Цисплатиновий комплекс та спосіб його одержання













Формула / Реферат
1. Спосіб одержання цисплатинового комплексу, що включає наступні етапи:
a) реакцію [PtA4]2- або його солі з L в розчиннику зі створенням [РtА3(L)]¯ і
b) реакцію [РtА3(L)]¯ з L' у другому розчиннику зі створенням цис-[PtA2(L')(L)],
де групи L і L' різні і кожна з них є аміном або заміщеним аміном, який пов'язаний з атомом Pt координаційним зв'язком через атом азоту і являє собою гетероциклічний амін або гетероароматичний амін, або має формулу NRR'R", де R, R' або R" незалежно один від одного вибрані з групи, що включає водень, заміщені або незаміщені, розгалужені, нерозгалужені або циклічні аліфатичні, арильні, неароматичні або ароматичні гетероциклічні групи; за умови, що тільки L' може являти собою NН3 і що щонайменше одна з груп L і L' являє собою заміщений гетероциклічний або гетероароматичний амін, і де групи А можуть бути однаковими або різними, і кожна з них являє собою галогенову або негалогенову відщеплювану групу.
2. Спосіб за п.1, що додатково включає етап перетворення А в А', де А' являє собою галогенову або негалогенову відщеплювану групу, відмінну від А.
3. Спосіб за п. 1, в якому на етапі а) [PtA4]2- знаходиться у формі тонкоподрібненої платинової солі.
4. Спосіб за п. З, в якому на етапі а) перший розчинник являє собою апротонний розчинник.
5. Спосіб за п. 4, в якому згаданий апротонний розчинник вибирають із групи, що включає ацетон, хлороформ, диметилацетамід, диметилформамід, дихлорметан, N-метилпіролідинон і тетрагідрофуран.
6. Спосіб за п. 4, в якому згаданий апротонний розчинник являє собою N-метилпіролідинон.
7. Спосіб за п. 1, в якому на етапі а) амін L в ході згаданого реакційного етапу додають малими порціями.
8. Спосіб за п. 1, в якому етап а) виконують в температурному діапазоні 30-100°С.
9. Спосіб за п. 8, в якому етап а) виконують в температурному діапазоні 40-70°С.
10. Спосіб за п. 9, в якому етап а) виконують при 50-60°С.
11. Спосіб за п. 1, в якому етап а) виконують при співвідношенні розчинника до платини менше ніж 6:1 (мл розчинника)/(ммоль платини).
12. Спосіб за п. 11, в якому етап а) виконують при співвідношенні розчинника до платини менше ніж 2:1,0 (мл розчинника)/(ммоль платини).
13. Спосіб за п. 1, в якому замісник в заміщеному гетероциклічному або гетероароматичному аміні стерично утруднює доступ атома Pt до ланцюга ДНК клітини.
14. Спосіб за п. 13, де клітина є пухлинною клітиною.
15. Спосіб за п. 11, в якому на етапі а) перший розчинник містить менше ніж 10 % мас. води.
16. Спосіб за п. 1, в якому на етапі b) другий розчинник являє собою водний розчинник.
17. Спосіб за п. 1, в якому на етапі b) другий розчинник являє собою комбінацію водного розчинника і органічного розчинника.
18. Спосіб за п. 1, в якому на етапі b) другий розчинник містить хлорид в діапазоні від 0,1 до 6 N.
19. Спосіб за п. 1, в якому етап b) виконують в температурному діапазоні 30-60°С.
20. Спосіб за п. 19, в якому етап b) виконують в температурному діапазоні 40-50°С.
21. Спосіб за п. 1, в якому етап b) виконують при рН від ~7 до 14.
22. Спосіб за п. 21, в якому етап b) виконують при рН від 7 до 12.
23. Спосіб за п. 22, в якому етап b) виконують при рН від ~8 до 10.
24. Спосіб за п. 1, в якому етап b) виконують при співвідношенні розчинника до платини, меншому або рівному ~5:1 (мл розчинника)/(ммоль платини).
25. Спосіб за п. 1, в якому етап b) виконують при молярному співвідношенні L' в формі вільної основи до платини від 3:1 до 1:1.
26. Спосіб за п. 2, в якому А' вибирають з групи, що включає галоген, гідроксигрупу, алкоксигрупу, карбоксигрупу, а також бідентатну карбоксигрупу, фосфонкарбоксигрупу, дифосфонатну групу або сульфатну групу.
27. Спосіб за п. 26, в якому А являє собою хлоридну групу.
28. Спосіб за п. 1, в якому L являє собою 2-піколін.
29. Спосіб за п. 1, в якому щонайменше одна з груп L і L' являє собою гетероциклічний амін або гетероароматичний амін, або має формулу NRR'R", де R, R' або R" незалежно один від одного вибрані з групи, що включає водень, заміщені або незаміщені, розгалужені, нерозгалужені або циклічні аліфатичні, арильні, неароматичні або ароматичні гетероциклічні групи.
30. Спосіб за п. 29, в якому L' являє собою NH3.
31. Спосіб за п. 30, в якому А являє собою хлоридну групу.
32. Спосіб за п. 1, в якому L являє собою
 ,
,
L' – NH3, А - Сl, a Y - ОН.
33. Спосіб за п. 1, в якому L являє собою
 ,
,
L' – NН3, А - Сl, a Y - ОН.
34. Спосіб за п. 1, в якому L являє собою
,
L' - NH3, А - Сl і ОАс, а Y - ОН.
35. Спосіб за п. 1, в якому L являє собою
,
L' - NHa, А - Сl і ОН, а Y - ОН.
36. Цисплатиновий комплекс формули Іb
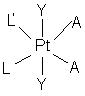 , Ib
, Ib
де L являє собою
,
L' - NH3, А - Сl і ОН, а Y - ОН.
37. Спосіб за п. 1 що додатково включає етап реакції цис-[РtА2(L')(L)], утвореного на етапі b), з Н2O2, якщо Y являє собою гідроксигрупу, або з галогеном, якщо Y являє собою галоген, зі створенням цисплатинового комплексу, що має формулу [PtA2Y2(L')(L)].
38. Спосіб за п. 37, що додатково включає етап перетворення А в А', де А' являє собою галогенову або негалогенову відщеплювану групу, відмінну від А.
39. Спосіб за п. 38, в якому А' вибирають з групи, що включає галоген, гідроксигрупу, алкоксигрупу, карбоксигрупу, а також бідентатну карбоксигрупу, фосфонкарбоксигрупу, дифосфонатну групу або сульфатну групу.
40. Спосіб за п. 39, в якому А являє собою хлоридну групу.
41. Спосіб за п. 1 що додатково включає спочатку утворення [PtA2OH2(L')(L)] шляхом реакції цис-[РtА2(L')(L)], утвореного на етапі b), з Н2O2, коли Y являє собою гідроксигрупу, а потім реакцію [PtA2OH2(L')(L)] з ацилюючим агентом з утворенням цисплатинового комплексу, що має формулу [PtA2Y2(L')(L)].
42. Спосіб за п. 41, що додатково включає етап перетворення А в А', де А' являє собою галогенову або негалогенову відщеплювану групу, відмінну від А.
43. Спосіб за п. 42, в якому А' вибирають з групи, що включає галоген, гідроксигрупу, алкоксигрупу, карбоксигрупу, а також бідентатну карбоксигрупу, фосфонкарбоксигрупу, дифосфонатну групу або сульфатну групу.
44. Спосіб за п. 43, в якому А являє собою хлоридну групу.
Текст
1. Спосіб одержання цисплатинового комплексу, що включає наступні етапи: a) реакцію [PtA4 ]2- або його солі з L в розчиннику зі створенням [РtА3(L)]¯ і b) реакцію [РtА3(L)]¯ з L' у другому розчиннику зі створенням цис-[PtA2(L')(L)], де групи L і L' різні і кожна з них є аміном або заміщеним аміном, який пов'язаний з атомом Pt координаційним зв'язком через атом азоту і являє собою гетероциклічний амін або гетероароматичний амін, або має формулу NRR'R", де R, R' або R" незалежно один від одного вибрані з групи, що включає водень, заміщені або незаміщені, розгалужені, нерозгалужені або циклічні аліфатичні, арильні, неароматичні або ароматичні гетероциклічні групи; за умови, що тільки L' може являти собою NН3 і що щонайменше одна з груп L і L' являє собою заміщений гетероциклічний або гетероароматичний амін, і де групи А можуть бути однаковими або різними, і кожна з них являє собою галогенову або негалогенову відщеплювану гр упу. 2. Спосіб за п.1, що додатково включає етап перетворення А в А', де А' являє собою галогенову або негалогенову відщеплювану груп у, відмінну від А. 2 (19) 1 3 71614 4 22. Спосіб за п. 21, в якому етап b) виконують при рН від 7 до 12. 23. Спосіб за п. 22, в якому етап b) виконують при N рН від ~8 до 10. , 24. Спосіб за п. 1, в якому етап b) виконують при L' - NHa, А - Сl і ОН, а Y - ОН. співвідношенні розчинника до платини, меншому 36. Цисплатиновий комплекс формули Іb або рівному ~5:1 (мл розчинника)/(ммоль плати, Y ни). L A 25. Спосіб за п. 1, в якому етап b) виконують при Pt молярному співвідношенні L' в формі вільної осноA ви до платини від 3:1 до 1:1. L Y 26. Спосіб за п. 2, в якому А' вибирають з групи, , Ib що включає галоген, гідроксигрупу, алкоксигрупу, де L являє собою карбоксигрупу, а також бідентатну карбоксигрупу, фосфонкарбоксигрупу, ди фосфонатну груп у або сульфатн у груп у. N 27. Спосіб за п. 26, в якому А являє собою хлори, дну гр уп у. L' - NH 3, А - Сl і ОН, а Y - ОН. 28. Спосіб за п. 1, в якому L являє собою 2-піколін. 