, D2О, d частей/млн.) соответствуе т спектру продукта, получаемого по примеру 5, за исключением того, что резонансный сигнал проявляется также при 4,31 частях/млн. (1 Н, s), что характерно для оксималоната. б) Повторяют процедуру, описанную в разделе а), с той лишь разницей, что после добавления первой порции раствора хлористого кальция (7,50 мл) добавляют еще одну порцию объемом 3,10 мл с концентрацией хлористого кальция 1000 моль/л. Содержание этанола в реакционной смеси доводят до 90 объемных процентов, добавляя 114 мл этанола, после чего продукт высушивают в токе азота до получения вещества в количестве 5,00 г (94,9%), в котором содержится 19,5 моль% соли кальция кальциевого комплекса (при расчете на общий макроцикл), а также динат-риевая соль кальциевого комплекса. Конечный продукт содержит 13,2 мас.% хлористого натрия, 7,35 мас.% воды и незначительное количество двунатриевого оксималоната. 1 Н-ЯМР-спектр продукта (200 мГц, D2O, d частей/млн.) соответствует спектру продукта, полученного в примере 5. Пример 7 Получение диаммония N,N'-бис(дикарбокси-метил)-1,4,10,13-тетраокса-7,16-диазациклоокта-декан кальциевого комплекса 0,5 мл воды, 2,04 мл раствора кальция в концентрации 0,998 моль/л и затем 30 мл безводного этанола добавляют к 0,554 г (1000 ммолям) тетра-натриевой соли, полученной в соответствии с примером 1б. Раствор упаривают приблизительно до трети первоначального объема и к оставшемуся его количеству добавляют этанол в количестве, достаточном для доведения его концентрации в растворе до 95-96% по объему. Вслед за этим раствор нагревают, перемешивают на протяжении 30 мин, оставшийся хлористый натрий отфильтровывают и промывают безводным этанолом. К объединенному фильтрату добавляют 0,554 г (1000 ммолей) тетранатриевой соли и 0,214 г (4000 ммолей) хлористого аммония, а затем воду в количестве, достаточном для растворения твердого материала. После упаривания раствора остаток обезвоживают, нагревая при 75-80°С и пониженном давлении. Получают 1, 452 г (98,1%) искомого продукта, содержащего 19,91 мас.% хлористого натрия и 7,33 мас.% воды. Очищенный от хлорида конечный продукт получают дальнейшей очисткой безводным этанолом. 1 Н-ЯМР-спектр свободного от хлорида и содержащего хлористый натрий продукт (200 мГц, D2O, d частей/млн.) идентичен: 3,90 (4 Н, широкий m); 3,73 (14 Н, широкий m); 2,92 (4 Н, широкий t); 2,72 (4 Н, широкий t). Пример 8 Получение кальциевой соли N,N'-бис(дикарб-оксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан кальциевого комплекса После нейтрализации 2,743 г (9,53 ммоля) 2-броммалоновой кислоты в 2,0 мл воды путем добавления отдельными порциями гидроокиси кальция в присутствии индикатора фенолфталеина в полученный раствор вводят 1000 г (3,81 ммоля) 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Реакционную смесь нагревают при 40°С, 45°С и, наконец, 50°С в течение, в общей сложности, 72 час, а затем до 60°С в течение 24 час, добавляя в нее отдельными порциями 0,85 г (11,47 ммоля) гидроокиси кальция при энергичном помешивании. После этого осадок, основную часть которого составляет оксималонат кальция, отфильтровывают и промывают тремя порциями воды по 4-5 мл каждая. Объединенный фильтрат упаривают при пониженном давлении, после чего из образующегося остатка дважды отгоняют по 35 мл метиленхлорида, получая твердый продукт, который подсушивают при 75-85°С и пониженном давлении. Таким образом, получают искомый продукт в количестве 2,710 г с выходом 89,7%, который содержит 31,5 мас.%. 35547 1 Н-ЯМР-спектр полученного продукта (200 мГц, D2O, d частей/млн.) соответствуе т спектру продукта, полученного в примере 5. ИК спектр (КВr, см -1): 2920, 2880 (m, g СН), 1615 (vs, g COO/as), 1450 (m, g COO/as). Другие характерные, но не идентифицированные частоты: 1355,1290 (m), 1250 (m), 1085 (vs).950 (m). Пример 9 Получение динатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан железистого (ІІ) комплекса, содержащей динатриевый оксималонат и хлористый натрий Используют 0,747 г (1,35 ммоля) продукта, полученного как описано в примере 1б, и 0,268 г (1,35 ммоля) тетрагидрида хлористого железа, из которых по способу, представленному в примере 4, получают требуемый продукт. В отличие от ранее описанного процесса предотвращают окисление двухвалентного железа до трехвалентного, осуществляя реакцию в атмосфере азота. Получают искомый продукт с выходом 91,0% в количестве 0,835 г, при содержании в нем хлористого натрия 17,16 мас.