37. Спосіб за п. 1 що додатково включає етап реа29. Спосіб за п. 1, в якому щонайменше одна з кції цис-[РtА2(L')(L)], утвореного на етапі b), з Н2O2, груп L і L' являє собою гетероциклічний амін або якщо Y являє собою гідроксигрупу, або з галогегетероароматичний амін, або має формулу ном, якщо Y являє собою галоген, зі створенням NRR'R", де R, R' або R" незалежно один від одного цисплатинового комплексу, що має формулу вибрані з групи, що включає водень, заміщені або [PtA2 Y2(L')(L)]. незаміщені, розгалужені, нерозгалужені або циклі38. Спосіб за п. 37, що додатково включає етап чні аліфатичні, арильні, неароматичні або аромаперетворення А в А', де А' являє собою галогенову тичні гетероциклічні групи. або негалогенову відщеплювану гр уп у, відмінну 30. Спосіб за п. 29, в якому L' являє собою NH3. від А. 31. Спосіб за п. 30, в якому А являє собою хлори39. Спосіб за п. 38, в якому А' вибирають з групи, дну гр уп у. що включає галоген, гідроксигрупу, алкоксигрупу, 32. Спосіб за п. 1, в якому L являє собою карбоксигрупу, а також бідентатну карбоксигрупу, фосфонкарбоксигрупу, ди фосфонатну груп у або сульфатн у груп у. N 40. Спосіб за п. 39, в якому А являє собою хлори, дну гр уп у. L' – NH 3, А - Сl, a Y - ОН . 41. Спосіб за п. 1 що додатково включає спочатку 33. Спосіб за п. 1, в якому L являє собою утворення [PtA2OH2(L')(L)] шляхом реакції цисN [РtА2(L')(L)], утвореного на етапі b), з Н2O2, коли Y являє собою гідроксигрупу, а потім реакцію [PtA2OH 2(L')(L)] з ацилюючим агентом з утворенN ням цисплатинового комплексу, що має формулу , [PtA2 Y2(L')(L)]. L' – NН3, А - Сl, a Y - ОН . 42. Спосіб за п. 41, що додатково включає етап 34. Спосіб за п. 1, в якому L являє собою перетворення А в А', де А' являє собою галогенову або негалогенову відщеплювану гр уп у, відмінну від А. N 43. Спосіб за п. 42, в якому А' вибирають з групи, , що включає галоген, гідроксигрупу, алкоксигрупу, L' - NH 3, А - Сl і ОАс, а Y - ОН. карбоксигрупу, а також бідентатну карбоксигрупу, 35. Спосіб за п. 1, в якому L являє собою фосфонкарбоксигрупу, ди фосфонатну груп у або сульфатн у груп у. 44. Спосіб за п. 43, в якому А являє собою хлоридну гр уп у. Дана заявка має пріоритет попередньої патентної заявки США сер. №60/128,939 від 13.04.1999p., яка включена в дану заявку у вигляді посилання. Даний винахід відноситься до області лікарських засобів на основі платини. Зокрема, він відноситься до вдосконаленого способу отримання амінних комплексів платини загальної формули (Іа) або (Іb): де L і L’ можуть бути однаковими або різними, 5 71614 6 при умові, що L' може являти собою NH3, але L не танії 2137198А описаний синтетичний спосіб може являти собою NH3; і отримання [PtX2(L)(L')], де L і L' являють собою кожна з груп L і L’ є аміном або заміщеним ліганди, зв'язані через азот аміну, і L L' (Rochon, аміном, який пов'язаний з атомом Pt координаційF.D.; Kong, P.-C. UK patent GB2137198A (1984) and ним зв'язком через атом азоту і являє собою гетеRochon, F.D.; Kong, P.-C. UK patent GB4533502 роциклічний амін або гетероароматичний амін або (1985)). Цей спосіб відомий в технології, і подромає формулу NRR'R", де R, R' або R" незалежно биці цього синтетичного способу опубліковані один від одного вибрані з групи, що включає во(Courtot, P.; Rumin, R.; Peron, Α.; Girault, J.P.J. день, заміщені або незаміщені розгалужені, нерозOrganornetallic Chem. 1978, 145, 343-357 and галужені або циклічні аліфатичні, арильні, неароRochon, F.D.; Kong, P.-C, Can. J. Chem. 1986, 64, матичні або ароматичні гетероциклічні групи; і 1894-1896). На Фіг.2 показаний цей спосіб для переважно L являє собою заміщений амін, де за[PtCl2(L)(L')] як приклад. При отриманні з K2[PtCl4] місник стерично утр уднює доступ атома Pt до ланспосіб, описаний в патенті США 4,533,502 і патенті цюга ДНК клітини, переважно пухлинної клітини; і Великобританії 2137198А, включає 4 етапи і ізолягрупи А можуть бути однаковими або різними і цію трьох проміжних продуктів. Олігомерний проявляють собою галоген або відщеплювану групу, міжний продукт представлений сполукою [PtLI2] x таку як гідроксигрупа, алкоксигрупа, карбоксигруде x=2-4; можливі також багатоолігомерні різновипа, і можуть бути однаковими або різними або ди. Загальний вихід з K2[PtCl4] в цьому патенті не утворювати двозубчасту карбоксигрупу, фосфонбув указаний. У способі використовуються іони карбоксигрупу, дифосфонатну гр упу або сульфатсрібла і йоду і утворюються відходи, забруднені ну гр уп у; і сполуками срібла і йоду. Υ являє собою галоген, гідроксигрупу, карбокВ даному патенті проміжною речовиною є сигрупу, карбамінову груп у або складноефірну [PtCl3L]-, в якому L являє собою амін, відмінний від груп у вугільної кислоти. NH3. Отримання [PtCl3L]- з розбавленого розчину У патентах США № 4,329,299 і 5,665,771 опиK2[PtCl4] в диметилформаміді (ДМФ), де L являє сані сполуки платини і їх корисність у якості протисобою піридин і похідні піридину, вже було описапухлинних засобів. Ці два патенти описують споно (Rochon, F.D.; Kong, P.-C. Can. J. Chem. 1978, луки платини, які охоплюють комплекси формули 56, 441-445 and Rochon, F.D.; Beauchamp, A.L.; цис-[PtA2(L')(L)] і ц, т, ц-[PtA2 Y2(L')(L)], де А - відBensimon, 3. Can. J. Chem. 1996, 74, 2121-2130). щеплювана група, така як галоген, гідроксигрупа Отримання [PtCl3L]- в розчинниках, відмінних від або карбоксигрупа, L - амін, зв'язаний через атом ДМФ або Н2О, або з іншими амінами, ніж піридин і азоту, і L' - амоній або заміщений амін. Спосіб похідні піридину, не описано. Синтез K[PtCl 3L] в отримання цих комплексів, описаних в патентах, ДМФ, як описано в літературі, виконували при 65відомий в даній технології (Hydes, P.C. патент 80°С, і вихід ізольованого продукту коливався від США 4,329,299 (1982); Murrer, В.А. US Patent 40% до 90% в залежності від похідного піридину. 5,665,771 (1997); Braddock, P.D.; Connors, T.A.; При синтезі [PtCl3L]- у ДМФ можуть утворюватися Jones, M; Khokhar, A.R.; Melzak, D.H.; Tobe, M.L. реакційноздатні або нестабільні комплекси платиChem.-Biol. Interactions 1975, 11, 145-161; and ни з ДМФ, які можуть утруднюва ти подальші реакGiandomenico, С.М.; Abrams, M.J.; Murrer, B.A.; ції або розкладатися з утворенням нерозчинних Vollano, J.F.; Rheinheimer, M.I.; Wyer, S.B.; Bossard, чорних платинових домішок. Наприклад, в Can. J. G.E.; Higgins (III), J.D. Ingorg. Chem. 1995, 34, 1015Chem. 1978, 56, 441 (також див. Chemical Abstracts 1021). Цей спосіб показаний на Фіг.1 для синтезу V.89 (July 1978), Abstract No. 35686), Rochon et al., цис-[PtCl2(NH3)(L)] і ц, т, ц-[PtCl2(OH)2(NH3)(L)] як описано осадження нерозчинної чорної речовини приклади. Синтез UHC-[PtCl 2(NH3)(L)] з вихідного при розчиненні К[РtCl3(2,6-диметилпіридину)] у матеріалу, який повсюдно випускається і часто водному розчині. Також описано, що під час ізолявикористовується, K2[PtCl4] включає чотири етапи, ції К[РtCl3(4-метилтридину)] і К[РtCl3(піридин)] була а синтез ц, т, ц-[PtCІ 2(OH)2(NH3)(L)] вимагає п'яти отримана масляниста паста, що містить етапів. Синтез цих комплексів способом, відомим в [РtCl2(ДМФ)(похідна піридину)], і інші домішки. технології, дає низький загальний вихід. У патенті Приклади комплексів [РtCl2(ДМФ)L] описані в літеСША №4,329,299 описаний загальний вихід з ратурі (Kong, P.-C; Rochon, F.D.; Can. J. Chem. K2[PtCl4] менше ніж 8%, а в патенті США 1979, 57, 682-684; Rochon, F.D.; Kong, P.-C; №5,665,771 і в літературі (Khokhar et al. And Melanson, R. Can. J. Chem. 1980, 58, 97-101; and Giandomenico et al.) описані загальні виходи 20Rochon, F.D.; Melanson, R.; Doyon, M.; Butler, I.S. 30%. Низький загальний вихід зумовлений велиInorg. Chem. 1994, 33, 4485-4493). кою кількістю етапів в способі, а також трудністю У Chemical Abstracts, V.126 (April 1997), здійснення і низькою ефективністю конверсії Abstract No. 194433 and Inorg. Chem. (1997), [PtCl2(NH3)2] в [PtCl 3(NH3)]-, яка вимагає викорис36:854-861 описано використання [Рt(цтання платинового каталізатора, який дорого кошС6Н11NH2)I2 ]2 в якості початкової речовини для тує. Синтез K[PtCl 3(NH3)] з [PtCl2(NH3)2] також не утворення Pt(NH3)(ц-C6H11NH2)Cl2 (Фіг.2). Однак ця дуже стійкий, і забезпечення великомасштабного реакція включала в себе розщеплення зв'язку Ptвиробництва K[PtCl3(NH3)] з [PtCl 2(NH3)2] із задовіA-Pt гр упою NH3 після утворення проміжної речольною якістю важкодосяжне. Вищеописаний спосіб вини з тими ж замісниками. вимагає використання іонів срібла і йоду, і в реВ Chemical Abstracts, V.108 (June 1988), зультаті утворюються відходи, забруднені сполуAbstract No. 215224 and Inorg. Chim. Acta (1988), ками срібла і йоду. 143:81-7 описана конверсія [Pt(Cl)4]2- в проміжну В патенті США 4,533,502 і патенті Великобриречовину [Pt(Cl)3NH3]1-, але не описано приєднан 7 71614 8 ня заміщеного циклічного аміну до цієї проміжної речовини. Посилання на вказані документи приведені не з метою визнання того, що все ви щеописане являє собою досягнення колишньої відповідної технології. Всі твердження відносно дат або змісту цих документів засновані на інформації, доступній защо включає наступні етапи: явникам, і не залишають сумнівів відносно правиa) реакцію [PtA4]2- або його солі з L в розчинльності дат і змісту цих документів. Крім того, всі нику зі створенням [PtA3(L)]-; документи, що згадуються тут, включені в дану b) реакцію [PtA3(L)]- з L' у другому розчиннику заявку у всій їх повноті у вигляді посилань. Зокрезі створенням цис-[PtA2(L')(L)]; ма, дана заявка підтверджує пріоритет попереc) у випадку, коли цисплатиновий комплекс дньої патентної заявки США сер. №60/128,939, має загальну формулу Ib, a Y являє собою гідрозареєстрованої 13 квітня 1999p., яка включена в ксигрупу або галоген, реакцію цис-[PtA2(L')(L)], дану заявку у вигляді посилання. утвореного на етапі b), з Н2О2, якщо Υ являє соДаний винахід надає спосіб отримання циспбою гідроксигрупу, або з галогеном, якщо Υ являє латинового комплексу загальної формули Іа або Іb собою галоген, зі створенням ц, т, ц-[PtA2 Y2(L')(L)]; і d) у випадку, коли цисплатиновий комплекс має загальну формулу Ib, a Y являє собою карбоксигрупу, карбамінову груп у або складноефірну груп у вугільної кислоти, спочатку створення [PtA2OH 2(L')(L)] з [PtA2(L')(L)] шля хом реакції цисщо включає наступні етапи: [PtA2(L')(L)], утвореного на етапі b), з Н2О2, згідно з 2a) реакцію [PtA4] або його солі з L в розчинетапом с), а потім реакцію [PtA2OH2(L')(L)] з ацинику зі створенням [PtA3(L)]-; люючим агентом зі створенням [PtA2 Y2(L')(L)]; і b) реакцію [PtA3(L)]- з L' у другому розчиннику е) перетворення А в А', де А' являє собою газі створенням цис-[PtA2(L')(L)]; логенову або негалогенову відщеплювану гр упу, c) у випадку, коли цисплатиновий комплекс відмінну від А; має загальну формулу Ib, a Y являє собою гідроде групи L і L' різні і кожна з них є аміном або ксигрупу або галоген, реакцію цис-[РtА2(L')(L)], заміщеним аміном, який пов'язаний з атомом Pt утвореного на етапі b), з Н2О2, якщо Υ являє сокоординаційним зв'язком через атом азоту і являє бою гідроксигрупу, або з галогеном, якщо Υ являє собою гетероциклічний амін або гетероароматичсобою галоген, зі створенням ц, т, ц-[PtA2 Y2(L')(L)]; ний амін або має формулу NRR'R", де R, R' або R" і незалежно один від одного вибрані з групи, що d) у випадку, коли цисплатиновий комплекс включає водень, заміщені або незаміщені розгамає загальну формулу Ib, a Υ являє собою карбоклужені, нерозгалужені або циклічні аліфатичні, сигрупу, карбамінову груп у або складноефірну арильні, неароматичні або ароматичні гетероцикгруп у вугільної кислоти, спочатку створення лічні групи; за умовою, що тільки L' може являти [PtA2OH 2(L')(L)] з [PtA2(L')(L)] шля хом реакції циссобою NH3 і що щонайменше одна з груп L і L' яв[PtA2(L')(L)], утвореного на етапі b), з Н2О2, згідно з ляє собою заміщений гетероциклічний або гетероетапом с), а потім реакцію [PtA2OH2(L')(L)] з ациароматичний амін; і люючим агентом зі створенням [PtA2 Y2(L')(L)]; і де групи А можуть бути однаковими або різде групи L і L' різні і кожна з них є аміном або ними, і кожна з них являє собою галогенову або заміщеним аміном, який пов'язаний з атомом Pt негалогенову відщеплювану груп у. координаційним зв'язком через атом азоту і являє Винахід також надає цисплатиновий комплекс собою гетероциклічний амін або гетероароматичформули Іb ний амін або має формулу NRR'R", де R, R' або R" незалежно один від одного вибрані з групи, що включає водень, заміщені або незаміщені розгалужені, нерозгалужені або циклічні аліфатичні, арильні, неароматичні або ароматичні гетероциклічні групи; за умовою, що тільки L' може являти собою NH3 і що щонайменше одна з груп L і L’ явде L являє собою ляє собою заміщений гетероциклічний або гетероароматичний амін; і де групи А можуть бути однаковими або різними, і кожна з них являє собою галогенову або L' являє собою NH3, А являє собою СІ і ОН, a негалогенову відщеплювану груп у. Y являє собою ОН. Даний винахід також надає спосіб отримання Даний винахід описує більш ефективний і екоцисплатинового комплексу загальної формули Іа номічний спосіб отримання платинових комплексів або Іb вигляду цис-[PtA2(L')(L)] (формула Іа) і ц, т, ц[PtA2 Y2(L')(L)] (формула Іb) безпосередньо з недорогого і платинового вихідного матеріалу, що повсюдно випускається, переважно тетрагалоплати 9 71614 10 ніту, такого як [PtCl4]2- або [PtBr4] 2-, тичні гетероцикличні групи; і переважно L являє де: собою заміщений амін, де замісник стерично струL і L' можуть бути однаковими або різними, за днює доступ атома Pt до ланцюга ДНК клітини, умовою, що L' може являти собою NH3, але L не переважно пухлинної клітини. Приклади такої заможе являти собою NH3; і міщеної групи L або L’ включають (але не обмекожна з груп L і L' є аміном або заміщеним жуються цим): алкіламіни, які можуть включати аміном, який пов'язаний з атомом Pt координаційметиламін; диметиламін; трибутиламін; діізопропіним зв'язком через атом азоту і являє собою гетеламін; ариламіни, які можуть включати анілін, тороциклічний амін або гетероароматичний амін або луїдин, амінонафтален і аміноантрацен; гетероцимає формулу NRR'R", де R, R' або R" незалежно клічні аміни, які можуть включати піперидин, один від одного вибрані з групи, що включає вопіперазин і піролідин; і гетероароматичні аміни, які день, заміщені або незаміщені розгалужені, нерозможуть включати: піридин, піразоли, імідазоли, галужені або циклічні аліфатичні, арильні, неарооксазоли, ізооксазоли; піримідин і піразин. Існують матичні або ароматичні гетероциклічні групи; і також інші замісники, відомі фахівцям в цій обласпереважно L являє собою заміщений амін, де заті, які легко можуть зрозуміти, що ці інші замісники місник стерично утр уднює доступ атома Pt до ланможуть бути використані в даному винаході спосоцюга ДНК клітини, переважно пухлинної клітини; і бом, який з'ясується з даним описом. групи А можуть бути однаковими або різними і Зокрема, наприклад, у випадку заміщених циявляють собою галоген або відщеплювану групу, клічних амінів замісником може бути нижчий алкіл таку як гідроксигрупа, алкоксигрупа, карбоксигруабо алкоксигрупа з 1-4 атомів вуглецю (особливо па, і можуть бути однаковими або різними або метил або метокси-), галоген (особливо хлор або утворювати двозубчасту карбоксигрупу, фосфонбром) або арил (особливо бензил). Сам замісник карбоксигрупу, дифосфонатну гр упу або сульфатможе бути заміщеним нижчим алкілом або галогену гр уп у; і ном. Вираз "нижчий алкіл" означає алкільну групу, Υ являє собою галоген, гідроксигрупу, карбокщо містить 1-6 атомів вуглецю. Циклічний амін сигрупу, карбамінову груп у або складноефірну може нести інші замісники, або суміжні з коордигруп у вугільної кислоти. наційним атомом азоту, або розташовані в іншому В одному з варіантів здійснення спосіб по дамісці кільця. Інші замісники включають електрононому винаходу переважний для отримання сполуакцепторні або електронодонорні замісники, такі ки формули Іа. як нітро- і алкокси наприклад, метокси-. Якщо цикТерміни, що використовуються тут, відповідалічний амін являє собою кільцеву систему, що ють їх загальноприйнятим значенням і з даного злилася, де кільце, що злилося, являє собою ароопису повинні бути цілком зрозумілі фахівцям. Для матичне кільце в положеннях 2 і 3 циклічного амібільшої ясності терміни також можуть мати спеціну, інши х замісників не потрібно, хоча замісник альні значення, які будуть зрозумілі по їх викорисможе бути присутнім. Можна також передбачити танню в контексті. Наприклад, ліганд - це іон або можливість використання даного винаходу для молекула, яка пов'язана з атомом або іоном метаотримання транс-ізомерів. У переважному варіанті лу і яка розглядається як створююча з ним коваздійснення даний винахід використовують для лентний зв'язок. "Однозубчастий" означає той, що отримання цис-ізомерів. має одне положення, за допомогою якого можуть Для ілюстрації винаходу як приклади викорисутворюватися ковалентні, або координаційні, зв'ятаний синтез цис-[РtСl2(NH3)(L)] і ц, т, цзки з металом. "Двозубчастий" означає той, що [PtCl2(OH)2(NH3)(L)] з [PtCl4]2-. Споріднені сполуки має два положення, за допомогою яких можуть цис-[PtBr2(NH3)(L)] і ц, т, ц-[PtBr 2(OH)2(NH3)(L)] таутворюватися ковалентні, або координаційні, зв'якож можуть бути отримані подібним чином з зки з металом. Даний винахід переважно відно[PtBr4] 2-. ситься до утворення монозубчастих координаційДля отримання цис-[PtCl2(NH3)(L)] вдосконаних зв'язків амінів L і L' через атом азоту з Pt. Далі, лений спосіб використовує два етапи, де перший "стерично утр уднений" використовується в загальетап являє собою конверсію суспензії або конценноприйнятому значенні. "Стерично утр уднений трованого розчину [PtCl4]2- в [PtCl 3L]- в апротонних амін" тут означає амінний компонент, який, внаслірозчинниках. Другий етап перетворює суспензію док