%. 1 Н-ЯМР-спектр конечного продукта не поддается определению из-за присутствия парамагнитных ионов железа. ИК-спектр (КВr, см -1): 2910, 2880 (m, g CH), 1630 (vs, g COO/as), 1400 (s, g COO/s). Другие характерные, но не идентифицированные частоты: 1355 (m), 1330 (m), 1100 (s), 930 (m). Пример 10 Получение динатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана цинкового комплекса Используют 0,735 г (1,32 ммоля) продукта, полученного, как описано в примере 1б, и 0,180 г (1,32 ммоля) безводного хлористого цинка, из которых по способу, представленному в примере 4, получают 0,89 г требуемого продукта с выходом 97,5%, который содержит 16,92 мас.% хлористого натрия. 1Н-ЯМР-спектр (200 мГц, D20, d частей/млн.): 3,6-4,2 (18 Н, широкая система полос t); 3,10 (8 Н, широкая t). Пример 11 Получение трехнатриевой соли N,N'-бис(ди-карбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана 1,72 г раствора соляной кислоты в концентрации 1,048 моля/л добавляют к раствору 1000 г (1,804 моля) тетранатриевой соли, полученной, как описано в примере 1б, в 5,0 мл воды в условиях о хлаждения (0-5°С). После этого раствор упа-ривают, остаток обезвоживают метиленхлоридом и подсушивают при 60°С и пониженном давлении. Получают 1,069 г конечного продукта с выходом 96,7%, который содержит 9,5 мас.% хлористого натрия. 1 Н-ЯМР-спектр (200 мГц, D2O, d частей/млн.): 4,18 (2 Н, s); 3,79 (16 Н, m, широкая); 3,29 (8 Н, широкая s). Спектр продукта в D2О в присутствии NaOD соответствуе т спектру, приведенному в примере 1б. ИК-спектр (КВr, см -1): 2940, 2850 (m, g С-Н), 1655, 1605 (vs, g COO/as), 1400 (m, g COO/s). Другие характерные, но не идентифицированные частоты: 1345 (s), 1320 (s), 1120( s), 1100 (s), 930 (m). Пример 12 Получение трехнатриевой соли N-дикарб-оксиметил)-N'-(1,1'-дикарбоксиэтил)-1,4,10,13-тет-раокса7,16-диазациклооктадекана После нейтрализации 3,003 (15,25 ммоля) бромметилмалоновой кислоты в 0,5 мл воды добавлением раствора гидроокиси натрия в концентрации 8,360 моль/л при 0-5°C в присутствии индикатора фенолфталеина в раствор добавляют 2000 г (7,62 ммоля) 1,4,10,13-тетраокса-7,16-ди-азациклооктадекана. Полученную смесь оставляют на 8-10 дней при 20-25°С, добавляя порциями 1,82 мл (15,25 ммоля) раствора гидроокиси натрия концентрацией 8,360 моль/л. После окончания реакции смесь перемешивают в течение 30 мин при 55-60°С, а затем упаривают. Сухой остаток экстрагируют несколькими порциями метиленхлорида общим объемом 25-30 мл. После упаривания экс-тракта остаток обрабатывают эфиром и твердый осадок отфильтровывают. Оставшийся материал (после экстракции эфиром) содержит небольшое количество 1,4,10,13-тетраокса-7,16-диазацикло-октадекана, который можно удалить, разводя продукт метиленхлоридом и осаждая эфиром. Таким способом получают очищенную двунатриевую соль N'-(1,1'-дикарбоксиэтил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана, содержащую 13,81 мас.% бромида натрия (промежуточного). Выход конечного продукта составляет 1,390 г (37,2%). После упаривания эфирного экстракта получают 1,159 г очищенного 1,4,10,13-тетраокса-7,16диазациклооктадекана. Таким образом, конечный выход продукта достигает 38,5%. 1 Н-ЯМР-спектр промежуточного соединения (200 мГц, D2О, d частей/млн.): 3,66 (16 Н, m); 2,82 (4 H, t); 2,72 (4 H, t); 1,36 (3 Н, s). Используя это промежуточное соединение, получают двузамещенное производное по описанному ниже способу. После нейтрализации 0,483 г (2,64 ммоля) 2-броммалоновой кислоты в 0,5 мл воды посредством добавления раствора гидроокиси натрия в концентрации 8,360 моль/л в присутствии индикатора фенолфталеина добавляют 1,002 г (2,04 ммоля) вышеописанного промежуточного соединения, содержащего 13,81 мас.% бромида натрия. После этого реакционную смесь оставляют на 72 час при 3045°С, а затем еще на 24 час при 50-60°С, добавляя порциями 0,32 мл (2,64 ммоля) раствора гидроокиси натрия в концентрации 8,369 моль/л. После окончания реакции раствор упаривают, а остаток высушивают 35547 при 75-80°С и пониженном давлении. Сухой осадок экстрагируют безводным этанолом. Этанольный экстракт упа-ривают досуха при пониженном давлении и высушивают. Получают 1,323 г двойной соли, содержащей 2,06 моля бромида натрия с выходом 83,0%. Продукт, свободный от бромида, можно получить посредством экстракции двойной соли 96% по объему этанолом, как описано в примере 1б. В результате образуется 0,532 г требуемого вещества с выходом 54,5%. 1 Н-ЯМР и ИК-спектры двойной соли практически идентичны спектрам четырехнатриевой соли. ИК-спектр (КВr, см -1): 2960, 2870 (m, g С-Н), 1645, 1600 (vs, g СОО/as), 1405, 1440 (m, g COO/s). Другие характерные, но не идентифицированные частоты: 1355 (s), 1315 (s), 1095 (s), 930 (m). 1 Н-ЯМР-спектр (200 мГц, D2О, d частей/млн.): 3,89 (1 Н, s); 3,68 (16 Н, m); 2,92 (4 H, t), 2,78 (4 H, t), 1,41 (3 Н, s). Пример 13 Получение тетранатриевой соли, двойной соли натрия бромида N-дикарбоксиметил-N'-(1,1'дикарбоксипропил-1,4,10,13-тетраокса-7,16-диаза-циклооктадекана Получение осуществляют по способу, описанному в примере 12, используя 1,608 г (7,62 ммоля) 2бромэтилмалоновой кислоты и 1000 г (3,81 ммоля) 1,4,10,13-тетраокса-7,16-диазацикло-октадекана, что дает 0,517 г (26,2%) двунатриевой соли N-(1,1'-дикарбоксипропил)-1,4,10,13-тетраок-са-7,16диазациклооктадекана, которая содержит 15,61 мас.% бромида натрия. После упаривания эфирного экстракта получают 0,508 г 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Таким образом, истинный выход искомого продукта составляет 53,3%. 1 Н-ЯМР-спектр промежуточного соединения (200 мГц, D2O, d частей/млн.): 3,68 (16 Н, m); 2,88 (4 H, t); 2,82 (4 H, t), 1,84 (2 H, q), 0,90 (3 Н, t). Искомая двойная соль, содержащая 3,10 моля бромида натрия, образуется с выходом 52,7% в количестве 0,475 г при использовании 0,517 г (1,00 ммоля) вышеуказанного промежуточного соединения, содержащего 15,61 мас.% бромида натрия и 0,229 г (1,20 ммоля) 2-броммалоновой кислоты. 1 Н-ЯМР-спектр исходной двойной соли (200 мГц, D2O, d частей/млн.): 3,88 (1 Н, s); 3,67 (16 Н, m); 2,91 (4 H, t), 2,86 (4 H, t), 1,84 (2 H, q), 0,88 (3 H, t). Пример 14 Получение четырехнатриевой соли N-ди-карбоксиметил)-N'-(бензил-дикарбоксиметил)-1,4, 10,13тетраокса-7,16-диазациклооктадекана, содержащей двунатриевый оксималонат. Используют процедуру, описанную в примере 12, с той разницей, что реакцию осуществляют в смеси воды с этанолом (1:1, объемное соотношение), в которую добавляют 2,081 г (7,62 ммоля) 2-бромо-2бензилмалоновой кислоты и 1000 г (3,81 ммоля) 1,4,10,13-тетраокса-7,16-диазацикло-октадекана. Таким образом, получают 0,372 г N-(бензил-дикарбоксиметил)-1,4,10,13-тетраокса-7, 16-диазациклооктадекана, содержащего 11,21 мас.% бромида натрия (промежуточного соединения) с выходом 17,4%. После упаривания эфирного экстракта получают 0,714 г 1,4,10,13-тетраокса-7,16-диазациклооктадекана. Таким образом, истинный выход достигает 60,9%. 1 Н-ЯМР-спектр промежуточного соединения (200 мГц, D2O, d частей/млн.): 7,45 (2 Н, d); 7,29 (3 Н, m); 3,68 (12 Н, m), 3,57 (4 H, t), 3,47 (2 H, s), 2,78 (8 H, m). Искомую двойную соль, содержащую 0,42 моля двунатриевого оксималоната, получают, используя 0,372 г (0,662 ммоля) вышеуказанного промежуточного соединения, содержащего 11,21 мас.% бромида натрия и 0,161 г (0,880 моля) 2-броммалоновой кислоты. Для получения конечного продукта сначала проводят экстракцию мети-ленхлоридом, а затем абсолютным этанолом, что дает 0,432 искомой соли с выходом 91,4%. 