його розміру або маси утруднює або перешкоабо концентрований розчин [PtCl3L]- в цисджає обертанню або іншій функції або властивості [PtCl2(NH3)(L)] в розчині гідроксиду амонію. У порібудь-якого іншого компонента платинових комплевнянні з синтетичними способами, що використоксів, що описуються тут. Способи по даному винавуються в цей час, вдосконалений спосіб включає ходу переважно використовують для отримання меншу кількість етапів синтезу, меншу кількість сполук, описаних в патенті США 5,665,771 (зокрепродуктів, що ізолюються, вимагає менших об'ємів ма, стерично утруднених амінів, визначених форекологічно шкідливих розчинників, виробляє менмулою Іа в патенті '771), зміст якого, в тому числі ше відходів, забруднених металами, і забезпечує визначення описаних в ньому груп-замісників, отримання цис-[РtСl2(NН3)(L)] з підвищеним загавключені в дану заявку у вигляді посилання. Вираз льним виходом. Спосіб також не вимагає викорис"заміщений" тут означає відносно L і L’ як пов'язатання іонів срібла і йоду і не виробляє відходів, них через азот гетероциклічних амінів або гетерозабруднених сполуками срібла і йоду. Всі етапи ароматичних амінів, що група-замісник незалежно способу стійкі, відтворюються і постійно вироблявибрана з групи, яка включає водень, заміщені або ють продукти однакової якості. незаміщені розгалужені, нерозгалужені або цикліПерший етап вдосконаленого способу являє чні аліфатичні, арильні, неароматичні або аромасобою реакцію [PtCl4]2- з аміном L у відповідних 11 71614 12 умовах в першому розчиннику зі створенням Can. J. Chem.1978, 56, 441-445 and Rochon, F.D.; [PtCl3L]-. Звичайно використовують калієву сіль Beauchamp, A.L.; Bensimon, С. Can. J. Chem. 1996, [PtCl4]2-, що найбільш широко випускається. Однак 74, 2121-2130). Для синтезу K[PtCl3L] в якості ілюсінші солі [PtCl4]2- також можуть бути використані. тративного прикладу використаний синтез Відповідні умови тут означають такі умови реакції, K[PtCl 3(2-піколіну)] для порівняння опублікованого які сприяють описаній і заявленій хімічній реакції. способу зі способом по винаходу. У оп ублікованоЗокрема, даний винахід передбачає, що такі відму способі ізоляція K[PtCl3(2-піколіну)] вимагає повідні умови включають (але не обмежуються двох окремих етапів, під час кожного з яких розцим): температур у; рН; концентрацію реагентів; чинники випаровують при зниженому тиску. Для міру збовтування; дисперсність реагентів і інші випаровування ДМФ при зниженому тиску потріумови, що полегшують хімічні реакції, які описубен нагрів при 40°С. У крупномасштабному промиються. Однак інші відповідні умови, відомі фахівсловому синтезі випаровування розчинників при цям в даній області, також можуть вплинути на зниженому тиску, особливо з нагрівом, являє соетапи хімічних реакцій, що описуються. Для полебою і трудомістку операцію, яка дорого коштує. У гшення розчинення K2[PtCl4] переважно викориснашій переважній операції синтез і ізоляція тати тонкодисперсний порошок K2[PtCl4]. ПереваK[PtCl 3(2-піколіну)] не вимагає випаровування розжно, щоб порошок K2[PtCl4] мав розмір ~240мкМ. чинників і не вимагає перенесення речовини з одБільш переважно, щоб порошок K2[PtCl4] мав розного розчинника в іншій. Спосіб по винаходу більш мір ~100мкМ. У процесі реакції 1-1,3екв. аміну реаефективний і краще пристосований для великомагує з 1екв. K2[PtCl4 ]. Більш переважно використансштабного промислового виробництва цієї сполуня 1-1,2екв. аміну. Найбільш переважно ки. Обидва способи забезпечують отримання використання 1,05-1,15екв. аміну з 1екв. K2[PtCl4]. К[РіС13(2-піколіну)] з порівнянним виходом і якісВикористання високих еквівалентів аміну збільшує тю. Дані інфрачервоного і ЯМР-спектра K[PtCl3(2швидкість реакції, але може також збільшити утвопіколіну)], отриманого способом, відомим в колишрення побічних продуктів і знизити вихід реакції. ній технології, і способом по винаходу, показані на Крім того, амін L додають до реакційної суміші Фіг.4 і 5. Синтез [PtCl3L]- в інших апротонних розмалими порціями протягом певного періоду часу. чинниках, таких як ацетон, хлороформ, дихлормеПереважно, амін додають двома або більше рівтан і N-метилпіролідинон, також продемонстрованими порціями, або, більш переважно, чотирма ний в даному винаході. або більше порціями. Координаційний зв'язок молекул розчинника з Реакція може проводитися при температурі Pt, що спричиняє створення реакційноздатних або близько 30-100°С, але більш переважно виконання нестабільних сполук платини, ускладнює здійсреакції при температурі близько 40-70°С. Найнення способу по винаходу. В опублікованому більш переважно використання температурного способі (Rochon, F.D.; Kong, P.-C. Can. J. Chem. діапазону 50-65°С. Загалом, чім вище реакційна 1978, 56, 441-445) описано [PtCl2(ДМФ)(похіднє температура, тим більше швидкість реакції між пірідину)] і інші домішки в синтезі [PtCl3(похідного [PtCl4]2- і аміном. Однак висока реакційна темперапірідину)]. В даному винаході ми приводимо темтура може збільшити створення побічних продуктів пературний діапазон, в якому створення небажаабо допустити створення реакційноздатних і нених сполук, таких як [РtСl2(ДМФ)(похідне пірідину)], стабільних платинових домішок. У розчинниках, мінімізовано. Створення чорного осаду під час здатних до створення координаційних зв'язків з ізоляції продукту є показником присутності реакатомами металів, таких як ДМФ, температура реаційноздатних платинових домішок. При викорискції ~60°С може сприяти утворенню комплексів Pt з танні синтезу K[PtCl3(2-niколіну)] в диметилфоррозчинником, які будуть розкладатися або перемаміді, як приклад, при проведенні синтезу нижче шкоджати проведенню наступного етапу способу. ~за 60°С утворення чорних осадів не спостерігаРеакцію проводять в апротонних розчинниках. ється. У найбільш переважному режимі перший Переважно, щоб розчинник містив менше ~за 25% реакційний етап проводять при температурі реакводи, але більш переважно вміст води менше ~за ції ~50-65°С. Однак даний спосіб включає будь-яку 10%. Найбільш переважно вміст води менше ~за температуру, при якій утворення небажаних спо3%. Реакція може проводитися в апротонних розлук або домішкових продуктів реакції, таких як чинниках, таких як ацетон, хлороформ, дихлорме[РtСl2(ДМФ)(похіднє пірідину)], мінімізовано (10(?) також може привести до зниження вихоутворювати двозубчасту карбоксигрупу, фосфонду через підвищене утворення побічних мультикарбоксигрупу, дифосфонатну гр упу або сульфатамінних сполук платини. ну груп у; і Υ являє собою галоген, гідроксигрупу, Реакцію проводять при концентрації 1г карбоксигрупу, карбамінову груп у або складноефіK[PtCl 3L] на 3-10мл розчинника. Концентрація 1г рну гр упу вугільної кислоти. K[PtCl 3L] на 4-8мл розчинника більш переважна, а Для комплексів 1а або 1b в технології відомі концентрація 1г K[PtCI3L] на 5-7мл розчинника способи перетворення ліганда А в іншу віщеплюнайбільш переважна. Несподівано виявилося, що вану гр упу (гр упи), таку як галоген, гідроксигрупу, проведення реакції при високій концентрації сприалкоксигрупу або монозубчасту карбоксигрупу, або яє ефективному створенню продукту з високим двозубчасту карбоксигруппу, або двозубчасту фовиходом. Реакція може проводитись при набагато сфонкарбоксигрупу, або двозубчасту фосфонатну більш низькій концентрації, але вихід був низьким груп у, або двозубчасту сульфатн у груп у. Приклади через утворення побічних продуктів. Більш високі таких трансформацій показані в Рівнянні 1 і Рівоб'єми розчинників і більш низька концентрація нянні 2. Можуть бути також розглянуті багато які також вимагають видалення великих кількостей інші перестановки і комбінації конверсій відщепекологічно шкідливих розчинників і відходів. Перелюваних гр уп, здатні привести до утворення кориважно провести цю реакцію в суворо водних розсних комплексів. Спосіб отримання проміжних речинах. Однак поєднання органічних і водних розчовин по винаходу може бути корисний для чинників також можливо. Другий розчинник може отримання всіх ци х сполук. містити хлорид в концентрації близько 0,1-6N. ЗоРівняння 1. Спосіб отримання комплексу форкрема, даний спосіб передбачає, що другий реакмули Іа, в якому дві відщеплювані групи А являють ційний етап 1b) проводять при співвідношенні розсобою галогени і відрізняються одна від одної. чинника до платини £5:1 (мл розчинника)/(ммоль платини). Другий етап способу проводять в діапазоні співвідношення NH3/Pt приблизно від 3 до 7. Співвідношення NH3/Pt приблизно від 4 до 6 переважно, а від 4,5 до 5,5 - найбільш переважно. Даний спосіб передбачає, що другий реакційний етап Рівняння 2. Конверсія обох лігандів А (де А = 1b проводять при молярному