1 Н-ЯМР-спектр конечного продукта (200 мГц, D2O, d частей/млн.): 7,43 (2 Н, d); 7,29 (3 Н, m); 3,90 (1 Н, s), 3,64 (16 Н, широкая), 3,34 (2 Н, s), 2,91 (4 H, t), 2,80 (4 H, t). Пример 15 Получение четырехнатриевой соли N, N'-бис (1,1'-дикарбоксиэтил)-1,4,10,13-тетраокса-7,16-диазациклооктадекана После нейтрализации 3,11 г (14,79 ммоля) бромметилмалоновой кислоты, разведенной в 1,0 мл, путем добавления раствора гидроокиси натрия в концентрации 8,360 молб/л при 0-5°C в присутствии индикатора фенолфталеина добавляют 3,52 г (6, 565 ммоля) двунатриевой соли N-(1,1'-икарбоксиэтил-1,4,10,13тетраокса-7,16-ди-азациклооктадекана, содержащей 18,6 мас.% бромида натрия (промежуточного). Затем реакционную смесь нагревают до 20-25°С и оставляют на 10-12 дней, добавляя порциями 1,77 мл (14,79 ммоля) раствора гидроокиси натрия в концентрации 8,360 моль/л. Перед окончанием реакции смесь в течение 30 мин прогревают при 55-60°С и упаривают. Сухой остаток экстрагируют несколькими порциями метиленхлорида общим объемом 45-50 мл. Экстракт упаривают, остаток обрабатывают эфиром, преципитат отфильтровывают и высушивают. Таким способом получают 2,08 г (4 ,29 ммоля) промежуточного соединения, содержащего 14,0 мас.% бромида натрия, который можно использовать при приготовлении следующей порции продукта. Твердый материал, оставшийся после экстракции метиленхлоридом, высушивают при 75-80°С и пониженном давлении. После экстракции сухого остатка 55-60 мл безводного этанола полученный экстракт упаривают до сухого остатка при пониженном давлении и высушивают до образования 3,90 г двойной соли, содержащей 78,2 мас.% бромида натрия при выходе 22,2%. Расчетный выход промежуточного продукта, используемого в реакции, составляет 64,3%. 35547 1 Н-ЯМР-спектр двойной соли (200 мГц, D2О, d частей/млн.): 3,67 (8 Н, s); 3,64 (8 Н, t); 2,73 (8 Н, t), 1,36 (6 H, s). Продукт, не содержащий бромида натрия, получают из 3,90 г двойной соли, содержащей 78,2 мас.% бромида натрия, которую экстрагируют 96% (по объему) этанолом, как описано в примере 1б. Этим способом получают 0,489 г требуемого продукта с выходом 57,5%. После упаривания, высушивания и повторной экстракции безводным этанолом получают двойную соль, содержащую 80-90 мас.% бромида натрия (из фильтрата с 96%-ным этанолом). Эту двойную соль можно использовать при получении следующей партии продукта. 1 Н-ЯМР-спектр искомого продукта (200 мГц, D2O, d частей/млн.): 3,70 (16 Н, широкая т); 2,71 (8 Н, коалесцентная t); 1,34 (6 H, расширенная s). Пример 16 Получение двойной соли бромида натрия тет-ранатриевой соли N,N'-бис(1,1-дикарбоксиметил)1,4,10,13-тетраокса-7,16-диазациклооктадекана. Используют процедуру, описанную в примере 15. Реакцию осуществляют с 0,362 г (1,324 ммоля) 2бромо-2-бензилмалоновой кислоты и 0,372 г (0,662 ммоля) однозамещенного промежуточного соединения, содержащего 11,21 мас.% бромида натрия в смеси этанола с водой при объемном соотношении 1:1. Таким путем получают 0,085 г двойной соли, содержащей 4,2 моля бромида натрия, с выходом 11,1%. В ходе описанной процедуры получают 0,296 г промежуточного продукта, который содержит 11,21 мас.% бромида натрия. Таким образом, расчетный выход для используемого в реакции промежуточного продукта составляет 54,2%. 1 Н-ЯМР-спектр искомого продукта (200 мГц, D2O, d частей/млн.): 7,64 (4 Н, d); 7,45 (6 H, m); 3,65 (12 H, m), 3,52 (4 H, t), 3,31 (4 H, s), 2,68 (8 H, t). Пример 17 Показатели острой токсичности продуктов, получаемых согласно примерам 1-16, определяли на лабораторных мышах и крысах по методике, приведенной ниже. Растворы, содержащие тестируемые соединения в различных концентрациях, готовили на основе физиологического раствора или 5%-ного (по весу) раствора глюкозы. Активные соединения в разных концентрациях вводили в кровяное русло животных, применяя для этой цели медленные, на протяжении 35 мин, инъекции. Для тестирования каждой дозировки использовали группы мышей Суисс и крыс Уистар по 6-10 особей в каждой. После инъекций наблюдения за животными продолжались в течение 30 дней. Показатели LD50/30, т.е дозы, вызывавшие гибель 50% животных на протяжении 30 дней, определяли по числу умерших в течение этого периода мышей и крыс с использованием пробит-анализа, как описано в (D.J. Finney: Probit Analysis (2nd ed.) Cambridge University Press (1952)). Эти показатели выражали в ммоль/кг веса тела. Результаты оценки токсичности соединений, полученных согласно настоящему изобретению суммированы в колонке В таблицы. Таблица Основные характеристики продуктов Пример № А 1/а 1/b 2 3/а 3/b 4 5 6/а 6/b 7 8 9 10 11 12 13 14 15 16 Острая токсичность LD50/30 ммоль/кг В 0,327 0,379 0,368 0,333 0,272 0,211 более 2,5 более 3,0 более 3,0 более 2,5 более 1,5 0,250 0,091 0,111 0,305 0,302 0,422 0,450 0,470 Пример 18 F фактор Показатель Е1 С 1,8 2,8 3,3 2,3 1,7 2,3 3,7 3,9 4,2 1,7 1,6 1,7 2,3 2,6 1,5 2,1 1,4 1,3 1,3 D 5,40 9,66 11,10 12,30 8,90 7,30 145,00 158,00 160,00 65,00 58,00 8,25 3,20 2,90 7,90 13,30 7,60 6,80 7,10 35547 Ниже описан способ оценки стимулирующего действия соединений, получаемых согласно примерам 116, на выведение радиоактивных изотопов из организма мышей. В брюшную полость животных вводили радиоактивные стронций (85SrCl2) или церий (144СеСІ3) с активностью от 37 до 74 кВк (1-2 мкКи). Затем животных подразделяли на группы по 5-10 особей каждая. Спустя 30-60 мин после введения радиоизотопов одной группе животных внутривенно инъекцировали активное соединение в количестве, обеспечивающем его концентрацию в организме от 50 до 100 мкмоль/кг веса тела. Животным контрольной группы аналогичным образом вводили (стерильный физиологический раствор или 5%-ный раствор глюкозы), не содержащий тестируемый препарат. Количество радиоактивности в организме определяли сразу после введения изотопа, а затем повторяли измерения ежедневно или через каждые два или три дня в особом аппарате, сконструированном для установления радиоактивности в теле мелких животных. Полученные результаты сравнивали с фоновой активностью с самого первого дня и рассчитывали так называемую задержку или временную корреляцию с уче том остаточной радиоактивности в организме. Изменения содержания радиоактивности в организме животных со временем (в днях) показаны на фиг. 1, на которой по ординате отложена задержка введенной радиоактивности (в процентах от ее исходного количества), а по абсциссе период после введения радиоактивности. Из рисунка следует, что скорость выведения стронция-85 после его введения в брюшную полость животных контрольной группы была невысокой: в течение первого дня выводилось только 15%, в течение 4 дней 25%, а на протяжении 7 дней 30% общего количества радиоактивности. В последующем скорость экскрекции еще более замедлялась. С другой стороны, после однократного введения активного соединения, полученного, как описано в примере 13, в дозе 100 мкмоль/кг в течение первого дня выводилось 40% всего количества радиоактивности, в течение 4 дней 65%, а в течение 7 дней 67%. Еще более впечатляющие результаты были получены при использовании соединения, полученного, как описано в примере 5. Выведение радиоактивности в указанные сроки составляло в этом случае соответственно 81, 84 и 85%. На основании статистического анализа вышеуказанным методом было установлено, что кривые задержки активных соединений согласно настоящему изобретению описываются двухкомпонентной нисходящей экспоненциальной функцией. Факторы F и показатели Е1, характеризующие продукты, получаемые согласно настоящему изобретению, представлены в колонках С и D таблицы. Очевидно, что эффективность отдельных препаратов с точки зрения их стимулирующего действия на выведение радиоактивности из организма подопытных животных значительно отличается, особенно если основываться при ее оценке на показателях Е1. По нашему мнению, соединения, имеющие показатель Е1 между 0 и 5, являются слабоактивными, соединения с показателями Е1 от 5 до 50 могут оцениваться как обладающие средней эффективностью, а соединения, имеющие показатели Е1 от 50 до 100 или более высокие, должны рассматриваться как высокоэффективные для данной цели препаратов. Пример 19 а) Кривые задержки радиоактивности, представленные на фиг. 2, иллюстрируют выведение радиоактивного стронция, который вводили внут-ритра хейно в легкие крыс линии Уистар после внутрибрюшинной