співвідношенні L' в галоген) для утворення нової сполуки, в якій обидформі вільної основи до платини приблизно від 3:1 ві групи А однакові і утворюють двозубчасту кардо 1:1. Великий надлишок NH3 знижує час реакції, боксильну гр упу. але може збільшити утворення побічних мультиамінних/амінних сполук платини. З цис-[PtA2(NH3)(L)] комплекс ц, т, ц[PtA2(OH) 2NH3)(L)] може бути отриманий шляхом реакції суспензії цис-[PtA2(NH3)(L)] з перекисом водня. З ц, т, ц-[PtA2(OH) 2(NH3)(L)] можуть бути Приведений вище загальний опис винаходу отримані інші комплекси Pt(IV) формули ц, т, цбуде більш зрозумілим при розгляді наступних [PtA2 Y2(NH3)(L)], де Υ являє собою галоген, гідроприкладів, які дані лише для ілюстрації і не обмексигрупу, карбоксигрупу, карбамінову групу або жують даний винахід, якщо не вказане інше. складноефірну групу вугільної кислоти, на відміну На Фіг.1 показаний синтез цис-[РtСl2(NH3)(L)] і від випадку, коли обидві групи Υ являють собою ц, т, ц-[PtX2 Y2(NH3)(L)] через проміжну речовину гідроксигрупу, з використанням способів, відомих в K[PtCl 3NH3]. технології. На Фіг.2 показаний синтез [PtCl2(L)(L')] через Приклади, показані для ілюстрації отримання проміжний олігомер [PtI2(L)] x. цис-[PtCl2(NH3)(L)] і ц, т, ц-[PtA2(OH)2(NH3)(L)], моНа Фіг.3 показаний синтез [PtA2(L')(L)] спосожуть бути також використані для отримання сполук бом по даному винаходу. загальної формули цис-[РtА2(L)(L')] і ц, т, цНа Фіг.4 показані дані інфрачервоного спектра [PtA2 Y2(L)(L')], де L і L' можуть бути однаковими і спектра ядерного магнітного резонансу для або різними, при умові, що L' може являти собою [PtCl2(NH3)(2-піколіну)], отриманого способом по NН3, але L не може являти собою NH 3; і кожна з винаходу. Фіг.4 А інфрачервоний спектр груп L і L' є аміном або заміщеним аміном, який [PtCl2(NH3)(2-піколіну)], отриманого способом по зв'язаний з атомом Pt координаційним зв'язком винаходу. Фіг.4В - спектр ЯМР 195Pt для через атом азоту і являє собою гетероциклічний [PtCl2(NH3)(2-піколіну)], отриманого способом по амін або гетероароматичний амін або має формувинаходу. Фіг.4С - спектр ЯМР 1Н для лу NRR'R", де R, R' або R" незалежно один від (PtCl2(NH3)(2-піколіну)], отриманого способом по одного вибрані з групи, що включає водень, замівинаходу. щені або незаміщені розгалужені, нерозгалужені На Фіг.5 показані дані інфрачервоного спектра або циклічні аліфатичні, арильні, неароматичні або і спектра ядерного магнітного резонансу для ароматичні гетероциклічні групи, і переважно L [PtCl2(NH3)(2-піколіну)], отриманого способом, віявляє собою заміщений амін, де замісник стеричдомим в технології, показаним на Фіг.1. Фіг.5 А но утр уднює доступ атома Pt до ланцюга ДНК кліінфрачервоний спектр [PtCl2(NH3)(2-піколіну)], тини, переважно пухлинної клітини. Групи А моотриманого способом, відомим в технології, пока 15 71614 16 заним на Фіг.1. Фіг.5В - спектр ЯМР 195Pt для Приклад 1) Синтез K[PtCl 3(2-піколіну)] в N[PtCl2(NH3)(2-піколіну)], отриманого способом, віметилпіролідиноні домим в технології, показаним на Фіг.1. Фіг.5С K2[PtCl4] розтерли в дуже тонкодисперсний спектр ЯМР 1Н для [PtCl2(NH3)(2-піколіну)], отрипорошок за допомогою ступки і товкачика. 3,5047г маного способом, відомим в технології, показаним (8,443ммоль) K2[PtCl4] вмістили в 25-мл круглона Фіг.1. донну колбу і додали 6-7мл безводного NПриклади метилпіролідинону (NMP). 0,8648г (9,286ммоль) 2У приведених нижче прикладах сполуки аналіпіколіну вмістили в 3-4мл NMP і розділили на 5 зували методами спектроскопії ЯМР 1Н і 195Рt’ рівних порцій. Першу порцію 2-піколіну додали до елементарного аналізу і HPLC. Спектри ЯМР заплатинової суміші. Суміш повністю занурили в 60писували на спектрометрі Braker Avance 300 (ЯМР градусну (°С) масляну баню і перемішували при 1 Н і 195Рt) в ДМФ і порівнювали зі спектрами ета1200об/хв. Наступні порції 2-піколіну додавали лонних сполук, синтезованих способами, відомими через 30-35 хвилинні інтербали. Міра додавання 2в те хнології. Елементарний аналіз (%С, %Н, %N) піколіну становила 20% кожні 30-35хв. Після додавиконували з використанням аналізатора Perkin вання останньої порції реакційну суміш залишили Elmer 2400 or Carlo Erba 1108. Процентний вміст для протікання реакції ще на 50-60хв. У кінці реакСІ визначали шляхом титрування нітратом срібла. ції реакційний розчин був жовтогарячого кольору. Дві методики HPLC (аніонну і катіонну методики Реакційному розчину дали охолодитися до навкоHPLC) використали для аналізу сполук, представлишньої температури. До реакційного розчину долених в нижченаведених прикладах. Для аніонної дали 100мл метиленхлориду при навколишній методики HPLC час утримання [PtCl2(NH3)(2температурі. Додавання метиленхлориду викликапіколіну)] становить 3 хвилини. Час утримання в ло осадження K[PtCl3(2-піколіну)] і KСI. Осад зіHPLC синтезованих сполук порівнювали згодом брали шляхом вакуумної фільтрації з використанутримання еталонних сполук, отриманих спосоням склоподібної фрити і промили бом, відомим у технології. Робочі умови аніонної і метиленхлоридом (3x5мл), а потім діетиловим катіонної методик HPLC наступні: ефіром (3x5мл). Осад висушили під вакуумом при Катіонна методика HPLC: навколишній температурі протягом 16-24 годин і Колонка: Hichrom-RPB, 5мкм, 100мм x 4,6мм, зважили. Вихід: 3,8440г (86,8%). Аналітичний 100Ε, сірий. №HIRPB3374 склад розрахунковий (виявлений) для Рухлива фаза:А: 0,02М Н3РО4 (99,999 %, С6H7N1СІ3KРt·KPt·1,2K1СI 1: 13,74 (13,54); Η, 1,35 Aldrich 34524-5), 5мМ гексансульфонової кислоти (1,39); Ν, 2,67 (2,59); СІ, 28,51 (28,32). Спектр ЯМР 1 (Sigma 39705-9), відрегульованої до рН 2,7 концеН (300МГц, ДМФ): 9,12 (d, 1 пірідин Н); 7,90 (t, 1 нтрованим NaOH пірідин Н); 7,61 (d, 1 пірідин Н); 7,40 (t, 1 пірідин Н); В: метанол (Fisher, марки HPLC) 3,40 (s, 3 метил Н). Спектр ЯМР 195Pt (300МГц, Градієнт: 0хв. 95%А 5%В ДМФ-d 6): відповідає спектру Я МР 195Pt K[PtCl 3(26хв. 95%А 5%В піколіну)], отриманому способом, відомим в техно20хв. 50%А 50%В логії. HPLC: (аніонна методика): час утримання 25хв. 50%А 50%В відповідає часу утримання для еталонної сполуки. 25,01хв. 95%А 5%В Приклад 2) Синтез K[PtCl 3(2,6-лутидину)] в Nметилпіролідиноні K2[PtCl4] розтерли в дуже тонкодисперсний Загальний час пропускання: 35,01хв. порошок за допомогою ступки і товкачика. 1,9427г Швидкість течії: 1,0мл/хв (4,68ммоль) K2[PtCl4] вмістили в 15-мл круглодонТемпература: 25°С ну колбу і додали 4мл безводного NMP. 0,5501г Детектор: DAD @ 267нм (5,13ммоль) 2,6-лутидину вмістили в 3-4мл NMP і Уприскування: 10мкл розділили на 5 рівних порцій. Першу порцію 2,6Аніонна методика HPLC: лутидину додали до платинової суміші. Суміш поКолонка: Hichrom-RPB С8/С18, 5мкм, 100мм x вністю занурили в 60-градусну (°с) масляну баню і 4,6мм, 100Ε, сер. №HIRPB3265 перемішували при 1200об/хв. Наступні порції 2,6Рухлива фаза: А: 0,02 М Н3РО4 (99,999%, лутидину додавали через 30-35 хвилинні інтерваAldrich 45228-9), 5мМ тетрабутиламонійгалогенсули. Міра додавання 2,6-лутидину становила 20% льфату (Sigma 39683-4), відрегульованого до рН кожні 30-35хв. Загальний час реакції становив 24 2,5 концентрованим NaOH години. У кінці реакції реакційний розчин був жовВ: метанол (Fisher, марки HPLC) тогарячого кольору. Реакційному розчину дали Градієнт: 0хв. 95%А 5%В охолодитися до навколишньої температури. До 5хв. 95%А 5%В реакційного розчину додали 200мл метиленхло22хв. 65%А 35%В риду при навколишній температурі. Додавання 23хв. 50%А 50%В метиленхлориду викликало осадження K[PtCl3(2,628хв. 50%А 50%В лутидину)] і KСI. Осад зібрали шляхом вакуумної 30хв. 95%А 5%В фільтрації з використанням склоподібної фрити і промили метиленхлоридом (3x5мл), а потім діетиЗагальний час пропускання: 40хв. ловим ефіром (3x5мл). Осад висушили під вакууШвидкість течії: 1,0мл/хв мом при навколишній температурі протягом 16-24 Температура: 35°С годин і зважили. Вихід: 2,1415г (84,7%). АналітичДетектор: DAD при 230нм ний склад розрахунковий (виявлений) для Уприскування: 15мкл C7H9N1Cl3KPt·1,24K1Cl1: С, 15,57 (15,40); Η, 1,68 Приклади 1-9 описують 1-й етап способу. 17 71614 18 (1,72); Ν, 2,59 (2,60); СІ, 27,83 (27,70). Спектр ЯМР ний склад розрахунковий (виявлений) для 1 Н (300МГц, ДМФ-d6): 7,6 (t, пірідин Η); 7,28 (d, 2 C7H9N1Cl3KPt·0,1K2[PtCl 4]: С, 17,19 (17,20); Η, 1,85 пірідин Η); 3,51 (s, 3 метил Н); 3,43 (s, 3 метил Η). (1,90); Ν, 2,86 (2,935); СІ, 24,64 (24,61). Спектр Приклад 3) Синтез K[PtCl 3(2-піколіну)] в димеЯМР 1Н (300МГц, ДМФ-d6): 7,6 (t, пірідин Η); 7,28 тилформаміді при 50°С (d, 2 пірідин Η); 3,51 (s, 3 метил Η); 3,43 (s, 3 метил K2[PtCl4] розтерли в дуже тонкодисперсний Η). порошок за допомогою ступки і товкачика. 