инъекции соединения, описанного в примере 6б. Характер верхней кривой на фиг. 2 свидетельствует о том, что радиоактивный стронций слабо выводится из организма контрольных животных, которым инъекцировали растворитель, не содержащий тестируемого соединения. На протяжении нескольких дней после радиоактивного загрязнения из организма выводилось не более 3035% исходного количества радиоактивности. В то же время уровень радиоактивного загрязнения организма в целом уменьшался с 90% (в контроле) до 20% у животных, которым вводили однократно 50 мкмоль/кг веса тела соединения согласно настоящему изобретению (средняя кривая) или которым вводили его дважды на протяжении суток после загрязнения с 3-часовым интервалом (нижняя кривая). Быстрое удаление радиоизотопа продолжалось в течение всего периода наблюдений и достигало 8890% при однократном введении препарата и до 94-96% после его двукратного введения. Важно подчеркнуть, что в ходе эксперимента радиоактивный стронций практически отсутствовал в костной ткани животных, которым вводили одно из соединений в соответствии с настоящим изобретением (по данным обследования после забоя в конце эксперимента). Остаточная радиоактивность в мягких тканях и печени составляла 5-10%, тогда как в контроле, основная часть метки (65-70%) задерживалась в костях. Аналогичные результаты были получены при внутривенных инъекциях тестируемых соединений или при их введении в подкожную соединительную ткань. б) Согласно настоящему изобретению, соединения, усиливающие выведение радиоактивности, испытывали также на способность стимулировать экскрецию други х радиоактивных металлов, в частности церия-144, который относится к группе редкоземельных элементов. В данном примере иллюстрируется выведение 144CeCl3 после его введения в легкие крыс линии Уистар как функция времени, прошедшего после однократной дозы или после повторного введения с интервалом в 24 часа (см. фиг. 3). Полученные результаты показывают, что соединения, согласно настоящему изобретению, можно использовать для усиления выведения из организма радиоактивных материалов с относительно слабой растворимостью в биологических жидкостях (при загрязнении легких). К концу эксперимента, т.е. на 30-й день, у контрольных животных в легких сохранялось до 40% исходного количества радиоактивности, тогда как у животных, получавши х однократную внутрибрюшинную инъекцию одного из активных соединений согласно изобретению, этот показатель не превышал 14%. Общая задержка радиоактивности под воздействием тестируемого соединения спустя 60 мин уменьшилась до 5,6% и продолжала уменьшаться на протяжении следующих суток. Ее резкое снижение в самом начале эксперимента и замедление в более поздние сроки 35547 были, по-видимому, обусловлены характером растворимости радиоактивного загрязняющего материала и особенностями элиминирования в составе комплекса. Пример 20 В примерах 17-18 описано действие соединений согласно настоящему изобретению на удаление радиоизотопов после их введения в кровяное русло, брюшную полость и подкожную соединительную тканью. С точки зрения терапевтической эффективности у людей и, что более важно, с точки зрения быстрой и эффективной защиты большего количества населения представлялось важным показать, что соединения согласно настоящему изобретению можно использовать для ускорения удаления радиоактивных изотопов и при других п утя х и х введения в организм. С этой целью были проведены эксперименты на крысах линии Уистар. Части животных в брюшную полость вводили радиоактивный стронций, а спустя 60 мин им внутритрахейно вводили активные соединения, полученные согласно настоящему изобретению. В качестве таких соединений использовали препараты с показателем Е1, более 100, которые показаны в таблице. После введения на протяжении 30 дней определяли общую радиоактивность организма крыс. При анализе задержки радиоактивности оказалось, что в случае их построения обычным способом соединения в соответствии с настоящим изобретением обладали высокой эффективностью с точки зрения элиминирования метки после их вдыхания в форме порошка или аэрозоля. У контрольных животных задержка достигала 91%, у крыс, которым вводили препарат согласно настоящему изобретению, она в первый же день уменьшилась до 15%, а на третий день не превышала 10%. Это наблюдение подтвердилось после расчета показателя Е1 (164) и фактора F (7, 9) (сравни с таблицей). В дополнительных экспериментах изучали возможность адсорбции активных соединений в соответствии с изобретением с поверхности эпителиальных тканей. На спине крыс выбривали участок размером 3х3 см. После введения стронция в их легкие внутри-трахейным способом (в условиях анестезии) определили общий уровень радиоактивности в организме. После этого раствор соединения в соответствии с изобретением наносили на подготовленный участок кожи и закрывали последний липким пластырем. На основании ежедневных измерений общего уровня радиоактивности в организме установлено, что соединение согласно настоящему изобретению обладало способностью усиливать экскрекцию радиоактивности также и при действии через кожу. Исходя из результатов измерений, рассчитали фактор F и показатель Е1, которые оказались равными соответственно 105 и 110. Полученные экспериментальные результаты показывают, что активные соединения, полученные в соответствии с изобретением, можно использовать в составе лекарственных средств в форме таблеток для помещения под язык, свечей, растворимых в пищеварительном тракте, драже, капсул или накожного пластыря. 35547 Фиг. 1 35547 Фиг. 2 35547 Фиг. 3
ДивитисяДодаткова інформація
Назва патенту англійською1,4,10,13-tetraoxa-7,16-diazacyclooctadecane derivatives, disodium salt of n,n1-bis(dicarbomethyl)-1,4,10,13-tetraoxa -7,16-diazacyclooctadecane -calcium complex and compositions on their basis for elimination of metal ions in living organism
Автори англійськоюJozsef Emri, Bela Gyori, Zoltan Kovacs, Erno Brucher, Sztanyik Laszlo, Laszlo Varga, Odon Kiraly, Bela Kanyar
Назва патенту російськоюПроизводные 1,4,10,13-тетраокса-7,16-диазациклооктадекана, динатриевая соль n,n1-бис(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-диазациклооктадекан-кальциевого комплекса и композиции на их основе для выведения из живого организма ионов металлов, которые его поражают
Автори російськоюЭМРИ Йожеф, ДЕРИ Бейла, КОВАЧ Золтан, БРЮХЕР Эрне, СТАНИК Ласло, ВАРГА Ласло, КИРАЙ Йоден, КАНЯР Бейла
МПК / Мітки
МПК: C07F 15/00, C07D 273/00, A61K 33/06, A61K 31/395, A61P 43/00, C07F 3/00, A61P 39/02
Мітки: динатрієва, основі, біс(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-діазациклоокта-декан-кальцієвого, 1,4,10,13-тетраокса-7,16-діазациклооктадекану, комплексу, похідні, виведення, сіль, композиції
Код посилання
<a href="https://ua.patents.su/14-35547-pokhidni-141013-tetraoksa-716-diazaciklooktadekanu-dinatriehva-sil-nn1-bisdikarboksimetil-141013-tetraoksa-716-diazaciklookta-dekan-kalciehvogo-kompleksu-ta-kompozici-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні 1,4,10,13-тетраокса-7,16-діазациклооктадекану, динатрієва сіль n,n1 -біс(дикарбоксиметил)-1,4,10,13-тетраокса-7,16-діазациклоокта-декан-кальцієвого комплексу та композиції на їх основі для виведення з ж</a>
Динатрієва сіль 2,2-біс(3сульфоксипропілокси)фенілетанону, як фотоініціатор радикальної полімеризації

Номер патенту: 27572
Опубліковано: 15.09.2000
Автори: Лапшин Олександр Валерійович, Мізюк Володимир Леонідович, Шибанов Володимир Вікторович, Барановська Оксана Євстахіївна
МПК: C07C 309/00, G03F 7/028, C07C 49/20
Мітки: радикальної, полімерізації, фотоініціатор, динатрієва, сіль, 2,2-біс(3сульфоксипропілокси)фенілетанону
Текст:
...- СН2 - СН2 - SO3Na II — С- ОСН 2 - СН 2 - СН 2 - SO 3 Na (2) см гю СМ 27572 За счет особенностей химического строения заявленное соединение растворяется в воде, обеспечивая хорошую совместимость со всеми элементами водорастворимых ФПК Благодаря этому улучшаются эксплуатационные качества готовых фогомолимерных материалов Пример 1. В колбу, емкостью 0,5 л, снабженную насадкой Дина-Старка и эффективным обратным холодильником помещают...