2,6461г Приклад 5) Синтез [PtCl 3(2-піколіну)]- в ацето(6,375ммоль) K2[PtCl4] вмістили в 25-мл круглоні, дихлорметані або хлороформі донну колбу і додали 6мл безводного ДМФ. 1,0040г (2,419ммоль) K2[PtCl4] вмістили в 250,6233г (6,693ммоль) 2-піколіну вмістили в 3-4мл мл круглодонну колбу і додали 1мл ацетону. 0,67г NMP і розділили на 5 рівних порцій. Першу порцію (2,4 моль) тетрабутиламонійхлориду розчинили в 2-піколіну додали до платинового розчину. Реак2мл ацетону і додали до розчину K2[PtCl4]. 0,2783г ційну суміш занурили в 50-градусну (°с) масляну (2,988ммоль) 2-піколіну розчинили в 2мл ацетону і баню і залишили для протікання реакції приблизно додали до платинового розчину. Реакційну суміш на 120хв. У кінці реакції реакційний розчин був нагріли до 60°С. K2[PtCl4] поступово розчинився жовтогарячого кольору. Реакційному розчину дали протягом години і перетворився в більш розчинну охолодитися до навколишньої температури. До тетрабутиламонієву сіль [PtCl4]2-. Реакційний розреакційного розчину додали 100мл хлороформу чин перемішували при 50°С протягом 16 годин. У при навколишній температурі. Додавання хлорокінці реакції реакційний розчин був жовтогарячого форму викликало осадження K[PtCl3(2-піколіну)] і кольору. Реакційний розчин профільтрували для KСI. Осад зібрали шляхом вакуумної фільтрації з видалення KСI і видалили ацетон при зниженому використанням склоподібної фрити і промили метиску, отримавши масло жовтогарячого кольору, тиленхлоридом (3x5мл), а потім діетиловим ефівідповідне продукту [PtCl3(2-піколін)]1-. Спектр ром (3x5мл). Осад висушили під вакуумом при ЯМР 1Н (300МГц, ДМФ): 9,0 (d, 1 пірідин Η); 7,8 (t, навколишній температурі протягом 16-24 годин і пірідин Η); 7,45 (d, 1 пірідин Η); 7,25(t, 1 пірідин Η); зважили. При розчиненні продукту у водному роз3,20 (s, 3 метил Н). Спектр ЯМР 195Pt (300МГц, чині чорного осаду не спостерігалося. Вихід: ДМФ-d 6): відповідає еталону. 2,8565г (84%). Аналітичний склад розрахунковий Ідентичну методику використали для отри(виявлений) для С 6Н7N1СI3KРt·1,3K1СII: С, 13,58 мання [PtCl3(2-піколіну)]1- в хлороформі або дих(13,65); Н, 1,33 (1,31); N, 2,67 (2,64); СІ, 28,73 лорметані в якості розчинників. Спектр ЯМР 1Н: 1 (28,78). Спектр ЯМР Н (300МГц, ДМФ): 9,12 (d, 1 відповідає еталону. пірідин Η); 7,90 (t, пірідин Η); 7,61 (d, 1 пірідин Η); Для ізоляції [PtCl3(2-піколіну)]1- у вигляді каліє7,40 (t, 1 пірідин Η); 3,40 (s, 3 метил Η). Спектр вої солі жовтогаряче масло розчинили в 2мл меЯМР 195Pt (300МГц, ДМФ-d6): відповідає спектру танолу. Додали ацетат калію, розчинений в метаЯМР 195Рt K[PtCl 3(2-піколіну)], отриманому спосонолі, викликавши осадження K[PtCl3(2-піколіну)]. бом, відомим в технології. HPLC: (аніонна методиОсад висушили під вакуумом при навколишній ка): час утримання відповідає часу утримання для температурі протягом 16-24 годин і зважили. Виеталонної сполуки. хід: 0,5762г (55%). Приклад 4) Синтез K[PtCl3(2,6-лутидину)] в Приклад 6) Синтез [PtCl 3(трибутиламіну)]- з тедиметилформаміді при 50°С трабутиламонію тетрахлорплатинату в ацетоні K2[PtCl4] розтерли в дуже тонкодисперснии 0,2715г (0,33ммоль) тетрабутиламонію тетрапорошок за допомогою ступки і товкачика. 1,0900г хлорплатинату розчинили в ацетоні. 0,1323г (2,62ммоль) K2[PtCl4] вмістили в 15-мл круглодон(0,7135ммоль) трибутиламіну додали до платинону колбу і додали 2-3мл безводного ДМФ. 0,3078г вого розчину. Реакційний розчин нагрівали при (2,87ммоль) 2,6-лутидину вмістили в 1-2мл ДМФ і 60°С протягом ночі. Реакційний розчин профільтрозділили на 5 рівних порцій. Першу порцію 2,6рували для видалення KСI і видалили ацетон при лутидину додали до платинової суміші. Суміш позниженому тиску, отримавши масло жовтогарячого вністю занурили в 50-градусну (°с) масляну баню і кольору, відповідне продукту перемішували при 1200об/хв. Наступні порції 2,6[PtCl3(трибутиламін)]2-. Спектр ЯМР 195Pt (300МГц, лутидину додавали через 30-35 хвилинні інтерваДМФ-d 6): відповідає еталону. Для ізоляції ли. Міра додавання 2,6-лутидину становила 20% [PtCl3(трибутиламіну)]2- у вигляді калієвої солі жовкожні 30-35хв. Загальний час реакції становив 72 тогаряче масло розчинили в 2мл метанолу. Додагодини. У кінці реакції реакційний розчин був жовли ацетат калію, розчинений в метанолі, викликатогарячого кольору. Реакційному розчину дали вши осадження K[PtCl3(трибутиламіну)]. Осад охолодитися до навколишньої температури і провисушили під вакуумом при навколишній темперафільтрували. До реакційного розчину додали турі протягом 16-24 годин і зважили. Вихід: 0,1577г 100мл метиленхлориду при навколишній темпера(64%). турі. Додавання метиленхлориду викликало осаПриклад 7) Синтез K[PtCl3(2,5дження K[PtCl3(2,6-лутидину)]. Осад зібрали шлядиметилпіразину)] в N-метилшролідиноні (NMP) хом вакуумної фільтрації з використанням K2PtCl4 розтерли в дуже тонкодисперсний посклоподібної фрити і промили метиленхлоридом рошок за допомогою ступки і товкачика. 1,0724г (3x5мл), а потім діетиловим ефіром (3x5мл). Осад (2,58ммоль) K2PtCl4 вмістили в 10-мл круглодонну висушили під вакуумом при навколишній темпераколбу і додали ~5мл NMP. Реакційну суміш перетурі протягом 16-24 годин і зважили. При розчимішали при 700об/хв і колбу занурили в масляну ненні продукту у водному розчині чорного осаду не баню при 65°С. 0,3196г (2,96ммоль) 2,5спостерігалося. Вихід: 0,6815г (53,1%). Аналітичдиметилпіразину змішали з ~1мл NMP. Близько 4 19 71614 20 рівних порцій розчину 2,5-диметилпіразину додали 6,819г (12,50ммоль) K[PtCl3(2-піколіну)] 1,5KСI до реакційної суміші з 30-хвилинними інтервалавмістили в 25-мл круглодонну колбу і додали 10мл ми. Після останнього додавання реакційну суміш 2,5N розчину KСI. 8,2688г (63,12ммоль) амоній залишили для протікання реакції на 60хв. і потім ацетату тригідрату розчинили в 25мл 2,5N розчину охолодили до навколишньої температури. До реаамонію гідроксиду і додали до платинової суміші, кційної суміші додали 150мл метиленхлориду. Дощо перемішується. Загальний об'єм реакційної давання метиленхлориду викликало осадження суміші становив ~35мл. Жовтогарячу суміш занупродукту. Осад зібрали шляхом вакуумної фільтрили в 45-градусну (°С) масляну баню і перемішурації з використанням склоподібної фрити і провали 1 годину в темряві при швидкості >1000об/хв. мили метиленхлоридом (3х30мл), а потім діетилоЖовтогаряча суміш поступово перетворилася в вим ефіром (3x10мл). Осад висушили під жовту. Жовтий осад зібрали шляхом вакуумної вакуумом при навколишній температурі протягом фільтрації з використанням склоподібної фрити і 16 годин і зважили. Вихід: 1,0507г (66,3%). Аналіпромили водою (2x5мл) і ацетоном (3x5мл). Осад тичний склад розрахунковий (виявлений) для висушили під вакуумом при навколишній темпераC6H8N2Cl3KPt·2,2KСI: С, 11,73 (11,50); Н, 1,31 турі протягом 16-24 годин і зважили. Вихід: 3,8996г (1,50); N, 4,56 (4,27); СІ, 30,14 (29,86). Спектр ЯМР (83%). Аналітичний склад розрахунковий (виявле1 Н (300МГц, ДМФ-d7): 9,11 (s, 1 піразин Η); 8,68 (s, ний) для C6H1ON2Cl2Pt: С, 19,16 (19,25); Η, 2,68 1 піразин Η); 3,31 (s, З метил Η); 2,68 (s, 3 метил (2,72); Ν, 7,45 (7,43); СІ, 18,85 (18,81). Спектр ЯМР 1 Н). Н (300МГц, ДМФ-d6): 9,19 (d, 1 пірідин Η); 8,03 (t, 1 Приклад 8) Синтез K[PtCl3(4,6пірідин Η); 7,15 (d, 1 пірідин Η); 7,51 (t, 1 пірідин Н); диметилпіримідину)] в Ν ΜΡ 4,39 (bs, 3 NH3 Η); 3,34 (s, 3 метил Η). Спектр ЯМР 195 K2PtCl4 розтерли в дуже тонкодисперсний поРt (300МГц, ДМФ-d6): відповідає спектру ЯМР 195 рошок за допомогою ступки і товкача. 0,5277г Рt R[PtCl 2(NH3)(2-піколіну)], отриманому спосо(1,27ммоль) K2PtCl4 вмістили в 15-мл круглодонну бом, відомим в технології. HPLC (катіонна методиколбу і додали ~3мл NMP. Реакційну суміш перека): час утримання відповідає часу утримання для мішали при високій швидкості і колбу занурили в еталонної сполуки. масляну баню при 65°С. 0,1549г (1,43ммоль) 4,6Приклад 11) Синтез [PtCl2(NH3)(2,6-лутидину)] диметилпіримідину змішали з ~1мл NMP. Розчин у водному розчині 2,5-диметилпіразину додавали до реакційної су1,7412г (3,224ммоль) K [PtCl3(2,6-лутидину)] міші приблизно чотирма рівними порціями з 301,24 KСI вмістили в 25-мл круглодонну колбу і дохвилинними інтервалами. Після останнього додадали 3мл 2,5N розчину KСI. 1,3478г (17,48ммоль) вання реакційну суміш залишили для протікання амонію ацетату розчинили в 6,4мл 2,5N розчину реакції на 60хв. і потім охолодили до навколишамонію гідроксиду і додали до платинової суміші, ньої температури. Реакційну суміш погасили що перемішується. Загальний об'єм реакційної ~80мл метиленхлориду, що викликало осадження суміші становив ~9,5мл. Жовтогарячу суміш занутвердої речовини. Осад зібрали шляхом вакуумної рили в 45-градусну (°С) масляну баню і перемішуфільтрації з використанням склоподібної фрити і вали 40 годин в темряві при швидкості >1000об/хв. промили метиленхлоридом (3x30мл), а потім діеЖовтогаряча суміш поступово перетворилася в тиловим ефіром (3x10мл). Осад висушили під важовту. Жовтий осад зібрали шляхом вакуумної куумом при навколишній температурі протягом 16 фільтрації з використанням склоподібної фрити і годин і зважили. Вихід: 0,4353г (76,3%). Спектр промили водою (2x5мл) і ацетоном (3x5мл). Осад ЯМР 1Н (300МГц, ДМФ-d7): 9,58 (s, 1 піримідин Η); висушили під вакуумом при навколишній темпера7,65 (s, 1 піримідин Η); 3,32 (s, 3 метил Η); 2,65 (s, турі протягом 16-24 годин і зважили. Вихід: 0,9791г 3 метил Η). (78%). Спектр ЯМР 1Н (300МГц, ДМФ-d6): 7,87 (t, 1 Приклад 9) Синтез [РtCl3(діізопропіламіну)] з лутидин Η); 7,49 (d, 2 лутидин Н); 4,28 (bs, 3 NH3 тетрабутиламонію тетрахлорплатинату в ацетоні Η); 3,49 (s, 6 метил Η). Спектр ЯМР 195Pt (300МГц, 0,7961г (0,9687ммоль) тетрабутиламонію тетДМФ-d 6): відповідає еталону. Аналітичний склад рахлорплатинату вмістили в 25-мл круглодонну розрахунковий (виявлений) для C7H12N2Cl2Pt: C, колбу і додали 8мл ацетону. 