Сульфатна сіль n,n-диметил-2-[5-(1,2,4-триазол-1ілметил)-1н-індол-3-іл]етиламіну та/або її сольвати, фармацевтична композиція на її основі, спосіб одержання фармацевтичної композиції і спосіб лікування

Номер патенту: 27908
Опубліковано: 16.10.2000
Автори: Гіблін Александр Річард, Олів Кароль, Матасса Віктор Гіліо, Бейкер Раймонд, Пітт Кендал Джордж, Стріт Леслі Джозеф, Сторі Девід Едвард
МПК: A61K 31/41, A61P 25/06, C07D 403/06, A61K 31/4196, C07D 521/00, A61K 31/415, A61K 9/20, A61P 43/00, A61P 25/04, A61K 31/00, A61K 9/08
Мітки: сульфатна, n,n-диметил-2-[5-(1,2,4-триазол-1ілметил)-1н-індол-3-іл]етиламіну, сольвати, основі, композиції, лікування, фармацевтична, композиція, одержання, спосіб, фармацевтично, сіль
Текст:
...Композиции для интраназального введения в общем случае могут быть представлены в форме жидкости или сухого порошка. Хорошие композиции для интраназального введения должны быть достаточно стабильными - химически и физически, быть равномерно распределены в точно измеренных дозах даже после длительного хранения с возможными колебаниями температур в пределах от 0 до 40°С. Соответственно активный ингредиент должен быть совместим с...
Похідні 3-циклоалкілпропен-2-аміду, їх таутомерні форми та їх солі, що мають протизапальну активність, спосіб їх одержання та фармацевтична композиція на їх основі
Номер патенту: 27328
Опубліковано: 15.09.2000
Автор: Куо Елізабет Енн
МПК: C07C 255/31
Мітки: солі, одержання, таутомерні, активність, основі, похідні, мають, композиція, 3-циклоалкілпропен-2-аміду, фармацевтична, протизапальну, форми, спосіб
Текст:
...фармакологическими свойствами. В частности, они проявляют противовоспалительное действие Они ингибируют, с одной стороны, воспалительные явления, вызванные раздражающими агентами, а с другой стороны, тормозят реакции запаздывающей суперчувствительности, препятствуя активации иммунных клеток специфическим антигеном. Эти свойства иллюстрированы ниже в опытной части. Эти свойства позволяют использовать новые производные...
Похідне тетрагідробензимідазолу або його фармацевтично прийнятна сіль, що проявляють активність антагоніста 5-нт3-рецептора, та фармацевтична композиція на його основі

Номер патенту: 27290
Опубліковано: 15.09.2000
Автори: Токуо Коіде, Дзунйа Охморі, Кейдзі Міята, Акіра Матсухіса, Ісао Янагісава, Мітсуакі Охта, Такесі Сузукі
МПК: C07D 403/08, C07D 403/12, A61K 31/40, C07D 235/06, C01B 7/00, A61K 31/415
Мітки: похідне, тетрагідробензимідазолу, сіль, основі, проявляють, прийнятна, 5-нт3-рецептора, фармацевтична, композиція, активність, антагоніста, фармацевтично
Текст:
...%: C 71,77; H 6,13; N 15,13. Масс-спектр (Е1): m/z; 358 (M+). Примеp 17. Гидрохлорид N-[(4,5,6,7- тетрагидробензимидазол-5-ил)карбонил]-фенотиазина Физико-химические свойства: Т.пл. 268 – 270оС. Элементный анализ для C20H17N3О × HCl × 0,5 х х х H2O: Рассчитано, %: C 61,14; H 4,87; N 10,69; Cl 9,02. Найдено, %: C 61,15; H 4,64; N 10,60; Cl 8,59. Масс-спектр (Е1): m/z; 347 (M+, сво бодное соединение). Пример 18....
Сіль, яка утворена ранітидином та комплексом вісмуту з карбоновою кислотою, спосіб її одержання, фармацевтична композиція на її основі

Номер патенту: 26669
Опубліковано: 12.11.1999
Автор: Клітроу Джон Уотсон
МПК: C07D 307/52
Мітки: комплексом, яка, спосіб, утворена, кислотою, сіль, одержання, вісмуту, композиція, основі, ранітидином, фармацевтична, карбоновою
Формула / Реферат:
1. Соль, образованная ранитидином и комплексом висмута с карбоновой кислотой, или сольват такой соли, где карбоновая кислота выбрана из лимонной или винной кислот, обладающая терапевтической активностью.2. Соль по п.1, где она представляет собой N-[2-[[[5-[(диметиламино)метил]-2-фуранил]метил]тио]этил]-N'-метил-2-нитро-1,1-этендиамин-2-гидрокси-1,2,3-пропантрикарбоксилат висмута (3+) и ее сольваты.3. Соль по п.1, где она...
Попередній патент: Спосіб приготування муфти, що термоусаджується, із наповнених епоксидних полімерів
Наступний патент: Спосіб профілактики затримки розвитку плода
Випадковий патент: Спосіб відновлення функціонального стану м'язового корсету обличчя