0,1699г (1,679ммоль) 21,55 (21,70); Η, 3,10 (3,13); Ν, 7,18 (7,07); СІ, 18,17 діізопропіламіну розчинили в 2мл ацетону і додали (18,28). до платинового розчину. Реакційний розчин зануПриклад 12) Синтез [PtCl2(NH3)(2,5рили в масляну баню при 60°С і перемішували диметилпіразину)] у водному розчині протягом 60 годин. Червоний розчин [PtCl4]- пере0,5325г (0,8665ммоль) K [PtCl3(2,5творився в жовтогарячий [PtCl3(діізопропіламін)]-, диметилпіразину)] 2,2 KСI вмістили в 15-мл кругщо було підтверджено спектроскопією ЯМР 195Pt. лодонну колбу і додали 1,0мл 2,5Μ розчину KСI. [PtCl3(діізопропіламін)]- може бути використаний 0,335г (4,35ммоль) амонію ацетату розчинили в для отримання [PtCl2(NH3)(діізопропіламіну)] без1,75мл 2,5Μ (4,38ммоль) розчину амонію гідроксипосередньо, без ізоляції, як калієва або тетрабуду і додали до платинової суміші, що перемішутиламонієва сіль. Спектр ЯМР 195Pt (300МГц, ДМФється. Загальний об'єм реакційної суміші становив 195 d7) відповідає спектру ЯМР Рt ~9,5мл. Жовтогарячу суміш занурили в 45[PtCl3(діізопропіламіну)]-, отриманому способом, градусну (°С) масляну баню. Через 15хв. суміш відомим в технології. набула жовте забарвлення. Через 1 годину суміш Приклади 10-18 показують другий етап спосоохолодили до навколишньої температури і жовтий бу. осад зібрали шляхом вакуумної фільтрації з викоПриклад 10) Синтез [PtCl2(NH3) (2-піколіну)] у ристанням склоподібної фрити. Осад промили водному розчині водою (2x10мл) і ацетоном (1x5мл) і висушили під 21 71614 22 вакуумом при навколишній температурі. Спектр розчину KСI. Суспензію навантажили в 45ЯМР 1H (300МГц, ДМФ-d6): 9,16 (s, 1 піразин Η); градусну (°С) масляну баню і перемішували при 8,80 (s, 1 піразин Η); 4,70 (bs, 3 ΝΗ3 Η); 3,26 (s, 3 ~1000об/хв. Через п'ять хвилин до реакційної суметил Η); 2,69 (2, 3 метил Н). міші додали розчин, що містить 0,1704г 40% меПриклад 13) Синтез [РtСl2(NН3)(2-піколіну)] в тиламіну (2,19ммоль) і 1мл води. Величина рН N-метилпіролідиноні/водному розчині розчину становила 12. Нагрівання припинили че1,84г (14,0ммоль) амонію ацетату тригідрату рез 1 годину загального часу реакції. Реакційну розчинили в 4,63мл 2,9N амонію гідроксиду. Водсуміш охолодили до навколишньої температури. ний розчин додали до 2,68ммоль [РtСl3(2Блідо-жовтий осад зібрали шляхом вакуумної фіпіколіну)]- в 2,5мл Ν-метилпіролідинону. Реакційльтрації з використанням склоподібної фрити і ний розчин перемішували при 45°С протягом 80хв. промили водою (2x20мл) і ацетоном (3x20мл). Утворився жовтий осад. Осад зібрали шляхом Осад висушили під вакуумом при навколишній вакуумної фільтрації з використанням склоподібтемпературі протягом 16 годин. Аналітичний склад ної фрити і промили водою (2x5мл) і ацетоном розрахунковий (виявлений) для C7H12N2Cl2Pt: С, (3x5мл). Осад висушили під вакуумом при навко21,55 (21,73); Η, 3,10 (3,09); Ν, 7,18 (7,14); СІ, 18,17 лишній температурі протягом 16-24 годин і зважи(18,20). Спектр ЯМР 1Н (300МГц, ДМФ-d6): 9,24 (d, ли. Вихід: 0,3391г (34%). Аналітичний склад розра1 пірідин Η); 8,06 (t, 1 пірідин Η); 7,75 (d, 1 пірідин хунковий (виявлений) для C6H1ON2Cl2Pt: C, 19,16 Η); 7,55 (t, 1 пірідин Η); 5,22 (bs, 2 метиламін Η), (19,22); Η, 2,68 (2,69); Ν, 7,45 (7,23); СІ, 18,85 3,35 (s, 3 метил Н 2-піколіну); 2,45 (t, 3 метил Η (18,83). Спектр ЯМР 1H (300МГц, ДМФ): 9,2 (d, 1 метиламіну). пірідин Η); 8,0 (t, 1 пірідин Η); 7,2 (d, 1 пірідин Η); Приклад 17) Синтез [PtCl2(27,5 (t, 1 пірідин Н); 3,4 (s, 3 метил Η). Спектр ЯМР піколіну)(NH(СН3)2])( у водному розчині)] 195 Pt (300МГц, ДМФ): відповідає спектру ЯМР 195Pt 0,5459г (1,26ммоль) K[PtCl3(2-піколіну)] вміс[РtСl2(NH3)(2-піколіну)], отриманому способом, тили в 15-мл круглодонну колбу і додали 1,5мл відомим в технології. HPLC (катіонна методика): 2,5Μ розчину KСI. Суспензію занурили в 45час утримання відповідає часу утримання для градусну (°С) масляну баню і перемішували при еталонної сполуки. високій швидкості. Через п'ять хвилин до реакційПриклад 14) Синтез [PtCl2(NH3)(2-піколіну)] в ної суміші додали розчин, що містить 0,1426г 40% диметилформаміді/водному розчині диметиламіну (1,27ммоль) і ~1мл води. Величина [PtCl2(NH3)(2-піколін)] отримували в диметилрН розчину становила 12. Через 1 годину реакції формаміді/водному розчині, як описано в Прикладі нагрівання припинили і реакційну суміш о холодили 13). Аналітичний склад розрахунковий (виявлений) до навколишньої температури. Жовтий осад зідля C6H1ОN2Cl2Pt: C, 19,16 (19,30); Η, 2,68 (2,62); брали шляхом вакуумної фільтрації з використанΝ, 7,45 (7,18); СІ, 18,85 (18,59). Спектр ЯМР 1Н ням склоподібної фрити і промили водою (2x20мл) (300МГц, ДМФ): 9,1 (d, 1 пірідин Н); 8,1 (t, 1 пірідин і ацетоном (2x10мл). Осад висушили під вакуумом Н); 7,3 (d, 1 пірідин Н); 7,4 (t, 1 пірідин Н); 3,4 (s, 3 при навколишній температурі протягом 16 годин. метил Н). Спектр ЯМР 195Pt (300МГц, ДМФ-d6): Аналітичний склад розрахунковий (виявлений) для відповідає спектру ЯМР 195Pt [РtСl2(NН3)(2C8H14N2Cl2Pt: С, 23,77 (24,00); Η, 3,48 (3,49); Ν, піколіну)], отриманому способом, відомим в техно6,93 (6,80); СІ, 17,54 (17,63). Спектр ЯМР 1Н логії. (300МГц, ДМФ-d6): 9,31 (d, 1 пірідин Η); 8,09 (t, 1 Приклад 15) Синтез пірідин Η); 7,78 (d, 1 пірідин Η); 7,58 (t, 1 пірідин Η); [РtСl2(NН3)(діізопропіламіну)] в ацетоні/водному 6,06 (bs, 1 ΝΗ Η), 3,37 (s, 3 метил Н піколіну); 2,76 розчині 6мл 2,5N розчину амонію гідроксиду дода(d, 3 метил Η диметиламіну); 2,70 (d, 3 метил Η ли до [PtCl3(NH3)(діізопропіламіну]- (~2,69ммоль) в диметиламіну). 2,5мл ацетону. Величина рН розчину становила Приклад 18) Синтез [PtCl2(2-піколіну)(NBu3)] у 12. Реакційний розчин перемішували при 45°С водному розчині протягом 48 годин. Утворився жовтий осад. Осад 0,6289г (1,45ммоль) K[PtCl3(2-піколіну)] вмісзібрали шляхом вакуумної фільтрації з використили в 15-мл круглодонну колбу і додали 1,0мл танням склоподібної фрити і промили водою 2,5Μ розчину KСI. Суспензію занурили в 45(2X5мл) і ацетоном (3x5мл). Осад висушили під градусну (°С) масляну баню і перемішували 5хв. вакуумом при навколишній температурі протягом при високій швидкості. До жовтогарячої реакційної 16-24 годин. Аналітичний склад розрахунковий суміші додали 0,2735г (1,47ммоль) трибутиламіну, (виявлений) для C6H18N2Cl2Pt·0,095C6H3ON2Cl2Pt: розчиненого в 1,0мл води. Величина рН розчину С, 20,00 (19,98); Η, 4,90 (4,89); Ν, 7,16 (7,12); СI, становила 12. Через 1 годину нагрівання припини18,11 (17,93). Спектр ЯМР 1Н (300МГц, ДМФ-d6): ли. Після охолоджування до навколишньої темпе4,5 (bs, 1 діізопропіламін Η); 3,9 (bs, 3 NH 3 Η); 3,3 ратури осади зібрали шляхом вакуумної фільтрації (m, 2 метил Η в діізопропіламіні); 1,7 (d, 6 метил Η з використанням склоподібної фрити. Зібрану твев дііизопропіламіні); 1,5 (d, 6 метил Η в діізопропірду речовину висушили під вакуумом при навколаміні). Спектр ЯМР 195Pt (300МГц, ДМФ): відповілишній температурі. Спектр ЯМР1Н (300МГц, 195 дає спектру ЯМР Рt ДМФ-d 7): 9,14 (d, 1 пірідин Η); 7,90 (t, 1 пірідин Η); [PtCl2(NH3)(діізопропіламіну)], отриманому спосо7,60 (d, 1 пірідин Η); 7,42 (t, 1 пірідин Η); 3,41 (s, 3 бом, відомим в технології. метил Η 2-піколіну); 3,28 (m, 2 метилен Η трибутиПриклад 16) Синтез [PtCl2(2-піколіну)(NH2СН3)] ламіну); 1,10 (t, 3 метил Η трибутиламіну). у водному розчині Приклади 19-23 показують додаткові етапи 0,5055г (1,17ммоль) K[PtCl3(2-піколіну)] вмісспособу. тили в 15-мл круглодонну колбу і додали 1мл 2,5Μ Приклад 19) Синтез ц, т, ц-[PtCl2(ОН)2(NH3)(2 23 71614 24 піколіну)] рації і промили діетиловим ефіром. Продукт вису5,0мл води і 5,0мл 30% Н2О2 додали до сушили під вакуумом при навколишній температурі спензії 3,142г ZD0473 в 15-20мл гептану. Цю суміш протягом ночі. Вихід: 1,318г (96%). Аналітичний перемішували і нагрівали до ~80°С протягом 2 склад розрахунковий (виявлений) для годин. Суміш охолодили до кімнатної температури С10Н16N2CI 2О 4Рt: 3, 24,30 (24,32); Н, 3,26 (3,15); N, і потім перемішували 1 годину в крижаній бані. 5,67 (5,66); СІ, 14,35 (14,29). Яскраво-жовту тверду речовину зібрали шляхом вакуумної фільтрації і промили водою і метанолом. Продукт висушили під вакуумом при навколишній температурі протягом ночі. Вихід: 2,975г (87%). Аналітичний склад розрахунковий (виявлений) для C6H12N2Cl2O2Pt: C, 17,57 (17,67); Η, 2,95 (2,93); Ν, 6,83 (6,79); СІ, 17,29 (17,38). Приклад 20) Синтез ц, т, ц[PtCl2(ОН)2(NH3)(2,3-диметилпіразину)] 2,5мл води і 3,5мл 30% Н2О2 додали до суспензії 1,6731г цис-[PtCl2(NH3)(2,3диметилпіразину)] в 10мл гептану. Цю суміш перемішували і нагрівали до ~80°С протягом 2 годин. Суміш охолодили до кімнатної температури і потім перемішували 1 годину в крижаній бані. Яскравожовту тверду речовину зібрали шляхом вакуумної фільтрації і промили водою і метанолом. Продукт висушили під вакуумом при навколишній температурі протягом ночі. Вихід: 1,1341г (62%). Аналітичний склад розрахунковий (виявлений) для C6H13N3Cl2O2Pt: С, 16,95 (16,81); Η, 3,08 (3,12); Ν, 9,88 (9,66); СІ, 16,68 (16,44). Приклад 21) Синтез [РtСi(ОН)3(NH3)(2піколіну)] 0,246г LiOH Н2О розчинили в 5мл води. 2,402г ц, т, ц-[PtCl 2(ОН) 2(NH3)(2-піколіну)] суспендували в цьому розчині. Суміш перемішували протягом ночі при навколишній температурі. Тверда жовта речовина за ніч поступово розчинилася. рН розчину відрегулювали до 7. Розчинник видалили при зниженому тиску, отримавши тверду жовту речовину. Для видалення результуючого LiCl тверду речовину перемішували в 10мл етанолу протягом 30 хвилин. Суміш центрифугували і надосадову рідину декантували. Цей процес промивання повторювали, поки хлорид літію не був видалений. Продукт висушили під вакуумом при навколишній температурі протягом ночі. Вихід: 1,209 (50%). Аналітичний склад розрахунковий (виявлений) для C6H13N2CIO 3Pt·2Η2Ο·0,12LiCl: С, 16,65 (16,45); Η, 3,96 (4,04); Ν, 6,47 (6,75); СІ, 9,17 (9,47). Приклад 22) Синтез [РtСl(ОАс)3(NH3)(2піколіну)] 0,352г [РtСl(ОН)3(NH3)(2-піколіну)] додали малими порціями до 1,1мл оцтового ангідриду при 0°С. Цю суміш перемішували з великою швидкістю при навколишній температурі. Через 3 дні тверда речовина перейшла в розчин. Продукт висушили під вакуумом при навколишній температурі протягом ночі. Вихід: 0,314г (70%). Аналітичний склад розрахунковий (виявлений) для C12H19N2CIO 6Pt: С, 27,83 (27,93); Η, 3,70 (3,66); Ν, 5,41 (5,34); СІ, 6,85 (7,00). Приклад 23) Синтез [PtCl2(ОАс)2(NH3)(2піколіну)] 1,367г ц, т, ц-[PtCl 2(ОН)2(NH3)(2-піколіну)] додали малими порціями до 3,1мл оцтового ангідриду при 0°С. Цю суміш перемішували з великою швидкістю при кімнатній температурі. Через 4 дні тверду речовину зібрали шляхом вакуумної фільт 25 71614 26 Даний винахід був описаний за допомогою прямого опису і за допомогою Прикладів. Як вказаний вище, Приклади дані лише для ілюстрації і не обмежують даний винахід ні в якій мірі. Крім того, фахівцеві в тій області, до якої даний винахід відноситься, з вивчення опису і формули винаходу буде зрозуміло, що існують еквіваленти заявлених аспектів даного винаходу. Автори винаходу вважають ці еквіваленти включеними в область даного винаходу. 27 Комп’ютерна в ерстка О. Гапоненко 71614 Підписне 28 Тираж 37 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюCis-platinum complex and a method for the preparation thereof
Назва патенту російськоюЦисплатиновый комплекс и способ его получения
МПК / Мітки
МПК: C07D 213/60, C07D 239/28, A61P 35/00, C07D 241/14, C07F 15/00
Мітки: одержання, спосіб, цисплатиновий, комплекс
Код посилання
<a href="https://ua.patents.su/14-71614-cisplatinovijj-kompleks-ta-sposib-jjogo-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Цисплатиновий комплекс та спосіб його одержання</a>
Платиновий комплекс, спосіб його одержання, комплекс включення платинового комплексу, спосіб його одержання та фармацевтична композиція

Номер патенту: 56315
Опубліковано: 15.05.2003
Автори: ТУРЯНЕК Ярослав, МІСТР Адольф, ПОУЛОВА Анна, МЕЛЬКА Мілан, ЗАЛУСКА Дана, ЖАК Франтішек
МПК: A61P 35/00, C07F 15/00, C07C 211/38, A61K 31/282
Мітки: включення, комплексу, композиція, комплекс, фармацевтична, платинового, платиновий, спосіб, одержання
Формула / Реферат:
1. Платиновий комплекс зі ступенем окиснення IV формули (І)(I),де Χ являє собою атом галогену, В незалежно один від одного являють собою атом галогену, гідроксильну групу або карбоксилатну групу, що містить від 1 до 6 атомів вуглецю, і А являє собою групу -NH2-R, де R є трициклічною вуглеводневою частиною, що містить від 10 до 14 атомів вуглецю, яка...
Платиновий комплекс, спосіб його одержання, комплекс включення платинового комплексу, спосіб його одержання та фармацевтична композиція

Номер патенту: 56314
Опубліковано: 15.05.2003
Автори: ЖАК Франтішек, МЕЛЬКА Мілан, ЗАЛУСКА Дана, ТУРЯНЕК Ярослав, ПОУЛОВА Анна, МІСТР Адольф
МПК: C07F 15/00, A61K 31/282, A61P 35/00
Мітки: одержання, комплекс, спосіб, включення, платинового, композиція, комплексу, платиновий, фармацевтична
Формула / Реферат:
1. Платиновий комплекс зі ступенем окиснення II формули (І)(I),де Χ являє собою атом галогену, А являє собою групу -NH2-R, де R є трициклічною вуглеводневою частиною, що містить від 10 до 14 атомів вуглецю, яка може бути необов'язково заміщена на трициклічному кільці однією або двома алкільними групами, що мають від 1 до 4 атомів вуглецю.2....
Мінерально-органічний комплекс та спосіб його одержання

Номер патенту: 25560
Опубліковано: 17.05.2004
Автор: Михайличенко Борис Валентинович
МПК: A61P 43/00, A23L 1/052, A23L 1/302, A23L 1/056, A23L 1/308, A23L 1/30
Мітки: мінерально-органічний, спосіб, комплекс, одержання
Формула / Реферат:
1. Мінерально-органічний комплекс, що містить мінеральну й органічну складові, який відрізняється тим, що як мінеральну складову використовують мінералізат, одержаний із продуктів тваринного або рослинного походження, а як органічну - ліганди, наприклад, вітаміни, амінокислоти і/або лікарські засоби, і/або харчові добавки, стійкі в середовищі з pH 3 - 7,4.2. Комплекс за п.1, який відрізняється тим, що як продукти для одержання...
Комплекс включення 3-морфоліносидноніміну, або його солей, або його таутомірного ізомеру, що має антиішемічний ефект, спосіб їх отримання, фармацевтична композиція, спосіб її одержання та спосіб лікування ішемі

Номер патенту: 27242
Опубліковано: 15.08.2000
Автори: Ховат Агнеш, Хорват Габор, Хермец Іштван, Сенте Лайош, ГААЛ Йожеф, Сейтлі Йожеф, Мункачі Ірейн, Вікмон Марія, МАРМАРОШІ Каталін
МПК: A61P 9/08, A61P 9/10, A61K 31/4245, A61K 31/41, C07D 271/04, C08B 37/16, A61K 47/40, C08B 37/00
Мітки: ефект, лікування, композиція, отримання, таутомірного, має, 3-морфоліносидноніміну, включення, одержання, солей, комплекс, ізомеру, спосіб, антиішемічний, фармацевтична, ішемії
Текст:
...разложения можно также проследить с помощью тонкослойной хроматографии Интенсивность пятна SIN 10, выявленного при Rf 0,36 в буфере с рН 6,4, отличалась заметно даже после хранения в продолжение недели, в буфере с рН 7 и 7,6 после хранения в течение 1 дня наблюдалось ощутимое различие в интенсивности пятна SIN 1 при Rf 0,04 В буфере с рН 7 все еще заметен был неизмененный SIN 1, тогда как при рН 7,6 он практически не определялся Равновесие...
Поліпептид, glp – 1-молекулярний комплекс на його основі, фармацевтична композиція (варіанти ) та спосіб одержання glp – 1-молекулярного комплексу

Номер патенту: 44696
Опубліковано: 15.03.2002
Автори: Гоффманн Джеймс Артур, Гелловей Джон Еллісон
МПК: A61K 33/30, A61P 5/50, A61K 38/26
Мітки: одержання, 1-молекулярного, фармацевтична, варіанти, поліпептид, композиція, 1-молекулярний, основі, комплексу, комплекс, спосіб
Формула / Реферат:
1. Полипептид формулы:.R1 -X-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Y-Gly-Gln-Ala-Ala-Lys-Z-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-R2,где:R1 выбран из группы, включающей в себя L-гистидин, D-гистидин, дезамино-гистидин, 2-амино-гистидин, -гидрокси-гистидин, гомогистидин, альфа-фторометил-гистидин и альфа-метил-гистидин;Х...
Попередній патент: Спосіб одержання фармацевтичного препарату перорального введення з уповільненим вивільненням толперизону (варіанти)
Наступний патент: Спосіб виготовлення світлоповертальних елементів
Випадковий патент: Газовий колектор агломашини